P1100276

Ponieważ prędkość migracji substancji po chronmtogramie jest charakterystyczna dla rodzaju substancji i w danych warunkach jest wielkością stalą, wprowadzono wielkości pomocnicze, które ją bliżej określają:
ji — stosunek odległości pasma substancji mierzonej od powierzchni kolumny, o którą w określonym przedziale czasu przesunie się. do odpowiedniego obniżenia fazy ruchomej w kolumnie chromatograficznej.
|
T* | ||||
|
W/ | ||||
|
- |
h | |||
|
y>; |
w. | |||
|
//// | ||||
|
X> | ||||
|
r///. | ||||
|
'/// |
V
Rys. 31.3. Wartość R i Rr substancji: a) dla kolumny. b)dln bibuły
Rf - stosunek odległości, o którą przesunie się plama substancji do odległości, o którą w tym samym czusie przesunie się czoło rozpuszczalnika mierzone od startu (rys. 31.3).
31.3. TEORIA CHROMATOGRAFII PODZIAŁOWEJ
Przy opracowywaniu teorii chromatografii podziałowej wychodzi się z podstaw przedstawionych w poprzednich rozdziałach, według których głównym czynnikiem określającym zachowanie się substancji rozdzielanych na kolumnie lub na bibule, jest ich współczynnik podziału charakterystyczny dla danego układu faz i dla danych warunków eksperymentalnych.
Dla wszystkich metod chromatograficznych obowiązuje podstawowe prawo zachowania ilości substancji. Ilość substancji, która przeszła w procesie rozdzielczym t jednej fazy do drugiej jest dla pierwszej fazy ubytkiem a dla drugiej — przyrostem, a suma zmian ilości substancji równa się zeru. Ten warunek zastosowali Martin i Syngc w swojej interpretacji procesu chromatograficznego. Za prace te otrzymali nagrodę Nobla w 1952 r.
Przy ilościowym opracowaniu ogólnego procesu rozdzielania w chromatografii podziałowej uwzględnia się dwa podstawowe założenia.
1. Substancja w fazie ruchomej pozostaje nieustannie w równowadze z sub-
iUflcft * fe&e stacjonarnej, tj. w każdej chwili ustala się stosunek ukich aę*ń iibiUmcji w obu fazach, jak tego wymaga funkcja podziału, a więc rac uwzględnia yę wpływu prędkości przepływu fazy ruchomej.
2. Pominięcie dyfo/ji. Jeżeli w każdym miejscu w roztworze jest inne stężenie $\ib*tancjt. dyfuzja te różnice w stężeniach zawsze wyrównuje. Tik jest również i niejednakowymi stężeniami substancji w różnych miejscach kolumny chromano pificacj. Dla uproszczenia wywodu (wstępnie) nie uwzględnia się wyrównywania stężeń przez dyfuzję,
Wyobraźmy sobie, Ze mamy długi walec, kolumnę chromatograficzną, napełnioną obojętnym nośnikiem, na którym jest osadzona faza nieruchoma. Przez tę furę przecieka odpowiednio dobrana faza ruchoma, która praktycznie nie miesza się z fazą pierwszą. Substancja chromatografowana przechodzi z roztworu przepływającego do fazy stacjonarnej. Następnie substancja rozpuszcza się w fn/ie ruchomej, jest przenoszona dalej i zatrzymywana w dalszych warstwach kolumny chroniło-gnfencj.
Kolumnę chromatograficzną można podzielić na hipotetyczne, cząstkowe warstwy, charakteryzowane jako półki efektywne.
Pojęcie półki efektywnej opisuje rzeczywiste stosunki na kolumnie chroailo-pficznej tylko formalnie. Całkowicie dokładny opis jest złożony nawet dla bardzo uproszczonych przypadków.
Głównym czynnikiem, który określa zachowanie się substancji na chromało-gramie jest ich współczynnik podziału zdefiniowany prawem NerusUt. W chrcmato-grali podziałowej na podstawie współczynnika podziału można wnioskować o wartości R albo Rr. Dla kolumnowej chromatografii podziałowej ta zależność jest określona równaniem
Ai+KAt
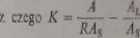
gdzie; A * AL- AS+A,;A - całkowita powierzchnia przekroju kolumny ; AL - powierzchnia przekroju kolumny przypadająca na fazę tuchomą, ;1S - powierzchnia przekroju kolumny przypadająca na fazę stacjonarną. A\ - powierzchnia przekroju kolumny przypadająca na obojętny nośnik.
Dla bibułowej chromatografii podziałowej zależność między współczynnikiem podziału a wartością Rf określa się zależnością


Jeżeli dochodzi do zetknięcia fazy zawierającej substancję z drugą fazą, substancja dyfcndujc z jednej fazy do drugiej, dopóki spadek stężenia w fazie pierwszej i wzrost stężenia w fazie drugiej me ustali wartości stężeń lak, aby ich słowittk liczbowo był zgodny zc współczynnikiem podziału K.
Pracę związaną z przejściem jednego mola substancji t jednej fazy o stężeniu r, do drugiej fazy o stężeniu c3 określa się zależnością
lV-RT]n-±- » RTbK Ci
II
Wyszukiwarka