![]()
Metale i ich stopy
Metale charakteryzują się wiązaniem metalicznym. Układy wieloskładnikowe złożone z więcej niż jednego pierwiastka, charakteryzujące się przewagą wiązania metalicznego tworzą stopy metali.
Metale i ich stopy cechują następujące własności:
1) dobre przewodnictwo cieplne i elektryczne; 2) opór elektryczny zwiększa się z podwyższeniem temperatury; 3) połysk metaliczny, polegający na odbijaniu promieni świetlnych od wypolerowanych powierzchni; 4) plastyczność (zdolność do trwałych odkształceń pod wpływem przyłożonych naprężeń).
Metale otrzymuje się z rud, będących najczęściej tlenkami. Procesy metalurgiczne polegają zwykle na redukcji prowadzącej do ekstrakcji metalu z rudy oraz na rafinacji, usuwającej z metalu pozostałe zanieczyszczenia. Elementy metalowe zwykle wykonywane są metodami odlewnicz., przeróbki plastycznej lub obróbki skrawaniem, a często także metalurgii proszków. Własności metali i stopów są kształtowane metodami obróbki cieplnej, a powierzchnia elementów metalowych często jest uszlachetniana metodami inżynierii powierzchni, zwiększającymi m.in. odporność na korozję lub odporność na zużycie.
Najczęściej używanymi spośród materiałów metalowych są stale, czyli stopy żelaza z węglem i innymi pierwiastk., a także stopy odlewnicze żelaza, tzn. staliwa i żeliwa.
Liczną grupę stosowanych materiałów metalowych stanowią również metale nieżelazne i ich stopy.
Polimery
Materiały organiczne, złożone ze związków węgla. Polimery są tworzone przez węgiel, wodór oraz Si, N, O, F Polimery są makrocząsteczkowymi i powstają w wyniku połączenia wiązaniami kowalencyjnymi w łańcuchy wielu grup atomów zwanych monomerami jednego lub kilku rodzajów. Charakteryzują się: 1) małą gęstością; 2)izolacyjnymi własnościami cieplnymi i elektrycznymi; 3) słabo odbijają światło i zwykle są przezroczyste; 4) nie nadają się do pracy w podwyższonej temperaturze.
Zaletą polimerów jest łatwość formowania z nich wyrobów oraz niski koszt wytwarzania.
Ceramiki i szkła
Związki nieorganiczne (najczęściej tlenki, azotki, węgliki) o jonowych i kowalencyjnych wiązaniach międzyatomow., będące najczęściej izolatorami (bardzo słabo przewodzą prąd elektryczny) mają niską udarność i plastyczność, lecz dużą twardość i wytrzymałość na ściskanie, są odporne na działanie wysokich temperatur. Ceramiki są zazwyczaj polikrystaliczne, szkła są amorficzne.
Kompozyty
Są połączeniami dwóch lub więcej odrębnych i nierozpuszczających się w sobie faz, z których każda odpowiada innemu podstawowemu materiałowi inżynierskiemu, zapewniającymi lepszy zespół własności i cech strukturalnych, od właściwych dla każdego z materiałów składowych oddzielnie.
Wiązanie jonowe (heteropolarne) - polega na elektrostatycznym przyciąganiu się jonów odmiennego znaku. Jony przyciągają się w myśl prawa Coulomba z siłą wprost proporcjonalną do ładunku (elektrowartościowości) i odwrotnie proporcjonalną do kwadratu odległości. Cechy: 1) w stałym stanie skupienia tworzą sieci krystaliczne, w których występują jony dodatnie i ujemne; 2) w stanie stopionym i w roztworach przewodzą prąd elektryczny, a w stanie stałym są złymi przewodnikami elektryczności; 3) na ogół mają wysokie temperatury wrzenia i topnienia.
Wiązanie atomowe (homeopolarne, kowalencyjne) - polega na istnieniu wiążących par elektronów należących jednocześnie do dwóch sąsiadujących ze sobą atomów. Zewnętrzne powłoki atomów zachodzą wzajemnie na siebie, przez co niektóre elektrony są wspólną własnością obu atomów.
Wiązanie metaliczne - gdy pierwiastek przechodzi ze stanu pary w stan ciekły lub stały, to słabo związane z jądrem atomu elektrony walencyjne przestają należeć do poszczególnych atomów i stają się elektronami swobodnymi, stanowiącymi wspólną własność wszystkich atomów. Sieci krystaliczne metali są uporządkowanym zbiorem jonów dodatnich, tzw. rdzeni atomowych metalu pogrążonych jak gdyby w gazie elektronowym, który je cementuje.
Wiązania siłami van der Waalsa - Siłami van der Waalsa nazywamy siły wzajemnego przyciągania się cząsteczek. Siły te działają między cząsteczkami substancji gazowych i ciekłych, jak również między cząsteczkami w sieciach krystalicznych molekularnych.
Układy krystalograficzne i typy sieci przestrzennych
F - ściennie centrowana, C - centrowana na podstawach, I - przestrzennie centrowana, P - prymitywna
regularny (a=b=c, α=β=χ=90°) - P, I, F;
tetragonalny (a=b≠c, α=β=χ=90°) - P, I;
rombowy (a≠b≠c, α=β=χ=90°) - P, I, F, C;
trygonalny (romboedryczny) (a=b=c, α=β=χ≠90°) - P;
heksagonalny (a=b≠c, α=β=90°, χ=120°) - P;
jednoskośny (a≠b≠c, α=β=90°, χ≠90°) - P, C;
trójskośny (a≠b≠c, α≠β≠χ≠90°) - P.
Metale krystalizują wyłącznie w pierwszych pięciu układach krystalograficznych. Większość metali krystalizuje w układach krystalograficznych charakteryzujących się wysoką symetrią zapełnienia sieci przestrzennej atomami, w szczególności w sieciach:
A1 - ściennie (płasko) centrowanej układu regularnego
A2 - przestrzennie centrowanej układu regularnego
A3 - heksagonalnej o gęstym ułożeniu atomów
Własności metali, a szczególnie podatność na odkształc. plastyczne, w dużej mierze zależą od typu sieci przestrzen.
Wady budowy krystalicznej
Wady budowy krystalicznej w istotny sposób wpływają na własności wytrzymałościowe i plastyczne metali. Dążenie do ograniczenia wad budowy krystalicznej jest technicznie b. trudne. Stosuje się więc metodę umacniania metali przez znaczne zwiększenie gęstości wad budowy krystalicznej, co można osiągnąć przez stosowanie stopów metali o strukturze polikrystalicznej, w wyniku rozdrobnienia ziarn, wydzielania faz o dużej dyspersji, a także przez zgniot wskutek odkształcenia plastycznego na zimno. Osiągnięciu tego celu sprzyjają więc procesy technologiczne odlewania, obróbki plastycznej i obróbki cieplnej.
Defekty punktowe
Do nich należą wakanse, tj. wolne węzły w sieci krystal., oraz atomy międzywęzłowe, które zajęły pozycje w lukach opuszczając węzły sieci na skutek drgań cieplnych. Wodór, bor, tlen, azot i węgiel są uprzywilejowane do zajmowania przestrzeni międzywęzł. ze względu na małą średnicę. Obecność zarówno wakansów, jak i atomów międzywęzł., powoduje wokół nich lokalne odkształcenie sieci przestrz. kryształu, zwane odpowiednio kontrakcją lub ekspansją.
Mechanizmy tworzenia wad punktowych: Liczba wad punktowych budowy krystalicznej jest funkcją temperatury Podwyższeniu temperatury towarzyszy wzrost amplitudy drgań cieplnych, co ułatwia opuszczenie przez rdzenie atomowe pozycji w węźle sieci krystalicznej. Wyróżnia się tu dwa mechanizmy:
defekt Schottky'ego - polega na przemieszczaniu się atomu w miejsce sąsiadującego wakansu w wyniku czego powstaje wakans w innym miejscu sieci.
defekt Frenkla - polega na przemieszczeniu się rdzenia atomowego z pozycji węzłowej do przestrzeni międzywęzłowej.
Wzrost gęstości defektów powoduje wzrost oporności, wzrost twardości i obniżenie plastyczności.
![]()
n - ilość wakansów, N - liczba atomów, A - stała proporcjonalności, T - temp. w Kelvinach, k - Boltzmana, Ew - energia potrzebna do wytworzenia wakansu
Źródło Franka - Reada (rozmnażanie się dyslokacji)
Pod działaniem sił zewnętrznych w krysztale zachodzi odkształcenie plastyczne, w czasie którego z kryształu wydostają się na zewnątrz dyslokacje. Jest to możliwe dzięki rozmnażaniu się dyslokacji zgodnie z mechanizmem Franka - Reada. Źródłem jest utwierdzona w punktach D i D1 dodatnia dyslokacja krawędziowa, której linia leży na płaszczyźnie łatwego poślizgu. Pod działaniem naprężeń stycznych linia dyslokacji przesuwa się i wygina tworząc kształt półkola. Wtedy oprócz dyslokacji krawędziowej przy obu punktach utwierdzenia powstają odcinki dyslokacji śrubowych. Dyslokacje te pod działaniem tego samego naprężenia zaczynają przemieszczać się na zewnątrz (od siebie). W ten sposób powstają dwie spirale, a te mają już krótkie odcinki linii prostopadłych do kierunku działania naprężeń i wektora Burgersa. Są to już dyslokacje krawędziowe ujemne, przemieszczające się w kierunku przeciwnym niż dyslokacja dodatnia. Spirale w miarę dalszego rozszerzania stykają się z sobą i powstaje zamknięta pętla. Pętla ta poszerza się, a po wyjściu z kryształu jedna jego część jest przesunięta względem drugiej. Pozostałe części spirali odtwarzają sytuację początkową i cały proces może rozpocząć się od nowa. Takie powstawanie nowych dyslokacji mogłoby trwać bardzo długo. W rzeczywistych kryształach pętle mogą zatrzymywać się na przeszkodach i utrudniać ruch dyslokacji powstałych później. W wypadku zatrzymania wielu dyslokacji działanie źródła może ustać. Osiągnięte zostaje stadium, w którym opór przeciw dalszym odkształ- ceniom znacznie wzrasta. Przyczyną jest zwiększenie się gęstości dyslokacji, które blokują się wzajemnie.
Defekty liniowe (dyslokacje)
krawędziowa - stanowi krawędź ekstrapłaszczyzny, tj. półpłaszczyzny sieciowej umieszczonej między nieco rozsuniętymi płaszczyznami sieciowymi kryształu o budowie prawidłowej. Dyslokacje krawędziowe leżące w płaszczyznach najgęściej obsadzonych atomami będących płaszczyznami poślizgu, przemieszczają się pod działaniem naprężenia stycznego. Powstanie dyslokacji krawędziowej można sobie wyobrazić zakładając pewną ściśliwość kryształu, dzięki której przemieszczenie górnej części kryształu, wynoszące na brzegowej jeden odstęp międzyatomowy, w miarę oddalania się od tej płaszczyzny będzie malało, aż wreszcie zanika. Poślizg zachodzi zatem nie na całej płaszczyźnie łatwego poślizgu, ale tylko na jej części (poślizg niejednorodny). Dyslokacja krawędziowa już pod działaniem niewielkich naprężeń łatwo zmienia swoje położenie, a po wyjściu z kryształu tworzy na przeciwległej powierzchni stopień. W zależności od położenia dodatkowej półpłaszczyzny dyslokacje mogą być dodatnie (⊥) lub ujemne (T). Dodatnie jeśli półpłaszczyzna znajduje się nad płaszczyzną poślizgu, ujemne - odwrotnie. Wielkością charakterystyczną dla dyslokacji jest wielkość zaburzenia sieci krystalicznej jakie ona wywołuje, a dokładniej energia związana z tym zaburzeniem. Jako miarę tego zaburzenia przyjęto wektor Burgersa. Wyznacza się go za pomocą tzw. konturu Burgersa (obiegu składającego się z jednakowej liczby odstępów sieciowych w każdym kierunku). Jego długość określa wielkość zaburzenia w dyslokacji krawędziowej. Jest prostopadły do linii dyslokacji, a jego zwrot jest zgodny z kierunkiem. W czasie ruchu dyslokacje krawędziowe mogą na swojej drodze napotkać inne defekty sieciowe. Gdy krawędź półpłaszczyzny napotka atom w pozycji międzywęzłowej, wydłuża się ona o jeden odstęp sieciowy - zmieniając tym samym płaszczyznę poślizgu. Gdy napotka wakans, krawędź skróci się o jeden odstęp sieciowy. Dyslokacje mogą się przesuwać aż do przeszkody (koniec materiału, granica ziarna, granica obcofazowa). Jeśli po tej samej płaszczyźnie poślizgu przesuwać się będzie, druga dyslokacja o tym samym znaku, to zatrzyma się ona w takiej odległości od poprzedniej, by wzrastające zaburzenie sieci krystalicznej i związane z nim naprężenie równoważyły się z naprężen. zewnętrznymi. Następna z kolei dyslokacja jednoimienna przemieszczająca się po tej samej płaszczyźnie poślizgu zatrzyma się w większej odległości od poprzedniej, gdyż jest odpychana już przez dwie dyslokacje poprzednie. Podobnie będzie z dyslokacjami następnymi, a powstałe zgrupowanie zatrzymanych dyslokacji nazywane jest spiętrzeniem dyslokacji. Obce wtrącenia, drobne wydzielenia innych faz, gęsta sieć granic ziarn w polikryształach, wszystko to utrudnia swobodę ruchu dyslokacji i zwiększa odporność materiału na odkształcen. plastyczne. Materiał staje się mniej plastyczny, twardszy i wzrasta jego wytrzymałość; całokształt zmian-umocnienie.
śrubowa - defekt liniowy struktury krystalicznej spowodowany przemieszczeniem części kryształu wokół osi, zwanej linią dyslokacji śrubowej. Wektor Burgersa dyslokacji śrubowej jest skierowany równolegle do jej lini. Dyslokacje śrubowe występują wtedy, gdy na materiał działają naprężenia tnące skierowane przeciwnie. Pod działaniem tych naprężeń dyslokacje śrubowe przemieszczają się. Dyslokacja krawędziowa przemieszcza się w kierunku działania naprężenia, natomiast linia dyslokacji śrubowej przemieszcza się w głąb kryształu, prostopadle do kierunku działania naprężenia stycznego. Dyslokacje śrubowe mogą być prawo- lub lewoskrętne.
mieszana - dyslokacja o dowolnej orientacji wektora Burgersa względem linii dyslokacji (nierównoległy i nie-prostopadły)
umacnianie - metoda zmian właściwości poprzez:
dodawanie dodatkowych pierwiastków stopowych
wzrost gęstości defektów (dyslokacji) w materiale - najczęściej używana metoda (przeróbka plastyczna)
zmianę wielkości naprężenia tnącego
Defekty płaskie
granice ziarn - powierzchnie oddzielające dwa ziarna różniące się orientacją głównych osi krystalograficznych (w metalach), w stopach technicznych także składem chemicznym. Granice wąskokątowe (kąt dezorientacji: 6-10°) charakteryzują się budową dyslokacyjną. Płaszczyzny atomowe w pobliżu styku kończą się w taki sposób, jak w dyslokacjach krawędziowych. Taką nachyloną granicę wąskokątową można uważać zatem za zbiór równoległych dyslokacji krawędziowych ułożonych jedna nad drugą. Odległość D między liniami sąsiednich dyslokacji zależy od kąta dezorientacji i można ją wyznaczyć uwzględniając łuk wyznaczony przez kąt skręcania θ na okręgu o promieniu D. Gdy odstęp między atomami na kierunku prostopadłym do granicy, równy wektorowi Burgersa jest dużo mniejszy niż D, wówczas wektor![]()
jest bardzo bliski długości omawianego łuku. Można więc napisać ![]()
, a stąd: D = b/θ. Kąt dezorientacji może być również kątem o jaki obrócono względem siebie przyległe ziarna. Jeśli kąt ten jest niewielki, to granica taka stanowi ścianę przecinających się podobnie jak w sieci rybackiej linii dyslokacji śrubowych. Granica taka nazywana jest granicą skręconą. Granice szerokokątowe - charakteryzują się dużym kątem (>10°) dezorientacji krystalicznej ziarn, na styku których powstają. Budowa tych granic jest b. złożona i nie w pełni zbadana. Sądzi się, że na granicach ziarn powstaje strefa miejsc koincydentnych, tj. jednoczesnych, tworzących supersieć przestrzenną, nakładającą się na sieć przestrzenną sąsiadujących ze sobą ziarn. Parametr supersieci miejsc koincydentnych jest wielokrotnością parametru sieci ziarn. W strefie granicy ułożenie atomów charakterystyczne dla wnętrza ziarn jest zaburzone. Granica szerokokątowa nie jest przy tym płaska, lecz zawiera liczne dyslokacje oraz protuzje, tj. wybrzuszenia i występy. Szczególnym przypadkiem granic szerokątowych są granice bliźniacze. Tworzą się one przy ściśle określonej orientacji ziarn, gdy płaszczyzna granicy staje się płaszczyzną symetrii. Na granicy takiej zachodzi zatem pełne dopasowanie (koherencja) sieci obu ziarn. Niemal zupełny brak zaburzeń w prawidłowym rozmieszczeniu atomów sprawia, że energia takiej granicy jest małą i wynosi 3-10% energii granic szerokokątowych. Gdy granica bliźniacza odchyli się o mały kąt od płaszczyzny idealnego dopasowania, wtedy dzieli się ona na strefy, w których dopasowanie jest dobre i strefy w których dopasowanie uzyskuje się kosztem niewielkich odkształceń sprężystych (przesunięć atomów poza położenia równowagi) lub okresowo powtarzających się dyslokacji. Pociąga to za sobą zwiększenie się energii takiej granicy, a gdy kąt odchylenia wzrasta, prowadzi to do osiągnięcia takich wartości energii, jaką wykazują granice szerokokątowe.
Wyraźna granica plastyczności Granicą plastyczności jest nazywane naprężenie niezbędne do zapoczątkowania makroskopowego odkształcenia plastycznego we wszystkich ziarnach. Dolna granica plastyczności (wyraźna granica plastyczności) zwiększa się wraz ze zmniejszeniem wielkości ziarn, zgodnie z równaniem Halla - Petcha:
![]()
gdzie: σ0 - naprężenie uplastyczniające (siła potrzebna do rozpoczęcia ruchu dyslokacji), k - stała materiałowa, d - średnica ziarn.
Błędy ułożenia
Są to zaburzenia w kolejności ułożenia najgęściej upakowanych płaszczyzn atomowych. Granice między płytkami o odmiennej strukturze są koherentne i choć skład chemiczny jest taki sam, to odmienna struktura krystaliczna jest powodem różnic energii. Obecność płytki o sieci A3 w krysztale o sieci A1 powoduje zwiększenie energii układu o wartości (w J/m3): ΔF = FA3 - FA1, przy czym FA3 i FA1 są odpowiednio energiami kryształów o sieci A3 i A1. Jeżeli różnicę ΔF pomnożymy przez grubość powstałej warstewki, to otrzymamy energię przypadającą na jednostkę powierzchni błędu ułożenia (w J/m2): γ=ΔF⋅g.
Błędy ułożenia powstają b. łatwo wtedy, gdy ich energia jest mała. Powstają one albo podczas krystalizacji, albo wskutek rozpadu dyslokacji jednostkowych na dyslokacje częściowe. Energia powstałego błędu ułożenia działa w kierunku zmniejszenia odległości między dyslokacjami częściowymi i to tym bardziej, im większa jest ta energia. Odległość między dyslokacjami częściowymi jest zatem proporcjonalna do 1/γ. Wartość tej energii decyduje o zachowaniu się materiałów podczas obróbki plastycznej, a współczyn. umocnienia rośnie w miarę zmniejszania się γ.
Energia granic ziarn
W idealnej sieci krystalicznej odległości między atomami są takie, że energia całkowita takiego układu jest najmniejsza. Energia granic ziarn σ wynika w dużej mierze z tego, że odległości między atomami leżącymi na granicy uległy zmianie. Ponieważ zaburzenie w rozmieszczeniu atomów leżących na granicy zwiększa się ze wzrostem kąta dezorientacji, zwiększa się też energia granic ziarn. Energia ta odnoszona jest do jednostki powierzchni granicy i wyrażana jest w J·m-2. W wypadku granic wąskokątowych, ich energia σ równa jest sumie energii dyslokacji, jakie tworzą tę granicę. Ponieważ energia dyslokacji odnoszona jest do jednostki długości, więc im większa jest gęstość dyslokacji na granicy, tym większa jest energia. Liczba dyslokacji na jednostce długości granicy wynosi 1/D = θ/b i z tego σ ∼ 1/b·θ·Ed, przy czym Ed jest energią jednostki długości dyslokacji. Dążność do zmniejszenia rozciągłości granic ziarn, a tym samym energii całkowitej polikryształu powoduje, że w odpowiednio wysokich temperaturach granice ziarn zaczynają się przemieszczać. Jest to uwarunkowane jednak możliwością zmiany przez atomy zajmowanych położeń. Kierunek ruchu granic ziarn jest taki, że towarzyszy temu zmniejszanie się energii całego układu. Przejawia się to przede wszystkim w tym, że granice krzywoliniowe prostują się, c w układzie przestrzennym jakim jest polikryształ jest równoznaczne ze zmianą powierzchni ukształtowanych przypadkowo w płaszczyzny.
Na styku trzech ziarn tej samej fazy dążność do osiągnięcia stanu równowagi prowadzi do zmiany kątów dwuściennych, tak aby spełniony był warunek:
![]()
gdzie γ12, γ23, γ31 są odpowiednimi napięciami powierzchn., a α1, α2, α3 kątami dwuściennymi między granicami.
Zakładając, że są to granice szerokokątowe można przyjąć, jednakowe napięcia powierzchniowe, czyli γ12 = γ23 = γ31, a stąd kąty też będą jednakowe i równe 120°. Stąd wniosek, że stabilne mogą być tylko granice między takimi ziarnami, które będą bryłami wypełniającymi dokładnie przestrzeń, a jednocześnie przecięte dowolną płaszczyzną utworzą siatkę granic w postaci sześcioboków foremnych o kątach narożnych równych 120°. Najbardziej zbliżone do takich brył są czternastościany.
Wszystkie ziarna mające w przekroju mniej niż sześć boków będą zanikać. W wysokich temperaturach w poli-kryształach zachodzi samorzutny proces zmniejszania się ziarn, przez co pozostałe zwiększają swoje wymiary. Krótko proces ten nazywany jest rozrostem ziarn. Z punktu widzenia własności mechanicznych rozrost ziarn i spowodowana tym zjawiskiem gruboziarnistość jest niekorzystna. Wiadomo, że granice ziarn blokują ruch dyslokacji i im drobniejsze jest ziarno, tym większa jest granica plastyczności. Zwiększa się również udarność. Przechodzenie atomów z jednego ziarna do drugiego może jednak ustać, jeśli na granicy znajdzie się jakaś obca faza, np. wtrącenie żużla, tlenek metalu lub inny związek. Jeśli takich wtrąceń jest wiele, to nawet w wysokich temperaturach rozrost ziarn zostaje zahamowany. Prędkość ruchu granic ziarn mogą też zmniejszyć obce atomy, jakie ulokują się na nich w następstwie ich ruchu. Szczególnie skuteczne będą atomy wykazujące mniejszą ruchliwość w porównaniu z atomami osnowy. Wynika z tego, że im bardziej czysty jest metal, tym większa jest jego skłonność do gruboziarnistości.
Granice międzyfazowe
Fazą nazywamy jednorodną część układu, oddzieloną od reszty układu powierzchnią graniczną, po przekroczeniu której własności zmieniają się w sposób nieciągły. Różnica w składzie chemicznym, typie sieci krystalicznej i wzajemnej orientacji powoduje na ogół występowanie na takiej granicy licznych zaburzeń w prawidłowym rozmieszczeniu atomów. Podobieństwo do budowy szerokokątowych granic ziarn wyjaśnia duże wartości energii tych granic międzyfazowych, nazywanych granicami niekoherentnymi.
Granice koherentne. Mimo odmiennych sieci krystalicz. może się zdarzyć, że w sieciach tych istnieją takie płaszczyzny atomowe, na których rozmieszczenie atomów jest identyczne. Jeżeli wzajemna orientacja obu tych faz będzie taka, że zetkną się one z sobą tymi płaszczyznami, to na granicy międzyfazowej brak jest jakichkolwiek nieprawidłowości w rozmieszczeniu atomów. Energia granic koherentnych jest więc mała. Całkowite sprzężenie sieci krystalicznych obu sąsiadujących ze sobą faz jest jednak możliwe tylko wtedy, gdy płaszczyzny atomowe obu faz są do siebie równoległe i tak zorientowane, aby kierunki rzędów atomowych się pokrywały. Spełnione muszą zatem być następujące warunki:
(hkl)α || (h1k1l1)β,
[uvw]α || [u1v1w1]β,
odstępy między atomami wzdłuż powyższych kierunków powinny być takie same.
Rzadko zdarza się jednak, by na płaszczyźnie styku dwóch faz istniała idealna zgodność odstępów między atomami. Gdy różnice te są małe, wówczas wzajemne dopasowanie może być osiągnięte przez odkształcenie sprężyste obu sieci. Wielkość tego odkształcenia zależy od tego jak duża jest niezgodność w rozmieszczeniu atomów na płaszczyźnie granicznej. Energia tego odkształcenia zależy natomiast od stałych sprężystości obu faz i powoduje wzrost energii takiej koherentnej granicy międzyfazowej.
Gdy niezgodność w rozmieszczeniu atomów na płaszczyźnie styku obu faz jest za duża, aby mogła być skompensowana samym tylko odkształceniem sprężystym, a rozmiary cząstek drugiej fazy są znaczne, wówczas na granicach międzyfazowych pojawiają się dyslokacje. Dyslokacje te tworzą sieć o oczkach tym mniejszych, im większe jest niedopasowanie. Siatka dyslokacji przyczynia się do zmniejszenia energii granicy i składa się na nią wtedy energia odkształceń sprężystych (między dyslokacjami) i energia dyslokacji. Mówimy wtedy, że granica jest półkoherentna. Dążność do zmniejszenia energii całkowitej układu, jakim jest polikryształ zbudowany z dwóch lub więcej faz przejawia się też w postaci dążności do zmniejszenia energii całkowitej wszystkich granic. Gdy obie fazy mogą utworzyć koherentną granicę międzyfazową, wówczas przyjmują one kształt płytek, gdyż całkowite sprzężenie sieci krystalicznych obu faz możliwe jest tylko na granicach płaskich. W razie niekoherentnej granicy międzyfazowej dążność do zmniejszenia energii całkowitej przejawia się występowaniem drugiej fazy postaci kulistej.
Wydzielenia drugiej fazy na granicach ziarn przyjmują natomiast kształt zapewniający równowagę napięć powierzchniowych. Rozpatrując styk trzech ziarn z których jedno jest inną fazą z warunków równowagi otrzymamy zależność: γαα = 2γαβ ⋅cos(Φ/2), w której: γαα jest napięciem powierzchniowym na granicy ziarn tej samej fazy, γαβ - napięciem powierzchniowym na granicy międzyfazowej, a Φ - kątem dwuściennym krawędzi ziarna fazy β. Wartość kąta Φ zależy zatem od stosunku napięć powierzchniowych, gdyż cos(Φ/2) = γαα / 2γαβ.
Jeżeli granica fazowa jest niekoherentna, to przy małej wartości napięcia powierzchniowego na granicy między ziarnami tej samej fazy może być spełniona zależność γαβ » γαα, a wtedy cos(Φ/2) ≈ 0. Wtedy kąt Φ ≈ 180° i wydzielenia fazy β przyjmują kształt kulisty. W miarę jak będzie malało napięcie granicy międzyfazowej lub wzrastało napięcie granicy ziarn, kąt dwuścienny Φ będzie malał i zmieniać się będzie kształt wydzieleń fazy β.
W granicznym wypadku, gdy kąt ten będzie równy zeru, faza β utworzy wokół ziarn fazy α cienką błonkę. Z energetycznego punktu widzenia korzystniejsze jest bowiem zastąpienie powierzchni granic ziarn fazy α dwoma powierzchniami międzyfazowymi α - β o energii powierzchniowej mniejszej niż połowa wartości energii granic ziarn.
Energia swobodna
Energia swobodna układu jest określona równaniem:
F = E - TS,
w którym: E - energia wewnętrzna układu, S - entropia, T - temperatura bezwzględna.
Energia wewnętrzna jest sumą energii kinetycznej Ek i energii potencjalnej Ep wszystkich cząstek tworzących ten układ. Energię potencjalną należy rozumieć jako energię wzajemnego oddziaływania, a energię kinetyczną jako energię drgań cieplnych cząstek układu. Stan układu określa się przez podanie jego pewnych własności makroskopowych, takich jak temperatura T, ciśnienie p i objętość V. Energia wewnętrzna jest funkcją tych parametrów układu, czyli: E = Ep + Ek = E(p, V, T). Entropia jest drugą własnością układu określającą jego równowagę. Jest, podobnie jak energia wewnętrzna, proporcjonalna do liczby cząstek wchodzących w skład układu. S = k⋅ln w. Entropia charakteryzuje stopień uporządkowania układu i mierzy tym samym prawdopodobieństwo trwałości danego stanu, przy czym stany o większej entropii są bardziej prawdopodobne. Zarówno energia wewn. jak i entropia zmieniają się z temperaturą, natomiast zmiana energii swobodnej z temp. może być określona następująco: Jeżeli przy stałym ciśnieniu p, temperatura układu wzrośnie od T do T + ΔT, to zmianę energii swobodnej określa różniczka:
DF = dE - TdS - SdT, a stąd:
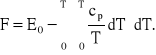
Z powyższego równania wynika, że energia swobodna maleje ze wzrostem temperatury szybciej, im większe jest ciepło właściwe układu. Zmniejszanie się energii swobodnej rozpoczyna się od punktu określającego energię wewnętrzną układu w temperaturze zera bezwzględnego. W temperaturze tej energia wewnętrzna jest równa energii potencjalnej i zależy od sposobu rozmieszczenia atomów. Ma ona inną wartość dla stanu krystalicznego, a inną dla rozmieszczenia bezładnego - charakterystycznego dla stanu ciekłego. Energia potencjalna układu z nieuporządkowanymi atomami jest większa niż energia układu z atomami uporządkowanymi. Czyli: E0L > E0S. Jak wynika z omawianego równania wraz ze zmianą temperat. energie swobodne cieczy będą malały zależnie od wartości ciepła właściwego tych faz. Ponieważ również cpL > cpS, więc krzywe energii swobodnych opadają niejednakowo i przetną się w jakimś punkcie o temperaturze T0, w którym energie swobodne obu faz są jednakowe, czyli FL = FS. Temperatura T0 jest temperaturą równowagi, a w tym wypadku jest to teoretyczna temperatura krzepnięcia.
Każdy ze sposobów rozmieszczenia atomów w przestrzeni charakteryzuje w temperaturze zera bezwgl. inna energia wewn. E0, inne też będą ciepła właściwe. W rezultacie każdemu sposobowi rozmieszczeń tych samych atomów odpowiada inna krzywa obrazująca zmianę energii swobodnej z temperaturą. W danych warunkach, tj. w danym zakresie temperatur trwała jest ta sieć krystaliczna (takie rozmieszczenie atomów), której energia swobodna jest najmniejsza. Przecięcie się w jakiejś temperaturze krzywej energii swobodnej, odpowiadającej jednemu sposobowi rozmieszczenia atomów, z bardziej stromo opadającą krzywą energii swobodnej charakteryzującą drugi sposób rozmieszczenia atomów wskazuje, że w tej temperaturze zajdzie przemiana, której istotą będzie zmiana typu sieci krystalicznej. Przemiana taka nazywana jest przemianą alotropową, a różniące się typem sieci krystalicznej odmiany tego samego pierwiastka nazywamy odmianami alotropowymi. Ogólnie można stwierdzić, że w niskich temperaturach trwałe są układy o małej energii potencjalnej, czyli układy krystalizujące w sieciach o dużej liczbie koordynacji. W wyższych temperaturach trwałe mogą być układy o większej energii (sieci krystaliczne o mniejszej liczbie koordynacji), jeśli tylko ich entropia jest większa. Wskutek tego ze wzrostem temperatury trwałe stają się układy mniej zwarte, o mniejszym uporz. atomów.
Krystalizacja
Krystalizacją nazywamy powstawanie kryształów podczas przechłodzenia substancji ze stanu termodynamicznie mniej trwałego w stan bardziej trwały. W wypadku substancji czystych kryształy mogą powstawać z przechłodzonej pary lub z cieczy, a w stanie stałym - z nietrwałych odmian alotropowych. W substancjach złożonych jakimi są stopy, nowe kryształy mogą powstawać także z przesyconych roztworów stałych. Podczas krystalizacji zachodzi zmiana wielu własności fizycznych (gęstości, ciepła właściwego, rozszerzalności cieplnej, przewodności elektrycznej itp.) i rejestracja tych zmian w zależności od temperatury pozwala określić dokładnie temperatury, w których proces ten zachodzi. W substancjach czystych zmiany te zachodzą skokowo, w stałej temperaturze, a w stopach - w zakresie temperatur. Do rozpoczęcia krzepnięcia jest konieczne przechłodzenie niezbędne do wystąpienia różnicy energii swobodnych i im jest ono większe, tym większe jest prawdopodobieństwo rozpoczęcia się tej przemiany.
Występowanie przechłodzeń wskazuje, że krzepnięcie nie jest procesem przebiegającym niezwłocznie po pojawieniu się różnicy energii swobodnych. Można to wyjaśnić tym, że poziomy energii swobodnych obu stanów - ciekłego i stałego - są przedzielone barierą o większej energii. Wysokość bariery oddzielającej dwa stany - metastabilny i stabilny - jest czynnikiem decydującym o szybkości przemiany, gdyż zmienić stan mogą tylko atomy dysponujące odpowiednim nadmiarem energii. Liczba takich atomów w układzie metastabilnym jest natomiast tym mniejsza, im większa jest nadwyżka energii konieczna do pokonania bariery energetycznej. Istnienie podczas krzepnięcia bariery energetycznej powoduje, że przemiana ta przebiega stopniowo. Ilość energii jaką należałoby doprowadzić do układu, by wszystkie atomy jednocześnie przekroczyły barierę energetyczną, byłaby bowiem b. duża Barierę tę może przekroczyć niewiele atomów i stąd w czasie krzepnięcia tworzą się w cieczy b. drobne cząstki fazy stałej, które następnie rosną przez przyłączanie z cieczy coraz większej liczby atomów. Te drobne cząstki krystaliczne są nazywane zarodkami krystalizacji (krytycznymi). Do tego aby zaszło skrzepnięcie cieczy konieczne jest więc:
przechodzenie cieczy,
powstanie w cieczy zarodków krystalizacji,
rośnięcie tych zarodków i powstanie krystalitów.
Zarodkowanie jednorodne (homogeniczne)
Zarodkowanie jednorodne zachodzi w cieczy metalicznej całkowicie jednorodnej, a w jego wyniku powstaje zupełnie jednorodna faza stała. Zarodki w kształcie kul, utworzone w dowolnych miejscach w całej objętości cieczy z zespołów bliskiego uporządkowania, osiągają promień krytyczny R* przy znacznym przechłodzeniu, czyli znacznym obniżeniu temperatury poniżej temperatury równowagi TR. Szybkość zarodkowania jest b. mała, po czym zwiększa się dopóty, dopóki przechłodzenie nie osiągnie wartości krytycznej, by następnie gwałtownie się zmniejszyć.
Pojawienie się poniżej temperatury krzepnięcia cząstki fazy stałej powoduje zmniejszenie się energii swobodnej układu proporcjonalnie do jej objętości V i różnicy energii swobodnych -ΔFV = FS - FL, ale z drugiej strony energia swobodna układu zwiększa się wskutek powstania wraz z tą cząstką powierzchni międzyfazowej ciecz-ciało stałe. Spowodowany tym przyrost energii swobodnej układu jest proporcjonalny do pola tej powierzchni S i jej energii σLS. W rezultacie zmianę energii swobodnej układu można przedstawić w postaci zależności: ΔF = -V(ΔFV) + SσLS. Znak minus wskazuje, że energia układu maleje, a znak plus, że rośnie. Zakładając że uporządkowane zgrupowanie atomów jest sześcianem o krawędzi a, poprzednie równanie przyjmie postać: ΔF = -a3(ΔFV) + 6a2σLS.
![]()
stąd
![]()
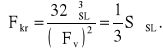
Z powyższego równania wynika, że rozmiar zarodka krytycznego zależy od swobodnej energii powierzchni międzyfazowej σSL i różnicy energii swobodnych fazy stałej i ciekłej. Ponieważ energia powierzchni międzyfazowej σSL praktycznie nie zmienia się z temperaturą, a ΔFV rośnie wraz z przechłodzeniem, przeto o rozmiarze zarodka krytycznego decyduje wielkość przechłodzenia ΔT. Im większe jest zatem przechłodzenie, tym mniejszy jest zarodek krytyczny i tym mniejsza jest energia ΔFkr potrzebna do jego utworzenia. Ponieważ w cieczy znajdują się ugrupowania atomów o różnych rozmiarach, więc przy większych przechłodzeniach, gdy rozmiary zarodków krytycznych są mniejsze, więcej takich ugrupowań stanie się zarodkami krystalizacji. Oznacza to, że liczba zarodków krystalizacji zwiększa się ze stopniem przechłodzenia.
Zarodkowanie niejednorodne (heterogeniczne)
Praca potrzebna do utworzenia zarodka krytycznego wynosi 1/3 nakładu energii na utworzenie jego powerzchni granicznej. Obrazowo można to sformułować następująco: zysk na energii swobodnej, wynikający ze zmiany sześciennej cząsteczki cieczy w ciało stałe, wystarcza na utworzenie 4 z jego sześciu ścian. Energię potrzebną do utworzenia pozostałych 2 ścian powierzchni granicznej musi układ dostarczyć sam kosztem fluktuacji energetycznych, tj. lokalnych, krótkotrwałych odchyleń energii od wartości średnich. Gdyby jednak jedna z tych ścian miała mniejszą energię powierzchniową, wówczas praca potrzebna do utworzenia zarodka zdolnego do rośnięcia bez zwiększania energii układu byłaby mniejsza. Stałoby się to możliwe, gdyby powierzchnią tą zarodek stykał się nie z cieczą, ale z fazą stałą, np. z już wykrystalizowanymi tlenkami, azotkami itp. związkami lub ze ściankami naczynia. Cząstki takie spełniają rolę podkładek, na których rozpoczyna się krystalizacja. Są to zatem swego rodzaju katalizatory zarodkowania, których aktywność jest tym większa, im mniejsza jest energia powierzchni styku z krystalizującym ciałem, czyli spełniony jest warunek σPS < σSL. Indeksy P, S i L oznaczają podkładkę, ciało stałe i ciecz, a σPS oznacza energię powierzchni granicznej między ciałem obcym a powstałym kryształem itd. Stosunki energetyczne na powierzchniach styku wymienionych faz określa kąt zwilżania θ, jaki ustala się między nimi. Warunkiem równowagi jest by: σLP = σPS + σSL⋅cosθ, stąd:
![]()
Między fazami krystalicznymi a cieczą granica jest zawsze niekoherentna i można przyjąć, że σLP ≈ σSL, natomiast między 2-oma fazami krystalicznymi może na powierzchni granicznej wystąpić pełne lub częściowe sprzężenie sieci krystalicznych, dzięki czemu σPS może mieć wartości znacznie mniejsze niż σSL. Powoduje to następnie zmiany kąta zwilżania θ. Jak zmiana kąta zwilżania wpływa na pracę ΔFkr*, potrzebną do utworzenia niejednorodnego zarodka krytycznego, można określić posługując się znanymi już zależnościami. Ostatecznie:
ΔFkr* = ΔFkrf(cosθ).
Na zarodkowanie, a tym samym na strukturę skrzepniętych metali można wpływać przez wprowadzenie do nich w stanie ciekłym, substancji zapewniających powstanie odpowiednio aktywnych cząstek, np. tlenków, azotków, węglików itp. Substancje te są wprowadzane w praktyce przemysłowej zwykle w niewielkich ilościach i zmieniają przebieg krystalizacji. Substancje te nazwano modyfikatorami, a całokształt związanych z tym czynności - modyfikowaniem.
Wzrost kryształów
Analizując strukturę atomową powierzchni międzyfazowej ciecz-ciało stałe zakłada się, że ciało stałe znajdują się w równowadze i dlatego powierzchnia ta ma taką strukturę, która w danych warunkach zapewnia najmniejszą energię swobodną. Rzeczywista struktura powierzchni międzyfazowej jest wynikiem dwóch przeciwstawnych tendencji. Energia atomu zmniejsza się bowiem wraz ze wzrostem liczby najbliższych sąsiadów i liczba ta jest największa na powerzchni gładkiej najgęściej upakowanej. Entropia powierzchni rośnie natomiast ze wzrostem chropowatości - nieuporządkowania. Względny udział obu tych czynników decyduje o energii swobodnej powierzchni międzyfazowej. Kiedy przeważa czynnik entropii, wtedy zmiana energii swobodnej spowodowana przejściem atomu z cieczy do kryształu jest w zasadzie niezależna od miejsca jego przyłączenia. W rezultacie powierzchnia jest chropowata. Jeśli jednak nad entropią przeważa czynnik energii, silnie zależny od położenia atomu, to atomy zajmują te miejsca na powierzchni, w których mają one najwięcej sąsiadów.
Kinetyka krystalizacji
Liczba zarodków krystalizacji powstających w cieczy, jak i liniowa szybkość ich wzrostu są głównymi parametrami, decydującymi o kinetyce krystalizacji. W rzeczywistych warunkach krystalizacji zarówno liczba zarodków powstających w jednostce czasu i w jednostce objętości, jak i liniowa szybkość wzrostu, zależą od przechłodzenia. Z rozważań teoretycznych oraz doświadczalnych wynika, że w miarę wzrostu przechłodzenia szybciej rośnie szybkość wzrostu kryształów niż liczba zarodków. Dla otrzymania po skrzepnięciu budowy drobnoziarnistej konieczne jest szybkie chłodzenie, zapewniające odpowiednio duże przechłodzenie. Ogólnie obowiązuje zasada, że rozmiar ziarn jest wprost proporcjonalny do szybkości wzrostu, a odwrotnie proporcjonalny do liczby zarodków.
Krystalizacja dendrytyczna
Przechłodzenia występujące podczas krystalizacji cieczy decydują także o ukształtowaniu powierzchni międzyfazowej ciecz-ciało stałe w skali makroskopowej. W warunkach równowagi powierzchnia jest płaska i nieruchoma. Aby spowodować ruch tej powierzchni i wzrost kryształów, musi na niej wystąpić przechłodzenie i im będzie ono większe, tym szybciej przemieszczać się będzie granica między ciałem stałym i cieczą. Ukształtowanie powierzchni międzyfazowej zależy natomiast od rozkładu temperatury w cieczy. Możliwe są tu dwa wypadki: dodatni lub ujemny gradient temperatury. Kiedy gradient temperatury jest dodatni, wówczas utajone ciepło wydzielane na powierzchni międzyfazowej jest odprowadzane wyłącznie przez fazę stałą. Szybkość odprowadzanie tego ciepła decyduje o szybkości przemieszczania się powierzchni międzyfazowej. Zahamowanie tego przepływu powoduje wzrost temperatury powierzchni, a jeśli osiągnięta zostanie temperatura równowagi T0, to krystalizacja ustanie. Każda wypukłość jaka powstanie przypadkowo na powierzchni granicznej sięgając głębiej w ciecz przestanie rosnąć i powierzchnia pozostanie płaska. W razie ujemnego gradientu w cieczy, ciepło utajone jakie wydziela się na powierzchni międzyfazowej może być oprowadzane zarówno przez ciało stałe, jak i przez ciecz. Ruch powierzchni nie jest już kontrolowany samym tylko odprowadzaniem ciepła przez fazę stałą. W takich warunkach przypadkowe wypukłości na powierzchni granicznej sięgają do cieczy bardziej przechłodzonej i wierzchołki tych wypukłości rosną szybciej. Płaska powierzchnia międzyfazowa przestaje być trwała i pojawiają się na niej liczne wypukłości. Wypukłości te sięgając głębiej rosną coraz szybciej, ale szybkość ta kontrolowana jest wydzielaniem się większej ilości ciepła utajonego, zmniejszającego przechłodzenie. Płaskie ścianki rosnących wypukłości też nie są trwałe i na nich też pojawiają się podobne wypukłości, podrzędne w stosunku do pierwszych. Proces ten przy bardzo dużych przechłodzeniach może się powtarzać, a kryształy przyjmują charakterystyczny kształt, nazywany dendrytami a taki sposób krystalizacji - krystalizacją dendrytyczną.
Dendryty mogą pojawić się nie tylko na wewnętrznej powierzchni skrzepniętej warstwy metalu, ale i wewnątrz przechłodzonej cieczy. Przechłodzenia konieczne do powstania zarodków jednorodnych lub na mało aktywnych cząstkach obcych, są znacznie większe niż przechłodzenia powodujące ruch powierzchni międzyfazowej. Zarodek powstaje zatem w cieczy silnie przechłodzonej, a jego płaskie powierzchnie międzyfazowe są niestabilne. Powstają wtedy dendryty, a w miarę tego jak przechłodzenia są większe, to gałęzie dendrytów pojawiają się częściej i ich drzewiasty charakter uwypukla się jeszcze bardziej.
Budowa krystaliczna wlewków
Większość technicznie ważnych metali otrzymuje się w postaci ciekłej i dlatego musi się je odlewać z formy, w celu otrzymania po skrzepnięciu albo wyrobów o pożądanym kształcie, albo bloków zwanych wlewkami, które przeznacza się do dalszej obróbki plastycznej. W formie ciekły metal zaczyna krzepnąć, przy czym jeśli była to forma metalowa (kokila lub wlewnica), a jej ścianki były zimne, to warstwa cieczy przyległa do jej ścian ulega silnemu przechłodzeniu. W takich warunkach w warstwie tej powstaje bardzo duża ilość bezładnie zorientowanych zarodków krystalizacji. W rezultacie zewnętrzna warstwa odlewu jest bardzo drobnoziarnista i nazywana jest strefą kryształów zamrożonych.
Dalsze krzepnięcie polega na wzroście tych kryształów, które stykają się z cieczą. Rosnące kryształy zaczynają sobie przeszkadzać we wzroście wszerz i w głąb cieczy. W rezultacie powstają podłużne kryształy zwane słupkowymi. Pozostałe ziarna, ze względu na ich dość regularny kształt nazwano równoosiwymi. Zdarza się, że nie występuje w odlewach i wlewkach strefa kryształów równoosiowych, a kryształy słupkowe rosnące od przeciwległych ścian stykają się z sobą w środku odlewu - to transkrystalizacja.
Budowa krystaliczna odlewu ma duży wpływ na jego własności wytrzymałościowe i technologiczne. Jest ona szczególnie ważna we wlewkach, które poddawane są obróbce plastycznej. Szczególnie niepożądana jest transkrystalizacja, gdyż na styku dwóch stref kryształów słupkowych spójność jest znacznie obniżona.
W większości wypadków krzepnięciu metali towarzyszy zmniejszenie objętości o 3-6% i tylko bizmut, gal, krzem oraz german zwiększają swoją objętość. Na skutek skurczu w górnej środkowej części odlewów i wlewków tworzy się jama skurczowa. Jej kształt zależy w dużej mierze od kształtu formy. Ponieważ część wlewka zawierająca jamę skurczową musi być przed obróbką plastyczną odcięta, jamy głębokie są niepożądane.
Przemiany alotropowe
W niektórych metalach, już po skrzepnięciu, a więc w stanie stałym zachodzą dalsze przemiany. Są to przemiany alotropowe, a ich istotą jest zmana sposobu rozmieszczenia atomów w przestrzeni. Przemiany alotropowe są spotykane w metalach, a w niektórych z nich między temperaturą krzepnięcia a temperaturą otoczenia jest ich kilka. Poszczególne odmiany alotropowe tego samego pierwiastka oznacza się kolejnymi literami alfabetu greckiego, poczynając od odmiany trwałej w najniższych temperaturach, przy czym literę umieszcza się obok symbolu chemicznego pierwiastka, np. Tiα lub α-Ti.
Odmiany alotropowe różnią się między sobą typami sieci krystalicznych i jest regułą, że odmiany trwałe w niskich temperaturach krystalizują w sieciach bardziej zwartych, o większej liczbie koordynacji (żelazo jest pod tym względem wyjątkiem). Zmiana sieci krystalicznej pociąga za sobą całkowitą zmianę własności chemicznych, fizycznych i mechanicznych. Zmiana tych własności zachodzi w temperaturze przemiany skokowo i dlatego temperatury te można określać śledząc zmianę wybranych własności wskutek zmiany temperatury, np. zmianę rozszerzalności cieplnej, oporności elektrycznej itp. Bardzo często określa się temperatury przemian alotropowych podobnie ja temperatury krzepnięcia, gdyż na krzywych grzania lub chłodzenia pojawiają się w tych temperaturach przystanki, związane z efektem cieplnym przemian nazywanym utajonym ciepłem przemiany. Przemiany alotropowe są odwracalne, ale temperatury tych przemian są podczas chłodzenia niższe, a podczas grzania - wyższe niż temperatury równowagi wyznaczone na wykresach energii swobodnej. Różnica między temperaturą przemiany wyznaczoną podczas chłodzenia i grzania występuje nawet przy bardzo powolnej zmianie temperatury, a zjawisko to nazywane jest histerezą cieplną.
Przemiana alotropowa rozpoczyna się dopiero przy pewnym przechłodzeniu, kiedy faza β jest już nietrwała. Powyżej temperatury T1 trwała jest natomiast faza α i jej cząstki pojawiają się w fazie β. Po osiągnięciu rozmiarów krytycznych cząstki te stają się zarodkami i rosnąc kosztem atomów fazy β powodują jej zanik. Mechanizm wzrostu zarodków fazy α przypomina w dużej mierze rozrost ziarn, gdyż atomy fazy β przemieszczają się poprzez granicą międzyfazową i przyłączane są do rosnących zarodków i ziarn fazy trwalszej. Koniecznym warunkiem takiego mechanizmu przemiany jest znaczna ruchliwość cieplna atomów, jaka występuje dopiero w dostatecznie wysokich temperaturach. Ruchliwość atomów jest podstawowym warunkiem dyfuzji i dlatego przemiany, podczas których fazy trwalsze rosną kosztem faz metastabilnych w wyniku niezależnego od siebie ruchu pojedynczych atomów na odległości większe niż ich średnica atomowa nazywamy przemianami dyfuzyjnymi. Zmiana całkowitej energii swobodnej układu spowodowana powstaniem cząstki fazy α o objętości V w fazie β jest określona równaniem: ΔF = -VΔFv + Sσαβ+VZ, w którym ΔFv = Fα - Fβ, S jest polem powstałej powierzchni międzyfazowej a σαβ - energią swobodną tej powierzchni zależną w dużej mierze od wzajemnej orientacji krystalograficznej sieci obu faz, decydującej o koherentności lub jej braku, Z jest energią odkształcenia związanego z przemianą fazy β w jednostkę fazy α. Pamiętając o tym, że stałe sprężystości są tym większe, im niższa jest temperatura, można się spodziewać, że energia ta będzie duża, gdy temperatury przemian są niskie. W przemianach alotropowych do powstania zarodka krytycznego konieczne są większe przechłodzenia, gdyż różnicę energii swobodnych ΔFV zmniejsza wydatek energii na odkształcenie fazy macierzystej zarodka. W przemianach alotropowych zarodki powstają w fazie metastabilnej wszędzie tam, gdzie ich powstawanie jest ułatwione. Uprzywilejowane są pod tym względem granice ziarn, a szczególnie krawędzie i naroża ziarn, tj. miejsca styku odpowiednio 3 lub 4 ziarn. Występujące w tych miejscach duże zaburzenia w rozmieszczeniu atomów stwarzają dogodne warunki do ich przegrupowania, mniejszy jest też wydatek energii na utworzenie granicy międzyfazowej. Im zatem bardziej drobnoziarnista jest faza wyjściowa, tym bardziej rozwinięta jest powierzchnia granic ziarn, więcej jest naroży i krawędzi ziarn i tym więcej może powstać zarodków. Dążność do zmniejszenia całkowitej energii swobodnej układu powoduje, że zarodki rosnące w fazie metastabilnej przyjmują kształt i orientację zapewniającą możliwie najmniejszą energię powierzchniową σαβ. Wydzielenia nowo powstałej fazy mają dlatego często kształt płytek usytuowanych w taki sposób, by na płaszczyznach styku występowało możliwie najdokładniejsze sprzężenie sieci krystalicznych obu faz. W razie b. dużych prechłodzeń lub wtedy, gdy temperatury przemian są niskie, może się zdarzyć, że cieplny ruch atomów praktycznie ustaje. W tych warunkach przemiana alotropowa dyfuzyjna jest niemożliwa i przemiana musi przebiegać bezdyfuzyjnie. Z energetycznego punktu widzenia przemiana ta jest mniej wygodna i aby mogła zajść konieczna jest duża różnica energii swobodnych. Nowy typ sieci krystalicznej powstaje w wyniku jednoczesnego, skoordywanego ruchu wszystkich atomów leżących na określonych płaszczyznach atomowych. Kierunek tego ruchu jest ściśle określony, a odległości jakie atomy przebywają względem atomów płaszczyzny sąsiedniej są jednakowe i stanowią ułamek odległości między atomami. W ten sposób atomy nie zmieniając swego otoczenia, w uporządkowany sposób zajmują nowe położenia, właściwe dla sieci krystalicznej fazy trwałej. Przy takim sposobie przegrupowania atomów, kryształy nowej fazy mają przeważnie kształt płytek, a powierzchnie międzyfazowe są w przeważającej mierze koherentne. Płytki te przecięte płaszczyzną szlifu mają postać igieł.
Przemiany alotropowe mogą być wywołane także zmianą ciśnienia.
Przemiany magnetyczne
Niektóre metale charakteryzuje duża przenikalność magnetyczna oraz nieliniowa zależność stanu namagnesowania od natężenia zewnętrznego pola magnetycznego, przy czym własności magnetyczne zostają zachowane także po usunięciu tego zewnętrznego pola. Metale te nazywane są ferromagnetykami i charakteryzują się takimi własnościami magnetycznymi jak żelazo. Oprócz żelaza własności ferromagnetyczne wykazują jeszcze kobalt, nikiel, a także niektóre metale ziem rzadkich, jak: gadolin (Gd), dysproz (Dy), erb (Er), holm (Ho), terb (Tb) i tul (Tu). Ferromagnetykami są także stopy i związki chemiczne wymienionych metali oraz niektóre stopy metali nieferromagnetycznych, np. Mn-Bi, Cr-Te,itp.
Każdy ferromagnetyk traci swoje własności magnetyczne w charakterystycznej dla niego temperaturze nazywanej punktem Curie. W odróżnieniu od przemian alotropowych przemiana magnetyczna:
nie wywołuje zmiany innych włsności fizycznych poza własnościami elektrycznymi, magnetycznymi i cieplnymi,
nie pociąga za sobą przekrystalizowania, tj. tworzenia się nowych ziarn wskutek zmiany sieci krystalicznej,
przebiega bez histerezy, zawsze w tej samej temp-rze, niezależnie od szybkości chłodzenia lub grzania.
Stopy
Stopy dzielimy na: 1) mieszaniny, 2) roztwory, 3) związki i fazy międzymetaliczne.
Gdy siły oddziaływania między atomami tego samego składnika stopu są większe niż oddziaływanie między atomami niejednakowymi, wówczas w stopie wystąpią obok siebie kryształy składników, tworząc mieszaninę.
Roztwory
Mimo jednorodności roztwory są substancjami złożonymi, składającymi się w najprostszym wypadku z 2 składników. Jeden z tych składników występujący w przewadze jest rozpuszczalnikiem, a drugi jest rozpuszczany. W ciekłych stopach metalicznych rozpuszczalnikiem jest metal, w którym mogą się rozpuszczać metale lub niemetale. Aby zachować charakter metaliczny, w roztworach występujących w stopach metalicznych muszą przeważać metale. Jeśli jednorodność charakteryzująca roztwór ciekły zostanie zachowana po skrzepnięciu, to otrzymuje się roztwór stały. Jest to wynik utrzymania się rozpuszczalności składników stopu także w stanie stałym. Cechą charakterystyczną roztworów stałych jest zachowanie sieci krystalicznej rozpuszczalnika. Do sieci tej jest wbudowana dowolna lub ograniczona liczba atomów składnika rozpuszczonego.
Roztwory stałe, w których jednorodność występuje w całym zakresie stężeń od 100% jednego składnika do 100% składnika drugiego są nazywane roztworami stałymi ciągłymi, a rozpuszczalność całkowitą lub nieograniczoną. W stopach metali całkowitą rozpuszczalność spotyka się nie często, zwykle drugi składnik rozpuszcza się w ilości ograniczonej. Roztwory takie są nazywane roztworami stałymi granicznymi lub nieciągłymi. Stwierdzono dwa sposoby rozmieszczenia atomów składnika rozpuszczonego w sieci krystalicznej rozpuszczalnika i na tej podstawie podzielono roztwory stałe na:
różnowęzłowe lub substytucyjne, nazywane tak ze względu na to, że atomy składnika rozpuszczonego zastępują atomy rozpuszczalnika w węzłach jego sieci krystalicznej.
międzywęzłowe, w których atomy składnika rozpuszczonego zajmują wolne przestrzenie między atomami, czyli luki międzywęzłowe w sieci krystalicznej rozpuszczalnika.
Warunki konieczne do powstania roztworu ciągłego:
tworzące ten roztwór substancje muszą tworzyć roztwór różnowęzłowy,
sieci obu składników muszą być izomorficzne (krystalizować w tym samym typie sieci),
różnica promieni atomów składników tworzących nie może być zbyt duża (max. różnica - 15%).
składniki tworzące muszą mieć podobny charakter elektrochemiczny (elektroujemny lub elektrododatni).
pierwiastki muszą znajdować się blisko siebie w tablicy Mendelejewa (podobna wartościowość).
Jeżeli choć jeden z tych warunków nie zostanie spełniony otrzymany roztwór będzie roztworem granicznym.
Gdy między atomami tworzącymi fazę międzymetaliczną występują obok wiązań metalicznych również wiązania silniejsze, uniemożliwiające powstanie roztworów międzywęzłowych lub różnowęzłowych, może powstać roztwór stały nazywany pustowęzłowym lub substrakcyjnym. W roztworze tym atomy składnika znajdującego się w nadmiarze zajmują swoje normalne położenia, ale wtedy nie wszystkie węzły zajmowane przez drugi składnik są obsadzone. Roztwory pustowęzłowe stwierdzono np. w węglikach tytanu TiC, tantalu TaC, wanadu VC i innych, gdy występuje w nich wyłącznie nadmiar atomów metali, a nie są obsadzone węzły przynależne atomom węgla.
Roztwory stałe uporządkowane
Roztwory stałe różnowęzłowe i międzywęzłowe, podstawowe i wtórne, mogą być nieuporządkowane lub uporządkowane w zależności od tego, czy atomy składnika rozpuszczonego są rozmieszczone bezładnie, czy zajmują ściśle określone miejsca w sieci rozpuszczalnika. Czynnikiem sprzyjającym uporządkowaniu roztworów stałych są nieco większe siły przyciągania między atomami niejednakowymi w porównaniu z siłami przyciągania między atomami jednakowymi. W niskich temperaturach zmniejszenie energii swobodnej mimo małych wartości entropii (mała liczba rozmieszczeń. W rezultacie energia swobodna nieuporządkowanych roztworów stałych jest mniejsza niż roztworów uporządkowanych. Istnieje zatem pewna stała temperatura, w której następuje w roztworze stałym przemiana nieporządek — porządek. Temperatura ta jest tym wyższa, im większe są siły wzajemnego oddziaływania między niejdnakowymi atomami. W czasie chłodzenia w temperaturze przemiany nieporządek — porządek atomy składników przemieszczają się w taki sposób, by określone węzły sieci krystalicznej zajmowały atomy rozpuszczalnika, a pozostałe atomy składnika rozpuszczonego. Wynika z tego, że pełne uporządkowanie jest możliwe tylko dla ściśle określonych stosunków ilościowych składników. W stopach dwuskładnikowych, w sieciach typu regularnego, uporządkowanie występuje przy stężeniach odpowiadających wzorom stechiometrycznym A3B, AB i AB3, czyli przy stosunkach 1:1, 1:3 lub 3:1.
Po uporządkowaniu wyraźnie wzrasta przewodnictwo elektryczne oraz twardość, a maleją własności plastyczne.
Fazy międzymetaliczne
W stopach metali spotyka się dużo różnorodnych faz, często o skomplikowanych strukturach krystalicznych. Fazy te można podzielić na grupy zależnie od tego jakie czynniki decydują o ich powstaniu i trwałości. Czynnikami tymi mogą być:
stężenie elektronowe,
określony stosunek średnic atomowych,
pozycje składników w układzie okresowym pierwiast.,
wartościowość (małe znaczenie).
Spotyka się następujące fazy międzymetaliczne:
elektronowe,
najgęstszego ułożenia (fazy Lavesa),
międzywęzłowe,
o strukturach złożonych.
Fazy elektronowe
Fazy te krystalzują w tych samych typach sieci i występują zawsze przy określonych stężeniach elektronowych. Ponieważ stężenia elektronowe e/a decydują powstaniu i budowie tych faz nazwano je fazami elektronowymi. Poszczególne rodzaje faz elektronowych oznaczono symbolami β, γ i ε, przy czym występują one tylko w stopach jakie tworzą z sobą składniki grupy I ze składnikami grupy II. Są to:
Grupa I: Cu, Ag, Au oraz Feγ, Co, Ni, Rh, Pt i Pd.
Grupa II: Be, Mg, Zn, Cd, Hg, Al, Ga, In, Si, Ge, Sn i Sb.
Zgodnie z empiryczną regułą podaną przez Hume-Rothery'ego fazy β pojawiają się w stopach wymienionych pierwiastków przy stężeniu elektronowym 3/2 (21/14), fazy γ przy stężeniu 21/13, a fazy ε przy stężeniu 7/4 (21/12). Pamiętając, że stężenie elektronowe jest stosunkiem ilości elektronów swobodnych do ilości atomów w stopie, wzory stechiometryczne faz elektronowych można obliczyć układając dwa równania z dwoma niewiadomymi:
X⋅wI + Y⋅wII = e,
X + Y = a,
w których X i Y są niewiadomymi liczbami atomów składników, wI i wII są natomiast wartowościowościami tych składników, podczas gdy e i a oznaczają odpowiednio liczbę elektronów wartościowości oraz liczbę atomów w wybranej fazie elektronowej. Zależnie od stężenia elektronowego fazy elektronowe mają charakterystyczne struktury krystaliczne. Faza β krystalizuje najczęściej w sieci A2, rzadziej A3 i tylko w nielicznych wypadkach w złożonej sieci typu manganu-β, która jest siecią układu regularnego, ale w jednej komórce zawiera 20 atomów. Fazy γ krystalizują w bardzo złożonej sieci przestrzennej układu regularnego, ale w komórce mieszczą się aż 52 atomy. W fazach ε występujących przy stężeniu elektronowym 7/4, charakterystyczną siecią krystaliczną jest sieć A3.
Fazy Lavesa
Czynnikiem decydującym o powstaniu tych faz jest stosunek średnic atomowych tworzących je składników, a cechą charakterystyczną - bardzo zwarte wypełnienie przestrzeni atomami. Fazom tym przypisuje się ogólny wzór stechiometryczny AB2, ale wartościowość składników nie ma tu większego znaczenia. We wzorze AB2 składnik A ma atomy o większej średnicy, a składnik B - o mniejszej, jednak występująca między nimi różnica nie może być większa niż 20-30%. Idealny stosunek promieni atomowych rA/rB wynosi 1,225, ale rzeczywiste różnice są większe.
Aczkolwiek czynnikiem warunkującym powstanie faz Lavesa jest określona różnica średnic atomowych, jednak podobnie jak w fazach elektronowych nie bez wpływu jest stężenie elektronowe. Stężenie to decyduje o typie struktury tych faz i jeśli wynosi ono 1,33-1,75, to powstają fazy typu MgCu2 (ułożenie typu ABCABC...), gdy stężenie wynosi 1,78-1,95 - fazy typu MgNi2 (ABACABAC…), natomiast dla stężenia 1,81-2,05 - fazy MgZn2 (ABAB…). Fazy Lavesa występują w stopach żarowytrzymałych, a ich drobne wydzielenia zwiększają żarowytrzymałość.
Fazy międzywęzłowe
Fazy międzywęzłowe są drugą po fazach Lavesa grupą faz międzymetalcznych, w których czynnikiem warunkującym ich powstanie jest rozmiar tworzących je atomów. Tworzą te fazy metale przejściowe z pierwiastkami o małych promieniach atomowych. Są to wodór, bor, węgiel i azot. Dzięki małym rozmiarom atomy tych pierwiastków mogą zajmować luki między atomami metali tworzących sieć krystaliczną. Hägg wykazał, że fazy te powstają wtedy gdy: rx/rm. < 0,59. Powstałe struktury są stosunkowo proste, gdyż atomy metalu przejściowego zajmują węzły jednej ze znanych sieci krystalicznych, np. A1, A3, rzadziej A2 lub prymitywnej sieci heksagonalnej. Sieć ta przeważnie nie jest identyczna z siecią w jakiej krystalizuje ten metal w stanie czystym. Luki międzyatomowe zajmują natomiast atomy niemetalu. Jeżeli np. atomy niemetalu zajmują w sieci A1 środek sześcianu i środki krawędzi to faza ma wzór stechiometryczny MX. W razie zajęcia luk znajdujących się w środku małych sześcianów, powstałych przez podzielenie komórki A1 na osiem części (luki tetraedryczne), faza międzwęłowa będzie miała wzór MX2.
Fazom międzywęzłowym można przypisać proste wzory stechiometryczne M4X, M2X, MX i MX2, przy czym najczęściej spotyka się fazy M2X i MX.
Fazy międzywęzłowe mają interesujące własności fizyczne. Węgliki i azotki są b. twarde, b. trudno topliwe i wykazują dobrą przewodność elektryczną oraz znaczną odporność chemiczną. Niektóre z nich, np. NbC, NbN, ZrN itp. są nadprzewodnikami o stosunkowo wysokiej temperaturze przejścia w stan nadprzewodności.
Fazy międzywęzłowe, a szczególnie węgliki, mają duże znaczenie praktyczne i znalazły zastosowanie jako składniki twardych spieków oraz stopów używanych do wyrobu narzędzi.
Krzepnięcie stopów w warunkach nierównowagi
Kryształy mają budowę warstwową, a największe stężenie składnika trudniej topliwego występuje w środku, podczas gdy w warstwach zewnętrznych w większym stężeniu występuje składnik łatwiej topliwy. Gdy jest zwiększona szybkość chłodzenia, może początkowo wystąpić znaczne przechłodzenie i krystalizacja dendrytyczna. Przechłodzenie zanika pod koniec krzepnięcia wskutek wydzielania się znacznych ilości utajonego ciepła krystalizacji. Rozmieszczenie składników na przekroju ziarna odzwierciedla jednak dendrytyczny charakter krystalizacji i dlatego występującą niejednorodność składu chemicznego nazwano segregacją dendrytyczną. Niejednorodność składu chemicznego kryształów pociąga za sobą niejednorodność własności, która jest zwykle niepożądana. Segregację dendrytyczną i jej następstwa można usunąć przez długotrwałe wyżarzanie odlewów w wysokich temperaturach, w których atomy wykazują znaczną ruchliwość i szybko zachodzi dyfuzja.
Drugim czynnikiem ułatwiającym ujednorodnienie jest drobne ziarno odlewu, gdyż krótkie są wtedy drogi dyfuzji.
Gdy stop krzepnie w formie metalowej (we wlewnicy), wówczas w odlewie (we wlewku) może wystąpić segregacja nazywana strefową. Krzepnięcie rozpoczyna się od ścianek formy (wlewnicy), przy których krzepną kryształy bogatsze w składnik trudniej topliwy. Wszelkie zanieczyszczenia i składnik łatwiej topliwy gromadzą się i krzepną w centralnej części odlewu (wlewka). Odległość stref odlewu (wlewka) różniących się znacznie składem chemicznym jest w tym wypadku b. duża, a usunięcie tej segregacji przez wyżarzanie ujednorodniające jest praktycznie niemal niemożliwe. W rezultacie segregację tę można stwierdzić nawet w gotowych wyrobach walcowanych lub kutych, jakie otrzymuje się z wlewków.
Jeszcze inny rodzaj segregacji może wystąpić wtedy, gdy dwa składniki stopu różnią się wyraźnie ciężarem właściwym, a w dodatku wzajemnie nie rozpuszczają się w stanie stałym lub rozpuszczalność ta jest mała. Powstające podczas krzepnięcia kryształy mają wtedy skład zbliżony do czystych składników i różnią się wyraźnie ciężarem właściwym od ciekłego stopu. Jeśli są one cięższe, to opadają na dno formy, a jeśli są lżejsze, to wypływają na powierzchnię stopu. Prowadzi to do znacznych różnic w składzie chemicznym odlewu, a segregacja taka jest grawitacyjną. Ze względu na to, że różnice składu chemicznego występują na znacznych odległościach, usunięcie segregacji jest praktycznie niemożliwe. Sprzyja segregacji powolne chłodzenie i dlatego można jej zapobiegać przez szybkie chłodzenie. Inny sposób zapobiegania segregacji polega na dodawaniu do stopu trzeciego składnika, który tworzy ze składnikami podstawowymi fazę międzymetaliczną, krystalizującą najwcześniej w postaci kryształów iglastych.
Oziębienie stopu w wodzie lub oleju powoduje całkowite zahamowanie zarodkowania, a w temperaturze normalnej otrzymuje się przesycony roztwór stały α. Szybkie chłodzenie stopu, w celu zapobieżenia wydzielaniu się z jednorodnego roztworu stałego nadmiaru jednego ze składników nazwano przesycaniem.
Ten samorzutny proces, w którego wyniku z przesyconego roztworu stałego następuje wydzielanie się faz metastabilnych lub równowagowej fazy βII nazwano starzeniem.
Rafinacja metodą strefowego topienia
Naturalną skłonność roztworów do segregacji w czasie krystalizacji od dawna wykorzystywano do oczyszczania substancji chemicznych z różnych zanieczyszczeń. Do rafinowania metali i półprzewodników Pfann opracował metodę topienia strefowego, dzięki której możliwe jest otrzymywanie tych materiałów w postaci bardzo czystej. Metoda polega na topieniu wąskiej strefy i przesuwanie jej przez całą długość wlewka. Powtarzając wielokrotnie ten zabieg zwiększamy efekt oczyszczania bez potrzeby przecinania wlewka i odrzucania bardziej zanieczyszczonej części. Po zakończeniu rafinacji odcina i odrzuca się końcową część wlewka, a pozostałą część można ujednorodnić przez przesuwanie strefy stopionego metalu na przemian z jednego końca na drugi. W rezultacie otrzymany materiał jest znacznie mniej zanieczyszczony niż materiał wyjściowy. Metodą tą można otrzymać materiały b. czyste, o czystości większej niż 99,99%.
Stale
Stale niestopowe to stale w których stężenie każdego z pierwiastków jest mniejsze od poniższych wartości (%):
Al - 0,1; B - 0,0008; Bi - 0,1; Cr - 0,3; Zr - 0,05; Co - 0,1; Si - 0,5; Lantanowce - 0,05; Mn - 1,65; Cu-0,4; Mo - 0,08; Ni - 0,3; Nb - 0,06; Pb - 0,4; Se - 0,1; Te - 0,1; Ti - 0,05; V - 0,1; W - 0,1; inne - 0,05.
Ze względu na sumaryczne stężenie pierwiastków stale stopowe dzieli się na:
niskostopowe, w których stężenie jednego pierwiastka (oprócz węgla) nie przekracza 2%, a suma pieriwastków łącznie nie przekracza 3,5%,
średniostopowe, w których stężenie jednego pierwiastka przekracza (oprócz C) 2% lecz nie przekracza 8%, lub suma pierwiastków łącznie nie przekracza 12%,
wysokostopowe, w których stężenie jednego pierwiastka przekracza 8%, a suma pierwiastków łącznie nie przekracza 55%.
Według stopnia odtlenienia można wyróżnić następujące rodzaje stali:
stal nieuspokojoną, w której przy krzepnięciu we wlewnicy dochodzi do reakcji węgla z rozpuszczonym tlenem, a tworzący się w wyniku tego tlenek węgla uchodzi ze stali wywołując jej gotowanie się,
stal półuspokojoną, w której stężenie rozpuszczonego tlenu obniżono tak, aby przy jej krzepnięciu we wlewnicy dochodziło jedynie do ograniczonej reakcji węgla z O2,
stal uspokojoną, w której przed odlaniem do wlewnicy nie dochodzi do reakcji tlenu z węglem, a stal po wlaniu do wlewnicy zachowuje się spokojnie.
Wpływ węgla na własności stali węglowych
Dominujący wpływ na strukturę i własności stali węglowych wywiera węgiel. W miarę podwyższania stężenia tego pierwiastka w stali zmniejszeniu ulega udział miękkiego i plastycznego ferrytu w strukturze stali, a zwiększeniu - udział twardego i kruchego cementytu. Z tego względu stale o większym stężeniu węgla wykazują większą twardość, wytrzymałość na rozciąganie Rm i granicę plastyczności Re. Zwiększenie stężenia węgla powoduje przy tym jednoczesne zmniejszenie własności plastycznych i ciągliwości stali, a w szczególności - wydłużenia A i przewężenia Z określanych w próbie rozciągania, oraz udarności KC określanej w próbie zginania udarowego. Stężenie węgla decyduje również o własnościach technologicznych stali. Przy większych stężeniach węgla stal cechuje się większym współczynnikiem liniowej rozszerzalności cieplnej i mniejszą przewodnością cieplną, co zwiększa naprężenia cieplne i skłonność do pęknięć w trakcie obróbki cieplnej. Zwiększone stężenie węgla pogarsza podatność stali na obróbkę plastyczną na zimno i na gorąco. Węgiel o stężeniu 0,25% zdecydowanie pogarsza również spawalność stali. Stale niskowęglowe o stężeniu węgla mniejszym od 0,25% - ze względu na dużą ciągliwość - wykazują gorszą skrawalność niż stale o strukturze perlitycznej. Stale nadeutektoidalne - ze względu na znaczny udział kruchego i twardego cementytu - charakteryzuje również obniżona skrawalność.
W stalach przedeutektoidalnych stwierdzono doświadczalnie prostą zależność między twardością HB a wytrzymałością na rozciąganie Rm:
![]()
W zakresie od 0,0 do 0,8% wzrost zawartości węgla w stali powoduje wzrost wytrzymałości na rozciąganie o ok. 500 MPa. Można zatem określić orientacyjną wytrzymałość na rozciąganie stali węglowych w zależności do zawartości węgla z prostego wzoru:
Rm =300 + (600 %C) MPa.
Wpływ domieszek i zanieczyszczeń na własności stali
Domieszki występują w stalach niestopowych jako pozostałość z procesu metalurgicznego wytapiania stali, a także w wyniku przetapiania złomu stalowego. Mangan działa korzystnie, tworząc siarczek MnS o wyższej temperaturze topnienia od siarczku żelaza. Powoduje jednak niekorzystny rozrost ziarn w czasie obróbki cieplnej i plastycznej na gorąco. Krzem powoduje korzystne obniżenie stężenia gazów w stali lanej i przeciwdziała segregacji fosforu i siarki. Fosfor i siarka stanowią zanieczyszczenia niekorzystne. Stale zawierające fosfor charakteryzują się podwyższoną temperaturą przejścia w stan kruchości, skłonnością do kruchości na niebiesko, gruboziarnistości i segregacji. Siarka tworzy siarczki MnS o temperaturze topnienia 1620°C i FeS, ciekłe już w 1000°C, co powoduje kruchość stali na gorąco, zwłaszcza podczas obróbki plastycznej. Siarka o dużej skłonności do segregacji pogarsza spawalność i wytrzymałość stali na zmęczenie. Wodór działa zdecydowanie szkodliwie, powodując powstawanie płatków śnieżnych, odwęglenia, pęcherzy gazowych oraz segregacji fosforu. Azot powoduje zmniejszenie plastyczności i kruchości na niebiesko, a także zwiększenie skłonności stali do starzenia, szczególnie po zgniocie. Tlen powoduje zmniejszenie własności wytrzymałościowych oraz plastycznych stali. Z tego względu jest b. istotne
odtlenianie kąpieli stalowej.
Wpływ wtrąceń niemetalicznych
W stalach występują również wtrącenia niemetaliczne, stanowiące nieciągłości w osnowie metalicznej stali, co niekorzystnie wpływa na własności mechaniczne stali. Wtrącenia niemetaliczne mogą być:
endogeniczne - takie jak siarczki, tlenki i krzemiany, powstające w ciekłej stali podczas procesu stalowniczego,
egzogeniczne, do których zalicza się głównie cząstki materiałów ogniotrwałych stanowiących wyłożenie pieca, rynien spustowych i kadzi, dostające się z zewnątrz do ciekłej stali.
Przemiana perlit-austenit
Perlit jest mieszaniną ferrytu i cementytu przeważnie o budowie płytkowej, która przemienia się w austenit podczas grzania w temperaturze A1 - 727°C. Przemiana ta może zajść dopiero po przegrzaniu, a więc w temperaturze Ac1 > A1, gdyż dopiero wtedy wystąpi konieczna do uruchomienia jej różnica energii swobodnych. Im różnica ta będzie większa, tym większa będzie dążność układu do przejścia w stan bardziej trwały, tj. w austenit. Przemiana perlitu w austenit jest przemianą dyfuzyjną zarówno dlatego, że rozpoczyna się od zarodków, które następnie rosną, jak i dlatego, iż jednocześnie zachodzi dyfuzja węgla, którego rozpuszczalność w austenicie jest większa niż w ferrycie. W temperaturach nieco wyższych niż A1 austenit zawiera ok. 0,77% C i prawdopodobieństwo jego powstania wewnątrz płytki ferrytu jest małe. Wymagałoby to zarodkowania jednorodnego oraz znacznej fluktuacji składu chemicznego, gdyż zarodki muszą mieć znacznie więcej węgla niż ferryt. Bardziej prawdopodobne, gdyż wymaga mniejszego nakładu energii, jest zarodkowanie niejednorodne na granicy międzyfazowej ferryt-cementyt, a poza tym w miejscu tym zarodek łatwiej może osiągnąć niezbędną zawartość węgla. Powstałe zarodki austenitu rosną następnie kosztem obu faz, z którymi się stykają, a więc ferrytu i cementytu. Powoduje to jednak inną zawartość węgla w austenicie przylegającym do ferrytu i inną w austenicie przylegającym do cementytu.
Wzrost powstałych ziarn austenitu prowadzi do ich wzajemnych zetknięć, ale nawet po całkowitym zaniku ferrytu austenit jest niejednorodny, gdyż w miejscach występowania ferrytu zawartość węgla jest mniejsza niż w miejscach, w których występował uprzednio cementyt. Okazuje się nawet, że resztki cementytu występują w austenicie jeszcze wtedy, gdy ferrytu już nie ma. W miarę wzrostu temperatury lub upływu czasu resztki te rozpuszczają się, a w austenicie zachodzi następnie dyfuzja węgla i wyrównanie jego składu chemicznego. To ujednorodnenie austenitu zachodzi stosunkowo najwolniej. Jako, że od chwili pojawienia się zarodka do powstania jednorodnego austenitu upływa pewien okres, w którym wzrasta temperatura, łatwo wywnioskować, że im większa jest szybkość grzania, tym większa jest różnica temperatur między początkem a końcem przemiany perlitu w austenit.
Podczas austenityzowania dąży się zwykle do otrzymania możliwie jednorodnego austenitu, gdyż ma to z kolei duży wpływ na procesy zachodzące podczas chłodzenia. Nie należy jednak przypuszczać, że korzystne jest z tego względu długotrwałe wygrzewanie lub znaczne podwyższanie temperatury ponad temperatury przemian, prowadzi to bowiem do niepożądanego rozrostu ziarn austenitu. Austenit bezpośrednio po powstaniu z perlitu jest drobnoziarnisty, gdyż podczas grzania powstaje duża liczba zarodków, które stają się ziarnami. Liczba tych zarodków jest tym większa, im większa jest szybkość grzania. Rozrost ziarn austenitu przebiega samorzutnie, gdyż powoduje to zmniejszenie energii swobodnej układu. Proces ten hamują nie rozpuszczone cząstki innych faz i dlatego widoczne są one zwykle na granicach ziarn. Wynika to z tego, że cząstki takie utrudniają lokalnie przemieszczanie się atomów z jednego ziarna do drugiego, a poza tym przemieszczenie się granicy poza cząstkę wymaga odtworzenia nowego jej fragmentu. Dlatego stale, w których znajduje się duża ilość b. drobnych, trudno rozpuszczalnych w austenicie cząstek wykazują małą skłonność do rozrostu ziarn. W tych stalach rozrost ziarn może się rozpocząć (często b. szybki) dopiero wtedy, gdy cząstki te rozpuszczą się. Stale o małej skłonności do rozrostu ziarn są nazywane drobnoziarnistymi, w odróżnieniu od stali gruboziarnistych, w których rozrost ziarn rozpoczyna się bezpośrednio po powstaniu austenitu.
Stale drobnoziarniste wykazują większą udarność i granicę plastyczności niż stale gruboziarniste.
W zwykłych stalach węglowych cząstkami hamującymi rozrost ziarn są rozdrobnione tlenki i azotki aluminium, jakie znajdują się w stalach odtlenianych za pomocą tego pierwiastka. W stalach stopowych rolę cząstek hamujących ruch granic ziarn spełniają trwałe, trudno rozpuszczalne węgliki i azotki, jakie tworzą niektóre pierwiastki stopowe, np. tytan, wanad itp.
Staliwo
Staliwem nazywamy stal w postaci lanej, nieobrobionej plastycznie. Odlewy staliwne uzyskują ostateczny kształt po skrzepnięciu w formach, przeważnie piaskowych i podlegać mogą różnym operacjom technologicznym z wyjątkiem obróbki plastycznej.
Własności mechaniczne staliw w porównaniu ze stalami obrobionymi plastycznie o takiej samej zawartości węgla są gorsze tylko w zakresie własności charakteryzujących ciągliwość i plastyczność, natomiast wytrzymałość na rozciąganie i twardość jest niemal taka sama. Gorsze własności plastyczne oraz mniejsza udarność staliwa są następstwem zachowania pierwotnej struktury, którą można usunąć tylko przez obróbkę plastyczną. Własności mechaniczne i spawalność staliw są wyższe niż żeliw. W porównaniu z żeliwami staliwa mają jednak gorszą lejność, są wrażliwe na działanie karbu, posiadają skłonność do pęcherzy. Wadą staliw jest także mały uzysk.
Składnikami strukturalnymi występującymi w staliwie węglowym są ferryt i perlit. W zależności od sposobu i szybkości chłodzenia odlewu w stanie surowym w staliwie węglowym może wystąpić tzw. struktura globulityczna o okrągłych ziarnach lub struktura Widmannstättena. Struktura Widmanstättena charakteryzuje się iglastą budową ferrytu osnowie perlitu i ma niekorzystny wpływ na własności mechaniczne staliwa. W celu usunięcia niekorzystnych struktur pierwotnych, niejednorodności składu chemicznego, a także naprężeń odlewniczych, odlewy staliwne poddaje się obróbce cieplnej, głównie wyżarzaniu ujednorodniającemu lub normalizującemu, a także wyżarzaniu odprężającemu. Odlewy staliwne można również hartować, a także obrabiać cieplno-chemicznie, stosując zasady podobne jak przy obróbce cieplnej elementów stalowych o zbliżonym składzie chemicznym.
Żeliwa
Żeliwami nazywamy stopy żelaza z węglem zawierające ponad 2% węgla. Od tej zwartości węgla (2%) pojawia się w tych stopach nie spotykany w stalach składnik mikrostruktury - ledeburyt, w którego skład wchodzi cementyt. Węgiel w żeliwie może występować zarówno w postaci wiązanej (cementyt) jak i wolnej (grafit). W zależności od postaci w jakiej występuje węgiel rozróżnia się żeliwa: szare, w których węgiel występuje w postaci grafitu; białe, w których węgiel jest związany w cementycie, połowiczne, w których występuje zarówno grafit, jak i cementyt (powyżej 0,8% ogółu węgla w żeliwie).
Strukturę żeliwa stanowi osnowa metaliczna, którą może być ferryt, perlit lub ich mieszaniny, ewentualnie z cementytem i wtrąceniami niemetalicznymi a także grafit o różnej wielkości i kształcie. Grafit jest b. miękki, a jego wytrzymałość jest bliska zeru. Grafit może się tworzyć przy krzepnięciu z cieczy jako płatkowy na skutek przemiany eutektoidalnej austenitu lub w wyniku rozpadu cementytu w żeliwie białym poddanym długotrwałemu wyżarzaniu w temperaturze nieznacznie niższej od solidusu. W stopach eutektycznych grafit wydziela się z cieczy w postaci b. drobnych płatków w eutektyce grafitowej. Grube płatki grafitu pierwotnego wydzielają się w czasie krzepnięcia żeliw nadeutektycznych.
Grafit powoduje zmniejszenie własności wytrzymałościowych żeliwa i zmianę niektórych innych własności, a szczególnie: działa jako karb wewnętrzny, stanowiąc nieciągłości w metalu; zmniejsza skurcz odlewniczy; polepsza skrawalność; zwiększa własności ślizgowe; sprzyja tłumieniu drgań.
Według Polskich Norm żeliwa dzielimy na:
szare - PN-92/H03101
sferoidalne (grafit w postaci kulek) - PN-92/H03123
ciągliwe (grafit kłaczkowy) - PN-92/H83221
stopowe - PN-92/H83144
Żeliwa szare dzielimy na:
1. zwykłe - charakteryzuje się niską wytrzymałością, dobrą skrawalnością oraz małą odpornością na zużycie ścierne. Twardość i wytrzymałość żeliwa szarego zwiększa się w miarę zwiększania udziału perlitu w strukturze. Żeliwa zwykłe dzielmy na: ferrytyczne 100, ferrytyczno-perlityczne 150, perlityczne 200
2. modyfikowane - bezpośrednio przed odlaniem do kąpieli metalowej o temp. ok. 1400°C, w rynnie spustowej lub kadzi, dodaje się ok. 0,1-0,5% sproszkowanego modyfikatora, najczęściej żelazo-krzemu, wapnio-krzemu lub aluminium. Żeliwa te są perlityczne (250, 300, 350). Żeliwo sferoidalne wykazuje b. dobre własności - zarówno wytrzymałościowe jak i plastyczne. Uzyskuje się je w wyniku modyfikowania żeliwa o tendencji krzepnięcia jako szare, lecz o b. małym stężeniu siarki i fosforu. Jako modyfikatorów używa się ceru lub magnezu. W wyniku tego zabiegu technologicznego grafit występuje w tych żeliwach w postaci kulistej. Żeliwa sferoidalne można podzielić na: ferrytyczne (350-22, 400-18, 400-15, 450-10), ferrytyczno-perlityczne (508-7), perlityczno-ferrytyczne (600-3), perlityczne (700-2), po obróbce cieplnej (800-2, 900-2).
Żeliwo ciągliwe jest otrzymywane z żeliwa białego w wyniku wyżarzania grafityzującego. Podczas tej operacji cementyt ulega rozpadowi o wydziela się tzw. węgiel żarzenia w postaci kłaczkowatych skupień. Żeliwo ciągliwe charakteryzuje się dobrymi własnościami wytrzymałościowymi i plastycznymi. W zależności od parametrów procesu technologicznego żeliwo ciągliwe można podzielić na: białe (W), czarne (B), perlityczne (P).
Przemiana perlityczna (austenit-perlit)
Przemiana perlityczna zachodzi po ochłodzeniu austenitu nieznacznie Ar1. W jej wyniku z austenitu powstaje mieszanina eutektoidalna złożona z płytek ferrytu i cementytu zawana perlitem. Siłą pędną przemiany perlitycznej jest różnica energii swobodnej austenitu i mieszaniny ferrytu i cementytu. Przemiana perlityczna jest przemianą dyfuzyjną związaną z przegrupowaniem atomów węgla i zachodząca przez zarodkowanie oraz rozrost zarodków. Zarodkowanie perlitu odbywa się heterogenicznie, czyli w sposób uprzywilejowany, na cząstkach cementytu, płytkach ferrytu, a w jednorodnym austenicie - na granicach ziarn tej fazy. Wzrost płytki cementytu bogatej w węgiel powoduje znaczne zmniejszenie stężenia węgla w austenicie do wartości Cα umożliwiającej powstanie płytki ferrytu. W wyniku ograniczonej rozpuszczalności węgla w ferrycie jego nadmiar wzbogaca austenit w pobliżu utworzonej płytki ferrytu, umożliwiając tworzenie kolejnej płytki cementytu. Proces kolejnego dobudowywania płytek trwa aż do wyczerpania się austenitu. Przemiana perlityczna przebiega również przez wzrost czołowy utworzonych wcześniej płytek.
Utworzone kolonie perlitu mają kształt kulisty, gdyż szybkości dobudowania nowych płytek i ich rozrostu czołowego są jednakowe. W wyniku dyfuzji węgla do płytek cementytu następuje zróżnicowanie składu chemicznego austenitu przed frontem przemiany. Grubość płytek cementytu jest ok. siedmiokrotnie mniejsza od grubości płytek ferrytu. Przy stałej temperaturze, grubości płytek każdej z faz perlitu są prawie stałe i nie zależą od wielkości i jednorodności austenitu, z którego powstają. Obniżaniu temperatury przemiany towarzyszy zmniejszanie się odległości między płytkami perlitu.
Szybkość przemiany perlitycznej zależy od szybkości zarodkowania i szybkości wzrostu. Funkcja szybkości zarodkowania perlitu od czasu jest nieliniowa i wykazuje okres inkubacyjny. Przy małym przechłodzeniu szybkość zarodkowania jest b. mała, a szybkość rozrostu - duża. W związku z tym w opisanych warunkach tworzą się nieliczne jedynie kolonie perlitu o b. dużych wymiarach.
Przemiana bainityczna
Przemiana bainityczna łączy w sobie cechy przemiany bezdyfuzyjnej i dyfuzyjnego przemieszczania węgla. Zachodzi przy przechłodzeniu stali do temperatury w zakresie ok. 450-200°C. W wyniku przemiany powstaje bainit, będący mieszaniną ferrytu przesyconego węglem i dyspersyjnych węglików. Zarodkowanie bainitu rozpoczyna dyfuzyjne przemieszczanie węgla w austenicie do granic ziarn i dyslokacji. Zarodkami przemiany są miejsca ubogie w węgiel, utworzone w pobliżu granic ziarn i dyslokacji. Zróżnicowanie składu chemicznego austenitu wymaga pewnego czasu inkubacji, niezbędnego do zainicjowania przemiany bainitycznej. W obszarach przechłodzonego austenitu o małym stężeniu węgla i wysokiej temperaturze Ms zachodzi bezdyfuzyjna przemiana martenztenzytyczna. W obszarach austenitu o dużym stężeniu węgla następuje jednocześnie dyfuzyjny proces wydzielania b. drobnych cząstek cementytu o dużej dyspersji. W wyniku tego tworzą się nowe obszary niskowęglowego austenitu, ulegające następnie bezdyfuzyjnej przemianie martenzytyczej, podczas dalszego chłodzenia następuje wydzielanie cementytu oraz węglika ε, a osnowa staje się ferrytem przesyconym węglem. Rozrost bainitu jest kontrolowany szybkością dyfuzji węgla w austenicie, a nie szybkością przemiany martenzytycznej.
W zależności od temperatury przechłodzenia rozróżnia się: bainit górny i bainit dolny. Bainit górny składa się z ziarn przesyconego węglem ferrytu o nieregularnych kształtach z nieregularnymi wydzieleniami węglików oraz z austenitu szczątkowego. Bainit dolny składa się z przesyconego węglem ferrytu o postaci listwowej, zbliżonego do martenzytu, płytkowych węglików w równoległych rzędach, ściśle zorientowanych względem listew ferrytu oraz z austenitu szczątkowego.
Przemiana martenzytyczna
Przemiana martenzytyczna jest przemianą bezdyfuzyjną i zachodzi przy dużym przechłodzeniu austenitu do temperatury M.s, początku tej przemiany, w przypadku dużej liczby stali mniejszej nawet od ok. 200°C, przy chłodzeniu z szybkością większą od krytycznej vk. W wyniku tej przemiany powstaje martenzyt, czyli przesycony roztwór węgla w żelazie α. Przemiana martenzytyczna zachodzi pod warunkiem ciągłego obniżania temperatury w zakresie od temperatury początku przemiany Ms, do temperatury M.f - jej końca. Wartości temperatury M.s i M.f zależą od skladu chemicznego austenitu i obniżają się ze zwiększeniem stężenia węgla w austenicie oraz wszystkich niemal dodatków stopowych z wyjątkiem Al i Co.
Przemiana martenzytyczna rozpoczyna się od utworzenia w austenicie embrionów, tj. zarodków pierwotnych, którymi mogą być błędy ułożenia, źródła Franka-Reada, defekty sieciowe w austenicie, w tym pętle dyslokacji i równolegle ułożone dyslokacje śrubowe. Zarodkami przemiany martenztycznej są embriony, które przekraczają wielkość krytyczną. Z upływem czasu przemiany następuje autokataliza polegająca na przyspieszeniu zarodkowania. Tworzące się zarodki odkształcają bowiem otaczającą osnowę i sprzyjają powstawaniu nowych zarodków.
Podczas przemiany martenzytycznej następuje skoordynowane przemieszczenie atomów bez zmiany sąsiadujących atomów dziedziczonych z austenitu. Wszystkie atomy podlegają małym przemieszczeniom o ułamek odległości międzyatomowej względem atomów sąsiednich. W wyniku tego następuje charakterystyczny relief powierzchni austenitu, związany z nachyleniem powierzchni odpowiadającym każdej płytce lub listwie martenzytu. Granice ziarn martenzytu są położone wzdłuż nieodkształconej i nie ulegającej obrotowi płaszczyzny austenitu, zwanej płaszczyzną habitus. Płaszczyzna habitus jest zwykle zbliżona do {225}γ - w przypadku powstawania martenzytu listwowego lub {259}γ - w przypadku tworzenia się martenzytu płytkowego.
Przemiana martenzytyczna może przebiegać:
atermicznie przez tworzenie się kryształów martenzytu z austenitu w ciągu 10-7 s bez aktywacji cieplnej,
wybuchowo w kilku wybuchach zachodzących wyłącznie poniżej 0°C,
izotermicznie, zwłaszcza gdy temperatura M.s jest wysoka, a przemiana jest funkcją czasu i zależy od małej szybkości zarodkowania aktywowanego cieplnie. Przemianę martenzytyczną ułatwiają naprężenia rozciągające i odkształcenie plastyczne. Temperatura początku przemiany martenzytycznej austenitu odkształconego plastycznie w temperaturze niższej od temperatury rekrystalizacji jest oznaczana M.d i jest wyższa od temperatury M.s stali nieodkształconej plastycznie przed rozpoczęciem przemiany.
W wyniku przemiany martenzytycznej w stalach mogą utworzyć się dwa rodzaje martnzytu: listwowy i płytkowy.
Martenzyt listwowy powstaje we wszystkich niemal stopach żelaza z pierwiastkami stopowymi i cechuje się dużą gęstością dyslokacji wewnątrz kryształów wykazujących najczęściej komórkową podstrukturę dyslokacyjną. Pojedynczy kryształ ma kształt listwy o szerokości 0,1-3 μm i w stosunku wymiarów ok. 1:7:30. Listwy tworzące się w kierunku <111〉α martenzytu układają się równolegle względem siebie tworząc tzw. pakiety. Sąsiednie listwy wykazują granice niskokątowe lub bliźniacze, natomiast pakiety między sobą tworzą granice szerokokątowe, nierzadko o wzajemnej orientacji bliźniaczej. Martenzyt płytkowy powstaje w nielicznych stopach żelaza w ściśle określonych zakresach stężeń pierwiastków stopowych. Kryształy martenzytu mają kształt płytek zbliżonych do soczewek o powierzchni w różnym stopniu nieregularnej w zależności od stopnia zbliźniaczenia. Wielkość płytek jest zróżnicowana, gdyż powstają niezależnie. Wzrost płytki kończy się na wadach struktury austenitu lub na sąsiednich, wcześniej utworzonych płytkach martenzytu. Płytki martenzytu mogą być zbliźniaczone całkowicie lub częściowo. W martenzycie częściowo zbliźniaczonym środkowa część płytki powstaje w postaci drobnych, równoległych bliźniaków przemiany zwanych midribem. Zewnętrzna część płytki tworzy się w wyniku poślizgu.
Objętość właściwa martenzytu jest o ok. 3% większa od objętości właściwej austenitu, w wyniku czego w nieprzemienionym austenicie powstają silne naprężenia ściskające, hamujące lub całkowicie zatrzymujące dalszą przemianę. Przemiana nie przebiega więc w całej objętości i dlatego po jej zakończeniu w strukturze stali pozostaje pewna ilość austenitu, zwanego austenitem szczątkowym. W wyniku stabilizacji, w przypadku wytrzymania izotermicznego podczas chłodzenia w zakresie między temperaturami Ms i Mf, udział austenitu szczątkowego w stali zwiększa się. Udział austenitu szczątkowego wzrasta również wraz ze zwiększeniem stężenia węgla w stali.
Wyżarzanie
Wyżarzanie to operacja zwykłej obróbki cieplnej polegająca na nagrzaniu stali do określonej temperatury, wygrzaniu w tej temperaturze i studzeniu w celu uzyskania struktur zbliżonych do stanu równowagi.
Wyżarzanie ujednorodniające polega na nagrzaniu stali do temperatury 1050-1200°C o ok. 100-200°C niższej od temperatury solidusu, wygrzaniu długotrwałym w tym zakresie temperatury i następnym studzeniu. Celem tej operacji, stosowanej głównie dla wlewków stalowych, jest ograniczenie niejednorodności składu chemicznego, spowodowanej mikrosegregacją, a w części także likwidacją. Wyżarzanie rekrystalizujące polega na nagrzaniu metalu uprzednio odkształconego plastycznie na zimno do temperatury wyższej od temperatury rekrystalizacji, wygrzaniu w tej temperaturze i chłodzeniu z dowolną szybkością. Wyżarzanie rekrystalizujące, często stosowane jako międzyoperacyjne podczas walcowania lub ciągnienia metali na zimno, usuwa umocnienie zgniotowe, powodując zmniejszenie twardości i wytrzymałości oraz zwiększenie ciągliwości metalu, co umożliwia dalszą obróbkę plastyczną na zimno. Wyżarzanie odprężające polega na nagrzaniu stali do temperatury niżej od Ac1, wygrzaniu w tej temperaturze i następnym powolnym studzeniu. Celem tej operacji jest usunięcie naprężeń odpowiednio odlewniczych, spawalniczych, cieplnych oraz spowodowanych obróbką plastyczną. Wyżarzanie odprężające prawie nie wiąże się z wprowadzaniem zmian strukturalnych. Zakres temperatury i czasu wyżarzania odprężającego jest szeroki; parametry te zależą od rodzaju materiału oraz przyczyn wywołujących naprężenia. W przypadku odlewów staliwnych temperatura może wynosić ok. 650°C. W temperaturze do 150°C jest wykonywane odprężanie stabilizujące, które ma na celu zapewnienie niezmienności wymiarowej oraz zmniejszenie naprężeń własnych. Odprężanie samorzutne - tzw. sezonowanie - zachodzi w temperaturze pokojowej, w czasie wynoszącym kilka lub kilkanaście miesięcy, a niekiedy nawet kilka lat. Wyżarzanie normalizujące polega na nagrzaniu stali do temperatury o 30-50°C wyższej od Ac3, wygrzaniu w tej temperaturze i następnym studzeniu w spokojnym powietrzu. Operacja ma na celu uzyskanie jednorodnej struktury drobnoziarnistej, a przez to polepszenie własności mechanicznych stali. Jest stosowana do stali niestopowych konstrukcyjnych i staliwa - często przed dalszą obróbką cieplną - w celu ujednolicenia struktury. Wyżarzanie zupełne, stosowane do stali stopwych, polega na nagrzaniu stali do temperatury o 30-50°C wyżej od Ac3, ACcm (linia GSE), wygrzaniu w tej temperaturze i następnym b.wolnym chłodzeniu w zakresie temperatury między Ac3 i ACcm, a Ac1. Dalsze studzenie może odbywać się w powietrzu. Wyżarzanie izotermiczne, będące odmianą wyżarzania zupełnego, polega na nagrzaniu stali do temperatury o 30-50°C wyżej od Ac1, wygrzanu w tej temperaturze, szybkim ochłodzeniu do temperatury nieco niższej od Ac1, wytrzymaniu izotermicznym w tej temperaturze aż do zakończenia przemiany perlitycznej i następnym chłodzeniu w powietrzu. Wyżarzanie izotermiczne jest stosowane w przypadku obróbki cieplnej stali stopowych, które po wyżarzaniu zupełnym wykazują zbyt dużą twardość. Odmianą wyżarzania izotermicznego jest patentowanie drutów, polegające na wygrzewaniu w temperaturze 900-1100°C, chłodzeniu izotermicznym w 500-550°C i następnie obróbce plastycznej na zimno. Wyżarzanie sferoidyzujące (zmiękczanie), polega na nagrzaniu stali do temperatury zbliżonej do Ac1, wygrzaniu w tej temperaturze, b. wolnym chłodzeniu do temperatury ok. 600°C i następnie dowolnym chłodzeniu do temperatury otoczena. Wygrzewanie może się odbywać w temperaturze nieco wyższej lub nieco niższej od Ac1 (przy zmianach temperatury w zakresie ± 20°C wokół Ac1), a także z wytrzymaniem izotermicznym poniżej temperatury Ac1, po uprzednim krótkim wygrzewaniu w temperaturze wyższej od Ac1. W wyniku operacji wyżarzania sferoidyzującego strukturę stali stanowi cementyt kulkowy, tzw. sferoidyt, w osnowie ferrytu. Struktura taka zapewnia niewielką twardość, dobrą skrawalność oraz dobrą podatność na odkształcenie plastyczne w czasie obróbki plastycznej na zimno. Jest także prawidłową strukturą stali nadeutektoidalnych przed następnym hartowaniem.
Odpuszczanie
Odpuszczanie polega na nagrzaniu stali zahartowanej do temperatury niższej od Ac1, wygrzaniu w tej temperaturze i ochłodzeniu do temperatury pokojowej. W zależności od temperatury odpuszczanie może być: niskie, średnie, wysokie. Odpuszczanie niskie jest wykonywane w temperaturze 150-200°C i stosowane głównie do narzędzi, sprężyn, sprawdzianów. Celem tej operacji jest usunięcie naprężeń hartowniczych z zachowaniem dużej twardości, wytrzymałości i odporności na ścieranie. Odpuszczanie średnie, odbywające się w temperaturze 250-500°C, jest stosowane do sprężyn, resorów, matryc i innych części maszyn. W wyniku tej operacji twardość stali ulega wprawdzie niewielkiemu zmniejszeniu, lecz zostają zachowane duża wytrzymałość i sprężystość. Odpuszczanie wysokie, wykonywane w temperaturze wyższej od 500°C, lecz niższej od Ac1, ma na celu osiągnięcie możliwie dobrych własności plastycznch stali. Stosowane jest między innymi dla elementów maszyn, od których wymagana jest wysoka granica plastyczności Re.
Struktury stali odpuszczonych. W wyniku odpuszczania niskiego uzyskuje się strukturę martenzytu niskoodpuszczonego, który w stalach węglowych jest mieszaniną martenzytu tetragonalnego z dyspersyjnymi węglikami typu ε oraz austenitu szczątkowego. Martenzyt średnioodpuszczony cechuje się małym odkształceniem tetragonalnym oraz dyspersyjnymi wydzieleniami cementytu. Martenzyt wysokoodpuszczony nie jest przesycony węglem i charakteryzuje się b. małą gęstością dyslokacji, stając się podobny do ferrytu. Występują w nim natomiast wydzielenia cementytu, w dużej mierze skoagulowane. W stalach stopowych wydzielają się węgliki stopowe, ulegające również koagulacji w wyższych temperaturach odpuszczania. Po wysokim odpuszczaniu w strukturze stali stopowych nie ma już austenitu szczątkowego, lub jego udział jest niewielki.
Hartowanie i niskie odpuszczanie wykonane łącznie są utwardzaniem cieplnym. Hartowanie wysokie i odpuszczanie stanowią łącznie tzw. ulepszanie cieplne. Miarą ulepszania cieplnego jest stosunek Re : Rm.
Przemiany w stali podczas odpuszczania
Podczas wygrzewania w temperaturze niższej od A1 i studzenia stali uprzednio zahartowanej zachodzą w niej liczne przemiany fazowe. Należą do nich: rozkład martenzytu, przemiana austenitu szczątkowego w fazę α, wydzielanie węglika ε i cementytu, a w stalach stopowych również innych węglików, koagulacja węglików wydzielonych we wcześniejszych stadiach odpuszczania. Siłą pędną przemian podczas odpuszczania jest różnica energii swobodnej między metastabilnym martenzytem a mieszaniną faz złożoną z ferrytu i węglików. W zależności od warunków odpuszczania, głównie zaś temperatury, można wyróżnić kilka stadiów tego procesu, w których przeważa jedna z przemian. Śledzenie przemian podczas odpuszczania umożliwiają badania dylatometryczne. Pierwsze stadium odpuszczania stali węglowych - w zakresie ok. 80-200°C - jest związane z rozkładem martenzytu i wydzielaniem w nim węglika ε są koherentne z osnową. Wydzielanie węglika ε wywołuje zmniejszenie stężenia węgla w martenzycie. Zmniejszenie stopnia przesycenia martenzytu węglem powoduje z kolei zmniejszenie tetragonalności martenzytu i tworzenie się martenzytu o sieci regularnej, zwanego martenzytem odpuszczonym.
Odpuszczanie w stadium drugim - w zakresie ok. 200-
300°C - wywołuje przemianę austenitu szczątkowego w martenzyt odpuszczony według mechanizmu zbliżonego do przemiany bainitycznej.
Trzecie stadium odpuszczania - w zakresie ok. 300-400°C - polega na rozpuszczaniu się węglika ε w osnowie i niezależnym wydzielaniu się cementytu.
W kolejnym stadium odpuszczania zachodzi koagulacja cząstek tej fazy, związana ze zwiększaniem się niektórych cząstek cementytu i rozpuszczaniem cząstek małych. Towarzyszy temu sferoidyzacja, polegająca na przyjmowaniu przez cząstki cementytu kształtu kulistego. W wyniku tego uzyskuje się strukturę martenzytu wysokoodpuszczonego (zwanego niekiedy sorbitem), złożonego z b. drobnych kulistych cząstek cementytu w osnowie ferrytycznej.
Odpuszczanie w temperaturze wyższej od ok. 600°C powoduje koagulację cementytu, której towarzyszą procesy zdrowienia i rekrystalizacji osnowy. W wyniku tego uzyskuje się strukturę sferoidytu, tj. cementytu kulkowego w osnowie ferrytu o twardości mniejszej od 300 HB.
Naprężenia własne i wady hartownicze
W wyniku procesów technologicznych, głównie obróbki cieplnej, obróbki plastycznej, odlewania lub spawania, w elementach metalowych mogą występować naprężenia własne, niezależnie od przyłożonych naprężeń zewnętrznych. W zależności od zasięgu działania naprężenia własne można podzielić na: naprężenia I rodzaju, obejmujące cały przedmiot i powodujące głównie zmiany wymiarów, a także odkształcenia lub pęknięcia związane z obróbką cieplną, naprężenia II rodzaju, występujące w ziarnach, napr. III rodzaju, istniejące w sieci przestrzennej kryształu. Rozkład i wartość naprężeń własnych zależą m.in. od wielkości i kształtu przedmiotu, szybkości chłodzenia, gatunku, struktury, hartowności, przewodnictwa cieplnego i współczynników rozszerzalności temperaturowej stali.
Ze względu na przyczyny powstawania naprężenia własne i wywołane przez nie odkształcenia możemy podzielić na:
naprężenia cieplne, powodowane nierównomiernymi dylatacyjnymi zmianami wymiarów w wyniku gradientu temperatury podczas grzania i chłodzenia oraz naprężenia strukturalne, wywoływane przemianami fazowymi i związanymi z tym zmianami objętości właściwej faz.
Pęknięcia hartownicze
Pęknięcia powstają w wyniku hartowania wtedy, gdy naprężenia rozciągające są większe od wytrzymałości. Pęknięcia hartownicze powstają w temperaturze niższej od M.s, głównie w czasie chłodzenia. Skłonność do pęknięć zwiększa się wraz ze wzrostem stężenia węgla w stali, podwyższeniem temperatury hartowania i zwiększeniem szybkości chłodzenia w zakresie między temperaturami początku i końca przemiany martenzytycznej. Przyczyną pęknięć hartowniczych może być również występowanie karbów, gwałtownej zmiany przekroju lub miejscowych zagłębień i występów. Pęknięcia należą do wad obróbki cieplnej, które nie mogą być usunięte. Przeciwdziałanie tworzeniu tych wad polega na unikaniu karbów, hartowaniu z możliwie najniższej temperatury, łagodnym chłodzeniu w zakresie między temperaturami początku i końca przemiany martenzytycznej.
Hartowanie powierzchniowe
Hartowanie powierzchniowe polega na szybkim nagrzaniu warstwy wierzchniej przedmiotu do temperatury hartowania i następnie szybkim chłodzeniu. Hartowanie powierzchniowe umożliwia ograniczenie nagrzewania do cienkiej warstwy powierzchniowej i to jedynie w miejscach, które powinny być obrobione cieplnie. Nie wywołuje więc dużych naprężeń i odkształceń cieplnych. Hartowanie powierzchniowe umożliwia automatyzację i mechanzację procesów technologicznych obróbki cieplnej. W zależności od sposobu nagrzewania można wyróżnić następujące rodzaje hartowania powierzchniowego: indukcyjne, płomieniowe, kąpielowe, kontaktowe, elektrolityczne. Podczas hartowania indukcyjnego grzanie odbywa się prądem elektrycznym indukowanym w obrabianym cieplnie przedmiocie przez zmienne pole magnetyczne. Pole magnetyczne jest wytwarzane przez wzbudnik I, tj. cewkę zasilaną prądem wytwarzanym przez generatory prądu zmiennego. Chłodzenie może być wykonywane przez zanurzenie przedmiotu w kąpieli chłodzącej lub natrysk cieczy chłodzącej bezpośrednio we wzbudniku. Indukcyjnie są hartowane zwykle wałki, koła zębate, zawory, rolki, sworznie, prowadnice i inne przedmioty, często b. drobne.
Obróbka cieplno-chemiczna stali
Obróbka cieplno-chemiczna jest dziedziną obróbki cieplnej obejmującą zespół operacji i zabiegów umożliwiających zmianę składu chemicznego i struktury warstwy powierzchniowej stopu (a przez to zmianę własności obrabianych elementów) w wyniku zmian temperatury i chemicznego oddziaływania ośrodka. Obróbka cieplno-chemiczna polega zatem na zamierzonej dyfuzyjnej zmianie składu chemicznego warstwy powierzchniowej elementów metalowych w celu uzyskania odpowiednich ich własności użytkowych. Celem obróbki cieplno-chem. jest wytworzenie warstw powierzchniowych o zwiększonej odporności na ścieranie i zużycie trybologiczne, o zwiększonej odporności korozyjnej i erozyjnej, często zwiększenie odporności elementów na zmęczenie lub poprawa niektórych własności fizycznych powierzchni. Podczas dowolnego sposobu obróbki cieplno-chemicznej przebiegają jednocześnie trzy procesy fizykochemiczne:
1) powstawanie aktywnych atomów w środowisku otaczającym przdmiot obrabiany, 2) adsorpcja zachodząca na styku przedmiotu ze środowiskiem, 3) dyfuzja zachodząca w materiale obrabianego przedmiotu.
Aktywne atomy jednego lub kilku pierwiastków nasyconych powstają w wyniku reakcji dysocjacji, wymiany lub redukcji. Ten proces składowy obróbki cieplno-chemicznej jako jedyny może przebiegać w sposób odizolowany od pozostałych. Umożliwia to uzyskanie, np. aktywnych gazów, w specjalnych generatorach, niezależnie od reakcji przebiegających w metalu podczas obróbki cieplno-chemicznej.
Adsorpcja polega na osadzaniu się wolnych atomów, z fazy gazowej lub ciekłej, na granicy fazy stałej w postaci warstewki o grubości jednego atomu. Przyczyną adsorpcji są siły przyciągania atomów ośrodka gazowego lub ciekłego przez nienasycone rdzenie atomów występujące na powierzchni metalu. Zjawisko to jest nazywane adsorpcją fizyczną. Adsorpcja chemiczna jest związana z tworzeniem warstewki fazy międzymetalicznej złożonej z atomów metalu i ośrodka adsorbowanego.
Dyfuzja jest aktywowanym cieplnie procesem zachodzącym wskutek ruchu atomów w sieci przestrzennej metalu w kierunku wyrównania stężenia składników. Warunkiem przebiegu dyfuzji jest rozpuszczalność w stanie stałym pierwiastka nasycającego w metalu osnowy. Ogólnie można stwierdzić, że procesy dyfuzji są zależne od temperatury, czasu i gradientu stężenia dyfundujących pierwiastków. Od czynników tych zależy zatem grubość i struktura warstw powierzchniowych otrzymanych w wyniku obróbki cieplno-chemicznej. Prawa Ficka opisują następujące zależności: Pierwsze prawo Ficka określa strumień dyfuzji J składnika nasycającego:
![]()
gdzie: D - współczynnik dyfuzji, dc/dx - gradient stężenia pierwiastka dfundującego, Q - energia aktywacji dyfuzji, k - stała Boltzmanna, T - temperat. w skali bezwzględnej, D0 - stała, zależna od struktury krystalicznej metali.
Drugie prawo Ficka opisuje zależność rozkładu stężenia składnika od czasu (τ - czas procesu):
![]()
Dyfuzja w roztworach różnowęzłowych przebiega głównie za pośrednictwem mechanizmu wakansowego, natomiast w roztworach międzywęzłowych - międzywęzłowego. Mechanizm międzywęzłowy jest charakterystyczny dla dyfuzji m.in. C, N, B w stopach żelaza. W zależności od energii aktywacji różne są drogi łatwej dyfuzji, która przebiega: wzdłuż powierzchni metalu (najłatwiej), wzdłuż granic ziarn (trudniej), wewnątrz ziarn (najtrudniej).
Nawęglanie
Nawęglanie polega na nasycanu warstwy powierzchniowej stali w węgiel podczas wygrzewania obrabianego przedmiotu w ciągu określonego czasu w ośrodku zawierającym węgiel atomowy. Nawęglanie odbywa się w temperaturze 900-950°C. O grubości warstwy nawęglonej, która zwykle osiąga 0,5-2 mm, decyduje czas nawęglania, który dobiera się tak, aby skład fazowy warstwy powierzchniowej odpowiadał strukturze stali eutekoidalnej
Istnieją następujące metody nawęglania: w ośrodkach stałych, w roztopionych solach, gazowe, w złożach fluidalnych, próżniowe oraz jonizacyjne.
Twardość stali węglowej i chłodzonej w powietrzu wynosi ok. 250-300 HB, a jej własności mechaniczne są stosunkowo niskie ze względu na rozrost ziarn zachodzący w czasie procesu. W celu poprawienia własności stal nawęgloną poddaje się dalszej obróbce cieplnej; w szczególności dąży się do: 1) otrzymania struktury drobnolistwowego martenzytu z węglikami w postaci ziarnistej w warstwie powierzchniowej, 2) zwiększenia twardości stali na powierzchni do ok. 60 HRC, 3) zapewnienia znacznej ciągliwości, odporności na dynamiczne działanie obciążeń oraz wymaganych własności wytrzymałściowych w nie nawęglonym rdzeniu.
Obecnie stosuje się cztery sposoby obróbki cieplnej przedmiotów nawęglonych:
hartowanie bezpośrednie,
hartowanie bezpośrednie z ochładzaniem (zmniejsza wrażliwość powstawania pęknięć),
hartowanie pojedyncze z normalizowaniem (ochładzanie w spokojnym powietrzu),
hartowanie podwójne (od rdzenia do nawierzchni).
Nawęglanie z następnym hartowaniem i niskim odpuszczaniem zapewnia dużą twardość powierzchni obrobionych elementów, dużą odporność na ścieranie i naciski powierzchniowe, znaczną wytrzymałość zmęczeniową. Rdzeń stali po takich operacjach obróbki cieplno-chemicznej i cieplnej wykazuje dużą ciągliwość, sprężystość i odporność na dynamiczne działanie obciążeń. W celu zapewnienia wymienionych własności nawęglanie jest stosowane m.in. w procesach technologicznych kół zębatych i z wielowypustami, wałków rozrządu, sworzni tłokowych i kulistych, pierścieni i wałków łożysk tocznych o dużych wymiarach.
Azotowanie
Azotowanie polega na nasycanu warstwy powierzchniowej stali azotem podczas wygrzewania obrabanego przedmiotu przez określony czas w ośrodku zawierającym wolne atomy azotu. Operacja ta jest wykonywana w temperaturze niższej od Ac1. Azotowanie może być: krótkookresowe, gdy czas jego trwania wynosi od kilkunastu minut do kilku godzin, długookresowe - gdy wynosi kilkadziesiąt godzin. Przy stałej temperaturze azotowania struktura warstw wierzchnich obrabianej stali zależy od czasu tej operacji. Azotowanie przyczynia się do: zwiększenia twardości i odporności na ścieranie, podwyższenia zmęczeniowej wytrzymałości powierzchni, zwiększenia odporności na korozję. Istnieją następujące metody azotowania: w proszkach, gazowe, jonizacyjne i w złożach fluidalnych. Azotowanie jest ostatnią operacją w procesie technologicznym. Dlatego azotowaniu poddaje się przedmioty zahartowane i odpuszczone, najkorzystniej w temperaturze wyższej od temperatury azotowania. W niektórych przypadkach operacja odpuszczania może być połączona z operacją azotowania. Ze względu na małą grubość warstwy azotowanej przedmioty azotowane nie mogą być poddane szlifowaniu.
Azotowanie jest stosowane do elementów ze stali węglowych i stopowych, konstrukcyjnych i narzędziowych narażonych podczas pracy na zużycie ścierne, do elementów narażonych na korozję w środowisku wodnym lub wilgotnej atmosfery, a także do narzędzi skrawających. Azotowaniu poddaje się liczne elementy silników i pomp wytwarzanych w przemyśle lotniczym, okrętowym i motoryzacyjnym, np. wały korbowe, korbowody, koła zębate, wałki, tłoki, cylindry.
Stale stopowe i ich obróbka cieplna
Stal stopowa to stal do której wprowadzono pierwiastki w celu poprawienia jej własności. Oznaczenia: N - nikiel, H - chrom (stale konstrukcyjne), C - chrom (narzędziowe), W - wolfram, M - molibden, F - wanad (konstrukcyjne), V - wanad (pozostałe), G - mangan, S - krzem, K- kobalt, J - aluminium, T - tytan, Cu - miedź, B - bor.
Dodatki stopowe wprowadzane są do stali w celu:
spowodowania określonych zmian strukturalnych,
zwiększenia własności wytrzymałościowych i polepszenia niektórych własności chemicz. lub fizycznych,
zwiększenia hartowności,
polepszenia efektywności i ułatwienia obróbki cieplnej.
Pierwiastki: Co, Mn, Ni, zwane austenitotwórczymi, w stopach żelaza powodują rozszerzenie obszaru występowania roztworu stałego γ. Przy odpowiednio dużych stężeniach tych pierwiastków jednofazowa struktura austenityczna występuje w całym zakresie poniżej temperatury solidusu. Pierwiastki zwane ferrytotwórczymi,np. Cr, V, Al, Si, Ti, Mo, W, ograniczają w stopach zelaza występowanie roztworu stałego γ. Przy dostatecznie dużym stężeniu tych pierwiastków w stanie stałym w całym zakresie temperatury (od temperatury pokojowej do solidusu) występuje więc struktura roztworu stałego α. Wszystkie pierwiastki stopowe przesuwają do mniejszego stężenia węgla położenie punktu eutektoidalnego oraz punktu E granicznej rozpuszczalności węgla w austenicie. Przesunięcie linii SE do mniejszego stężenia węgla, powodowane przez wszystkie niemal pierwiastki stopowe, sprzyja wydzielaniu węglików wtórnych przy stężeniach węgla mniejszych niż w stalach węglowych. Niemal wszystkie pierwiastki stopowe rozpuszczone w roztworach stałych w sposób znaczący decydują o przebiegu przemian strukturalnych zachodzących podczas obróbki cieplnej. W szczególności wpływ ten dotyczy procesów tworzenia i ujednorodniania austenitu w czasie nagrzewania stali, kinetyki przemian tej fazy podczas chłodzenia oraz związanej z tym hartowności stali. Pierwiastki przejściowe cechujące się dużym powinowactwem chemicznym do węgla, szczególnie Cr, W, Mo, V, Ti, Nb oraz Fe, tworzą w stalach fazy międzywęzłowe: węgliki o strukturach złożonych, tj. M3C, M23C6, M7C3, M6C, oraz węgliki o strukturach prostych MC i M2C. Podczas obróbki cieplnej węgliki ulegają rozpuszczeniu w austenicie, wpływając na zmianę własności tej fazy, głównie na hartowność i kinetykę przemian austenitu przechłodzonego. Zróżnicowana rozpuszczalność węglików w austenicie decyduje o wielkości ziarna i składzie chemicznym austenitu. Nie rozpuszczone węgliki utrudniają bowiem rozrost ziarn austenitu. Dopiero po nagrzaniu stali do odpowiednio wysokiej temperatury, nierzadko zbliżonej do temperatury solidusu, następuje zwykle rozpuszczenie znacznej liczby węglików, czemu towarzyszy znaczny rozrost ziarn austenitu.
Stale konstrukcyjne
Stale konstrukcjne stopowe są stosowane w budownictwie oraz w budowie maszyn i urządzeń pracujących w zakresie temperatury od ok. 25 do 300°C, w środowiskach o niewielkim działaniu korozyjnym. Kryterium doboru stali stanowią najczęściej podstawowe własności mechaniczne. W przypadku obciążeń statycznych, kryterium stanowi granica plastyczności Re lub granica sprężystości Rsp - gdy niedopuszczalne jest odkształcenie plastyczne konstrukcji. W przypadku obciążeń zmęczeniowych jako kryterium przyjmuje się granicę zmęczenia Zg.
Własności stali konstrukcyjnych stopowych zależą od stężenia węgla i pierwiastków stopowych. Główne znaczenie Cr, Mn, Si, Ni, Mo i innych dodaków stopowych w stalach konstrukcyjnych polega na polepszeniu hartowności stanowiącej podstawowe kryterium doboru tych stali. Uzyskanie struktury martenzytycznej na założonym przekroju elementu zapewnia po obróbce cieplnej wydatne zwiększenie własności mechanicznych. Pierwiastki węglikotwórcze, takie jak np. Cr, V, Mo, W, zwiększają także twardość i odporność na ścieranie w wyniku tworzenia węglików, wpływają na zmniejszenie wielkości ziarn, przeciwdziałają kruchości odpuszczania (Mo, W) oraz powodują polepszenie wielu innych własności technologicznych i użytkowych stali. W celu uzyskania najkorzystniejszych własności stale konstrukcyjne stopowe należy stosować w stanie obrobionym cieplnie lub po innych procesach technologicznych zapewniających wykorzystanie pozytywnego działania pierwiastków stopowych.
Znakowanie: Gatunek stali konstrukcyjnych stopowych jest określany, zgodnie z PN, znakiem stali, składającym się z: 1) liczby 2-cyfrowej na początku znaku, określającej przybliżone stężenie węgla w stali w setnych częściach procentu, 2) litery lub kilku liter określających dodatki stopowe, 3) liczb całkowitych, podawanych po znaku literowym poszczególnych pierwiastków, określających przybliżone stężenie dodatków stopowych w stali w procentach, gdy ich zawartość jest nie mniejsza od ok. 2%.
Dodanie na końcu litery A oznacza wyższą jakość stali. Stale łożyskowe są oznaczane literą Ł, następnie literą H oraz liczbą określającą średnie stężenie chromu w dziesiętnych częściach procentu.
Stale spawalne
Większość elementów konstrukcyjnych stosowanych do budowy m.in. konstrukcji przemysłowych, mostów, do zbrojenia betonu, na rury do rurociągów, jest łączona przez spawanie lub zgrzewanie. Elementy tych konstrukcji, o ile wymaga się od nich większej wytrzymałości niż uzyskiwana przez stale konstrukcyjne węglowe, wykonuje się z niskostopowych stali konstrukcyjnych spawalnych o podwyższonej wytrzymałości. Stale te powinny charakteryzować się dobrą spawalnością, dużymi wartościami wytrzymałości Rm, granicy plastyczności Re oraz niską temperaturą przejścia w stan kruchy. Wymagane własności tych stali zapewnia odpowiedni dobór składu chemicznego, a także technologia wytapiania, obróbki plastycznej i obróbki cieplnej.
Skład chemiczny stali spawalnych podlega ograniczeniom i jest dobierany z uwzględnieniem wartości równoważnika węgla obliczanego wg wzoru:
![]()
gdzie: C, Mn, Cr, Mo, V, Ni, Cu - odpowiadają stężeniu masowemu tych pierwiastków w stali. W przypadku gdy CE ≤ 0,45%, stale są spawalne bez żadnych ograniczeń. Stale o większym równoważniku węgla wymagają podgrzewania przed spawaniem, regulowanego chłodzenia albo wyżarzania po spawaniu.
Poza wpływem na spawalność, pierwiastki stopowe oddziałują na własności mechaniczne stali spawalnych o strukturze ferrytycznej. Własności wytrzymałościowe Rm, Re i twardość są podwyższane głównie przez Mn i Si, które jednak obniżają udarność przy stężeniach większych odpowiednio od ok. 1,2 i 0,8%. Nikiel, zwiększając własności wytrzymałościowe, zwiększa także ciągliwość. Dodatki Ni i Mn, nawet przy niewielkich stężeniach powodują znaczne obniżenie temperatury przejścia w stan kruchy. W przeciwieństwie do węgla. Dodatki Cr, W, Mo, polepszając hartowność w stalach ulepszanych cieplnie, powodują znaczne zmniejszenie udarności stali ferrytycznych. W stalach ferrytyczno-perlitycznych podstawowymi pierwiastkiem stopowym jest Mn przy stężeniu ok. 2%, a w stalach ulepszanych cieplnie zawierających po ok. 0,5 do 0,8% Cr, Ni i Mo - mangan jest dodawany w stężeniu ok. 1%. Do stali o podwyższonej wytrzymałości są wprowadzane również mikrododatki Al, V, Nb, Zr, Ti, N, po kilka setnych części procentu. Mikrododatki tworzą dyspersyjne wydzielenia węglików i azotków, hamujące rozrost ziarn austenitu i rekrystalizację podczas obróbki cieplnej, a także - w strefie wpływu ciepła - podczas spawania. Wpływa to na obniżenie temperatry przejścia w stan kruchy i powoduje dodatkowe umocnienie stali. Mikrododatek B przy stężeniu kilku tysięcznych części procentu wpływa głównie na zwiększenie hartowności stali ulepszanych cieplnie.
Przykładowe stale: 09G2, 18G2A, 18G2ACu, 18G2AV.
Stale do nawęglania
Stale do nawęglania cechuje dobra skrawalność, odporność na przegrzanie, mała skłonność do odkształceń podczas obróbki cieplnej, hartowność dostosowana do przekroju, obciążeń i cech geometrycznych wykonanych z nich elementów konstrukcyjnych oraz wysokie własności wytrzymałościowe nawęglonej powierzchni i duża ciągliwość rdzenia. Stale stopowe do nawęglania charakterzują się małym stężeniem C, nie przekraczającym 0,25% i zwykle dodatkiem 1-2% Cr. W zależności od gatunku zawierają także Mn, Ni, Mo,niekiedy V, W lub Ti. Dodatki Cr, Mn i Ni w stalach tej grupy zwiększają hartowność, zapewniają wymagane własności wytrzymałościowe rdzenia, a także zmniejszają naprężenia hartownicze w warstwie nawęglonej. Stężenie wymienionych pierwiastków jest ograniczane, gdyż powodują one obniżenie temperatury Ms, co wpływa na zwiększenie udziału austenitu szczątkowego, decydującego o zmniejszeniu twardości warstwy nawęglonej. Najniższe własności wytrzymałościowe rdzenia wykazują stale chromowe i chromowo-manganowe. Mangan powoduje niepożądany rozrost ziarn, czemu przeciwdziała dodatek Ti
W przypadku gdy są wymagane b. duże własności plastyczne rdzenia i wysokie własności wytrzymałościowe, stosuje się stale chromowo-manganowo-molibdenowe. Molibden sprzyja drobnoziarnistości, natomiast nikiel zwiększa plastyczność rdzenia. Najwyższe własności wytrzymałościowe i wysokie własności plastyczne rdzenia wykazują więc stale chromowo-niklowe, korzystnie z dodatkiem Mo, a szczególnie W. Przykładowe stale: 15H, 16HG, 18HGM, 22HMN.
Stale do azotowania
Niektóre elementy konstrukcyjne, takie jak np. korbowody, wały korbowe, koła zębate, tuleje, cylindry powinny się cechować b. twardą i odporną na ścieranie warstwą wierzchnią w części współpracującej z innymi elementami oraz rdzeniem o dużej wytrzymałości przy możliwie dużej ciągliwości. Twardość warstwy wierzchniej, jak i wytrzymałość rdzenia powinny być większe od uzyskiwanych w wyniku nawęglania i obróbki cieplnej elementów ze stali konstrukcyjnych do nawęglania. Własności takie zapewniają stale konstrukcyjne stopowe do azotowania poddane ulepszaniu cieplnemu, a następnie azotowaniu. Skład chemiczny stali do azotowania jest zbliżony do stali konstrukcyjnych do ulepszania cieplnego i jest tak dobrany, aby poza zapewnieniem dużej hartowności oraz odporności na kruchość odpuszczania, pierwiastki stopowe tworzyły także dyspersyjne i twarde azotki podczas nasycania warstwy wierzchniej azotem. Stale te mają najczęściej dodatki Cr, Mo oraz Al, chociaż podane wymagania spełniają także w mniejszym stopniu średniowęglowe stale chromowe i chromowo-molibdenowe z dodatkiem V lub Ti. Obróbka cieplna części maszyn przeznaczonych do azotowania polega na hartowaniu i wysokim odpuszczaniu, po którym następuje szlifowanie wykończające powierzchni przewidzianych do nasycenia azotem i samo azotowanie w temperaturze niższej od temperatury uprzedniego odpuszczania. Przykładowe stale: 38HMJ, 33H3MF, 25H3M.
Stale do ulepszania cieplnego
Stale konstrukcyjne do ulepszania cieplnego mają średnie stężenie węgla ok. 0,25-0,5% decydujące o własnościach wytrzymałościowych oraz dodatki stopowe, których głównym celem jest nadanie stali określonej hartowności. Większość tych stali jest niskostopowa o łącznym stężeniu pierwiastków stopowych nie przekraczającym 3%, są również stale średniostopowe o łącznym stężeniu dodatków stopowych 3-5% oraz nieliczne o stężeniu pierwiastków stopowych przekraczającym 5%. Najtańsze stale z tej grupy jako główny pierwiastek zwiększający hartowność zawierają Mn, który jednak sprzyja rozrostowi ziarn austenitu podczas hartowania i związanemu z tym obniżeniu ciągliwości stali, zwłaszcza przy stężeniu większym od ok. 1,8%. Mangan może być częściowo zastępowany przez Si, który sprzyjając drobnoziarnistości, wpływa na zwiększenie granicy sprężystości. Stal manganowe są stosowane na wały, osie i śruby, a manganowo-krzemowe - na części narażone na ścieranie. Głównym dodatkiem stopowym w stalach konstrukcyjnych do ulepszania cieplnego jest chrom. Zwiększając hartowność i opóźniając przemiany martenzytu podczas odpuszczania, chrom zapewnia stalom dużą wytrzymałość i ciągliwość. Stale chromowe są stosowane na silnie obciążone wały, osie, korbowody, przekładnie zębate, śruby i inne części maszyn o niedużych przekrojach. Podobne zastosowanie, przy możliwości wykonania elementów o większym przekroju, mają stale Cr-Mn i Cr-Mn-Si, w których chrom znacznie zwiększa hartowność. Stale te po odpuszczaniu wymagają chłodzenia w oleju, ze względu na możliwość wystąpienia kruchości odpuszczania. Nikiel również bardzo intensywnie zwiększa hartowność stali konstrukcyjnych do ulepszania cieplnego, obniżając jednocześnie temperaturę przejścia w stan kruchy i zwiększając ciągliwość. Stale chromowo-niklowe w stanie ulepszonym cieplnie charakteryzują się wysokimi własnościami wytrzymałościwymi oraz dużą udarnością i plastycznością. Wykazują jednak tendencje do kruchości odpuszczania. W celu przeciwdziałania temu zjawisku do stali chromowo-niklowych dodaje się ok. 0,2% Mo, który powoduje dodatkowo zwiększenie hartowności stali.
Założone własności elementów konstrukcyjnych ze stali do ulepszania cieplnego uzyskuje się po obróbce cieplnej polegającej na hartowaniu i wysokim odpuszczaniu. Temperatura odpuszczania mieści się w zakresie 500-650° i może być tym wyższa, im większe jest w stali stężenie węgla i pierwiastków węglikotwórczych. Podwyższenie temperatury odpuszczania stali o określonym składzie chemicznym powoduje zmniejszenie własności wytrzymałościowych i zwiększenie własności plastycznych w związku z intensyfikacją procesu rozpadu martenzytu. Po ulepszaniu cieplnym strukturę stali stanowi martenzyt wysokoodpuszczony. Niektóre elementy konstrukcyjne, od którch wymaga się dużej wytrzymałości i dopuszcza się ich obniżoną udarność, można wykonać ze stali o stężeniu węgla 0,4-0,5% i zastosować po zahartowaniu niskie odpuszczanie. Poza ulepszaniem cieplnym, niektóre stale można poddawać hartowaniu izotermicznemu. Przykładowe stale: 30G2, 38HA, 35HGS, 40HM, 45HN2A, 34HNM.
Stale sprężynowe
Materiał stosowany na elementy sprężyste powinien cechować się b. dobrymi własnościami sprężystymi, tzn. wysoką granicą sprężystości Rsp oraz dużą wartością stosunków tej wielkości do granicy plastyczności Re i wytrzymałości na rozciąganie Rm. Pożądana jest duża wytrzymałość na zmęczenie przy ograniczonych wymaganiach dotyczących własności plastycznych. Stale sprężynowe zawierają ok. 0,5-07% C, od którego przede wszystkim zależą własności wytrzymałościowe i granica sprężystości. Podstawowym pierwiastkiem stopowym w tych stalach jest Si, najintensywniej zwiększający Rsp, Re i Rm. Stale sprężynowe krzemowe cechują się jednak małą hartownością i z tego względu są stosowane na sprężyny o niewielkich przekrojach. Większą hartownością charakteryzują się stale sprężynowe manganowe. Dodatek Cr jeszcze intensywniej zwiększa hartowność i odporność stali na odpuszczanie, hamując jednocześnie relaksację naprężeń. Podobny wpływ wywiera V, bardziej węglikotwórczy od Cr, zapewniając stalom sprężynowym drobnoziarnistość, zmniejszając szybkość przemian martenzytu i zmian własności wytrzymałościowych podczas odpuszczania oraz ograniczając skłonność stali do odwęglenia. Stale chromowo-wanadowe stosuje się na najsilniej obciążone sprężyny, w tym także o dużych przekrojach. Sprężyny ze stali chromowo-wanadowych, a także ze stali chromowo-krzemowych mogą pracować w temperaturze do ok. 300°C, natomiast z pozostałych - w temperaturze nie przekraczającej 150°C.
Obróbka cieplna sprężyn i resorów polega na austenityzowaniu w temperaturze 800-870°C i hartowaniu w oleju lub wodzie zależnie od gatunku zastosowanej stali, a następnie średnim odpuszczaniu w temperaturze 380-520°C. Po takiej obróbce cieplnej sprężyny mają strukturę martenzytu odpuszczonego o wymaganych własnościach mechanicznych. Przykład stali: 45S, 50S2, 65G, 50HF.
Stale na łożyska toczne
Stale konstrukcyjne stosowane do wytwarzania elementów łożysk tocznych powinny cechować się b. dużą twardością, jednorodnością struktury, wysokim stopniem czystości oraz hartownością, zapewniającą uzyskanie struktury martenzytycznej bez austenitu szczątkowego w całym przekroju. Stale te w stanie obrobionym cieplnie są odporne na ścieranie, wykazują dużą wytrzymałość zmęczeniową i statyczną oraz odpowiednią ciągliwość. Dużą twardość i odporność na ścieranie zapewnia stalom łożyskowym węgiel o stężeniu ok. 1%. Wysoka czystość i jednorodna struktura bez skupień i pasmowej segregacji węglików gwarantują dużą wytrzymałość zmęczeniową. Wymaganą hartowność stale te osiągają dzięki dodatkowi ok. 1,5 % Cr. W przypadku dużych wymiarów elementów łożysk są stosowane stale, które oprócz Cr zawierają Si i Mn, zapewniające większą hartowność.
Łożyska toczne są wytwrzane, a więc i obrabiane cieplnie, w specjalistycznych zakładach produkcyjnych. Wytwarza się je z półwyrobów hutniczych wyżarzonych zmiękczająco, o jednorodnej strukturze drobnoziarnistego sferoidalnego cementytu w ferrytycznej osnowie. Elementy łożysk tocznych hartuje się z temperatury 820-840°C w oleju, po czym odpuszcza w temperaturze 180°C przez 1-2 h. Po takiej obróbce cieplnej stale łożyskowe mają strukturę niskoodpuszczonego martenzytu drobnolistwowego z równomiernie rozmieszczonymi drobnymi ziarnami cementytu stopowego i wykazują twardość przekraczającą 62 HRC. Przykładowe stale: ŁH15, ŁH15SG.
Stale niestopowe narzędziowe
Stale narzędziowe niestopowe wysokowęglowe znalazły zastosowanie na proste narzędzia tnące do drewna, papieru i tworzyw sztucznych, takie jak pilniki i proste narzędzia rolnicze - np. kosy lub zęby bron. Stale o mniejszym stężeniu węgla są stosowane na proste narzędzia tnące, np. piły, dłuta oraz narzędzia pracujące udarowo, jak młotki, przecinaki i cechowniki. Stale wysokowęglowe, po odpowiedniej obróbce cieplnej, charakteryzują się głównie dużą twardością, a stale o niższym stężeniu węgla - nieco większą ciągliwością. Stale narzędziowe niestopowe cechują się ponadto małą hartownością i małą skłonnością do rozrostu ziarna austenitu. Są produkowane jako głęboko lub płytko hartujące się. Stale płytko hartujące się są stosowane na narzędzia o średnicy mniejszej od 20 mm, a głęboko hartujące się - na narzędzia grubsze.
W celu uzyskania wymaganych własności stale narzędziowe niestopowe poddaje się hartowaniu z temperatury 760-800°C z chłodzeniem w wodzie i odpuszczaniu w temperaturze 180-300°C z wygrzaniem przez 2 h. Temperatura odpuszczania jest dobierana w zależności od wymaganej twardości i ciągliwości narzędzia i zwykle nie przekracza 200°C. Podwyższanie temperatury odpuszczania powoduje szybkie zmniejszanie twardości i odporności na ścieranie stali niestopowych. Przykłady stal: N9E, N11E (płytko), N9, N11 (głęboko).
Stale narzędziowe stopowe szybkotnące
Stale szybkotnące są stosowane głównie na wieloostrzowe narzędzia skrawające, często narzędzia wykrojnikowe, a także na narzędzia do obróbki plastycznej na zimno i gorąco. Stale te wykazują dużą twardość i odporność na ścieranie w temperaturze do ok. 600°C. Wymagane własności, zwłaszcza b. dużą hartowność oraz efekt twardości wtórnej, uzyskuje się przez odpowiednią kombinację stężenia C i takich pierwiastków stopowych, jak Cr, W, Mo i V, a w wielu gatunkach dodatkowo - Co. Oprócz optymalizacji składu chemicznego, na zwiększenie własności stali szybkotnących obrobionych cieplnie duży wpływ wywierają czynniki metalurgiczne, decydujące o kształcie i położeniu węglików pierwotnych. Ledeburytyczna siatka węglików pierwotnych uzyskana po wykrystalizowaniu wlewka z tych stali jest rozbijana podczas obróbki plastycznej, najkorzystniej kucia, częściej walcowania. Od stopnia przerobu podczas obróbki plastycznej zależy segregacja węglików, która jest głównie uwarunkowna pierwotną strukturą wlewka. Koniecznością jest zatem produkcja wlewków nie zawierających segregatów eutektyki, na co wpływa głównie kształt wlewnic, obniżona temperatura odlewania, chłodzenie wewnętrzne oraz modyfikacja, powodująca rozdrobnienie węglików i równomierne rozmieszczenie eutektyki. Stopień segregacji węglików ulega również zmniejszeniu w wyniku zastosowania rafinacji stali szybkotnących przez przetapianie próżniowe lub elektrożużlowe, związane również ze zwiększeniem czystości stali. Uzyskanie pożądanych własności stali szybkotnących zależy ponadto od prawidłowo wykonanej obróbki cieplnej. Stale szybkotnące są dostarczane w stanie zmiękczonym. Zapewnia to dobrą obrabialność mechaniczną stali. Narzędzia wykonane metodami obróbki skrawaniem poddaje się obróbce cieplnej polegającej na hartowaniu i wysokim odpuszczaniu. Strukturę stali szybkotnących uzyskaną w wyniku hartowania w optymalnych warunkach stanowi martenzyt listwowy i w ok. 20% austenit szczątkowy oraz węgliki nie rozpuszczone w roztworze stałym podczas austenityzowania. Optymalne własności w stanie odpuszczonym wykazują stale szybkotnące zahartowane tak, że wskaźnik wielkości ziarn austenitu mieści się w zakresie od 10 do 14 wg Snydera-Graffa.
Przykładowe stale: sw18, sk5.
Stale narzędziowe stopowe do pracy na gorąco
Stale narzędziowe stopowe do pracy na gorąco są stosowane na narzędzia pracujące w zakresie temperatury 250-700°C. Skład chemiczny tych stali oraz ich obróbka cieplna zapewniają wysoką wytrzymałość, twardość i odporność na ścieranie w wysokiej temperaturze pracy. W stalach tych stężenie węgla jest ograiczone do ok. 0,3-06%. Wynika to z konieczności zapewnienia tym stalom wymaganej odporności na zmęczenie cieplne i obciążenia dynamiczne. Głównymi pierwiastkami stopowymi są Cr, W, Mo i V, powodujące efekt twardości wtórnej podczas odpuszczania. Powierzchnie narzędzi do pracy na gorąco są narażone na b. częste zmiany temperatury w wyniku cyklicznego kontaktu z odkształcanym materiałem, nagrzanym do wysokiej temperatury w wyniku cyklicznego kontaktu z odkształcanym materiałem, nagrzanym do wysokiej temperatury, i chłodzenia po zakończeniu obróbki plastycznej. Wywołuje to cykliczne zmiany naprężeń w warstwie powierzchniowej i w konsekwencji - zmęczenie cieplne. W wyniku tego zjawiska na powierzchniach długo pracujących narzędzi tworzy się siatka pęknięć. W przypadku nakładania się zjawisk charakterystycznych dla zmęczenia cieplnego i naprężeń zewnętrznych, spowodowanych np. tarciem lub naciskami powierzchniowymi, występuje tzw. zmęczenie cieplno-mechaniczne, przyspieszające zużywanie się narzędzi. Większą odporność na zmęczenie cieplne wykazują stale wysokostopowe otrzymane przez przetapianie elektrożużlowe lub w próżni. Zadawalającą żaroodporność stali narzędziowych stopowych do pracy na gorąco zapewnia Cr i Si. Stale narzędziowe do pracy na grąco dostarcza się w stanie zmiękczonym, zapewniającym dobrą obrabialność i jednorodny rozkład węglików w osnowie ferrytu. Narzędzia wykonane przez obróbkę skrawaniem poddaje się obróbce cieplnej składającej się z hartowania i wysokiego odpuszczania. Przykładowe stale: WCL, WWV, WNLV.
Stale narzędziowe stopowe do pracy na zimno
Stale narzędziowe stopowe do pracy na zimno są stosowane na narzędzia nie osiągające w czasie pracy temperatury wyższej niż 200°C. Stale stopowe do pracy na zimno w porównaniu ze stalami narzędziowymi niestopowymi wykazują podwyższoną hartowność, powodowaną głównie zwiększonym stężeniem Mn, Cr, a w niektórych gatunkach - także W, V i Ni. Dodatki stopowe, zwłaszcza V, Cr i W, wpływają na tworzenie w stalach narzędziowych węglików stopowych sprzyjających uzyskwaniu dużej odporności stali na ścieranie. Pierwiastki te powodują również wysoką skrawność stali narzędziowych stopowych i opóźniają rozpad martenzytu oraz spadek twardści podczas odpuszczania, w porównaniu do charakterystycznego dla stali niestopowych. W przpadku odpowiednio małego stężenia węgla w obecności takich pierwiastków stopowych, jak Cr, W i Si, niektóre stale narzędziowe stopowe wykazują zwiększoną ciągliwość, co umożliwia ich stosowanie na narzędzia narażone na udarowe działanie obciążeń. Od stali narzędziowych stopowych do pracy na zimno wymaga się przede wszystkim dużej twardości i odporności na ścieranie. Dlatego poddaje się je hartowaniu i niskiemu odpuszczaniu. Stale narzędziowe stopowe do pracy na zimno w stanie zahartowanym wykazują strukturę martenzytu listwowego z austenitem szczątkowym i węglikami nie rozpuszczonymi podczas austenityzowania, równomiernie rozmieszczonymi w osnowie. Natomiast powierzchnia narzędzi wykonywanych ze stali narzędziowych do pracy na zimno w czasie obróbki cieplnej powinna być zabezpieczona przed utlenianiem i odwęglaniem. Przykłady stali: NC4, NCV1, NC11, NZ2.
Stale żaroodporne
Żaroodporność to odporność stopu na działanie czynników chemicznych, głównie powietrza oraz spalin i ich agresywnych składników w temperaturze wyższej niż 600°
Żaroodporność jest ściśle związana ze skłonnością stali do tworzenia zgorzeliny. Zgorzelina powinna stanowić ciągłą warstwę, dokładnie przylegającą do metalicznego rdzenia, co utrudnia dyfuzję utleniacza i jonów metalu. Wymagania te spełniają niskowęglowe stale o jednofazowej strukturze ferrytu lub austenitu, o dużym stężeniu chromu i niklu oraz dodatkowo krzemu i aluminium. Chrom jest podstawowym pierwiastkiem zwiększającym żaroodporność stali. Dodatek ok. 5% Cr zapewnia odpowiednią żaroodporność w temperaturze 600-650 °C. Zwiększenie stężenia tego pierwiastka powoduje wzrost żaroodporności do ok. 1100° przy stężeniu ok. 30% Cr w stali. Dodatki Si i Al, mimo analogicznego wpływu na żaroodporność, są dodawane w ograniczonym stężeniu - odpowiednio ok. 3 i 2,5% ze względu na niekorzystny wpływ na własności plastyczne stali i obniżanie podatności na obróbkę plastyczną. Dodatki V i Mo wywierają niekorzystny wpływ na żaroodporność stali. Tlenki wanadu bowiem łatwo ulegają stopieniu, natomiast tlenki molibdenu utleniają się. Nikiel nie jest samodzielnie stosowany, gdyż nie zwiększa żaroodporności stali. Przykłady stali: H6S2, 2H17.
Stale żarowytrzymałe
Żarowytrzymałością jest nazywana odporność stopu na odkształcenia, z czym wiąże się zdolność do wytrzymywania obciążeń mechanicznych w wysokiej temperaturze - powyżej 600°C. Żarowytrzymałość w temperaturze wyższej od 600°C jest uzależniona głównie od odporności na pełzanie. Dużą żarowytrzymałość wykazują stale o strukturze austenitycznej - ze względu na mniejsze współczynniki dyfuzji niż w ferrycie, o znacznej wielkości ziarn i z dyspersyjnymi wydzieleniami faz, głównie na granicach ziarn. Nikiel przy stężeniu 9%, w obecności ok. 18% Cr, powoduje tworzenie trwałej struktury austenitycznej, co decyduje o zwiększeniu żarowytrzymałości stali. Żarowytrzymałość podwyższają pierwiastki stopowe zwiększające energię wiązania atomów sieci roztworu stałego, a więc podwyższające temperaturę topnienia i rekrystalizacji, do których należą Mo, W, V, Co, a także Ti, Cr i Si. Żarowytrzymałość jest ponadto zwiększana w wyniku umocnienia zgniotowego oraz utwardzania dyspersyjnego. Natomiast obniżenie żarowytrzymałości następuje wskutek poligonizacji i rekrystalizacji stali uprzednio odkształconej plastycznie na zimno oraz koagulacji wydzieleń faz. Stężenie węgla w tych stalach - ze względu na zapewnienie odpowiedniej spawalności - jest ograniczone do ok. 0,2%. Przykłady stali: H18N9S, H23N18
Stale zaworowe
Szczególną grupę stali żarowytrzymałych, używanych na zawory w slnikach spalinowych, stanowią stale zaworowe. Charakteryzują się one dużą odpornością na korozję w atmosferze spalin, w temperaturze do ok. 750°C. Odporność tę zapewniają główne dodatki Si i Cr, stąd nazwa tych stali - silchromy. Dużą twardość i odporność na ścieranie zapewnia im stosunkowo duże stężenie węgla - 0,4-0,6%. Ponieważ stale o strukturze austenitycznej wykazują większą wytrzymałość w wysokiej temperaturze, niż stale o strukturze perlitycznej, niektóre gatunki mają duże stężenie Cr i Ni. Częściowo Ni może być zastępowany tańszym Mn, również austenitotwórczym. Dodatki W i Mo powodują zwiększenie żarowytrzymałości i rozdrobnienie ziarn. W stalach o strukturze perlitycznej dodatki te powodują zwiększenie odporności na odpuszczanie i przeciwdziałają kruchości odpuszczania. Stale krzemowo-chromowe o strukturze perlitycznej poddaje się hartowaniu z temperatury 1010-1060°C i odpuszczaniu w temperaturze 700-790°C z chłodzeniem w wodzie, co zapobiega kruchości odpuszczania. Strukturę stali obrobionej cieplnie stanowi martenzyt wysokoodpuszczony. Stale o strukturze austenitycznej poddaje się przesycaniu z temperatury 1050-1170°C, z chłodzeniem w wodzie, i starzeniu w temperaturze 700-750°C. W wyniku tej obróbki otrzymuje się strukturę austenitu z dyspersyjnymi wydzieleniami węglików M6C i M23C6 oraz węglikoazotków. Przykłady stali: H9S2, 4H14N14W2M.
Stale odporne na korozję
Stale odporne na korozję obejmują trzy grupy:
Stale trudno rdzewiejące, o odporności na korozję jedynie nieznacznie większej od stali węglowych, zawierają 0,1% C oraz dodatki 1-3% pasywującego Cr i ok. 0,5% Cu, tworzącej na powierzchni warstewkę pasywującą złożoną z siarczanów i węglanów miedzi. Do stali tych są wprowadzane także w niewielkich stężeniach P, Al i Ni. Stale te znajdują zastosowanie głównie jako stale spawalne pracujące w środowisku atmosfery przemysłowej oraz morskiej. Stale wysokochromowe o strukturze ferrytycznej ferrytyczno-martenzytycznej lub martenzytycznej są odporne głównie na korozję chemiczną, w tym na utlenianie w atmosferze powietrza, wody naturalnej i pary wodnej w niskiej i podwyższonej temperaturze, na działanie zimych roztworów alkalicznych, rozcieńczonych kwasów i soli, z wyjątkiem chlorków i jodków, oraz na działanie ropy naftowej i jej par, paliw, olejów, alkoholi, a także środków spożywczych. Stale chromowo-niklowe i chromowo-niklowo-manganowe, o strukturze austenitycznej są odporne głównie na korozję elektrochemiczną w środowisku kwasów nieorganicznych i organicznych, związków azotu, roztworów soli i agresywnych środków spożywczych. Przykłady stali: 0H13, 1H13, 4H14, 0H17T, 3H17M.
Stopy aluminium
Stosunkowo niewielkie własności wytrzymałościowe aluminium można zwiększyć - nawet kilkakrotnie - przez wprowadzenie pierwiastków stopowych oraz obróbkę cieplną stopów. W porównaniu ze stalmi stopy aluminiowe charakteryzują się znacznie mniejszą masą, a w niskiej temperaturze - większą udarnością. Najogólniej - ze względu na sposób wytwarzania - stopy aluminium dzieli się na: odlewnicze i do obróbki plastycznej. Niektóre z tych stopów mogą być stosowane zarówno jako odlewnicze jak i przeznaczone do obróbki plastycznej. Odlewnicze stopy aluminium są przeważnie stopami wieloskładnikowymi o dużym stężeniu - od 5 do 25% - pierwiastków stopowych, głównie Cu, Si, Mg i Ni lub ich różnych zestawień. Charakteryzują się dobrą lejnością i często małym skurczem odlewniczym. Stopy do obróbki plastycznej zawierają znacznie mniej, bo ok. 5%, pierwiastków stopowych, zwykle Cu, Mg, Mn, niekiedy także Si, Zn, Ni, Cr, Ti lub Li. Niektóre z tych stopów są stosowane w stanie zgniecionym lub po wyżarzaniu rekrystalizującym, a część jest poddawana obróbce cieplnej polegającej na utwardzaniu wydzielinowym.
Stopy aluminium z krzemem
Aluminium tworzy z krzemem układ z eutektyką, występujący przy stężeniu 12,6% Si i dwoma roztworami stałymi granicznymi o rozpuszczalności składników zmniejszającej się wraz z obniżaniem temperatury. Roztwór α (Si w Al) wykazuje sieć regularną typu A1. Aluminium w temperaturze eutektycznej rozpuszcza się w Si w b. niewielkim stężeniu - ok. 0,07%, a w temperaturze pokojowej nie wykazuje niemal zupełnie rozpuszczalności w Si. Podstawową grupę stopów Al z Si stanowią stopy odlewnicze zwane siluminami o stężeniu 4-30% Si. Krzem, jako podstawowy składnik tych stopów, zapewnia dobrą rzadkopłynność oraz lejność i mały skurcz odlewniczy. Siluminy mogą być również stopami wieloskładnikowymi. Zawierają wówczas dodatki Cu, Mg i Mn, zwiększające wytrzymałość. Dodatek Cu oraz Mg umożliwia utwardzanie wydzielinowe stopów wieloskładnikowych Al z Si, w wyniku wydzielania faz CuAl2 lub Mg2Si. Dodatek Cu pogarsza jednak odporność na korozję, która z kolei poprawia dodatek ok. 1% Ni. Dodatek 0,5% Mn przeciwdziała ujemnemu wpływowi domieszek Fe tworzących wydzielenia Fe2Si2Al9 oraz Fe3Si2Al12 znacznie zmniejszające ciągliwość stopu. Siluminy eutektyczne i nadeutektyczne wykazujące znaczną żarowytrzymałość są stosowane na wysoko obciążone tłoki silników spalinowych. Z siluminów podeutektycznych wytwarza się silnie obciążone części dla przemysłu okrętowego i elektrycznego, pracujące w podwyższonej temperaturze i w wodzie morskiej. Wieloskładnikowe stopy Al z Si są stosowane m.in. na głowice silników spalinowych oraz inne odlewy w przemyśle maszynowym. Stopy Al z niewielkim dodatkiem - ok. 1% Si - są przeznaczone do obróbki plastycznej, na średnio obciążone elementy konstrukcji lotniczych i pojazdów mechanicznych oraz elementy głębokotłoczne i kute o złożonych kształtach.
Stopy miedzi
Mosiądze. Mosiądze dwuskładnikowe jako stopy Cu i Zn - ze względu na skład fazowy dzieli się na: 1) jednofazowe - o strukturze roztworu α i stężeniu od 2 do 39% Zn, 2) dwufazowe - o strukturze mieszaniny α+β i stężeniu od 39 do 45% Zn. Mosiądze jednofazowe cechuje b. duża plastyczność, co umożliwia stosowanie ich na wyroby głęboko tłoczone i obrabiane plastycznie na zimno. Duża plastyczność w podwyższonej temperaturze umożliwia ich obróbkę plastyczną na gorąco. Dodatek Zn do 30% zwiększa plastyczność oraz wytrzymałość mosiądzu. Wytrzymałość mosiądzów zawierających ok. 30 do 45% Zn zwiększa się przy znacznym zmniejszeniu plastyczności. Mosiądze w znacznym stopniu umacniają się w wyniku zgniotu. W zależności od stopnia gniotu mogą być dostarczane w różnym stanie. Przy większych stopniach gniotu jest stosowane międzyoperacyjne wyżarzanie rekrystalizujące mosiądzów w temperaturze 500-580°C. Mosiądze charakteryzują się dobrą odpornością na korozję, szczególnie atmosferyczną i w wodzie morskiej. Odporność mosiądzów na korozję zwiększa się wraz ze wzrostem stężenia Cu. Najczęściej spotykanymi rodzajami korozji mosiądzów jest odcynkowanie oraz korozja naprężeniowa, zwana pękaniem sezonowym mosiądzów.
Odcynkowanie zachodzi w mosiądzach dwufazowych o stężeniu Zn przekraczającym 20% zanurzonych w elektrolitach zawierających Cl. W elektrolitach takich Cu oraz Zn przechodza do roztworu, z którego Cu wytrąca się w postaci gąbczastej, co wzmaga korozję. Odcynkowanie nie powoduje zmian kształtu korodującego przedmiotu, lecz wpływa na znaczne obniżenie własności wytrzymałościowych mosiądzu.
Pękanie sezonowe jest międzykrystaliczną korozją naprężeniową mosiądzów jedno- lub dwufazowych, obrobionych plastycznie na zimno i poddanych działaniu ośrodka zawierającego amoniak. Temu rodzajowi korozji można zapobiegać przez wyżarzanie odprężające w temperaturze 200-300°C. Własności mosiądzów dwuskładnikowych są polepszane przez wprowadzenie dalszych dodatków stopowych. Należą do nich Si, Al, Sn, Pb, Fe, Mn, Ni i As, dodawane pojedynczo lub w różnych zestawieniach, zwykle o łącznym stężeniu nie przekraczającym 4%. Dodatki te z wyjątkiem niklu rozpuszczają się w roztworze stałym i zmieniają strukturę mosiądzu podobnie jak cynk. Dodawane pierwiastki stopowe powodują zwiększenie wytrzymałości i odporności mosiądzów na korozję.
Wieloskładnikowe mosiądze odlewnicze zwykle cechuje dobra odporność na korozję i ścieranie oraz dobre własności wytrzymałościowe przy obciążeniach statycznych. Stosuje się je głównie na armaturę, osprzęt, łożyska, śruby okrętowe i elementy maszyn.
Brązy cynowe
Techniczne stopy Cu z Sn mają zazwyczaj strukturę roztworu α. Duży zakres temperatury krystalizacji brązów o strukturze α sprzyja jednak ich skłonności do segregacji. Z tego powodu w stopach chłodzonych w warunkach rzeczywistych, nawet przy niewielkim stężeniu Sn, oprócz niejednorodnej fazy α tworzą się fazy, które w warunkach równowagi występują przy większym stężeniu Sn. Segragacja może być w pewnym stopniu usunięta przez długotrwałe wyżarzanie ujednorodniające w ciągu 24 h w temperaturze 700-750°C. Brązy cynowe wykazują dobrą odporność na korozję, szczególnie w środowisku atmosfery przemysłowej i wody morskiej. Odporność ta ulega polepszeniu wraz ze zwiększeniem stężenia Sn, lecz do wartości nie większej od zapewniającej wystąpienie struktury dwufazowej, decydującej o ułatwieniu korozji. Brązy cynowe o strukturze jednorodnego roztworu α cechuje duża plastyczność i z tego względu mogą być obrabiane na zimno, podobnie jak stopy o niejednorodnej strukturze α, zawierające więcej niż 4% Sn. W praktyce do obróbki plastycznej przeznaczone są brązy cynowe zawierające do 8% Sn, choć obrabia się je źle, przy dużej skłonności do pęknięć. W stanie obrobionym plastycznie na zimno brązy cynowe charakteryzują się dużymi własnościami mechanicznymi, co ułatwia stosowanie ich w przemyśle chemicznym, papierniczym i okrętowym, m.in. na elementy aparatury kontrolno-pomiarowej, siatki, sprężyny, tulejki, łożyska ślizgowe.
W celu polepszenia niektórych własności oraz zaoszczędzenia Sn są produkowane brązy cynowe wieloskładnikowe zawierające oprócz Sn dodatki Zn lub Pb. Dodatek Zn przeciwdziała segregacji brązów cynowych przez zmniejszenie zakresu temperatury krystalizacji fazy α, sprzyjając ujednorodnieniu ich własności mechanicznych i zwiększeniu własności wytrzymałościowych. Cynk jest dobrym odtleniaczem i poprawia lejność tych stopów. Ołów, nie tworzący roztworów, polepsza skrawalność brązu cynowego, zmniejsza współczynnik tarcia i korzystnie wpływa na szczelność odlewów, jednak przy większym stężeniu powoduje pogorszenie własności mechanicznych.
Brązy aluminiowe
Jednofazowe brązy aluminiowe zawierają do 8% Al i ze względu na duża plastyczność mogą być obrabiane plastycznie na zimno i na gorąco. Brązy o składzie eutektoidalnym można odkształcać plastycznie wyłącznie na gorąco w temperaturze, w której istnieje struktura jednofazowa β. Brązy aluminiowe wykazują dobrą odporność na korozję w środowisku wody morskiej i kwasów utleniających, dzięki pasywacji i tworzeniu się warstewki Al2O3 na ich powierzchni. Charakteryzują się dużymi własnościami mechanicznymi w temperaturze pokojowej i podwyższonej oraz wysoką odpornością na ścieranie. Własności mechaniczne brązów aluminiowych zwiększają dodatki stopowe, głównie Fe, Mn, Ni. Pierwiastki Fe i Ni powodują podwyższenie własności wytrzymałościowych i odporności na ścieranie w wyniku działania modyfikującego i sprzyjania drobnoziarnistości stopów. Mangan rozpuszcza się w roztworze αCu w stężeniu do ok. 10%, powodując znaczne zwiększenie odporności tych stopów na korozję i ścieranie oraz własności mechanicznych.
Brązy krzemowe
Techniczne stopy Cu z Si wykazują strukturę jednofazową roztworu α, a stężenie Si w brązach krzemowych dwuskładnikowych nie przekracza 3-4%. Jednofazowa struktura zapewnia brązom krzemowym duże własności plastyczne, przy czym Si powoduje zwiększenie ich odporności na korozję . Praktyczne zastosowanie znalazły głównie brązy krzemowe wieloskładnikowe zawierające dodatki Mn, Fe, Zn, Ni, niekiedy także Co i Cr. Pierwiastki Mn, Zn i Ni, występujące w roztworze, silnie zmniejszają rozpuszczalność Si w fazie α, przy czym Mn i Ni zwiększają wytrzymałość i odporność na korozję, natomiast Zn polepsza lejność. Własności mechaniczne najbardziej poprawia Fe. Pierwiastek ten prawie wcale nie rozpuszcza się w roztworze α, wchodząc w skład faz międzymetalicznych FeSi i Fe3Si. Z tego względu Fe jest wprowadzane wyłącznie do brązów krzemowych odlewniczych. Brązy krzemowe charakteryzują się dużymi własnościami mechanicznymi w temperaturze pokojowej i podwyższonej do ok. 300°C, dużą wytrzymałością zmęczeniową i dobrymi własnościami ślizgowymi. Cechuje je ponadto duża odporność na korozję, dobra lejność i skrawalność.