
1. Lipidy 36
Odparować z nich rozpuszczalnik i zanalizować je w argentacyjnej chromato-
grafii cienkowarstwowej (zob. s. 33).
Uzyskane frakcje nasyconych, jedno- i dwunienasyconych lipidów rezorcyno-
lowych rozdzielić na homologi różniące się długością łańcuchów alifatycz-
nych w wysokociśnieniowej chromatografii cieczowej na fazach odwróco-
nych stosując kolumnę wypełnioną hydrofobowym żelem krzemionkowym
(Si 60 RP-18) i elucję mieszaniną metanolu z wodą (96:4). Detekcja UV przy
280 nm. Poszczególne homologi zbierać do oddzielnych naczyń, odparować
z nich rozpuszczalnik i rozpuścić w określonej ilości metanolu. Oznaczyć stę-
żenie lipidów rezorcynolowych metodą z Fast Blue B (zob. rozdział dotyczący
metod ilościowych). Przechowywać w 0
0
C.
1.3. Oznaczanie składu
reszt kwasów tłuszczowych
w glicerydach i fosfolipidach
Aby stwierdzić, jakiej długości reszty acylowe występują w badanych przez nas
lipidach, należy przeprowadzić hydrolizę tych lipidów w celu uwolnienia kwasów
tłuszczowych, które następnie zostaną poddane analizie chromatograficznej. Po-
nieważ najbardziej popularną metodą analizy kwasów tłuszczowych jest chroma-
tografia gazowa, konieczne jest przeprowadzenie kwasów tłuszczowych w lotne
pochodne, zwykle estry metylowe. Najczęstszym postępowaniem umożliwiają-
cym równoczesną hydrolizę glicerydów i fosfolipidów i ich modyfikację jest al-
koholiza w środowisku kwaśnym. Uzyskane estry metylowe mogą być analizo-
wane bezpośrednio w chromatografii gazowej lub też w chromatografii cienko-
warstwowej (szczególnie jakościowo).
1.3.1. Transestryfikacja lipidów
(wg Christie W. W., w: Handbook of chromatography (red. Mangold H. K.)
CRC Press, Boca Raton, 1984, vol. 1. s. 33-46)
Odczynniki i roztwory:
heksan;
5% HCl w bezwodnym metanolu (A) lub 1% stężony kwas siarkowy w absolut-
nym metanolu (B) lub 14% trójfluorek boru w metanolu (C);
5% NaCl;
4% węglan potasu;
bezwodny siarczan sodu w substancji.

1.3. Oznaczanie składu reszt kwasów tłuszczowych 37
Sprzęt:
mikrostrzykawki;
zakręcane probówki reakcyjne;
łaźnia wodna.
Postępowanie:
• l ml roztworu benzenowego zawierającego 50-100 mg lipidów umieścić w za-
kręcanej probówce i dodać 3 ml odczynnika modyfikującego (A, B lub C).
• Zakręcić probówkę i umieścić w temperaturze 60°C na 30 min.
• Po ochłodzeniu próbki dodać 5 ml 5% roztworu NaCl i po wymieszaniu
ekstrahować mieszaninę 3-krotnie 5 ml porcjami n-heksanu.
• Ekstrakty heksanowe połączyć, przemyć 10 ml 4% roztworu węglanu potasu,
a następnie osuszyć bezwodnym siarczanem sodu.
• Osuszone ekstrakty zagęścić przez odparowanie części rozpuszczalnika np.
strumieniem azotu i użyć do dalszej analizy.
1.3.2. Rozdział estrów metylowych
kwasów tłuszczowych
w cienkowarstwowej chromatografii argentacyjnej
(wg Ackman R. G., w: Handbook of chromatography (red. Mangold H. K.)
CRC Press, Boca Raton, 1984, vol. 1. s. 95-240)
Materiały, odczynniki i roztwory:
płytki do chromatografii cienkowarstwowej pokryte żelem krzemionkowym;
10% azotan srebrowy w 50% metanolu;
heksan;
eter etylowy;
roztwór 2',7'-dichlorofluoresceiny (zob. s. 45).
Sprzęt:
komora chromatograficzna;
suszarka;
lampa UV.
Postępowanie:
• Przygotowane płytki zanurzyć w płaskim naczyniu do 10% roztworu azotanu
srebrowego, wytrząsać delikatnie przez 2-3 min, odsączyć od nadmiaru roz-
tworu i wysuszyć na powietrzu przez 5 min, a następnie w 70°C przez 30 min.
• Roztwór estrów metylowych w heksanie nanieść na płytki w postaci kresek
i chromatogram rozwijać jednokrotnie lub dwukrotnie (po rozwinięciu wysuszyć
płytkę i ponownie rozwinąć chromatogram w tym samym kierunku i w tym sa-
mym układzie rozpuszczalników) w układzie heksan/eter etylowy (95:5).
1. Lipidy
38
Po rozwinięciu chromatogramu płytki wysuszyć i w celu detekcji plam spry-
skać roztworem dichlorofluoresceiny, oglądać chromatogramy pod lampą UV
(366 nm). Poczynając od frontu rozpuszczalnika kolejność migracji jest nastę-
pująca: nasycone, jedno- i dwunienasycone estry kwasów tłuszczowych. Moż-
na również zaobserwować rozdzielanie izomerów cis i trans (izomery trans
migrują szybciej od izomerów cis). Stosując płytki do chromatografii prepara-
tywnej można wydzielić poszczególne grupy estrów w ilości wystarczającej
do ich analizy w chromatografii gazowej.
1.3.3. Analiza estrów metylowych
kwasów tłuszczowych w chromatografii gazowej
Sprzęt:
chromatograf gazowy z detektorem FID
4
i rejestratorem;
butle z gazami: helem, wodorem i powietrzem;
kolumna kapilarna z fazą stacjonarną SE-54;
mikrostrzykawka do nanoszenia próbek.
Postępowanie:
• Ustawić następujące parametry pracy chromatografu:
kolumna 25 m x 0.25 mm, gaz nośny He, ciśnienie 0.08-0.09 MPa,
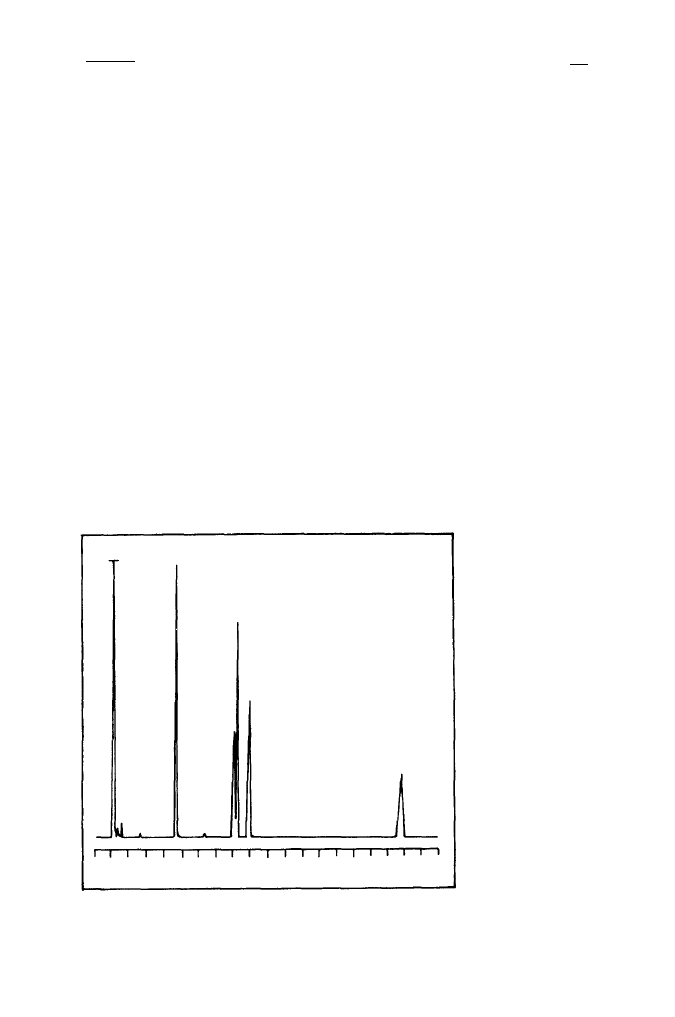
Rys. 2. Rozdział
mieszaniny estrów
metylowych kwasów
tłuszczowych
w kapilarnej
chromatografii
gazowej na kolumnie
z SE-54
4
Flame Ionization Detector - detektor płomieniowo-jonizacyjny.
18:
1
18:
3
18:
0
20:
0
0
min
5 10 15 20
16:0
18:
2

1.4. Metody ilościowe analizy lipidów 39
detektor płomieniowo-jonizacyjny; H
2
, 0.014 MPa; O
2
, 0.1 MPa;
temperatura komory nastrzykowej - 200°C,
temperatura detektora - 270°C,
rozdział izotermiczny w temperaturze 190°C,
czułość wzmacniacza 1x10
9
lub 3 x l0
9
,
wielkość próbki l μl, dzielnik l :50
• Po ustabilizowaniu termicznym i elektronicznym aparatu
wstrzyknąć do ko-
mory nastrzykowej l μl roztworu heksanowego estrów metylowych kwasów
tłuszczowych; zaznaczyć moment wstrzykiwania na rejestratorze.
• Śledzić proces rozdziału przez 30 min.
• W celu porównania i identyfikacji frakcji przeprowadzić rozdział mieszaniny
standardowej metylowanych kwasów tłuszczowych.
Na Rys. 2 przedstawiono typowy chromatogram wzorców.
• Określić ilościowy skład reszt acylowych w analizowanej mieszaninie.
1.4. Metody ilościowe analizy lipidów
1.4.1. Oznaczanie lipidów całkowitych
metodą wanilinową
Odczynniki i roztwory:
kwas siarkowy stężony;
0.01 M wanilina w 11.5 M kwasie o-fosforowym (do 115 ml 85% kwasu ortofo-
sforowego dodać 20 ml wody), odczynnik trwały przez rok;
wzorzec - 700 mg cholesterolu rozpuścić w 100 ml metanolu.
Podstawą oznaczania jest reakcja fosfowaniliny z wiązaniem nienasyconym. Re-
akcję dodatnią dają również ketony, wyższe alkohole, związki z aktywną grupą
metylenową, hydrazyny i niektóre leki.
Postępowanie:
• W suchej probówce umieścić próbę badaną (50 μl) i dodać do niej 2 ml kwasu
siarkowego, po ostrożnym wymieszaniu ogrzewać przez 20 min we wrzącej
łaźni wodnej, ochłodzić, podobnie postąpić z próbą wzorcową używając 50 μl
roztworu cholesterolu.
•
Pobrać do probówek po 0. l ml powyższych hydrolizatów (próby badanej lub
wzorca), dodać 4 ml odczynnika wanilinowego, wymieszać i odstawić na 20
min.
• Odczytać absorbancje próby badanej oraz wzorcowej przy 525 nm wobec
próby ślepej odczynnikowej zawierającej zamiast hydrolizatu 0.1 ml stężone-

1. Lipidy 40
go kwasu siarkowego. Odczyty powinny być dokonane w ciągu 30 min od
zakończenia reakcji.
Obliczenia:
μg lipidu w
próbie =
1.4.2. Oznaczanie
cholesterolu całkowitego
(wg Courchaine A. J., Miller W. H., Stein D. B. Jr., Clin. Chem. 5 (1959) 609-613)
Produkty reakcji cholesterolu z silnym kwasem dają barwne kompleksy z solami
żelazowymi.
Odczynniki i roztwory:
kwas octowy;
odczynnik żelazowy - 4 ml FeCl
3
(2.5 g FeCl
3
x 6 H
2
O rozpuścić w 100 ml 85%
kwasu ortofosforowego) uzupełnić ostrożnie do 50 ml stężonym kwasem siarko-
wym; odczynnik jest trwały do utraty klarowności;
wzorzec - 10 mg cholesterolu rozpuścić w 100 ml kwasu octowego.
Postępowanie:
• Próbkę lipidów zawierającą do 100 μg cholesterolu umieścić w suchej pro-
bówce i odparować z niej rozpuszczalnik.
• Do pozostałości dodać l .5 ml stężonego kwasu octowego i l ml odczynnika
żelazowego.
• Po dokładnym wymieszaniu odczekać 10 min i odczytać absorbancję próby
przy 550 nm wobec ślepej odczynnikowej.
• Ilość cholesterolu w próbie określić z krzywej kalibracyjnej sporządzonej dla
20-100 μg cholesterolu.
1.4.3. Oznaczanie
glikolipidów
metodą fenolową
(wg Roughan P. G., Batt R. D., Anal. Biochem. 22 (1968) 74-88)
Stężone kwasy odwadniają reszty cukrowe gliko- i galaktolipidów do oksyme-
tylenofurfurali, które z fenolem dają barwne produkty.
Odczynniki i roztwory:
stężony kwas siarkowy;
5% (w/v) roztwór fenolu w wodzie;
wzorzec cukru - 2.5 mM roztwór wodny galaktozy.

l .4. Metody ilościowe analizy lipidów 41
Postępowanie:
• Do l ml roztworu fenolu wstrzyknąć próbkę chloroformowego roztworu lipi-
dów (do 50 μl).
•
Dodać ostrożnie (!) 4 ml stężonego kwasu siarkowego, wymieszać na mikro-
wytrząsarce i ostawić do ostygnięcia do temperatury pokojowej.
• Odczytać absorbancję próby przy 485 nm wobec próby ślepej odczynnikowej.
Przy 500 nmolach galaktozy absorbancja sięga 0.7 A.
• Oznaczyć zawartość cukru w próbie w oparciu o krzywą kalibracyjną wyko-
naną dla 100-500 nmoli galaktozy (40-200 μl).
1.4.4. Oznaczanie fosforu w fosfolipidach
Fosfolipidy ogrzewane z kwasem nadchlorowym
ulegają destrukcji dając CO
2
,
H
2
O oraz PO
43-
. W środowisku kwaśnym ortofosforan tworzy z molibdenianem
amonowym fosforomolibdenian amonowy. Związek ten pod wpływem czynni-
ków redukujących ulega redukcji do mieszanych tlenków molibdenu, tzw. błękitu
molibdenowego (Mo
2
O
5
x MoO
3
), którego ilość określana jest kolorymetrycznie.
Metoda I
(wg Bartlett G. R., J. Biol. Chem. 234 (1959) 466-469)
Odczynniki i roztwory:
molibdenian amonowy;
stężony kwas siarkowy;
dwusiarczyn sodowy Na
2
S
2
O
5
;
siarczyn sodowy Na
2
SO
3
;
kwas l -amino-2-naftaleno-4-sulfonowy;
70% kwas nadchlorowy.
Roztwory robocze:
• Rozpuścić 2.2 g molibdenianu w 20 ml 98% kwasu siarkowego (traktując
stężony jako 100%) stosując ciągłe mieszanie przez 10 min, uzupełnić wodą
destylowaną do 1000 ml.
• Odczynnik Fiske-Subbarowa: 27.36 g dwusiarczynu sodowego oraz l g siar-
czynu sodowego rozpuścić w 200 ml wody destylowanej, dodać 0.5 g kwasu
aminonaftalenosulfonowego i mieszać przez 15 min w temperaturze ok. 40°C,
odstawić na noc w ciemne miejsce, a następnie przesączyć do ciemnej butelki.
Odczynnik trwały przez ok. 8 tygodni w 4°C.
• Roztwór standardowy: 275.7 mg KH
2
PO
4
rozpuścić w 250 ml wody. 10 ml
tego roztworu uzupełnić do 250 ml wodą; l ml zawiera 10 μg P
i
.

1. Lipidy 42
Postępowanie:
• Próbkę lipidów uwolnić od rozpuszczalników przez ogrzanie kilka min
w 120°C.
• Dodać 300 μl kwasu nadchlorowego, wymieszać, przykryć kulką szklaną
i umieścić na 30 min w bloku grzejnym (180-190°C).
• Ochłodzić i dodać 3 ml molibdenianu, zmieszać.
• Dodać 120 μl odczynnika Fiske-Subbarowa i zmieszać.
• Przykryć kulką szklaną i ogrzewać we wrzącej łaźni wodnej przez 15 min.
• Ochłodzić i odczytać absorbancję próby przy 830 nm (barwa stabilna przez 24
godz.). 90 nmoli P
i
daje absorbancję ok. 0.7.
• Oznaczyć ilość fosforu w próbie posługując się wykonaną krzywą kalibracyj-
ną (10-90 nmoli P
i
). Etap spalania może być tu pominięty.
Metoda II
(wg Rouser G., Siakotos A. N., Fleischer S., Lipids l (1966) 85-86)
Roztwory:
70% kwas nadchlorowy;
2.5% wodny roztwór molibdenianu amonowego;
10% wodny roztwór kwasu askorbinowego (na świeżo!);
standardowy roztwór fosforanu jw.; krzywa kalibracyjną jest liniowa do 10 μg P
i
.
Postępowanie:
• Próbkę lipidów umieścić w suchej probówce, odparować rozpuszczalnik i do-
dać 0.9 ml kwasu nadchlorowego.
• Przykryć probówkę kulką szklaną i spalać 30 min w 180°C.
• Ochłodzić i dodać 5 ml wody spłukując przy okazji ścianki probówki.
• Dodać l ml molibdenianu, l ml kwasu askorbinowego oraz 2 ml wody, do-
kładnie wymieszać.
• Przykryć kulką i ogrzewać we wrzącej łaźni wodnej przez 5 min.
• Ochłodzić w strumieniu zimnej wody.
• Odczytać absorbancję klarownego supernatantu przy 825 lub 797 nm wobec
ślepej odczynnikowej (w przypadku oznaczeń frakcji z TLC ślepą stanowić
będzie podobna jak w próbie badanej ilość żelu, lecz zdrapana z miejsca,
w którym nie wykazano fosfolipidów).
• Odczytać ilość fosforu w próbie posługując się krzywą kalibracyjną.

l .4. Metody ilościowe analizy lipidów 43
1.4.5. Oznaczanie fosfolipidów w zawiesinach wodnych
(np. w liposomach)
(wg Hallen R. M„ J. Biochem. Biophys. Methods 2 (1980) 251-255)
Metoda opiera się na tworzeniu rozpuszczalnych w chloroformie kompleksów
fosfolipidów z błękitem molibdenowym. Metoda jest niewrażliwa na dodatki
mogące być obecne w zawiesinie, takie jak 5% etanol, 0.05 M CaCl
2
, 0.1 M NaCl,
0.5% dezoksycholan sodowy, 0.01 M fosforan sodowy, 0.5% albumina surowicy,
0.5% cholesterol.
Odczynniki i roztwory:
chloroform;
metanol;
roztwór roboczy:
A. 5 g molibdenianu amonowego rozpuścić w 200 ml 75% kwasu siarkowego;
B. 3 g molibdenianu amonowego rozpuścić w 50 ml stężonego kwasu solne-
go, a następnie mieszać przez 30 min na mieszadle magnetycznym z 2 ml rtęci.
Przesączyć i dodać powoli do roztworu A. Uzyskany ciemnoniebieski roztwór
(C) pozostawić do ostygnięcia i przelać do ciemnej butelki. Odczynnik trwały
w temperaturze pokojowej co najmniej 3 miesiące.
Postępowanie:
• 2 ml zawiesiny fosfolipidów w roztworze wodnym (liposomów) zawierającej
nie więcej niż l μmoli lipidu umieścić w zakręcanej probówce i dodać l ml
roztworu C.
• Wymieszać i ogrzewać przez 2 min we wrzącej łaźni wodnej.
• Przestudzić próbę przez 5 min, a następnie dodać 3.5 ml mieszaniny chloro-
formu z metanolem (9:1).
• Zakręcić probówkę i dokładnie wymieszać fazy przez jej odwracanie.
• Odstawić probówkę na 30 min dla rozdzielenia faz.
• Pobrać fazę chloroformową (dolna) i odczytać absorbancję niebieskiego kom-
pleksu przy 760 nm wobec chloroformu jako ślepej odczynnikowej. Krzywa
kalibracyjną jest liniowa w zakresie 0.05-1.0 A, co odpowiada stężeniu do 0.6
μmola lipidu.
1.4.6. Oznaczanie
lipidów rezorcynolowych
(wg Tłuścik F., Kozubek A., Mejbaum-Katzenellenbogen W., Acta Soc. Bot. Polon. 50
(1981)645-651)
Metoda opiera się na tworzeniu barwnych związków dwuazowych w wyniku
reakcji barwników dwuazonionych z fenolami. Lipidy rezorcynolowe dają swoi-
ste fioletowoczerwone związki, których ilość oznaczana jest kolorymetrycznie.
Wyszukiwarka
Podobne podstrony:
Oznaczanie składu ziarnowego kruszyw Metoda przesiewania, Budownictwo
Analizy - Gleba cwiczenia, OZNACZENIE SKŁADU GRANULOMETRYCZNEGO GLEBY
Utlenianie kwasów tłuszczowych i ketogeneza
UTENIANIE KWASÓW TŁUSZCZOWYCH
Reakcje utleniania kwasów tłuszczowych i triacylogriceroli
tluszcze glicerydy
oznaczanie składu wina, Technologia żywnosci i Żywienie człowieka, 4 SEMESTR, Analiza żywności
Sprawozdanie-oznaczenie skladu granulometrycznego, niezbędnik rolnika 2 lepszy, Gleboznawstwo, spraw
Oznaczenie skladu granulometrycznego nasze, Ochrona gleb
Oznaczanie składu mechanicznego gleby
8 Dieta niskocholesterolowa O kontrolowanej zawartości kwasów tłuszczowych
Oznaczenie składu ziarnowego kruszyw mineralnych
kwasy i pochodne Reakcja kwasów tłuszczowych z roztworem bromu i roztworem manganianu(VII) potasu
Tłuszcze glicerydy
PN EN 933 1 2000 Badania geometrycznych wl kruszyw Oznaczanie skladu ziarnowego Metoda przesiewan
więcej podobnych podstron