
FARMACEUTYCZNE STANDARDY
SPORZÑDZANIA MIESZANIN
DO ˚YWIENIA POZAJELITOWEGO
POLSKIE TOWARZYSTWO FARMACEUTYCZNE
SEKCJA ˚YWIENIA DO I POZAJELITOWEGO
2009

2
FARMACEUTYCZNE STANDARDY SPORZÑDZANIA
MIESZANIN DO ˚YWIENIA POZAJELITOWEGO
Opracował zespół w składzie:
mgr farm. El˝bieta Balcerzak
Szpital Kliniczny Przemienienia Paƒskiego Uniwersytetu Medycznego w Poznaniu
dr n. farm. Krystyna Chmal-Jagiełło
Wojewódzki Szpital Specjalistyczny im. L. Rydygiera w Krakowie
dr n. farm. Maria Ciszewska-J´drasik
Zakład Farmacji Stosowanej Warszawski Uniwersytet Medyczny
mgr farm. Alina Górecka
Szpital Kliniczny im. Heliodora Âwi´cickiego Uniwersytetu Medycznego w Poznaniu
mgr farm. Joanna Jackowska-Janda
Uniwersytecki Szpital Dzieci´cy w Krakowie
dr n. farm. Hanna Jankowiak-Gracz
Szpital Kliniczny Przemienienia Paƒskiego Uniwersytetu Medycznego w Poznaniu
mgr farm. Stanisława Krystynowicz
Uniwersytecki Szpital Dzieci´cy w Krakowie
mgr farm. Anna Łohynowicz
Szpital Kliniczny im. Karola Jonschera Uniwersytetu Medycznego w Poznaniu
mgr farm. Krystyna Malinger
Ginekologiczno-Poło˝niczy Szpital Kliniczny Uniwersytetu Medycznego w Poznaniu
mgr farm. Gra˝yna Rembowska
Szpital Kliniczny im. Heliodora Âwi´cickiego Uniwersytetu Medycznego w Poznaniu
mgr farm. Ewa Tobolska-Klimek
Pomorskie Centrum Traumatologii Wojewódzki Szpital Specjalistyczny
im. Mikołaja Kopernika w Gdaƒsku
mgr farm. Bo˝enna Tondys
Centralny Szpital Kliniczny MSWiA w Warszawie
Konsultacja naukowa:
prof. dr hab. Edmund GrzeÊkowiak
prof. nadzw. dr hab. Edmund Sieradzki
Redakcja:
mgr farm. Ilona Anisimowicz
ZATWIERDZONE PRZEZ
POLSKIE TOWARZYSTWO FARMACEUTYCZNE SEKCJ¢ ˚YWIENIA DO I POZAJELITOWEGO
Rok wydania 2009

3
S¸OWO WST¢PNE
Zawód farmaceuty szpitalnego zaliczany jest bez wàtpienia do grupy zawodów zaufa-
nia publicznego, w stosunku do których społeczeƒstwo oczekuje spełnienia pewnej misji.
Wdra˝ane obecnie globalne strategie farmakoterapeutyczne sprawiajà, ˝e współczesny
farmaceuta szpitalny to nie tylko wybitny technolog postaci leku, profesjonalnie przy-
gotowany analityk w dziedzinie oceny jakoÊci i biodost´pnoÊci leku, aktywny w odniesie-
niu do pacjentów i innych członków zespołu terapeutycznego uczestnik opieki
zdrowotnej, ale tak˝e wysokiej klasy członek interdyscyplinarnego zespołu terapeuty-
cznego, współodpowiedzialny za optymalizacj´, bezpieczeƒstwo i skutecznoÊç przyj´-
tych procedur terapeutycznych.
Kliniczny charakter współczesnej farmacji szpitalnej przejawia si´ tak˝e w uczestnicze-
niu farmaceutów szpitalnych w monitorowaniu działaƒ niepo˝àdanych, badaniach klini-
cznych leków, terapeutycznym monitorowaniu st´˝eƒ leków czy wykorzystywaniu
znajomoÊci farmakokinetyki w ustalaniu schematów dawkowania.
Szczególnà aktywnoÊcià zawodowà farmaceutów szpitalnych jest równie˝ indywiduali-
zacja dawkowania, realizowana w opracowywaniu i wdra˝aniu standardów jakoÊ-
ciowych w farmacji onkologicznej, standardów sporzàdzania mieszanin do ˝ywienia
pozajelitowego czy wprowadzania systemów unit-dose.
Wymienione wy˝ej aktywnoÊci zawodowe współczesnego farmaceuty szpitalnego Êwiad-
czà dobitnie nie tylko o swoistej słu˝bie na rzecz pacjentów, ale tak˝e o realizacji
społecznej misji, której nadrz´dnym celem jest dobro i zdrowie pacjenta.
Z tym wi´kszym zadowoleniem nale˝y przyjàç i zaakceptowaç „Farmaceutyczne
standardy sporzàdzania mieszanin do ˝ywienia pozajelitowego” opracowane przez
zespół farmaceutek szpitalnych, reprezentujàcych oÊrodki akademickie w Gdaƒsku,
Krakowie, Poznaniu i Warszawie.
Przejawem wielkiej odpowiedzialnoÊci zawodowej Autorek niniejszego opracowania jest
nie tylko przytoczenie obowiàzujàcych podstaw prawnych, regulujàcych zasady
sporzàdzania mieszanin do ˝ywienia pozajelitowego w aptece szpitalnej, prezentacja
wymogów dotyczàcych pomieszczeƒ do ich przygotowywania, standaryzacji procesów
sporzàdzania i kontroli preparatów, klasyfikacji i utylizacji odpadów, ale tak˝e przed-
stawienie problemów dotyczàcych tzw. domowego ˝ywienia pozajelitowego.
Nale˝y stwierdziç, ˝e prezentowane przez Autorki poglàdy w tym wzgl´dzie całkowicie
korelujà ze stanowiskiem Prezydium Naczelnej Izby Lekarskiej z dnia 13 lutego 2009
i doskonale wpisujà si´ w poszukiwanie konstruktywnych, prawno-technologiczno-
-logistycznych rozwiàzaƒ dla dobra pacjenta.
Prof. dr hab. Edmund GrzeÊkowiak
Krajowy Konsultant ds. Farmacji Szpitalnej

4
FARMACEUTYCZNE STANDARDY SPORZÑDZANIA
MIESZANIN DO ˚YWIENIA POZAJELITOWEGO
S¸OWO WST¢PNE
Dynamiczny rozwój leczenia ˝ywieniowego i wielka ró˝norodnoÊç zastosowaƒ w prak-
tyce klinicznej wymaga wysoko specjalistycznej wiedzy w zakresie zastosowaƒ oraz
produkcji preparatów do ˝ywienia pozajelitowego i dojelitowego.
Zgodnie z Prawem farmaceutycznym w odniesieniu do aptek szpitalnych, usługà
farmaceutycznà jest mi´dzy innymi sporzàdzanie leków do ˝ywienia pozajelitowego.
Aktualnie na Êwiecie produkcja leków w przemyÊle czy w aptekach powinna odbywaç
si´ zgodnie z wytycznymi Dobrej Praktyki Wytwarzania. Jednym z istotnych elementów
Dobrej Praktyki Wytwarzania jest standaryzacja procesów produkcji.
W skład zespołu, który podjàł si´ opracowania farmaceutycznych standardów
sporzàdzania mieszanin do ˝ywienia pozajelitowego, wchodzà znakomici farmaceuci
pracujàcy w ró˝nych oÊrodkach akademickich w Polsce. Aktywnie uczestniczà oni w kra-
jowych i mi´dzynarodowych zjazdach, których wiodàcà tematykà jest zagadnienie
˝ywienia pozajelitowego, prezentujàc tam swoje wyniki i doÊwiadczenia w tym zakresie.
Koncepcja opracowanych standardów sporzàdzania mieszanin do ˝ywienia pozajeli-
towego jest logiczna i chronologicznie obejmuje wszystkie etapy zwiàzane z wy-
twarzaniem produktów leczniczych spełniajàcych wymagania farmakopei, ich kontrole
oraz utylizacj´.
Na szczególnà uwag´ zasługuje umieszczenie w opracowaniu schematu organiza-
cyjnego pomieszczeƒ przeznaczonych do sporzàdzania mieszanin do ˝ywienia poza-
jelitowego. Opracowanie to nabiera szczególnego znaczenia w sytuacji, kiedy
organizowane sà nowe pracownie w szpitalach.
Opracowanie standardów sporzàdzania mieszanin do ˝ywienia pozajelitowego
w neonatologii daje r´kojmi´, ˝e stosowanie mieszanin w praktyce klinicznej u noworod-
ków urodzonych przedwczeÊnie b´dzie bezpieczne i efektywne klinicznie.
Poszczególne rozdziały zostały opracowane zgodnie z wymaganiami Dobrej Praktyki
Wytwarzania. Zawarte treÊci pozwalajà na prawidłowe sporzàdzanie mieszanin do
˝ywienia pozajelitowego przeznaczonych dla chorych dorosłych, pediatrycznych i neona-
tologicznych.
W oparciu o opracowane standardy sporzàdzania mieszanin do ˝ywienia poza-
jelitowego powinny odbywaç si´ systematyczne szkolenia farmaceutów, lekarzy i piel´g-
niarek, a tak˝e powinny one byç uwzgl´dnione w programach kształcenia studentów
na Wydziałach Farmaceutycznych.
Przestrzeganie opracowanych i obowiàzujàcych schematów znacznie ułatwi prac´,
zminimalizuje ryzyko popełniania bł´dów, a przez to poprawi bezpieczeƒstwo
stosowania mieszanin do ˝ywienia pozajelitowego w praktyce klinicznej.
Prof. nadzw. dr hab. Edmund Sieradzki
Kierownik Zakładu Farmacji Stosowanej Warszawski Uniwersytet Medyczny

5
SPIS TREÂCI
0
1. Wprowadzenie..............................................................................................6
0
2. Definicje i okreÊlenia .....................................................................................7
0
3. Podstawy prawne ........................................................................................10
0
4. Pomieszczenia do sporzàdzania mieszanin do ˝ywienia pozajelitowego (˚P) ...15
0
5. Badania przestrzeni pracy oraz personelu.....................................................18
0
6. Personel......................................................................................................21
0
7. Przygotowanie pracownika do pracy w warunkach aseptycznych....................23
0
8. Przygotowanie produktów leczniczych oraz wyrobów medycznych
przed wprowadzeniem ich do aseptycznego boksu........................................24
0
9. Recepta ......................................................................................................25
10. Etykieta.......................................................................................................26
11. Ogólne zasady sporzàdzania mieszanin do ˚P .............................................27
12. Sporzàdzanie mieszanin metodà grawitacyjnà ..............................................30
13. Sporzàdzanie mieszanin za pomocà biurety..................................................32
14. Sporzàdzanie mieszanin w neonatologii .......................................................35
15. Sporzàdzanie mieszanin w workach gotowych do u˝ycia (RTU) ......................38
16. Sporzàdzanie mieszanin za pomocà pomp sterowanych komputerowo ..........44
17. Zalecane Êrodki ostro˝noÊci podczas sporzàdzania,
przechowywania i podawania mieszanin do ˚P.............................................46
18. Kontrola procesu sporzàdzania mieszanin do ˚P...........................................47
19. Utylizacja....................................................................................................50
20. Szkolenia ....................................................................................................52
21. Domowe ˝ywienie pozajelitowe....................................................................53
22. PiÊmiennictwo .............................................................................................58

6
FARMACEUTYCZNE STANDARDY SPORZÑDZANIA
MIESZANIN DO ˚YWIENIA POZAJELITOWEGO
1. WPROWADZENIE
˚ywienie pozajelitowe (˚P) jest obecnie rutynowo stosowanà metodà leczenia chorych,
którzy nie mogà od˝ywiaç si´ drogà przewodu pokarmowego. Jednym z najistotniejszych
czynników decydujàcych o skutecznoÊci ˚P jest poda˝ choremu kompletnej, stabilnej
mieszaniny od˝ywczej o składzie dostosowanym do jego indywidualnych potrzeb. O jakoÊci
mieszaniny decydujà przede wszystkim warunki i sposób jej wykonania. Podstawowe
znaczenie ma równie˝ odpowiednio wyszkolony, kompetentny personel farmaceutyczny
przygotowany do realizacji powierzonych mu zadaƒ.
Sporzàdzanie mieszanin do ˝ywienia pozajelitowego musi odbywaç si´ w aptece szpitalnej,
zgodnie z obowiàzujàcymi aktami prawnymi i opracowanymi procedurami. Daje to
gwarancj´ uzyskania produktu o po˝àdanej i powtarzalnej jakoÊci.
Dynamiczny rozwój leczenia ˝ywieniowego, wprowadzenie go do codziennej praktyki
w wielu szpitalach wpłyn´ły na zwi´kszenie aktywnoÊci farmaceutów szpitalnych w zakresie
realizacji usługi farmaceutycznej, jakà jest sporzàdzanie mieszanin do ˚P. Zaistniała potrzeba
opracowania uaktualnionych, dostosowanych do obecnych wymagaƒ, farmaceutycznych
standardów sporzàdzania mieszanin do ˚P tj. dokumentów, które w uporzàdkowany sposób
opisujà jakie czynnoÊci, w jakiej kolejnoÊci, przez kogo i w jakich warunkach majà byç
wykonywane. Zatwierdzone przez Krajowego Konsultanta ds. Farmacji Szpitalnej standardy
mogà ułatwiç rozmowy kierowników aptek z dyrekcjami szpitali na temat dostosowania
pomieszczeƒ i wyposa˝enia aptek szpitalnych do obowiàzujàcego Prawa farmaceutycznego.
Celem standardów jest pomoc farmaceutom w organizacji pracowni ˚P oraz prawidłowym
sporzàdzaniu mieszanin do ˚P, przeznaczonych dla chorych dorosłych, pediatrycznych
i neonatologicznych.
Ka˝da pracownia ˚P, w oparciu o Farmaceutyczne Standardy, powinna opracowaç własne,
dostosowane do lokalnych warunków pisemne procedury i instrukcje. Dobra organizacja
pracy, ÊciÊle zwiàzana z przestrzeganiem standardów i procedurpost´powania, daje
gwarancj´ bezpieczeƒstwa oraz pewnoÊci, ˝e ka˝dy preparat b´dzie sporzàdzony w prawi-
dłowy sposób, a dodatkowo skraca czas pracy i przynosi oszcz´dnoÊci ekonomiczne. Poprzez
dobrà organizacj´ pracy mo˝na osiàgnàç zadowalajàcy standard Dobrej Praktyki
Wytwarzania nawet przy ograniczonej powierzchni, wyposa˝eniu oraz kosztach.
Bezpieczeƒstwo, które w Unii Europejskiej stawiane jest wy˝ej ni˝ jakoÊç, musi byç gwaran-
tem naszej pracy na rzecz chorych. Mo˝emy osiàgnàç je poprzez: standaryzacj´
sporzàdzania mieszanin do ˚P, kontrol´ i walidacj´ warunków wytwarzania oraz wiedz´,
którà musimy nabyç, aby byç partnerem dla lekarza i pacjenta.
W niniejszym opracowaniu przedstawiamy: podstawy prawne sporzàdzania mieszanin do
˚P w aptece szpitalnej, schemat pracowni ˚P, wymagane badania przestrzeni pracy oraz
personelu, ró˝ne metody sporzàdzania mieszanin – zasady post´powania na poszcze-
gólnych etapach pracy, kontrol´ procesu, utylizacj´ odpadów. Poszczególne tematy zostały
opracowane w oparciu o wytyczne Dobrej Praktyki Wytwarzania, obowiàzujàce akty
prawne, piÊmiennictwo, a tak˝e wieloletnie doÊwiadczenia własne.

7
WPROWADZENIE
2. DEFINICJE I OKREÂLENIA
All-in-One – metoda ˝ywienia pozajelitowego systemem jednego pojemnika, w którym
mieszane sà wszystkie substancje od˝ywcze.
Aseptyka – post´powanie zapobiegajàce lub niedopuszczajàce do zaka˝enia.
Działanie takie ma zapewniç jałowoÊç produktu leczniczego.
Chory leczony ˝ywieniem pozajelitowym lub dojelitowym – osoba, która z powodu
braku mo˝liwoÊci poda˝y substancji od˝ywczych drogà naturalnà w iloÊci wystarczajàcej
do utrzymania przy ˝yciu, wymaga całkowitego lub suplementarnego podawania sub-
stancji od˝ywczych do˝ylnie, przez zgł´bnik lub przetok´ od˝ywczà.
Czàstki – obiekty stałe o wymiarach w przedziale 0,1–10 μm (np. czàstki emulsji tłusz-
czowej, czàstki mechaniczne w powietrzu, płynach).
CzystoÊç mikrobiologiczna – nieobecnoÊç drobnoustrojów w iloÊci wi´kszej ni˝
dopuszczona dla danej klasy czystoÊci.
Data wa˝noÊci – data, przed upływem której dany produkt leczniczy i wyrób medyczny
spełnia wszystkie ustalone wymagania, a po upływie której nie mo˝e byç stosowany.
Dezynfekcja – zabicie lub inaktywacja drobnoustrojów chorobotwórczych i ich form
przetrwalnikowych za pomocà Êrodków fizycznych lub chemicznych.
Dobra Praktyka Wytwarzania (Good Manufacturing Practice) – system zapewnienia
jakoÊci, obejmujàcy zasady prawidłowego post´powania podczas wytwarzania i kon-
troli produktów leczniczych.
Etykieta – nalepka lub napis zawierajàcy informacje o zawartoÊci opakowania.
Filtr HEPA (High-Efficiency Particulate Air Filter) – filtr absolutny, słu˝àcy do oczyszcza-
nia powietrza, zatrzymujàcy w 99,9997% czàstki o Êrednicy 0,3 μm i wi´ksze, zapew-
niajàcy nawiew laminarny jałowego powietrza.
JałowoÊç – nieobecnoÊç zdolnych do ˝ycia drobnoustrojów.
Klasa czystoÊci powietrza – okreÊlona liczba czàstek mechanicznych i ˝ywych drobno-
ustrojów w jednostce obj´toÊciowej powietrza.
Laminarny przepływ jałowego powietrza – przepływ liniowy, jednokierunkowy

8
FARMACEUTYCZNE STANDARDY SPORZÑDZANIA
MIESZANIN DO ˚YWIENIA POZAJELITOWEGO
jałowego powietrza, o jednostajnej pr´dkoÊci; mo˝e byç pionowy lub poziomy.
Lek recepturowy – produkt leczniczy sporzàdzony w aptece na podstawie recepty
lekarskiej.
Mieszanina do ˝ywienia pozajelitowego – mieszanina wszystkich substancji od˝yw-
czych o ÊciÊle okreÊlonym składzie, podawana pozajelitowo.
Mieszanina do ˝ywienia pozajelitowego „na zapas” – mieszanina przygotowana na
kilka dni przed planowanym podaniem pacjentowi, przechowywana w obni˝onej
temperaturze.
Monitorowanie – systematyczne wykonywanie, w sposób ciàgły lub w regularnych
odst´pach czasu, z góry zaplanowanych pomiarów w celu stwierdzenia, czy wymagane
warunki sà cały czas spełniane.
Najgorszy przypadek – sposób przeprowadzenia walidacji polegajàcy na udowod-
nieniu, ˝e nawet w najgorszych mo˝liwych warunkach prowadzenia procesu wytwarzany
jest produkt spełniajàcy wymagania z góry ustalonej jakoÊci.
NiezgodnoÊci – niepo˝àdane oddziaływania wyst´pujàce pomi´dzy poszczególnymi
składnikami mieszaniny do ˝ywienia pozajelitowego w tzw. fazie farmaceutycznej,
tj. w czasie przygotowywania, przechowywania i podawania.
Obszar krytyczny – obszar znajdujàcy si´ wewnàtrz strefy przestrzeni czystej, w którym
wykonywane sà czynnoÊci aseptyczne obarczone najwi´kszym ryzykiem ska˝enia.
Pomieszczenie czyste – pomieszczenie o okreÊlonej i kontrolowanej iloÊci
zanieczyszczeƒ czàstkami stałymi i drobnoustrojami, zbudowane i u˝ytkowane w taki
sposób, aby ograniczyç wprowadzanie, powstawanie i gromadzenie si´ zanieczyszczeƒ.
Procedura – opis trybu post´powania lub sposobu wykonania czynnoÊci zwiàzanych
z wytwarzaniem, badaniem, stosowaniem lub dystrybucjà produktów leczniczych czy
wyrobów medycznych.
StabilnoÊç – zdolnoÊç do zachowania przez produkt wymaganych cech bàdê właÊci-
woÊci w zakresie ustalonych granic, po przechowywaniu przez okreÊlony czas
w odpowiednich warunkach.
Strefa czysta – wszystkie pomieszczenia czyste, odizolowane od innych pomieszczeƒ
wspólnym systemem Êluz.

9
DEFINICJE I OKREÂLENIA
Âluza – zamkni´ta przestrzeƒ przeznaczona do przemieszczania si´ ludzi lub materia-
łów z pomieszczeƒ o ró˝nych klasach czystoÊci powietrza.
Test media fill – test aseptycznego napełniania; symulowany wlew po˝ywki wykony-
wany w celu walidacji i kontroli aseptycznego procesu wytwarzania.
Walidacja procesu – uzyskanie udokumentowanych dowodów na to, ˝e okreÊlony pro-
ces pozwala, z wysokim stopniem zaufania, wytwarzaç w sposób powtarzalny produkt
spełniajàcy wymagania o ustalonej z góry jakoÊci.
WartoÊç CAN (Critical agregation number) – krytyczne st´˝enie elektrolitów, które mo˝e
wywołaç agregacj´ czàstek emulsji tłuszczowej; dla stabilnych mieszanin CAN <600.
WczeÊniak – pacjent neonatologiczny, według definicji WHO, noworodek urodzony
przedwczeÊnie – po 22. tygodniu cià˝y, a przed ukoƒczeniem 37. tygodnia cià˝y (t.c.).
WczeÊniactwo skrajne – noworodki urodzone przed ukoƒczeniem 32. t.c.: noworodki
z małà masà ciała (LBW) <2500 g; z bardzo małà masà ciała (VLBW) <1500 g; ze
skrajnie małà masà ciała (ELBW) <1000 g.
˚ywienie pozajelitowe domowe – dotyczy pacjentów, którzy nie wymagajà dalszej hos-
pitalizacji, w stanie zdrowia umo˝liwiajàcym bezpieczne leczenie w warunkach
domowych bez koniecznoÊci stałej kontroli metabolicznej i całodobowej obserwacji,
natomiast z powodu braku mo˝liwoÊci poda˝y substancji od˝ywczych w iloÊci wystar-
czajàcej do utrzymania przy ˝yciu drogà naturalnà, wymagajàcych całkowitego lub
suplementarnego, długotrwałego podawania substancji od˝ywczych do˝ylnie.
˚ywienie pozajelitowe immunomodulujàce – podawanie êróde∏ białka i energii oraz
elektrolitów, witamin, pierwiastków Êladowych, wody drogà do˝ylnà z dodaniem sub-
stancji modulujàcych działanie układu immunologicznego: glutaminy i/lub kwasów
tłuszczowych omega 3 z osobnych opakowaƒ w dawce nie mniejszej ni˝ 1 ml/kg
m.c./na dob´ Dipeptivenu i/lub Omegavenu.
˚ywienie pozajelitowe kompletne – podawanie êródel białka i energii oraz elektro-
litów, witamin, pierwiastków Êladowych i wody drogà do˝ylnà.
˚ywienie pozajelitowe niekompletne (cz´Êciowe) – podawanie êróde∏ białka i ener-
gii, elektrolitów, tiaminy i wody drogà do˝ylnà. Leczenia takiego nie wolno prowadziç
przez okres dłu˝szy ni˝ 10–14 dni, u chorych wyniszczonych oraz u krytycznie chorych.

10
FARMACEUTYCZNE STANDARDY SPORZÑDZANIA
MIESZANIN DO ˚YWIENIA POZAJELITOWEGO
3. PODSTAWY PRAWNE
Ka˝da nowa aktywnoÊç farmaceuty wymaga znajomoÊci aktów prawnych, które
okreÊlajà sposób jej tworzenia, organizacji i wykonywania. Przedstawione ustawy i roz-
porzàdzenia majà fundamentalne znaczenie dla prawidłowego tworzenia i prowa-
dzenia pracowni ˝ywienia pozajelitowego.
Zalecamy zapoznanie si´ z pełnym tekstem poni˝szych aktów prawnych.
I. Ustawa Prawo farmaceutyczne z dnia 6 wrzeÊnia 2001 r. z póêniejszymi zmianami
(DZ. U. z 2008 r. Nr 45, poz. 271 – tekst jednolity)
Art. 86. 3. W odniesieniu do aptek szpitalnych usługà farmaceutycznà jest równie˝:
1) sporzàdzanie leków do ˝ywienia pozajelitowego;
Art. 90. Przy wykonywaniu w aptece czynnoÊci fachowych mogà byç zatrudnieni
wyłàcznie farmaceuci i technicy farmaceutyczni w granicach ich uprawnieƒ zawodowych.
Art. 91. 1. Technik farmaceutyczny posiadajàcy dwuletnià praktyk´ w aptece w pełnym
wymiarze czasu pracy, mo˝e wykonywaç w aptece czynnoÊci fachowe polegajàce na
sporzàdzaniu, wytwarzaniu, wydawaniu produktów leczniczych i wyrobów medycznych
z wyjàtkiem produktów leczniczych majàcych w swoim składzie:
1) substancje bardzo silnie działajàce okreÊlone w Urz´dowym Wykazie
Produktów Leczniczych dopuszczonych do obrotu na terytorium Rzeczy-
pospolitej Polskiej,
2) substancje odurzajàce,
3) substancje psychotropowe grupy I-P oraz II-P – okreÊlone w odr´bnych
przepisach.
2. Technik farmaceutyczny, o którym mowa w ust.1, mo˝e równie˝ wykonywaç czyn-
noÊci pomocnicze przy sporzàdzaniu i przygotowywaniu preparatów leczniczych,
o których mowa w art. 86 ust. 3 pkt 1–4 oraz pkt 6.
II. Rozporzàdzenie Ministra Zdrowia z dnia 26 wrzeÊnia 2002 r. w sprawie wykazu
pomieszczeƒ wchodzàcych w skład powierzchni podstawowej i pomocniczej apteki
(DZ. U. z 2002 r. Nr 161, poz. 1338)
§ 3. 1. W skład powierzchni podstawowej lokalu apteki szpitalnej wchodzà pomiesz-
czenia wymienione w art. 1 pkt 1, 2 i 4–8, a tak˝e:
1) pomieszczenie do jałowego przygotowania leków;
§ 3. 2. Je˝eli w aptece szpitalnej wykonywane sà leki do ˝ywienia pozajelitowego i do-
jelitowego, leki cytostatyczne, koncentraty do hemodializy i dializy otrzewnowej lub
płyny infuzyjne, to w skład powierzchni podstawowej wchodzà ponadto:
1) pracownia do przygotowywania płynów infuzyjnych z laboratorium kontroli
jakoÊci;

11
PODSTAWY PRAWNE
§ 3. 3. Rodzaj i liczba pomieszczeƒ powinny wynikaç z rodzaju wykonywanych przez
aptek´ czynnoÊci, a tak˝e wykonywanych Êwiadczeƒ zdrowotnych przez placówk´,
w której apteka jest utworzona.
III. Rozporzàdzenie Ministra Zdrowia z dnia 30 wrzeÊnia 2002 r. w sprawie
szczegółowych wymogów, jakim powinien odpowiadaç lokal apteki (DZ. U. z 2002 r.
Nr 171, poz. 1395)
§ 2. 1. Materiały budowlane i wykoƒczeniowe zastosowane w lokalu apteki muszà speł-
niaç wymagania obowiàzujàce w odniesieniu do lokalu zakładu opieki zdrowotnej.
§ 2. 2. Instalacje znajdujàce si´ w aptece powinny byç wykonane zgodnie z odr´bnymi
przepisami.
§ 6. 1. Poszczególne pomieszczenia w aptece:
1) wyposa˝a si´ w odpowiednie urzàdzenia wentylacyjne, zapewniajàce
minimum 1,5-krotnà wymian´ powietrza w ciàgu godziny;
2) rozplanowuje si´ w sposób zapewniajàcy prawidłowà organizacj´ pracy,
bezpieczeƒstwo oraz bezkolizyjnoÊç komunikacyjnà;
3) zabezpiecza si´ przed dost´pem osób nieuprawnionych.
§ 6. 3. Pomieszczenia apteki, w których sà sporzàdzane, wydawane i przechowywane
produkty lecznicze oraz wydawane i przechowywane wyroby medyczne, muszà byç
wyposa˝one w urzàdzenia eliminujàce nadmierne nasłonecznienie.
§ 8. 2. Je˝eli apteka sporzàdza leki w warunkach aseptycznych, to podstawowe jej
wyposa˝enie stanowià ponadto:
1) lo˝a z nawiewem laminarnym do przygotowania leków w warunkach asep-
tycznych, umieszczona w oddzielnym pomieszczeniu lub izbie recepturowej;
§ 9. 2. Je˝eli w aptece sà sporzàdzane leki do ˝ywienia pozajelitowego i dojelitowego,
leki cytostatyczne, koncentraty do hemodializy i dializy otrzewnowej lub płyny infuzyjne,
aptek´ wyposa˝a si´ ponadto odpowiednio w aparatur´ słu˝àcà do:
1) sporzàdzania leków do ˝ywienia pozajelitowego;
IV. Rozporzàdzenie Ministra Zdrowia z dnia 18 paêdziernika 2002 r. w sprawie pod-
stawowych warunków prowadzania apteki (DZ. U. z 2002 r. Nr 187, poz. 1565)
§ 6. 1. Leki recepturowe i leki apteczne sporzàdzane sà w aptece na podstawie:
1) leki recepturowe:
a) recepty lekarskiej,
b) odpisu recepty lekarskiej;
§ 7. 1. Opakowanie leku recepturowego, leku aptecznego i produktu leczniczego
homeopatycznego musi byç zaopatrzone w etykiet´ aptecznà.

12
FARMACEUTYCZNE STANDARDY SPORZÑDZANIA
MIESZANIN DO ˚YWIENIA POZAJELITOWEGO
§ 7. 2. Etykieta apteczna, o której mowa w ust.1, zawiera:
1) adres apteki oraz jej nazw´, o ile apteka jà posiada;
2) skład leku, z zastrze˝eniem ust. 8
1
;
3) sposób u˝ycia leku lub produktu;
4) dat´ sporzàdzenia leku lub produktu.
§ 7. 4. Etykieta apteczna dla leku recepturowego sporzàdzanego w aptece szpitalnej
poza danymi, o których mowa w ust. 2, zawiera nazw´ oddziału, działu lub innej
komórki organizacyjnej, dla której lek jest przeznaczony.
§ 7. 5. Etykieta apteczna dla leku aptecznego sporzàdzanego w aptece ogólnodost´-
pnej, produktu leczniczego homeopatycznego, o którym mowa w § 6 ust. 2 pkt 2, lub
leku recepturowego przeznaczonego do iniekcji lub infuzji sporzàdzanego w aptece
szpitalnej, poza danymi okreÊlonymi w ust. 2, zawiera dodatkowo numer kolejnej serii
sporzàdzanego leku lub produktu.
§ 7. 7. Etykiet´ aptecznà dla leku recepturowego przeznaczonego do iniekcji lub infuzji,
sporzàdzanego w aptece szpitalnej, nale˝y oznakowaç:
1) napisem czarnym na niebieskim tle otoczonym czarnà obwódkà – je˝eli
w skład leku wchodzà Êrodki bardzo silnie działajàce;
2) napisem czerwonym na białym tle otoczonym czerwonà obwódkà – je˝eli
w skład leku wchodzà Êrodki silnie działajàce;
3) napisem czarnym na białym tle otoczonym czarnà obwódkà – je˝eli
w skład leku wchodzà inne Êrodki.
§ 7. 9. Etykiety apteczne, o których mowa w ust. 3–5, nale˝y oznakowaç:
4) danymi o sposobie stosowania, a w przypadku leków do iniekcji – tak˝e
informacjami o sposobie podawania leku.
§ 9. 1. Apteka musi prowadziç ewidencj´ sporzàdzanych w aptece leków receptu-
rowych, leków aptecznych i produktów leczniczych homeopatycznych.
§ 9. 2. Ewidencja sporzàdzanych w aptece leków recepturowych zawiera:
1) dat´ i czas przyj´cia recepty do realizacji;
2) dat´ i czas sporzàdzenia leku recepturowego;
3) numer kontrolny recepty;
4) imi´, nazwisko i podpis osoby sporzàdzajàcej.
§ 9. 4. Ewidencj´ sporzàdzanych w aptece leków aptecznych oraz produktów
homeopatycznych, o których mowa w § 6 ust. 2, prowadzi si´ w formie ksià˝ki
laboratoryjnej, która zawiera:
1
Ust. 8 dotyczy leków homeopatycznych

13
PODSTAWY PRAWNE
1) nazw´, postaç farmaceutycznà, dawk´;
2) rodzaj, iloÊç oraz seri´ substancji wyjÊciowych;
3) podstaw´ sporzàdzenia leku aptecznego lub produktu leczniczego
homeopatycznego, o której mowa w § 6;
4) dat´ sporzàdzenia oraz numer serii;
5) iloÊç sporzàdzonego leku aptecznego lub produktu homeopatycznego
i iloÊç opakowaƒ z okreÊleniem zawartoÊci opakowania jednostkowego;
6) termin wa˝noÊci;
7) imi´, nazwisko i podpis osoby sporzàdzajàcej.
§ 9. 5. Ewidencja, o której mowa w ust. 2 i 3, mo˝e byç prowadzona w formie elektro-
nicznej.
V. Rozporzàdzenie Ministra Zdrowia z dnia 17 sierpnia 2009 r. (DZ. U. z 2009 r.
Nr 135, poz. 1114) zmieniajàce rozporzàdzenie w sprawie wymagaƒ Dobrej Prak-
tyki Wytwarzania (GMP) z dnia 1 paêdziernika 2008 r. (DZ. U. z 2008 r. Nr 184, poz.
1143)
2
OkreÊla zasady zarzàdzania miejscem wytwarzania oraz procesem wytwarzania pro-
duktów leczniczych poprzez polityk´ jakoÊci.
Rozporzàdzenie opisuje:
• szczegółowe wymagania dla Êrodowiska pracy do wytwarzania produktów leczniczych
jałowych (np. klasy czystoÊci powietrza itd.);
• zasady monitorowania jakoÊci procesu wytwarzania;
• wymagania dotyczàce prawidłowego przygotowania pracowników uczestniczàcych
w procesie wytwarzania produktów leczniczych;
• zasady walidacji Êrodowiska pracy, procedur procesu wytwarzania, personelu i pro-
duktu koƒcowego;
• zasady prowadzenia dokumentacji procesu wytwarzania produktów leczniczych
jałowych oraz warunki dopuszczenia produktu do u˝ycia.
VI. FARMAKOPEA POLSKA VII, VIII
3
Rozdział 2. 6. Biologiczne metody badania.
Badanie jałowoÊci stosuje si´ do substancji, preparatów lub produktów, które zgodnie
z wymaganiami Farmakopei muszà byç jałowe.
Rozdział opisuje szczegółowo dopuszczone metody badania (metod´ sàczków mem-
branowych i metod´ bezpoÊredniego posiewu), ich walidacj´ oraz zasady pobierania
prób, obserwacji i interpretacji uzyskanych wyników.
VII. Zarzàdzenie Prezesa Narodowego Funduszu Zdrowia z dnia14 paêdziernika 2008
r. nr 82/2008 w sprawie okreÊlenia warunków zawierania i realizacji umów
w rodzaju Êwiadczenia zdrowotne kontrolowane odr´bnie
2, 3
Oznacza informacj´ autorów o zakresie regulacji aktów prawnych, istotnych dla pracowni ˝ywienia poza-
jelitowego, nie jest cytatem z aktu prawnego.

14
FARMACEUTYCZNE STANDARDY SPORZÑDZANIA
MIESZANIN DO ˚YWIENIA POZAJELITOWEGO
Załàcznik nr 4
Opis Êwiadczenia
˚ywienie pozajelitowe w warunkach domowych
2. Warunki wykonania
2. 5. Konieczne i niezb´dne warunki lokalowe dla udzielenia Êwiadczenia
• apteka szpitalna (docelowo z pracownià ˝ywienia pozajelitowego),
• poradnia dla chorych ˝ywionych pozajelitowo w warunkach domowych,
• ciàgły kontakt telefoniczny z zespołem leczàcym.
2. 6. Minimalne kwalifikacje pracowników fachowych udzielajàcych Êwiadczeƒ
1) lekarze – specjaliÊci w dziedzinie medycyny, w której program szkolenia do
uzyskania specjalizacji obejmuje ˝ywienie pozajelitowe i dojelitowe
(np. anestezjologia, chirurgia ogólna, pediatria) – w łàcznym wymiarze czasu
pracy odpowiadajàcym czasowi pracy oÊrodka;
2) piel´gniarka – przynajmniej jedna zatrudniona w wymiarze odpowiadajàcym
łàcznemu czasowi pracy oÊrodka;
3) mgr farmacji;
2. 7. Konieczne umiej´tnoÊci i doÊwiadczenie zawodowe pracowników fachowych
udzielajàcych Êwiadczeƒ (certyfikaty)
Certyfikat ukoƒczenia kursu z zakresu ˝ywienia pozajelitowego i dojelitowego
zatwierdzony przez Centrum Medyczne Kształcenia Podyplomowego w porozumieniu
z Polskim Towarzystwem ˚ywienia Pozajelitowego i Dojelitowego (lekarz, piel´gniarka,
farmaceuta).

15
POMIESZCZENIA DO SPORZÑDZANIA
MIESZANIN DO ˚P
4. POMIESZCZENIA DO SPORZÑDZANIA
MIESZANIN DO ˚P
Z uwagi na sposób sporzàdzania mieszanin do ˚P (mieszanie jałowych składników bez
sterylizacji produktu koƒcowego), pomieszczenia muszà odpowiadaç standardom
pomieszczeƒ przeznaczonych do pracy aseptycznej.
I. POMIESZCZENIA
Zespół pomieszczeƒ przeznaczonych do sporzàdzania mieszanin powinien stanowiç
zamkni´ty kompleks, odizolowany od reszty apteki.
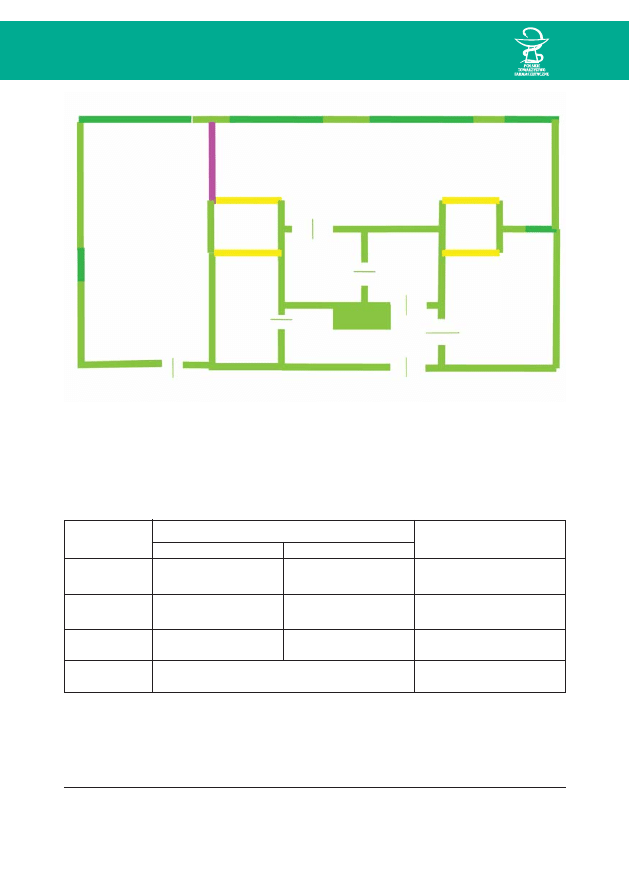
W skład kompleksu wchodzà (ryc. 1):
• boks aseptyczny,
• Êluzy osobowe tzw. personalne ( II i III),
• Êluzy materiałowe (A i B),
• magazyn produktów leczniczych i wyrobów medycznych,
• magazyn gotowego produktu,
• pomieszczenie administracyjne,
• przedsionek (I).
Podłogi w całym kompleksie powinny byç wyło˝one łatwo zmywalnà wykładzinà
zachodzàcà na Êciany (bez listew przypodłogowych). Âciany oraz sufity powinny byç
gładkie (przystosowane do łatwego zmywania). Połàczenia Êcian, podłóg i sufitów
powinny byç zaokràglone. Wszystkie drzwi prowadzàce do boksu powinny byç szczelne
(najlepiej drzwi automatyczne o zsynchronizowanym mechanizmie otwierania). W kom-
pleksie powinien funkcjonowaç system wentylacji nawiewno-wywiewnej z układem
filtrów mikrobiologicznych.
1. Boks aseptyczny – w boksie powinno panowaç nadciÊnienie. Ró˝nica ciÊnieƒ
pomi´dzy sàsiadujàcymi pomieszczeniami powinna wynosiç 10–15 paskali (kon-
trolowane poprzez system czujników), co gwarantuje, ˝e przy otwarciu drzwi
zanieczyszczenia z zewnàtrz nie dostanà si´ do boksu. W pomieszczeniach aseptycznych
zaleca si´ 20-krotnà wymian´ powietrza na godzin´, temperatur´ w granicach
18–23°C, wilgotnoÊç wzgl´dnà 50%±10%. Boks przeznaczony do pracy aseptycznej
powinien zapewniaç klas´ czystoÊci powietrza A lub B. Je˝eli w boksie uzyskano klas´
czystoÊci powietrza B, niezb´dne jest zamontowanie w nim lo˝y z laminarnym przepły-
wem jałowego powietrza zapewniajàcej klas´ czystoÊci A (tabl. 1).
2. Dwie Êluzy osobowe (II, III) tzw. personalne, ka˝da o powierzchni minimum
2–3 m
2
. W pierwszej cz´Êci tzw. „brudnej” powinna znajdowaç si´ umywalka, poje-
mniki z mydłem, papierowymi r´cznikami i płynem dezynfekcyjnym do ràk oraz szafka

16
FARMACEUTYCZNE STANDARDY SPORZÑDZANIA
MIESZANIN DO ˚YWIENIA POZAJELITOWEGO
do pozostawienia odzie˝y ogólnoaptecznej. Cz´Êç druga tzw. „czysta” powinna byç
zaopatrzona w dozownik z płynem dezynfekcyjnym do ràk oraz szafk´ z odzie˝à jałowà
(fartuch, czepek, maseczka, ochraniacze na buty i r´kawice jednorazowe).
Zaleca si´ wytyczenie dwóch oddzielnych dróg: dla osób wchodzàcych do boksu
(z cz´Êci czystej Êluzy) i wychodzàcych z boksu (do cz´Êci brudnej Êluzy).
3. Dwie Êluzy materiałowe – A i B powinny byç zaopatrzone w szczelne, otwierane
naprzemiennie drzwi, wykonane z materiału zapewniajàcego łatwe zmywanie i dezyn-
fekcj´. Âluza A słu˝y do podawania do boksu produktów leczniczych i wyrobów
medycznych niezb´dnych do sporzàdzania mieszanin. Âluza B – do przekazywania
sporzàdzonych mieszanin do magazynu gotowego produktu.
4. Magazyn produktów leczniczych i wyrobów medycznych – powinny znajdowaç si´
w nim regały i chłodziarki z urzàdzeniem do monitorowania temperatury, przezna-
czone do przechowywania zapasów oraz wyciàg, pod którym dokonuje si´ dezynfekcji
produktów leczniczych przed wprowadzeniem ich do Êluzy materiałowej.
5. Magazyn gotowego produktu – połàczony z boksem Êluzà materiałowà,
wyposa˝ony w regały i chłodziarki z urzàdzeniem do monitorowania temperatury,
przeznaczone do przechowywania gotowych mieszanin.
6. Pomieszczenie administracyjne przeznaczone do opracowywania recept, wypo-
sa˝one w komputery i szafy do przechowywania dokumentów .
Pomi´dzy pomieszczeniem administracyjnym a boksem powinien byç mo˝liwy
kontakt wzrokowy (szyba) oraz g∏osowy (intercom).
7. Przedsionek (I) prowadzàcy do Êluz personalnych, magazynu produktów leczniczych
i wyrobów medycznych oraz magazynu gotowego produktu. W pomieszczeniu tym
powinny znajdowaç si´ szafki z odzie˝à i obuwiem ogólnoaptecznym oraz wydzielone
miejsce ze Êrodkami przeznaczonymi do utrzymania czystoÊci w tej cz´Êci kompleksu.
II. PRZYGOTOWANIE BOKSU ASEPTYCZNEGO DO PRACY
Przed rozpocz´ciem pracy wszystkie powierzchnie powinny byç umyte i zdezynfekowane
odpowiednim Êrodkiem według wewn´trznej procedury szpitala (ogólny plan dezyn-
fekcji szpitala). Do dezynfekcji powierzchni w klasie czystoÊci A zaleca si´ stosowaç
preparaty jałowe. Podłogi boksu aseptycznego i Êluz powinny byç codziennie zmywane
i dezynfekowane (przed rozpocz´ciem pracy i po zakoƒczeniu pracy). Pozostałe
powierzchnie (Êciany, sufit) powinny byç myte i dezynfekowane przynajmniej raz
w tygodniu. Ârodek u˝ywany do dezynfekcji powinien byç zmieniany według procedury
szpitala. Przed rozpocz´ciem pracy w boksie nale˝y uruchomiç przepływ jałowego
powietrza według instrukcji/procedury. Preparaty do dezynfekcji nale˝y przechowywaç
w czystych, jałowych pojemnikach.
BOKS ASEPTYCZNY
POMIESZCZENIE
ADMINISTRACYJNE
MAGAZYN
PRODUKTÓW
LECZNICZYCH
I WYROBÓW
MEDYCZNYCH
MAGAZYN
GOTOWEGO
PRODUKTU
A
B
III
II
I
17
POMIESZCZENIA DO SPORZÑDZANIA
MIESZANIN DO ˚P
Tab. 1. Klasy czystoÊci powietrza w przestrzeni aseptycznej podczas pracy
Klasa
czystoÊci
A
B
C
D
Limit ska˝enia
mikrobiologicznego
4
<1
5
50
100
Limit liczby czàstek w 1 m
3
powietrza
0,5 μm 5 μm
3520 20
352000 2900
3520000 29000
nieokreÊlony
Ryc. 1. Podstawowy schemat organizacyjny pomieszczeƒ przeznaczonych do sporzàdzania
mieszanin do ˚P
A – Êluza materia∏owa produktów leczniczych i wyrobów medycznych
B – Êluza materia∏owa gotowego produktu
I – przedsionek
II, III – Êluzy osobowe
4
Metoda sedymentacyjna

18
FARMACEUTYCZNE STANDARDY SPORZÑDZANIA
MIESZANIN DO ˚YWIENIA POZAJELITOWEGO
5. BADANIA PRZESTRZENI PRACY
ORAZ PERSONELU
Przestrzeƒ pracy podlega kontrolnym badaniom technicznym i mikrobiologicznym.
I. BADANIA TECHNICZNE
Zaleca si´ stałe monitorowanie oraz walidacj´ ni˝ej wymienionych parametrów.
Cz´stotliwoÊç wykonywanych badaƒ powinna byç zgodna z wewn´trznymi procedurami.
1. Wentylacja i filtry Hepa – nadzór nad pracà systemu prowadzi wyznaczony
pracownik techniczny szpitala.
2. Komory laminarne – kontrolowane i konserwowane przez autoryzowany serwis.
Badania obejmujà: okreÊlenie integralnoÊci filtrów, szybkoÊci laminarnego przepływu
powietrza (zalecana szybkoÊç od 0,36 do 0,54 m/s), czystoÊci powietrza (liczby
i wielkoÊci czàstek).
• Filtr wst´pny w komorze – kontrolowany przez pracowników apteki.
3. Temperatura i wilgotnoÊç powietrza
• Codzienny nadzór pełnià pracownicy apteki szpitalnej.
• Zapisy temperatury i wilgotnoÊci powinny byç odnotowane w dokumentacji.
Komentarz
Raporty z badaƒ technicznych powinny byç przechowywane w dokumentacji pracowni
przez okres 5 lat.
II. BADANIA MIKROBIOLOGICZNE
Badania obejmujà:
• okresowe badanie czystoÊci przestrzeni pracy,
• walidacj´ aseptycznego procesu sporzàdzania mieszanin,
• rutynowà kontrol´ mikrobiologicznà.
1. Okresowe badanie czystoÊci przestrzeni pracy
Obejmuje badania mikrobiologiczne czystoÊci powietrza i powierzchni.
• Badania zaleca si´ wykonywaç jeden raz w miesiàcu, jednak nie rzadziej ni˝ dwa
razy w roku, a tak˝e po ka˝dej wymianie filtrów Hepa lub awarii.
• Badania wykonuje si´ podczas pracy „w działaniu” oraz „w spoczynku”.
5
Wyniki badania powinny potwierdzaç klas´ czystoÊci pomieszczenia.
1. 1. Badanie mikrobiologiczne czystoÊci powietrza
• Badanie okreÊla liczb´ drobnoustrojów w 1 m
3
powietrza.
• Metody badania:
• metoda sedymentacyjna (półiloÊciowa),
• metoda pobierania obj´toÊciowych prób powietrza (iloÊciowa).
5
Stan „w spoczynku” – stan uzyskany po 15–20 min. oczyszczania powietrza, po zakoƒczeniu operacji, gdy
w pomieszczeniu nie ma ju˝ ludzi.

19
• Wykonanie badania – zgodnie z wytycznymi Dobrej Praktyki Wytwarzania, według
Farmakopei Polskiej i wewn´trznej procedury szpitala.
1. 2. Badanie mikrobiologiczne czystoÊci powierzchni
• Kontroli podlegajà powierzchnie: stoły, podłogi, Êciany, powierzchnie urzàdzeƒ.
• Metoda badania – metoda płytek kontaktowych.
• Wykonanie badania – zgodnie z wytycznymi Dobrej Praktyki Wytwarzania, według
Farmakopei Polskiej i wewn´trznej procedury szpitala.
Komentarz
Raporty z badaƒ mikrobiologicznych czystoÊci przestrzeni pracy powinny byç prze-
chowywane w dokumentacji pracowni przez okres 5 lat.
2. Walidacja aseptycznego procesu sporzàdzania mieszanin
Walidacja obejmuje:
• przeprowadzenie testu media fill,
• badanie jałowoÊci mieszaniny testowej,
• badanie czystoÊci mikrobiologicznej przestrzeni pracy i powietrza.
2. 1. Przeprowadzenie testu media fill
Kontrola aseptycznego wytwarzania polega na wykonaniu symulowanego wlewu
po˝ywki – testu media fill. Badanie to ma na celu wykazanie, ˝e procedury
wytwarzania dajà gwarancj´ jałowoÊci produktu koƒcowego. Badanie powinno
naÊladowaç, tak dokładnie jak to mo˝liwe, rutynowy proces aseptycznego
wytwarzania i obejmowaç wszystkie nast´pujàce po sobie etapy pracy.
Badanie powinno byç wykonywane przed rozpocz´ciem rutynowej pracy oraz
powtarzane po ka˝dej istotnej modyfikacji urzàdzenia lub procedury. Liczba
pojemników napełnianych po˝ywkami powinna byç wystarczajàca do wiary-
godnej oceny. Test media fill jest metodà kontroli Êrodowiska produkcyjnego,
sprz´tu oraz personelu. OkreÊla jakoÊç procesu wytwarzania preparatów.
W ramach walidacji wst´pnej zalecane jest trzykrotne wykonanie testu, w warun-
kach najgorszego przypadku. Badania powtórne zaleca si´ wykonywaç 2 razy
w roku.
2. 2. Badanie jałowoÊci mieszaniny testowej według Farmakopei Polskiej.
Badanie wykonywane w pomieszczeniu o udokumentowanej czystoÊci mikrobio-
logicznej – klasa B, pod nawiewem laminarnym zapewniajàcym klas´ czystoÊci A.
2. 3. Kontrola czystoÊci mikrobiologicznej powierzchni i powietrza w klasie A i B przed
i po pracy według pkt. II 1. 1. i 1. 2.
3. Rutynowa kontrola mikrobiologiczna
• Podczas ka˝dego cyklu pracy – wykonanie tzw. Êlepej próby, która jest modyfikacjà
testu media fill; napełnienie ostatniego worka w cyklu produkcyjnym 5% glukozà,
0,9% NaCl lub produktami leczniczymi do ˝ywienia pozajelitowego, w sposób
BADANIA PRZESTRZENI PRACY ORAZ PERSONELU

20
FARMACEUTYCZNE STANDARDY SPORZÑDZANIA
MIESZANIN DO ˚YWIENIA POZAJELITOWEGO
naÊladujàcy proces sporzàdzania mieszanin, i przekazanie worka do badania
jałowoÊci.
• Okresowo (cz´stotliwoÊç badania według wewn´trznej procedury) – badanie
jałowoÊci mieszanin do ˚P (rozdział 18. „Kontrola procesu sporzàdzania mieszanin
do ˚P”).
III. BADANIA PERSONELU
Badania personelu obejmujà:
1.badanie czystoÊci mikrobiologicznej r´kawiczek personelu sporzàdzajàcego
mieszaniny
• metoda odciskowa zgodnie z wytycznymi Dobrej Praktyki Wytwarzania, według
Farmakopei Polskiej i wewn´trznej procedury szpitala
2. badanie personelu na nosicielstwo Staphylococcus aureus (wymaz z jamy nosowej
i gardła)
• zaleca si´ przeprowadzenie badania jeden raz na rok.
Komentarz
Wszystkie raporty z badaƒ powinny byç przechowywane w dokumentacji pracowni przez
okres 5 lat.
IV. DZIAŁANIA INTERWENCYJNE
W przypadku przekroczenia dopuszczalnych limitów nale˝y, zgodnie z opracowanymi
procedurami, podjàç działania korygujàce, które obejmujà:
1. analiz´ bł´dów,
2. sprawdzenie prawidłowoÊci i toku przeprowadzonego badania (czy był
zgodny z procedurà),
3. wdro˝enie działaƒ naprawczych,
4. powtórne przeprowadzenie badania.

21
6. PERSONEL
Przy sporzàdzaniu leków jałowych w warunkach aseptycznych mogà byç zatrudnieni
wyłàcznie farmaceuci i technicy farmaceutyczni w granicach ich uprawnieƒ
zawodowych.
Zaleca si´, aby minimum dwie uprawnione osoby – operator i pomocnik – pracowały
jednoczeÊnie w boksie aseptycznym.
1. Wszyscy pracownicy powinni znaç zasady Dobrej Praktyki Wytwarzania.
2. Zakres obowiàzków i odpowiedzialnoÊci powinien byç w pełni zrozumiały i okreÊlony
w formie pisemnej w postaci procedur i instrukcji.
3. Pracownicy muszà odbyç szkolenia, których skutecznoÊç powinna byç weryfikowana.
Szkolenia – ustawiczne i specjalizacja zawodowa pracowników – majà zapewniç
personelowi wiedz´ teoretycznà i umiej´tnoÊci praktyczne.
4. Nale˝y opracowaç i przestrzegaç programy zachowania higieny, odpowiednie do
zakresu wytwarzania. Programy zachowania higieny obejmujà w szczególnoÊci
procedury dotyczàce kontroli zdrowia, higieny i odzie˝y roboczej pracowników.
5. Pracownicy bioràcy udział w wytwarzaniu produktów sterylnych powinni byç
poinstruowani o obowiàzku zgłaszania wszystkich przypadków, które mogà stanowiç
dodatkowe êródło zanieczyszczenia mikrobiologicznego.
6. Wszyscy pracownicy muszà byç poddani badaniom lekarskim. Po wst´pnym bada-
niu lekarskim powinny byç przeprowadzane nast´pne badania, w tym badanie na
nosicielstwo Staphylococcus aureus (wymaz z jamy nosowej i gardła), z cz´stotli-
woÊcià według wewn´trznej procedury szpitala. Powinny byç podj´te kroki zapew-
niajàce, aby ˝adna osoba chora na chorob´ zakaênà lub majàca otwarte zmiany na
odkrytej powierzchni ciała nie mogła byç dopuszczona do wytwarzania produktów
leczniczych.
7. Jedzenie, picie, ˝ucie, przechowywanie po˝ywienia, napojów w pomieszczeniach
aseptycznych jest zabronione.
8. Zmiana odzie˝y i mycie powinno przebiegaç zgodnie z pisemnymi procedurami, celem
zminimalizowania zanieczyszczenia odzie˝y stosowanej w pomieszczeniach czystych,
a tak˝e celem zapobiegania wprowadzaniu zanieczyszczeƒ do pomieszczeƒ czystych.
PERSONEL

22
FARMACEUTYCZNE STANDARDY SPORZÑDZANIA
MIESZANIN DO ˚YWIENIA POZAJELITOWEGO
9. W pomieszczeniach czystych nie powinno si´ nosiç zegarków, bi˝uterii ani stosowaç
makija˝u. Nale˝y unikaç lakieru do paznokci. Obowiàzuje bezwzgl´dny zakaz
noszenia tipsów.
10. Rodzaj odzie˝y i jej jakoÊç powinny byç dostosowane do rodzaju procesu i klasy
czystoÊci miejsca pracy.
11. W pomieszczeniach klasy A/B nakrycie głowy powinno całkowicie przykrywaç włosy
na głowie, brod´ i wàsy. Twarz powinna byç osłoni´ta maskà ochronnà. Pracownik
powinien byç ubrany w jałowy fartuch ochronny z długimi r´kawami zakoƒczonymi
Êciàgaczami.
12. Nale˝y nosiç wyjałowione, bezpudrowe r´kawice gumowe lub plastikowe (dla osób
uczulonych na lateks) oraz wyjałowione lub zdezynfekowane obuwie. R´kawice
nale˝y naciàgnàç na mankiety r´kawów. Zaleca si´ zmian´ r´kawic w trakcie
sporzàdzania mieszanin do ˚P.
13. Odzie˝ ochronna nie powinna byç potencjalnym êródłem włókien lub czàstek.
14. Zewn´trzna odzie˝ nie powinna byç wnoszona do przebieralni prowadzàcych do
boksu. Czysta jałowa odzie˝ ochronna powinna byç dostarczona ka˝demu
pracownikowi w Êluzie „czystej” na ka˝dy cykl pracy.
15. Odzie˝ przeznaczona do noszenia w pomieszczeniach czystych powinna byç prana
i chroniona tak, aby nie powodowaç gromadzenia si´ dodatkowych zanieczyszczeƒ.
CzynnoÊci te powinny przebiegaç zgodnie z pisemnymi procedurami. Wskazane
jest pranie odzie˝y w oddzielnych pralniach.
16. Pracownicy techniczni, studenci odbywajàcy szkolenia, ekipy remontowe, okresowo
przebywajàcy w pomieszczeniach aseptycznych, muszà wczeÊniej otrzymaç
wskazówki, szczególnie dotyczàce higieny osobistej i zalecanej odzie˝y ochronnej.
Osoby takie muszà pozostawaç pod Êcisłym nadzorem personelu fachowego
apteki.

23
7. PRZYGOTOWANIE PRACOWNIKA DO PRACY
W WARUNKACH ASEPTYCZNYCH
I. CZYNNOÂCI WYKONYWANE W ÂLUZIE „BRUDNEJ”
1. Zmieniç obuwie, zdjàç odzie˝ zewn´trznà, zegarek i bi˝uteri´.
2. Umyç r´ce zgodnie z wewn´trznà procedurà apteki.
3. Po umyciu r´ce osuszyç papierowym r´cznikiem.
4. R´ce zdezynfekowaç alkoholowym Êrodkiem antyseptycznym z dozownika, według
wewn´trznej procedury apteki.
Na granicy Êluzy
6
„brudnej” i Êluzy „czystej” zało˝yç jałowe lub zdezynfekowane
obuwie.
II. CZYNNOÂCI WYKONYWANE W ÂLUZIE „CZYSTEJ”
1. Zało˝yç sterylnà, jednorazowà odzie˝ ochronnà według kolejnoÊci:
• czepek,
• maska chirurgiczna,
• fartuch/komplet odzie˝y z długimi r´kawami zakoƒczonymi mankietem.
2. Wykonaç dezynfekcj´ dłoni alkoholowym Êrodkiem antyseptycznym z dozownika.
III. CZYNNOÂCI WYKONYWANE W BOKSIE
1. WejÊç do boksu, nie dotykajàc r´kami przedmiotów.
2. W lo˝y zało˝yç jałowe r´kawice chirurgiczne.
Komentarz
1. Nale˝y u˝ywaç r´kawic bezpudrowych i odzie˝y bezpyłowej.
2. Zaleca si´ zmian´ r´kawic w trakcie pracy, co 1,5 godziny lub cz´Êciej, w za-
le˝noÊci od potrzeb. Dopuszcza si´ dezynfekcj´ r´kawic Êrodkiem do tego prze-
znaczonym.
3. W boksie zaleca si´ montowanie dozowników z Êrodkiem do dezynfekcji r´kawic.
4. W Êluzie „brudnej” zaleca si´ montowanie dozowników z Êrodkiem ochronnym
do ràk.
PRZYGOTOWANIE PRACOWNIKA DO PRACY
W WARUNKACH ASEPTYCZNYCH
6
Granica pomi´dzy Êluzà „brudnà” i Êluzà „czystà” – miejsce umowne; mogà to byç ∏awka, linia lub drzwi.

24
FARMACEUTYCZNE STANDARDY SPORZÑDZANIA
MIESZANIN DO ˚YWIENIA POZAJELITOWEGO
8. PRZYGOTOWANIE PRODUKTÓW LECZNICZYCH
ORAZ WYROBÓW MEDYCZNYCH PRZED
WPROWADZENIEM ICH DO ASEPTYCZNEGO
BOKSU
I. ODPOWIEDZIALNOÂå
Za wykonanie procedury odpowiedzialni sà wyznaczeni pracownicy apteki szpitalnej:
1. farmaceuci i technicy farmaceutyczni za prawidłowe skompletowanie produktów
leczniczych oraz wyrobów medycznych, sprawdzenie szczelnoÊci i dezynfekcj´
opakowaƒ
2. personel pomocniczy za prawidłowe mycie opakowaƒ z produktami leczniczymi
potrzebnymi do sporzàdzenia mieszanin do ˝ywienia pozajelitowego oraz zawie-
szek do butelek.
II. SPOSÓB POST¢POWANIA
1. Na podstawie protokołu sporzàdzenia mieszaniny do ˚P nale˝y skompletowaç pro-
dukty lecznicze oraz wyroby medyczne.
2. Nale˝y wizualnie oceniç opakowania preparatów (butelki, fiolki, ampułki, worki).
3. Sprawdziç szczelnoÊç opakowaƒ z tworzyw sztucznych przez naciÊni´cie.
4. Umyç opakowania płynów infuzyjnych oraz zawieszki do butelek.
5. Zdezynfekowaç Êrodkiem dezynfekcyjnym, ustalonym według wewn´trznych proce-
dur szpitala: butelki (nie zapominaç o dolnej powierzchni), fiolki, ampułki, zawieszki
do butelek, osłonki, opakowania zewn´trzne igieł, strzykawek, kompresów, worków
i innych potrzebnych wyrobów medycznych.
6. Wprowadziç zdezynfekowane produkty lecznicze oraz wyroby medyczne do asepty-
cznego boksu przez Êluz´ materiałowà.
Komentarz
1. Nale˝y zwracaç uwag´ na zgodnoÊç z protokołem serii i daty wa˝noÊci produktów
leczniczych oraz wyrobów medycznych.
2. Zaleca si´ stosowanie płynów infuzyjnych w opakowaniach z podwójnymi portami.
3. Do pobierania płynów zaleca si´ stosowanie igieł o Êrednicy 0,8–1,2 mm.
4. Zaleca si´ stosowanie akcesoriów z filtrami do aseptycznego pobierania produktów
leczniczych.
5. Dezynfekcj´ opakowaƒ produktów leczniczych i wyrobów medycznych zaleca si´
wykonywaç pod wyciàgiem.

25
9. RECEPTA
Recepta lekarska musi byç wypisana na formularzu szpitalnym maszynowo, elektro-
nicznie lub r´cznie (czytelnie).
Recepta musi zawieraç nast´pujàce dane:
• nazw´ szpitala i oddziału, dla którego została wystawiona;
• imi´ i nazwisko pacjenta, jego dat´ urodzenia lub PESEL, mas´ ciała (w kg);
• skład mieszaniny – nazwy handlowe produktów leczniczych podane w mili-
litrach, z dodanym „procentem na dren” (procentowe zwi´kszenie iloÊci,
ka˝dego sk∏adnika mieszaniny o obj´toÊç pozostajàcà w drenie) w przy-
padku recept neonatologicznych lub pediatrycznych;
Zaleca si´ dodatkowo podanie na recepcie dawek elektrolitów w milimolach, amino-
kwasów, w´glowodanów, lipidów w gramach.
• całkowità obj´toÊç koƒcowà;
• drog´ podania;
• czas podania – szybkoÊç wlewu w ml/godz.;
• podpis i pieczàtk´ lekarza;
• dat´ wystawienia recepty.
Farmaceuta:
• kontroluje skład recepty pod wzgl´dem merytorycznym;
• sprawdza, czy nie b´dà wyst´powały niezgodnoÊci, czy skład preparatów,
pH, zakresy st´˝eƒ sà prawidłowe;
• zatwierdza recept´ podpisem;
• w przypadku pojawienia si´ wàtpliwoÊci wyjaÊnia je ze zlecajàcym lekarzem;
Poprawki muszà byç potwierdzone na recepcie podpisem i pieczàtkà lekarza.
•
oblicza iloÊç opakowaƒ preparatów niezb´dnych do sporzàdzenia
mieszaniny;
• ustala sposób wykonania mieszaniny (kolejnoÊç dodawania składników);
• zgodnie z procedurà przygotowuje protokoły wykonania, zawierajàce
informacje o składnikach mieszaniny, oraz etykiety.
Sprawdzona recepta musi byç wpisana do dokumentacji pracowni i nast´pnie
przekazana do wykonania.
Nazwisko pracownika przygotowujàcego mieszanin´ musi byç umieszczone na kopii
recepty.
RECEPTA

26
FARMACEUTYCZNE STANDARDY SPORZÑDZANIA
MIESZANIN DO ˚YWIENIA POZAJELITOWEGO
10. ETYKIETA
Etykiet´ nale˝y oznakowaç czarnym napisem na białym tle otoczonym czarnà obwódkà.
Na etykiecie powinny si´ znajdowaç nast´pujàce dane:
1. nazwa apteki szpitalnej;
2. pracownia ˝ywienia pozajelitowego – numer telefonu;
3. dane pacjenta: imi´, nazwisko, data urodzenia/wiek lub PESEL;
4. nazwa oddziału;
5. skład mieszaniny:
• aminokwasy, glukoza, lipidy w gramach;
• elektrolity w milimolach;
• pierwiastki Êladowe, witaminy w mililitrach;
• pozostałe składniki (np. insulina w j.m.);
6. obj´toÊç całkowita mieszaniny w mililitrach;
7. droga podania;
8. data i godzina sporzàdzenia mieszaniny;
9. termin wa˝noÊci;
10. uwagi dodatkowe:
• warunki przechowywania (od +2°C do +8°C);
• przed podłàczeniem doprowadziç do temperatury pokojowej;
• wykonano w warunkach aseptycznych;
11. czas podania – szybkoÊç wlewu w ml/godzin´.
Opakowanie zewn´trzne (osłonka chroniàca przed Êwiatłem) powinno byç oznakowane
dodatkowo etykietà zawierajàcà wszystkie powy˝sze dane.

27
11. OGÓLNE ZASADY SPORZÑDZANIA
MIESZANIN DO ˚P
I. OGÓLNE ZASADY SPORZÑDZANIA
1. Dodawanie produktów leczniczych z fiolek i ampułek do płynów infuzyjnych.
Zastosowanie właÊciwej techniki mieszania ma istotne znaczenie dla stabilnoÊci
mieszaniny.
Reguła dodawania elektrolitów, pierwiastków Êladowych, fosforanów, witamin,
leków do płynów infuzyjnych:
Preparaty wapnia
§
aminokwasy bez fosforanów ewentualnie glukoza
Preparaty magnezu
§
aminokwasy lub glukoza
Pierwiastki Êladowe
§
aminokwasy
Preparaty fosforanów
§
glukoza o najwy˝szym st´˝eniu, ewentualnie aminokwasy
z dodatkiem fosforanów
Elektrolity: sód, potas
§
dowolny płyn
Witaminy rozpuszczalne
§
emulsja tłuszczowa
w wodzie i tłuszczach
Cymetydyna, ranitydyna
§
glukoza
Insulina
§
worek z mieszaninà
Dla mieszanin pediatrycznych
Preparaty fosforanów
§
glukoza
Elektrolity: sód, potas,
§
aminokwasy pediatryczne (I cz´Êç)
preparaty magnezu,
pierwiastki Êladowe
Preparaty wapnia
§
aminokwasy pediatryczne (II cz´Êç)
Witaminy rozpuszczalne
§
woda (w przypadku mieszaniny bez emulsji tłuszczowej)
w wodzie
Witaminy
§
emulsja tłuszczowa
Preparaty wapnia, magnezu, fosforanów nale˝y dostrzykiwaç oddzielnie do płynów
infuzyjnych.
OGÓLNE ZASADY SPORZÑDZANIA
MIESZANIN DO ˚P

28
FARMACEUTYCZNE STANDARDY SPORZÑDZANIA
MIESZANIN DO ˚YWIENIA POZAJELITOWEGO
Nie wolno do tego samego pojemnika dostrzykiwaç preparatów wapnia i fosforanów
nieorganicznych!
Nie wolno do tego samego pojemnika dostrzykiwaç preparatów chlorku wapnia
i siarczanu magnezu!
2. Przetaczanie płynów infuzyjnych do worka:
• nale˝y zachowaç prawidłowà kolejnoÊç podłàczania i przetaczania płynów
infuzyjnych: glukozy, aminokwasów, emulsji tłuszczowej;
• nie przetaczaç tym samym przewodem preparatów z fosforanami i wap-
niem;
• nale˝y pami´taç, by fosforany znalazły si´ w maksymalnym rozcieƒczeniu;
• przed przetoczeniem do nich płynu infuzyjnego z preparatem wapnia.
3. Dodawanie produktów leczniczych z fiolek i ampułek do napełnionego worka
KolejnoÊç dostrzykiwania jest nast´pujàca:
• preparaty fosforanów;
• elektrolity (sód, potas);
• preparaty magnezu;
• preparaty wapnia;
• pierwiastki Êladowe;
• pozostałe składniki (np. cymetydyna, ranitydyna, preparaty glutaminy);
• witaminy;
• insulina.
II. METODY SPORZÑDZANIA MIESZANIN
1. Grawitacyjna.
2. Za pomocà biurety.
3. Manualna dla pacjentów neonatologicznych.
4. Aktywacja i uzupełnianie worków gotowych do u˝ycia (RTU).
5. Za pomocà pomp.
III. SPOSÓB POST¢POWANIA DLA WSZYSTKICH METOD
1. Recept´ w przezroczystej, zdezynfekowanej osłonce umieÊciç w widocznym miejscu.
2. Produkty lecznicze i wyroby medyczne ustawiç na blacie roboczym lo˝y, zgodnie z re-
ceptà.
3. Zdjàç z butelek i fiolek plastikowe kapsle zabezpieczajàce; metalowe kapsle zdjàç
przy pomocy wysterylizowanego przyrzàdu do otwierania.
4. Zdezynfekowaç Êrodkiem ustalonym według wewn´trznych procedur szpitala
powierzchni´ korków, miejsce do wkłucia w butelkach plastikowych, ampułki.

29
5. Ampułki i fiolki z preparatami otwieraç pojedynczo, zdecydowanym ruchem przy
u˝yciu jałowego kompresu.
Staraç si´, aby ampułki stały jak najkrócej otwarte.
6. ZawartoÊç ampułek i fiolek pobieraç podpierajàc strzykawk´ na dłoni trzymajàcej
ampułk´ (zmniejszone ryzyko aspiracji powietrza i zanieczyszczenia igły).
Uwaga! Nie dotykaç palcami igły i tłoka! W przypadku dotkni´cia igłà blatu, dłoni
itp. zało˝yç nowà, jałowà igł´.
7. Zmieniajàc ka˝dorazowo rodzaj pobieranego produktu leczniczego, nale˝y u˝yç
nowej, jałowej igły i strzykawki.
8. Po dostrzykni´ciu produktów leczniczych do płynów infuzyjnych ka˝dorazowo
wymieszaç zawartoÊç butelki i obserwowaç, czy nie ma zmian w klarownoÊci i bar-
wie.
9. Podczas napełniania worka płynami infuzyjnymi oraz ka˝dorazowo po dostrzykni´-
ciu produktów leczniczych z fiolek i ampułek do napełnionego worka, wymieszaç
zawartoÊç przez uciskanie.
10. Obserwowaç, czy w trakcie napełniania worka płynami nie nast´puje zm´tnienie
w fazie wodnej, a po przetoczeniu emulsji tłuszczowej, czy powstaje jednorodna
mieszanina.
11. Po sporzàdzeniu mieszaniny nakleiç etykiet´ na worek oraz na osłonk´.
12. Sprawdziç zgodnoÊç wypisania etykiety z receptà.
13. Na worek z mieszaninà nało˝yç osłonk´ chroniàcà przed Êwiatłem.
OGÓLNE ZASADY SPORZÑDZANIA
MIESZANIN DO ˚P

30
FARMACEUTYCZNE STANDARDY SPORZÑDZANIA
MIESZANIN DO ˚YWIENIA POZAJELITOWEGO
12. SPORZÑDZANIE MIESZANIN METODÑ
GRAWITACYJNÑ
I. SPOSÓB POST¢POWANIA
1. Dodawanie produktów leczniczych z fiolek i ampułek do płynów infuzyjnych
• Przeanalizowaç skład mieszaniny i ustaliç, które produkty lecznicze dostrzyknàç do
płynów infuzyjnych, a które do napełnionego worka.
• Oznaczyç butelki, do których nale˝y dostrzyknàç preparaty wapnia, magnezu,
fosforanów, zgodnie z regułà dodawania i zasadami przedstawionymi w rozdziale
11. „Ogólne zasady sporzàdzania mieszanin do ˚P”.
• CzynnoÊç wykonaç przed zdj´ciem kapsli zabezpieczajàcych z płynów infuzyjnych,
fiolek.
• Ponownie sprawdziç, czy rodzaj, st´˝enie i obj´toÊç przygotowanych produktów
leczniczych sà zgodne z receptà.
• Pobieraç produkty lecznicze z ampułek, fiolek w iloÊciach zgodnych z receptà
i dostrzykiwaç do odpowiednich płynów infuzyjnych zgodnie z regułà dodawania
i zasadami przedstawionymi w rozdziale 11. „Ogólne zasady sporzàdzania
mieszanin do ˚P”.
2. Przetaczanie płynów infuzyjnych do worka
• Blat roboczy lo˝y spryskaç Êrodkiem dezynfekcyjnym i przetrzeç jałowym kompre-
sem, w celu usuni´cia opiłków szkła.
• Worek o odpowiedniej pojemnoÊci wyjàç aseptycznie z opakowania na granicy
lo˝y, uło˝yç przy płynach infuzyjnych.
• Dokr´ciç połàczenie przewodów napełniajàcych z workiem, otworzyç odpo-
wietrzniki, zamknàç zaciski na przewodach.
• Kolejno zdejmowaç z przewodów napełniajàcych osłony zabezpieczajàce i wbijaç
koƒcówk´ przewodów w butelki z płynami infuzyjnymi.
• Butelki powiesiç na stojaku, otworzyç zaciski na przewodach.
• Przetoczyç zawartoÊç butelek w iloÊci zgodnej z receptà, zachowujàc prawidłowà
kolejnoÊç przetaczania przedstawionà w rozdziale 11. „Ogólne zasady sporzàdza-
nia mieszanin do ˚P”.
• Podczas napełniania worka mieszaç zawartoÊç przez uciskanie.
• Po przetoczeniu płynów worek trzeba odpowietrzyç. W tym celu nale˝y: otworzyç
zaciski przewodów do pustych butelek, unieÊç worek do pozycji pionowej i uciskaç,
a˝ całe powietrze zostanie z niego wypchni´te do pustych butelek; zamknàç zacisk
na worku, nie zwalniajàc ucisku na worek, zamknàç zaciski na przewodach.
• Zdjàç ze stojaka butelki wraz z przewodami.
• Wyjàç z opakowania nakr´tk´ zamykajàcà worek.
Uwaga! Nie dotykaç wewn´trznej powierzchni!
• Jednym ruchem r´ki odkr´ciç nakr´tk´ łàczàcà worek z przewodami i nało˝yç

31
nakr´tk´ zamykajàcà, dokr´ciç.
• Nakleiç etykiet´ na worek ze sporzàdzonà mieszaninà oraz na osłonk´.
• Ponownie sprawdziç zgodnoÊç wypisania etykiety z receptà.
• Na worek z mieszaninà nało˝yç osłonk´ chroniàcà przed Êwiatłem.
W przypadku, gdy skład mieszaniny nie pozwala dostrzyknàç wszystkich produktów
leczniczych do płynów infuzyjnych oraz w razie koniecznoÊci dodania insuliny, nale˝y je
podaç bezpoÊrednio do worka, przez kanał do wstrzykni´ç (port) post´pujàc w sposób
opisany w pkt. 3. Dodawanie produktów leczniczych z fiolek, ampułek do napełnionego
worka.
3. Dodawanie produktów leczniczych z fiolek i ampułek do napełnionego worka
• Zdjàç zatyczk´ zabezpieczajàcà z portu do wstrzykni´ç.
• Zdezynfekowaç miejsce dostrzykiwania, w przypadku gdy powierzchnia membrany
kanału do wstrzykni´ç nie jest aseptycznie zabezpieczona.
Uwaga! Nie ma potrzeby odka˝ania w przypadku, gdy powierzchnia membrany
jest zabezpieczona koƒcówkà, którà usuwa si´ przez złamanie.
• Produkty lecznicze pobieraç z ampułek, fiolek w iloÊci zgodnej z receptà,
pojedynczo, zmieniajàc igł´ i strzykawk´ dla ka˝dego produktu.
• Wkłuç igł´ w port tak, by nie przekłuç jego Êcian i wstrzykiwaç pobrane z ampułek,
fiolek produkty lecznicze w kolejnoÊci przedstawionej w rozdziale 11. „Ogólne
zasady sporzàdzania mieszanin do ˚P”.
Uwaga! W przypadku mo˝liwoÊci utrzymania portu w pozycji pionowej,
pozostawiç igł´ w porcie i dostrzykiwaç przez nià kolejne produkty lecznicze.
W przypadku worka le˝àcego na blacie, ka˝dorazowo wkłuwaç si´ nowà igłà
– mo˝liwoÊç wycieku mieszaniny.
• Ka˝dorazowo po dostrzykni´ciu produktu leczniczego do worka wymieszaç za-
wartoÊç przez uciskanie.
• W przypadku widocznych Êladów preparatu witaminowego w porcie, wstrzyknàç
kilka mililitrów 0,9% Natrium chloratum lub Aqua pro inj. w celu przepłukania
kanału.
• W razie koniecznoÊci odpowietrzyç worek, uciskajàc tak, by powietrze wypchnàç
przez igł´ wkłutà w port do wstrzykni´ç, nast´pnie wyjàç igł´.
• Miejsce wkłucia zdezynfekowaç.
• Zało˝yç zatyczk´ zabezpieczajàcà port.
• Nakleiç etykiet´ na worek ze sporzàdzonà mieszaninà oraz na osłonk´, zgodnie
z receptà.
• Ponownie sprawdziç zgodnoÊç wypisania etykiety z receptà.
• Na worek z mieszaninà nało˝yç osłonk´ chroniàcà przed Êwiatłem.
SPORZÑDZANIE MIESZANIN
METODÑ GRAWITACYJNÑ
32
FARMACEUTYCZNE STANDARDY SPORZÑDZANIA
MIESZANIN DO ˚YWIENIA POZAJELITOWEGO
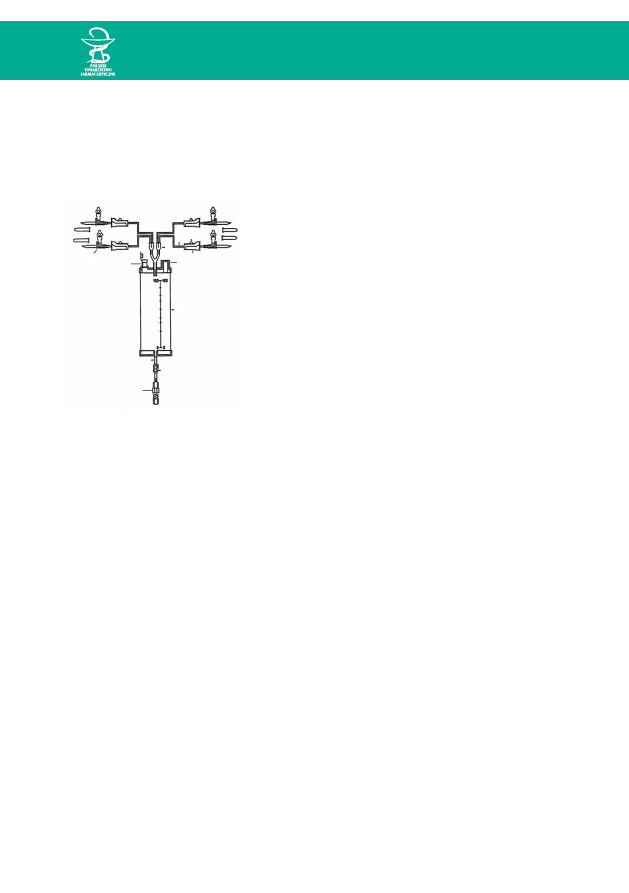
13. SPORZÑDZANIE MIESZANIN ZA POMOCÑ
BIURETY
Metoda stosowana w neonatologii i pediatrii, gdy niemo˝liwe jest uzyskanie właÊci-
wego składu za pomocà tradycyjnej metody grawitacyjnej.
I. SPOSÓB POST¢POWANIA
1. Przygotowanie potrzebnych produktów leczniczych i wyrobów medycznych
• Biuret´ wyjàç aseptycznie z opakowania, zamknàç wszystkie zaciski (3,9/10) na
przewodach doprowadzajàcych (8) i przewodzie wyprowadzajàcym (2), zamo-
cowaç biuret´ na stojaku.
• Zdjàç osłonk´ z koƒcówki przewodu napełniajàcego i wkłuç w zdezynfekowany
korek odpowiedniego produktu leczniczego, zawiesiç butelk´ preparatu na sto-
jaku; analogicznie podłàczyç nast´pne produkty lecznicze.
• Wyjàç aseptycznie z opakowania worek odpowiedniej pojemnoÊci, oznaczyç
i połàczyç z biuretà (5).
• Wyjàç z opakowania i wprowadziç do lo˝y opisany worek „zbiorczy” – słu˝àcy do
zbierania produktów leczniczych, u˝ywanych do przepłukiwania przewodów przed
sporzàdzeniem właÊciwej mieszaniny.
• Otworzyç zatyczk´ odpowietrzajàcà (4) w biurecie (1) i kanały odpowietrzajàce
(11) w przewodach doprowadzajàcych (8).
2. Przetaczanie produktów leczniczych do worka
• Napełniç biuret´ (1) odpowiednià obj´toÊcià glukozy, zamknàç zacisk (9/10) prze-
wodu doprowadzajàcego (8).
• Dodaç odpowiednià obj´toÊç preparatu fosforanów przez port do wstrzykni´ç (6)
w biurecie (1).
• Napełniç worek zawartoÊcià biurety poprzez otwarcie zacisku (3) na przewodzie
wyprowadzajàcym (2) biurety.
Ryc. 2. Schemat biurety
1
2
3
4
5
6
7
8
9
10
11

33
• Zamknàç zacisk (3) na przewodzie wyprowadzajàcym (2) biurety.
• Je˝eli istnieje taka potrzeba, napełniç ponownie biuret´ (1) glukozà i w ten sam
sposób przetoczyç do worka.
• Otworzyç zacisk (9/10) na przewodzie doprowadzajàcym (8) z aminokwasem
i napełniç biuret´ odpowiednià obj´toÊcià, zamknàç zacisk (9/10) – poprzez port
do wstrzykni´ç (6) biurety dodaç według recepty odpowiednie obj´toÊci
elektrolitów, pierwiastków Êladowych.
• Otworzyç zacisk (3) przewodu wyprowadzajàcego (2) biurety i przetoczyç zawartoÊç
biurety do worka (w razie potrzeby napełniç biuret´ ponownie aminokwasem
i powtórzyç procedur´ przetaczania).
• Do ostatniej partii aminokwasu dodaç preparat wapnia, płyn przetoczyç.
Uwaga! Nie łàczyç preparatów fosforanów i wapnia w jednym przetoczeniu.
• Wymieszaç składniki w worku poprzez lekkie uciskanie, zamknàç zacisk (3) na
przewodzie wyprowadzajàcym (2) biurety.
• Otworzyç zacisk (9/10) na przewodzie doprowadzajàcym (8) z wodà i napełniç
biuret´ odpowiednià obj´toÊcià, zamknàç zacisk (9/10) na przewodzie dopro-
wadzajàcym (8), otworzyç zacisk (3) na przewodzie wyprowadzajàcym (2)
biurety i przetoczyç wod´ do worka, zamknàç zacisk (3); witaminy rozpuszczalne
w wodzie dodaç przez port do wstrzykni´ç (6).
• W przypadku koniecznoÊci dodania do mieszaniny emulsji tłuszczowej, dodawaç
jà zawsze jako ostatnià [przez port doprowadzajàcy (6) biurety dodaç witaminy],
przetoczyç do worka, zawartoÊç wymieszaç poprzez uciskanie.
• Worek odpowietrzyç.
• Zamknàç zacisk na przewodzie doprowadzajàcym worka; odłàczyç worek od
biurety; zamknàç przewód dołàczonym do worka jałowym korkiem.
• Nakleiç etykiet´ na worek ze sporzàdzonà mieszaninà oraz na osłonk´, zgodnie
z receptà.
• Ponownie sprawdziç zgodnoÊç etykiety z receptà.
• Na worek z mieszaninà nało˝yç osłonk´ chroniàcà przed Êwiatłem.
3. Sporzàdzanie mieszaniny z wi´cej ni˝ czterech składników lub sporzàdzanie
nast´pnej mieszaniny za pomocà tej samej biurety z innych składników
• Odłàczyç worek ze sporzàdzanà mieszaninà (5).
• Dokr´ciç worek „zbiorczy” (5).
• Wkłuç koƒcówk´ przewodu doprowadzajàcego (8) w nowy produkt leczniczy,
zawiesiç butelk´ na stojaku.
• Otworzyç zacisk (9/10) na przewodzie doprowadzajàcym (8).
• Przetoczyç do biurety (1) ok. 20 ml płynu celem przepłukania.
• Zamknàç zacisk (9/10) na przewodzie doprowadzajàcym (8).
• Otworzyç zacisk (3) na przewodzie wyprowadzajàcym (2) biurety i przelaç płyn do
worka „zbiorczego”.
• Zamknàç zacisk (3) na przewodzie wyprowadzajàcym (2) biurety.
SPORZÑDZANIE MIESZANIN
ZA POMOCÑ BIURETY

34
FARMACEUTYCZNE STANDARDY SPORZÑDZANIA
MIESZANIN DO ˚YWIENIA POZAJELITOWEGO
• Odłàczyç worek „zbiorczy” od biurety (5), zakr´ciç go korkiem.
• Podłàczyç ponownie worek ze sporzàdzanà mieszaninà lub nowy worek i prze-
toczyç wymaganà obj´toÊç płynu.
Mieszaniny bez emulsji tłuszczowej sporzàdzaç w pierwszej kolejnoÊci. Je˝eli zachodzi
koniecznoÊç sporzàdzenia mieszaniny beztłuszczowej po preparacie z emulsjà tłusz-
czowà – wymieniç biuret´ na nowà.
35
14. SPORZÑDZANIE MIESZANIN
W NEONATOLOGII
I. OGÓLNE ZASADY
Spełnienie wymagaƒ ˝ywieniowych noworodka, zwłaszcza wczeÊniaka i opracowanie
dla niego stabilnej mieszaniny jest bardzo trudne. W neonatologii mamy do czynienia
z tworzeniem małych obj´toÊciowo mieszanin, zawierajàcych du˝e st´˝enia elektrolitów,
znacznie wi´ksze ni˝ u starszych dzieci, co sprzyja wyst´powaniu niezgodnoÊci.
W celu utrzymania stabilnoÊci fizykochemicznej mieszaniny stosuje si´ w neonatologii
system dwóch pojemników (Two In One). System ten zapewnia oddzielnà poda˝
emulsji tłuszczowej eliminujàc ryzyko jej rozkładu.
I pojemnik – mieszanina roztworów glukozy, aminokwasów, elektrolitów, fosforanów,
pierwiastków Êladowych.
II pojemnik – mieszanina emulsji tłuszczowej z witaminami rozpuszczalnymi w wodzie
i w tłuszczach.
Do poda˝y mieszanin do ˝ywienia pozajelitowego stosuje si´ infuzyjne pompy
obj´toÊciowe i strzykawkowe. Rodzaj zastosowanej pompy determinuje wybór
opakowania bezpoÊredniego dla sporzàdzanej mieszaniny.
Przy sporzàdzaniu mieszanin dla pacjentów neonatologicznych nale˝y uwzgl´dniç
tzw. + % na dren.
Mieszaniny dla pacjentów neonatologicznych nale˝y sporzàdzaç metodami opisanymi
w rozdziałach 13. i 16., z zachowaniem ogólnych zasad sporzàdzania (rozdział 11.).
W przypadku, gdy jest to niemo˝liwe, mieszaniny nale˝y sporzàdzaç manualnie,
wykorzystujàc dost´pne systemy bezigłowe (łàczniki, kraniki trójdro˝ne).
II. SPOSÓB POST¢POWANIA
1. CzynnoÊci wst´pne i ogólne zasady post´powania – jak podano w rozdziale 11.
„Ogólne zasady sporzàdzania mieszanin do ˚P”.
2. Sporzàdzanie mieszanin w strzykawkach
• „Strzykawk´ opakowanie” wyjàç aseptycznie z opakowania na granicy lo˝y.
• Na „strzykawk´ opakowanie” nakr´ciç łàcznik lub kranik trójdro˝ny.
• Do opakowaƒ z płynami infuzyjnymi (worek, butelka) podłàczyç odpowiednie
akcesoria do aseptycznego pobierania.
• Napełnianie „strzykawki opakowania” odbywa si´ według ogólnej metody miesza-
nia, rozpoczynajàc od glukozy o najwy˝szym st´˝eniu.
• Pobieraç produkty lecznicze z ampułek i fiolek, butelek i worków zmieniajàc igł´
i strzykawk´ dla ka˝dego preparatu.
• Przykr´ciç strzykawk´ z pobranym preparatem do łàcznika lub kranika przy
„strzykawce opakowaniu” i przetoczyç pobranà obj´toÊç płynu.
SPORZÑDZANIE MIESZANIN
W NEONATOLOGII

36
FARMACEUTYCZNE STANDARDY SPORZÑDZANIA
MIESZANIN DO ˚YWIENIA POZAJELITOWEGO
Nale˝y pami´taç, aby na koƒcu przetoczyç pewnà iloÊç ( 1–2 ml) preparatu wys-
t´pujàcego w mieszaninie w wi´kszej obj´toÊci (glukoza, aminokwasy, woda), celem
wypchni´cia ewentualnych pozostałoÊci innych składników z łàcznika.
• Za ka˝dym razem po dodaniu kolejnego składnika mieszanin´ wymieszaç przez
ostro˝ne potrzàsanie strzykawkà.
• Z gotowej mieszaniny usunàç powietrze przez przesuni´cie tłoka w gór´.
• Strzykawk´ zamknàç jałowym koreczkiem lub przedłu˝aczem bursztynowym.
• Nakleiç etykiet´ na strzykawk´ i osłonk´.
• Ponownie sprawdziç zgodnoÊç wypisanej etykiety z receptà.
• Na strzykawk´ nało˝yç osłonk´ chroniàcà przed Êwiatłem.
3. Sporzàdzanie mieszanin w workach
Stosuje si´ tylko wtedy, gdy nie ma mo˝liwoÊci sporzàdzenia mieszaniny za pomocà
biurety lub pomp sterowanych komputerowo. KolejnoÊç post´powania jest nast´-
pujàca:
• worek wyjàç aseptycznie z opakowania;
• zamknàç przewód doprowadzajàcy w worku za pomocà nakr´tki dołàczonej do
opakowania;
• zamknàç zacisk na przewodzie doprowadzajàcym;
• produkty lecznicze przetaczaç do worka przez kanał do wstrzykni´ç (port) za po-
mocà strzykawek;
• zdezynfekowaç miejsce dostrzykiwania, w przypadku gdy powierzchnia membrany
kanału do wstrzykni´ç nie jest aseptycznie zabezpieczona;
Uwaga! Nie ma potrzeby odka˝ania w przypadku, gdy powierzchnia membrany
zostaje odsłoni´ta przez złamanie koƒcówki.
• glukoz´, aminokwasy, preparaty z ampułek i fiolek dodawaç do worka pojedynczo,
pobierajàc je z opakowaƒ za pomocà oddzielnych strzykawek;
• wkłuç igł´ do kanału do wstrzykni´ç tak, aby nie uszkodziç jego Êcian i jako pier-
wszà dostrzyknàç glukoz´ o najwy˝szym st´˝eniu; od∏àczyç strzykawk´, pozosta-
wiajàc igł´ w miejscu wkłucia;
• w przypadku dodawania preparatów do worka le˝àcego na blacie nie zostawiaç
igły w porcie, z powodu mo˝liwoÊci wycieku płynu, lecz ka˝dorazowo wkłuwaç si´
w port nowà igłà;
• pobraç zlecone iloÊci pozostałych preparatów i dostrzyknàç je do worka zgodnie
z ogólnà regułà mieszania;
• ka˝dorazowo po dostrzykni´ciu poszczególnych preparatów wymieszaç zawartoÊç
worka przez uciskanie;
• obserwowaç, czy nie wyst´pujà zmiany w wyglàdzie: zm´tnienie, zabarwienie, krys-
talizacja i inne;
• odpowietrzyç worek, wyciskajàc powietrze przez wkłutà w port igł´;

37
• wyjàç igł´ z portu do wstrzykni´ç, miejsce wkłucia zdezynfekowaç i zało˝yç zatyczk´
zabezpieczajàcà;
• na worek i osłonk´ nakleiç etykiet´;
• ponownie sprawdziç zgodnoÊç etykiety z receptà;
• na worek z mieszaninà nało˝yç osłonk´ chroniàcà przed Êwiatłem.
SPORZÑDZANIE MIESZANIN
W WORKACH GOTOWYCH DO U˚YCIA (RTU)
15. SPORZÑDZANIE MIESZANIN W WORKACH
GOTOWYCH DO U˚YCIA (RTU)
Sporzàdzanie mieszanin RTU to aktywacja i uzupełnianie worków dwu-/trójkomorowych
przez dodanie witamin, pierwiastków Êladowych, brakujàcych elektrolitów, emulsji
tłuszczowej (w przypadku worków dwukomorowych) [kompletne ˚P], ewentualnie przez
dodanie jedynie witaminy B1 [niekompletne ˚P].
Zaleca si´, aby aktywacja i uzupełnianie worków odbywało si´ w aptece szpitalnej.
I. OGÓLNE ZASADY
Sporzàdzajàc mieszaniny w workach RTU nale˝y przestrzegaç zasad aseptyki pracy
oraz wytycznych producenta dotyczàcych aktywacji, uzupełniania i przechowywania
mieszanin.
1. Do worków RTU mo˝na dostrzykiwaç preparaty uzupełniajàce, tylko pod warun-
kiem ˝e przebadano ich stabilnoÊç i nieprzekroczone sà graniczne, dopuszczalne
przez producenta st´˝enia.
2. Nale˝y zawsze liczyç st´˝enia łàcznie z tym, co jest ju˝ w worku, a nie tylko to, co
dodawane jest przez kanał do wstrzykni´ç, aby po suplementacji dopuszczalne dawki
nie były przekroczone.
3. Zadaniem osoby sporzàdzajàcej mieszanin´ RTU jest bardzo dokładna kontrola
dawek preparatów podanych w zleceniu i po przeliczeniu ich – porównanie
z wytycznymi producenta.
II. ZASADY SPORZÑDZANIA MIESZANIN RTU W WORKACH DWUKOMOROWYCH
KolejnoÊç mieszania:
1. Aktywacja worka przez wymieszanie zawartoÊci dwóch komór – Glu + AA
¨
2. Dostrzykiwanie preparatów pierwiastków Êladowych i elektrolitów
¨
3. Przetoczenie do worka emulsji tłuszczowej
¨
Dostrzykiwanie witamin
38
FARMACEUTYCZNE STANDARDY SPORZÑDZANIA
MIESZANIN DO ˚YWIENIA POZAJELITOWEGO
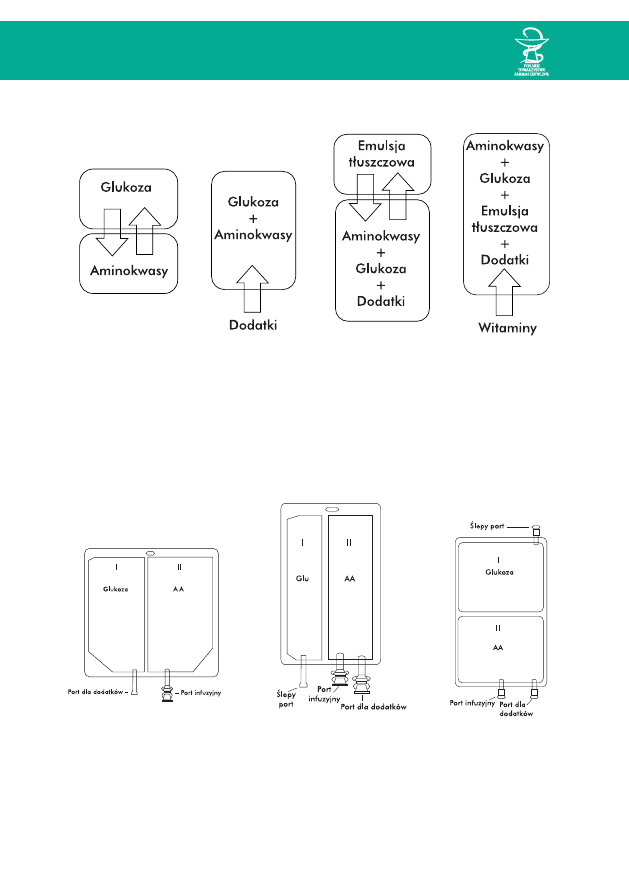
39
SPORZÑDZANIE MIESZANIN
W WORKACH GOTOWYCH DO U˚YCIA (RTU)
Ryc. 3. Ogólny schemat sporzàdzania mieszanin do ˚P w workach dwukomorowych
Sporzàdzanie mieszanin w workach dwukomorowych odbywa si´ według ogólnego
schematu (ryc. 3). Pewne ró˝nice w sposobie post´powania w przypadku systemów:
Aminomix, Clinimix, NuTRIflex zwiàzane sà z innym układem komór w workach.
Ryc. 4. Schemat worka Aminomix Ryc. 5. Schemat worka Clinimix Ryc. 6. Schemat worka
NuTRIflex
1 2 3 4
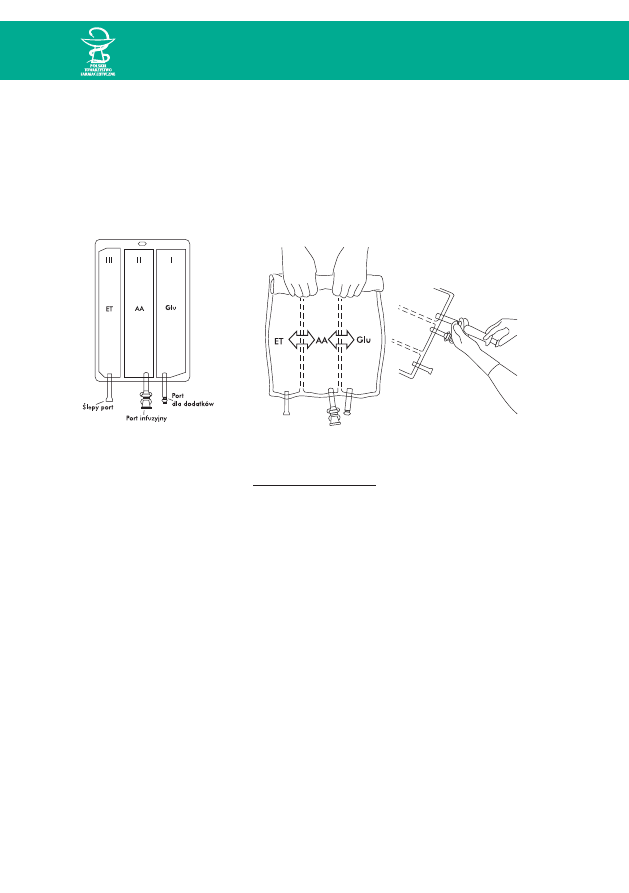
40
FARMACEUTYCZNE STANDARDY SPORZÑDZANIA
MIESZANIN DO ˚YWIENIA POZAJELITOWEGO
III. ZASADY SPORZÑDZANIA MIESZANIN RTU W WORKACH TRÓJKOMOROWYCH
W przypadku worków trójkomorowych zasada sporzàdzania mieszanin do ˚P zale˝y od
układu komór w worku i wytycznych producenta.
System Multimel
Ryc. 7. Schemat worka Multimel Ryc. 8. Aktywacja i uzupełnianie worka Multimel
KolejnoÊç mieszania:
1. Aktywacja worka przez wymieszanie zawartoÊci trzech komór – Glu + AA + ET
¨
2. Dostrzykiwanie preparatów pierwiastków Êladowych, elektrolitów, witamin.
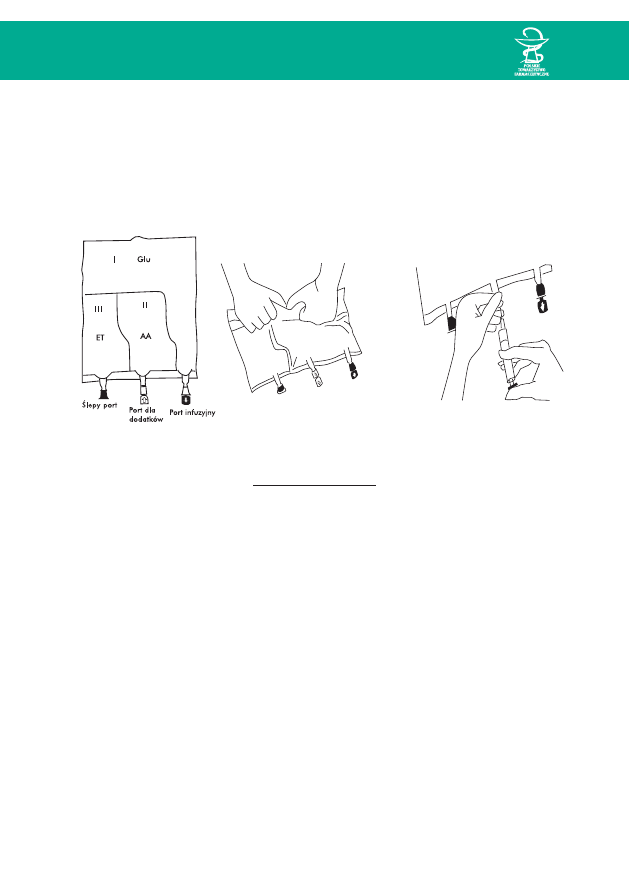
41
System Kabiven
Ryc. 9. Schemat worka Kabiven
Ryc. 10. Aktywacja i uzupełnianie worka Kabiven
KolejnoÊç mieszania:
1. Aktywacja worka przez wymieszanie zawartoÊci trzech komór: Glu + AA + ET
¨
2. Dostrzykiwanie preparatów pierwiastków Êladowych, elektrolitów, witamin.
SPORZÑDZANIE MIESZANIN
W WORKACH GOTOWYCH DO U˚YCIA (RTU)
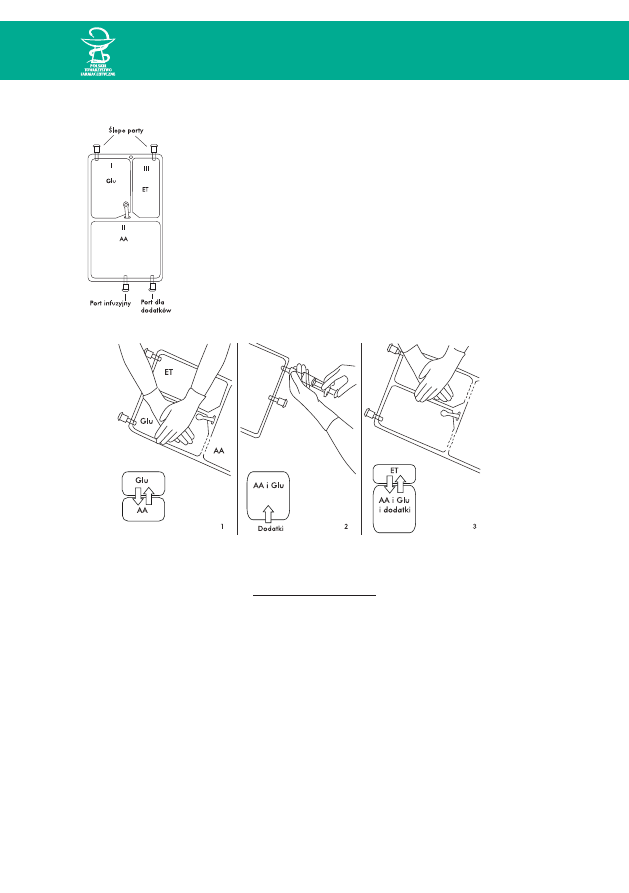
42
FARMACEUTYCZNE STANDARDY SPORZÑDZANIA
MIESZANIN DO ˚YWIENIA POZAJELITOWEGO
System NuTRIflex Lipid
Ryc. 11. Schemat worka NuTRIflex Lipid
Ryc. 12. Aktywacja i uzupełnianie worka NuTRIflex Lipid
KolejnoÊç mieszania:
1. Przetoczenie roztworu glukozy z komory I do komory II z roztworem aminokwasów
Glu + AA
¨
2. Dostrzykiwanie do mieszaniny (Glu + AA) elektrolitów i pierwiastków Êladowych
¨
3. Przetoczenie emulsji tłuszczowej z komory III do komory II z mieszaninà
(Glu + AA + elektrolity + pierwiastki Êladowe)
¨
Dodanie witamin do mieszaniny.

43
IV. SPOSÓB POST¢POWANIA
1. CzynnoÊci wst´pne i ogólne zasady – jak podano w rozdziale 11. „Ogólne zasady
sporzàdzania mieszanin do ˚P”.
2. Sporzàdzanie mieszanin
• Wyjàç aseptycznie z opakowania gotowy worek.
• Połàczyç komory worka poprzez rozerwanie przegrody oddzielajàcej składniki
(wg instrukcji producenta – ryc. 3, 8, 10, 12).
• CałoÊç dokładnie wymieszaç.
• Zdezynfekowaç miejsce dostrzykiwania Êrodkiem dezynfekcyjnym, ustalonym
według wewn´trznych procedur szpitala.
• W przypadku gdy powierzchnia membrany kanału do wstrzykiwaƒ jest zabezpie-
czona koƒcówkà – usunàç jà przez złamanie.
Uwaga! JeÊli powierzchnia membrany portu zostaje odsłoni´ta przez złamanie
zatyczki, to nie ma potrzeby jej odka˝ania.
• W przypadku dodawania do worka dwukomorowego emulsji tłuszczowej – nale˝y
wykonaç t´ czynnoÊç za pomocà dołàczonego do worka zestawu (według zaleceƒ
producenta).
• ZawartoÊç ampułek i fiolek (elektrolity, pierwiastki Êladowe, witaminy) dodawaç
pojedynczo, oddzielnà strzykawkà do worka przez kanał do wstrzykni´ç.
• Wbiç igł´ do portu tak, aby nie uszkodziç jego Êcian i jako pierwszy preparat
dostrzyknàç fosforany; usunàç strzykawk´, pozostawiajàc igł´ w miejscu wkłucia.
Uwaga! W przypadku dodawania preparatów do worka le˝àcego na blacie, igły
nie zostawiaç w porcie (mo˝liwoÊç wycieku płynu), lecz ka˝dorazowo wkłuwaç
si´ nowà igłà do portu.
• Dokładnie wymieszaç zawartoÊç worka przez jego uciskanie.
• Pobieraç zlecone iloÊci pozostałych produktów leczniczych – ka˝dy za pomocà innej
strzykawki z igłà; igł´ zdjàç i przez wkłutà igł´ (pozostawionà w porcie) dostrzy-
kiwaç pozostałe składniki według kolejnoÊci przedstawionej w rozdziale 11.
• W przypadku widocznych Êladów preparatu witaminowego w porcie, wstrzyknàç
kilka ml 0,9% NaCl lub Agua pro inj. w celu jego przepłukania.
• Po ka˝dorazowym dostrzykni´ciu poszczególnych preparatów wymieszaç całoÊç.
• Odpowietrzyç worek (wyciskajàc powietrze przez wkłutà igł´).
• Wyjàç igł´ z kanału do wstrzykni´ç, miejsce wkłucia zdezynfekowaç.
• Nakleiç etykiet´ na worek z mieszaninà.
• W razie koniecznoÊci zabezpieczyç worek ciemnà folià i nakleiç drugà etykiet´.
SPORZÑDZANIE MIESZANIN
W WORKACH GOTOWYCH DO U˚YCIA (RTU)

44
FARMACEUTYCZNE STANDARDY SPORZÑDZANIA
MIESZANIN DO ˚YWIENIA POZAJELITOWEGO
16. SPORZÑDZANIE MIESZANIN ZA POMOCÑ
POMP STEROWANYCH KOMPUTEROWO
Ka˝de urzàdzenie słu˝àce do sporzàdzania mieszanin do ˚P musi posiadaç deklaracj´
zgodnoÊci CE.
W chwili obecnej w Polsce dost´pne sà dwa typy pomp sterowanych komputerowo:
1. Mieszalnik MicroMacro-BAXA
2. Multicomp II – FRESENIUS
Za pomocà tych mieszalników mo˝na sporzàdziç mieszaniny z dokładnoÊcià:
1. Mieszalnik MicroMacro-BAXA:
• 12 êródeł do 0,2 ml.
2. Multicomp II – FRESENIUS:
• 6 pomp głównych do 10 ml,
• 3 dodatkowych pomp strzykawkowych do 1 ml.
I. SPOSÓB POST¢POWANIA
1. CzynnoÊci wst´pne – w pomieszczeniu administracyjnym pracowni.
1. 1. Wprowadzenie danych pacjenta i składu mieszaniny z recepty do programu
komputerowego.
Program komputerowy:
• pozwala sprawdziç limity st´˝eƒ składników, kontroluje graniczne st´˝enia
niezgodnych ze sobà jonów;
• oblicza osmolarnoÊç mieszaniny (okreÊla drog´ podania), liczb´ kalorii
białkowych i pozabiałkowych, zawartoÊç elektrolitów, szybkoÊç wlewu;
• pozwala na archiwizacj´ danych pacjenta i mieszanin do ˚P.
1. 2. Wydruk dokumentów dla indywidualnego pacjenta (lista potrzeb, etykieta,
skład).
1. 3. Kontrola zgodnoÊci treÊci etykiety z receptà.
2. Sporzàdzanie mieszanin zgodnie z instrukcjà producenta dla ka˝dego typu pomp
3. Raport powykonawczy (produkcyjny) zawiera nast´pujàce dane:
• nr serii mieszaniny,
• imi´ i nazwisko pacjenta,
• dat´ i godzin´ przygotowania mieszaniny,
• kolejnoÊç mieszania oraz obj´toÊç dodawanych składników,
• całkowità obj´toÊç planowanà oraz zmierzonà mieszaniny,
• wag´ teoretycznà i rzeczywistà mieszaniny oraz procentowà ró˝nic´ pomi´dzy nimi.

45
4. Kontrola procesu wytwarzania
4.1. Kontrola wagowa
Po wprowadzeniu recept indywidualnych drukowany jest skład i protokoły dla ka˝dej
mieszaniny, a po sporzàdzeniu – protokoły wykonania mieszaniny.
Urzàdzenie potwierdza liczb´ pobranych składników:
• informujàc na bie˝àco o zaistniałych przekroczeniach liczbowych poszczególnych
składników (dopuszczalny błàd ± 3%) – Multicomp II – FRESENIUS
lub
• porównujàc teoretyczny ci´˝ar z rzeczywistym ci´˝arem wykonanej mieszaniny
(dopuszczalny błàd ± 3%) – MicroMacro-BAXA
4. 2. Raporty koƒcowe
Po zakoƒczeniu sporzàdzania mieszanin drukowany jest raport dzienny. Jest to
wydruk zawierajàcy nast´pujàce dane:
• wszystkie czynnoÊci zwiàzane z wykonaniem mieszaniny,
• zu˝ycie wszystkich preparatów w danym dniu,
• numery serii i daty wa˝noÊci preparatów.
SPORZÑDZANIE MIESZANIN ZA POMOCÑ POMP
STEROWANYCH KOMPUTEROWO

46
FARMACEUTYCZNE STANDARDY SPORZÑDZANIA
MIESZANIN DO ˚YWIENIA POZAJELITOWEGO
17. ZALECANE ÂRODKI OSTRO˚NOÂCI PODCZAS
SPORZÑDZANIA, PRZECHOWYWANIA
I PODAWANIA MIESZANIN DO ˚P
1. Przed sporzàdzeniem mieszaniny do ˚P nale˝y sprawdziç dokładnie jakoÊç
stosowanych preparatów (klarownoÊç, homogennoÊç – emulsje tłuszczowe!, szczelnoÊç
opakowaƒ).
2. Nale˝y ÊciÊle przestrzegaç zaleceƒ dotyczàcych warunków i czasu przechowywania
mieszanin.
3. Przed podaniem mieszaniny choremu nale˝y sprawdziç godzin´ jej sporzàdzenia.
4. Mieszaniny do ˚P nie mo˝na podaç choremu, je˝eli:
• przewidywany czas – od sporzàdzenia do koƒca wlewu – jest dłu˝szy ni˝ 24 godz.
w temperaturze pokojowej (je˝eli wlew trwa dłu˝ej ni˝ 24 godz., nale˝y go przer-
waç i zniszczyç pozostałoÊç mieszaniny);
Niedopuszczalna jest praktyka sporzàdzania mieszanin do ˚P w cyklu
48-godzinnym (dwa dni), tzn. przetaczania mieszaniny w ciàgu >24–48 godz.
• temperatura przechowywania mieszaniny spadła poni˝ej wymaganych para-
metrów, tj. temp. +2°C;
• pojawiło si´ w niej zm´tnienie lub osad (mieszaniny bez tłuszczu);
• nastàpiła zmiana zabarwienia (kolor inny ni˝ zwykle Êwiadczàcy o zachodzàcych
reakcjach chemicznych w mieszaninie);
• nastàpiła zmiana homogennoÊci mieszaniny (rozwarstwienie, „kłaczkowanie”);
• nastàpiło uszkodzenie worka (nawet niewidoczne, np. przekłucie igłà).

47
18. KONTROLA PROCESU SPORZÑDZANIA
MIESZANIN DO ˚P
Proces sporzàdzania mieszanin do ˚P musi przebiegaç zgodne z wymaganiami
Dobrej Praktyki Wytwarzania oraz podlegaç systematycznej ocenie. Ocena mo˝liwa
jest tylko przez kontrol´ wszystkich etapów pracy. Nie mo˝e opieraç si´ wyłàcznie na
badaniu produktu koƒcowego. Jedynie kontrola całego procesu daje gwarancj´
wymaganej i powtarzalnej jakoÊci badanych produktów.
I. OGÓLNE ZASADY
1. Sporzàdzanie
• Sporzàdzanie mieszanin do ˚P musi byç prowadzone wyłàcznie w aptece, która
posiada odpowiednie pomieszczenie i sprz´t oraz odpowiednio przeszkolony
personel, pracujàcy pod nadzorem farmaceuty.
• Farmaceuta jest odpowiedzialny za wykonanie zlecenia lekarskiego; musi okreÊliç,
czy skład recepty nie budzi zastrze˝eƒ dotyczàcych stabilnoÊci, czy dobór
produktów leczniczych, zakresy st´˝eƒ sà prawidłowe (iloczyn CaxP, wartoÊç CAN);
w przypadku jakichkolwiek wàtpliwoÊci farmaceuta musi poinformowaç
o tym lekarza zlecajàcego i zaproponowaç zmian´ receptury, je˝eli to mo˝liwe.
• Musi byç okreÊlana zgodnoÊç fizykochemiczna mieszanin do ˚P.
• Sporzàdzanie mieszanin musi przebiegaç zgodnie z napisanymi i zatwierdzonymi
procedurami.
• Sporzàdzanie mieszanin do ˚P musi byç prowadzone zgodnie z zasadami asep-
tyki pracy.
• Proces aseptyczny musi byç walidowany przez symulowany wlew po˝ywki (test
aseptycznego napełniania – media fill).
• Etykietowanie sporzàdzonych mieszanin musi byç zgodne z obowiàzujàcymi
przepisami.
• Mieszaniny do ˚P muszà byç przechowywane, zgodnie z wymaganiami, w tem-
peraturze +2°C do +8°C i muszà byç chronione przed dost´pem Êwiatła.
• Transport mieszanin do ˚P musi odbywaç si´ w opakowaniach, które chronià
worek i jego zawartoÊç przed zmianami, w obni˝onej, kontrolowanej temperaturze.
2. Termin przydatnoÊci do u˝ycia
• Termin wa˝noÊci = przydatnoÊci do u˝ycia produktów koƒcowych okreÊlany jest na
podstawie wyników badaƒ stabilnoÊci, piÊmiennictwa itp.
• Mieszaniny do ˚P muszà zachowywaç stabilnoÊç fizycznà, chemicznà i mikro-
biologicznà przez okres nie krótszy ni˝ 24 godz. w temperaturze pokojowej, tj. od
momentu sporzàdzenia do koƒca wlewu.
• Wykonanie dokładnych badaƒ i okreÊlenie na ich podstawie stopnia trwałoÊci
pozwala na przechowywanie mieszanin przez dłu˝szy okres.
KONTROLA PROCESU SPORZÑDZANIA
MIESZANIN DO ˚P

48
FARMACEUTYCZNE STANDARDY SPORZÑDZANIA
MIESZANIN DO ˚YWIENIA POZAJELITOWEGO
• Kontrola ka˝dego składu mieszaniny do ˚P nie jest mo˝liwa – wskazane jest wi´c
opracowanie kilku standardowych składów i okreÊlenie ich trwałoÊci w czasie,
a nast´pnie okreÊlenie zakresów st´˝eƒ substancji uzupełniajàcych, które bez-
piecznie mo˝na dodaç do zbadanych mieszanin; takie opracowanie umo˝liwi
sporzàdzanie bez ryzyka niezgodnoÊci wielu stabilnych mieszanin, o składzie dos-
tosowanym do indywidualnych potrzeb leczonych chorych; jest to szczególnie
bezwzgl´dnie wymagane w przypadku przygotowywania mieszanin o dłu˝szym
terminie wa˝noÊci – „na zapas”, na kilka dni przed przewidywanym terminem
podania choremu.
• Przebadane mieszaniny muszà byç przechowywane w obni˝onej temperaturze
(+2°C do +8°C) przez okreÊlony badaniami czas, a nast´pnie po doprowadzeniu
do temperatury pokojowej (3–6 godz. w zale˝noÊci od obj´toÊci worka) podawane
choremu w czasie do 24 godz.
• Z powodu niskiej stabilnoÊci, witaminy oraz insulina powinny byç dodawane do
mieszaniny bezpoÊrednio przed podłàczeniem worka choremu.
3. Kontrola produktów koƒcowych
Analiza preparatów koƒcowych musi byç przeprowadzana systematycznie, według
okreÊlonych procedur.
• Rodzaj przeprowadzonych badaƒ uzale˝niony jest od liczebnoÊci serii.
Serià jest okreÊlona liczba preparatów koƒcowych uznawanych za jednorodne.
• 1 seria = 1worek, je˝eli jego skład jest specyficzny lub 1 seria = kilka
worków o tym samym składzie (do kontroli fizyko-chemicznej).
• 1 seria = kilka worków sporzàdzonych w takich samych warunkach, przez
ten sam personel (do kontroli jałowoÊci).
• Pobieranie prób powinno byç reprezentatywne dla serii. Musi byç wykonywane
według precyzyjnie napisanych procedur.
• Do kontroli jałowoÊci – dla serii liczniejszych – zalecane jest pobieranie trzech
worków: napełnianych na poczàtku, w Êrodku i na koƒcu serii; dla mniejszych serii
nale˝y pobraç co najmniej jeden worek i wybraç właÊnie ten ostatni.
• Pobrane próby powinny byç przechowywane do czasu ewentualnej kontroli.
Warunki przechowywania muszà byç takie same jak dla sporzàdzonych mieszanin
do ˚P.
• Kontrola jałowoÊci mieszanin do ˚P nie jest próbà „blokujàcà” dla zwolnienia serii;
zwolnienie jest zawsze przed wynikiem kontroli mikrobiologicznej, co tym
wa˝niejszym czyni system zapewnienia jakoÊci i kontroli Êrodowiska.
• Wszystkie sporzàdzone mieszaniny muszà byç poddawane kontroli jakoÊciowej,
chodzi tu szczególnie o kontrol´ wizualnà oraz, je˝eli to mo˝liwe, kontrol´ masy
(seria = 1 worek). Sprawdzana powinna byç zgodnoÊç danych na etykiecie ze zlece-
niem (receptà) oraz zgodnoÊç sposobu wykonania z obowiàzujàcà procedurà.
• Wszystkie dokumenty dotyczàce kontroli produktu koƒcowego muszà byç załà-
czone do raportu serii.

49
• Raport koƒcowy serii powinien zawieraç: dokumenty potwierdzajàce warunki asep-
tycznego wytwarzania oraz kontroli produktu koƒcowego, zlecenie lekarskie oraz
nazwy handlowe, nr serii, daty wa˝noÊci u˝ytych produktów leczniczych.
II. SPOSÓB POST¢POWANIA
1. Rutynowa kontrola mikrobiologiczna
• Kontrola czystoÊci mikrobiologicznej powietrza w klasie czystoÊci A – podczas
ka˝dego cyklu pracy – wykonanie tzw. Êlepej próby, która jest modyfikacjà testu
media fill; napełnienie ostatniego worka w cyklu 5% glukozà, 0,9% NaCl lub pro-
duktami leczniczymi do ˝ywienia pozajelitowego, w sposób naÊladujàcy proces
sporzàdzania mieszaniny do ˚P i przekazanie worka do badania jałowoÊci
• Okresowo (cz´stotliwoÊç badania wg wewn´trznej procedury) badanie jałowoÊci
mieszanin do ˚P – pobieranie próby według pkt. I. 3.
2. Kontrola jakoÊciowa (seria = 1 worek)
• Ka˝da mieszanina do ˚P przed wydaniem z apteki musi byç poddana kontroli
wizualnej. Zalecana jest równie˝ kontrola masy.
3. Kontrola wydania z apteki
• Wydawanie mieszanin do ˚P z apteki musi przebiegaç zgodnie z opracowanymi
wewn´trznymi procedurami.
• Zawsze przed wydaniem mieszaniny z apteki ponownie musi byç przeprowadzona
kontrola wizualna oraz zgodnoÊç etykiety z receptà.
• Osoba odbierajàca sporzàdzone mieszaniny musi potwierdziç ten fakt czytelnym
podpisem wraz z datà i godzinà odbioru.
• Fakt wydania mieszaniny powinien byç zaznaczony w dokumentacji apteki (ksi´ga
ewidencyjna).
• Recepta, wraz z protokołem wykonania mieszaniny, powinna byç przechowywana
w dokumentacji apteki.
4. Kontrola podłàczenia
Podczas podawania mieszanin do ˚P muszà byç przestrzegane zalecenia dotyczàce
warunków i czasu przetaczania. Wyznaczeni pracownicy apteki powinni okresowo
(zgodnie z opracowanymi wewn´trznymi procedurami) kontrolowaç warunki
przechowywania oraz sposób i czas przetaczania mieszanin.
KONTROLA PROCESU SPORZÑDZANIA
MIESZANIN DO ˚P

50
FARMACEUTYCZNE STANDARDY SPORZÑDZANIA
MIESZANIN DO ˚YWIENIA POZAJELITOWEGO
19. UTYLIZACJA
Odpady o charakterze szpitalnym zawsze oceniano jako bardziej niebezpieczne ni˝
komunalne. Główne ryzyko zwiàzane z ich powstawaniem wià˝e si´ z mo˝liwoÊcià
ska˝enia Êrodowiska patogenami i bakteriami chorobotwórczymi.
Problemy ich unieszkodliwiania wià˝à si´ zatem przede wszystkim z koniecznoÊcià
przeciwdziałania ska˝eniom biologicznym (epidemiologicznym) potencjalnie przez nie
wywoływanym.
Przy braku właÊciwie zorganizowanych systemów kontroli, ograniczaniu segregacji,
odpady szpitalne stanowiłyby bardzo zró˝nicowanà mieszank´ wszelkich typów Êmieci
– od typowych odpadków komunalnych (˝ywnoÊç, opakowania), poprzez toksyczne
chemikalia (leki, odczynniki), a koƒczàc na odpadach zainfekowanych biologicznie
(narz´dzia, opatrunki, odpady pooperacyjne).
I. PODSTAWY PRAWNE
Ustawa o odpadach z dnia 27 kwietnia 2001 r. z póên. zm. (tekst jednolity, Dz. U.
z 2007 r. Nr. 39, poz. 251)
• Art. 3 ust. 3 pkt 5. Odpady medyczne – odpady powstajàce w zwiàzku
z udzielaniem Êwiadczeƒ zdrowotnych oraz prowadzeniem badaƒ
i doÊwiadczeƒ naukowych w zakresie medycyny.
• Art. 42. Zabrania si´ poddawania odzyskowi odpadów medycznych. W sto-
sunku do tej kategorii dopuszczalne jest wyłàcznie unieszkodliwianie.
Rozporzàdzenie Ministra Ârodowiska z dnia 27 wrzeÊnia 2001 r. w sprawie katalogu
odpadów (Dz. U. z 2001 r., Nr 112, poz. 1206)
Rozporzàdzenie Ministra Zdrowia z dnia 23 sierpnia 2007 r. w sprawie szczegółowego
sposobu post´powania z odpadami medycznymi (Dz. U. z 2007 r. Nr 162, poz. 1153)
II. KLASYFIKACJA ODPADÓW
1. Odpady niebezpieczne
• Odpady zakaêne sà to odpady niebezpieczne, które zawierajà ˝ywe mikroorga-
nizmy lub ich toksyny, o których wiadomo lub co do których istniejà wiarygodne
podstawy, ˝e wywołujà choroby zakaêne u ludzi lub innych ˝ywych organizmów.
Do tej kategorii zalicza si´ mi´dzy innymi zu˝yte, zainfekowane materiały opa-
trunkowe, zu˝yty sprz´t jednorazowego u˝ytku, zu˝yte narz´dzia chirurgiczne i za-
biegowe, odpady z oddziałów i szpitali zakaênych oraz chorób płuc (w tym
pozostawione przez chorych resztki konsumpcyjne), kultury bakteryjne, zwłoki
i szczàtki zwierzàt laboratoryjnych, preparaty aktywne biologicznie np. szczepionki
i inne materiały podejrzane o kontakt z materiałem zakaênym, szczàtki ludzkie,
martwe płody, tkank´ pooperacyjnà i posekcyjnà, pojemniki na krew i konserwanty
do niej itp.
Gromadzi si´ je w workach koloru czerwonego.

51
• Odpady specjalne sà to odpady niebezpieczne, które zawierajà substancje
chemiczne, o których wiadomo lub co do których istniejà wiarygodne podstawy, ˝e
wywołujà choroby niezakaêne u ludzi lub innych ˝ywych organizmów albo mogà
byç êródłem ska˝enia Êrodowiska. Do tej kategorii zaliczono chemikalia, w tym od-
czynniki chemiczne zawierajàce substancje niebezpieczne, leki cytotoksyczne
i cytostatyczne, odpady amalgamatu dentystycznego, uszkodzone termometry
rt´ciowe itp.
Gromadzi si´ je w workach koloru ˝ółtego.
2. Odpady pozostałe
Sà to odpady medyczne nieposiadajàce właÊciwoÊci niebezpiecznych.
Do tej kategorii zaliczono chemikalia, w tym odczynniki chemiczne niezawierajàce sub-
stancji niebezpiecznych, leki inne ni˝ cytostatyki, zu˝yty sprz´t i materiały medyczne,
które nie miały kontaktu z materiałem zakaênym, puste fiolki, opakowania po lekach.
III. SPOSÓB POST¢POWANIA Z ODPADAMI POZOSTAŁYMI
1. Z odpadami pozostałymi post´puje si´ w sposób przewidziany dla odpadów komu-
nalnych.
Gromadzi si´ je w workach koloru niebieskiego.
2. Odpady medyczne o ostrych koƒcach i kraw´dziach (zu˝yte igły, ampułki) zbiera si´
w sztywnych, odpornych na działanie wilgoci, mechanicznie odpornych na przekłu-
cie bàdê przeci´cie pojemnikach jednorazowego u˝ycia. Pojemniki te powinny
znajdowaç si´ w miejscach powstawania odpadów. Nale˝y je wymieniaç na nowe nie
rzadziej ni˝ co 48 godz. Pojemniki mogà byç wypełnione nie wi´cej ni˝ 2/3 ich
obj´toÊci.
3. Dopuszcza si´ zbieranie odpadów okreÊlanych jako pozostałe do pojemników
wielokrotnego u˝ycia.
4. Pojemniki i worki powinny byç zaopatrzone w naklejk´ zawierajàcà widoczne
oznaczenie miejsca pochodzenia odpadów, rodzaju zawartoÊci, dat´ zamkni´cia
oraz dane identyfikacyjne osoby, która go dokonała.
UTYLIZACJA

52
FARMACEUTYCZNE STANDARDY SPORZÑDZANIA
MIESZANIN DO ˚YWIENIA POZAJELITOWEGO
20. SZKOLENIA
I. RODZAJE SZKOLE¡
Szkolenia teoretyczne i szkolenia praktyczne – według wewn´trznych procedur.
1. Wst´pne – dla ka˝dego pracownika na ka˝dym stanowisku przed podj´ciem
samodzielnej pracy (zapoznanie si´ z procedurami oraz z praktycznym wykonaniem
obejmowanych obowiàzków).
2. Ciàgłe – dla wszystkich pracowników po wprowadzeniu zmian w procedurach.
II. PLAN SZKOLE¡
1. Dla nowych pracowników przyst´pujàcych do pracy.
2. Dla pracowników rotujàcych si´.
3. Dla pracowników stałych.
III. SYSTEM OCENY
1. Kontrola poznanej wiedzy.
2. Kontrola wykonania.
3. Analiza b∏´dów.
IV. DOKUMENTACJA
1. Plan szkoleƒ.
2. Lista uczestników szkolenia.
3. ZaÊwiadczenie zawierajàce temat i czas szkolenia, podpis szkolonego i szkolàcego.

53
DOMOWE ˚YWIENIE POZAJELITOWE
21. DOMOWE ˚YWIENIE POZAJELITOWE
˚P pacjentów domowych budzi wiele emocji w Êrodowisku lekarzy i farmaceutów szpi-
talnych, ze wzgl´du na brak jasno sprecyzowanych przepisów ustawowych:
• brak spójnoÊci Prawa farmaceutycznego z wytycznymi NFZ i rozporzàdzeniami
Ministra Zdrowia;
• brak systemu przekazywania mieszanin od wytwórcy (apteka szpitalna) do pacjenta;
• brak jednoznacznych zapisów dotyczàcych odpowiedzialnoÊci za mieszanin´ na
poszczególnych etapach dystrybucji.
Przedstawiamy:
1) Opini´ GIF z 8 maja 2009 r. – pismo GIF-N-0741/21-1/MEK/09 do Dyrektora
Wojewódzkiego Szpitala Specjalistycznego im. M. Kopernika w Łodzi
W odpowiedzi na Pana pismo z dnia 25.02.09 znak NO-066/05/09, dotyczàce mo˝li-
woÊci przygotowywania w ramach Apteki Szpitalnej worków do ˝ywienia pozajelitowego
dla pacjentów ˝ywionych w warunkach domowych zgodnie z procedurà NFZ, uprzejmie
informuj´, i˝ zgodnie z przepisem art. 87 ust. 1 pkt 2 ustawy z dnia 6 wrzeÊnia 2001 r.
Prawo Farmaceutyczne (DZ. U. 2008. 45. 271 z póên. zm.) – apteki szpitalne zaopatrujà
oddziały szpitalne lub inne nie wymienione z nazwy zakłady przeznaczone dla osób,
których stan zdrowia wymaga udzielania całodobowych lub całodziennych Êwiadczeƒ
zdrowotnych wykonywanych w tym zakładzie lub jednostce organizacyjnej wchodzàcej
w skład zakładu.
Ponadto zgodnie z art. 106 ust. 3 w/w ustawy apteka szpitalna mo˝e zaopatrywaç
w leki inne zakłady opieki zdrowotnej przeznaczone dla osób, których stan zdrowia
wymaga udzielania całodobowych lub całodziennych Êwiadczeƒ zdrowotnych
w odpowiednim stałym pomieszczeniu, nie posiadajàce aptek, na podstawie umowy
zawartej przez uprawnione do tego podmioty, pod warunkiem ˝e nie wpłynie to
negatywnie na prowadzenie podstawowej działalnoÊci apteki.
W ramach prowadzonych działaƒ Główny Inspektor Farmaceutyczny zwrócił si´ do Krajowego
Konsultanta w dziedzinie Farmacji Szpitalnej o zaj´cie stanowiska w powy˝szym zakresie.
W swojej odpowiedzi Krajowy Konsultant przedstawił wàtpliwoÊci dotyczàce sposobu
dystrybucji oraz rzeczywistych warunków przechowywania transportu preparatów do
˝ywienia pozajelitowego, sporzàdzonych w aptekach szpitalnych oraz ich iloÊci
zwłaszcza w przypadku czternastodniowej terapii – w aspekcie braku precyzyjnego
okreÊlenia trwałoÊci tych preparatów oraz ustawowej odpowiedzialnoÊci farmaceuty za
jakoÊç wytworzonych produktów leczniczych.
Ze wzgl´du na fakt, i˝ do kompetencji ustawowych Głównego Inspektora Farma-
ceutycznego nie nale˝y sposób ordynowania leków przez lekarza pacjentom leczonym
w szpitalu bàdê pacjentom pozostajàcym w tzw. opiece domowej po zakoƒczonej
hospitalizacji zwrócono si´ do Naczelnej Rady Lekarskiej z proÊbà o zaj´cie stanowiska
w przedmiotowej sprawie.

54
FARMACEUTYCZNE STANDARDY SPORZÑDZANIA
MIESZANIN DO ˚YWIENIA POZAJELITOWEGO
W stanowisku z dnia 13.02.09 nr 10/09/P-V Prezydium Naczelnej Rady Lekarskiej
stwierdza, i˝ prowadzenie ˝ywienia pozajelitowego w warunkach domowych jest
wskazane i po˝àdane ze wzgl´du na komfort pacjenta. Ponadto podkreÊlono, i˝ zgod-
nie z obowiàzujàcymi przepisami w/w leki muszà byç przygotowywane przez apteki
szpitalne, które w obecnym stanie prawnym nie mogà Êwiadczyç usług na rzecz
indywidualnych pacjentów. Dodatkowo zwrócono uwag´ na fakt zagro˝eƒ dla
pacjentów, jakie mogà wynikaç z powodu braku uregulowania warunków Êwiadczenia
przez NFZ tzw. „leczenia ˝ywieniowego pozajelitowego w warunkach domowych”.
W zwiàzku z wàtpliwoÊciami, jakie zostały wyra˝one przez Naczelnà Rad´ Lekarskà,
Główny Inspektor Farmaceutyczny w dniu 02.03.09 zwrócił si´ do Prezesa Narodowego
Funduszu Zdrowia z proÊbà o zaj´cie stanowiska w powy˝szej sprawie.
Ze stanowiska przedstawionego przez Prezesa Narodowego Funduszu Zdrowia
w piÊmie z dnia 24.04.2009 wynika, ˝e Êwiadczenie „leczenie ˝ywieniowe pozajelitowe
w warunkach domowych” jest kontynuacjà terapii szpitalnej ˝ywienia pozajelitowego.
Âwiadczenie to w dalszym ciàgu przebiega pod nadzorem Zespołu ˚ywieniowego
szpitala wykonujàcego powy˝sze Êwiadczenie. Dodatkowo dystrybucjà sprz´tu i leków
do ˝ywienia pozajelitowego powinien zajmowaç si´ szpital dostarczajàcy bezpoÊrednio
do domu pacjenta mieszaniny i sprz´t.
Reasumujàc, zgodnie z obowiàzujàcymi przepisami produkty do ˝ywienia pozajeli-
towego przygotowywane w aptekach szpitalnych, przekazywane sà do Zespołów
˚ywieniowych na oddziały szpitalne, skàd w zale˝noÊci od decyzji lekarza dostarczane
pacjentowi.
Za warunki przechowywania (w tym transportu) powy˝szych mieszanin po opuszczeniu
apteki szpitalnej oraz nadzór nad stosowaniem odpowiedzialnoÊç ponosi oddział, pod
opiekà którego pozostaje pacjent po hospitalizacji, w ramach kontynuacji Êwiadczenia
leczenia ˝ywienia pozajelitowego poza zakładem opieki zdrowotnej.
2) Opini´ GIF z 9 sierpnia 2009 r. – pismo GIF-N/0741/56/MP/09 do Dyrektora ds.
Medycznych Szpitala Klinicznego Przemienienia Paƒskiego UM w Poznaniu
(…) Dystrybucja leków sporzàdzanych w aptekach szpitalnych do pacjentów pozosta-
jàcych pod opiekà oddziałów szpitalnych, powinna byç regulowana i nadzorowana
przez kierownictwo ZOZ-u. OdpowiedzialnoÊç za takie produkty, a szczególnie ich
zastosowanie, od chwili przyj´cia na oddział spoczywa w pełni na lekarzu zlecajàcym.
3) Opini´ eksperta Serwisu Prawo i Zdrowie Wolters Kluwer Polska (Gazeta Farma-
ceutyczna 6/2009)
Pytanie do eksperta:
Czy sà przepisy, które jasno okreÊlajà, ˝e płyny infuzyjne mogà byç przygotowywane na
oddziale szpitalnym, a nie w aptece szpitalnej?
Kto ma uprawnienia do ich przygotowywania?
55
Odpowiedê eksperta:
Apteki szpitalne, zgodnie z ustawà z dnia 6 wrzeÊnia 2001 r. – Prawo farmaceutyczne
(tekst jedn.: Dz. U. z 2008 r. Nr 45, poz. 271; dalej jako u.p.f.), zaopatrujà w leki i płyny
infuzyjne oddziały szpitalne lub inne nie wymienione z nazwy zakłady, przeznaczone dla
osób, których stan zdrowia wymaga udzielania całodobowych lub całodziennych
Êwiadczeƒ zdrowotnych wykonywanych w zakładzie lub jednostce organizacyjnej
wchodzàcej w skład zakładu (art. 87 ust. 1 pkt 2 u.p.f).
Apteki szpitalne Êwiadczà usługi farmaceutyczne okreÊlone w art. 86 ust. 2 u.p.f. ,
a ponadto Êwiadczà usługi farmaceutyczne (art. 86 ust. 3 u.p.f.), obejmujàce:
• sporzàdzanie leków do ˝ywienia pozajelitowego
• sporzàdzanie leków do ˝ywienia dojelitowego
• przygotowywanie leków w dawkach dziennych, w tym leków cytostatycznych
• wytwarzanie płynów infuzyjnych
• organizowanie zaopatrzenia szpitala w produkty lecznicze i wyroby medyczne
• przygotowywanie roztworów do hemodializy i dializy dootrzewnowej
• udział w monitorowaniu działaƒ niepo˝àdanych leków
• udział w badaniach klinicznych prowadzonych na terenie szpitala
• udział w racjonalizacji farmakoterapii
• współuczestniczenie w prowadzeniu gospodarki produktami leczniczymi i wyrobami
medycznymi w szpitalu.
Poprzez powy˝szy zapis ustawodawca jednoznacznie okreÊlił miejsce przygotowywania
płynów infuzyjnych oraz leków cytostatycznych.
4)
Wymogi NFZ dotyczàce kontraktu ze szpitalem na ˝ywienie pozajelitowe
w warunkach domowych (Załàcznik nr 4 do Zarzàdzenia Prezesa NFZ nr 82/2008
z 14 paêdziernika 2008 r.)
Opis Êwiadczenia
˚YWIENIE POZAJELITOWE W WARUNKACH DOMOWYCH
minimalne kwalifikacje pra-
cowników fachowych udzie-
lajàcych Êwiadczeƒ
1) lekarze
2) piel´gniarka
3) mgr farmacji
Konieczne umiej´tnoÊci i do-
Êwiadczenie zawodowe pra-
cowników fachowych udzie-
lajàcych Êwiadczeƒ (certy-
fikaty)
Certyfikat ukoƒczenia kursu z zakresu ˝ywienia pozajelitowego i dojeli-
towego zatwierdzony przez Centrum Medyczne Kształcenia Podyplomowego
w porozumieniu z Polskim Towarzystwem ˚ywienia Pozajelitowego i Dojeli-
towego (lekarz, piel´gniarka i farmaceuta)
Warunki wykonania
2.
2.5
2.6
2.7
konieczne i niezb´dne wa-
runki lokalowe dla udzie-
lenia Êwiadczenia
apteka szpitalna (docelowo z pracownià ˝ywienia pozajelitowego),

56
FARMACEUTYCZNE STANDARDY SPORZÑDZANIA
MIESZANIN DO ˚YWIENIA POZAJELITOWEGO
5) Rozporzàdzenie Ministra Zdrowia z 29 sierpnia 2009 r. w sprawie Êwiadczeƒ gwaran-
towanych z zakresu ambulatoryjnej opieki specjalistycznej (Dz. U. z 2009 r. Nr 139,
poz. 1142)
Opis Êwiadczenia
˚YWIENIE POZAJELITOWE W WARUNKACH DOMOWYCH
Âwiadczenie dotyczy pacjentów, którzy z powodu braku mo˝liwoÊci poda˝y substancji
od˝ywczych w iloÊci wystarczajàcej do utrzymania przy ˝yciu drogà naturalnà, wyma-
gajà długotrwałego podawania substancji od˝ywczych w sposób inny ni˝ doustny.
˚ywienie pozajelitowe w warunkach domowych – polega na podawaniu substancji
od˝ywczych drogà do˝ylnà w domu chorego wraz z kompleksowà opiekà nad chorym
wynikajàcà z choroby podstawowej i prowadzonego leczenia, obejmujàcà:
1. przygotowanie chorego do leczenia w warunkach domowych
2. zapewnienie ciàgłego kontaktu z chorym
3. program badaƒ kontrolnych zgodnie z obowiàzujàcymi standardami
4. dostarczanie preparatów i sprz´tu, i niezb´dnych Êrodków opatrunkowych do domu
chorego
5. zapewnienie hospitalizacji w przypadku podejrzenia lub wystàpienia powikłaƒ oraz
innych wskazaƒ wymagajàcych leczenia szpitalnego.
˚YWIENIE POZAJELITOWE
W WARUNKACH DOMO-
WYCH
Personel:
1) lekarze – specjaliÊci w dziedzinie medycyny,
2) piel´gniarki,
3) mgr farmacji.
Nazwa Êwiadczenia
4
Warunki realizacji Êwiadczenia

57

58
FARMACEUTYCZNE STANDARDY SPORZÑDZANIA
MIESZANIN DO ˚YWIENIA POZAJELITOWEGO
22. PIÂMIENNICTWO
1. Barnett H. I., Pertkiewicz M., Cosslett A. i wsp. Sporzàdzanie mieszanin przeznaczonych do
˝ywienia pozajelitowego [w:] Podstawy ˝ywienia klinicznego. Sobotka L. (red.), PZWL,
Warszawa 2007, 261–265.
2. Chmal-Jagiełło K., Jankowiak-Gracz H., Kuêniar H. Standardy jakoÊciowe w farmacji onkolo-
gicznej. Polskie Towarzystwo Farmaceutyczne Sekcja Farmacji Onkologicznej, Warszawa 2008.
3. Ciszewska-J´drasik M. Zasady przygotowywania i uzupełniania mieszanin od˝ywczych do
˝ywienia pozajelitowego. Post´py ˚ywienia Klinicznego 2008, 3 (8), 12–19.
4. Ciszewska-J´drasik M. ˚ywienie pozajelitowe. Cz´Êç I, II, III. Farmacja Polska 2008, 64 (24),
1063–1089.
5. Ciszewska-J´drasik M., Pertkiewicz M. Mieszaniny do ˝ywienia pozajelitowego. Standardy
post´powania i zalecenia dla farmaceutów. PZWL, Warszawa 2004.
6. Corriol O., Crauste-Manciet S., Arnaud P. i wsp. Zalecenia dotyczàce sporzàdzania mieszanin
do ˝ywienia pozajelitowego. Post´py ˚ywienia Klinicznego 2007, 2 (4), 17–39.
7. Pertkiewicz M., Cosslett A., Muhlebach S. StabilnoÊç mieszanin przeznaczonych do ˝ywienia poza-
jelitowego [w:] Podstawy ˝ywienia klinicznego. Sobotka L. (red.), PZWL, Warszawa 2007, 266–270.
8. Pertkiewicz M., Korta T. i wsp. Standardy ˝ywienia pozajelitowego i ˝ywienia dojelitowego.
PZWL, Warszawa 2005.
9. Pertkiewicz M., Orawczyk T., Korta T. Definicje i opisy leczenia ˝ywieniowego przekazane do
Narodowego Funduszu Zdrowia. Post´py ˚ywienia Klinicznego 2007, 2 (4), 4–8.
10. ASPEN Special Report. Safe Practices for Parenteral Nutrition Formulations. JPEN 2004, 28, 39–70.
11. ASHP Guidelines on Quality Assurance for Pharmacy-Prepared Sterile Products. Am. J. Health-
Syst. Pharm 2000, 57 ,1150–1169.
12. PIC/S Guide to good practices for the preparation of medicinal products in healthcare
establishments. Pharmaceutical Inspection Convention and Pharmaceutical Inspection
co-operation scheme – PIC/S. October 2008. www.picscheme.org
13. USP {797} Guidebook to Pharmaceutical Compounding – Sterile Preparations. United States
Pharmacopeia 31 – National Formulary 26. Suppl. 2. 2008.
14. Farmakopea Polska VII i VIII.
15. Ustawa Prawo farmaceutyczne z 6 wrzeÊnia 2001 r. z póêniejszymi zmianami (DZ. U.
z 2008 r. Nr 45, poz. 271 – tekst jednolity).
16. Załàcznik do Rozporzàdzenia Ministra Zdrowia z 17 sierpnia 2009 r. w sprawie wymagaƒ
Dobrej Praktyki Wytwarzania, Aneks I. (DZ. U. z 2009 r. Nr 135, poz. 1114.)

59
NOTATKI

60
FARMACEUTYCZNE STANDARDY SPORZÑDZANIA
MIESZANIN DO ˚YWIENIA POZAJELITOWEGO
Wyszukiwarka
Podobne podstrony:
Preparaty do żywienia pozajelitowego C2
Sporzadzenie kapieli do chromowania na bazie CrO3 z katalizatorem mieszanym Galwanizernie chromowa
pytania tple, materiały farmacja, materiały V rok, TPL, do zaliczenia, na kolokwium
Selenowa mieszanina do onaczania azotu
Wstęp do Żywienia Optymalnego
Żywienie pozajelitowe 3
zywienie pozajelitowe
cw sporządzanie mieszanek mąki
mieszarka do ciasta, Instrukcje BHP i Ppoż
Żywienie pozajelitowe W2, Materiały Dietetyka, PWSZ (Nina nevermind), Nowy folder, semestr VI (Nina
wykaz zagadnien do iv kolokwium 2011 2012, materiały farmacja, Materiały 3 rok, Farmakognozja, Do ko
FARMACJA2, STUDIA PŁ, TECHNOLOGIA ŻYWNOŚCI I ŻYWIENIA CZŁOWIEKA, ROK I, SEM 2, CHEMIA ANALITYCZNA I
FOLWYK8.DOC, Sporządzanie mieszanin nitrujących - zasady obliczeń. Przygotowanie poszczególnych kwas
istan Mieszarka do ciasta, BHP, Instrukcje-Stanowiskowe
23 Sporządzanie mapy do celów prawnych
więcej podobnych podstron