
1
Ćwiczenie 1
Oznaczanie składu gazu metodami chromatografii gazowej
I.
Celem ćwiczenia jest zapoznanie studentów z budową i zasadą działania
chromatografu gazowego oraz metodyką analiz składu gazu ziemnego z jego
zastosowaniem.
II.
Wprowadzenie
Chromatografia jest metodą rozdzielania składników jednorodnych mieszanin w
wyniku różnego ich podziału między fazę ruchomą i nieruchomą układu
chromatograficznego, przy czym fazą ruchomą może być gaz lub ciecz, natomiast
nieruchomą (stacjonarną) ciało stałe lub ciecz.
Poniższy rysunek 1.1 przedstawia istotę rozdzielania chromatograficznego
mieszaniny składającej się z dwóch składników: A i B
Rys.1.1. Schemat rozdzielania chromatograficznego mieszaniny dwóch składników – A i B
t = 0 – moment wprowadzenia mieszaniny do układu chromatograficznego,
t
1
, t
2
– wybrane czasy z całkowitego czasu przebiegu rozdzielania chromatograficznego,
t
3
– czas zakończenia rozdzielania składników mieszaniny, C – stężenie składnika (A lub B)
w fazie ruchomej (r) lub stacjonarnej (s)

2
Mieszaninę substancji A i B wprowadzono do fazy ruchomej w czasie, który przyjęto
za zerowy i od którego rozpoczął się proces rozdzielania składników. Składniki te w
różny sposób oddziałują z faza nieruchomą i ruchomą. Załóżmy, że składnik A oddziałuje
z fazą nieruchomą znacznie słabiej niż składnik B. Cząsteczki obu składników dzielą się
między obie fazy w różnych stosunkach, charakterystycznych dla tych składników.
Stosunki te można zapisać jako stałe podziału K dla składnika A i B:
Ar
As
A
C
C
K
=
;
Br
Bs
B
C
C
K
=
;
gdzie: C – stężenie składnika w fazie nieruchomej (s) i ruchomej (r).
Między liczbą cząsteczek związków chromatografowanych, obecnych w fazie
ruchomej i nieruchomej, ustala się równowaga dynamiczna z wielokrotnym
przechodzeniem tych cząsteczek z jednej fazy do drugiej. Ich przenoszenie wzdłuż
układu chromatograficznego jest możliwe tylko wtedy, gdy znajdują się w fazie
ruchomej. W czasie t
1
widoczny jest różny podział składników między obie fazy układu
chromatograficznego i rozdzielenie tych składników. Rozdzielenie składników jest
możliwe tylko wtedy, gdy ich stałe podziału różnią się między sobą (K
A
≠
K
B
). W czasie t
2
jeden ze składników został już wyniesiony z układu chromatograficznego i znajduje się
w fazie ruchomej, a w czasie t
3
oba składniki są już w fazie ruchomej poza zasięgiem
oddziaływania fazy stacjonarnej. Pasma stężeniowe składników po przejściu układu
chromatograficznego różnią się od pasma początkowego mieszaniny. Różnica polega na
poszerzeniu tych pasm i na przyjęciu kształtu krzywej Gaussa. Pasma te noszą nazwę
pików chromatograficznych.
Jeżeli faza ruchomą jest gaz, to chromatografia nosi nazwę gazowej, gdy faza
ruchową jest ciecz, wówczas chromatografia nazywana jest cieczową. Gdy faza
nieruchomą jest ciało stałe, wówczas chromatografię nazywa się adsorpcyjną. Jeżeli
faza nieruchomą jest ciecz, to chromatografia jest chromatografią podziałową. Faza
nieruchoma może być umieszczona w kolumnie lub na płaszczyźnie (tylko w
chromatografii cieczowej). W pierwszym przypadku chromatografia jest kolumnowa, w
drugim – planarna.
3
III.
Aparatura do chromatografii gazowej
Analizę za pomocą chromatografii gazowej wykonuje się przy użyciu
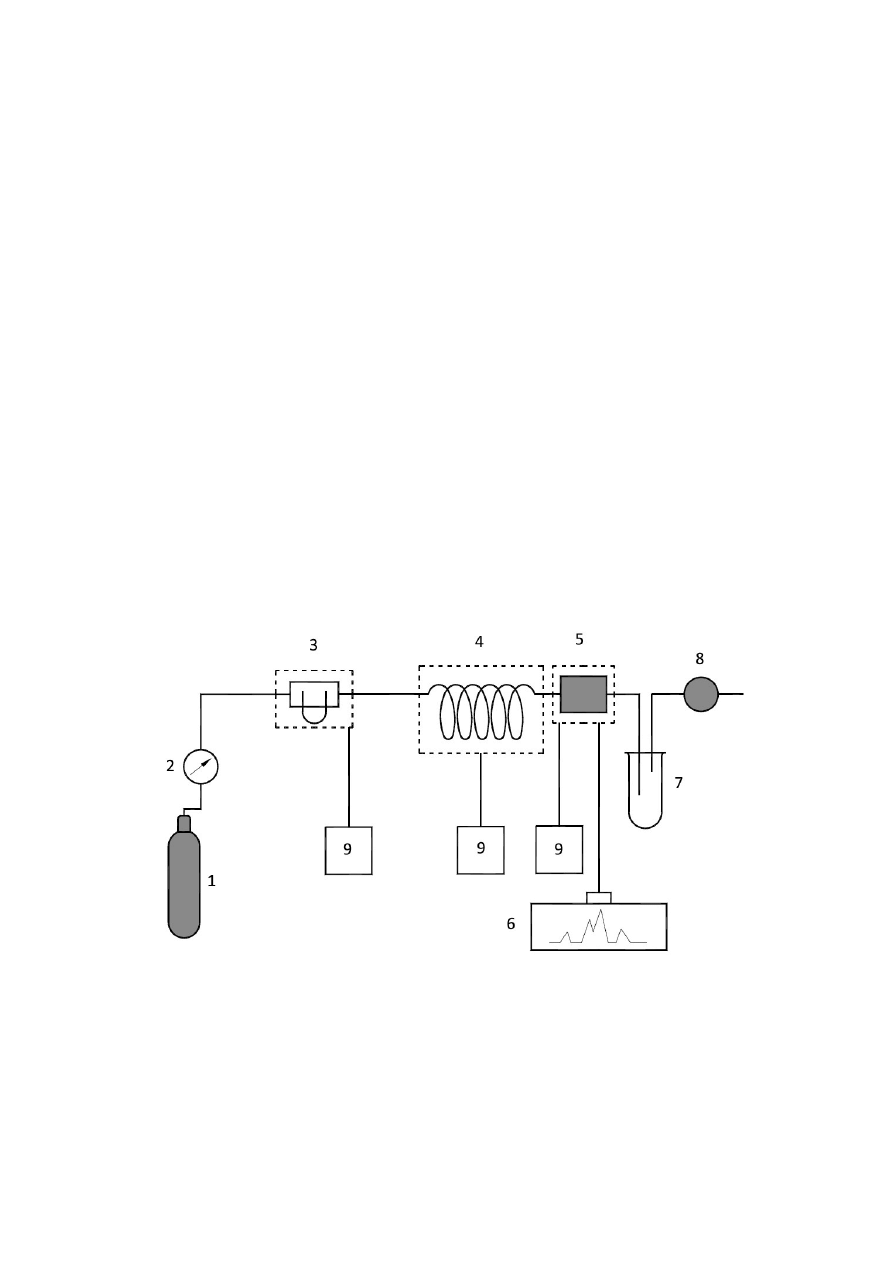
chromatografów gazowych. Schemat chromatografu gazowego przedstawia rys.1.2, a
zasada działania jego jest następująca: Gaz nośny ze zbiornika lub wytwornicy płynie
przez regulator przepływu, układ osuszania i odtleniania oraz przepływomierz do
dozownika, a następnie przez kolumnę i detektor do atmosfery. Kolumna jest
umieszczona w termostacie. Temperatura dozownika, kolumny i detektora jest
regulowana za pomocą regulatorów. Do dozownika próbkę wprowadza się strzykawką
(gazy, ciecze i roztwory ciał stałych) lub zaworem dozującym (gazy). Próbka ciekła
odparowuje w dozowniku i w strumieniu gazu nośnego jest przenoszona do kolumny. W
kolumnie następuje rozdzielenie składników próbki, które wynoszone z kolumny
trafiają do detektora, generując w nim sygnał elektryczny. Sygnały po wzmocnieniu
mogą być zapisywane na taśmie rejestratora w postaci pików (chromatogramu). We
współczesnych chromatografach do rejestracji chromatogramów i opracowywania
wyników stosuje się komputery.
Rys.1.2. Schemat chromatografu gazowego.
1 – zbiornik lub wytwornica gazu nośnego, 2 – kontrola i regulacja ciśnienia gazu nośnego,
3 – dozownik – komora nastrzykowa, 4 – kolumna, 5 – detektor, 6 – wzmocnienie sygnału i
rejestracja, 7 – ewentualne wyłapywanie frakcji, 8 – pomiar prędkości przepływu
9 – regulator temperatury dozownika, kolumny i detektora

4
a) Gazy nośne
Gaz nośny jest zasadniczym elementem chromatografu, wpływającym na wszystkie
etapy procesu chromatografii. Źródłem gazu nośnego jest zwykle butla zawierająca gaz
pod ciśnieniem. Umożliwia ona zasilanie aparatury gazem o stałym ciśnieniu,
uzyskiwanym za pomocą reduktora dwustopniowego. Niektóre gazy mogą być
wytwarzane w laboratorium. Wodór wytwarza się przy użyciu generatorów
elektrolitycznych, a azot i powietrze w wytwornicach napełnionych sitami
molekularnymi. Uzyskuje się w ten sposób gazy o czystości wymaganej w chromatografii
bez potrzeby ich przechowywania i transportowania w butlach.
Czystość gazu nośnego wpływa na pracę detektora i wypełnienia kolumn. W
przypadku wypełnień dotyczy to zarówno adsorbentów, które pod wpływem
zanieczyszczeń mogą ulegać dezaktywacji, jak i ciekłych faz stacjonarnych, które z kolei
mogą ulegać przemianom chemicznym i tracić swoje pierwotne właściwości. Dlatego gaz
nośny nie może zawierać zanieczyszczeń tj. tlenu, pary wodnej i węglowodorów.
Do osuszania gazów nośnych stosuje się sita molekularne lub żel krzemionkowy. Sita
molekularne oprócz wody pochłaniają też dwutlenek węgla, dwutlenek siarki,
dwutlenek azotu i chlorowodór. Sita molekularne i żel krzemionkowy stosuje się w
cylindrach metalowych lub szklanych, wyposażonych na końcach w krążki z porowatego
szkła lub metalu, w celu zapobieżenia przedostawania się cząstek materiału suszącego
do gazu nośnego. Pojemność osuszacza powinna być tak dobrana, żeby wystarczył on na
osuszenie gazu, co najmniej z jednej butli. Sita molekularne i żel krzemionkowy należy
aktywować przez wygrzewanie w temperaturze 300 – 350
o
C. W czasie osuszania gazu
osuszacz ma temperaturę otoczenia.
Do usuwania z gazu nośnego tlenu stosuje się najczęściej odtleniacze działające na
zasadzie chemisorpcji tlenu przez metale osadzone na nośnikach. Najczęściej stosuje się
odtleniacze miedziowe. Odtleniacz umieszcza się metalowym cylindrze i podgrzewa się
elektrycznie, przy użyciu drutu oporowego, do temperatury 180 – 200
o
C.
b)
Dozowniki i urządzenia dozujące
Stosując chromatografię gazową, można analizować substancje, które w
warunkach chromatografowania mają postać gazów lub par. Przyjmuje się, że są to
substancje gazowe oraz ciekłe i stałe, których temperatura wrzenia lub sublimacji (bez
rozkładu) nie przekracza 350 – 400
o
C. W chromatografie dozownik jest elementem

5
umożliwiającym wprowadzenie próbki w strumień gazu nośnego, który przenosi ją do
kolumny. Kolumna jest połączona z dozownikiem za pomocą krótkiego kapilarnego
łącznika. Dąży się do tego, aby połączenie dozownik-kolumna umożliwiało
wprowadzenie próbki bezpośrednio do kolumny. Próbka ciekła lub stała powinna w
dozowniku odparować w jak najkrótszym czasie i dlatego temperatura dozownika musi
być odpowiednio wysoka (20
o
C powyżej temperatury wrzenia najwyżej wrzącego
składnika próbki).
Próbki gazów dozuje się za pomocą strzykawek szklanych (ze szklanym tłokiem) lub
zaworami dozującymi. Zawory te mają wymienną pętlę dozowniczą o pojemności od
części mililitra do kilku mililitrów. Pętla ta jest połączona przewodem bezpośrednio z
instalacją lub zbiornikiem gazu, którego zawartość ma być analizowana. W zależności od
położenia dźwigni zaworu pętlę można napełniać analizowanym gazem lub gaz ten
włączać w strumień gazu nośnego i wprowadzać do kolumny chromatograficznej.
Próbka wprowadzana do kolumny nie może być większa od pojemności sorpcyjnej
kolumny, ponieważ jej przeładowanie prowadzi do powstawania szerokich,
niesymetrycznych pików i złego rozdzielenia składników próbki. Dotyczy to szczególnie
kolumn kapilarnych, w których próbka wprowadzona bezpośrednio mikrostrzykawką
lub zaworem dozującym jest zwykle większa od pojemności sorpcyjnej kolumny.
Próbki mogą być dozowane do chromatografu automatycznie z tego samego źródła
lub z zamkniętych fiolek. Dozowanie to jest zwykle bardziej powtarzalne, szybsze i mniej
kosztowne.
c) Kolumny
Kolumna jest sercem chromatografu gazowego, ponieważ w niej właśnie
następuje
rozdzielenie
mieszanin.
W
zależności
od
rozmiarów
kolumny
chromatograficzne możemy podzielić na:
•
zwykłe analityczne, pakowane, o średnicy wewnętrznej 2 – 6 [mm] i długości kilku
metrów (najczęściej 1 – 3 [m]),
•
mikropakowane o średnicy 0.8 – 1.2 [mm] i długości do kilkudziesięciu metrów,
•
kapilarne o średnicy 0.2 – 0.6 [mm] i długości do kilkudziesięciu metrów,
•
preparatywne o średnicy ponad 6 [mm] i długości kilku metrów,
•
mikrokapilarne o średnicy poniżej 0.1 [mm] i długości do kilkudziesięciu metrów.

6
Rys.1.3. Kapilarne kolumny krzemionkowe
Najczęściej stosowanymi kolumnami są kolumny kapilarne i pakowane. Kolumny
kapilarne mają postać zwoju, a wytwarza się je ze szkła lub z topionego kwarcu.
Kolumny kwarcowe pokryte są z zewnątrz warstwą tworzywa sztucznego lub
aluminium, co zwiększa ich wytrzymałość i trwałość mechaniczną. Faza stacjonarna w
kolumnach kapilarnych osadzona jest tylko na ściankach, w kolumnach pakowanych
natomiast jest nią wypełnienie w postaci cząstek adsorbentu najczęściej w fazie ciekłej.
Podstawowymi zaletami kolumn kapilarnych w porównaniu z pakowanymi jest zwykle
krótszy czas analizy niż przy użyciu kolumn pakowanych, możliwość uzyskania lepszych
efektów rozdzielania, małe zużycie gazu nośnego i fazy stacjonarnej oraz brak
konieczności stosowania nośnika. Natomiast wadą kolumn kapilarnych jest ich
trudniejsza preparatyka. W wyniku tego kolumny kapilarne zwykle są kupowane, a nie
przygotowywane samodzielnie.
d) Detektory
Detektor chromatograficzny jest urządzeniem przetwarzającym wielkość stężenia
rozdzielanych składników w gazie nośnym na sygnał elektryczny, który następnie
zapisywany jest na rejestratorach w postaci piku. Sygnał ten wywołany jest zmianami
fizycznych lub chemicznych własności mieszaniny wymywanych składników i gazu

7
nośnego. Określony detektor reaguje zwykle na jedną z wielu zmian własności,
wywołanych pojawieniem się w gazie nośnym wymywanego składnika.
Istnieją dwa kryteria podziału detektorów. Zgodnie z pierwszym detektory dzieli się
na całkowe i różnicowe. Detektory całkowe mierzą ilość wymytego składnika i dodają ją
do ogólnej ilości składników wymytych poprzednio. Ten typ detektora jest doskonały do
analiz ilościowych. Detektory różnicowe mierzą pewną właściwość fizyczną strumienia
gazu związaną ze stężeniem substancji lub wykrywają obecność substancji w sposób
bezpośredni. Według drugiego kryterium rozróżnia się detektory stężeniowe i detektory
masowe. Detektory stężeniowe mierzą zmiany własności fizycznych przepływającego
gazu i reagują na stężenie związku zawartego w strumieniu gazu nośnego. Sygnał
takiego detektora jest w każdej chwili proporcjonalny do stężenia składnika w gazie
nośnym. Detektory masowe mierzą ilość substancji bezpośrednio i reagują na ilość
wykrywanego związku napływającego do detektora.
Najczęściej stosowanymi detektorami w chromatografii gazowej są detektor cieplno-
przewodnościowy i płomieniowo-jonizacyjny.
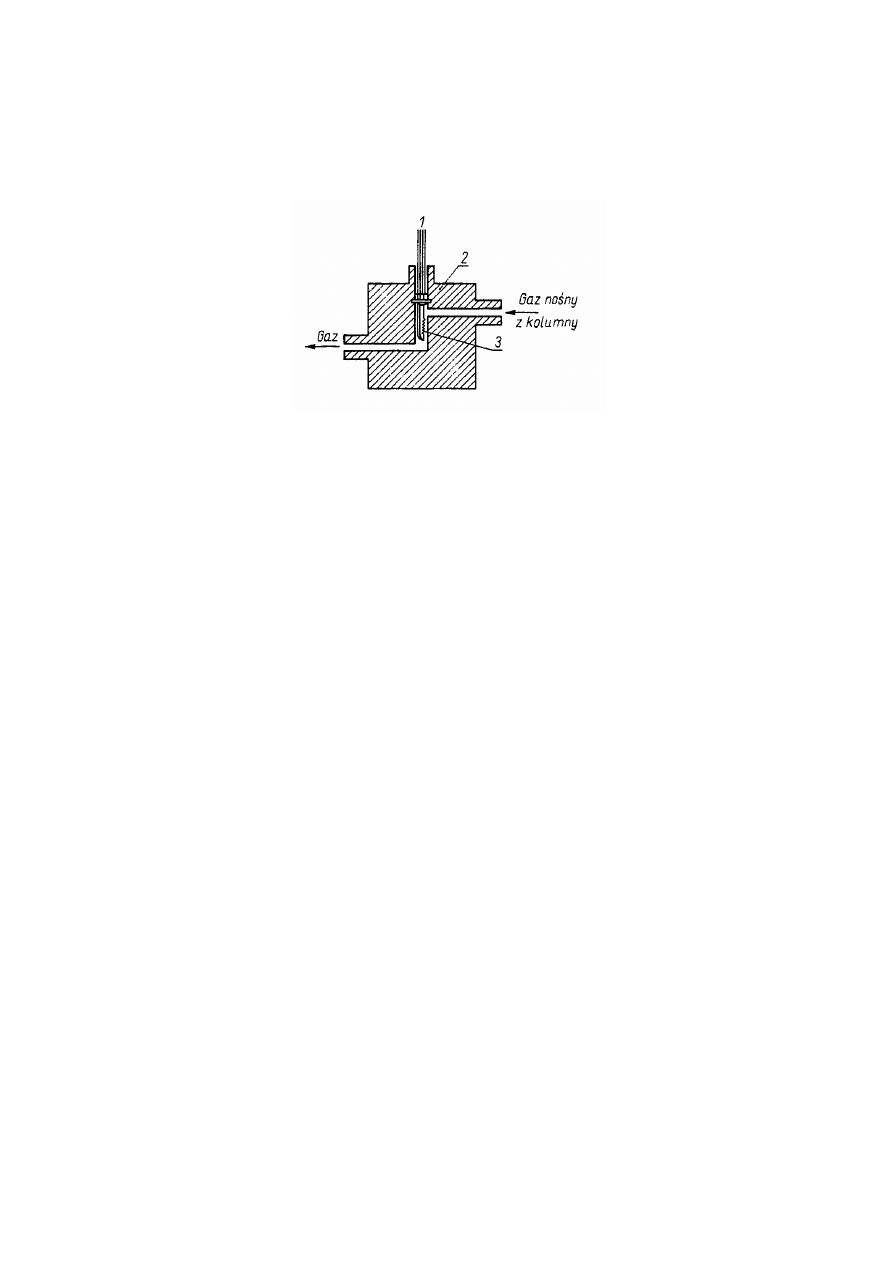
Detektor cieplno-przewodnościowy (Thermal Conductivity Detector – TCD)
W detektorze cieplno-przewodnościowym (katarometrze) czujnikiem jest spirala,
np. niklowa, wolframowa lub ze stopu platyny z irydem lub termistor. Cechą tych
czujników jest znaczna zmiana ich przewodności elektrycznej ze zmianą temperatury.
Dlatego też temperatura detektora musi być ustalona i utrzymywana z dokładnością do
dziesiątych części stopnia. Tak długo jak z kolumny wypływa tylko gaz nośny, tak długo
temperatura, a przez to i przewodność czujnika nie zmienia się i na taśmie rejestratora
rysowana jest linia prosta. Gdy z kolumny wraz z gazem nośnym wymywana jest
substancja chromatografowana o innym przewodnictwie cieplnym niż przewodnictwo
gazu nośnego, wówczas temperatura, a w wyniku tego i przewodność elektryczna
czujnika wzrasta lub maleje. W konsekwencji tego na taśmie rejestratora obserwuje się
odchylenie linii podstawowej w postaci piku trwające tak długo, jak długo substancja
wymywana z kolumny omywa czujnik.
Katarometry są powszechnie stosowanymi detektorami ze względu na ich prostotę i
dobrą powtarzalność pomiarów. Są detektorami stężeniowymi, a więc w pomiarach
ilościowych natężenie przepływu gazu nośnego musi być stałe. Czułość detektora jest
8
wprost proporcjonalna do trzeciej potęgi wielkości prądu zasilającego. Detektor ten
nadaje się do detekcji wszystkich gazów i par.
Rys.1.4. Schemat detektora cieplno-przewodnościowego
1 – przewód elektryczny, 2 – termostatowany blok metalowy, 3 – czujnik detektora
Detektor płomieniowo-jonizacyjny (Flame Ionization Detector – FID)
Detektor ten jest najbardziej rozpowszechnionym detektorem w chromatografii
gazowej. Zasada działania tego detektora oparta jest na zmianie oporu elektrycznego
płomienia wodorowego przy wprowadzeniu do niego śladowych ilości związków
organicznych, które w procesie utleniania wytwarzają jony. Wodór spalany w czystym
powietrzu lub w tlenie, nie tworzy prawie zupełnie jonów. Dlatego przewodnictwo
elektryczne płomienia wodorowego jest bardzo małe i prąd w obwodzie jest znikomo
mały. Kiedy natomiast w płomieniu wodorowym znajdą się cząstki substancji
organicznych, ulegają one jonizacji, w rezultacie czego przewodnictwo płomienia
znacznie wzrasta. W obwodzie, którego elementami są elektrody, powstaje prąd jonowy,
a jego wielkość zależy od ilości cząsteczek substancji organicznej, dochodzących do
płomienia w jednostce czasu.
Schemat detektora przedstawia rys.1.5. Składa się on z palnika wodorowego i z
układu elektrod połączonych z zasilaczem i układem pomiarowym. Ze względu na dużą
ilość wydzielanego ciepła w detektorze zarówno izolatory elektrod, jak i cały detektor
muszą być starannie izolowane. Do palnika detektora doprowadza się gaz nośny z
kolumny, wodór i powietrze.
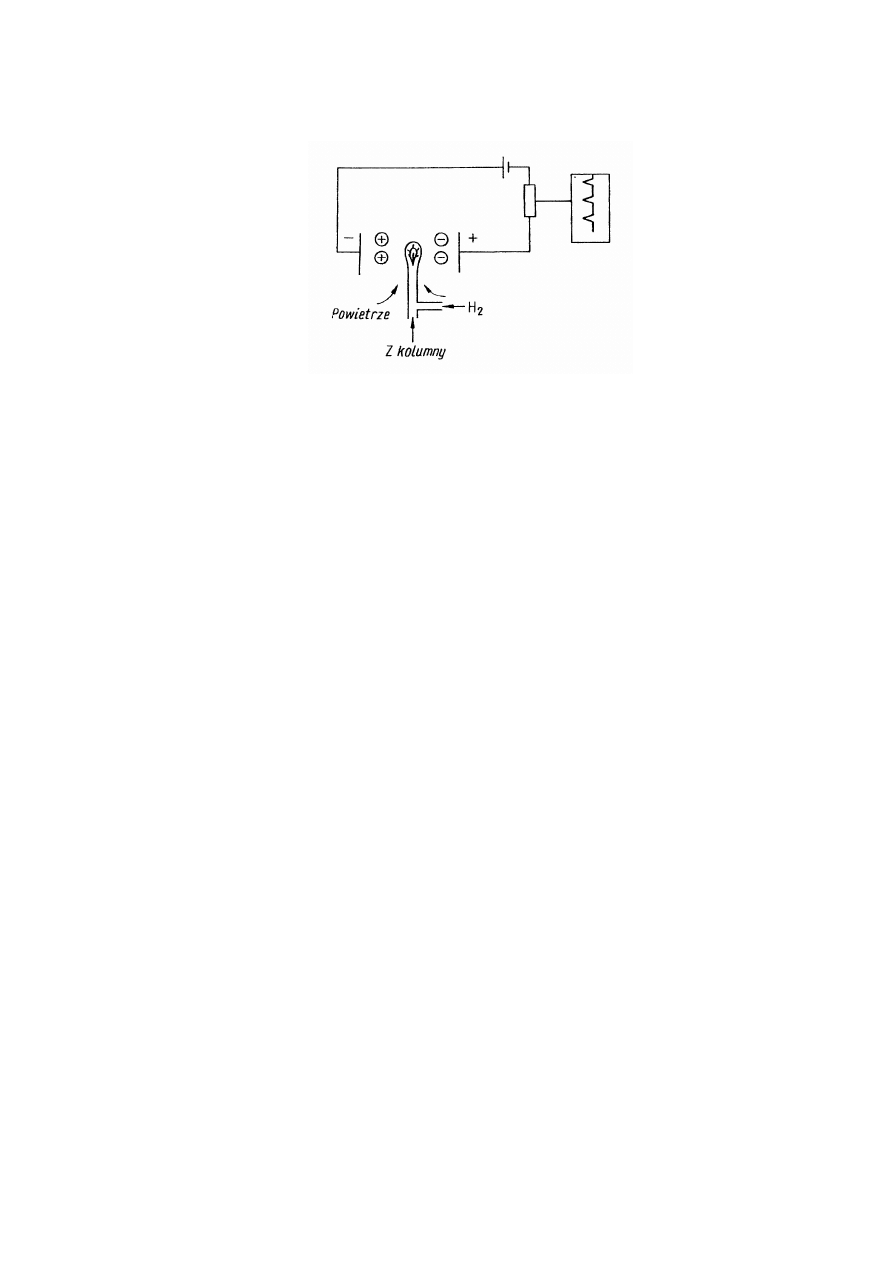
9
Rys.1.5. Schemat detektora płomieniowo-jonizacyjnego
Badane składniki opuszczające kolumnę chromatograficzną wprowadzane są do
palnika wodorowego, gdzie w procesie spalania cząsteczki tych składników ulegają
jonizacji. Powstałe jony zbierane są przez umieszczone w pobliżu palnika elektrody,
między którymi istnieje pole elektryczne. Wartość prądu jonizacyjnego zależy od składu
chemicznego badanych cząsteczek oraz od ich liczby. Detektor ten nadaje się szczególnie
do detekcji czystych węglowodorów. Jest mało czuły na gazy trwałe i takie związki jak:
H
2
S, SO
2
, CO.
IV.
Opracowywanie wyników analizy chromatograficznej
W celu właściwej interpretacji chromatogramu badanej substancji konieczna jest
znajomość podstawowych elementów chromatogramu i ich współzależność.
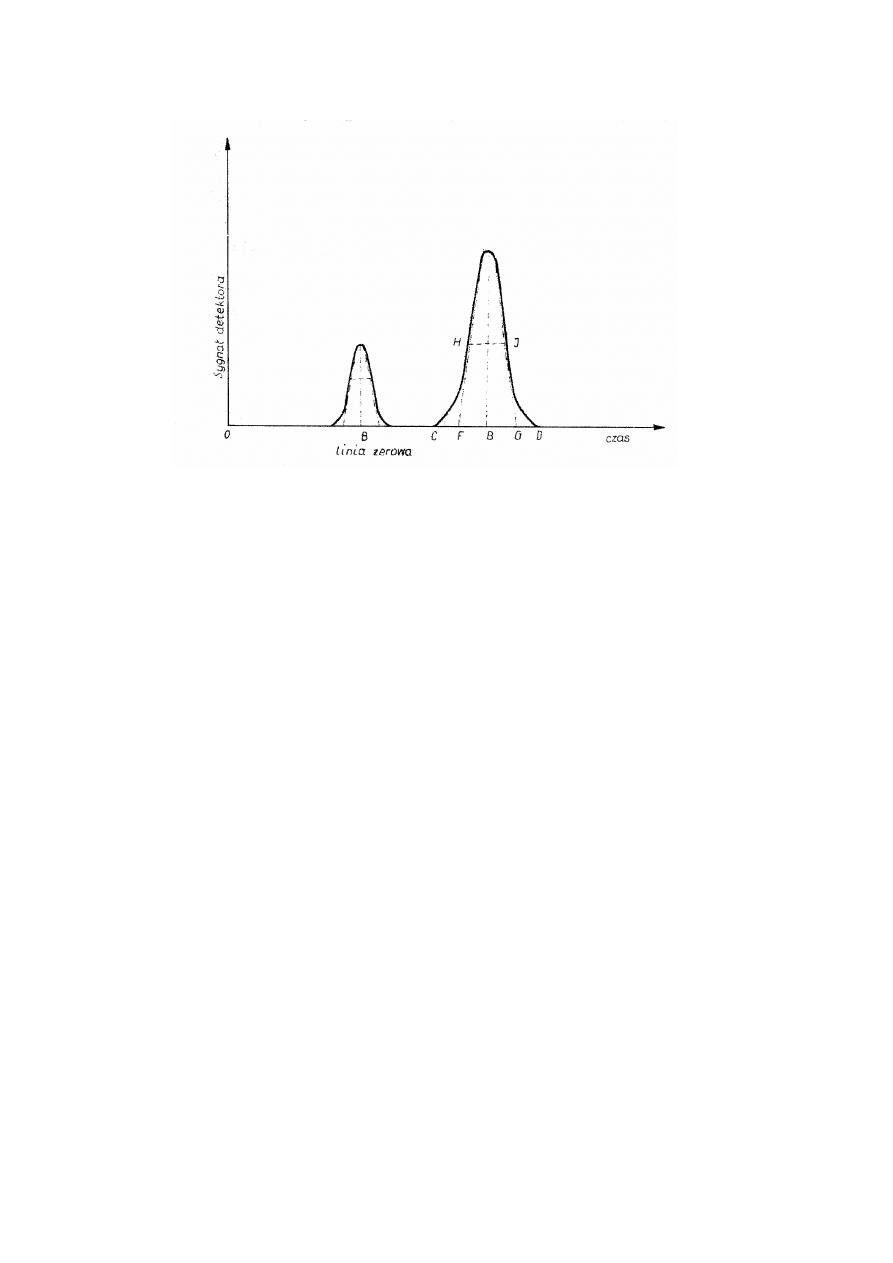
Teoretyczny chromatogram z zaznaczeniem typowych jego elementów przedstawiono
na rys.1.6.
Chromatogram jest to graficzne przedstawienie stężenia składnika wymywanego
(eluatu) w postaci symetrycznej fali wykazującej maksimum zwanej popularnie pikiem,
odpowiadające sygnałowi detektora w funkcji czasu, ujęte w układzie współrzędnych
prostokątnych. W celu prawidłowej interpretacji chromatogramu należy znać jego
następujące elementy i określenia:
•
linia zerowa – zwana też linią podstawy lub bazową, jest to linia obrazująca
wielkość wskazań detektora , wywołana przepływem samego gazu nośnego,
•
szum – przypadkowe zmiany linii zerowej wywołane własnościami układu
elektrycznego względnie zaburzeniami w przepływie gazu badanego,
10
Rys.1.6. Chromatogram i jego elementy
•
start – moment wprowadzenia próbki do układu rozdzielczego,
•
pik – linia wyznaczająca na chromatogramie sygnał detektora wywołany obecnością
składnika rozdzielczego w gazie nośnym wychodzącym z kolumny – krzywa CED,
•
podstawa piku – interpolowany odcinek linii podstawowej w przedziale wskazań
detektora, wywołanych obecnością rozdzielanego składnika w gazie nośnym
wpływającym do kolumny – linia CD,
•
wysokość piku – odległość między najwyższym punktem piku (maksimum) a jego
podstawą (mierzona prostopadle do linii zerowej) – odległość BE,
•
szerokość piku w połowie jego wysokości – odległość między bokami piku w
połowie jego wysokości (mierzona równolegle do linii zerowej) linia HJ,
•
powierzchnia piku – powierzchnia zawarta między linią piku a jego podstawą.
Inne wielkości częściowo możliwe do zmierzenia na chromatogramie to wielkości
charakteryzujące rozdział chromatograficzny:
•
czas retencji substancji – jest to czas od momentu zadozowania jej do kolumny do
momentu zarejestrowania maksimum piku odpowiadającego tej substancji. Tak
zdefiniowany czas zwany jest również całkowitym czasem retencji. Można napisać,
że całkowity czas retencji jest sumą czasu, w którym substancja oddziałuje z

11
wypełnieniem kolumny i czasu potrzebnego do przejścia tej substancji od dozownika
do detektora, gdyby takiego oddziaływania nie było,
•
martwy czas retencji – jest to czas, w którym substancja przebywa w połączeniach
kapilarnych między dozownikiem i kolumną oraz kolumną i detektorem. Czas
martwy retencji można najłatwiej wyznaczyć przez zadozowanie do kolumny
substancji, która nie oddziałuje z fazą stacjonarną lub oddziałuje bardzo słabo,
•
zredukowany czas retencji – czas retencji związany z przebywaniem substancji w
kolumnie tylko w wyniku jego oddziaływania tej substancji z wypełnieniem
kolumny,
•
objętość retencyjna składnika – jest to objętość gazu nośnego potrzebna do
wymycia tego składnika z kolumny. Objętość retencyjna wyraża się iloczynem czasu
retencji i objętościowej prędkości przepływu gazu nośnego,
•
względna objętość retencyjna składnika – jest to stosunek objętości retencyjnej
tego składnika do objętości do objętości retencyjnej substancji przyjętej za wzorzec,
•
względny czas retencji – stosunek czasu retencji badanego składnika do czasu
retencji substancji przyjętej za wzorzec.
a) Analiza jakościowa
Analizę jakościową rozdzielanych w kolumnie substancji można wykonywać
dwoma sposobami. Pierwszy polega na wykorzystaniu położenia na chromatogramie
piku odpowiadającego danej substancji, czyli na wykorzystaniu jej własności
retencyjnych. Według drugiego sposobu identyfikuje się składniki chromatografowanej
mieszaniny innymi metodami analizy chemicznej lub fizykochemicznej. Oprócz tych
dwóch podstawowych sposobów pewne informacje dotyczące rodzaju składników
analizowanej mieszaniny otrzymujemy z obserwacji ich zachowania się przy
zastosowaniu różnych detektorów i gazów nośnych. Należy przy tym uwzględnić
wiadomości dotyczące selektywności i specyficzności niektórych detektorów i to, że
obecny w próbce gaz nie może być wykryty wówczas, gdy jest on gazem nośnym.
Wykorzystując wielkości retencyjne przy pewnym już doświadczeniu, uzyskuje
się ogólne informacje dotyczące jakościowego składu analizowanej mieszaniny, bez
wykonywania pomiarów retencji. Jeżeli znane są właściwości wypełnienia
kolumnowego w stosunku do różnych substancji i z obserwacji otrzymanych
chromatogramów, można wyciągnąć wnioski dotyczące temperatury wrzenia, lotności,

12
masy
cząsteczkowej,
charakteru
chemicznego
poszczególnych
składników
analizowanych mieszanin.
W jednakowych warunkach chromatografowania substancja chromatografowana
ma zawsze taką samą retencję. Pod pojęciem warunków rozumie się rodzaj kolumny, jej
wypełnienie, temperaturę oraz rodzaj i przepływ gazu nośnego. Na tej podstawie można
identyfikować nieznaną substancję przez porównanie jej retencji z retencją wzorca.
Identyfikację składnika próbki wykonuje się w taki sposób, że po ustaleniu warunków
chromatografuje się bezpośrednio po sobie próbkę i wzorzec, a następnie porównuje się
retencję wzorca i identyfikowanego składnika. Jeżeli w chromatografowanej mieszaninie
na pewno znajduje się substancja, której miejsce na chromatogramie jest poszukiwane
za pomocą wzorca, to wystarczy użycie tylko jednej kolumny. Natomiast, jeżeli
poszukiwana substancja może być jedną z wielu, to jej identyfikację należy wykonać, na
co najmniej dwóch różnych kolumnach i w różnych temperaturach. Gdy wzorzec jest
substancją analizowaną, wówczas w każdych warunkach zarówno retencja wzorca, jak i
substancji analizowanej jest taka sama. Identyczność czasów retencji nie jest jednak
dowodem stuprocentowym, że obie substancje (identyfikowana i wzorcowa) są
identyczne. Wiele substancji może mieć taką samą retencję. Zastosowanie większej
liczby kolumn zwiększa prawdopodobieństwo pewnej identyfikacji.
Przy porównywaniu wielkości retencyjnych należy uwzględnić, że piki muszą być
symetryczne. Retencja tej samej substancji, gdy jej pik jest niesymetryczny, zmienia się
w zależności od wielkości próbki zadozowanej do kolumny. Im próbka jest większa, tym
krótszy jest czas retencji chromatografowanej substancji. Aby ustrzec się ewentualnych
różnic w czasach retencji spowodowanych wielkością próbki przy małej
niesymetryczności piku lub wynikających z różnych warunków chromatografowania,
wzorzec można dodać bezpośrednio do badanej mieszaniny, wskutek czego wielkość
piku substancji odpowiadającej wzorcowi zwiększy się w stosunku do pików
pozostałych składników mieszaniny.
b) Analiza ilościowa
Chromatografia gazowa jest jedną z nielicznych metod analitycznych, które
umożliwiają wykonanie analizy jakościowej i ilościowej złożonych mieszanin w jednym
procesie. Dokładność analizy ilościowej zależy od jakości chromatografu i związanej z
tym niezmienności warunków chromatografowania w czasie wykonywania analizy, od

13
rodzaju detektora i zakresu jego wskazań, od sposobu wykonywania analizy oraz od
sposobu zbierania danych do obliczeń.
O ilości substancji w mieszaninie można wnioskować na podstawie wielkości
odpowiadającego jej piku, gdyż wysokość i powierzchnia piku są proporcjonalne do
ilości oznaczanego składnika. Stosunek proporcjonalności jest różny dla poszczególnych
substancji i może się zdarzyć, że wielkość piku na chromatogramie dla jakiejś substancji
jest mniejsza, chociaż jej ilość w analizowanej próbce jest największa. Wynika to z różnej
odpowiedzi detektora względem różnych substancji. Wysokość piku wykorzystuje się do
oznaczeń ilościowych, gdy pik jest symetryczny i wąski. Przy wykorzystywaniu tego
parametru należy pamiętać, że jest on wrażliwy na czas trwania dozowania próbki i na
małe nawet zmiany prędkości gazu nośnego. Z punktu widzenia otrzymania
prawidłowych wyników analizy bardziej korzystne jest mierzenie powierzchni piku. W
zależności od sposobu wykonania, pomiar powierzchni piku może być niekiedy bardziej
kłopotliwy niż pomiar jego wysokości. Powierzchnie piku można mierzyć różnymi
sposobami. Najdogodniejszy jest automatyczny pomiar wielkości powierzchni piku przy
użyciu integratora lub komputera. W przypadku, gdy nie ma takiej możliwości, można
wykonać pomiar ręcznie. Spośród kilku sposobów wyznaczania powierzchni można
polecić obliczanie powierzchni symetrycznych pików przy założeniu, że powierzchnia ta
odpowiada powierzchni trójkąta.
Aby obliczyć powierzchnię piku symetrycznego, trzeba zmierzyć jego wysokość h,
i szerokość w połowie wysokości, w
0.5
, albo szerokość przy podstawie, w
p
. W pierwszym
przypadku powierzchnia piku S = hw
0.5
, a w drugim S = 0.5hw
p
. Gdy pik jest
niesymetryczny, wówczas S = h(w
0.15
+w
0.85
)/2, gdzie w
0.15
i w
0.85
są to szerokości piku
na 15 i 85% jego wysokości.
Istota chromatograficznej analizy ilościowej polega na porównywaniu wielkości
piku oznaczanego składnika z wielkością piku odpowiadającego znanej ilości tego
składnika w postaci substancji wzorcowej. Znając trzy wielkości oblicza się czwartą, tj.
masę lub stężenie analizowanej substancji w próbce.
14
V.
Przebieg ćwiczenia
Podstawowym elementem wyposażenia stanowiska badawczego jest chromatograf
gazowy firmy Hewlett-Packard serii 6890. W chromatografie tym zainstalowany jest
dozownik z automatyczną regulacją pneumatyki, kolumnę kapilarna (Carboxen 1006)
oraz detektor cieplno-przewodnościowy (TCD). Przed dozownikiem (komorą
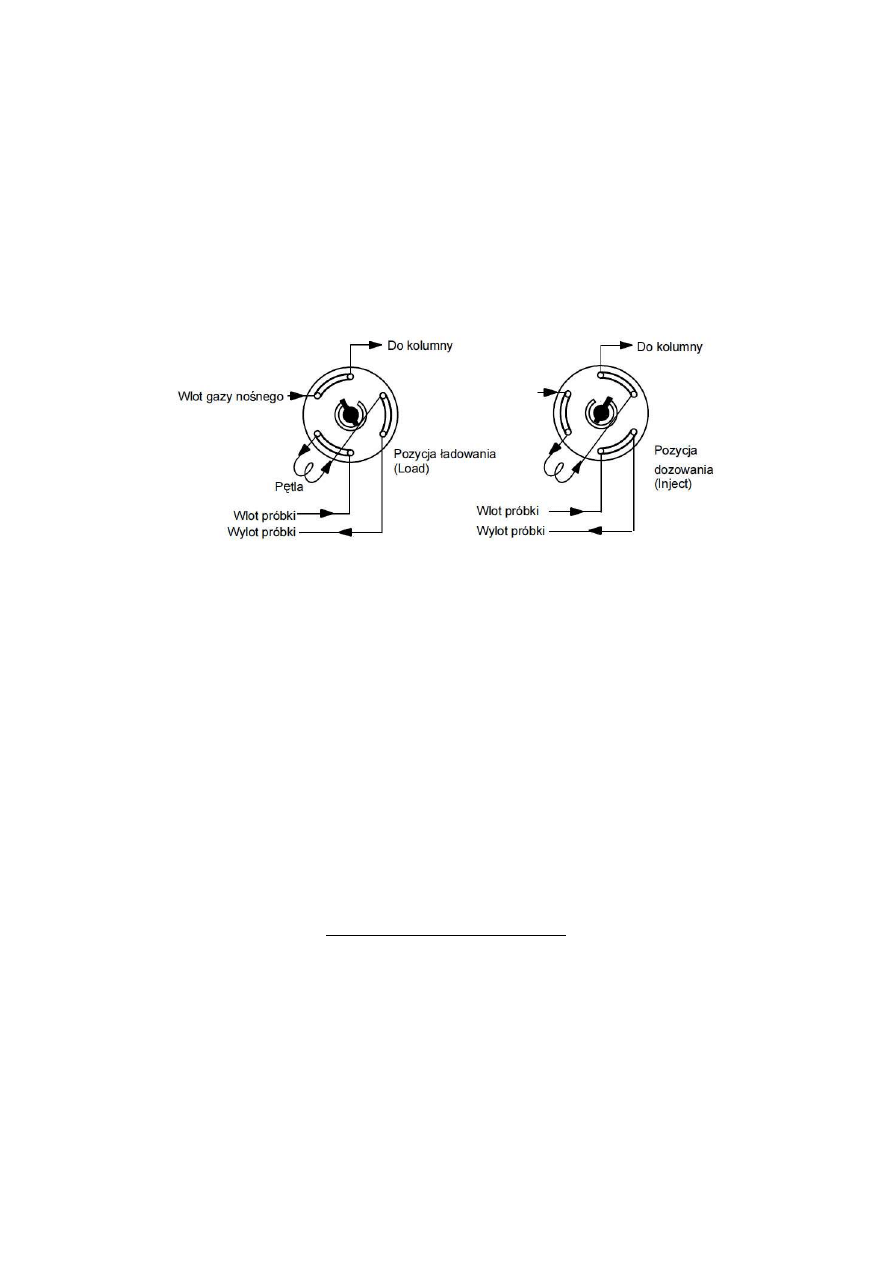
nastrzykową) zainstalowano zawór dozujący, którego zasadę działania objaśnia rys.1.7.
Rys.1.7. Zasada działania zaworu dozującego sześciodrożnego
Pozycja ładowania – pętla wypełniana jest próbką gazową, kolumna przepłukiwana jest
gazem nośnym.
Pozycja dozowania – zawartość napełnionej pętli zostaje wprowadzona do strumienia
gazu nośnego i następnie zostaje przeniesiona na kolumnę.
Stanowisko wyposażone jest również w butle ciśnieniowe zawierające powietrze
syntetyczne wykorzystywane do uruchamiania pneumatyki chromatografu oraz hel
stanowiący gaz nośny. Między butlami ciśnieniowymi a dozownikiem chromatografu
zainstalowano zespół oczyszczaczy gazu nośnego, wyłapujący takie zanieczyszczenia
jak: para wodna, węglowodory oraz przede wszystkim tlen.
Sposób wykonania ćwiczenia
1.
Przygotowanie chromatografu do analizy:
–
sprawdzić połączenie chromatografu ze zbiornikami gazów,
–
wczytać metodę (ustawienie parametrów pracy podzespołów stosownie do
przyjętego typu analizy),
–
przygotować próbkę do analizy (nastrzyk przez zawór sześciodrożny z butli
zawierającej analizowaną mieszankę gazową).

15
2.
Wykonanie analizy:
–
zadozować próbkę do komory nastrzykowej,
–
uruchomić chromatograf komendą START,
–
wykonać analizy dwóch mieszanin gazowych (wzorcowych).
3.
Analiza chromatogramu:
–
na podstawie odczytanych z chromatogramu wielkości (czas retencji, pole
powierzchni pod pikiem, wysokość piku, szerokość piku w połowie wysokości, itp.)
przyporządkować poszczególne piki składnikom mieszaniny gazowej,
–
wyznaczyć krzywe kalibracji dla poszczególnych składników mieszaniny gazowej (w
zależności od pola powierzchni pod pikiem lub ich wysokości).
VI. Bibliografia
[1].
Bonelii E. – “Basic Gas Chromatography”, Varian 1968,
[2].
Hill H.H. – “Detectors for Capillary Chromatography”, John Wiley & Sons 1992,
[3].
Jennings W. – “Sample Preparation for Gas Chromatographic Analysis, Huthig
1983,
[4].
Rodel W., Wolm G. – „Chromatografia gazowa”, Warszawa 1992,
[5].
Schupp O.E. – „Chromatografia gazowa”, Warszawa 1972,
[6].
Strugała A., Porada S. – „Ćwiczenia laboratoryjne z gazownictwa”, Kraków 1988,
[7].
Witkiewicz Z. – „Podstaway chromatografii”, Warszawa 1995.

16
Karta analizy składu gazu metodami chromatografii gazowej
Imię i nazwisko studenta: 1 ………………………………………………………………………….…………….
2 ………………………………………………………………………….…………….
3 ………………………………………………………………….…………………….
4 ……………………………….……………………………………………………….
Rok studiów: ………………………………………
Grupa: ……………..…………………………………
Data: …………………………………………………..
Godzina: …………………………………………….
Temperatura otoczenia: ……………………...
Ciśnienie otoczenia: ……………………………
Rodzaj gazu nośnego: hel
Rodzaj detektora: TCD
Typ kolumny chromatograficznej: Carboxen 1006 PLOT
Tab. Zestawienie wyników pomiaru
Mieszanina I
Mieszanina II
Składnik mieszaniny
CO
2
Ar
CO
2
Ar
Stężenie składnika w mieszaninie
wzorcowej [%]
Pole powierzchni pod pikiem dla
składnika mieszaniny
Wyszukiwarka
Podobne podstrony:
Analizy - Gleba cwiczenia, OZNACZENIE SKŁADU GRANULOMETRYCZNEGO GLEBY
PN EN 933 1 2000 Badania geometrycznych wl kruszyw Oznaczanie skladu ziarnowego Metoda przesiewan
Oznaczanie składu uziarnienia metoda areometryczna, Budownictwo studia, materiały budowalane
PN EN 933 1 2000 Badania geometrycznych wl kruszyw Oznaczanie skladu ziarnowego Metoda przesiewan
Oznaczanie składu ziarnowego kruszyw Metoda przesiewania, Budownictwo
cwiczenie 5 amylaza oznaczanie aktywnosci enzymu metoda kolorymetryczna 05 05 2014
Oznaczanie składu metodami chromatografii gazowej 1
Oznaczanie powierzchni właściwej i przybliżonego składu mineralnego metodą sorpcji pary wodnej
Oznaczanie składu metodami chromatografii gazowej, AKADEMIA GÓRNICZO - HUTNICZA
notatek pl oznaczenie laktozy w mleku metoda bertranda
Ćwiczenie 1 - oznaczanie stalej i stopnia dysocjacji, Biotechnologia PWR, Semestr 3, Chemia fizyczna
PN EN 1427 z 2009 Asfalty i lepiszcza asfaltowe Oznaczanie temperatury mięknienia Metoda Pierścień i
Oznaczanie cukrow prostych metoda Antronowa, Tż, Analiza żywności II, Sprawozdania
Kraking katalityczny – oznaczanie aktywności katalizatorów metodą UOP
chemia 11 2, Oznaczanie NaOH i Na2CO3 metodą Wardera
zabawy ruchowe, Zestaw cwiczen gimnastycz konspekt, ZESTAW ĆWICZEŃ GIMNASTYCZNYCH PROWADZONYCH NOWAT
więcej podobnych podstron