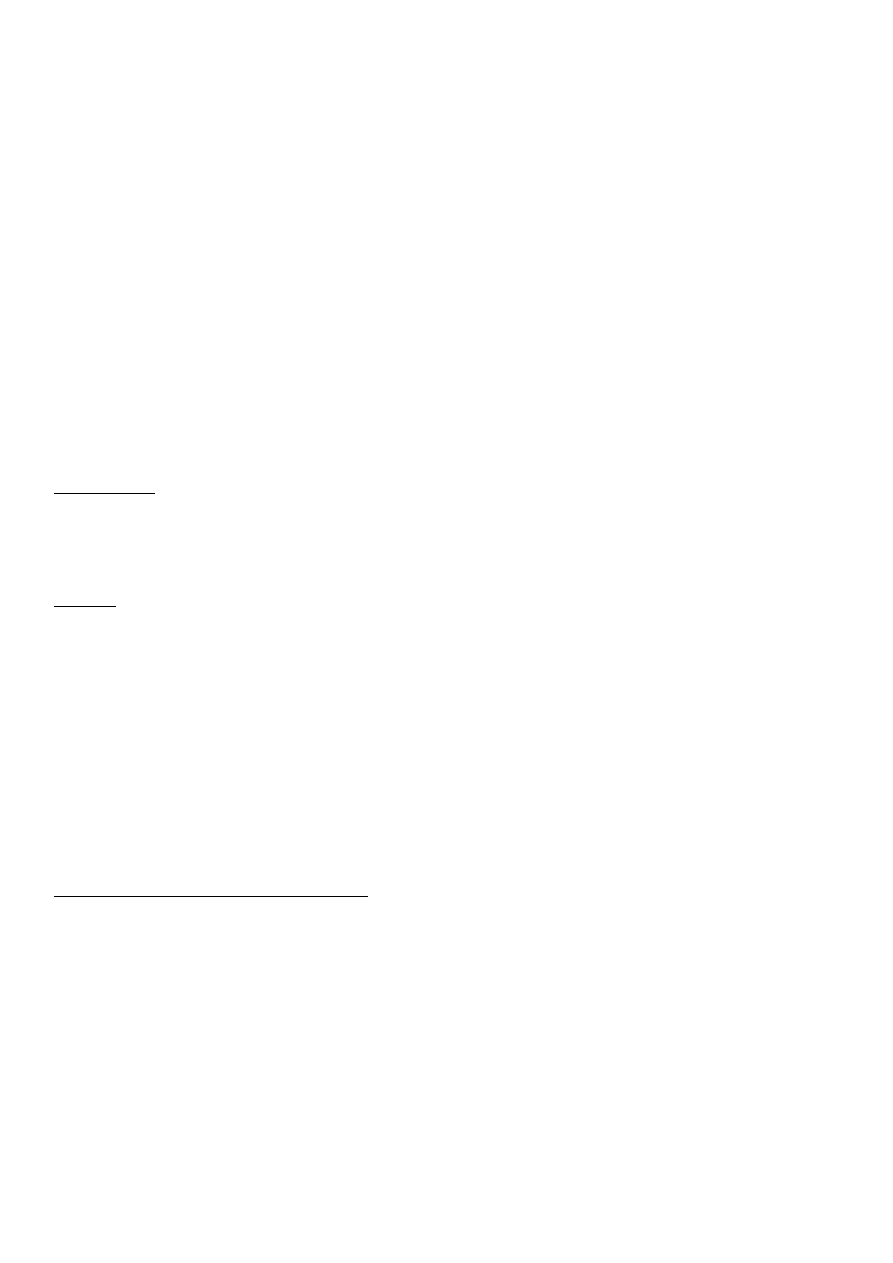
1. Definicja spektrofotometrii.
Technika instrumentalna, w której do celów analitycznych wykorzystuje się przejścia energetyczne zachodzące w cząsteczkach
spowodowane absorpcją promieniowania elektromagnetycznego w zakresie nadfioletu ( UV 200 – 380 nm), światła widzialnego(
UIS 380 – 750 nm), podczerwieni(IR powyżej 750 nm).
2. Jakie związki można oznaczać metodą spektrofotometrii.
Substancje organiczne (np. wiele związków posiadających wiązanie Π lub elektrony n, w tym węglowodory aromatyczne,
aldehydy, ketony, kwasy, aminy) i nieorganiczne wykazujące adsorpcję w nadfiolecie
Związki adsorbujące promieniowanie w zakresie widzialnym w tym barwne związki organiczne i barwne sole metali
Substancje których formy adsorbujące promieniowanie uzyskuje się na drodze reakcji chemicznej
3. Schemat blokowy spektrofotometru, rola poszczególnych elementów, zasada działania.
Źródło promieniowania ciągłego (lampa wolframowa, halogenowa)
monochromator (pryzmat, siatka dyfrakcyjna)
roztwór badany (uchwyt kuweta)
detektor (fotokomórka, fotopowielacz, fotodioda)
miernik
4. Rodzaje detektorów w spektrofotometrach.
► Detektory fotoelektryczne w spektrofotometrach UV-Vis:
1) fotokomórki
2) fotopowielacze (fotomnożniki)
3) fotodiody
Fotopowielacze
-są to układy, w których wykorzystuje się zjawisko wtórnej emisji elektronów
-fotopowielacze składają się z :
fotokatody
układu powielającego, składającego się z szeregu elektrod zwanych dynodami
anody
Fotodiody
-są detektorami, w których wykorzystuje się zjawisko fotoelektryczne wewnętrzne
-stosuje się w nich materiały półprzewodnikowe, w których pod wpływem absorbancji fotonów następuje wzbudzenie nośników
(elektronów) z pasma walencyjnego do pasma przewodnictwa
-fotodiody charakteryzują się:
dużą szybkością odpowiedzi
stabilnością wskazań i niskim poziomem szumów
dużą czułością, porównywalną z czułością fotokomórek
5. Jak działają fotokomórki, od czego zależy ich czułość.
FOTOKOMÓRKI
oparte na zjawisku fotoelektrycznym zewnętrznym
dostarczana przez fotony energia wyzwala e- z fotoczułej katody
uwolnione - są przyśpieszane w polu elektrycznym między katodą a anodą i wywołują prąd elektryczny w obwodzie
zewnętrznym
czułość fotokomórki zależy od materiału fotokatody
najczęściej jest stosowane rozwiązanie, w którym fotokatoda jest zbudowana z 3 warstw dobrze przewodzącego
materiału, np. Ag, warstwy półprzewodzącej, warstwy absorpcyjnej
Z reguły w spektrofotometrach UV-Vis są zamontowane 2 wymienione fotokomórki:
-
niebieska na zakresie UV do 650nm
-
czerwona
6. Rola i zalety fotodiod.
Rola - absorbowanie światła na powierzchni.
Zabsorbowane światło powoduje zbudzenie nośników,
są detektorami, w których wykorzystuje się zjawisko fotoelektryczne wewnętrzne
stosuje się w nich materiały półprzewodnikowe
DAD (detektor światła UV do widzialnego). Detektory DAD - SA to przyrządy o wielokanałowej detekcji umożliwiające szybka
odpowiedz i szeroki przedział liniowości wskazań
Zalety:
szybka odpowiedz,
wysoka czułość porównywalną z czułością fotokomórek
stabilność wskazań,

niski poziom szumów.
7. Analiza ilościowa metodą spektrofotometrii .
Polega na pomiarze absorbancji roztworu przy odpowiedniej długości fali i wykorzystaniu równania Lamberta-Beera:
A=ε
λ
I C
C– stężenie analitu w badanym roztworze, I – grubość warstwy absorbujacej, ε
λ
– współczynnik absorbancji przy długości fali λ
8. Prawo Lamberta - Beera.
Jeśli współczynnik absorpcji rozpuszczalnika jest równy zeru, to absorbancja wiązki promieniowania monochromatycznego
przechodzącej przez jednorodny roztwór jest wprost proporcjonalna do stężenia roztworu c i do grubości warstwy absorbującej l”.
9. Co to są grupy chromoforowe, auksochromowe
Chromofory, grupa chromoforowa, układ chromoforowy, ugrupowania atomowe o wiązaniach wielokrotnych, posiadające
pasma absorpcyjne w pobliżu 200 nm. Przykładami chromoforów są grupy: etylenowa =C=C=, karbonylowa =C=O, azowa -N=N-,
azometinowa -CH=N-, azoksy -N=N(→ O)-.
Na intensywność barwy związków wpływają również podstawniki zwane grupami autoksochromowymi. Mają one zdolność
przesuwania maksimum absorpcji w kierunku fal dłuższych, zjawisko to to efekt batochromowy -CH3, -NH2, -OH, -OCH3, -SH.
Auksochromy, grupy auksochromowe, grupy funkcyjne, które po wprowadzeniu do cząsteczki barwnika przesuwają pasmo
absorpcyjne chromoforu (efekt batochromowy lub hipsochromowy) i zmieniają jego natężenie (efekt hiperchromowy lub
hipochromowy), wskutek czego następuje zmiana barwy barwnika. Przykłady auksochromów: -NR
2
, -NHR, -NH
2,
-OH, -OCH
3
,
-OCOCH
3
, -NHCOCH
3
, -Br, -Cl.
10. Pojęcie chromatografii i mechanizm procesu chromatograficznego
jest to fizykochemiczna metoda rozdzielania z której składniki rozdzielane ulegają rozdzieleniu na 2 fazy:
stacjonarną (nieruchomą)
ruchomą (poruszającą się w określonym kierunku).
Jeżeli ruchomą fazą jest gaz mamy do czynienia z chromatografią gazową, jeżeli eluent- z chromatografią cieczową
Faza ruchoma porusza się wewnątrz kolumny natomiast faza stacjonarna jest osadzona na
wewnętrznych ściankach kolumny. Związki chemiczne z większym powinowactwem do fazy stacjonarnej są selektywnie
zatrzymywane przez nią i poruszają się wzdłuż kolumny znacznie wolniej. Związki z mniejszym powinowactwem do fazy
stacjonarnej poruszają się wzdłuż kolumny szybciej i tym samym opuszczają kolumnę, czyli eluują z kolumny, jako pierwsze.
Równowaga podziału pomiędzy fazy ma charakter dynamiczny, czyli cząsteczki substancji nieustannie przechodzą od fazy
ruchomej do stacjonarnej i z powrotem.
Mechanizm procesu chromatograficznego
Substancje charakteryzują się zróżnicowanym współczynnikiem retencji k.
11. Klasyfikacja metod chromatograficznych wg. stanu skupienia fazy ruchomej, stanu skupienia fazy stacjonarnej, natury
zjawisk
Stan skupienia fazy ruchomej:
chromatografia gazowa
chromatografia cieczowa
chromatografia fluidalna
Stanu skupienia fazy stacjonarnej:
Fazą stacjonarną może być ciecz na nośniku lub ciało stałe.
Można zatem wyróżnić chromatografię w układzie:
gaz- ciecz (gas- liquid chromatography GLC)
ciecz- ciecz ( liquid- liquid chromatography LLC)
gaz- ciało stałe (gas- solid chromatography GSC)
ciecz- ciało stałe (liguid- solid chromatography LSC)
12. Współczynnik retencji
Jest miarą czasu w jakim substancja X przebywa w fazie stacjonarnej w stosunku do czasu, w którym przebywa ona w fazie
ruchomej. Określa on ile razy dłużej dana substancja jest zatrzymywana przez fazę stacjonarną niż potrzebowałaby na przejście
przez kolumnę z prędkością poruszania się fazy ruchomej.
k= liczba moli substancji X w fazie stacjonarnej/liczba moli substanji X w fazie ruchomej
Związki charakteryzujące się różnymi współczynnikami retencji mogą zostać rozdzielone na kolumnie chromatograficznej
13. Chromatogram i sposób pomiaru podstawowych wielkości chromatograficznych
efekt rozdziału chromatograficznego jest wykreślony w postaci chromatogramu, który przedstawia wykres wskazań
sygnału uzyskanego w detektorze w funkcji objętości fazy ruchomej
zapis stężenia substancji w funkcji czasu na postać krzywej Gaussa i nosi nazwę piku (ang. peak- szczyt)
Rys. Chromatogram i sposób pomiaru podstawowych wielkości chromatograficznych.
t
M
- zerowy czas retencji- czas przebywania w kolumnie substancji, która nie oddziałuje z fazą
stacjonarną
t
R
- całkowity czas retencji danego składnika to czas liczony od momentu wprowadzenia próbki
(iniekcji) do piku, tzn. do momentu pojawienia się na wyjściu z kolumny max stężenia
wymywanego związku.
t’
R
- zredukowany czas retencji- różnica między całkowitym czasem retencji i zerowym czasem
retencji.
Sprawność kolumny chromatograficznej decyduje o tym, czy pik chromatograficzny jest
ostry czy też rozmyty. Korzystne jest, aby rozmycie było ograniczone, aby piki były wąskie,
gdyż wówczas będą lepiej rozdzielone. O sprawności kolumny decyduje liczba półek teoretycznych – im jest ich więcej tym
kolumna jest sprawniejsza (Rys. 3). Półka teoretyczna jest to objętość kolumny, w której zostaje osiągnięty stan równowagi
pomiędzy stężeniami substancji chromatografowanej w fazie ruchomej i w fazie stacjonarnej. Kolumna chromatograficzna składa
się z N półek teoretycznych.
L = N ´ H (7)
gdzie: L - długość kolumny chromatograficznej, H (lub inaczej WRPT) – wysokość równoważna półce teoretycznej, N - liczba półek
teoretycznych.
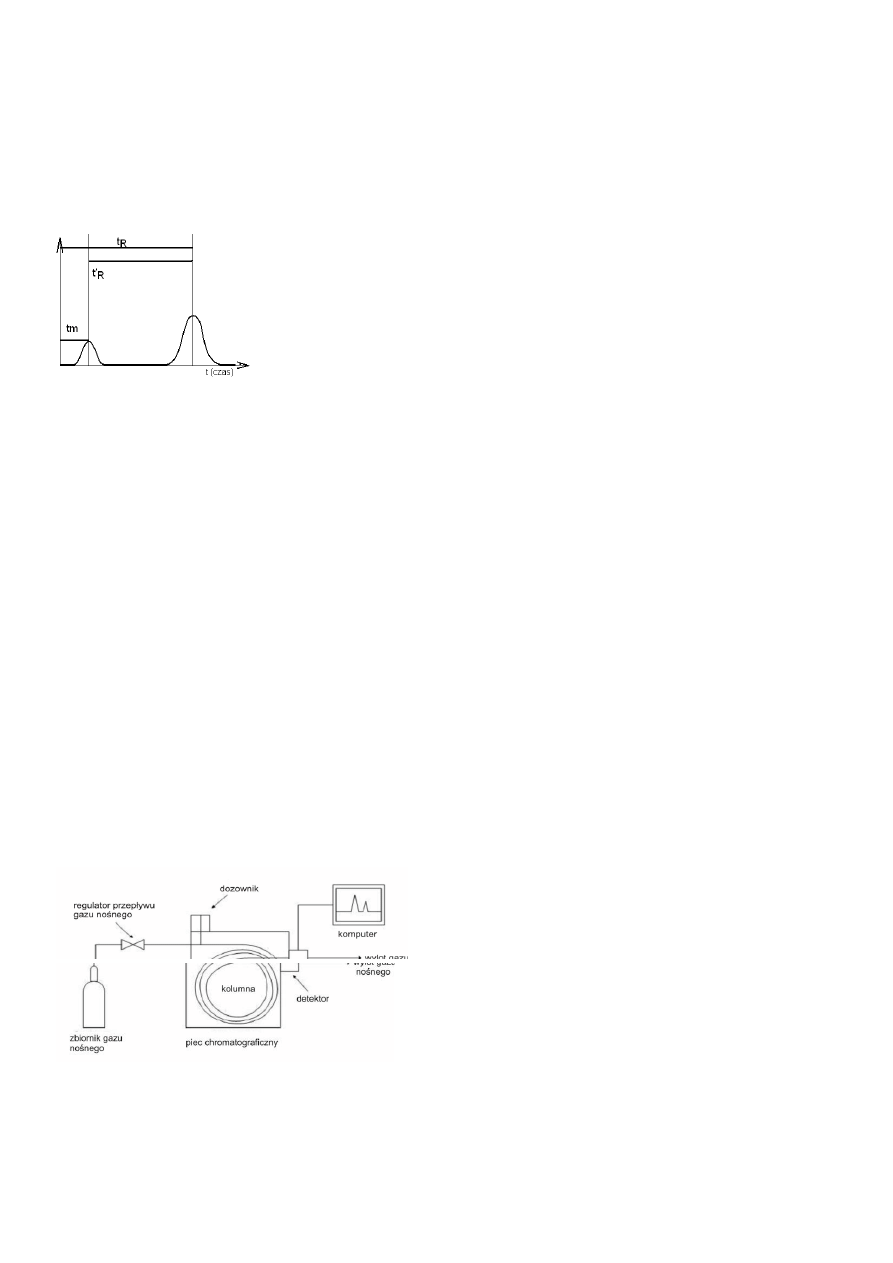
14. Budowa chromatografu gazowego (GC)
Składa się on z zbiorników z gazem, które to przepływają przez filtry, następnie przepływomierze (dławiki). W dozowniku jest
regulowana odpowiednia ilość gazu wtłaczana do kolumny chromatograficznej. Przed wtłoczeniem gaz jest standaryzowany.
Kolumna jest termostatowana. Na końcu kolumny jest palnik z detektorem. Rozdzielony gaz jest spalany a odebrany impulsy są
rejestrowane przez rejestrator.
Aparatura do chromatografii gazowej:
Gaz nośny (faza ruchoma) doprowadzony z butli płynie przez regulator przepływu dozownika, a następnie przez kolumnę
i detektor, skąd jest usuwany na zewnątrz do atmosfery;
Kolumna jest umieszczona w termostacie,
Temperatura dozownika, detektora i kolumny jest odpowiednio regulowana
Do dozownika wprowadza się próbkę, która po odparowaniu w dozowniku, miesza się ze strumieniem gazu nośnego i
następnie jest przenoszona do kolumny;
W kolumnie następuje rozdział chromatograficzny składników próbki, które opuszczają kolumnę wraz z gazem nośnym i
trafiają do detektora.;
Składniki próbki są monitorowane w detektorze generując w nim sygnał elektryczny;
Sygnały mogą być rejestrowane w komputerze lub rejestratorze;
W chromatografii gazowej fazę ruchomą stanowią gazy o małej gęstości, nieskiej lepkości w których współczynniki dyfuzji są duże.
Najczęściej jest to: wodór, azot, argon lub hel. Zadaniem gazu jest transport próbki przez dozownik, kolumnę i detektor.
15. Kolumna w procesie chromatografowania
W kolumnach chromatograficznych zachodzi właściwy proces chromatografowania i dlatego jej rodzaj ma decydujący wpływ na
jakość rozdzielania składników próbki czyli na wynik analizy chromatograficznej. Kolumna chromatograficzna jest nazywana
„sercem chromatografii”
16. Rodzaje kolumn w GC: różnice i zalety
Rozróżnia się następujące rodzaje kolumn (Rys. 11, 12):

pakowane (kolumny z wypełnieniem): analityczne, mikropakowane i preparatywne,
kolumny o przekroju otwartym – kapilarne.
Kolumny pakowane napełnione są fazą stacjonarną w całej swojej objętości. Ten rodzaj
kolumn jest najczęściej stosowany do preparatywnego otrzymywania niewielkich ilości czystych związków chemicznych.
Kolumny kapilarne pozwalają na osiągnięcie dużo wyższych sprawności niż kolumny
pakowane i obecnie większość analiz chromatograficznych wykonuje się przy zastosowaniu
kolumn kapilarnych. Fazy stacjonarne w kolumnach kapilarnych mogą być zarówno adsorbentami, jak i cieczami i mogą być
osadzone na ściankach kapilar w różny sposób.
Wyróżnia się następujące rodzaje kolumn kapilarnych:
WCOT (wall-coated open tubular) – kapilary ze ściankami wewnętrznymi pokrytymi nielotną ciekłą fazą stacjonarną. Jest
to najbardziej popularny typ kolumn,
PLOT (porous layer open tubular) – kapilary ze ściankami pokrytymi warstwą ziaren adsorbenta,
SCOT (support-coated open tubular) – ziarna wypełnienia pokryte są fazą ciekłą.
17. Fazy stacjonarne w kolumnach kapilarnych
Fazy stacjonarne w kolumnach kapilarnych mogą być zarówno adsorbentami, jak i cieczami i mogą być osadzone na ściankach
kapilar w różny sposób.
Ciekłe fazy stacjonarne w chromatografii gazowej to najczęściej:
fazy niepolarne: węglowodory będące dobrymi rozpuszczalnikami substancji niepolarnych (np. skwalan),
fazy średniopolarne: silikony. Są to siloksany o różnej masie cząsteczkowej. Najczęściej stosujemy: polidimetylosiloksan,
polimetylofenylosiloksan policyjanoalkilosiloksan,
fazy polarne: glikole polietylenowe (np. Carbowax) oraz estry,
fazy polarne oparte o kopolimery styrenu i diwinylobenzenu (np. PoraPLOT Q),
18. Sprawność kolumny chromatograficznej
Zależna od ilości półek chromatograficznych. O sprawności kolumny mówi WRPT (Wysokość Równoważna Półce
Chromatograficznej).Półka chromatograficzna jest to objętość kolumny w jakiej zostaje osiągnięty stan równowagi sub.
Chromatografowanej miedzy fazą stacjonarną i fazą ruch. Kolumna posiada N półek. Im ostrzejszy PIK (peak) tym lepsza
sprawność.
19. Detektory w GC i ich rola
substancje rozdzielone w kolumnie chromatograficznej są wykrywane przez detektor w miarę jak eluują z kolumny.
Detektor reaguje sygnałem elektrycznym na obecność śladów analizowanej substancji w gazie nośnym opuszczającym
kolumnę.
Dobry detektor powinien charakteryzować się:
- dużą czułością i wykrywalnością
- szerokim zakresem liniowości wskazań
- stabilnością wskazań i niskim poziomie „szumów” linii zerowej
- selektywnością lub uniwersalnością wskazań
- łatwością obsługi
- niskim kosztem
Detektory można podzielić na:
-> nieselektywne – detektory uniwersalne, reagują na wszystkie składniki próbki;
-> selektywne – reagują na pewną grupę związków, które mają podobne właściwości chemiczne lub fizyczne;
-> specyficzne – reagują na pojedynczy związek chemiczny, rzadko stosowane.
Detektor płomieniowo-fotometryczny FDP:
-jest detektorem selektywnym, służy do wykrywania związków zawierających siarkę i fosfor
-detektor ten wykorzystuje zjawisko emisji promieniowania (chemiluminescencja) występujące wówczas, gdy związki zawierające
S lub P spalają się w płomieniu wodorowo-tlenowym.
Detektor płomieniowo-jonizacyjny FID:
-jest najczęściej używanym detektorem
-detektor uniwersalny, czyli taki, który jest czuły na prawie wszystkie związki chemiczne;
-nie wykrywa obecności związków nieorganicznych oraz niektórych związków węgla: CO, CO
2
, CS
2
, HCOOH, COCl
2
.
Detektor wychwytu jonów ECD
-jest detektorem selektywnym
-zasada działania ECD polega na gwałtownym spadku natężenia prądu płynącego w komorze jonizacyjnej po wprowadzeniu do niej
substancji o dużym powinowactwie elektronowym.
-jest detektorem selektywnym charakteryzującym się dużą czułością na halogenozwiązki.
-jest bardzo przydatny do oznaczania chlorowcopestycydów.
20. Zasada działania FID
do detektora doprowadzane są z butli dwa gazy: wodór i powietrze
gaz nośny wypływający z kolumny jest mieszany z wodorem i kierowany przez dyszę do komory przez którą przepływa
powietrze
wodór spala się tworząc płomień wodorowy
składniki próbki doprowadzane są wraz z gazem nośnym do płomienia ulegają spaleniu.
powstający prąd jonowy jest wzmacniany i rejestrowany przez potencjometr.
sygnał z detektora jest proporcjonalny do liczby atomów węgla niezwiązanych z tlenem, a więc w przybliżeniu jest
proporcjonalny do masy substancji.
21. Kryteria wyboru fazy stacjonarnej w GC
Gazy o małej gęstości, niskiej lepkości, o wysoki współczynniku dyfuzji.
22. Analiza jakościowa w GC
Analiza jakościowa metodą GC (z wykładu)
Analiza jakościowa dotyczy identyfikacji pików odpowiadających poszczególnym składnikom próbki i opiera się na
wielkościach retencyjnych
Porównuje się czas retencji piku identyfikowanej substancji z czasem retencji piku wzorca.
23. Cechy idealnej metody ilościowej
wysoka precyzyjność i dokładność
możliwość wykonania szybkiej analizy próbki przy minimalnym koszcie, pracy manualnej i wymogów aparaturowych
niepodatność na interferencje zanieczyszczeń pochodzących od próbki i odczynników analitycznych
eliminacja możliwości utraty próbki i analitu podczas pracy lub analizy.
24. Metoda normalizacji wewnętrznej
Polega na wyznaczeniu udziału procentowego wszystkich substancji w próbce
Podstawowym warunkiem jest to, aby na chromatogramie znajdowały się piki wszystkich substancji obecnych w próbce
Zasada metody: po wykonaniu chromatogramu mierzy się powierzchnię poszczególnych pików i sumuje je
Suma wszystkich pików stanowi 100 %
Powierzchnia każdego piku w stosunku do powierzchni sumy wszystkich pików (sygnałów) odpowiada zawartości
procentowej analizowanych substancji w próbce:
%
i
= (A
i
/
A) 100
A
i
- powierzchnia piku oznaczonego składnika
Tak byłoby gdyby wskazania detektora dla wszystkich składników były równe.
Wiemy jednak, że różne detektory reagują różnym sygnałem na obecność określonych substancji.
Np wartość sygnału z detektora płomieniowo-jonizacyjnego zależy od składu chemicznego analizowanego związku, gdyż
od tego zależy liczba tworzących się w płomieniu wodorowym termojonów węgla.
w praktyce chromtograficznej wprowadzono poprawki czyli tzw współczynniki korekcyjne, które pozwalają na wzajemne
porównanie powierzchni pików zanalizowanych mieszanin.
jeżeli analizujemy związki zbliżone strukturalnie, nie jest konieczne wyznaczenie współczynników odpowiedzi
dla substancji o różnej budowie należy je wyznaczyć; w przeciwnym razie otrzymamy tylko szacunkowe wyniki.
f = m
s
Y
s
/m
w
Y
w
m
s
, m
w
– masy substancji badanej i wzorca
Y
s
, Y
w
- powierzchnie lub wysokości pików substancji i wzorca
f- współczynnik odpowiedzi
25. Metoda kalibracji bezwzględnej (wzorca zewnętrznego)
służy do określania ilości jednego lub kilku składników próbki, które są identyfikowane
dla każdego związku, którego ilość w próbce chcemy wyznaczyc, sporządza się kilka (3) roztworów o różnych stężeniach
i kilkukrotnie wykonuje się analizę chromatograficzną dla dla każdego z tych roztworów.
następnie oblicza się średnią powierzchnię lub wysokość piku i wykreśla się krzywą kalibracyjną, przedstawiającą
zależność powierzchni piku od stężenia substancji.
wynik analizy próbki o nieznanym stężeniu próbki odczytywany jest z krzywej.
krzywe kalibracyjne przedstawiają zależność powierzchni A i wysokości piku od ilości G analizowanego składnika.

26. Metoda wzorca wewnętrznego
Jest najpowszechniej stosowaną metodą analizy ilościowej z chromatografii gazowej
Ma ona 2 wersje:
o
wzorcem wewnętrznym jest substancja nieobecna w analizowanej próbce
o
wzorcem wewnętrznym jest analizowana substancja.
W pierwszym przypadku (jeśli wzorcem jest substancja nieobecna w analizowanej próbce):
o
Ściśle odmierzoną ilość wzorca dodaje się do dokładnie znanej ilości próbki
o
Wzorcem wewnętrznym jest substancja, której pik występuje blisko piku lub pików oznaczonych składników
W metodzie wzorca wewnętrznego, analitycy stosują związki, które przypominają badane anality w jak największym
stopniu i dodają je do próbki przed jej obróbką lub przygotowaniem do analizy
Wzorzec wewnętrzny powinien charakteryzować się właściwościami chemicznymi, chromatograficznymi zbliżonymi do
właściwości analitu
Wzorzec wewnętrzny powinien charakteryzować się znaną chemiczną strukturą, być dostępny o wysokiej czystości.
27. Pojęcie chromatografii cieczowej
rodzaj chromatografii gdzie fazą ruchomą jest ciecz a fazą stacjonarną jest ciało stałe (chromatografia absorpcyjna), rzadziej ciecz
osadzona na nośniku (chromatografia podziałowa).
► Fazy ruchome w chromatografii cieczowej jest czynnikiem aktywnym, a rodzaj i ilość rozpuszczalników ją stanowiących ma
istotny wpływ na proces chromatografowania.
fazy ruchome w chromatografii cieczowej stanowią pojedyncze rozpuszczalniki lub ich dwu lub więcej składowe
mieszaniny
faza ruchoma, którą wprowadza się do kolumny nosi nazwę eluentu
w kolumnie w skład fazy ruchomej oprócz eluentu mogą wchodzić także składniki rozdzielanej mieszaniny.
po rozdzieleniu składniki te są obecne w wycieku kolumny, który nosi nazwę eluatu
przy wyborze fazy ruchomej należy uwzględnić rodzaj i skład rozdzielanej mieszaniny, rodzaj zastosowanego wypełnienia
kolumny oraz rodzaj detektora.
Schemat budowy chromatografu cieczowego
1,2- zbiornik eluentów; 3- pompa; 4- manometr; 5-dozownik; 6- przedkolumna; 7- kolumna chromatograficzna; 8- termostat
kolumny; 9- przepływomierz; 10- detektor; 11- kolektor frakcji; 12- komputer
28. Klasyfikacja metod chromatografii cieczowej
chromatografię jonowymienną (IC)
chromatografię żelową
chromatografię powinowactwa
W zależności od rodzajów fazy stacjonarnej wyróżniamy:
Chromatografie ciecz- ciało stałe (LSC)
Chromatografie ciecz- ciecz (LLC)
29. Najistotniejsze cechy współczesnej chromatografii cieczowej
Wysoka sprawność
Dobra rozdzielczość
Duża szybkość procesu
Możliwość stosowanie wysokich ci
30. Podstawowe elementy chromatografu cieczowego i działanie (krótko)
zbiorniki eluent> pompa> manometr> dozownik > przedkolumna> kolumna chromatograficzna >termostat kolumny>
przepływomierz> detektor> kolektor frakcji> komputer. Działanie: ze zbiorników tłoczona jest faza stacjonarna, przez dozownik

dodawany jest eluent. Mieszanina trafia do kolumny chromatograficznej, często umieszczonej w termostacie. Zachodzi rozdział,
wynik jest wzmacniany i rejestrowany przez komputer w formie piku.
31. Co to jest eluent i eluat
eluent – faza ruchoma, mieszanina rozpuszczalników lub jeden rozpuszczalnik,
eluat – to co pozostaje po rozdzieleniu w wycieku z kolumny, składniki mieszaniny.
32. Detektor UV-Vis: jak działa, jakie związki wykrywa
działają na zasadzie pomiaru absorbancji promieniowania nadfioletowego UV lub nadfioletowego światła widzialnego uv/vis.
Stosuje je się do wykrywania związków zawierających wiązania nienasycone i grupy chromatoforowe, olefin, związków
aromatycznych i barwników.
33. Detektor fluorescencyjny: istota detekcji fluorescencyjnej, kiedy można w/w detektor zastosować.
istota detekcji fluorescencji – polega na pomiarze intensywności światka i określonej długości fali emitowanego przez wykrywany
związek w wyniku jego pobudzenia, światłem o wyższej energii (krótszej fali) niż emitowane (dłuższej). Kiedy można go stosować
– kiedy związek określany posiada naturalną zdolność do fluorescencji. Detektor ten spełnia swą role w zanieczyszczeniach w
żywności, polichlorowane di fenyle, akryloamid (w bardzo niskich stężeniach).
34. Co to jest układ faz normalnych (NP)
układ chromatograficzny w którym faza stacjonarna jest bardziej polarna niż faza ruchoma, a podstawowym zjawiskiem
decydującym o rozdzieleniu jest adsobrancja na powierzchni fazy stacjonarnej
35. Żel krzemionkowy: jego właściwości adsorpcyjne, chemiczna struktura powierzchni
Żel krzemionkowy jest materiałem amorficznym, porowatym i jako wypełnienie kolumn ma dużo zalet:
o
Posiada uziarnienie o pożądanej wielkości i odpowiednich rozmiarów porów
o
Określoną powierzchnię właściwą i dużą wytrzymałość mechaniczną
O właściwościach adsorpcyjnych żelu krzemionkowego
O właściwościach adsorpcyjnych żelu krzemionkowego i innych w/w tlenków decyduje rodzaj i ilość grup
hydroksylowych na powierzchni i ich dostępność
Szczególnie do grup OH adsorbują się cząsteczki rozdzielanych substancji.
Na powierzchni żelu krzemionkowego wyróżnia się trzy rodzaje miejsc aktywnych wynikających z obecności grup
hydroksylowych tj. wolne (a), bliźniacze (b) i tzw. zasocjowane (c).
Chemiczna struktura powierzchni jest związana z obecnością grup silanolowych lub siloksanowych oraz miejsc aktywnych
(wolnych, bliźniaczych, zasocjowanych).
36. Wiązane fazy stacjonarne w NP zasady tworzenia
polega na tym że centra aktywne tworzą grupy polarne, które są związane wiązaniem alkilo- lub arylosiloksanowm za
pośrednictwem grupy OH, obecnej w żelu krzemionkowym.
Najczęściej żel krzemionkowy jest modyfikowany związkami chemicznymi, posiadającymi następujące grupy: aminową,
nitrową, cyjanową, diolową, fenylową lub cyklodekstanami.
Można genralnie stwierdzić, że wziązane fazy stacjonarne stosuje się w układach faz normalnych do rozdzielenia tych
substancji, które jest bardzo trudno rozdzielić w układzie faz odwróconych (RP), albo gdy rozdzielanie w zamierzony
sposób nie jest możliwe w RP (rozdzielanie grupowe węglowodorów, lipidów, kwasów tłuszczowych).
37. Eluenty stosowane w układzie NP.
Ciecze stosowane jako faza ruchowa w chromatografii cieczowej, muszą spełniać określone warunki
Odpowiednia lepkość
Kompatybilność z detektorem
Dostępność o czystości wymaganej w chromatografii
Mała toksyczność i niski koszt.
Ze względu na pracę kolumny z detektorem w systemie ,,on-line”, konieczne jest by faza ruchoma nie dawała sygnału w
zakresie tych wartości parametru fizycznego, który jest wykorzystywany do detekcji rozdzielanych substancji.
Np. jeżeli pracujemy z detektorem UV-Vis nie należy stosować eluentu adsorbującego promienowanie w tym zakresie, co
cząsteczki rozdzielane.
Najbardziej przydatnym kryterium oceny przydatności rozpuszczalnika w absorpcyjnych układach faz normalnych jest
jego siła elucyjna i selektywność.
W celu ułatwienia wyboru właściwego rozpuszczalnika jako eluentu rozpuszczalniki ułożono w szeregi eluotropowe.
W przypadku polarnych faz stacjonarnych rozpuszczalniki ułożone wg rosnącej mocy eluowania (miarą której są indeksy
polarności) mają kolejność następującą:
o N-pentan<n.heksan<chloroform<tetrahydrofuran<pirydyna< etanol< metanol< woda < kwas octowy
Znajomość siły elucyjnej znacznie ułatwia dobór fazy ruchomej do rozdzielenia mieszanin określonych substancji.
Zazwyczaj dobiera się dwuskłądnikową fazę ruchomą stosując rozpuszczalnik o dużej sile elucyjnej (polarny) i
rozpuszczalnik niepolarny o małej sile elucyjnej (rozcieńczalnik eluentu)
38. Dlaczego jest ważna lepkość eluentów w NP.

Zbyt wysoka lepkość fazy ruchomej wymagałaby stosowania bardzo wysokich ciśnień.
Ponadto dla procesu chromatograficznego korzystne są możliwe niskie wartości współczynników dyfuzji, co jest sprzeczne
ze stosowaniem eluentu o znacznej lepkości.
39. Sposób doboru eluent oparty na indeksie polarności
kiedy rozdzielane substancje są słabo polarne eluent również powinien charakteryzować się niską polarnością, odwrotnie przy
wysokiej polarności substancji.
40. Wymień 6 przykładowych eluentów wg indeksów polarności
cykloheksan< tetra chlorek węgla< toluen< benzen< eter di etylowy< chloroform< tetrahydrofuran
41. Zastosowanie układów faz NP.
W chromatografii cieczowej dominuje obecnie stosowanie układów faz odwróconych
Generalnie można powiedzieć, chromatografię w układzie faz normalnych można stosować do rozdzielania związków
niejonowych i raczej nisko oraz średnio polarnych jako alternatywę dla układów faz odwróconych.
Wiele organicznych związków chemicznych jest lepiej rozpuszczalnych w organicznych rozpuszczalnikach niż w wodzie, czy
metanolu.
Są też substancje, któ®e mogą ulegać rozkładowi w środowisku wodny,
W konsekwencji, w takich przypadkach nie może być zastosowana faza ruchoma z dodatkiem wody i układ faz odwróconych
(RP),a układy faz normalnych (NP.) stanową oczywistą alternatywę.
42. Co to jest układ faz odwróconych RP
Układ faz odwróconych, to taki układ, w którym faza stacjonarna jest mniej polarna niż faza ruchoma.
Bardziej precyzyjnym określeniem układu faz odwróconych jest definicja:
o
Układy chromatograficzne, w których powierzchnia sorpcyjna fazy stacjonarnej wykazuje charakter hydrofobowy, a eluentem jest
mieszanina wody (buforu) i organicznego dodatku rozpuszczalnego w wodzie.
43. Otrzymywanie faz związanych na potrzeby RP
Otrzymuje się je w wyniku modyfikacji powierzchni sorbentów, najczęściej polarnych, nie polarnymi, albo średnio polarnymi
cząsteczkami fazy stacjonarnej, chemicznie związanymi z cząsteczkami powierzchni ,,nośnika”
Materiałem wyjściowym, powszechnie stosowanym jest żel krzemionkowy.
44. Mechanizm retencji w RP: rodzaje oddziaływań
Układ chromatograficzny składa się z trzech składników: fazy stacjonarnej, fazy ruchomej i mieszaniny substancji
poddawanych rozdzielaniu.
Fazę ruchomą w tym typie chromatografii stanowi mieszanina wody i rozpuszczalnika organicznego, mieszającego się z wodą.
Retencja substancji RP jest wypadkową wielu oddziaływań, do których należy:
Działanie sił Van der Vaalsa pomiędzy hydrofobową fazą stacjonarną a cząsteczkami substancji
Działanie sił elektrostatycznych pomiędzy cząsteczkami substancji rozdzielanej, zawierającej polarne grupy funkcyjne i
cząsteczkami fazy ruchomej.
Działanie sił Van pomiędzy cząsteczkami substancji rozdzielanej i fazy ruchomej.
Oddziaływanie elektrostatyczne pomiędzy cząsteczkami substancji rozdzielanej i fazy ruchomej, a powierzchniowymi grupami
–OH.
45. Kolejność elucji w RP
Substancje eluują w kolejności od najbardziej polarnych do niepolarnych, a w szeregach homologicznych od nisko- do
wysokocząsteczkowych.
W przypadku kwasów tłuszczowych, najpierw eluują krótkołańcuchowe nasycone KT, a na końcu długie WNKT,
46. Trzy rodzaje selektywności związane z oddziaływaniem substancji z fazą stacjonarną
Selektywność względem grupy metylenowej, zwana hydrofobową
Selektywność chemiczna, wynikająca z oddziaływań jonowych, dipolowych lub wiązań wodorowych z
powierzchniowymi grupami OH, ewentualnie tworzeniem kompleksów z metalami z warstwy powierzchniowej
Selektywność sferyczna, wynikająca ze struktury wypełnienia i kształtu cząsteczek substancji rozdzielanych.
47. Fazy ruchome w RP
są nimi mieszaniny wody i rozpuszczalników organicznych mieszających się z wodą: acetonitryl, metanol, tetrahyrofuran. Faza
ruchoma musi mieć odpowiednią siłę elucyjną, substancje nie powinny eluować zbyt szybko. Wsp. Retencji powinien zawierać w
zakresie od 1-10.
48. Do czego służy technika GC/MS i jej zaleta
Technika GC/MS polega na połączeniu chromatografu gazowego ze spektrometrem mas.
Technika ta pozwala na identyfikację związków chemicznych oraz ich mieszanin na podstawie widm masowych, nawet gdy związki
te znajdują się w śladowych ilościach. Chromatograf rozdziela analizowana próbę na pojedyncze związki chemiczne, które kolejno
są kierowane do spektrometru mas celem ich jednoznacznej identyfikacji.
Zaletą stosowania techniki GC/MS jest możliwość porównywania unikalnych widm masowych analizowanych związków z
ogólnoświatową biblioteką, co pozwala na identyfikację związków, w tym substancji niebezpiecznych, bez konieczności posiadania
wszystkich wzorców.
49. Przykłady zastosowań GC/MS
GC/MS – spektrometria mas
Zastosowania:
Biochemii – do pomiaru mas cząsteczkowych peptydów, białek, nukleotydów, a także oligosacharydów i lipidów.
ochrona środowiska,
kontrola antydopingowa,
farmakologia,
diagnostyka medyczna, np.: diagnostyce i monitorowaniu terapii chorób nerek i układu moczowego,
biotechnologii,
proteomice czyli badaniach nad identyfikacją proteomu (proteom - białkowa składowa kodowana przez genom)
W naukach o żywności do oznaczania akryloamidu lub innych substancji, w identyfikacji naturalnych substancji
barwiących
50. Budowa układu GC/MS (podstawowe elementy) (bez)
51. Cztery podstawowe elementy MS
W spektrometrii mas mierzy się stosunek masy jonów do ich ładunku. W ogólności spektrometria mas polega na:
1. Jonizacji
2. Przyspieszeniu jonów w polu elektrycznym
3. Separacji jonów (na przykład poprzez różne zakrzywienie ich torów w przyłożonym polu magnetycznym)
4. Detekcji jonów
Układ wprowadzania badanej próbki. Poszczególne układy różnią się zależnie od stanu skupienia analizowanej próbki jak również
metody jonizacji zastosowanej do analizy:
o
s
s
t
t
a
a
n
n
s
s
t
t
a
a
ł
ł
y
y – sondy z probówką, płytki (jonizacja typu EI, MALDI)
o
s
s
t
t
a
a
n
n
c
c
i
i
e
e
k
k
ł
ł
y
y – zawory wstrzykowe, pompy strzykawkowe, systemy HPLC, FPLC, systemy elektroforezy kapilarnej
(jonizacja typu ESI, MALDI)
o
s
s
t
t
a
a
n
n
g
g
a
a
z
z
o
o
w
w
y
y – układy chromatografii GC, komory próżniowe, systemy strzykawek gazoszczelnych (jonizacja typu EI, CI,
ICP)
Jonizator jest to urządzenie, w którym następuje jonizacja cząsteczek przy użyciu różnorodnych technik (EI, CI, ESI, MALDI), z
których część prowadzi do pękania wiązań chemicznych na skutek czego dochodzi do ich podziału na mniejsze fragmenty. Inne
techniki powodują tylko naładowanie cząsteczek bez ich fragmentacji.
Wiele metod jonizacji cząsteczek takich jak FAB, EI i LD prowadzi do fragmentacji cząsteczek chemicznych w trakcie jonizacji, co
powoduje, że różne spektrometry mogą generować różne widma dla tego samego związku chemicznego. Fragmentacja cząsteczek
może pomagać w analizie, gdy badany jest jeden związek chemiczny. Pomiar masy takiego związku często nie wystarcza do jego
identyfikacji, na którą pozwala analiza charakterystycznego wzoru fragmentacji takiego związku. W przypadku mieszanin wielu
związków chemicznych, wtórne reakcje między jonami pochodzącymi z różnych związków, uniemożliwiają praktyczną analizę
danych.
Analizator m/ z, w którym wcześniej powstałe jony ulegają rozdziałowi na podstawie stosunku ich masy do ładunku. W
spektrometrach mas stosowane są różne typy analizatorów masy np.: analizator czasu przelotu, sektor magnetyczny, sektor
elektryczny, pułapka jonowa, liniowa pułapka jonowa, analizator cyklotronowego rezonansu jonów.
Detektor - jego zadaniem w spektrometrze mas jest rejestracja jonów, przechodzących przez analizator. Obecnie stosuje sie
detektory przekazujące informację w postaci sygnałów elektrycznych. Sygnały te są we współczesnych spektrometrach mas
przetwarzane na postać cyfrową i dalej analizowane i przechowywane z wykorzystaniem komputerów. Można wyróżnić kilka
najczęściej stosowanych typów detektorów: powielacz elektronowy, detektor mikrokanalikowy, detektor fotopowielaczowy.
52. Na czym polega jonizacja elektronami
Jonizacja elektronami wykorzystywana jest w metodzie Spektometrii masowej (MS). Jest to uniwersalna technika analityczna,
zaliczana do metod spektroskopowych, której podstawą jest pomiar stosunku masy do ładunku elektrycznego danego
jonu.Podstawą działania każdego spektrometru, bez względu na konstrukcję, jest jonizacja cząsteczek badanej substancji, co
umożliwia przyspieszenie jej w polu elektrycznym w próżni. Jonizację próbki można przeprowadzić przez metody:
Electronimpact (EI)- ad.1
Jonizację chemiczną (CI)- ad.2
Electrospray (ESI)- ad.3
MALDI (desorpcja/jonizacja laserowa w matrycy)- ad.4
Zasada jonizacji elektronami:
Układ
wprowadzania
badanej próbki
Jonizator
Analizator m/z
Detektor
System rejestracji
danych
(komputer)

Heterogeniczny strumień jonów (dodatnich lub ujemnych) zostaje rozdzielony na szereg składowych, zależnie od stosunku masy do
ładunku (m/z).
W przypadku jonów naładowanych dodatnio, masa próbki mierzona w spektrometrze jest powiększona o masę protonu lub
protonów przyłączonych do cząsteczki analizowanej substancji.
Dla jonów naładowanych ujemnie masa próbki pomniejszona jest o masę protonu lub protonów oderwanych od cząsteczki
analizowanej substancji.
Istnieje możliwość oznaczenia m/z substancji nie jonizujących się poprzez dołączeniepodstawnika obdarzonego ładunkiem lub
podlegającego jonizacjilub poprzez utworzenie adduktów np. z sodem lub potasem.
Ad.1. Źródło jonów, którego integralnym elementem jest katoda. Po przyłożeniu napięcia emituje elektrony o ściśle określonej
energii, które zderzając się z cząsteczkami próbki wybijają elektron lub elektrony z ich orbit walencyjnych.
Ad.2. Jonizacja substancji następuje na skutek zderzeń z tzw. jonami pierwotnymi występującymi w źródle jonów (najczęściej są to
jony gazów obojętnych, metanu, izobutanu, amoniaku).
Ad.3.Próbka ulega jonizacji pod ciśnieniem atmosferycznym przy użyciu napięcia rzędu 2000-5000V.
Ad.4. Analizowaną substancję jonizuje się po jej uprzednim zmieszaniu z roztworem matrycy. Po odparowaniu rozpuszczalnika
próbkę naświetla się impulsami lasera, co powoduje wzbudzenie elektronów w matrycy. Jony, utworzone przez przeniesienie
protonu między wzbudzoną matrycą a analizowaną substancją, ulegają następnie desorpcji.
układ wprowadzania próbki
źródło jonów
analizator jonów
detektor jon
ów
analiza danych
53. Działanie analizatora kwadrupolowego
1.Jest jednym z typów analizatorów masy. Analizator ten jest zbudowany z czterech symetrycznie ułożonych równoległych prętów.
Działa jako filtr masy – w jednym momencie przepuszcza tylko jony o określonym stosunku masy do ładunku (m/z). Dzieje się to
dzięki przykładaniu do prętów prądu zmiennego o określonej częstotliwości i napięciu oraz napięcia stałego. Kwadrupol można
ustawić tak, aby przepuszczał jony o szerokim lub wąskim zakresie m/z. Jony przechodzące przez kwadrupol mogą być poddawane
dalszej analizie.
2.Zbudowany jest z czterech metalowych prętów, biegnących równolegle do przelatującej wiązki jonów. Jedna para prętów posiada
dodatni potencjał elektryczny [U
0
+U
m
cos(ωt)]i stanowi filtr dla jonów o wysokich masach, zaś druga posiada potencjał ujemny
-[U
0
+U
m
cos(ωt)] i jest filtrem dla jonów o masach niskich.Potencjały przyłożone do prętów składają się ze stałego napięcia (U
0
) i
napięcia zmiennego o częstotliwości radiowej (U
m
), co sprawia, że przelatujący jon jest naprzemiennie przyciągany lub odpychany.
Dla danej wartości potencjału tylko jony o określonym m/zprzejdą przez kwadrupol do detektora. Pozostałe jony nie uzyskają
stabilnej trajektorii i w związku z tym zostaną „odfiltrowane” poprzez zderzenia z rdzeniami kwadrupola, zobojętnione i
odpompowane przez układ próżniowy.
3.analizator kwadrupolowy (QMS, z ang. Quadruple Mass Spectrometry), który składa się z czterech elektrod w formie prętów
połączonych naprzeciwlegle i mających przeciwną polaryzację (polaryzacja zawiera dwie składowe: stałą U i zmienną V). Jony o
określonym stosunku m/z, przyspieszane pomiędzy źródłem jonów a analizatorem kwadrupolowym przez małe napięcie
przyspieszające (rzędu 5V), przyjmują trajektorię równoległą do osi prętów i są kierowane do detektora, natomiast pozostałe jony
uderzają w pręty kwadrupola i ulegają rozładowaniu.
54. Rola detektora masowego
Detektor masowy MSD. substancje wprowadza si
ę do komory próżniowej, gdzie jest ona jonizowana. Zjonizowane cząstki
rozpadaj
ą się na fragmenty, które są segregowane przez analizator masowy wg stosunku masy do ładunku i następnie są zbierane
przez kolektor. Na kolektorze fragmenty jon
ów tworzą sygnały elektryczne proporcjonalne do liczby jonów. Wszystkie te sygnały
s
ą następnie przetwarzane na widmo masowe. Można otrzymać całkowity chromatogram jonowy i widmo masowe (rozpad na
fragmenty
odcisk palca tej substancji), b
ędące podstawą do identyfikacji substancji. Widmo masowe jest charakterystyczne dla
ka
żdej substancji.
Zatem detektor masowy umo
żliwia otrzymanie chromatogramu jonowego oraz widma masowego, które są podstawą do
identyfikacji badanej substancji.
55. Pojęcie widma masowego oraz rodzaje jonów
Widmo masowe – widmo powstałe przez rozdzielenie w spektrometrze mas strumienia jonów według stosunku ich masy
do ładunku elektrycznego, w którym postawie szczególne linie opowiadają różnym masom.Na podstawie widm masowych ustalono
procentowe zawartości poszczególnych izotopów w pierwiastkach, a także precyzyjne wyznaczono masy jąder atomowych.
Rodzaje jonów:
Jon molekularny
Związki organiczne poddane działaniu strumienia elektronów o odpowiedniej energii
ulegają rozpadom, z których najprostszy polega na utracie przez cząsteczkę jednego elektronu
i utworzeniu jonu molekularnego (cząsteczkowego, masowego, macierzystego),
oznaczanego M+
Jony fragmentacyjne
Jon molekularny może ulegać dalszym rozpadom dając obojętne cząsteczki, rodniki
oraz dodatnio naładowane jony fragmentacyjne. Jony fragmentacyjne powstają zarówno

przez rozpad wiązań, jak i poprzez różnego rodzaju przegrupowania, związane z tworzeniem
nowych wiązań lub przeniesieniem atomu wodoru w obrębie cząsteczki
56. Identyfikacja MS
Spektrometria masowa jest nowoczesną techniką analityczną pozwalającą na dokładny pomiar masy. MS stała się konkurencyjna w
stosunku do degradacji Edmana jako metoda sekwencjonowania białek, umożliwiaidentyfikację modyfikacji posttranslacyjnych,
czy analizę związków endogennych występujących w bardzo niskich stężeniach. Technikami uzupełniającymi dla MS są
elektroforeza kapilarna (CZE) oraz wysokosprawna chromatografia cieczowa (HPLC). Stosowanie tych metod sprzężonych ze
spektrometrem dostarcza dodatkowych informacji i umożliwia identyfikację związków na jeszcze niższym poziomie stężeń.
MS znajduje również zastosowanie w dziedzinach takich jak ochrona środowiska, kontrola antydopingowa, farmakologia,
diagnostyka medyczna, biotechnologii czy ostatnio dynamicznie się rozwijającej proteomice czyli badaniach nad
identyfikacjąproteomu. (PROTEincomplement of the genOME - białkowa składowa
kodowana przez genom).
57. Pojęcie elucji gradientowej i czym się różni od elucji izokratycznej
polega na zmienianiu właściwości retencyjnych poprzez dodawanie w trakcie rozdzielania dodatkowych składników o wyższej sile
elucyjnej, do składnika o sile niskiej. Stosuje się ja do rozdzielenia substancji różniących się właściwościami retencyjnymi. Różnica
polega na zmianie siły eluencyjnej (wzroście) w elucji gradientowej natomiast izokratycznej jest ona stała, niezmienna.
58. Kiedy należy stosować elucję gradientową
Stosuje się ja do rozdzielenia substancji różniących się właściwościami retencyjnymi.
Wyszukiwarka
Podobne podstrony:
METODY BADANIA PRACY poziome 2
Metody badania antybiotykoopornoci
J Kossecki, Cele i metody badania przeszłości w różnych systemach sterowania społecznego
METODY BADANIA UKŁADU LIMFATYCZNEGO
Instrumentalne metody analizy
METODY BADANIA UKŁADU LIMFATYCZNEGO, Mieszanka Mareckiego
Interpretacja czynnikowa CPQ, psychologia, studia psychologia, semestr V, materiały gmail, Brachowic
podanie rodzica dziecka zameldowanego i zamieszkalego p, PWR, Zarządzanie, SEMESTR IV, Metody badani
Metody badania białek, Materiały - Biotechnologia
Testy psychologiczne - Metody badania klinicznego, PSYCHOLOGIA, Etyka zawodowa
13. Miareczkowanie amperometryczne, Technologia Chemiczna, Rok III, Semestr II, Instrumentalne metod
METODY BADANIA POWIERZCHNI KATALIZATORÓW TECHNIKI SKANINGOWE
Instrukcja 14 Badanie podstawowych kladow ste
instrumenty metodyczne
objawy zaburzeń percepcji słuchowej i metody badania percepcji słuchowej
1 Dydaktyka, pojęcia, metody badaniaid 9187 ppt
więcej podobnych podstron