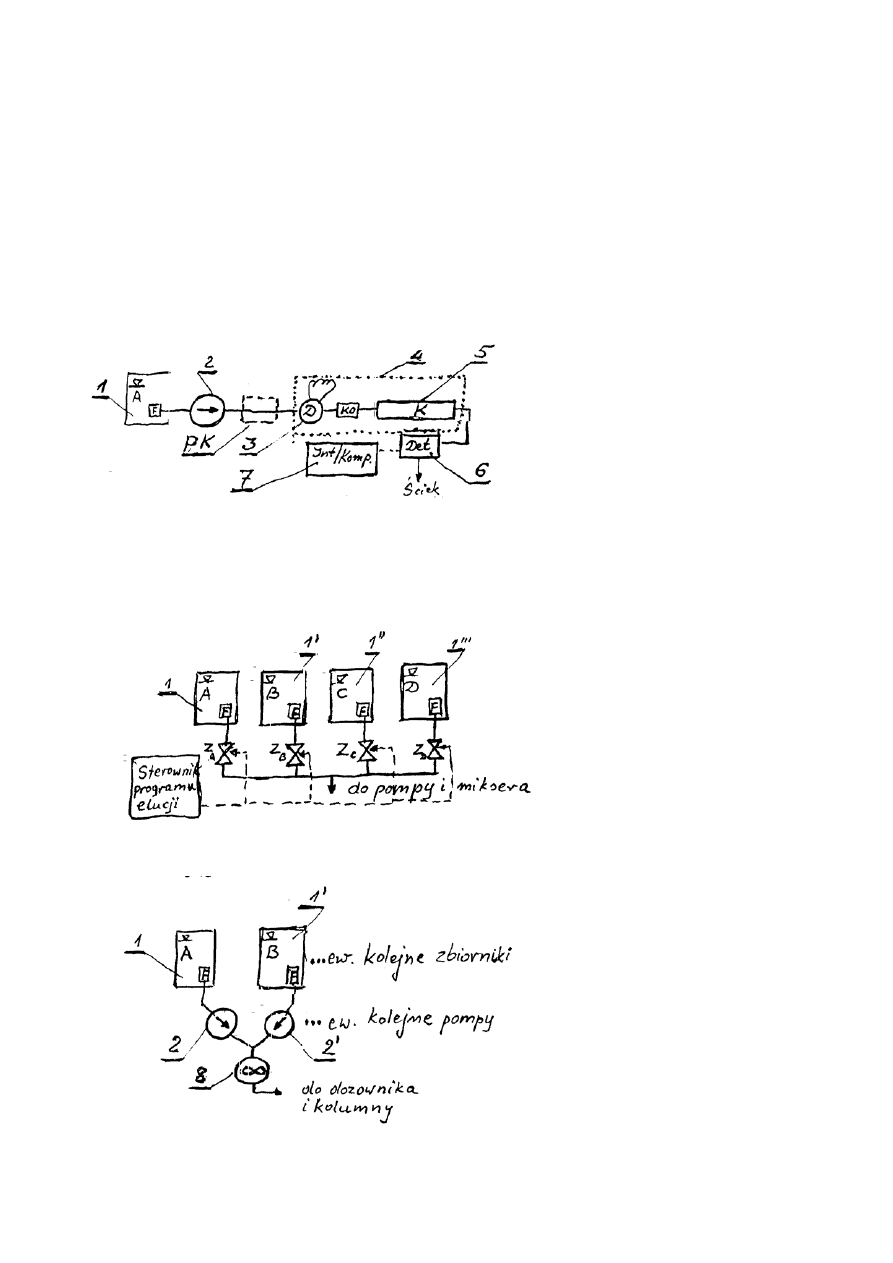
Aparatura HPLC
Informacje ogólne
Obecnie najważniejsze firmy produkujące chromatografy cieczowe oferują aparaty o
bardzo zbliżonej konstrukcji podstawowych modułów. Różnice polegają na jakości ma-
teriałów, jakości wykonania, zakresu możliwości funkcjonalnych oprogramowania i
wygody jego użytkowania, ale także niekiedy zakresu wyposażenia aparatu w detektory
różnego typu, a niekiedy także różne są własności użytkowe oferowanych detektorów.
Warto zwrócić uwagę, czy producent aparatury legitymuje się certyfikatem systemu
zapewnienia jakości w/g normy ISO 9001.
Najważniejsze moduły chromatografu cieczowego:
A) Aparat typu izokratycznego (stały skład eluenta)
B) Aparat typu gradientowego (programowanie składu zmian eluenta albo stały skład).
Różnica dotyczy tylko modułów 1 i 2. z rys. 1 (poniżej), tzn. system zasilania kolumny
eluentem. Systemy zasilania występują w aparatach różnego typu i różnej ceny w
dwóch opcjach:
a) z zastosowaniem zaworów proporcjonujących (2-4 zawory proporcjonujące) po
stronie ssącej pompy – opcja tańsza, lecz ma wady,
b) z zastosowaniem dwóch do czterech równolegle pracujących pomp o wzajemnie
zmienianej wydajności.
Znaczenie symboli na rys. 1- 3:
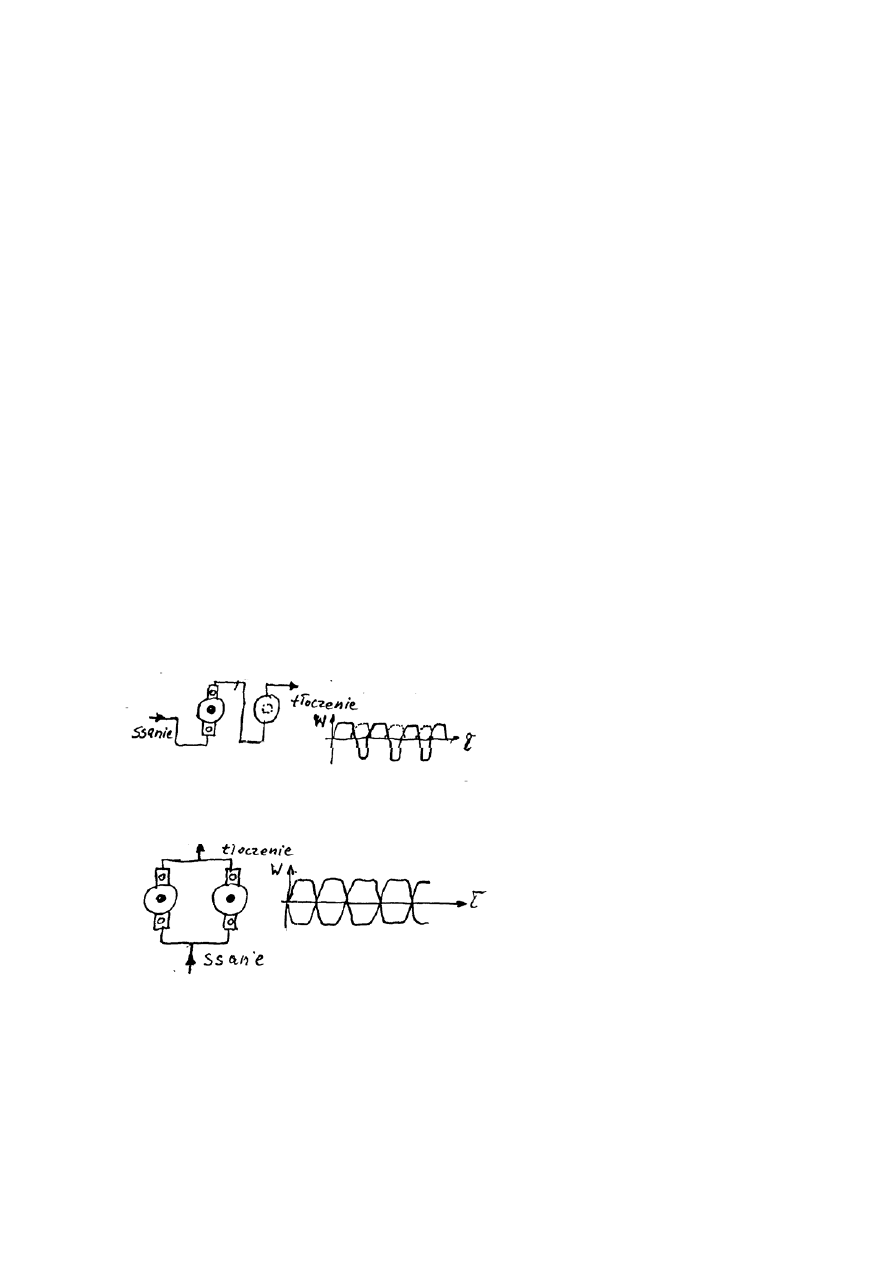
1, 1’, 1”, 1”’ – Butelka z eluentem lub składnikiem eluenta (A, B, C, D) albo specjalny
pojemnik eluenta z systemem odgazowania eluenta helem.
F
- filtr cieczy ze spieku porowatego.
2, 2’ - Pompa
na
preparatyw
ml
kolumnowa
ml
klasyczna
ml
min
/
100
1
min
/
0
,
1
01
,
0
"
"
min
/
10
1
,
0
−
−
−
⎪⎩
⎪
⎨
⎧
μ
3 (D) - zawór dozujący albo autosampler
Ko
- kolumna ochronna
(5) K - kolumna właściwa separacyjna
4
- komora termostatyczna
6
- detektor lub szeregowo połączone detektory
7
- integrator lub komputerowy system rejestracji i przetwarzania danych oraz ste-
rownik aparatu
8
- mikser (mieszalnik) statyczny lub dynamiczny
PK
- prekolumna; w celu nasycenia eluenta fazą stacjonarną, dla zabezpieczenia
właściwej kolumny przed „zdzieraniem” fazy stacjonarnej
Najważniejsze parametry i informacje na temat modułów składowych apa-
ratury chromatograficznej HPLC.
1. Pompy
Najczęściej tłokowe typu ssąco-tłoczącego jednogłowicowe, dwutłokowe: z jed-
nym tłokiem pracującycm jako pompa (zawory ssąco-tłoczące szafir, rubin) i z
drugim tłokiem o ruchu przesuniętym w fazie o 180
°, pracującym z wydajnością ½
(50%) wydajności tłoczenia tłoka czynnego – jako antypulsator. Wydajność naj-
częściej regulowana automatycznie w zakresie 0,1 – 10 ml/min., ciśnienie do
43Mpa. Bywają też stosowane pompy z dwiema lub trzema głowicami z zaworami
ssąco-tłoczącymi i tak napędzame są tłoki, aby wyeliminiwać pulsacje ciśnienia
(wydajności) cieczy.
Schemat ideowy pompy dwugłowicowej z jednym tłokiem czynnym i jednym tło-
kiem biernym
Schemat ideowy pompy dwugłowicowej z dwoma czynnymi tłokami
Inne urządzenia pompujące: pompy strzykawkowe, urządzenia bezpompowe zasi-
lane spręż. Ar lub N
2
(historyczne)
2. Zawór dozujący lub autosampler
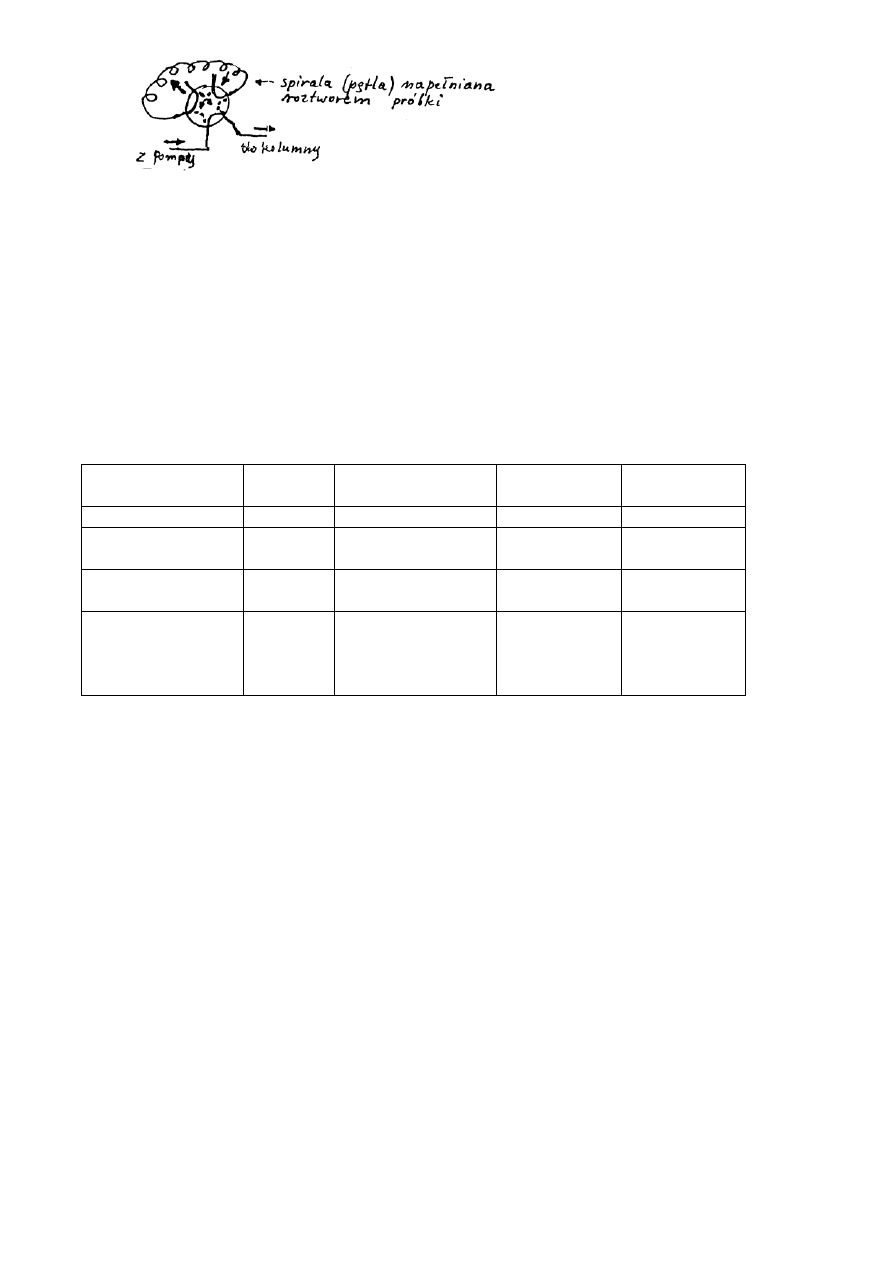
_____ - połączenie przewodów w czasie napełniania pętli („ładowanie”)
- - - - - - połączenie przewodów w czasie wprowadzania próby do kolumny
Często zawór Rheodyne Rh 7125.
Pojemności pętli:
"
"
5
20
klasyczne
l
l −
⎭
⎬
⎫
μ
μ
kolumny
l
μ
μ
−
1
⎭
⎬
⎫
l
l
μ
μ
1000
100
semipreparatywna chromatografia lub oznaczanie śladów substancji
3. Kolumny HPLC
mikro
-
kolumny
klasyczne kolumny
analityczne
semi-
preparatywne
średnice d
c
[mm]
0,5
4
8
długość L
c
[mm]
50
250
1000
najczęściej
250 mm
wielkość ziaren
sorbenta [um]
3; 5; 10
15; 25
40
liczba półek teore-
tycznych na 1 m
wypełnienia ko-
lumny
aaaaa
~1
tyś
4. Komora termostatyczna
Zakres temperatur
: pokojowa do 80
°C
lepiej
:
+4
°C do 80°C (biochromatografia)
Korzystne jest, aby termostatowany był również zawór dozujący. Trzeba też ter-
mostatować ciecz doprowadzaną do zaworu dozującego.
Uwagi użytkowe:
- warto co jakiś czas przepłukać przestrzeń za uszczelką w przypadku użycia bufora
jako eluenta.
- Pulsacyjna praca pompy może być spowodowana przylepianiem się zaworu ssące-
go do gniazda – oczyścić w acetonie, izopropanolu, wodzie, w detergencie w łaźni
ultradźwiękowej.
- Ważne, aby wszystkie filtry były drożne - przepłukiwać w przepływie w przeciw-
nym kierunku niż praca filtra, stosując wibracje ultradźwiękowe.
- Filtry mogą być zapychane przez bakterie – stosować NaN
3
lub inne substancje
bakteriobójcze jako dodatki do eluenta nietrującego dla bakterii.
Elucja gradientowa – praktyczne problemy stosowania
1. Problem „odpowietrzenia cieczy” – zapobieganie złej pracy pompy, wysokim szu-
mom detektora, utlenianiu katalitycznemu na sorbencie
Metody: He, ultradźwięki, próżnia, wrzenie, permeacja gazu przez membranę
2. Problem interakcji objętości podczas mieszania (urządzenia wysokociśnieniowe)
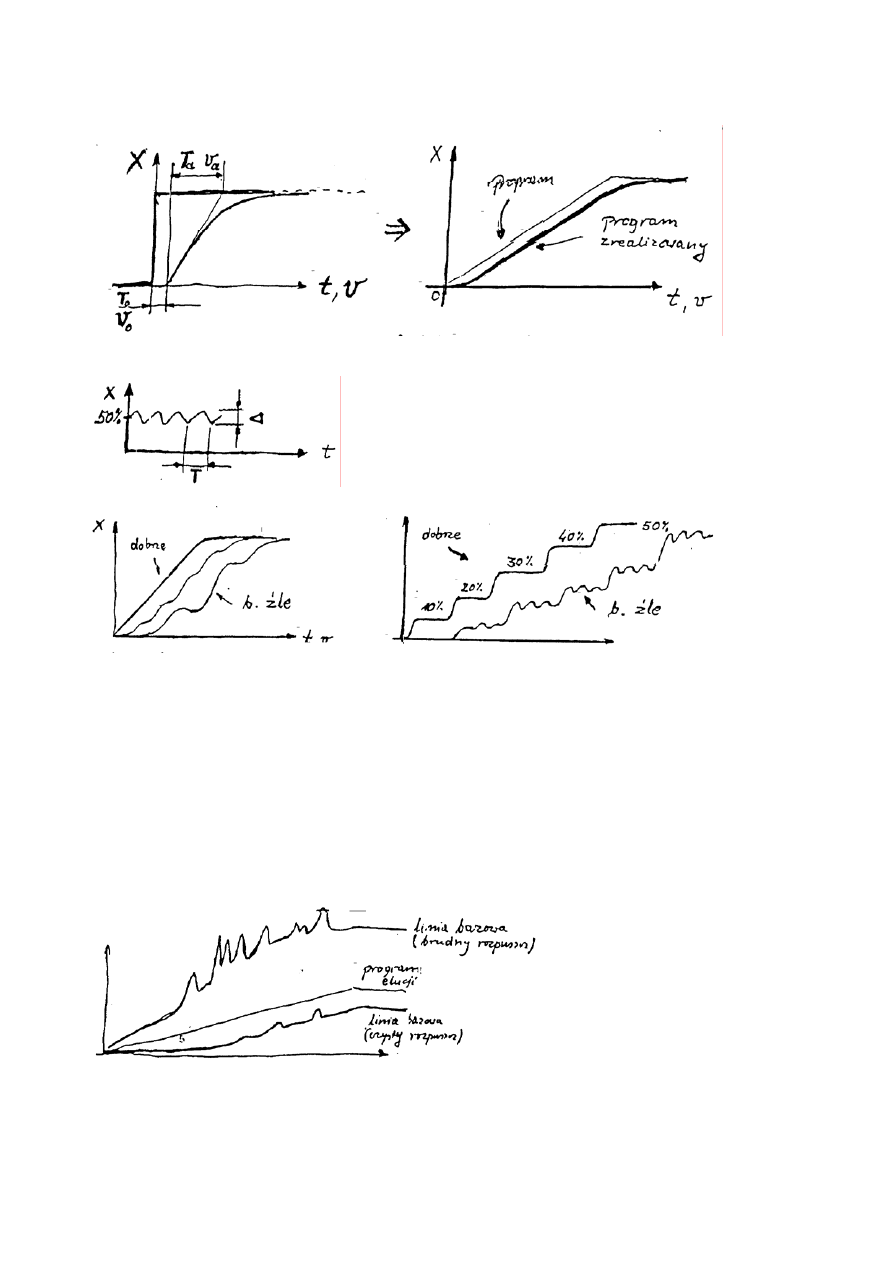
3. Zniekształcenia przebiegu programu w wyniku mieszania cieczy w elementach apa-
ratury (urządzenie z zaworami proporcjonującymi) i możliwość przeciwdziałania
skutkom; konieczne określenie zastępczej objętości mieszania i opóźnienia trans-
portowego aparatu
4. Kwestia optymalne objętości miksera
5. Problem interferencji pompa – zawory proporcjonujące
6. Zalecenia praktyczne
- Odłączyć kolumnę.
- określić eksperymentalnie
υ
o
,
υ
a
(test aparatury)
- sprawdzić realizację w praktyce linii prostej, gradientu skokowego oraz powta-
rzalność realizacji – w warunkach liniowej odpowiedzi detektora.
- zminimalizować zbędne objętości mieszania.
- możliwość stosowania „desynchronizacji pracy tłoków pompy i zaworów pro-
porcjonujących
-
7. Wpływ zanieczyszczeń eluenta na przebieg linii bazowej podczas „ślepej próby” i
możliwość odejmowania tego przebiegu (po przyłączeniu kolumny)
8. Metody oczyszczania rozpuszczalników: H
2
O, CH
3
OH, THF, Hexan – patrz Vogel
„Preparatyka organiczna”
Detekcja w LC
1. Zasady ogólne
- poziom szumów i pojęcie czułości detektora, np. 10
-3
AU/FS, tzn. poziom szumów
<10
-5
Jedn. Absorbancji: Cz=Syg./szum
- dryf detektora < 20% skali 100%, np.: 10
-4
AU/h
- zakres liniowości detektora, np. S
i
= k C
i
x
, x
∈ (0,98; 1,02)
- selektywność detektora s=(C
2
/C
1
) > 10
n
; im bardziej n>1 tym bardziej selektywny
detektor
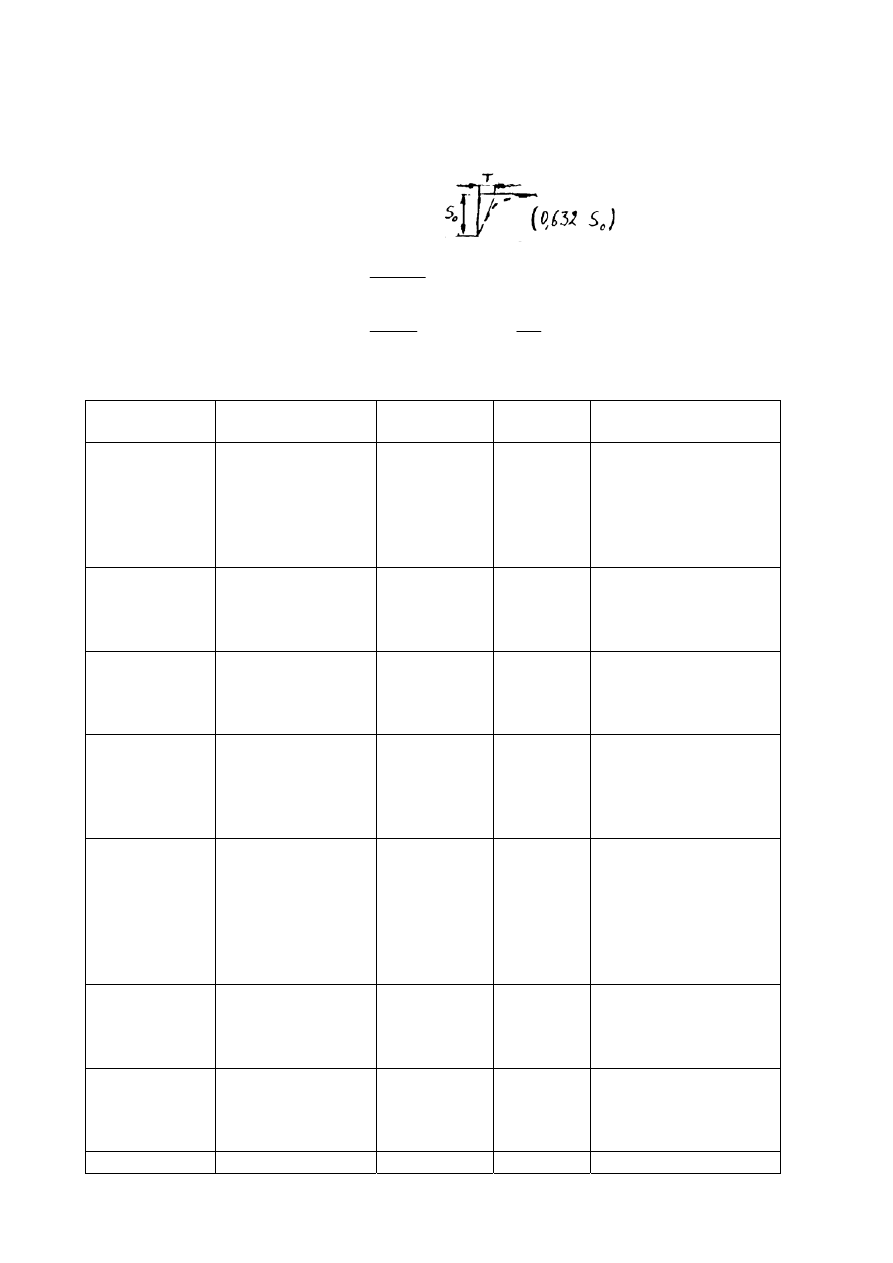
- stała czasowa detektora (T)
(
)
T
t
e
S
S
/
0
1
−
−
=
- detektor stężeniowy: S
s
= C
s
⋅C
i
⎥
⎦
⎤
⎢
⎣
⎡
ml
mg
mV
/
; ( S
s
= f(C
i
) );
- detektor masowy: S
m
= C
m
⋅W⋅C
i
⎥
⎦
⎤
⎢
⎣
⎡
S
mg
mV
/
; (
⎟
⎠
⎞
⎜
⎝
⎛
=
dt
dm
f
S
m
);
2. Zestawienie najważniejszych metod detekcji – cechy charakterystyczne
Typ Zakres
przydatności granica
ozna-
czalności
zakres li-
niowości
uwagi
UV-VIS
„klasyczny”
selektywny, substan-
cje aktywne w UV-
VIS (200-800nm)
lub odwrócona de-
tekcja UV, stęże-
niowy [jedn.absorb]
~0,2 ng/ml
~2*10
-5
Jedn.
Absorb.
10
3
- 10
4
Bardzo szeroki zakres
wykorzystania, najważ-
niejszy w HPLC
UV-VIS
„DAD”
190-800 nm
90-1024 fotoelemen-
tów
b.przydatny
~0,5 ng/ml
~5*10
-5
Jedn.Absor.
10
3
- 10
4
Szczególnie potrzebny
podczas doboru warun-
ków rozdzielania, infor-
macje jakościowe
Refraktometr
(„RI”)
„uniwersalny”
(GPC)
elucja gradientowa
wykluczona
~0,7
μg/ml
~2
⋅10
7
Jedn.
Refr.
3
⋅10
3
Typ
- refleksyjny,
- odchyleniowy,
- interferencyjny
Fluorescencyjny
(„FL”)
selektywny
dla substancjji flu-
oryzujących natural-
nie lub odpowied-
nich pochodnych
~0,8 pg/ml
10
3
- 10
4
PAH, katecholaminy,
leki, płyny fizjol., b.
czuły i łatwy w stoso-
waniu
Elektroche-
miczny
(„EC”)
- kulometryczny
(rzadko)
- amperometrycz-
ny (3 elektrody)
Selektywny tylko dla
łatwo redukowal-
nych lub utlen. sub.
~1 pg/ml
-
b. trudny w stosowaniu,
możliwość „zatrucia”
elektrod; b.czuły,
±1,2 V
Konduktome-
tryczny
jonoselektywny
do 0,2% róż-
nicy przewod-
nictwa sub-
stancji
10
5
detekcja jonów w chro-
matografie jonowym
Reakcyjna de-
tekcja
selektywny jak
fluore-
scencyjny lub
UV-VIS
bardzo szerokie zasto-
sowanie. Problem reak-
tora i rozmycia dodat-
kowego.
Radioaktywność selektywność -
do ok. 2000
konieczny scyntylator
ogromna rozkł/min.
Spektrom. ma-
sowy
uniwersalny i
b.czuły, jakościowy i
ilościowy
do 0,1 pg/ml
10
7
b. przydatny, kosztowny
Dyspersja masy i ocena kolumny
1. Dyspersja masy jako efekt niekorzystny lecz nieunikniony
2
'
2
'
2
1
2
1
1
4
1
N
k
k
Rs
⋅
⎟⎟
⎠
⎞
⎜⎜
⎝
⎛
+
⋅
⎟
⎠
⎞
⎜
⎝
⎛ −
=
α
α
2
H
2
L
N
c
=
2. Zjawiska powodujące dyspersję (najważniejsze)
a) wewnątrz kolumny ( w warstwie wypełnienia)
Profil tłokowy jako warunek podstawowy.
u
C
C
A
u
B
L
H
s
m
c
⋅
+
+
+
≅
≡
)
(
2
σ
równanie Van De-
emtera
↑
↑
↑
Dyfuzja
molekul.
Dyfuzja
wirowa
(mieszanie
strumieni)
Opory przenoszenia
masy w fazie ru-
chomej (m) i stacjo-
narnej (s)
b) „ poza kolumną” (poza warstwą wypełnienia)
- dozowanie:
τ
σ
+
=
12
2
2
i
v
V
;
2
2
2
v
L
S
σ
σ
=
c
;
- kapilary łączące
m
D
124
k
k
v
w
d
L
1
1
4
2
⋅
⋅
⋅
=
σ
- kuweta detektora i inne przestrzenie mieszające:
2
2
M
v
V
≅
σ
- „kieszenie” i inne „martwe” przestrzenie
m
v
D
w
x
2
2
2
⋅
≅
σ
-
c) niedostateczna dynamika układu detektor – rejestrator jako przyczyna „pozornej
dyspersji”
2
2
2
w
v
⋅
=
τ
σ
0
2
⇓
+
+
⇓
=
poz
ext
p
c
H
H
d
H
H
3. Oczekiwana sprawności kolumny (j. d
c
=4mm, w=1-1,5ml/min)
d
p
[
μm]
N
o
[1/m]
10
μm
15 – 20 tyś.
7
μm
30 – 35 tyś.
5
μm
50 – 60 tyś.
3
μm
100 – 120 tyś.
4. Warunki podczas testu kolumny
- odtwarzanie warunków testu kolumny
- test indywidualny
• proste substancje łatwo rozdzielające się
• liniowy zakres izotermy sorbcji
• mała objętość dozowania (V
i
< 20
μl ), d
c
=4 mm, L
c
> 100 mm
• dobra dynamika układu detektor - rejestratorr
5. Test pełny kolumny winien opisywać:
a) sprawność kolumny (N),
b) przepuszczalność kolumny (
Φ),
c) selektywność kolumny (k
’
,
α),
d) efekty pozakolumnowe (
)
2
ext
v
σ
e) asymetria pików (As
0,1
)
1. Sprawność kolumny – obliczanie na podstawie chromatogramu
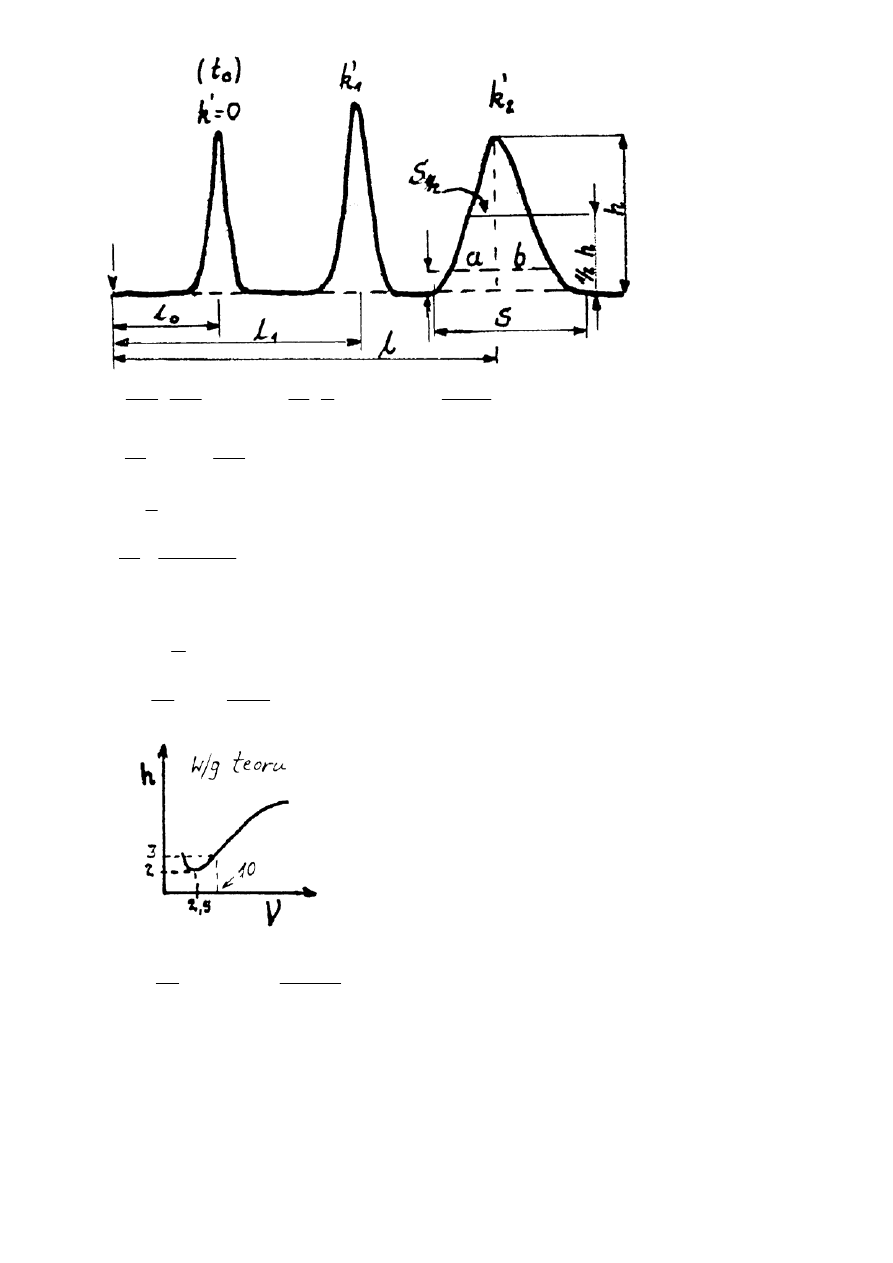
2
2
/
1
54
,
5
⎟
⎠
⎞
⎜
⎝
⎛
=
l
S
L
H
c
lub
2
16
⎟
⎠
⎞
⎜
⎝
⎛
=
l
S
L
H
c
lub
2
1
2
)
(M
L
H
c
μ
=
2
2
/
1
54
,
5
⎟⎟
⎠
⎞
⎜⎜
⎝
⎛
⋅
=
=
S
l
H
L
N
c
a
b
As
=
1
,
0
T
c
c
d
w
t
L
u
ε
⋅
⋅
Π
=
=
2
0
4
75
,
0
≅
T
ε
2. Sprawność teoretyczna
ν
ν
ν
⋅
+
⋅
+
=
C
A
B
h
teor
33
,
0
B=2, A=1, C=0,1
p
d
H
h
=
;
M
p
D
d
u
⋅
=
ν
3. Przepuszczalność
K
d
p
2
=
Φ
gdzie:
P
L
u
K
c
Δ
⋅
⋅
=
η
500 <
Φ < 2000 najczęściej 1000
4. Test dla efektów pozakolumnowych
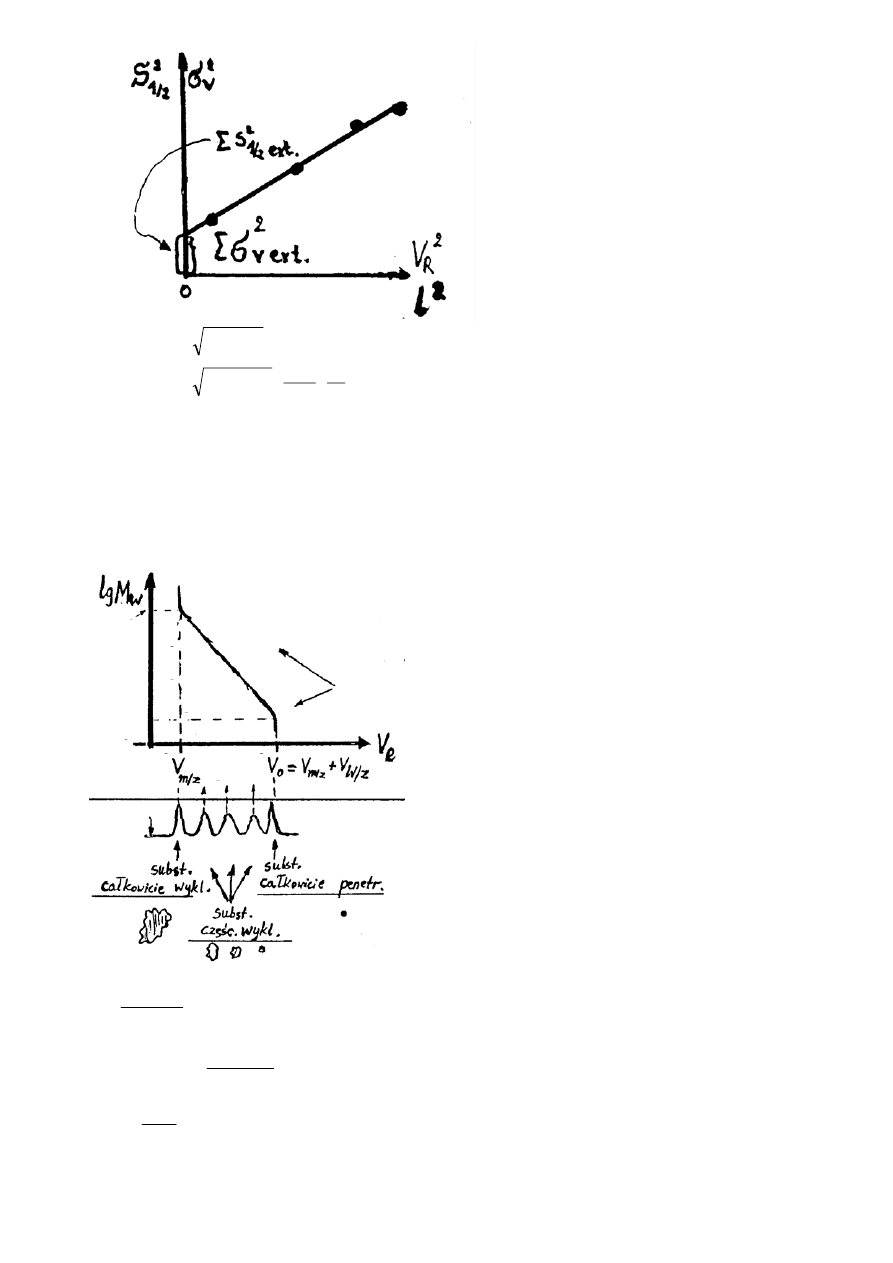
∑
≅
2
ext
v
ext
V
cak
σ
σ
t
ext
ext
V
w
S
cak
ν
σ
⋅
⋅
≅
∑
53
,
2
1
2
2
/
1
HPLC – Inne mechanizmy selektywności
SEC/GPC – wykluczanie molekularne – sito molekularne – chromatografia
żelowa
V
e
= V
m/z
+ K
d
(V
0
– V
m/z
)
w
e
M
d
dV
S
ln
=
- „pojemność” rozdzielcza kolumny GPC
np.:
3
2
/
1
1
1
⎥
⎥
⎦
⎤
⎢
⎢
⎣
⎡
⋅
−
≅
p
w
d
r
M
k
k
K
t
z
m
z
m
V
V
/
/
=
ε
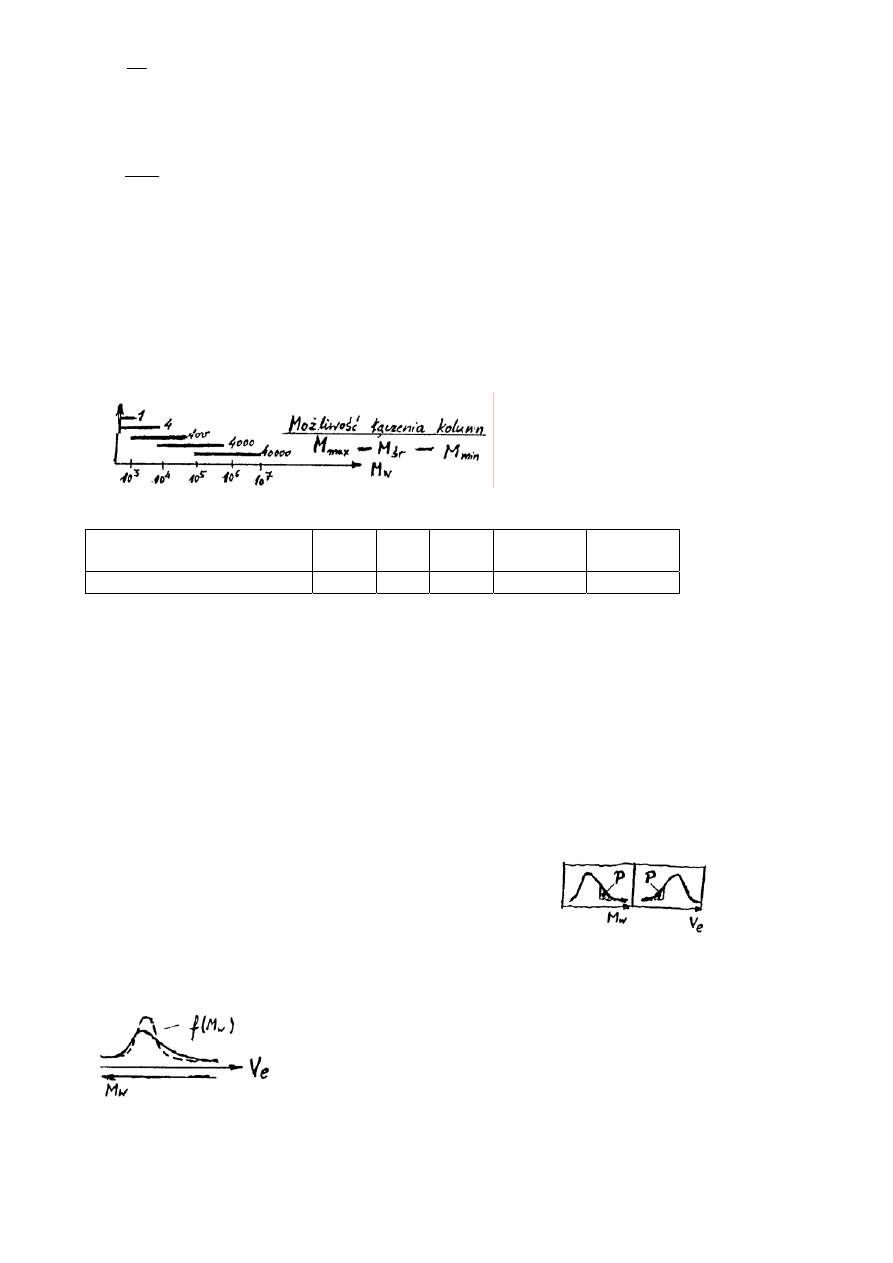
t
V
V
0
0
=
ε
4
/
2
c
c
t
L
d
V
⋅
⋅
Π
=
i
z
m
t
z
m
V
V
V
V
ε
⋅
−
+
=
)
(
/
/
0
t
z
w
i
V
V
/
=
ε
Rodzaje faz stacjonarnych (ogólnie):
- pęczniejące (wymagające przygotowania) – uwaga na
ΔP
- twarde
• liofilowe
• hydrofilowe – dezaktywacja np. glikol polietylenowy
Specyfika wykorzystania GPC – dla biopolimerów – białka, cukry, wirusy
Możliwość łączenia kolumn
Stosowane eluenty
Eluent toluen
THF
aceton
dwuchlo-
robenzen
DMFA
„Siła elucyjna” w/g Bartona
8,9
9,1
9,9
10,1
12,1
Problemy i zakłócenia, które trzeba brać pod uwagę:
- możliwość oddziaływań sorbcyjnych,
- pęcznienie i kurczenie się żelu pod wpływem różnych substancji
- ściśliwość żelu,
- b. wolna dyfuzja makromolekuł,
- wzajemne przeszkadzanie molekuł i zatykanie niektórych porów,
- polikondensacja makromolekół
- i inne
Ilościowe oznaczanie rozkładu mas cząsteczkowych – metodyka:
- kalibracja bezpośrednia – problem posiadania wzorców,
- kalibracja z wykorzystaniem polimerów polidyspersyjnych,
- kalibracja uniwersalna log( [
η]⋅M
w
) = f( V
e
)
Problem eliminacji wpływu dyspersji hydrodynamicznej podczas określania f(M
w
)
- dostępność specjalistycznego oprogramowania,
- wyspecjalizowane integratory
LLSD
Detekcja -: RI, UV, Lepkość,
Typy żeli handlowych:
Organiczne Nieorganiczne
(często modyfikowane)
• dekstranowe (Sephadex G10-G200, LM-20,
LH60, Sephacryl)
• agarozowe (Sepharosc, Biogel A, Ultragel A, …)
• akrylowe, metakrylowe (Biogel P, Ultragel ACA,
…)
• polistyrenowe (PS-DVB, TSK G1000-G7000,
PORAGEL 60-500, Lichrogel PS 1-4000)
• silikażele (Lichrospher SI 100 –4000,
TSK-GEL SW 2000-4000)
• szkła porowate – CPG 40-3000
Å
• alumina
Literatura
M.Berek, M.Dressler, M.Kubin, K.Marcinka „Chromatografia żelowa”, PWN, War-
szawa, 1989
Chromatografia jonowymienna IC
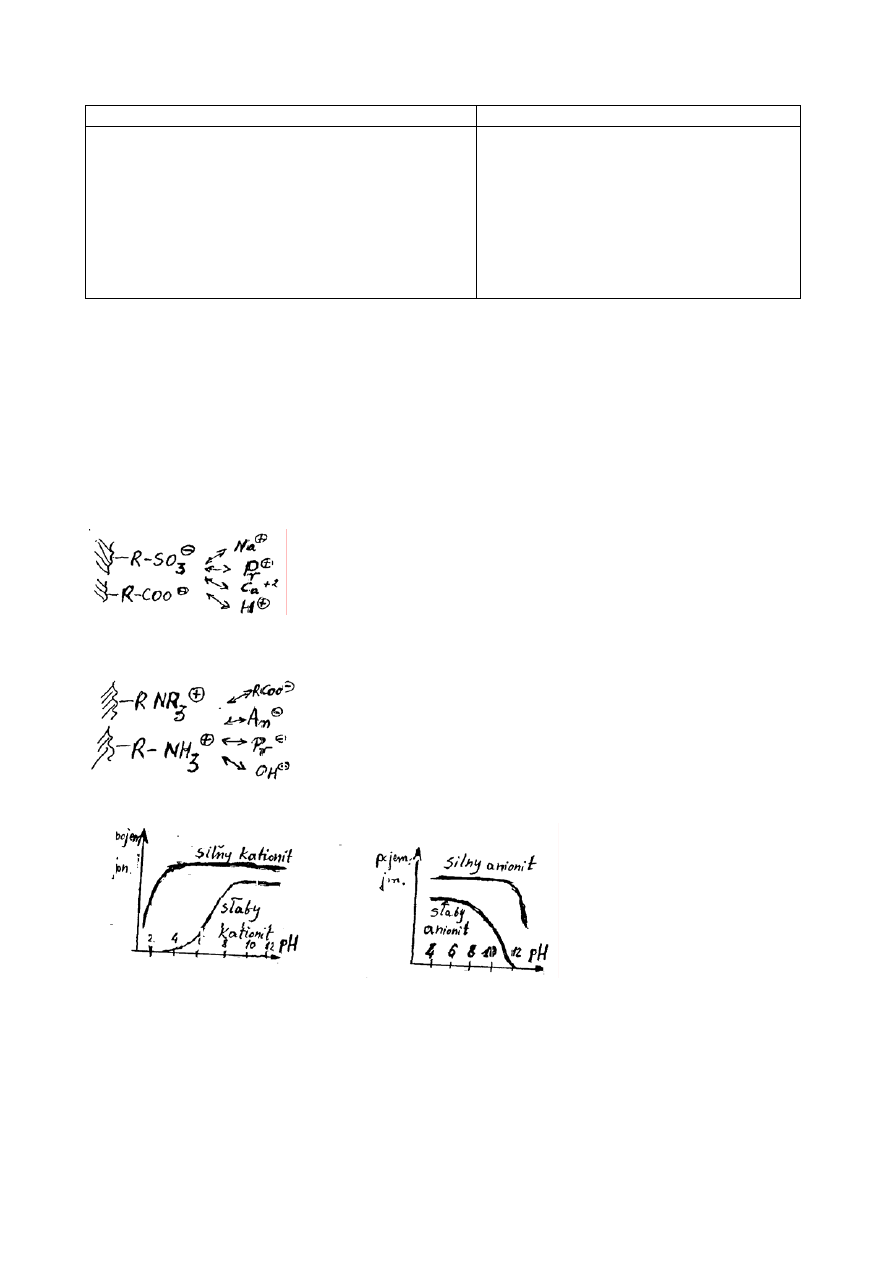
1. Typy wymieniaczy jonowych:
a) Kationit (kwas)
- mocny, np.:
- słaby, np.:
b) Anionit (zasada)
- mocny, np.:
- słaby, np.:
c) wymieniacze z dodatkowymi oddziaływaniami sorbcyjnymi
Szeregi w kierunku malejącej siły elucyjnej przeciwjonu:
• wymiana kationów: Ba
2+
, Pb
2+
, Sr
2+
, Ca
2+
, Ni
2+
, Cd
2+
, Cu
2+
, Co
2+
, Zn
2+
, Mg
2+
,
Mn
2+
, UO
2
2+
, Te
+
, Ag
+
, Cs
+
, Rb
+
, K
+
, NH
4
+
, H
+
, Li
+
• wymiana anionów: cytrynian, siarczan, szczawian, BO
3
-3
, NO
3
-
, PO
4
-3
, Br
-
,
SCN
-
, CN
-
, NO
2
-
, Cl
-
, HCOO
-
, CH
3
COO
-
, F
-
, OH
-
, ClO
-
.
2. Wpływa na retencję:
- typ wymieniacza jonowego,
- pH fazy ruchomej,
- siła jonowa fazy ruchomej,
- rodzaj przeciwjonu
3. Reguły ogólne
- wzrost siły jonowej przeciwjonu obniża V
R
,
- w przypadku wymiany kationu:
• wzrost pH obniża retencję; wyjątek: słabe wymieniacze kationów lepiej dyso-
cjujące przy wzroście pH
- w przypadku wymiany anionu
• spadek pH obniża retencję; wyjątek: słabe wymieniacze anionów lepiej dysocju-
jące przy niższym pH
- rodzaj przeciwjonu w/g następujących reguł podwyższa retencję próby:
• im wyższy ładunek przeciwjonu,
• im mniejsza średnica przeciwjonu,
• im łatwiej polaryzowalny przeciwjon
Zalecenia praktyczne
- wykorzystanie dla separacji najwyżej 5% pojemności,
- pH
≥ pK
s
+ 1,5 (zasady) lub pH
≤ pK
s
–1,5
- substancje
przeciwgrzybowe, przeciwgrzybiczne
: NaN
3
, kwas kapronowy, CCl
4
,
fenol,
- płukanie tłoka pompy !
Detekcja:
- przewodnictwo elektr. – konieczność stosowania supresji jonów !
- odwrotna detekcja UV
Alternatywy dla chromatografii jonowymiennej:
- chromatografia par jonowych C18, C8, C2
??
- cofanie dysocjacji słabych kwasów lub zasad
??
Przykłądy przeciwjonów
??
- kwas p-amini benzoesowy
Chromatografia powinowadztwa (affinity chromatography)
Etapy:
1) związanie liganda z nośnikiem,
2) sorbcja specyficzna cząsteczek P (powinowadztwo)
3) odszczepienie P
• czynnikiem o silniejszym powinowadztwie,
• zmiana pH,
• nadmiarowe stężenie jonów
4) reaktywacja sorbenta
Przykłady
♦ oznaczanie glukohemoglobiny HbGl – metoda kliniczna,
♦ przeciwciała z zastosowaniem Anty IgG – ligandów
♦ proteiny roślinne z zastosowaniem Concanavaliny A i inne.
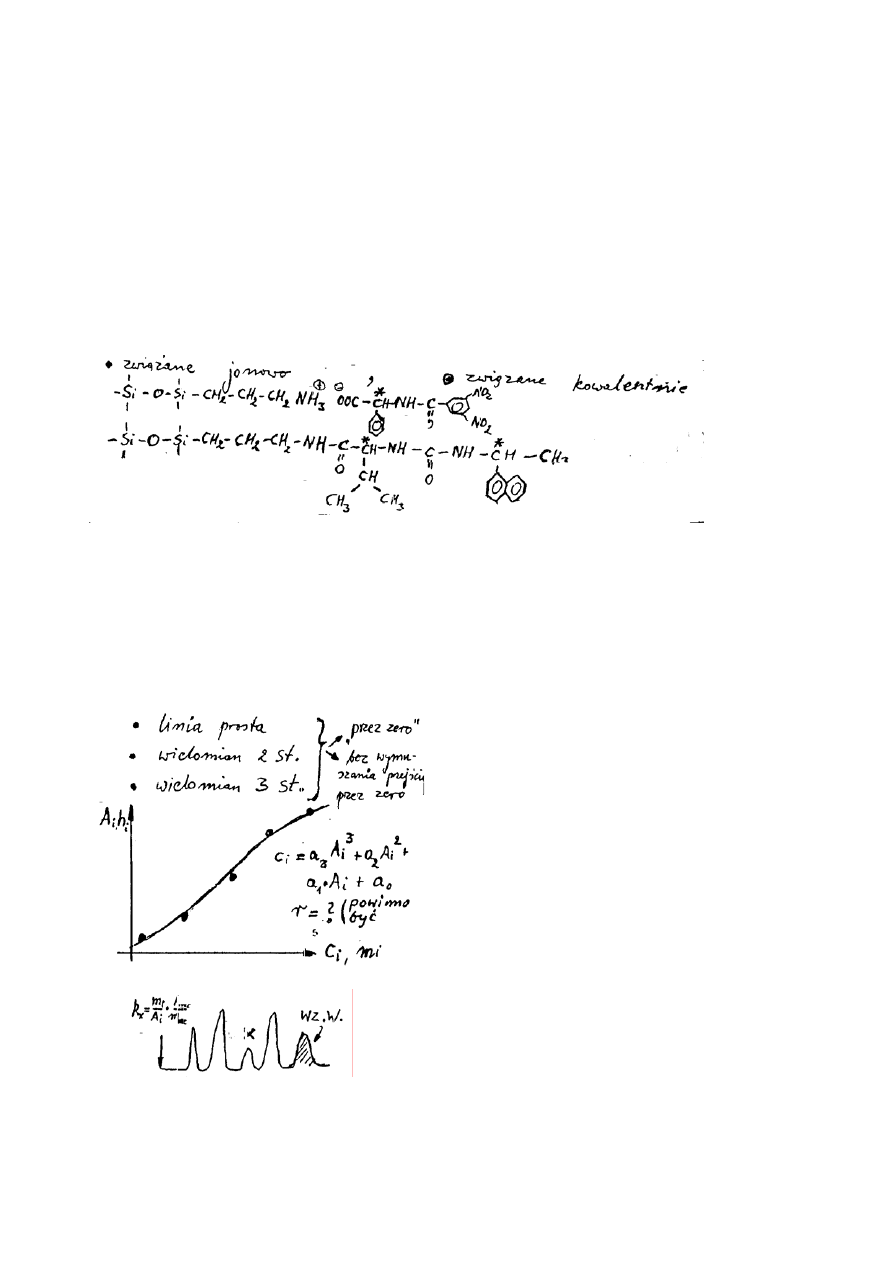
Rozdzielanie enancjomerów
Mechanizmy
- prekolumnowe lub wewnątrzkolumnowe tworzenie i separacja diastereoizomerów –
optycznie czynne dodatki do eluenta
- wykorzystanie oddziaływań
Π - Π, dipol – dipol, -OH
-…
H
+
, (optycznie czynne fazy
stacjonarne)
Typy faz stacjonarnych:
- kompleksy Cu
+2
– aminokwas; rozdzielanie: aminokwasy, dwupeptydy, 2 OH kwa-
sy karboksylowe
- polimery helicalne – trójacetyloceluloza, poli-triphenyl metyl metakrylan,
-
β-cyklodextryna związana na SiO
2
,
- fazy chiralne typu szczotkowego – tzw. fazy Pirkla, np. dinitrobenzoilofenyloglicy-
na, dinitrobenzoiloleucyna
związane jonowo
??
związane kowalencyjnie
??
-
Analiza ilościowa w chromatografii
???????
1. Metoda krzywej kalibracyjnej lub „standardu zewnętrznego”
2. Metoda wzorca wewnętrznego
Substancja (wzorzec) dodawana do próbki
– nie może występować w próbce,
– musi mieć podobne właściwości jak substancja analizowana
Zalety metody
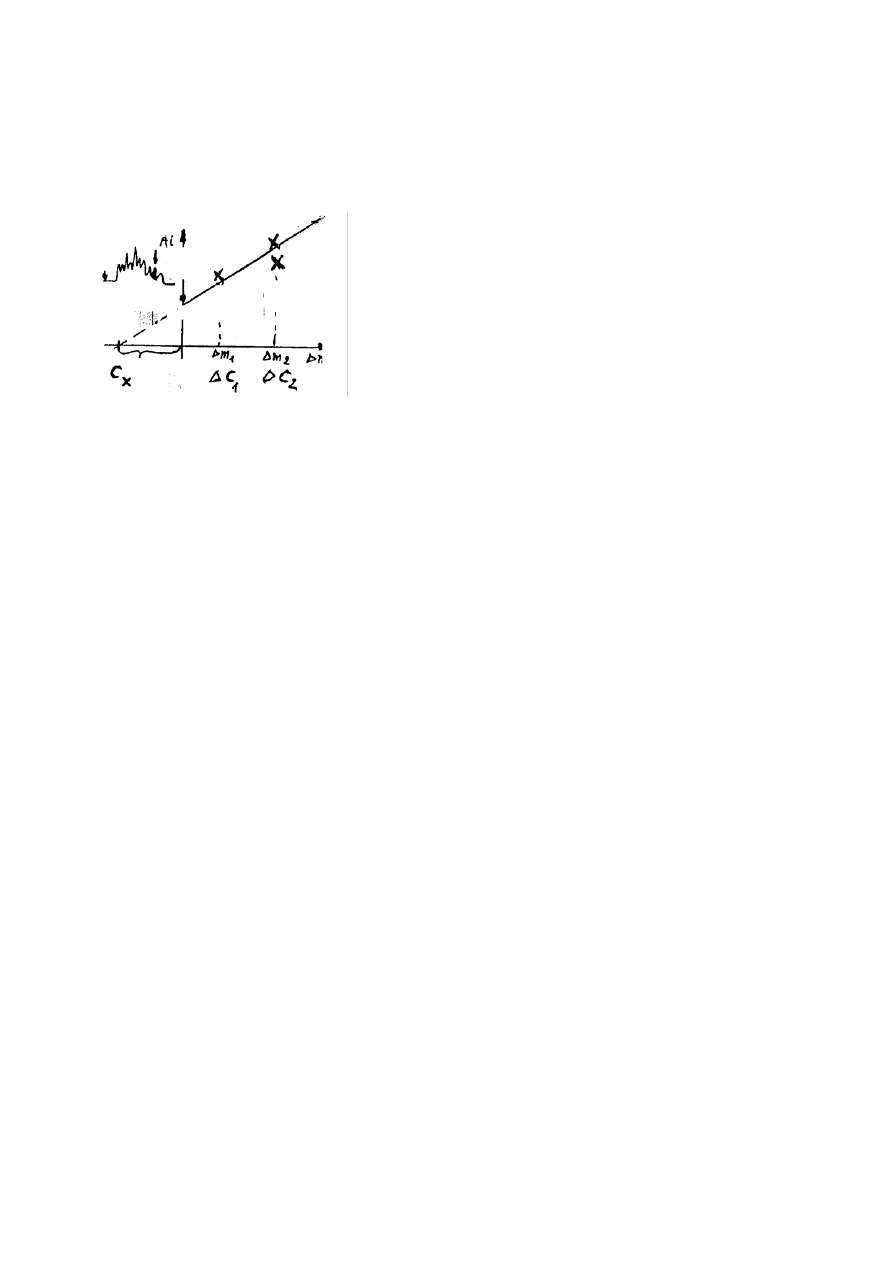
• nie jest konieczne precyzyjne dozowanie,
• możliwa złożona obróbka próby
Wady
• niekiedy trudno dobrać wzorzec,
• stężenie wzorca w próbce musi być dokładnie znane
3. Metoda dodatku wzorca („fortyfikacji”)
- jednokrotny dodatek – wymuszenie „zer – zero”
- dwukrotny dodatek – zróżnicowany - pewniejszy
Znaczenie symboli nie wyjaśnionych w tekście
Aparatura HPLC
d
c
-
średnica wewnętrzna kolumny;
L
c
-
długość wypełnienia w kolumnie;
w -
natężenie przepływu eluenta – objętościowo [ml/min];
d
p
-
wielkość ziaren wypełnienia;
X -
ułamek objętościowy składnika B w eluencie;
t -
czas;
υ -
objętość eluenta,
V
mix
-
objętość mieszania;
υ
0
-
opóźnienie transportowe [ml],
υ
a
-
zastępcza objętość mieszania w elementach chromatografu cieczowego,
Δ -
amplituda
wahań stężenia składnika B w eluencie,
C
z
-
czułość detektora;
S
i
, S
s
, S
m
, S
0
– sygnał detektora, odpowiednio: S
i
– w odpowiedzi na stężenie c
i
, S
s
–
detektora stężeniowego, S
m
– detektora masowego, S
0
– pełny sygnał odpowe-
idzi;
C
s
-
czułość detektora stężeniowego;
C
m
-
czułość detektora masowego;
m
- masa substancji wpływającej do detektora;
dm/dt - szybkość doprowadzania masy do detektora;
UV-VIS - detektor foroabsorbcjometryczny;
s -
selektywność detektora;
R
s ½
-
stopień rozdzielenia substancji 2/1 (pików 2/1);
α
- retencja względna,
k’
2
-
współczynnik retencji substancji II;
N
2
- liczba półek teoretycznych kolumny dla substancji 2;
d
p
-
średnica ziaren wypełnienia kolumny;
H -
wysokość wypełnienia równoważna półce teoretycznej (HETP);
H
c
- j.w., ale tylko dla samego wypełnienia (po odjęciu tzw. efektów pozakolum-
nowych);
u
- liniowa prędkość przepływu eluenta w kolumnie;
2
2
,
L
v
σ
σ
- liczone objętościowo (v) i liniowo (L) wariancje składowe rozkładu stężenia
wzdłuż kolumny,

L
- droga elucji;
C, B, A, C
m
, C
s
, - stałe dla konkretnej kolumny i równania;
Vi -
objętość dozowania;
S
c
- powierzchnia przekroju poprzecznego wypełnienia kolumny;
L
k
-
długość kapilary;
d
k
-
średnica kapilary;
w -
natężenie przepływu eluenta;
D
m
-
współczynnik dyfuzji molekularnej w eluencie
V
m
-
objętość mieszania;
X
- wymiar „znamienny” kolory;
τ -
stała czasowa detektora (0,632 amplitudy So osiągane jest po czasie
τ)
H
ext
-
wysokość równoważna półce teoretycznej od efektów pozakolumnowego
rozmycia;
H
prz
-
wysokość równoważna półce teoretycznej od złej dynamiki detektora;
N
0
- liczba półek teoretycznych kolumny w przeliczeniu na 1 m długości wypełnie-
nia;
d
c
-
średnica wypełnienia kolumny;
Φ -
przepuszczalność właściwa kolumny,
A
s0,1
-
współczynnik asymetrii piku w 0,1 wysokości;
S
1/2
-
szerokość piku w ½ wysokości;
S -
szerokość piku przy podstawie;
l -
odległość mierzona na chromatogramie (w warunkach elucji izokratycznej i
stałego natężenie przepływu eluenta);
μ
2
- drugi moment centralny piku jako krzywej rozkładu;
M
1
- pierwszy moment zwykły piku jako krzywej rozkładu (położenie środka cięż-
kości);
to
- czas retencji substancji niesorbowanej;
h
- tzw. zredukowana wysokość półki teoretycznej;
h
teor
-
wartość h otrzymana z równania Knoxa;
ν
- zredukowana prędkość przepływu eluenta;
η -
lepkość dynamiczna eluenta;
K -
przepuszczalność kolumny;
H
ext
-
udział efektów pozakolumnowych w H;
∑
2
ext
v
σ
- objętościowo liczona wariancja rozmycia pozakolumnowego stref substancji;
∑
2
2
/
1
ext
S
- udział rozmycia pozakolumnowgo w wartości kwadratu szerokości piku w
połowie wysokości;
F
c
- pole przekroju poprzecznego wypełnienia kolumny;
υ
t
-
szybkość przesuwu papieru rejestratora;
L
c
-
długość wypełnienia w kolumnie;
a, b
- część szerokości piku w 0,1 wysokości (patrz szkic);
V
R
-
objętośći retencji
HPLC Inne mechanizmy selektywności
Chromatografia żelowa (GPC/SEC)
V
e
-
objętość elucji (do maksimum albo do środka ciężkości piku
V
m/2
-
objętość międzyziarnowa wypełnienia;
V
0
-
objętość ”martwa” kolumny (objętość przebicia przez substancję o niskiej ma-
sie cząsteczkowej – wnikającej we wszystkie pory ziaren wypełnienia);
M
w
-
średnia masa cząsteczkowa polimeru (wagowo);
k, k
1
, A, B – stałe;

r
p
-
średni promień porów wypełnienia w ziarnach;
ε
0
-
porowatość całkowita kolumny;
ε
m/z
-
porowatość międzyziarnowa wypełnienia;
ε
i
-
porowatość wewnętrzna ziaren sorbenta;
V
w/z
-
objętość porów wewnątrz ziaren sorbenta;
V
t
-
objętość „pustej” (niewypełnionej) kolumny;
L
c
-
długość wypełnienia kolumny;
d
c
-
średnica wypełnienia w kolumnie – średnica kolumny;
M
min
, M
max
– odpowiednio: minimalna i maksymalna masa cząsteczkowa substancji
podlegających efektowi sita molekularnego w danej kolumnie;
M
śr
-
średnia masa cząsteczkowa polimeru;
η -
lepkość;
[
η] -
lepkość graniczna (roztworu nieskończenie rozcieńczonego polimeru);
P
- pole pod częścią krzywej rozkładu stężenia (piku);
LLSD - detektor laserowy światła rozproszonego;
THF -
tetrahudrofuran;
DMFA - di-metyloformamid;
Chromatografia jonowymienna
P
r
-
cząsteczki substancji rozdzielanych albo ich stężenie;
B -
przeciwjon;
pKs - pK kwasu lub zasady
Analiza ilościowa w chromatografii
A
i
- powierzchnia piku „i”;
h
i
-
wysokość piku „i”;
C
i
-
stężenie substancji „i”;
m
i
- masa substancji „i”;
A
wz
- powierzchnia piku wzorca wewnętrznego;
m
wz
- masa wzorca;
Δm
1/2
- masa dodatku wzorca;
m
x
- masa substancji oznaczanej;
C
x
-
stężenie substancji oznaczanej;
a
3
, a
2
, a
1
, a
0
– współczynniki wielomianu;
Document Outline
Wyszukiwarka
Podobne podstrony:
Aparatura sciaga mini
INSTRUKCJA OBSŁUGI APARAT KODAK EASYSHARE Z1285 PL
Aparat Golgiego, Szkoła, przydatne w szkole
zagadnienia - Karkowska, II rok, Aparatura
wyplyw cieczy ze zbiornika, Technologia chemiczna, 5 semestr, Podstawowe procesy przemysłu chemiczne
podbielska,elektroniczna aparatura medyczna, Elektrokardiograf Charakterystyka bloków
Aparat ustaleniowy
podbielska,elektroniczna aparatura medyczna, elektrokardiografia holterowska
Budowa i rozwój aparatu ruchu
Fg 4 Aparat trójosiowego ściskania
więcej podobnych podstron