
Development of a Polymeric Patch Impregnated with Naproxen
as a Model of Transdermal Sustained Release System
ANNA ARGEM´ı,
1
JEFFREY L. ELLIS,
2
JAVIER SAURINA,
1
DAVID L. TOMASKO
2
1
Department of Analytical Chemistry, University of Barcelona, Mart´ı i Franqu`es 1-11, 08028 Barcelona, Spain
2
William G. Lowrie, Department of Chemical and Biomolecular Engineering, The Ohio State University, Columbus, Ohio 43210
Received 29 April 2010; revised 6 July 2010; accepted 24 August 2010
Published online 16 September 2010 in Wiley Online Library (wileyonlinelibrary.com). DOI 10.1002/jps.22346
ABSTRACT: This paper describes the preparation and characterization of transdermal patches
impregnated with naproxen. A mixture of ethylene vinyl acetate and Eudragit
R
E100 (80:20,
w/w) is used as a polymeric matrix to obtain a thin membrane to be impregnated. Drug impreg-
nation is carried out under pressurized
CO
2
as a processing medium according to a two-step
procedure. The patch is first soaked at 1000 psi and 22
◦
C for 2 h, and then foamed as a result of
the rapid release of
CO
2
pressure in order to increase the porosity of the surface. Subsequently,
the naproxen solution is placed in contact with the membrane and then soaked in
CO
2
at 450
psi and 37
◦
C for 2.5 h to enhance the mass transfer of drug into the polymer matrix. The char-
acterization of the resulting samples by liquid chromatography, microscopy, and calorimetry
provides information on naproxen content and distribution. Patches synthesized in this way are
loaded with about 1% naproxen. The drug release and diffusion process through a membrane
have been studied chromatographically using a Franz diffusion cell. Results have shown that
a sustained delivery for more than 24 h is obtained. © 2010 Wiley-Liss, Inc. and the American
Pharmacists Association J Pharm Sci 100:992–1000, 2011
Keywords:
controlled delivery; transdermal drug delivery; processing; in vitro models; poly-
meric drug carrier; supercritical fluids
INTRODUCTION
An important pharmaceutical research field is fo-
cused on the development of new pharmaceutical
forms with improved bioavailability and stable dosage
by using clean technologies.
1–4
Processes carried out
under dense (pressurized) or supercritical carbon
dioxide result in an attractive alternative to those
involving traditional solvents, especially to overcome
the problems associated with toxicity and resid-
ual impurities.
5
Recent technological applications of
pressurized and supercritical fluids comprise extrac-
tion of natural products; removal of contaminants,
6,7
protein, and peptide fractionation
8
; and prepara-
tive supercritical fluid chromatography.
9,10
However,
apart from such type of industrial applications,
the potentiality of
CO
2
as a processing medium
in pharmaceutical particle engineering cannot be
Correspondence to: Anna Argem´ı (Telephone:
+34-934-034-445;
Fax:
+34-934-021-233; E-mail: annaargemi@ub.edu)
Journal of Pharmaceutical Sciences, Vol. 100, 992–1000 (2011)
© 2010 Wiley-Liss, Inc. and the American Pharmacists Association
underestimated
11,12
and following three main work-
ing topics are being investigated: (a) the preparation
of active compound powders with improved or mod-
ified therapeutic action, (b) the production of poly-
mers to be used as a matrix for drug impregnation,
and (c) the preparation of drug delivery systems with
enhanced bioavailability or sustained release char-
acteristics. Hence, multiple pharmaceutical applica-
tions have been reported including the preparation of
patches, sponges,
13
and catheters
14
with potential use
in tissue engineering and drug delivery.
Advantages gained from the use of supercritical
or dense
CO
2
include the excellent uniformity in the
distribution of the solute into the matrix, the reduc-
tion of process steps, and the simplicity of solvent
removal. As nonporous polymeric matrices exposed
to these fluids swell, the solute penetration through
the matrix is thus enhanced. In addition, the drug
entrapment can be carried out in a quick and easy
one-step procedure. As an example, Kazarian and
Martirosyan
15
described the impregnation of ibupro-
fen in polyvinylpyrrolidone (PVP), resulting in the
992
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 100, NO. 3, MARCH 2011

NAPROXEN-POLYMERIC PATCH FOR TRANSDERMAL SUSTAINED RELEASE SYSTEM
993
formation of a molecular dispersion of drug into the
matrix.
Although a wide variety of polymers have been
used in the past 40 years as drug carriers, recent
trends rely on water-soluble matrices such as PVP
or its copolymer with vinylacetate (PVP-VA 64).
16
Other polymers such as cellulose derivatives [e.g.,
ethylcellulose (EC), methylcellulose]
17,18
ethylene
vinyl acetate (EVA),
19,20
and pH-dependent polymers
such as Eudragit
R
E100 (Evonik Degussa, Essen,
Germany), (polymethacrylate copolymer)
18
are being
increasingly used.
This study is focused on the preparation and char-
acterization of a transdermal patch as a model sys-
tem of sustained released using pressurized
CO
2
as
a processing medium. Naproxen is the drug chosen
here for this development. Naproxen is a member of
the 2-arylpropionic acid family of nonsteroidal anti-
inflammatory drugs commonly used for the reduction
of mild-to-moderate pain, fever, inflammation, and
stiffness.
21
The US Food and Drug Administration
approved the use of naproxen sodium as an over the
counter drug in 1994. Analytical techniques, includ-
ing differential scanning calorimetry (DSC), confocal
fluorescence microscopy, and high-performance liq-
uid chromatography (HPLC), have been utilized for a
more rigorous characterization of naproxen samples.
MATERIALS AND METHODS
Materials
Sodium
hydrogenphosphate,
sodium
dihydro-
genphosphate,
formic
acid,
rhodamine
(5,6-
carboxytetramethylrhodamine),
and
naproxen
(99%) were purchased from Sigma–Aldrich (St.
Louis, Missouri). Methanol and methylene chloride
(HPLC grade, Merck, Darmstadt, Germany) were
used as solvents. Carbon dioxide (
CO
2
, 99.99 mol%
purity) was supplied by Praxair (Columbus, Ohio).
Polymers used were EC 20 cps from Keyser & Mackay
(Brussels, Belgium), PVP-VA 64 (molecular weight
=
45,000–70,000 g mol
−1
) from BASF (Ludwigshafen,
Germany), Eudragit
R
E100 (acrylic polymer, molecu-
lar weight
= 150,000 g mol
−1
) from Evonik Degussa
(Essen, Germany), and EVA (70 wt% of vinyl acetate)
from Sigma. Ultrapure water (Millipore, Milford,
Massachusetts) was used for the preparation of
aqueous solutions.
Analytical Instrumentation
The chromatographic system consists of an HPLC Ag-
ilent 1100 Series instrument equipped with a G1311A
quaternary pump, a G1379A degasser, a G1329B
standard autosampler (1200 Series), a G1315B diode-
array detector furnished with a 13-mL flow-cell,
a G1321A fluorescence detector, and an Agilent
Chem Station for data acquisition and analysis (Rev.
A 10.12), all of them from Agilent Technologies
(Waldbronn, Germany). The analytical column was a
reverse phase C
18
(Synergi Hydro-RP, Phenomenex,
150
× 4.6 mm
2
d.i., 4 µm particle size). Naproxen was
eluted isocratically with 10 mM of formic acid/formate
aqueous solution (pH 3.2)
+ MeOH (20/80, v/v) as a
mobile phase. The flow rate was maintained at 1 mL
× min
−1
and the injection volume was 20 µL. Ultra-
violet (UV) spectrophotometric detection was carried
out at 270 nm. Fluorescence detection was carried out
at 270 and 357 nm as excitation and emission wave-
lengths, respectively. A magnetic stirrer IKA
R
RCT
basic (Staufen, Germany) was used for controlling the
release conditions.
Standard solutions for calibration were prepared in
methanol in the concentration range from 5.2
× 10
−7
to 3.9
× 10
−5
M. For UV spectrophotometric detection,
a good linearity in the studied range was found with a
regression coefficient r
2
= 0.9989. Detection limit es-
timated for a signal-to-noise ratio of three was 1.8
×
10
−7
M. Repeatability expressed as relative standard
deviation (RSD in %) for the peak area was calcu-
lated from eight replicates at a concentration of 2.5
× 10
−6
M and was 2.1%. For fluorescence detection,
the linearity was found with a regression coefficient
r
2
= 0.9988. Detection limit was 1.1 × 10
−7
M and
repeatability was 1.5%. The chromatographic method
was used in both the determination of the impreg-
nation percentage and in the monitoring of the drug
release.
A differential scanning calorimeter (DSC-822e/400,
Mettler Toledo, Greifensee, Switzerland) was used to
determine melting and glass transition temperatures.
Thermograms were obtained at a heating rate of 10
◦
C
× min
−1
from 30
◦
C to 250
◦
C under a N
2
purge of
50 mL
× min
−1
.
A confocal microscope Leica TCS SPII (Leica Mi-
crosystems, Wetzlar, Germany) operating in both re-
flectance and fluorescence modes was used. Excita-
tion was at 351 and 364 nm using UV lasers and
reflectance, and emission intensities were recorded
in the range of 400—800 nm. The objective used
was a 10
× 0.3 N.A. HCPL FLUOTAR lens (Leica
Microsystems). Images were processed using the
ImageJ (NIH Image; www.rsb.info.nih.gov/ij) and
Photoshop 7.0 software (Adobe Corp., San Jose,
California). Transversal sections were taken every
2.4 µm.
Preparation of Working Solutions and Naproxen
Impregnated Patches
The working solution of rhodamine for preliminary
impregnation studies consisted of 1.5 mg dissolved in
100 mL of sodium phosphate buffer solution (PBS)
DOI 10.1002/jps
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 100, NO. 3, MARCH 2011

994
ARGEM´I ET AL.
at pH 6.2. Naproxen working solution consisted of
100 mg in 25 mL of PBS at pH 7.4.
Patch Synthesis.
Ten grams of EVA and 2.5 g of Eudragit
R
E100
were treated with 30 mL of CH
2
Cl
2
. The mixture
was left in an ultrasonic water bath (Unisonics ul-
trasonic cleaner FXP8) for 90 min (until polymers
were completely dissolved). This water bath sonica-
tor has an ultrasonic power of 50 W and provides a
frequency of 40 kHz. The resulting solution was cast
on a microporous Teflon film placed on a glass plate.
The Teflon film was used as a release agent because
polymer sticks on the glass. A thin layer of polymer
solution was obtained with the aid of an adjustable
thin film applicator (GARDCO, Paul N. Gardner Co.,
Pompano Beach, Florida). The solvent was allowed
to evaporate, first at ambient conditions, and then in
the vacuum oven (P
= 30 mmHg) at room temper-
ature overnight. The thin polymer layer was peeled
off and thickness was measured using an electronic
gauge Mitutoyo (Model 543-252B, Mitutoyo America
Corp., Aurora, Illinois) with an accuracy of
±0.5 µm.
Membranes with a thickness ranging from 231.5 to
242.4 µm were obtained. Finally, patches were cut in
square shape (4
× 4 cm
2
size, approx.) and were stored
at ambient conditions until further use.
Patch Impregnation Using
CO2
.
The procedure for impregnation of patches with
naproxen consisted of two steps as follows:
(1) Patch foaming process: First, the synthesized
patches (see patch synthesis section) were
treated with pressurized
CO
2
to induce the for-
mation of pores in the material. The polymeric
patch (placed on a piece of Teflon film) was lo-
cated inside a stainless steel high-pressure ves-
sel (Pressure Products Industries, Inc., Warmin-
ster, Pennsylvania). Another piece of Teflon film
and a piece of stainless steel were placed on
top of the patch. The system was pressurized
to 1000 psi and held at constant pressure for
2 h with an ISCO Syringe pump 500D (ISCO,
Lincoln, Nebraska). The temperature was main-
tained at 22
◦
C. The foaming process was initi-
ated with a rapid
CO
2
depressurization in 4 s
and the porous patch obtained was ready to be
impregnated with the drug.
(2) Impregnation process: 1000 µL of solution of
model (rhodamine, 1.5 mg in 100 mL PBS at pH
6.2) and active drug (naproxen, 0.1 g in 25 mL
PBS at pH 7.4) were dipped onto the porous ma-
trix surface of the patch. The high-pressure ves-
sel was then sealed airtight and heated with
a Peltier system up to the desired experimen-
tal temperature. A minimum of 15 min was
allowed to ensure thermal equilibrium. Then,
the 500D syringe pump was used to pressurize
the vessel with
CO
2
until the working pressure
was reached. The temperature and pressure
were held constant during this period. The sys-
tem was then depressurized over nearly 30 min
by slowly opening the purge valve. Finally, the
patch was dried at room temperature for 8 days
at least and stored in a sealed plastic bag until
future characterizations. The experimental pro-
cessing window for impregnation was explored
by experimental design. For this purpose, a full
factorial design with three factors (temperature,
pressure, and time) at two levels was utilized,
which corresponded to eight experiments.
Characterization Studies
Determination of the Percentage Impregnation.
The amount of naproxen entrapped in the polymeric
matrix was determined by HPLC. For this purpose,
about 30–40 mg of sample were dissolved in 10 mL of
CH
2
Cl
2
by ultrasonication for 1 h. Subsequently, the
solvent was evaporated under nitrogen current and
the dry residue was redissolved in 25 mL of methanol.
Twenty microliter of the resulting solution, previously
filtered through a 0.45 µm pore-size membrane, was
injected into the chromatograph.
Drug Release Studies.
The study of naproxen diffusion from the patches was
carried out using a Franz glass cell with 3.14 cm
2
of
diffusion area and a receptor chamber of 12 mL vol-
ume. Naproxen is soluble in the receptor medium at
a concentration of 5.2 mg mL
−1
. A sample amount of
25–60 mg was distributed on a synthetic Nylon mem-
brane of 0.45 µm pore size (Whatman Int., Maidstone,
Kent, UK) and placed in the donor chamber. Imme-
diately after that, the top of the cell cap was covered
with Parafilm
R
(Chicago, Illinois) to minimize evap-
oration during the test. The receptor chamber was
filled with 100 mM of PBS at pH 7.4 and the tem-
perature was kept at 32
± 0.5
◦
C. The solution in the
receptor chamber was stirred with the aid of a cylin-
drical magnetic stir bar at a constant rate of 70 rpm.
Aliquots of 300 µL withdrawn at preselected times of
1, 2, 4, 6, 8, and 24 h were analyzed by HPLC and,
immediately after, equal volumes of fresh temperate
PBS solution were added to the receptor chamber.
Sink conditions were well ensured by correcting any
volume losses, when necessary. At the end of the pro-
cess, the drug content remaining in the patch was
determined as indicated previously in section of the
percentage impregnation determination. Kinetic re-
leases were performed in triplicate.
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 100, NO. 3, MARCH 2011
DOI 10.1002/jps

NAPROXEN-POLYMERIC PATCH FOR TRANSDERMAL SUSTAINED RELEASE SYSTEM
995
RESULTS AND DISCUSSION
Polymer Selection
The patch flexibility is recognized as an important
characteristic to be investigated. Flexibility strongly
depends on the composition of polymers utilized in
the patch synthesis. Here, various polymer mixtures
were assayed in order to optimize material to be used
as a matrix to develop the transdermal patch. These
preliminary studies were carried out in a hydraulic
press at different temperatures.
A mixture of ethyl cellulose and Eudragit
R
E100
was first considered. EC 20 cps, an ethyl ether of cellu-
lose, is widely used as a film coating. It is a semicrys-
talline polymer with a glass transition temperature
of approximately 130
◦
C and a melting point of about
180
◦
C. Eudragit
R
E100 is soluble in acidic solutions
up to pH 5, then suitable for releasing naproxen at the
skin pH. These two polymers were treated under var-
ious experimental conditions of pressure and temper-
ature. When the working temperature was lower than
the melting temperature of ethyl cellulose, the poly-
mers did not melt, even working at higher pressures.
Melting at higher temperatures caused EC degrada-
tion. Thus, in any case, the characteristics of the final
products were found satisfactory.
PVP-VA 64 is used as a binder pharmaceutical
tablets; it simply passes through the body when taken
orally. PVP-VA 64 was here used as an adhesive pro-
viding successful adhesion, although the stiffness of
the produced material was too rigid.
As a consequence of this rigidity and EC decomposi-
tion, we chose other polymers that could provide suit-
able properties. Finally, a combination of EVA (70%
acetate content) together with Eudragit
R
E100 was
found to be a good candidate. EVA was selected as
a flexible polymeric component and Eudragit
R
E100
resulted in a suitable drug carrier. EVA is mainly
used in the materials field because of its properties
as an elastomer with a high flexibility and softness,
yet it can be processed like other thermoplastics. The
percentage of each polymer in the mixture was opti-
mized. The patch became more rigid when increasing
the Eudragit
R
E100 percentage due to the pres-
ence of higher methacrylate amounts. The compo-
sition finally chosen was 20% of Eudragit
R
/80% of
EVA (w/w).
Patch Impregnation Studies
Raw patches were obtained as thin membranes of
Eudragit
R
/EVA polymer mixtures according to pro-
cedure detailed in patch synthesis section. The patch
impregnation with model compounds was first stud-
ied using rhodamine to check visually the charac-
teristics of the resulting materials. The influence of
the foaming process as a preliminary treatment of
polymeric membranes was here investigated. Apart
from inducing the formation of pores on the matrix
surface, this stage allows the control of the final mor-
phology to be exploited in applications such as biolog-
ical scaffolds and drug delivery systems.
22
The foam-
ing process was developed for 2 h at 1000 psi and the
resulting membranes were treated with the dye in a
second process developed at 450 psi and 22
◦
C for 2 h.
Such process proved that unaltered membranes were
not a suitable support for impregnation. In contrast,
membranes pretreated with dense CO
2
were modified
superficially. As a result, a successful distribution of
rhodamine on the surface of the activated matrix was
obtained.
Conditions established from rhodamine assays
were adapted to the elaboration of naproxen patches.
The membrane treatment relied on the two-step pro-
cedure consisting of the foaming surface preactivation
followed by the drug impregnation. In this case, how-
ever, experimental impregnation conditions of the sec-
ond step were studied, being pressure, temperature,
and time (P, T, and t) as the variables to be considered.
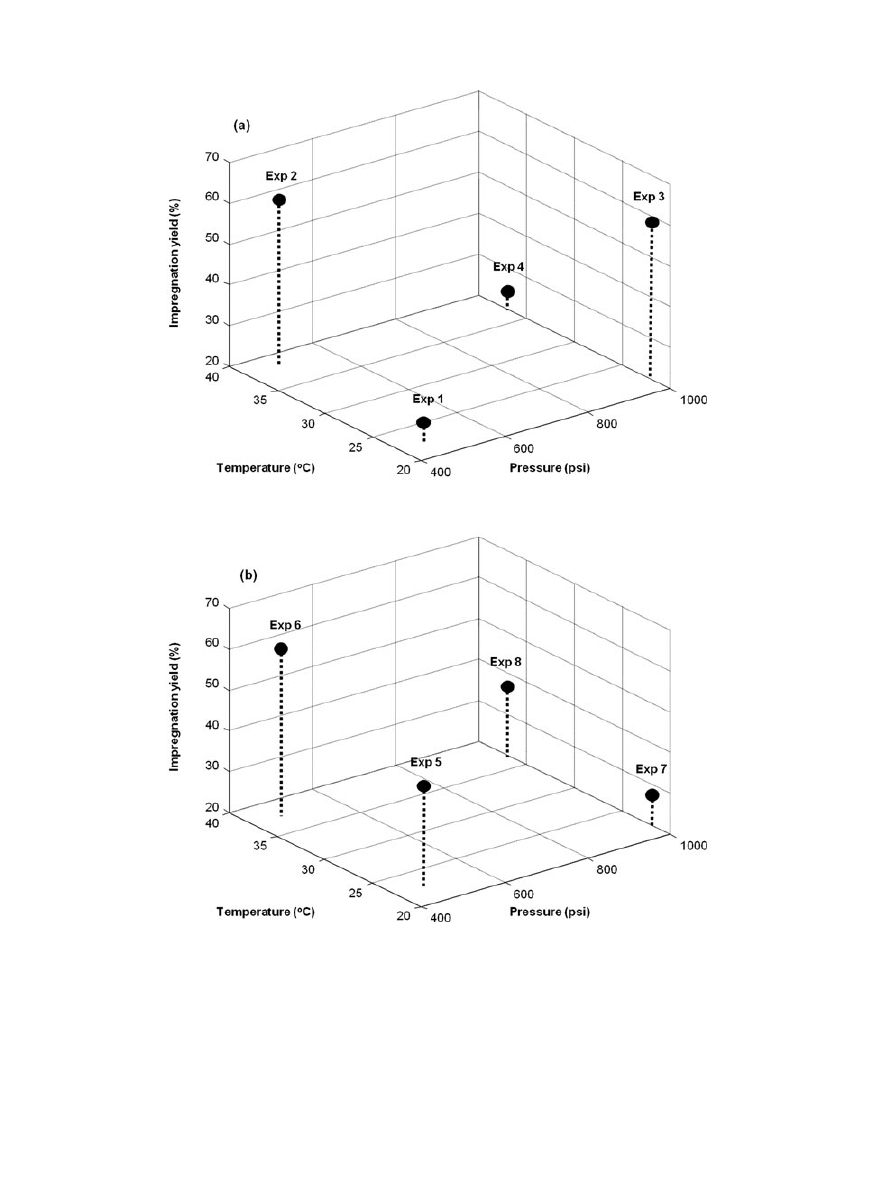
The highest impregnation yields corresponded to the
experiments 2 and 6 in Figure 1, which were per-
formed at low pressure and high temperature. Among
them, conditions of experiment 2 were more efficient
as the process time was shorter.
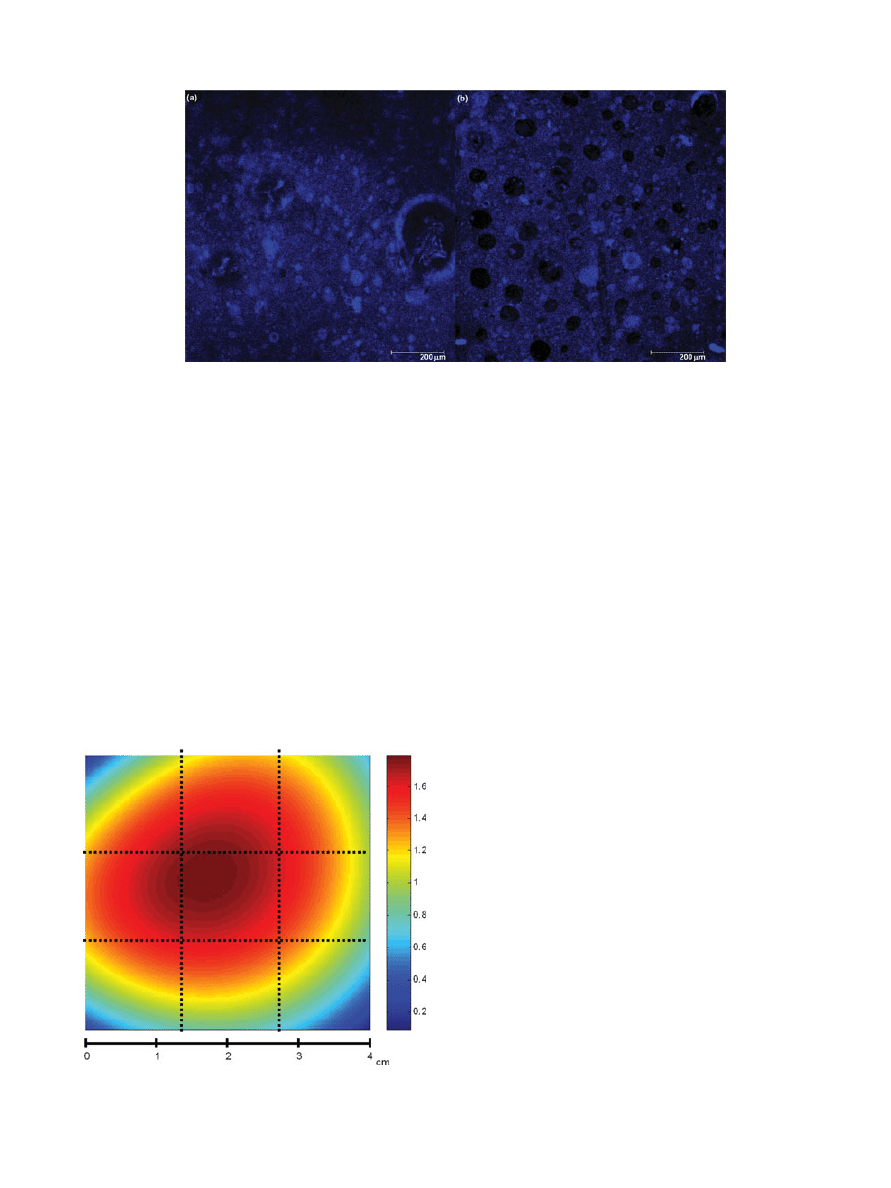
Confocal fluorescence microscopy was used to eval-
uate the morphology of a small section (1
× 1 mm
2
)
of the polymeric material after being in contact with
CO
2
. Different transversal sections of the sample
were studied to investigate the drug penetration in
the matrix. As an example, two pictures of sam-
ples obtained in experiments 3 and 5 are shown in
Figure 2. Porous cavities, which could also be observed
in Figure 2b, were generated during foaming process
and could be filled with the drug improving the im-
pregnation process. The pictures also showed the mi-
croscopic distribution of the drug and the heterogene-
ity of the samples. In addition, it was found that the
impregnation occurred not only superficially but also
in the depth.
Confocal fluorescence microscopy is a very useful
technique for surface profiling. In the pharmaceutical
industry, it was recommended to follow the manufac-
turing process of thin film pharmaceutical forms as
well as to control the quality and uniformity of the
drug distribution.
The homogeneity of naproxen contents was stud-
ied in more detail from a sample prepared at P
=
1000 psi, T
= 37
◦
C, and t
= 2.5 h. This was a sep-
arated sample, not included in the experimental de-
sign. The patch of 4
× 4 cm
2
was cut in nine equal
square pieces, which were analyzed chromatographi-
cally as described in the characterization studies sec-
tion. Naproxen contents in each piece (1.3
× 1.3 cm
2
,
approx.) were determined and an estimation of
DOI 10.1002/jps
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 100, NO. 3, MARCH 2011
996
ARGEM´I ET AL.
Figure 1.
Impregnation yields from the two-level three-variable experimental design. (a)
Processing time
= 2.5 h; (b) Processing time = 6 h.
macroscopic drug distribution is shown in Figure 3.
As 1000 µL aliquot of sample solution was poured in
the center of the patch, this area reasonably contained
a higher amount of naproxen. It was confirmed that
the drug impregnation in the center corresponded to
1.8% (w/w), whereas percentages were lower at the
corners (from 0.1% to 0.6%, approx.). Sections around
the central square piece contained intermediate drug
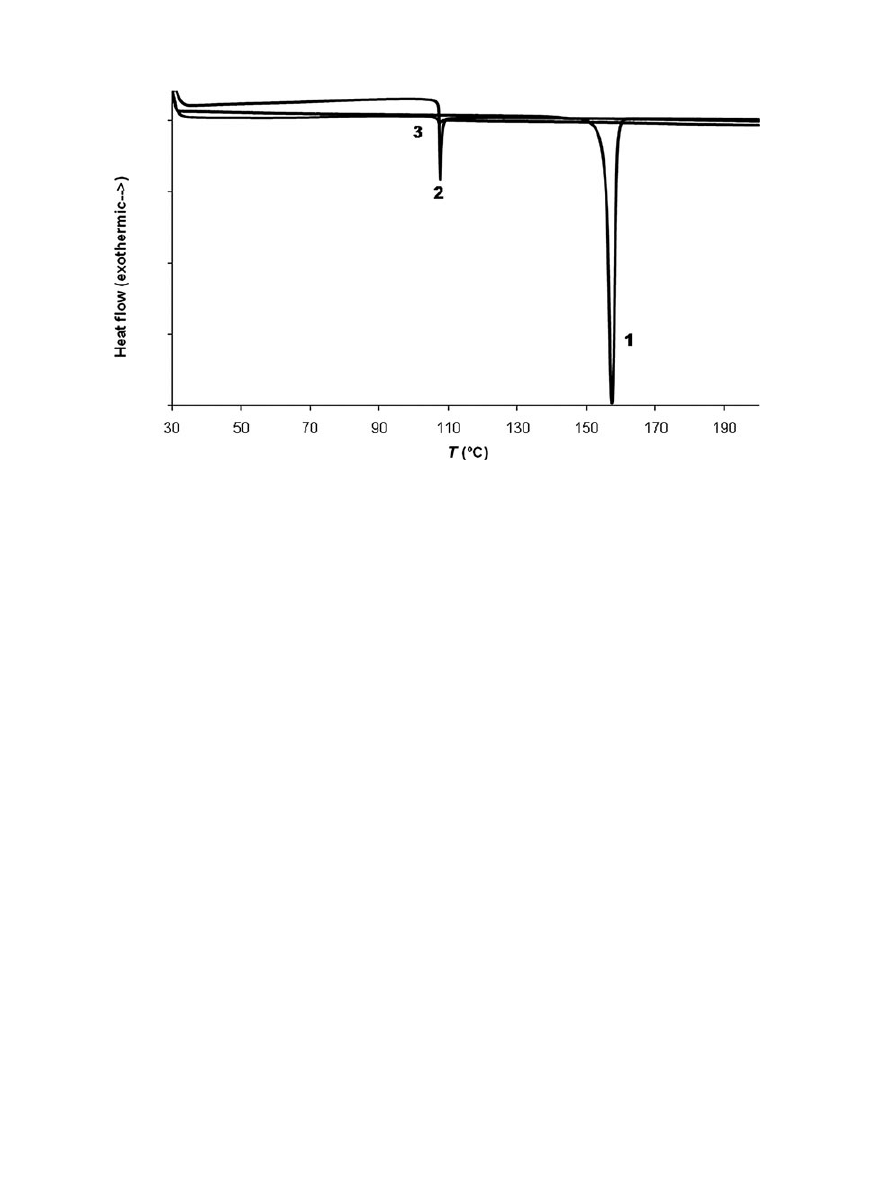
percentages (0.9%–1.4%, approx.).Differential scan-
ning calorimetry was used to study the thermal
properties of the
CO
2
-treated samples. Thermo-
grams of raw drug, unloaded polymeric patch, and
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 100, NO. 3, MARCH 2011
DOI 10.1002/jps
NAPROXEN-POLYMERIC PATCH FOR TRANSDERMAL SUSTAINED RELEASE SYSTEM
997
Figure 2.
Pictures obtained from fluorescence confocal microscopy in two dimensions. Pictures
of samples 3a and 5b measured at a certain depth. Scale bars shown.
drug–polymer impregnated patch (sample from ex-
periment 3) are compared in Figure 4. An endother-
mic peak corresponding to the melting temperature
of crystalline naproxen was clearly observed (T
m
=
154
◦
C). The peaks observed at 107
◦
C in both the un-
loaded polymeric patch and the drug–polymer im-
pregnated patch corresponded to the EVA melting
(typically occurring from 80
◦
C to 115
◦
C, depending on
the acetate content). Regarding Eudragit
R
, this is an
amorphous polymer with a glass transition tempera-
ture of 52
◦
C.
23
However, in these studies, no evidence
of glass transition was detected. As described else-
where, naproxen could decrease the T
g
of Eudragit
R
,
acting as a nonconventional plasticizer. This suggests
the existence of molecular interactions between the
polymer and the drug.
24
Figure 3.
Estimation of naproxen distribution in a 4
×
4 cm
2
patch.
Drug Release Monitoring
Preliminary studies of naproxen drug diffusion
through membranes were carried out with raw drug
in order to evaluate the influence of some experi-
mental conditions on the process. Various PBSs were
utilized as the receptor medium of the Franz diffu-
sion cell. In particular, 100 and 10 mM of PBS were
assayed; each one adjusted at pH 6.8 and 7.4. It
was concluded that naproxen diffused more rapidly
when the buffer concentration was 100 mM. This
fact was attributed to the higher buffering capac-
ity of the concentrated PBS. The pH of the PBS re-
ceptor solution affected the solubility and apparent
permeability as the analyte is a weakly ionizable
compound.
25
As a result, the diffusion/dissolution pro-
cess was faster at pH 7.4 than at pH 6.8. These re-
sults are in accordance with previous studies, which
measured naproxen permeability through synthetic
membranes.
26
Apart from chemical variables, the de-
sign characteristics of two kinds of Franz diffusion
cells were compared. One cell was designed with a
porous plate as a membrane support and the other
was constructed without a plate. It was evidenced that
the plate resulted in a physical obstacle, influencing
the diffusion process and the drug dissolution. As a
result, further evaluation of diffusion from patches
was carried out using a 100 mM PBS (at pH 7.4) and
working with a Franz cell without a plate to avoid any
physical hindrance.
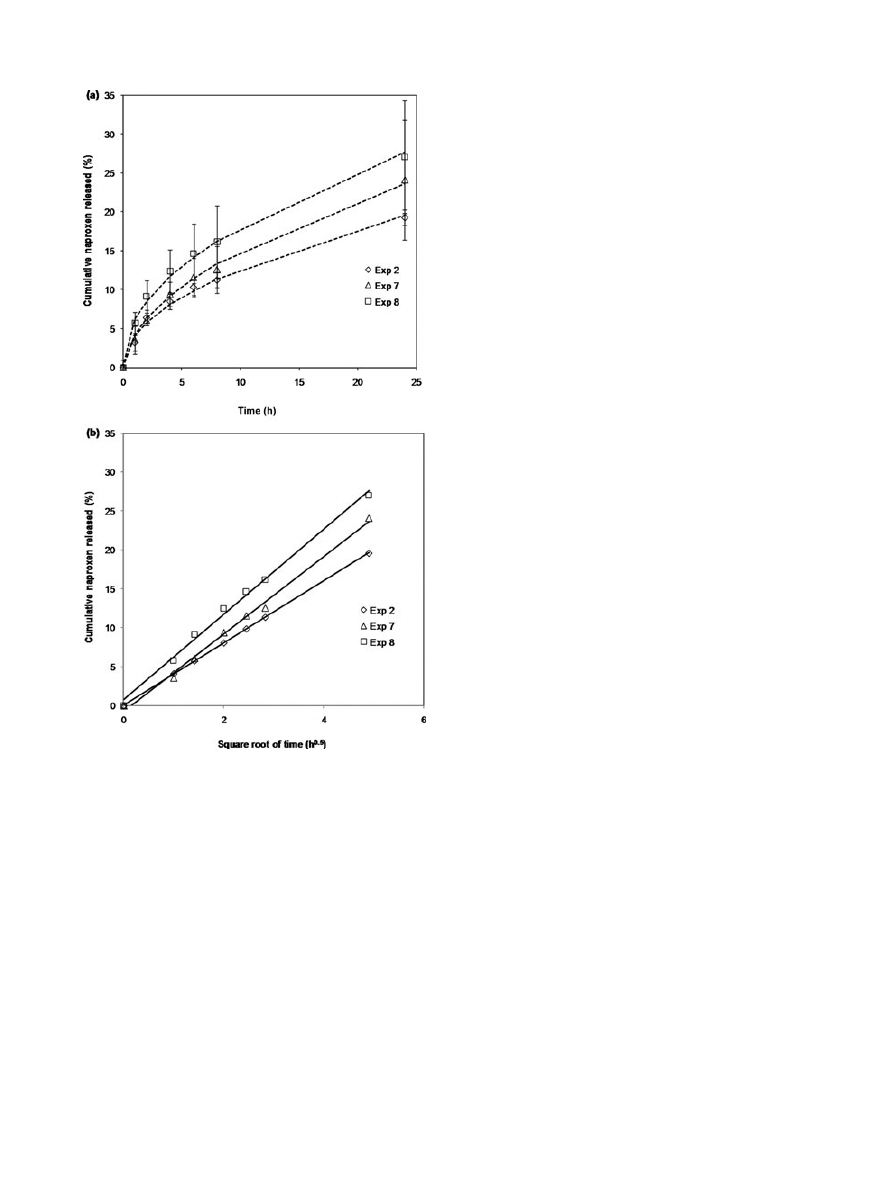
Raw naproxen dissolution tested in the receptor
medium shown a fast permeation rate (t
1/2
= 1.7 h).
Studies of naproxen diffusion from the patch through
a nylon membrane were performed for 24 h. Figure 5
depicts the concentration profiles of the analyte
expressed in cumulative naproxen released (w/w, in
percentage) as a function of time. Drug delivery ki-
netics of all samples under study showed an initial
faster drug release step in which about a 10% of drug
DOI 10.1002/jps
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 100, NO. 3, MARCH 2011
998
ARGEM´I ET AL.
Figure 4.
Result from differential scanning calorimetry (DSC) analysis. (1) Raw naproxen,
(2) unloaded patch, and (3) impregnated patch.
release in the first 6 h was obtained. After this pe-
riod, a more prolonged release process was observed.
For all samples belonging to the experimental design,
the percentage of naproxen release over 24 h ranged
from 15% to 25%. In addition, a more sustained drug
delivery from the patches was attained in comparison
with the kinetics of the raw naproxen dissolution, ob-
taining t
1/2
> 24 h (Fig. 5a). No lag times were found
in the concentration delivery profiles. This fact was a
proof that the delivery was controlled by the pristine
formulation, hence the polymeric matrix. Moreover,
there are no diffusion limitations due to the mem-
brane. The release profile followed the well-known
Higuchi model
27
for simple diffusion processes. Suc-
cessful correlation with the experimental data was
achieved. Figure 5b plots the cumulative released
amount in function of the square root of time. The de-
livery rates determined for all samples were from 3.8
to 5.5 wt%h
0.5
. The relationship between delivery and
the square root of time was associated with a mecha-
nism controlled by the polymeric matrix. In this way,
the longitudinal diffusion resistance was increased
with residence time through the outer zone of the
matrix (where a reduction of drug has happened) and
the border of water–matrix layer. Naproxen concen-
tration in the donor chamber was gradually decreased
and as a consequence the drug delivery. This trend ex-
plained the two different behaviors of the cumulative
naproxen profiles: release stage and sustained deliv-
ery to the skin.
The respective rates of release were determined
from the slopes of the regression lines in the Higuchi
plots, being higher for experiments 7 and 8 (5.0 and
5.5 wt%h
0.5
, respectively) than for experiment 2 (4.0
wt%h
0.5
). Experiments 7 and 8 corresponded to lower
impregnation yields and experiment 2 to a higher im-
pregnation yield. In conclusion, lower impregnation
of naproxen facilitated its diffusion and release.
CONCLUSIONS
The novelty in this work consisted in the preparation
of a naproxen transdermal patch by using pressurized
CO
2
. The device presented here seemed to be a promis-
ing alternative approach to conventional formula-
tions. Although various procedures were assayed
for the preparation of the membranes, including
melting processes in a press, the most successful
strategy relied on dissolution/evaporation of poly-
mers. It has been evidenced that the patch pretreat-
ment with dense
CO
2
creates a higher porous mate-
rial, which was further impregnated with naproxen.
After that, the membrane was efficiently loaded with
the drug at 450 psi and 37
◦
C for 2.5 h. Significant
advantages were gained from the use of dense
CO
2
as
a processing agent because it is a not toxic gas and is
easily removable by depressurization. Characteriza-
tion in vitro assays proved excellent results, such as
the naproxen delivery for a prolonged period of time,
at least 24 h. Future perspectives should be focused
on the processing of the patch, optimizing impregna-
tion working parameters and paying attention in new
materials. Furthermore, drug capacity loading should
be appropriately studied and if possible, improved.
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 100, NO. 3, MARCH 2011
DOI 10.1002/jps
NAPROXEN-POLYMERIC PATCH FOR TRANSDERMAL SUSTAINED RELEASE SYSTEM
999
Figure 5.
In vitro naproxen release kinetics in the Franz
diffusion cell in a phosphate buffer solution (PBS) at pH 7.4
at 32
± 0.5
◦
C by a circulating-water jacket while stirring at
70 rpm. Error bars represent the standard deviation based
on measurements in triplicate. (a) exp. 2, exp. 7, and exp. 8
(dashed lines correspond to Higuchi model correlation), (b)
Higuchi plots of exp. 2 (y
= 3.97 t
0.5
+ 0.13, R
2
= 0.992), exp.
7 (y
= 4.98 t
0.5
− 0.78, R
2
= 0.995), and exp. 8 (y = 5.49 t
0.5
+ 0.72, R
2
= 0.995).
ACKNOWLEDGMENTS
This paper has been supported by the Departa-
ment d’Educaci´o i Universitats de la Generalitat
de Catalunya i del Fons Social Europeu. A. Argem´ı
also gratefully acknowledges the financial support re-
ceived for a research grant BE2008 for a sojourn.
REFERENCES
1. Gorle BSK, Smirnova I, Arlt W. 2010. Adsorptive crystalliza-
tion of benzoic acid in aerogels from supercritical solutions. J
Supercrit Fluids 52(3):249–257.
2. Okamoto H, Sakakura Y, Shiraki K, Oka K, Nishida S, Todo
H, Iida K, Danjo K. 2005. Stability of chitosan-pDNA complex
powder prepared by supercritical carbon dioxide process. Int J
Pharm 290(1-2):73–81.
3. Sauceau M, Rodier E, Fages J. 2008. Preparation of inclusion
complex of piroxicam with cyclodextrin by using supercritical
carbon dioxide. J Supercrit Fluids 47(2):326–332.
4. T ¨
urk M, Hils P, Helfgen B, Schaber K, Martin HJ, Wahl
MA. 2002. Micronization of pharmaceutical substances by the
rapid expansion of supercritical solutions (RESS): A promising
method to improve bioavailability of poorly soluble pharma-
ceutical agents. J Supercrit Fluids 22(1):75–84.
5. Elvira C, Fanovich A, Fernandez M, Fraile J, San Roman J,
Domingo C. 2004. Evaluation of drug delivery characteris-
tics of microspheres of PMMA-PCL-cholesterol obtained by
supercritical-CO
2
impregnation and by dissolution–evapora-
tion techniques. J Controlled Release 99(2):231–240.
6. Marr R, Gamse T. 2000. Use of supercritical fluids for different
processes including new developments—a review. Chem Eng
Process 39(1):19–28.
7. Kawashima A, Watanabe S, Iwakiri R, Honda K. 2009. Re-
moval of dioxins and dioxin-like PCBs from fish oil by coun-
tercurrent supercritical CO
2
extraction and activated carbon
treatment. Chemosphere 75(6):788–794.
8. Winters MA, Frankel DZ, Debenedetti PG, Carey J, Devaney
M, Przybycien TM.
1999. Protein purification with vapor-
phase carbon dioxide. Biotechnol Bioeng 62(3):247–258.
9. Perrut M.
1994. Advances in supercritical-fluid chromato-
graphic processes. J Chromatogr A 658:292–313.
10. Majewski W, Valery E, Ludemann-Hombourger O. 2005. Prin-
ciple and Applications of Supercritical Fluid Chromatography.
J Liq Chromatogr Related Technol 28(7):1233–1252.
11. Jung J, Perrut M.
2001. Particle design using supercriti-
cal fluids: Literature and patent survey. J Supercrit Fluids
20(3):179–219.
12. Knez Z. 2004. High pressure process technology—Quo vadis?
Chem Eng Res Des 82(A12):1541–1548.
13. Lopez-Periago AM, Vega A, Subra P, Argemi A, Saurina J,
Garcia-Gonzalez CA, Domingo C.
2008. Supercritical CO
2
processing of polymers for the production of materials with
applications in tissue engineering and drug delivery. J Mater
Sci 43(6):1939–1947.
14. Greiner RW. 1991. Impregnation of catheters with pharma-
ceutical agents - by immersing in pharmaceutical agent satd.
soln., contacting at supercritical pressure and temp European
Patent Application
15. Kazarian SG, Martirosyan GG.
2002. Spectroscopy of
polymer–drug formulations processed with supercritical flu-
ids: In situ ATR-IR and Raman study of impregnation of
ibuprofen into PVP. Int J Pharm 232(1-2):81–90.
16. Weuts I, Kempen D, Decorte A, Verreck G, Peeters J, Brewster
M, Van Den Mooter G. 2005. Physical stability of the amor-
phous state of loperamide and two fragment molecules in solid
dispersions with the polymers PVP-K30 and PVP-VA64. Eur
J Pharm Sci 25(2-3):313–320.
17. Duarte ARC, Costa MS, Simplicio AL, Cardoso MM, Duarte
CMM. 2006. Preparation of controlled release microspheres
using supercritical fluid technology for delivery of anti-
inflammatory drugs. Int J Pharm 308(1-2):168–174.
18. Rowe RC, Sheskey PJ, Weller PJ. 2003. Handbook of pharma-
ceutical excipients; 4th ed. London: Pharmaceutical Press.
DOI 10.1002/jps
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 100, NO. 3, MARCH 2011

1000
ARGEM´I ET AL.
19. Devallencourt C, Marais S, Saiter JM, Labbe M, Metayer M.
2002. Study of transport of small molecules through ethylene-
co-vinyl acetate copolymers films. Part A: Water molecules.
Polym Test 21(3):253–262.
20. Marais S, Saiter JM, Davallencourt C, Nguyen QT, Metayer M.
2002. Study of transport of small molecules through ethylene-
co-vinyl acetate copolymers films. Part B: CO
2
and O-2 gases.
Polym Test 21(4):425–431.
21. Huskisson EC, Woolf DL, Balme HW, Scott J, Franklyn S.
1976. 4 New anti-inflammatory drugs—responses and varia-
tions. Br Med J 1(6017):1048–1049.
22. Jacobs MA, Kemmere MF, Keurentjes JTF. 2004. Foam pro-
cessing of poly (ethylene-co-vinyl acetate) rubber using super-
critical carbon dioxide. Polymer 45(22):7539–7547.
23. Verreck G, Decorte A, Li HB, Tomasko D, Arien A, Peeters
J, Rombaut P, Van Den Mooter G, Brewster ME.
2006.
The effect of pressurized carbon dioxide as a plasticizer and
foaming agent on the hot melt extrusion process and extru-
date properties of pharmaceutical polymers. J Supercrit Fluids
38(3):383–391.
24. Alvarez-Roman R, Ganem-Quintanar A, Quintanar-Guerrero
D. Proceedings—28th International Symposium on Controlled
Release of Bioactive Materials and 4th Consumer & Diver-
sified Products Conference, San Diego, CA, 2001, pp 1426–
1427.
25. Brain KR, Walters KA, Watkinson AC.
2002. Methods for
studying percutaneous absorption. Dermatological and Trans-
dermal Formulations 119:197–269.
26. Farinha A, Toscano C, Campos R, Bica A, Hadgraft J. 2003.
Permeation of naproxen from saturated solutions and commer-
cial formulations through synthetic membranes. Drug Dev Ind
Pharm 29(4):489–494.
27. Higuchi WI. 1962. Analysis of data on medicament release
from ointments. J Pharm Sci 51(8):802–804.
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 100, NO. 3, MARCH 2011
DOI 10.1002/jps
Wyszukiwarka
Podobne podstrony:
jps 21681
jps 21579
jps 22023
jps 22081
jps 22139
Viofor JPS jest aparatem do magnetostymulacji, FIZJOTERAPIA
jps 21484
jps 21792
jps 21755
jps 22267
jps, kryminologia
jps 22075
jps 21904
jps 21667
jps 21681
jps 21828
jps 22219
jps 21704
jps 21947
więcej podobnych podstron