
doi: 10.1111/j.1529-8817.2005.00199.x
ATM Gene Founder Haplotypes and Associated
Mutations in Polish Families with Ataxia-Telangiectasia
M. Mitui
1
, E. Bernatowska
2
, B. Pietrucha
2
, J. Piotrowska-Jastrzebska
3
, L. Eng
1
, S. Nahas
1
,
S. Teraoka
4,5
, G. Sholty
1
, A. Purayidom
1
, P. Concannon
4,5
and R. A. Gatti
1,
∗
1
Department of Pathology and Laboratory Medicine, The David Geffen School of Medicine at UCLA, Los Angeles, CA 90095–
1732, USA
2
Department of Immunology, The Children’s Memorial Health Institute, Warsaw, Poland
3
Department of Pediatrics, The Medical University of Bialystok, Bialystok, Poland
4
Department of Molecular Genetics, Benaroya Research Institute at Virginia Mason, Seattle, WA 98101, USA
5
Department of Immunology, University of Washington School of Medicine, Seattle, WA 98195, USA
Summary
Ataxia-telangiectasia (A-T) is an early onset autosomal recessive ataxia associated with characteristic chromosomal
aberrations, cell cycle checkpoint defects, cancer susceptibility, and sensitivity to ionizing radiation. We utilized
the protein truncation test (PTT), and single strand conformation polymorphism (SSCP) on cDNA, as well as
denaturing high performance liquid chromatography (dHPLC) on genomic DNA (gDNA) to screen for mutations
in 24 Polish A-T families. Twenty-six distinct Short Tandem Repeat (STR) haplotypes were identified. Three
founder mutations accounted for 58% of the alleles. Three-quarters of the families had at least one recurring
(shared) mutation, which was somewhat surprising given the low frequency of consanguinity in Poland. STR
haplotyping greatly improved the efficiency of mutation detection. We identified 44 of the expected 48 mutations
(92%): sixty-nine percent were nonsense mutations, 23% caused aberrant splicing, and 5% were missense mutations.
Four mutations have not been previously described. Two of the Polish mutations have been observed previously
in Amish and Mennonite A-T patients; this is compatible with historical records. Shared mutations shared the
same Single Nucleotide Polymorphism (SNP) and STR haplotypes, indicating common ancestries. The Mennonite
mutation, 5932 G
>T, is common in Russian A-T families, and the STR haplovariants are the same in both Poland
and Russia. Attempts to correlate phenotypes with genotypes were inconclusive due to the limited numbers of
patients with identical mutations.
Keywords: ATM mutations, Polish, Amish, Mennonite, Haplotypes
Introduction
Ataxia-telangiectasia (A-T; MIM # 208900) is an auto-
somal recessive, neurological disorder with a frequency
of 1/40 000–1/100 000 (Gatti, 2002). Cerebellar ataxia,
immunodeficiency, oculocutaneous telangiectasia, and
radiation sensitivity are characteristic findings in A-T
patients. These patients also have a greatly increased risk
∗ Correspondence to: Richard Gatti, The David Geffen School
of Medicine, Department of Pathology, Los Angeles, CA
90095-1732, Phone (310) 825-7618, Fax (310) 825-7618.
E-mail: rgatti@mednet.ucla.edu
of cancer (Gatti & Good, 1971; Swift et al. 1986). They
typically manifest premature aging, degeneration of the
cerebellum, thymus and gonads, growth retardation, and
telomere shortening (Gatti, 2002; Chun & Gatti 2004).
Carrier frequencies of ATM mutations have been esti-
mated as 1–1.8% and are proving significant with regard
to breast cancer susceptibility (Swift et al. 1987; Easton,
1994; Gatti et al. 1999; Concannon, 2002; Buchholz
et al. 2004).
A-T is caused by mutations in the Ataxia-
Telangiectasia Mutated gene (ATM) located at 11q23.1
(Gatti et al. 1988; Lange et al. 1995; Savitsky et al. 1995).
The ATM gene is over 150 kb in size and includes 62
C
University College London 2005
Annals of Human Genetics (2005) 69,657–664
657

Mitui et al.
coding exons, encoding a 13 kb main transcript, with
an open reading frame of 9168 bp (Uziel et al. 1996;
Platzer et al. 1997). The ATM protein is 370 kDa, is
found predominantly in the cell nucleus, and is a pro-
tein serine/threonine kinase (Shiloh, 2003; Bakkenist &
Kasdan, 2003).
A-T patients are typically compound heterozygotes
carrying unique mutations, and no “hot spots” in the
ATM gene have been found (Mitui et al. 2003). There-
fore, the entire gene must be screened to determine
the two disease-causing mutations for each patient. Our
strategy for ATM mutation screening has been to first
perform SNP and STR haplotyping (Mitui et al. 2003;
Coutinho et al. 2004), followed by PTT (Telatar et al.
1996; Den Dunnen et al. 1999), SSCP (Castellvi-Bel
et al. 1999) or dHPLC (Bernstein et al. 2003). Finally,
each relevant genomic region is sequenced to identify
the mutation.
Previous studies have shown that STR haplotyping
can greatly increase mutation detection in ethnic pop-
ulations by associating founder mutations with their
STR haplotypes (Uhrhammer et al. 1995; Telatar et al.
1998; Laake et al. 1998; Ejima et al. 1998; Campbell
et al. 2003; Mitui et al. 2003; Coutinho et al. 2004;
Babaei et al. 2005; Birrell et al. 2005). Haplotyping is
also useful for prenatal testing and occasionally for het-
erozygote identification within A-T families (Gatti et al.
1993). Herein we studied twenty-four Polish families
with A-T and found that three founder mutations re-
curred (were shared) in 58% of the families, and nine
recurring founder haplotypes accounted for 83% of
the families. Mutations were identified for all founder
haplotypes.
Materials and Methods
Subjects
Twenty-four unrelated A-T families from Poland com-
prised this study group. All patients displayed classical
A-T phenotypes. Lymphoblastoid cell lines (LCLs) were
established for most of the probands. The diagnosis was
confirmed by the absence of the ATM protein by im-
munoblotting and the finding of radiosensitivity by a
colony survival assay (Sun et al. 2002); at least one ATM
mutation was also identified for each proband. Family
WAR 49 included two affected sibs. Blood collection
followed approved Human Subject Protection protocols
in Poland and the United States.
Haplotype Analysis
STR haplotyping was used to first determine whether
founder mutations were present in the Polish popula-
tion, thus minimizing the number of mutations that
would have to be screened. As previously described
(Mitui et al. 2003), STR haplotypes were identified us-
ing four markers: S1819 (Rotman et al. 1994), NS22
(Udar et al. 1999), S2179 (Vanagaite et al. 1995), and
S1818 (Rotman et al. 1994). Markers NS22 and S2179
are located within the ATM gene; markers S1819 and
S1818 flank the gene within 1.4 cM on the proxi-
mal and distal ends, respectively. PCR amplified end-
radiolabelled fragments were run on 6% polyacrylamide
gel (National Diagnostics, Atlanta, Georgia) and com-
pared with a known control (CEPH1347-2) (Mitui et al.
2003). This control has allowed the allele sizes to be stan-
dardized so that haplotypes from various ethnic popu-
lations could be compared (Mitui et al. 2003; Coutinho
et al. 2004; Birrell et al. 2005). Haplotype phase was
defined using parents or by comparing haplotypes of
patients with the same mutation.
SNP haplotyping was carried out by SSCP (Castellvi-
Bel et al. 1999), using three SNP markers: IVS17-
56G
>C, 5557G>A, and IVS62-55T>C. These three
SNPs defined the three most common SNP haplotypes
across the ATM region (H2, H3 and H4), which encom-
pass 91% of SNP haplotypes worldwide (Thorstenson
et al. 2001; Campbell et al. 2003). The more uncom-
mon Haplotype H1 was also identified in two families.
Mutation Detection
Mutation screening was performed with PTT (Telatar
et al. 1996) followed by SSCP (Castelvi-Bel et al.
1999) and dHPLC (Bernstein et al. 2003). PTT de-
tects truncating mutations, such as nonsense mutations,
frameshifts caused by small insertions or deletions, or
aberrant splicing (Telatar et al. 1996; Teraoka et al.
1999). SSCP was used to further identify abnormal
regions in the cDNA of the ATM gene. Sequencing
changes were revealed by a measurable difference in
mobility through a gel, due to differences in the sec-
ondary structure of single stranded cDNA or genomic
DNA (Castellvi-Bel et al. 1999). This technique used 34
658
Annals of Human Genetics (2005) 69,657–664
C
University College London 2005
ATM Mutations in Polish Families
overlapping fragments to cover the gene, with each frag-
ment spanning
∼300 nt of coding sequence. dHPLC
was performed on samples that still had one mutation
unidentified after the PTT and SSCP screening. Candi-
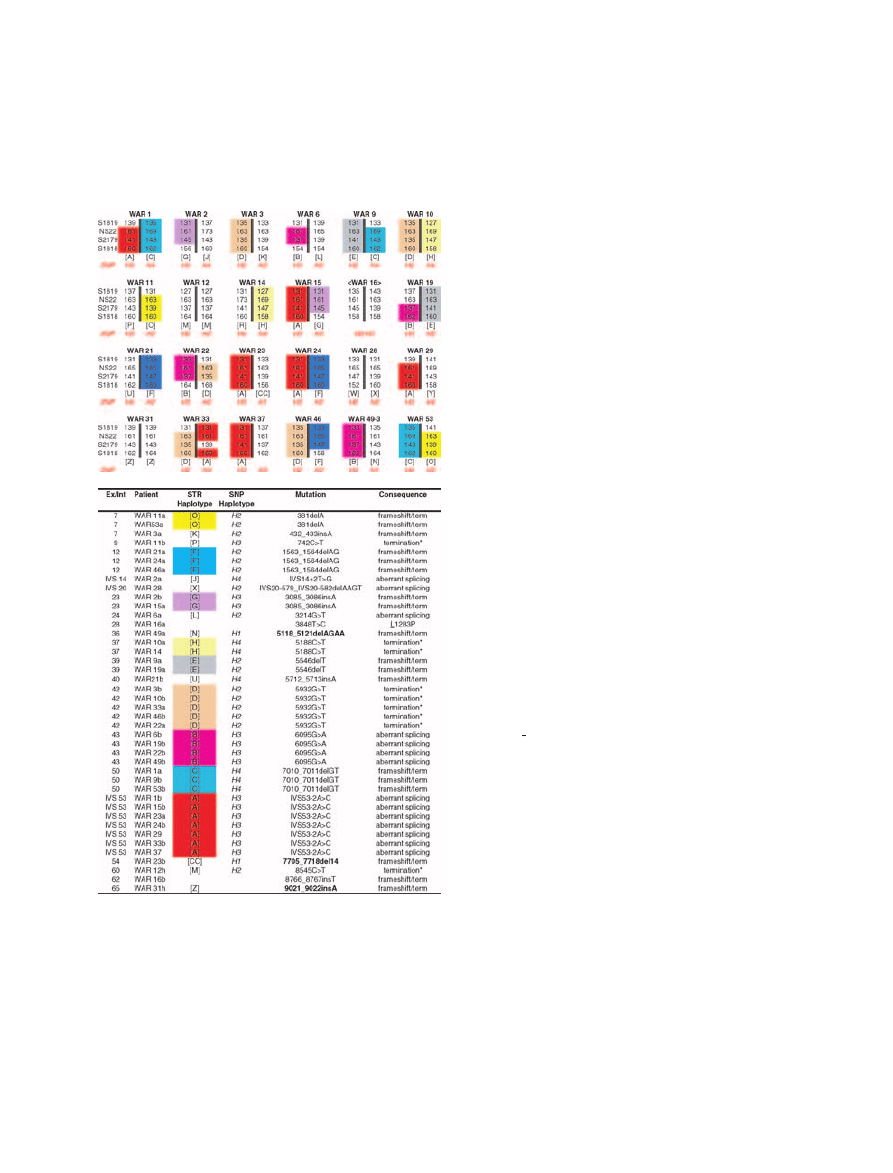
Figure 1
Haplotypes and mutations in 24 Polish A-T families.
A. Recurring haplotypes are colour shaded. The
indicate
that phase has not been defined. B. Mutations corresponding to
the affected haplotypes in A. Bold mutations have not been
previously reported. Conserved amino acids are underlined.
Superscripts: a, first allele; b, second allele; h: homozygous.
Asterisk denotes a primary premature termination codon
(PTC). Nucleotide numbering is based on +1 being the A of
the initiation start codon.
date regions were sequenced in both directions and the
mutated site identified using genomic DNA.
Results
Haplotypes
Twenty-six distinct STR haplotypes were observed in
24 ostensibly unrelated Polish families. The incidence
of homozygosity was very low; only two probands had
homozygous haplotypes and mutations, WAR 12 and
WAR 31 (Figure 1). Nine haplotypes were observed in
more than one family, encompassing 83% of the families.
The Polish A-T population proved to be more homo-
geneous than expected, as 83% of the families carried at
least one founder haplotypes (Figure 1A). Indeed, the
three most common haplotypes, [A], [B], and [D], oc-
curred in 14 of the 24 families (58%).
With few exceptions, STR haplotypes are con-
served between A-T patients with the same mutation
(Campbell et al. 2003). We observed this to be true
for markers NS22 and S2179. However, some variation
was found for S1819, and to a lesser degree S1818. This
most likely reflects the instability of STR markers or
could be due to polymerase slippage in some cases. Oc-
casional recombination outside the gene is also possible,
although recombination within the gene is very un-
common (Bonnen et al. 2000; Thorstenson et al. 2001).
Variant STR haplotypes (haplovariants) were observed
for some patients carrying the same mutation. For ex-
ample, haplotype [G] of WAR2 and WAR15, carried
the 3085 3086insA mutation. Another example can be
seen with WAR 31, where the S1818 alleles differ in a
family with consanguineous history. WAR33 was more
difficult to interpret: the mutation (IVS53-2A
>C) and
the H3 SNP haplotype remained unchanged as com-
pared to other patients (e.g., WAR 24), while the STR
haplotype was different for S2179, similar to changes
observed by Campbell et al. (2003). It was not possible
to define phase in WAR16.
SNP haplotypes H1, H2, H3, and H4 were identified
in the Polish population. SNP haplotype frequencies in
Poland were similar to previously described worldwide
SNP haplotype frequencies (Throstenson et al. 2001).
H2 was the most common haplotype (41%). H3 was
found in 33%; this is higher than the global frequency
of 12%. The H4 frequency was lower than expected
C
University College London 2005
Annals of Human Genetics (2005) 69,657–664
659

Mitui et al.
(20% vs 40%). H1 was seen in association with two
distinct STR haplotypes (WAR 23 and WAR 49) and
two distinct mutations. This haplotype is uncommon
outside of Africa (Thorstenson et al. 2001; Coutinho
et al. 2004).
Mutations
Mutations were detected in 44 of 48 alleles (92%). Of
21 different mutations, 15 were truncating, 5 aberrant
splicing, and 1 missense (Figure 1B). The most com-
mon mutation, IVS53-21
<C, a splicing mutation, was
found in 7 of 48 chromosomes (14.6%) and was asso-
ciated with Haplotype [A]. The second and third most
common mutations, 5932G
>T and 6095G>A, were
each found in 5 and 4 chromosomes (10% and 8%), in
association with Haplotypes [D] and [B], respectively.
Only three mutations were novel; however, our labora-
tory has also previously published other unique Polish
mutations (Telatar et al. 1998).
Mutation 5932G
>T results in GAA>TAA or prema-
ture termination condon. However, a small proportion
of the transcript is missing all of exon 42 (88nt), suggest-
ing that the mutation may also lead to aberrant splicing.
This mutation was observed in five of the families, on
three haplovariants. Two of the haplovariants have been
observed in Russian A-T families as well (Birrell et al.
2005). .
Genotypes versus Phenotypes
Serum alphafetoprotein (AFP) levels were elevated in all
24 patients. Similarly, all patients tested lacked detectable
intranuclear levels of ATM protein. None of the geno-
type/phenotype comparisons were significantly differ-
ent, due to the limited number of patients in each group
and the paucity of homozygous patients; none of the pa-
tients carrying haplotypes [A], [B] or [D] were homozy-
gous. A summary of the clinical dataset is presented in
Table 1.
Haplotype [A], carrying IVS53-2A
>C, was observed
in seven patients; none were homozygous. The average
age of onset of ataxia was 2.0 years and the average age
of onset of telangiectasia was 3.6 years. Of the seven
patients with this mutation, three were not yet confined
to a wheelchair (all were under 10 years old); the other
four patients were wheelchair bound but were older, av-
Table 1
Genotype/phenotype comparisons
Haplotype
A
B
D
All
Number of patients (N)
7
5
∗
5
25
∗
Male: Female
4:3
3:2
3:2
10:14
Ataxia onset (years old)
2.0
3.1
1.3
2 (0.9–7 yrs)
Progression of ataxia
†
1.7
1.4
2
1.6
Wheelchair (%N)
57
100 100 67
Wheelchair (years old)
15
14
12
14 (9–19 yrs)
Telangiectasia onset (years old) 3.6
6
3.4
4 (2–7 yrs)
Growth Retardation (%N)
57
20
50
50
Mental Retardation (%N)
43
20
20
29
Bronchiectasia (%N)
43
60
20
29
Cancer (%N)
0
0
0
8.7
IVIg therapy (%N)
57
80
60
50
AFP elevated (%N)
100 100 100 100
AFP (IU/ml)
276 107 147 208
IgM
↑ (%N)
‡
16
0
25
6
IgG
↓ (%N)
‡
0
43
25
25
IgA
↓ (%N)
‡
66
40
100 45
IgE
↓ (%N)
‡
80
100 67
96
†
Progression of ataxia 1 = slow, 2 = moderate, 3 = rapid
∗
The family of WAR 49 includes two affected children.
‡
Immunoglobulins were increased on at least two occasions.
eraging 15 years of age when they became wheelchair
dependent. No other phenotype differences were ap-
parent.
Haplotype [B], carrying 6095G
>A, affected five pa-
tients (two of whom were sibs in family WAR 49; only
one is shown in Fig 1). The average age of ataxia onset
was 3.1 years, with greater variation than for haplotype
[A]. Within this group, the WAR 49 sibs did not have
telangiectasia at ages 14 and 15. The average age of onset
for the other three patients was 6 years. All five patients
required a wheelchair by an average age of 14.
Haplotype [D], carrying 5932G
>T, affected 5 pa-
tients; the average age of ataxia onset was 1.3 years. This
average was almost identical to that of the Russian A-T
families (Birrell et al. 2005).
Discussion
Due to the large size of the ATM gene and the broad
spectrum of ATM mutations, mutation detection is not
yet cost-effective for establishing a diagnosis of A-T. In
this study, the diagnosis was confirmed by a lack of ATM
protein western blotting and radiosensitivity on by CSA,
in all patients. Serum AFP levels were elevated in all
660
Annals of Human Genetics (2005) 69,657–664
C
University College London 2005

ATM Mutations in Polish Families
Table 2
Mutation
Also found in
381delA
Iranian
742C
>T
Japanese
1563 1564delAG
Amish, Turkish, Italian,
German, Brazilian
IVS20–579˙IVS20–582delAAGT German, American-Hispanic
5188C
>T
Spanish
5712 5713insA
Phillippino, Turkish,
5932G
>T
Norwegian, Danish,
Mennonite,
American-Hispanic,
German, Russian
6095G
>A
Swedish, German, French
7010 7011delGT
English/Irish
IVS53-2A
>C
Danish, American-Hispanic,
Brazilian, Portuguese
8545C
>T
Italian
patients. Thus, these aspects of the A-T phenotype were
not influenced by genotype in any apparent way.
We observed that 58% of the A-T families in Poland
shared one of three founder mutations (Haplotype [A],
[B], and [D]), and 83% of the families carried at least
one of eight Polish founder haplotypes. We were sur-
prised to find this degree of genetic homogeneity, con-
sidering that Polish population migrations have not been
restricted by geographical features such as large bodies of
water or high mountain ranges. Nonetheless, our previ-
ous studies of ATM haplotypes and mutations strongly
suggest that shared, recurring mutations predate modern
ethnicities and nationalities (Campbell et al. 2003), and
STR haplotypes such as [A], [B], and [D] may reflect
influences on ancient migrations rather than on mod-
ern ones. Eleven Polish ATM mutations have also been
found in other ethnic groups (Table 2).
Splicing mutations comprised 23% of the mutations
found in this study, a proportion not unlike those in
previous studies (Teraoka et al. 1999; Mitui et al. 2003).
Splicing mutations typically involve the highly con-
served canonical 3
or 5
splice sites, as is the case for
IVS 53-2A
>C on Polish haplotype [A]. Three other
splicing mutations were noted on non-recurring Polish
haplotypes. IVS20–597delAAGT is a ‘masked’ muta-
tion that causes Type II splicing with pseudoexon for-
mation (Eng et al. 2004). The mutation occurs deep
within intron 20, and disrupts the U6 portion of a U1
snRNA binding site (Pagani et al. 2002). It has also been
observed in German, Turkish and Hispanic-American
patients (Mitui et al. 2003; Eng et al. 2004); the standard-
ized STR haplotypes of the Hispanic-American families
differ slightly from those of the Old World (Polish, Ger-
man, Turkish) A-T families (Eng et al. 2004), provid-
ing further evidence that many ATM mutations predate
STR haplotypes, but not the common SNP haplotypes
(Thorstenson et al. 2001; Campbell et al. 2003)
Most ATM mutations are associated with specific
STR and SNP haplotypes (Campbell et al. 2003; Mi-
tui et al. 2003; Eng et al. 2004). This held true with-
out exception for the SNP haplotypes associated with
Polish mutations. In general, this was also true for the
association of these mutations with STR haplotypes,
with two exceptions: in WAR33 [A][D] and WAR 19
[B][E]. WAR33 carries the Haplotype [A] mutation,
IVS53-2A
>C; however, the S2179 allele appears to have
changed from ‘141’ to ‘139’. The H3 SNP haplotype
background remains the same as that observed for all [A]
haplovariants in this study. Haplovariants were also ob-
served for the 5932 G
>T mutation on Haplotype [D]
(see below) and for the 6095G
>A mutation on Haplo-
type [B]. Both long and short forms of this Haplotype
[B] were observed (Fig 1A), with only a single allele
(S2179 ‘137’) shared by all four chromosomes (WAR
6, 19, 22, and 49–3). Taken together, these data suggest
that the longest variant (eg: WAR 49–3) is the older, an-
cestral haplotype for this mutation, although alternative
interpretations are possible.
The mutation on Haplotype [F], 1563 1564delAG, is
perhaps the most commonly observed ATM mutation
worldwide and always occurs on a SNP H
2
background.
It was observed in three Polish families in association
with SNP haplotype (H2), but with several STR haplo-
variants. As previously described (Campbell et al. 2003),
1563 1564delAG is associated with STR haplovariant
1 (in Turkish, Polish and Amish A-T patients), haplo-
variant 2 (in a Brazilian patient), and haplovariant 3 (in
Turkish and Italian patients). In all of these families, the
allele for S1818 was ‘160’, as is also observed in two of
the Polish families; however, in WAR 46, a new haplo-
variant 4 was defined by allele S1818 ‘158’ (instead of
‘160’). These findings are compatible with the historical
origins of the Amish of Pennsylvania (U.S.A) from Ger-
manic settlers, descendants of an Anabaptist movement
in northern Europe (1525–1536) (Hostetler, 1983a).
C
University College London 2005
Annals of Human Genetics (2005) 69,657–664
661

Mitui et al.
The mutation 5932G
>T, found on Polish Hap-
lotype [D], has also been observed in Mennonites,
another Germanic Christian sect of Anabaptist roots
that settled in Kentucky and Pennsylvania. In the early
1500s, Mennonites from the Netherlands and North
Germany migrated to the Vistula Detta (now Poland)
and later continued their migrations to Canada and the
United States (1873–74; 1922–30) (Hostetler, 1983b).
This mutation has also been found in A-T patients from
Denmark, England and Guatemala, always in associa-
tion with a SNP (H2) haplotype as previously described
(Campbell et al. 2003). It has recently been reported
to be the most common founder mutation (44%) in
a group of Russian A-T families, and the STR hap-
lotypes are the same (Birrell et al. 2005). Alleles at
S1819 vary as either ‘131’ or ‘135’, thereby defining
two haplovariants that are found in both Russia and
Poland.
We attempted to correlate clinical data with specific
mutations. Such genotype/phenotype correlations are
difficult to achieve unless several important criteria are
met: (1) the classical diagnosis must be confirmed on
a molecular basis to distinguish variant A-T pheno-
types from other phenotypically similar diseases; (2) a
sufficient number of patients must be homozygous for
a mutation so that the genotypic effects can be iso-
lated from other ATM alleles. Unless one postulates that
a heterozygous mutation will have a dominant inter-
fering effect (in which case one parent should man-
ifest symptoms), comparing compound heterozygous
patients who share only a single mutation is not likely to
reveal significant genotype/phenotype correlations. De-
spite this, a dominant interfering effect for at least one
ATM mutation (2546delSRI) has been demonstrated in
the mouse both in vivo and ex vivo, with some sugges-
tion that parent carriers of this mutation may manifest
an increased incidence of cancer (Concannon, 2002;
Scott et al. 2002; Spring et al. 2002). The Polish data
set contained only two homozygous patients (WAR 12
and WAR 31) and neither of these haplotypes was ob-
served in other families. Only Haplotypes [A], [D], and
[B] were observed repeatedly in 7, 5, and 4 patients, re-
spectively, and no significant genotype/phenotype cor-
relations were noted. The entire clinical dataset is in-
cluded in Table 1 so that it might later contribute
to a meta-analysis of a larger cohort of A-T patients.
This Table also suggests parameters for planning such
analyses.
Mutation detection for the Polish A-T population
was performed with the hope that it would be of as-
sistance in counselling Polish families for family plan-
ning, prenatal diagnosis, and identification of heterozy-
gote carriers. This information may also help in di-
agnosing A-T patients at a younger age by SNP and
STR haplotype prescreening for the eight recurring
Polish founder haplotypes. Certain types of muta-
tions (asterisked in the ‘consequence’ column of Fig-
ure 1B) may be amenable to therapeutic interven-
tion with aminoglycosides or other compounds (Lai
et al. 2004). Lastly, understanding the spectrum of ATM
mutations in Polish patients with A-T allows these
mutations to be sought in breast cancer and other
diseases.
References
Babaei, M., Mitui, M., Olson, E. R. & Gatti, R. A. ATM
haplotypes and associated mutations in Iranian patients with
A-T: recurring homozygosity without a founder haplotype.
Hum Genet In press.
Bakkenist, C. J. & Kastan, M. B. (2004) Initiating cellular stress
responses. Cell 118, 9–17.
Bernstein, J. L., Bernstein, L., Thompson, W. D., Lynch,
C. F., Malone, K. E., Teitelbaum, S. L., Olsen, J. H.,
Anton-Culver, H., Boice, J. D., Rosenstein, B. S.,
Borresen-Dale, A. L., Gatti, R. A., Concannon, P., Haile,
R. W., WECARE Study Collaborative Group. (2003)
ATM variants 7271T
>G and IVS10-6T>G among women
with unilateral and bilateral breast cancer. Br J Cancer 89,
1513–6.
Bernstein, J. L., Teraoka, S., Haile, R. W., Borresen-Dale, A-
L., Rosenstein, B. S., Gatti, R. A., Diep, A. T., Jansen, L.,
Atencio, D. P., Olsen, J. H., Bernstein, L., Teitelbaum, S. L.,
Thompson, W. D. & Concannon, P. J. (2004) Designing and
implementing quality control for multi-center screening of
mutations in the ATM gene among women with breast
cancer. Hum Mutat 21, 542–550, 2003.
Birrell, G. W., Kneebone, K., Nefedov, M., Nedfedova, E.,
Jartsev, M. N., Mitsui, M., Gatti, R. A. & Lavin, M. F.
(2005) ATM mutations, haplotype analysis and immuno-
logical status of Russian patients with ataxia telangiectasia.
Hum Genet In Press.
Bonnen, P. E., Story, M. D., Ashorn, C. L., Buchholz, T. A.,
Weil, M. M. & Nelson, D. L. (2000) Haplotypes at ATM
identify coding-sequence variation and indicate a region
of extensive linkage disequilibrium. Am J Hum Genet 67,
1437–51.
662
Annals of Human Genetics (2005) 69,657–664
C
University College London 2005

ATM Mutations in Polish Families
Buchholz, T. A., Weil, M. M., Ashorn, C. L., Strom, E. A.,
Sigurdson, A., Bondy, M., Chakraborty, R., Cox, J. D.,
McNeese, M. D. & Story, M. D. (2004) A Ser49Cys vari-
ant in the ataxia telangiectasia, mutated, gene that is more
common in patients with breast carcinoma compared with
population controls. Cancer 100, 1345–51.
Campbell, C., Mitui, M., Coutinho, G., Eng, L., Thorsten-
son, Y. & Gatti, R. A. (2003) ATM Mutations on Distinct
SNP and STR Haplotypes in Ataxia-Telangiectasia Patients
of Differing Ethnicities Reveal Ancestral Founder Effects.
Hum Mutat 21, 80–85.
Castellvi-Bel, S., Sheikhavandi, S., Telatar, M., Tai, L-Q.,
Hwang, M., Wang, Z., Yang, Z., Cheng, R. & Gatti, R. A.
(1999) New mutations, polymorphisms, and rare variants
in the ATM gene detected by a novel SSCP strategy. Hum
Mutat 14, 156–62.
Chun, H. H. & Gatti, R. A. (2004) Ataxia-telangiectasia, an
evolving phenotype. DNA Repair 3, 1187–1196.
Concannon, P. (2002) ATM heterozygosity and cancer risk.
Nat Genet 32, 89–90.
Coutinho, G., Mitui, M., Campbell, C., Costa Carvalho, B.
T., Nahas, S., Sun, X., Huo, Y., Lai, C. H., Thorstenson,
Y., Tanouye, R., Raskin, S., Kim, C. A., Llerena, J. Jr. &
Gatti, R. A. (2004) Five haplotypes account for fifty-five
percent of ATM mutations in Brazilian patients with ataxia
telangiectasia: seven new mutations. Am J Med Genet 126A,
33–40.
Den Dunnen, J. & Van Ommen, G-J. (1999) The Protein
Truncation Test: A Review. Hum Mutat 14, 95–102.
Easton, D. F. (1994) Cancer risks in A-T heterozygotes. Int J
Radiat Biol 66, S177–82.
Ejima, Y. & Sasaki, M. S. (1998) Mutations of the ATM gene
detected in Japanese ataxia-telangiectasia patients: possible
preponderance of the two founder mutations 4612del165
and 7883del5. Hum Genet 102, 403–8.
Eng, L., Coutinho, G., Nahas, S., Yeo, G., Tanouye, R.,
Babaei, M., Dork, T., Burge, C. & Gatti, R. A. (2004) Non-
classical Splicing Mutations in the Coding and Noncoding
Regions of the ATM Gene: Maximum Entropy Estimates
of Splice Junction Strengths. Hum Mutat 23, 67–76.
Gatti, R. A. (2002) Ataxia-Telangiectasia. In: The Genetic Ba-
sis of Human Cancer. Edition 2. (eds.B. Vogelstein, K. W.
Kinzler), pp.239–266. New York: Publ. McGraw-Hill.
Gatti, R. A. & Good, R. A. (1971) Occurrence of malignancy
in immunodeficiency diseases. A literature review. Cancer
28
, 89–98.
Gatti, R. A., Berkel, I., Boder, E., Braedt, G., Charmley,
P., Concannon, P., Ersoy, F., Foroud, T., Jaspers, N. G. J.,
Lange, K., Lathrop, G. M., Leppert, M., Nakamura, Y.,
O’Connell, P., Paterson, M., Salser, W., Sanal, O., Silver,
J., Sparkes, R. S., Susi, E., Weeks, D. E., Wei, S., White, R.
& Yoder, F. (1988) Localization of an ataxia-telangiectasia
gene to chromosome 11q22–23. Nature 336, 577–80.
Gatti, R. A., Peterson, K. L., Novak, J., Chen, X., Yang-
Chen, L., Liang, T., Lange, E. & Lange, K. (1993)
Prenatal genotyping of ataxia-telangiectasia. Lancet 342,
376.
Gatti, R. A., Tward, A. & Concannon, P. (1999) Cancer risk in
ATM heterozygotes: a model of phenotypic and mechanis-
tic differences between missense and truncating mutations.
Mol Genet Metab 68, 419–23.
Gatti, R. A., Becker-Catania, S., Chun, H. H., Sun, X., Mi-
tui, M., Lai, C. H., Khanlou, N., Babaei, M., Cheng, R.,
Clark, C., Huo, Y., Udar, N. C. & Iyer, R. K. (2001)
The pathogenesis of ataxia-telangiectasia. Learning from a
Rosetta Stone. Clin Rev Allergy Immunol 20, 87–108.
Hostetler, J. (1983a) Amish Life. Publ: Herald Press, Scottsdale,
AZ. Library of Cobgress, catalog card number 82, 83964.
Hostetler, J. (1983b) Mennonite Life. Publ: Herald Press,
Scottsdale, AZ. Library of Cobgress, catalog card number
82
, 83962.
Laake, K., Telatar, M., Geitvik, G. A., Hansen, R. O.,
Heiberg, A., Andresen, A. M., Gatti, R. & Borresen-Dale,
A. L. (1998) Identical mutation in 55% of the ATM alleles
in 11 Norwegian AT families: evidence for a founder effect.
Eur J Hum Genet 6, 235–44.
Lai, C. H., Chun, H. H., Nahas, S. A., Mitui, M., Gamo, K.
M., Du, L. & Gatti, R. A. (2004) Correction of ATM gene
function by aminoglycoside-induced read-through of pre-
mature termination codons. Proc Natl Acad Sci 101, 15676–
81.
Lange, E., Borrensen, A-L., Chen, X., Chessa, L.,
Chiplunkar, S., Concannon, P., Dandekar, S., Gerkin, S.,
Lange, K., Liang, T., McConville, C., Polakow, J., Por-
ras, O., Rotman, G., Sanal, O., Sheikhavandi, S., Shiloh,
Y., Sobel, E., Taylor, M., Telatar, M., Teraoka, S., Tolun,
A., Udar, N., Uhrhammer, N., Vanagaite, L., Wang, Z.,
Wapelhorst, B., Yang, H-M., Yang, L., Ziv, Y. & Gatti, R.
A. (1995) Localization of an ataxia-telangiectasia gene to a
∼500 kb interval on chromosome 11q23.1: linkage analysis
of 176 families in an international consortium. Am J Hum
Genet 57, 112–9.
Mitui, M., Campbell, C., Coutinho, G., Sun, X., Lai, C. H.,
Thorstenson, Y., Castellvi-Bel, S., Fernandez, L., Monros,
E., Tavares Costa, C. B., Porras, O., Fontan, G. & Gatti,
R. A. (2003) Independent mutational events are rare in
the ATM gene: Haplotype prescreening enhances mutation
detection rate. Hum Mutat 22, 43–50.
Pagani, F., Buratti, E., Stuani, C., Bendix, R., Dork, T. &
Baralle, F. E. (2002) A new type of mutation causes a splicing
defect in ATM. Nat Genet 30, 426–9.
Platzer, M., Rotman, G., Bauer, D., Uziel, T., Savitsky, K.,
Bar-Shira, A., Gilad, S., Shiloh, Y. & Rosenthal, A. (1997)
Ataxia-telangiectasia locus: sequence analysis of the 184 kb
of human genomic DNA containing the entire ATM gene.
Genome Research 7, 592–605.
C
University College London 2005
Annals of Human Genetics (2005) 69,657–664
663

Mitui et al.
Rotman, G., Vanagaite, L., Collins, F. S. & Shiloh, Y. (1994)
Three dinucleotide repeat polymorphisms at the ataxia-
telangiectasia locus. Hum Mol Genet 3, 2079.
Savitsky, K., Bar-Shira, A., Gilad, S., Rotman, G., Ziv, Y.,
Vanagaite, L., Tagle, D. A., Smith, S., Uziel, T., Sfez, S.,
Ashkenazi, M., Pecker, I., Frydman, M., Harnik, R., Patan-
jali, S. R., Simmons, A., Clines, G. A., Sartiel, A., Gatti,
R. A., Chessa, L., Sanal, O., Lavin, M. F., Jaspers, N. G.
J., Taylor, M. R., Arlett, C. F., Miki, T., Weissman, S. M.,
Lovett, M., Collins, F. S. & Shiloh, Y. (1995) A single ataxia-
telangiectasia gene with product similar to PI-3 kinase. Sci-
ence 268, 1749–1753.
Scott, S. P., Bendix, R., Chen, P., Clark, R., Dork, T. & Lavin,
M. F. (2002) Missense mutations but not allelic variants alter
the function of ATM by dominant interference in patients
with breast cancer. Proc Natl Acad Sci 99, 925–30.
Shiloh Y. (2003) ATM and related protein kinases: safeguard-
ing genome integrity. Nat Rev Cancer 3, 155–68.
Spring, K., Ahangari, F., Scott, S. P., Waring, P., Purdie, D.
M., Chen, P. C., Hourigan, K., Ramsay, J., McKinnon, P.
J., Swift, M. & Lavin, M. F. (2002) Mice heterozygous for
mutation in Atm, the gene involved in ataxia-telangiectasia,
have heightened susceptibility to cancer. Nat Genet 32, 185–
90.
Sun, X., Becker-Catania, S. G., Chun, H. H., Hwang, M.
J., Huo, Y., Wang, Z., Mitui, M., Sanal, O., Chessa, L.,
Crandall, B. & Gatti, R. A. (2002) Early diagnosis of ataxia-
telangiectasia using radiosensitivity testing. J Pediatr 140,
724–31.
Swift, M., Morrell, D., Cromartie, E., Chamberlin, A. R.,
Skolnick, M. H. & Bishop, D. T. (1986) The incidence and
gene frequency of ataxia-telangiectasia in the United States.
Am J Hum Genet 39, 573–83.
Swift, M., Reitnauer, P. J., Morrell, D. & Chase, C. L.
(1987) Breast and other cancers in families with ataxia-
telangiectasia. N Engl J Med 316, 1289–94.
Telatar, M., Wang, Z., Udar, N., Liang, T., Bernatowska-
Matuszkiewicz, E., Lavin, M., Shiloh, Y., Concannon, P.,
Good, R. A. & Gatti, R. A. (1996) Ataxia-telangiectasia:
mutations in ATM cDNA detected by protein-truncation
screening. Am J Hum Genet 59, 40–44.
Telatar, M., Teraoka, S., Wang, Z., Chun, H. H., Liang, T.,
Castellvi-Bel, S., Udar, N., Borresen-Dale, A-L, Chessa, L.,
Bernatowska-Matuszkiewicz, E., Porras, O., Watanabe, M.,
Junker, A., Concannon, P. & Gatti, R. A. (1998) Ataxia-
telangiectasia: identification and detection of founder mu-
tations in the ATM gene in ethnic populations. Am J Hum
Genet 62, 86–97.
Teraoka, S., Telatar, M., Becker-Catania, S., Liang, T.,
Onengut, S., Tolun, A., Chess, L., Sanal, O., Bernatowska,
E., Gatti, R. A. & Concannon, P. (1999) Splicing Defects
in the Ataxia-Telangiectasia Gene, ATM: Underlying Mu-
tations and Consequences. Am J Hum Genet 64, 1617–31.
Thorstenson, Y. R., Shen, P., Tusher, V. G., Wayne, T. L.,
Davis, R. W., Chu, G. & Oefner, P. J. (2001) Global anal-
ysis of ATM polymorphism reveals significant functional
constraint. Am J Hum Genet 69, 396–412.
Udar, N., Farzad, S., Tai, L-Q., Bay, J-O., Gatti, R. A.
(1999) NS22: A highly polymorphic complex microsatel-
lite marker within the ATM gene. Am J Med Genet 82,
287–9.
Uhrhammer, N., Lange, E., Porras, O., Naeim, A., Chen,
X., Sheikhavandi, S., Chiplunkar, S., Yang, L., Dandekar,
S., Liang, T., Patel, N., Teraoka, S., Udar, N., Calvo, N.,
Concannon, P., Lange, K. & Gatti, R. A. (1995) Sublocal-
ization of an ataxia-telangiectasia gene distal to D11S384 by
ancestral haplotyping in Costa Rican families. Am J Hum
Genet 57, 103–111.
Uziel, T., Savitsky, K., Platzer, M., Ziv, Y., Helbitz, T., Behls,
M., Boehm, T., Rosenthal, A., Shiloh, Y. & Rotman, G.
(1996) Genomic organization of the ATM gene. Genomics
33
, 317–320.
Vanagaite, L., James, M. R., Rotman, G., Savitsky, K., Var-
Shira, A., Gilad, S., Ziv, Y., Uchenik, V., Sartiel, A., Collins,
F. S., Sheffield, V. C., Richard, C. W. III, Weissenback, J. &
Shiloh, Y. (1995) A high-density microsatellite map of the
ataxia telangiectasia locus. Hum Genet 95, 451–4.
Received: 16 November 2004
Accepted: 14 April 2005
664
Annals of Human Genetics (2005) 69,657–664
C
University College London 2005
Wyszukiwarka
Podobne podstrony:
Population Based Estimates of Breast Cancer Risks Associated With ATM Gene Variants c 7271T4G and c
Kiermasz, Zuzanna Investigating the attitudes towards learning a third language and its culture in
Breast and other cancers in 1445 blood relatives of 75 Nordic patients with ataxia telangiectasia
A nonsense mutation (E1978X) in the ATM gene is associated with breast cancer
Functional and Computational Assessment of Missense Variants in the Ataxia Telangiectasia Mutated (A
Variants in the ATM gene associated with a reduced risk of contralateral breast cancer
Spectrum of ATM Gene Mutations in a Hospital based Series of Unselected Breast Cancer Patients
Variants in the ATM gene and breast cancer susceptibility
Cancer Risk According to Type and Location of ATM Mutation in Ataxia Telangiectasia Families
Mutations in the CgPDR1 and CgERG11 genes in azole resistant C glabrata
A recurrent mutation in type II collagen gene causes Legg Calvé Perthes disease in a Japanese family
RAD51C Germline Mutations in Breast and Ovarian Cancer Cases from High Risk Families
ATM POLYMORPHISM IVS6260GA IS NOT ASSOCIATED WITH DISEASE AGGRESSIVENESS IN PROSTATE CANCER
Risk of Cancer by ATM Missense Mutations in the General Population
Functional Origins of Religious Concepts Ontological and Strategic Selection in Evolved Minds
2008 4 JUL Emerging and Reemerging Viruses in Dogs and Cats
Angielski tematy Performance appraisal and its role in business 1
więcej podobnych podstron