Zespół Klinefeltera
1
Zespół Klinefeltera
Zespół Klinefeltera
Klinefelter's syndrome
Q98
Q98.0 Zespół Klinefeltera, kariotyp 47,XXY
Q98.1 Zespół Klinefeltera, mężczyźni z więcej niż dwoma chromosomami X
Q98.2 Zespół Klinefeltera, mężczyźni z kariotypem 46,XX
Q98.4 Zespół Klinefeltera, nie określony
Zespół Klinefeltera (ang. Klinefelter syndrome) – grupa chorób spowodowanych aberracją chromosomalną
polegającą na obecności przynajmniej jednego dodatkowego chromosomu X w części lub we wszystkich komórkach
organizmu mężczyzny.
Klasyczny zespół Klinefeltera występuje u mężczyzn o kariotypie 47,XXY i jest najczęstszą aneuploidią człowieka,
której częstość ocenia się na 1:500 noworodków płci męskiej
[1]
.
azoospermię, zwiększone stężenie gonadotropin i postępujące szkliwienie kanalików krętych jądra.
Historia
Zespół nazwę zawdzięcza Harry'emu Flitchowi Klinefelterowi, który opisał 9 przypadków pacjentów z dysgenezją
gonad, podwyższonymi poziomami gonadotropin w moczu, mikroorchidyzmem, eunuchoidyzmem, azoospermią i
ginekomastią w 1942 roku, razem z Fullerem Albrightem i Edwardem Conradem Reifensteinem
[2]
. Później okazało
się, że doniesienia o przypadkach zespołu miały miejsce wcześniej: w 1895 roku pacjenta z objawami zespołu opisał
Richard AltmannWikipedia:Weryfikowalność. Początkowo zespół Klinefeltera uważano za schorzenie
endokrynologiczne o niejasnej etiologii.
Po odkryciu ciałka Barra (chromatyny płciowej) zaczęto badać jej obecność w zespole Klinefeltera. Okazało się, że
zdecydowana większość chorych posiada obecną (dodatnią) chromatynę płciową, zaś w dużo mniej licznych
przypadkach tej chromatyny płciowej nie wykryto. Stąd powstał historyczny podział na: prawdziwy zespół
Klinefeltera dla chorych z dodatnią (obecną) chromatyną płciową i rzekomy zespół Klinefeltera – z ujemną
chromatyną płciową. Zauważono też, że w przypadku rzekomego zespołu Klinefeltera nie występują przypadki
ociężałości umysłowej a budowa ciała nie odbiega od normy
[3]
.
Postępy genetyki i wykrycie aberracji chromosomowych dało pogłębiony wgląd na patogenezę zespołu Klinefeltera
– chromosomalne podłoże zespołu odkryli Patricia A. Jacobs i J.A. Strong z Western General Hospital w Edynburgu
w 1959 roku u 24-letniego mężczyzny; było to pierwsze doniesienie o aneuploidii u człowieka
[4]
[5]
. Kariotyp
48,XXYY opisali pierwsi u 15-letniego chłopca Sylfest Muldal i Charles H. Ockey w 1960 roku
[6]
.
Obecnie można przyjąć, że historycznie określony rzekomy zespół Klinefeltera wiązać się może z przypadkiem
niezupełnej aberracji chromosomalnej (zob. mozaicyzm) lub nie odkrytymi jeszcze zaburzeniami genetycznymi.
W wyjątkowych przypadkach (przypadki kazuistyczne) stwierdza się niezaburzoną płodność w zespole Klinefeltera
– można to tłumaczyć prawidłowym kariotypem w gonadach.

Zespół Klinefeltera
2
Etiologia
Kariotyp 47,XXY
Zespół Klinefeltera w większości przypadków spowodowany jest
obecnością dodatkowego chromosomu X, rzadziej aberracje
chromosomalne wywołujące fenotyp zespołu mają postać innych
aberracji liczbowych (dwa, trzy, cztery dodatkowe chromosomy płci)
albo aberracjami strukturalnymi. Możliwe kariotypy są następujące:
• 47,XXY – większość (80%) przypadków
• 48,XXXY
• 48,XXYY
• 49,XXXXY
• 46,XY/47,XXY (mozaicyzm)- około 15%
• 46,XY i aberracje strukturalne.
bądź drugim podziale mejotycznym w gametogenezie, albo w podziale mitotycznym rozwijającej się zygoty. Zespół
XXY jako jedyna aberracja chromosomowa w dużej części (około 50%) jest spowodowany nondysjunkcją w
pierwszym podziale mejotycznym gamety męskiej. Prawdopodobieństwo zespołu Klinefeltera u dziecka wzrasta z
wiekiem matki, podobnie jak w zespole Downa, Edwardsa i Pataua
[7]
, przypadki spowodowane nondysjunkcją w
komórce macierzystej gamety męskiej wiążą się z kolei z zaawansowanym wiekiem ojca
[8]
.
Epidemiologia
Częstość zespołu Klinefeltera ocenia się na 1:500 męskiej populacji; inne aneuploidie chromosomu X są znacznie
rzadsze. Warianty o kariotypie 48,XXYY i 49,XXXY stwierdza się, odpowiednio, u 1:17.000 i 1:50.000
noworodków płci męskiej
[1]
. Zespół Klinefeltera na tle aneuploidii 49,XXXXY spotyka się u 1:85.000–1:100.000
noworodków. Mężczyźni o kariotypie 46,XX stanowią jeszcze mniejszą grupę pacjentów.
Objawy i przebieg
Objawy u pacjentów z kariotypem 47,XXY
Klasyczna postać zespołu Klinefeltera u mężczyzny z kariotypem 47,XXY jest związana z charakterystycznymi, ale
zmiennymi i dyskretnymi objawami. Chłopcy 47,XXY z reguły nie różnią się od rówieśników z kariotypem 46,XY.
Zespół Klinefeltera
3
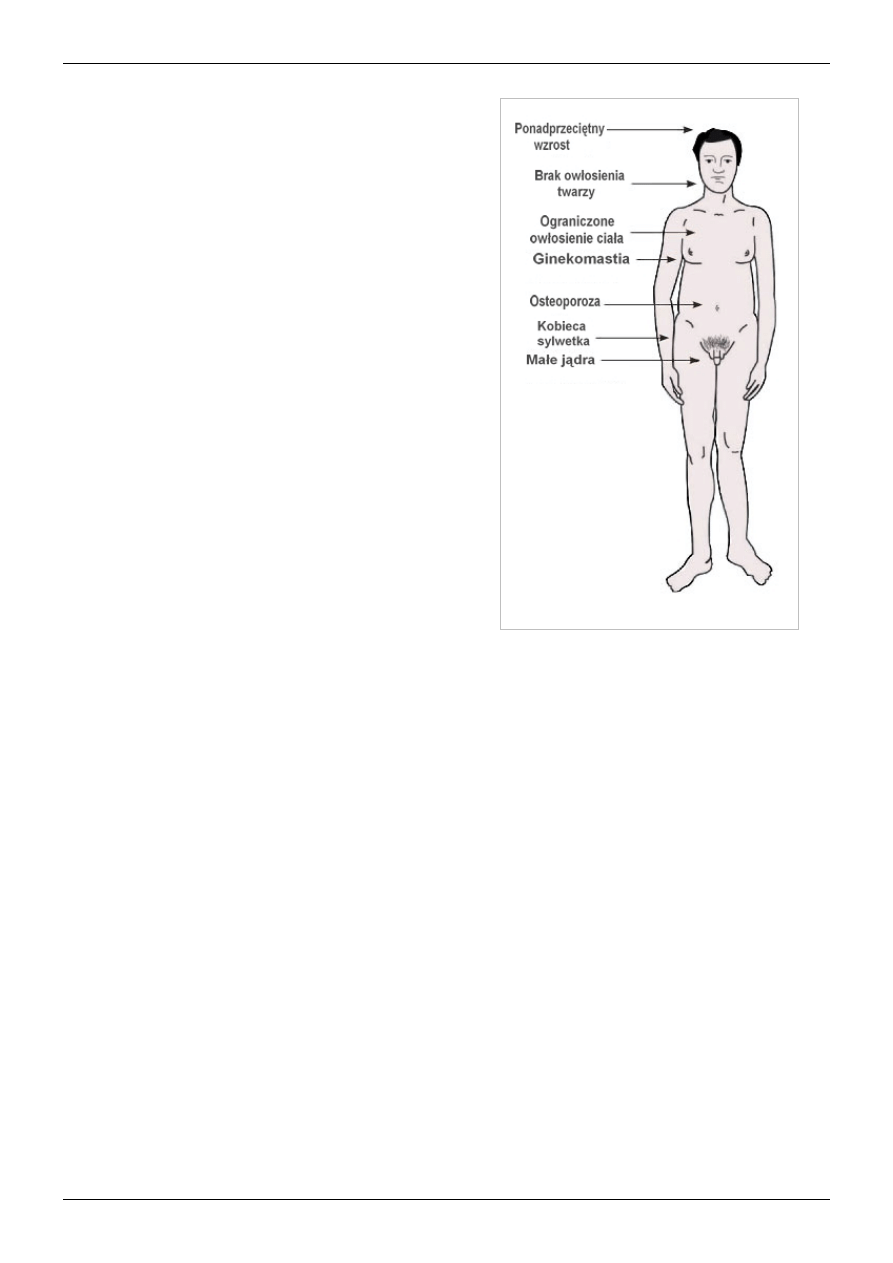
Główne objawy zespołu Klinefeltera
Charakterystyczne objawy zespołu to:
• wysoki wzrost: średni ostateczny wzrost chorych to 179,2 ±
6,2 cm
[1]
• słabiej rozwinięte mięśnie
• bardziej kobieca (gynoidalna) sylwetka, wynikająca z
charakterystycznej budowy klatki piersiowej i miednicy
• dłuższe niż przeciętnie kończyny górne i dolne
• cechy dysmorficzne: pacjenci z kariotypem 47,XXY nie mają
w zasadzie charakterystycznych cech dysmorficznych, które
ułatwiałyby rozpoznanie w okresie przedpokwitaniowym. U
około 40% mężczyzn XXY stwierdza się taurodontyzm
[9]
; dosyć częsta jest
• objawy radiologiczne: zmniejszenie kąta podstawy czaszki,
pogłębienie tylnego dołu czaszkowego, nadmiernie
upowietrznione zatoki klinowe, małe lub średnich rozmiarów
siodło tureckie z nadmiernie rozwiniętymi wyrostkami
przednimi i tylnymi, zmniejszenie grubości żuchwy na
wysokości jej kąta
[10]
[11]
• nieprawidłowy przebieg dojrzewania płciowego: dojrzewanie
pacjentów z zespołem Klinefeltera zaczyna się prawidłowo,
ale poziom testosteronu stopniowo maleje i drugorzędowe
cechy płciowe nie wykształcają się prawidłowo. Normalnie
rozwija się prącie
• u mężczyzn 47,XXY często (38-88%
[12]
[13]
) rozwija się obustronna, niebolesna ginekomastia i rzadko występuje
• obserwuje się zmniejszenie rozmiaru jąder, które dodatkowo mają nieprawidłowo twardą konsystencję; ich
objętość po ukończeniu pokwitania zwykle nie przekracza 10 ml
[12]
; powrózki nasienne, nasieniowody, gruczoł
krokowy i pęcherzyki nasienne nie wykazują odchyleń
• u około 27% pacjentów stwierdza się w dzieciństwie zaburzenia zstępowania jąder
[14]
• w gonadach nie zachodzi spermatogeneza, czego konsekwencją jest azoospermia i niepłodność, dotycząca blisko
100% pacjentów z zespołem Klinefeltera; objętość ejakulatu jest prawidłowa
• u około 70% pacjentów po 25. roku życia libido jest obniżone
[13]
, częste są problemy z erekcją
• rozmieszczenie owłosienia może być typowo męskie, zwykle jednak jest ono skąpe. Zarost na twarzy występuje u
jedynie około 20% pacjentów
[13]
.
Inteligencja pacjentów na ogół nie odbiega od normy; odnotowano zarówno dużo niższe, jak i dużo wyższe od
W badaniach laboratoryjnych stwierdza się obniżony poziom testosteronu <3,5 nmol/l (100 ng/dl), podwyższone
poziomy gonadotropin (FSH znacznie i LH > 15 j.m./l), SHBG, poziom estradiolu w osoczu jest powyżej normy dla
mężczyzn.
• po podaniu hCG brak zwiększenia lub nieznaczne zwiększenie stężenia testosteronu
• po podaniu GnRH lub klomifenu nadmierne wydzielanie LH i FSH przez przysadkę
Zespół Klinefeltera często przebiega z nadwagą i tendencją do otyłości; wiąże się też ze zwiększonym ryzykiem
upośledzonej tolerancji glukozy i cukrzycy
[15]
. Ryzyko raka sutka według jednych badań jest zwiększone i podobne
do ryzyka u kobiet
[16]
. U około 25% pacjentów rozwinie się osteoporoza
, według innych badań na większych
Zespół Klinefeltera
4
grupach pacjentów nie jest wyższe niż u normalnych kariotypowo mężczyzn
[17]
.
Objawy u pacjentów 48,XXYY i 48,XXXY
Objawy i przebieg u pacjentów z większą liczbą chromosomów X są dużo cięższe niż w klasycznym zespole
Klinefeltera. Tak jak w klasycznym wariancie 47,XXY występują u nich gynoidalna sylwetka, nieprawidłowo
rozwinięte drugorzędowe cechy płciowe, hipogonadyzm hipergonadotropowy, ginekomastia.
U pacjentów z kariotypem 48,XXYY nasilone są zaburzenia psychiczne, chorzy są zwykle nieśmiali, ale mogą być
kompulsywni i agresywni
[18]
. Pacjenci z kariotypem 48,XXXY mają cechy dysmorficzne i wady wrodzone:
• płaską nasadę nosa
• synostozę promieniowo-łokciową
Ich jądra i prącie są małe.
Objawy u pacjentów 49,XXXXY (zespół Fraccaro)
MRI głowy u niemowlęcia z kariotypem 47,XXY/48,XXXY/49,XXXXY
uwidaczniające niekomunikujące wodogłowie (A, B) i nieco hipoplastyczne ciało
modzelowate (C)[19] .
W przeciwieństwie do chorych z kariotypem
48,XXYY i 48,XXXY, pacjenci
49,XXXXY są niscy; mają nasilone cechy
dysmorficzne (hiperteloryzm, płaską nasadę
nosa, mongoidalne ustawienie szpar
powiekowych), mogą urodzić się z
rozszczepem podniebienia i języczka,
nierzadkie są wady wrodzone serca
(najczęściej przetrwały przewód Botalla),
synostoza promieniowo-łokciowa, stopa
wydrążona (pes cavus), kolana koślawe (genu valgum), hipotonia i wiotkość w stawach. Również występują
zaburzenia psychiczne. Fenotyp jest na tyle odrębny od fenotypu klasycznego zespołu Klinefeltera, że na określenie
tej rzadkiej aneuploidii wprowadzono termin zespołu Fraccaro
[20]
[21]
[22]
.
Wszyscy pacjenci z większą niż jeden dodatkową liczbą chromosomów X prezentują umiarkowane do ciężkiego
upośledzenie umysłowe: obliczono, że każdy dodatkowy chromosom X wiąże się z obniżeniem IQ o przeciętnie
15–16 punktów
[23]
.
Rozpoznanie i różnicowanie
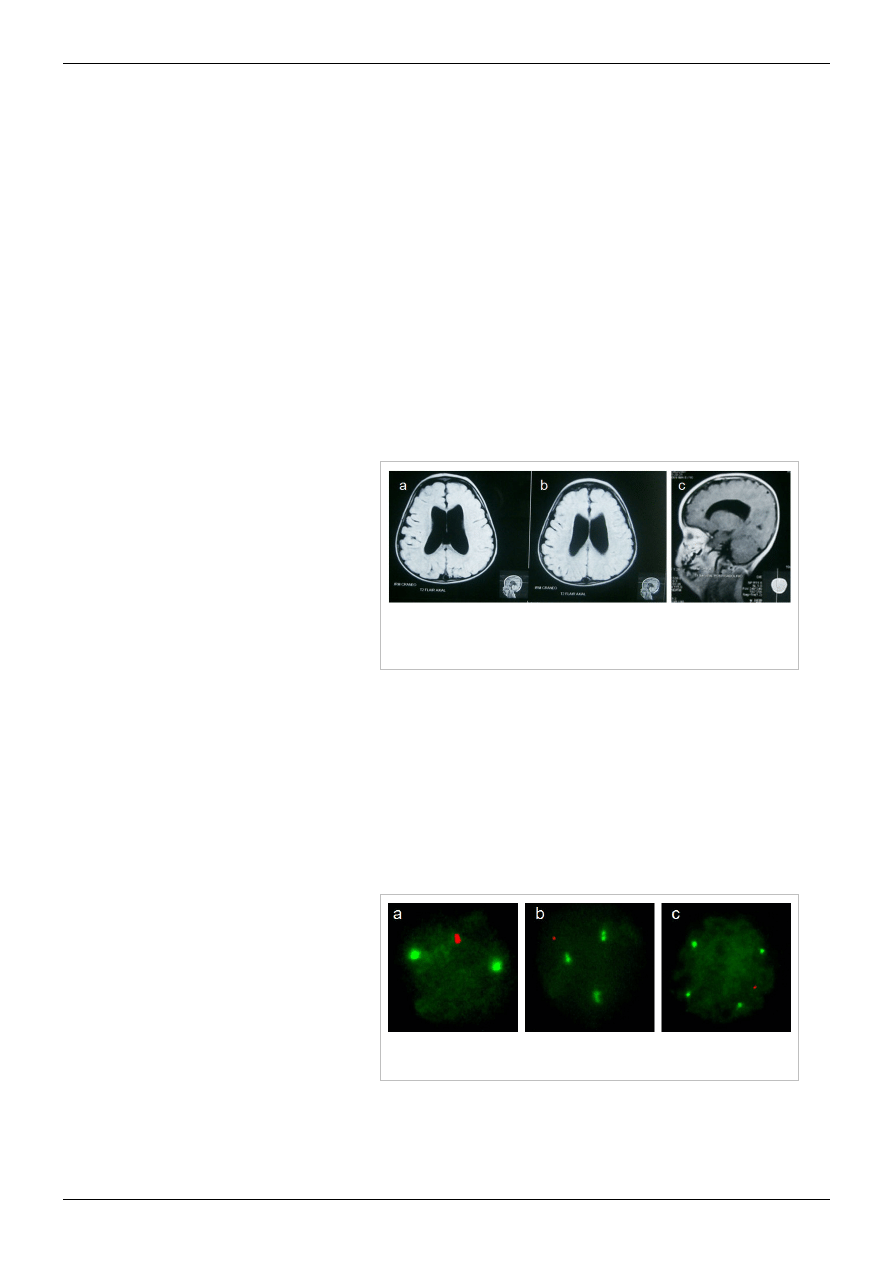
Analiza FISH na jądrach obwodowych limfocytów wykazała obecność kariotypów
XXY, XXXY i XXXXY (odpowiednio, A B i C)[19] .
Rozpoznanie zespołu Klinefeltera można
postawić jedynie na podstawie kariotypu.
Prawidłowy kariotyp zmusza do dalszej
diagnostyki – istnieje możliwość
mozaicyzmu komórek somatycznych (np.
46,XY/47,XXY) albo zespołu mężczyzny
XX. Inne jednostki chorobowe jakie należy
uwzględnić w różnicowaniu to związane z

Zespół Klinefeltera
5
Leczenie
Leczenie zespołu Klinefeltera polega na dożywotniej substytucji testosteronu w postaci enantatu testosteronu lub
pokwitania. Nie jest możliwe pobudzenie czynności kanalików plemnikotwórczych.
Powikłania
Pacjenci z zespołem Klinfeltera mają 50-krotnie zwiększone ryzyko wystąpienia nowotworu germinalnego
[24]
. Jest
nienasieniakowe, obecne we wcześniejszym wieku, rzadko występują w jądrach.
Bibliografia
• Visootsak J, Graham JM. Klinefelter syndrome and other sex chromosomal aneuploidies. „Orphanet Journal of
Rare Diseases”. 1. 42 (2006). doi:10.1186/1750-1172-1-42
• Lanfranco F., Kamischke A., Zitzmann M., Nieschlag E. Klinefelter's syndrome. „Lancet”. 07;364. 9430, ss.
273-83 (2004). doi:10.1016/S0140-6736(04)16678-6
. PMID 15262106.
• Krzysztof Boczkowski: Nieprawidłowości rozwoju płciowego. Warszawa: PZWL, 1971.
• Dobrzańska A, Ryżko J: Pediatria. Podręcznik do Państwowego Egzaminu Lekarskiego i egzaminu
specjalizacyjnego. Wrocław: 2005. ISBN 83-89581-25-6.
• Dueñas-Arias JE, Aguilar-Medina M, Arámbula-Meraz E, Valenzuela-Camacho JB, Vega-Solano A, Granados J,
Ramos-Payán R. 47,XXY/48,XXXY/49,XXXXY mosaic with hydrocephaly: a case report and review of the
literature. „J Med Case Reports”. 1. 94 (2007). PMID 17880714. DOI:10.1186/1752-1947-1-94
• Wattendorf DJ, Muenke M. Klinefelter syndrome
. „Am Fam Physician”. 72, ss. 2259-62 (2005). PMID
16342850.
• Jenö Molnár, Ferenc Szarvas: Andrologia. Warszawa: PZWL, 1984.
Linki zewnętrzne
• Artykuł z eMedicine
(
)
w bazie Who Named It
)
Przeczytaj zastrzeżenia dotyczące pojęć medycznych i pokrewnych w Wikipedii!
Przypisy
[1] Visootsak, J, Graham, JM. Klinefelter syndrome and other sex chromosomal aneuploidies (http:/
42).
„Orphanet Journal of Rare Diseases”. 1. 42 (2006). PMID 17062147.
[2] Klinefelter, HF Jr, Reifenstein, EC Jr, Albright, F. Syndrome characterized by gynaecomastia, aspermatogenesis without A-Leydigism and
increased excretion of follicle-stimulating hormone. „Journal of Clinical Endocrinology (Baltimore)”. 2, ss. 615-627 (1942).
[3] A. Krawczuk, Z. Janczewski w Endokrynologia Kliniczna pod red. W. Hartwiga PZWL 1984 r., Tom II; Z. Janczewski w: Seksuologia
Kliniczna pod red. K. Imielińskiego PZWL 1974
[4] Jacobs PA, Strong JA. A case of human intersexuality having possible XXY sex-determining mechanizm. „Nature”. 2, ss. 164-167 (1959).
PMID 13632697.
[5] Jacobs, PA. The William Allan Memorial Award address: human population cytogenetics: the first twenty-five years. „Am J Hum Genet”. 34.
5, ss. 689-698 (1982). PMID 6751075.
[6] Muldal S, Ockey CH. The "double male": a new chromosome constitution in Klinefelter's syndrome. „Lancet”. 276. 7147. Ss. 492-3.
[7] Morris JK, Alberman E, Scott C, Jacobs P. „European Journal of Human Genetics” (2007). DOI: 10.1038/sj.ejhg.5201956 (http:/

Zespół Klinefeltera
6
[8] Isabel Lorda-Sanchez, Franz Binkert, Marco Maechler, Wendy P. Robinson and Albert A. Schinzel. Reduced recombination and paternal age
effect in Klinefelter syndrome. „Human Genetics”. 89. 5.
[9] R. Steffen. Zahnanomalien als Hinweis zur Diagnose einer genetischen Erkrankung – ein Fallbericht. „Medizinische Genetik”. Ss. 432-434.
DOI: 10.1007/s11825-007-0047-x (http:/
[10] Kosowicz J. Zmiany radiologiczne czaszki w niedoczynności gonady męskiej. „Endokr Pol”. 19, ss. 276 (1968).
[11] Kosowicz J, Rzymski K. Radiological features of the skull in Klinefelter's syndrome and male hypogonadism. „Clin Radiol”. 26. 3, ss. 371-8
(1975). PMID 1201634.
[12] Smyth, CM, Bremner, WJ. Klinefelter syndrome (http:/
1309). „Archives of Internal
Medicine”. 158, ss. 1309-1314 (1998). PMID 9645824.
[13] Lanfranco F., Kamischke A., Zitzmann M., Nieschlag E. Klinefelter's syndrome. „Lancet”. 07;364. 9430, ss. 273-83 (2004).
doi:10.1016/S0140-6736(04)16678-6 (http:/
S0140-6736(04)16678-6). PMID 15262106.
[14] Disorders at the testicular level. W: Nieschlag E, Behre HM, Meschede D, Kamischke A: Andrology: male reproductive health and
dysfunction. Nieschlag E, Behre HM, Nieschlag S, eds. Berlin, Heidelberg, New York: Springer, 2000, ss. 143–76.
[15] Ota K, Suehiro T, Ikeda Y, Arii K, Kumon Y, Hashimoto K. Diabetes mellitus associated with Klinefelter’s syndrome: a case report and
review in Japan. „Int Med”. 41, ss. 842–47 (2002).
[16] Ian S Fentiman, Alain Fourquet, Gabriel N Hortobagyi. Male breast cancer. „Lancet”. 367. 9510, ss. 595-604 (2006).
doi:10.1016/S0140-6736(06)68226-3 (http:/
[17] Hasle H, Mellemgaard A, Nielsen J, Hansen J. Cancer incidence in men with Klinefelter syndrome. „Br J Cancer”. 71, ss. 416–20 (1995).
[18] Visootsak, J, Rosner, B, Dykens, E, Tartaglia, N, Graham, JM. Adaptive and Maladaptive Behavior of Males with Sex Chromosome
Aneuploidy. „J Investig Med”. 54, ss. 280 (2006). PMID 17497714.
[19] Dueñas-Arias JE, Aguilar-Medina M, Arámbula-Meraz E, Valenzuela-Camacho JB, Vega-Solano A, Granados J, Ramos-Payán R.
47,XXY/48,XXXY/49,XXXXY mosaic with hydrocephaly: a case report and review of the literature. „J Med Case Reports”. 1. 94 (2007). PMID
17880714. DOI: 10.1186/1752-1947-1-94 (http:/
[20] Dueñas-Arias JE, Aguilar-Medina M, Arámbula-Meraz E, Valenzuela-Camacho JB, Vega-Solano A, Granados J, Ramos-Payán R.
47,XXY/48,XXXY/49,XXXXY mosaic with hydrocephaly: a case report and review of the literature. „J Med Case Reports”. 1. 94 (2007). PMID
17880714. DOI: 10.1186/1752-1947-1-94 (http:/
[21] Hou JW. 49, XXXXY syndrome. „Chang Gung Med J”. 27, ss. 551-4 (2004). PMID 15508879.
[22] Peet J, Weaver DD, Vance GH: 49,XXXXY: a distinct phenotype. Three new cases and review. „J Med Genet”. 35, ss. 420-4 (1998). PMID
9610808.
[23] Linden, MG, Bender, BG, Robinson, A. Sex chromosome tetrasomy and pentasomy. „Pediatrics”. 96, ss. 672-682 (1995). PMID 7567329.
[24] Gregory G. Bebb, Frederic W. Grannis, Jr, Isaac B. Paz, Marilyn L. Slovak, Robert Chilcote. Mediastinal germ cell tumor in a child with
precocious puberty and Klinefelter syndrome (http:/
547). „Annals of Thoracic Surgery”.
66, ss. 547-548 (1998).
[25] http:/
[26] http:/
[27] http:/
[28] http:/
[29] http:/
[30] http:/

Źródła i autorzy artykułu
7
Źródła i autorzy artykułu
Zespół Klinefeltera Źródło: http://pl.wikipedia.org/w/index.php?oldid=21123401 Autorzy: 4C, Chrumps, Dawciu, Filip em, Iksnigo, Kauczuk, Kb, Kocio, Kpjas, Mario58, McMonster,
Miggawka, Mmalinosia, Mobile, Mpn, Mroman, Patrol110, Piastu, Rabidmoon, Red 81, Remedios44, Roo72, Sati, Skotos, SkyMaja, Stepa, Szwedowski, Thelarch, Tomasz O., Vuvar1, Wolas,
Zaltab, 26 anonimowych edycji
Źródła, licencje i autorzy grafik
Plik:Caduceus.svg Źródło: http://pl.wikipedia.org/w/index.php?title=Plik:Caduceus.svg Licencja: Public Domain Autorzy: User:Fadookie, User:Rama
Plik:47,XXY.jpg Źródło: http://pl.wikipedia.org/w/index.php?title=Plik:47,XXY.jpg Licencja: GNU Free Documentation License Autorzy: The cat
Plik:Klinefelter2.jpg Źródło: http://pl.wikipedia.org/w/index.php?title=Plik:Klinefelter2.jpg Licencja: GNU Free Documentation License Autorzy: M.Komorniczak, Rabidmoon
Plik:Klinefelter fraccaro syndrome.jpg Źródło: http://pl.wikipedia.org/w/index.php?title=Plik:Klinefelter_fraccaro_syndrome.jpg Licencja: Creative Commons Attribution 2.0 Autorzy:
Dueñas-Arias JE et al
Plik:Xxxxx.jpg Źródło: http://pl.wikipedia.org/w/index.php?title=Plik:Xxxxx.jpg Licencja: Creative Commons Attribution 2.0 Autorzy: Filip em, GeorgHH, Multichill
Plik:Star of life2.svg Źródło: http://pl.wikipedia.org/w/index.php?title=Plik:Star_of_life2.svg Licencja: Attribution Autorzy: User:Verdy p
Licencja
Creative Commons Attribution-Share Alike 3.0 Unported
http:/
Document Outline
Wyszukiwarka
Podobne podstrony:
cw4 Zespół Klinefeltera
ZESPÓŁ KLINEFELTERA
Zespół Klinefeltera, VI rok, Genetyka, Genetyka, Egzamin
Zespół Klinefeltera, oligofrenopedagogika - różne materiały i teksty
Zespół Klinefeltera
zespol klinefeltera przypadki
cw4 Zespół Klinefeltera
Klinefeltera zespół, studia, I ROK, Biomedyka
Zespół nerczycowy
9 RF ZEspól 0 Środki trwałe
Zespół kanału łokciowego i nerw pachowy (tryb edytowalny)
Zespoly paranowotworowe
Zespoly interdyscyplinarne
więcej podobnych podstron