
Laboratorium Bioinformatyka autor: Aleksandra Gruca
Zadnia
Strona 1 z 6
Celem laboratorium jest zapoznanie Państwa z metodami analizy danych pochodzących z
eksperymentów mikromacierzowych. W trakcie laboratorium zostanie przeprowadzona
analiza danych opisanych przez Goluba – są to dane chorych na dwa różne typy białaczki
– dane z mikromacierzy oznaczonych symbolem ALL to dane chorych na ostrą białaczkę
limfoblastyczną, natomiast mikromacierzy oznaczone symbolem AML pochodzą z badań
chorych na ostrą białaczkę szpikową. Wynikiem analiz będzie opisana (zanotowana) lista
50 genów, które najbardziej różnicują obydwa zbiory danych.
Opis danych :
http://www.broad.mit.edu/
cgi-bin/cancer/publications/pub_paper.cgi?mode=view&paper_id=43
Odnośnik do artykułu Goluba, w którym przedstawiono wyniki obliczeń dla danych
wykorzystywanych w trakcie laboratorium.
http://www.sciencemag.org/cgi/reprint/286/5439/531.pdf
Utwórz katalog roboczy i skopuj do niego skrypty
Normalize.R
i
getGOIDs.R
Uruchom program R.
Wykorzystując polecenie
setwd
lub z menu (File->Change Dir) zmień domyślny katalog
roboczy na katalog utworzony przez Ciebie
Pobierz oraz skopiuj do przestrzeni danych zbior golubEsets.
library(golubEsets)
data(Golub_Merge)
Jakiego typu jest to obiekt? Sprawdź jak wygląda struktura wczytanych danych –
polecenie
str(Golub_Merge).
Jakie dane można przechowywać w obiektach klasy
ExpressionSet?
Wyświetl informacje na temat wczytanej zmiennej poprzez podanie jej nazwy:
Golub_Merge
W tym wypadku zostanie wyświetlona tylko ogólna informacja o zmiennej
zadeklarowanej metodą show.
Wykonaj na danych funckcję featureNames:
featureNames(Golub_Merge)
Co otrzymałeś wyniku?
Preprocesing danych.
Aby znormalizować dane wykorzystaj funkcję
normalize.R
dostarczoną w ramach
laboratorium. Postaraj się zrozumieć działanie tej funkcji. Skomentuj w raporcie każdą
jej linijkę.
Za pomocą polecenia source wczytaj skrypt do przestrzeni roboczej R:
source("normalize.R")
Wykorzystaj skrypt do znormalizowania danych

Laboratorium Bioinformatyka autor: Aleksandra Gruca
Zadnia
Strona 2 z 6
expressedData =normalize(Golub_Merge)
expr.Golub=expressedData$expressedData
expr.ProbeSetsNames=expressedData$probeSetsNames
Aby w prosty sposób moc odwoływać się do grup danych dla typu białaczki AML oraz
ALL, wygodnie jest utworzyć zmienne zawierające numery indeksów dla obydwu grup
danych.
ALL_indexes =which(Golub_Merge$ALL.AML=="ALL")
AML_indexes =which(Golub_Merge$ALL.AML=="AML")
Ile było mikromacierzy dla białaczki typu AML w analizowanych danych, a ile
mikromacierzy dla białaczki typu ALL? Wykorzystaj funkcję length.
Wyznaczanie zbioru genów różnicujących pomiędzy dwoma typami białaczki:
W celu wyznaczenia genów różnicujących grupy białaczki zostanie wykorzystany prosty
test t. Rezultaty tego testu mogą zostać wykorzystane do wyznaczenia, które geny uległy
ekspresji w sposób istotny odbiegający od średniej. Takie geny mogą później zostać
wykorzystane jako geny różnicujące pomiędzy grupą AML oraz ALL
Przykładowy test t dla pierwszej sondy:
t.test(expr.Golub[1,ALL_indexes], expr.Golub[1,AML_indexes])
Sprawdź jak wygląda struktura zwracanej zmiennej - poleceniem str
Dla wykonywanych przez nas analiz interesująca jest wartość poziomu istotności testu
(p-value). W jaki sposób możemy otrzymać tą wartość?
Test dla każdej sondy macierzy może zostać przeprowadzony na dwa sposoby: z
wykorzystaniem pętli for lub też poprzez użycie funkcji
sapply
, która w poniższym
zapisie jest równoznaczna z pętlą for, lecz jest prostsza w zapisie:
expr.Golub.pVal<-sapply(1:nrow(expr.Golub), function(i)
t.test(expr.Golub[i,AML_indexes],expr.Golub[i,ALL_indexes])$p.value)
Mając tak wyliczony wektor p-value można sprawdzić jaka liczba sond rozróżnia
wskazane grupy na poziome istotności mniejszym od 5%. Podaj tą liczbę.
table(expr.Golub.pVal<0.05)
Ponieważ wykonana powyżej analiza jest wielokrotnym testowaniem konieczne jest
wprowadzenie odpowiedniej korekcji p-value uzyskanych w teście t. Wykorzystana
zostanie tu metoda oszacowania odsetka wyników fałszywie dodatnich (FDR)
Benjaminiego i Hochberga. Do tego celu zostanie wykorzystana funkcja
p.adjust
:
expr.Golub.pVal.bh=p.adjust(expr.Golub.pVal,method="BH")

Laboratorium Bioinformatyka autor: Aleksandra Gruca
Zadnia
Strona 3 z 6
Jaka liczba sond ma wartość p-value poniżej 5% dla wyników skorygowanych metodą
BH? Podaj otrzymaną wartość.
Przygotuj zmienną zawierającą raport z otrzymanych wyników:
W raporcie (zmiennej0 powinny znaleźć się następujące informacje:
- nazwy sond Affymetrixa - dla genów które uległy ekspresji (
results$AffyID)
- wartości poziomu istotności p (p-value) (
results$pVal)
- skorygowane wartości p-value (
results$pVal.bh)
- średnie wartości w całym przedziale danych (
results$A)
- krotność ekspresji (fold change) (
results$M)
- średnie wartości ekspresji dla danych ALL (
results$M.ALL)
- średnie wartości ekspresji dla danych AML (
results$M.AML)
Wyniki wpisz do zmiennej results – listy :
results=list()
results$AffyID<
results$pVal<-
results$pVal.bh<-
results$A<-
results$M<-
results$M.ALL<-
results$M.AML<-
Do obliczenia średnich wartości wykorzystaj funkcję
apply
. Funkcja
apply
wykonuje
na zadanej macierzy funkcję względem zadanego wymiaru. Obliczenie średniej dla
każdego z wierszy tablicy można wykonać następująco:
tmp.A<-apply(expr.Golub,1,mean)
Sprawdź strukturę otrzymanej zmiennej
str(tmp.A)
Ponieważ otrzymana zmienna posiada atrybuty nazwy, który w naszym przypadku jest
zbędny, należy go usunąć wykorzystując polecenie
unname
:
results$A<-unname(tmp.A)
Aby obliczyć wartość krotności M ekspresji (dla danych zlogarytmowanych) oblicz
różnicę średnich ekspresji w grupie ALL i AML. Usuń zbędne nazwy.
apply(expr.Golub[,AML_indexes],1,mean)-
apply(expr.Golub[,ALL_indexes],1,mean)
Na podstawie powyższych przykładów oblicz średnie wartości ekspresji w grupach AML
i ALL.
Ogranicz raport tylko do genów (sond) mających znamienną wartość p-value
skorygowaną (przy założeniu że za istotne uznamy geny dla których FDR<0.01,
uzyskana lista zawierać będzie wtedy 1% genów fałszywie dodatnich).
Aby osiągnąć ten cel posortuj wyniki.

Laboratorium Bioinformatyka autor: Aleksandra Gruca
Zadnia
Strona 4 z 6
Ponieważ najprościej wykonać sortowanie dla obiektu typu data.frame, przekonwertuj
zmienną results typiu lista na zmienną typu data frame (ramka danych):
results=as.data.frame(results)
Posortuj wszystkie kolumny ramki danych według kolumny
results$pVal.bh
res.sorted=results[order(results$pVal.bh),]
Zostaw tylko te wartości, dla których p-value po skorygowaniu jest mniejsza od 0.01
res.sorted<-res.sorted[res.sorted$pVal.bh<0.01,]
Adnotacja otrzymanych wyników
Dla tak otrzymanego zbioru danych dokonaj adnotacji.
Adnotacja będzie składać się z dwóch kroków. W pierwszym kroku przetłumacz nazwy
sond Affymetryxa na zrozumiałe dla biologów symbole genów. Najprościej zrobić to
wykorzystując gotowe biblioteki adnotacji przygotowanych dla każdego typu macierzy.
W jaki sposób można sprawdzić jakiego typu macierz została wykorzystana w
eksperymencie przeprowadzonym przez Goluba?
Biblioteki niezbędne do przeprowadzenia adnotacji sond:
library(annotate)
library(hu6800.db)
- biblioteka zawierająca adnotacje dla macierzy HG8600
Dopisanie symbolu genu do listy wynikowej obciętej do p.value<0.01 można uzyskać
stosując funkcję getSYMBOL. Funkcja ta wymaga podania parametrów: numeru sondy
(w tym przypadku jest to wektor numerów sond) oraz typu macierzy.
res.sorted
$AffyID =as.character(
res.sorted
$AffyID)
res.sorted
$Symbol=getSYMBOL(
res.sorted
$AffyID,”hu6800”)
W bazie danych Gene Ontology znajdują się terminy (GO Terms), które mogą być
wykorzystywane do adnotacji genów – czyli opisywania tych genów pod względem ich
funkcji iologicznej. Terminy te dzielą się na trzy podstawowe ontologie: Proces
Biologiczny, Komponent Komórkowy oraz Funkcja Molekularna.
Drugi krok adnotacji będzie polegał na pobraniu dla każdego genu jego zbioru
identyfikatorów GO dla Procesu Biologicznego, przetłumaczenie ich na terminy GO i
opisanie tymi terminami każdego genu. Do tego celu konieczne będzie wykorzystanie
biblioteki „GO”
Pobranie listy terminów GO dla wszystkich genów w zbiorze:
gg=getGO(res.sorted$AffyID,"hu6800")
Aby pobrać listy GO dla wszystkich sond znajdujących się w wekorze res.sorted$AffyID
wykorzystaj funkcję (getGOIDs):
source("getGOIDs.R",echo=TRUE)
GOIDsList= getGOIDs(res.sorted$AffyID)

Laboratorium Bioinformatyka autor: Aleksandra Gruca
Zadnia
Strona 5 z 6
Napisz skrypt który dla podanego wektora sond zwróci listy terminów GO
Może się przydać:
Lista wszystkich identyfikatorów GO dla pierwszej sondy:
oneGeneGOIDs= GOIDsList[[1]]
Pobranie opisu GO dla pierwszego terminu GO z listy oneGeneGOIDs -wszystkie
ontologie. Funkcja getGOdesc przyjmuje dwa argumenty: identyfikator GO oraz symbol
typu ontolgi który chcemy uzyskać (ANY – wszystkie ontologie, BP – proces
biologiczny)
GOdesc=getGOdesc(oneGeneGOIDs[1],"ANY")
Pobranie terminu GO dla pierwszego terminu GO z listy oneGeneGOIDs
goTerm=GOdesc[[1]]@Term
Wynikiem końcowym laboratorium (poza skryptem) powinien być plik tekstowy
zawierający listę dla 50 genów – kryteria doboru genów zostaną podane na zajęciach.
Każdy gen powinien znajdować się w osobnym wierszu, a wartości dla każdego genu
powinny być oddzielone znakiem tabulacji.
Dla każdego genu w pliku powinny znajdować się następujące informacje:
-nazwa sondy Affymetrixa
-symbol genu
-wartość p-value
-skorygowana wartość p-value
-krotność ekspresji
-lista terminów GO dla wszystkich genów – typ ontologii zostanie podany na zajęciach
Materiały
ź
ródłowe:
1.Gala G.: „Podstawy analizy danych uzyskanych technik
ą
mikromacierzy
oligonukleotydowych
wysokiej
g
ę
sto
ś
ci
Affymetrix”
w
ś
rodowisku
R/Bioconductor. Warsztaty Bioinformatyczne Projeku PBZ-MNiI-2/1/2005
Analiza Danych w Genomie Funkcjonalnej
2. http://www.r-project.org/
3. http://www.bioconductor.org/
Laboratorium Bioinformatyka autor: Aleksandra Gruca
Zadnia
Strona 6 z 6
Zadanie dodatkowe:
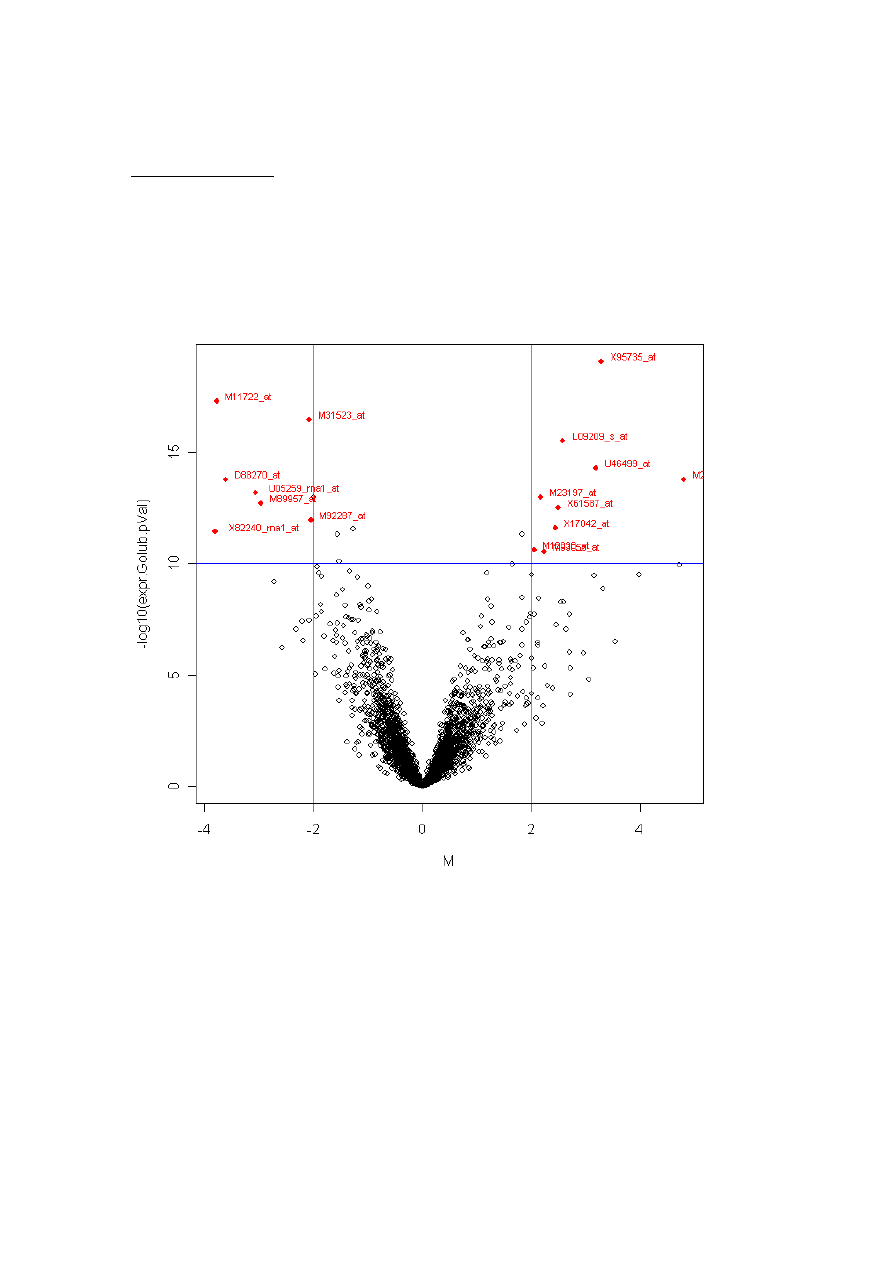
Dla otrzymanych wyników stworzyć wykres (volcano plot) rozrzutu pomiędzy wynikami
krotności ekspresji, a wartością poziomu istotności.
Geny dla których –log10 z poziomu istotności jest większy od 10 i równocześnie
absolutna wartość krotności ekspresji jest większa od 2 zaznaczyć oraz podpisać tak jak
na poniższym wykresie.
Informacje pomocnicze:
plot(M,-log10(expr.Golub.pVal))
– funkcja która narysuje wykres rozrzutu
wartości M oraz –log10 z poziomu istotności dla danych z laboratorium.
Do opisania wykresu wykorzystać funkcje:
lines
,
points
,
text
.
Aby znaleźć indeksy genów spełniające obydwa warunki (-log10 z poziomu istotności >
10 oraz wartość absolutna krotności ekspresji >2) wykorzystaj funkcję
which
oraz
funkcję
intersect.
Wyszukiwarka
Podobne podstrony:
Opisy programów-Zadanie
sko1-zad-programistyczne zadanie1
Opisy programów Zadanie
Podstawy programowania zadania
zadania if proste, Szkoła, Programowanie, C++, Zadania z IF
tp-zadania cz3, Szkoła, Programowanie, C++, Zadania z programowania cz.3
zadania lista 3, programowanie-zadania
ProgramowanieR Zadania
programowanie-zadania na kolo, Automatyka i robotyka air pwr, II SEMESTR, Programowanie w języku C
zadania lista 1, programowanie-zadania
zbi r5~1, wisisz, wydzial informatyki, studia zaoczne inzynierskie, podstawy programowania, zadania
Program 3, zadania III INF KASIS dzienne zaoczne
zbi rt~1, wisisz, wydzial informatyki, studia zaoczne inzynierskie, podstawy programowania, zadania
Bezhanov K A , i dr Programma i zadanija po teorii funkcij kompleksnogo peremennogo (3 kurs FRTK i F
Visual Studio 2013 Podrecznik programowania w C z zadaniami 2
Visual Studio 2013 Podrecznik programowania w C z zadaniami vs12pa 2
Visual Studio 2013 Podrecznik programowania w C z zadaniami
Visual Studio 2013 Podrecznik programowania w C z zadaniami
więcej podobnych podstron