
Acta Haematologica Polonica 2010, 41, Nr 4, str. 501–511
PRACA POGLĄDOWA – Review Article
EWA STUDNIAK, STANISŁAW ZAJĄCZEK
Rola genu P16 i delecji w obszarze 9p w powstawaniu i przebiegu biała-
czek oraz zespołów mielodysplastycznych
The role of the P16 gene and deletions in 9p region in the pathogenesis and
course of leukemia and myelodysplastic syndromes
Samodzielna Pracownia Cytogenetyczna
Zakład Patologii; Pomorska Akademia Medyczna
Kierownik: Prof. dr hab. Stanisław Zajączek
STRESZCZENIE
Inhibitory kinaz zależnych od cyklin (CDKIs), w tym gen P16
INK4A
należą do grupy uznanych supresorów nowotwo-
rów. Zmiany genu P16 w białaczkach, prowadzące do jego hemi- lub homozygotycznej inaktywacji, mogą być iden-
tyfikowane technikami o różnej rozdzielczości: stwierdzeniem w kariotypie delecji większego przyległego obszaru
w obrębie 9p21, delecją samego tylko genu uwidocznioną badaniem FISH, jak również poprzez zmiany w obrębie
genu wykrywane technikami molekularnymi. Zmiany wewnątrzgenowe rzadziej mają charakter mutacji punkto-
wych, a częściej polegają na jego metylacji. Wszystkie te zaburzenia wydają się dotyczyć głównie ostrych białaczek
limfoblastycznych, i mogą mieć znaczenie w wyjaśnieniu patomechanizmu i ocenie przebiegu tego typu białaczki.
W ostrych szpikowych i przewlekłych białaczkach oraz w zespołach mielodysplastycznych, zaburzenia te odnotowu-
je się rzadko i prawdopodobnie mają marginalne znaczenie. Dotychczasowe badania inaktywacji genu P16, niezależ-
nie od dokładności zastosowanej techniki i badanego typu białaczki, dają rozbieżne wyniki.
SŁOWA KLUCZOWE: Gen P16 – Białaczki – Delecje – Zmiany Ekspresji.
SUMMARY
Inhibitors of cyclin-dependent kinases (CDKIs), including gene P16
INK4A
belong to tumor suppressor genes. Changes
of the P16 gene in leukemias leading to hemi – or homozygous inactivation may be identified with different
techniques of resolution: finding the karyotype of adjacent larger deletions within the 9p21 region, deletion of the
P16 gene only shown by FISH technique as well as through changes in the gene detected with molecular techniques.
Intragenomic aberrations rarely exist as point mutations, more often involve its methylation. All of these changes
appear to primarily affect acute lymphoblastic leukemia, and may be important in clarification of the pathogenesis
and evaluation of the course of this type of leukemia. In acute myeloid, chronic leukemias and also myelodysplastic
syndromes, these conditions appear to be rare and probably have a marginal importance. Previous studies of
inactivation of P16, regardless of the accuracy of the technique used and type of leukemia, give divergent results.
KEY WORDS: P16 Gene - Leukemias – Deletions – Alterations of Expression.
Geny kontrolujące cykl komórkowy mają także działanie chroniące integralność genomu; niektóre
z nich są znane jako geny supresorowe nowotworów (TSGs = tumor suppressor genes). Ich mutacje
mogą uniemożliwić kontrolę błędów genetycznych w przebiegu replikacji DNA i być przyczyną wzro-
stu nowotworowego [1]. Supresorami nowotworów są m.in. inhibitory kinaz zależnych od cyklin (CD-
KIs): P15, P16, P18, P19, P21, P27, P57 oraz P53 i RB1; geny te w cyklu komórkowym odgrywają
regulacyjną rolę podczas przejścia z fazy G1 do fazy S [2]. Prawidłowy podział komórkowy odbywa się
dzięki zgodnie działającym cyklinom i kinazom cyklinozależnym (CDKs). Działanie kompleksu CDKs
jest pozytywnie regulowane przez cykliny, a negatywnie poprzez inhibitory kinaz cyklinozależnych na

M. KRAWCZYK-KULIŚ, S. KYRCZ-KRZEMIEŃ
502
różnych etapach cyklu komórkowego; zaburzenie tej aktywności może występować w przebiegu roz-
woju niemal każdego typu nowotworu, w tym białaczki [3].
"Rodzina INK4"
CDKIs podzielono na podstawie budowy na dwie grupy. Pierwsza z nich to tzw. "rodzina INK4", do
której należą: P14, P15, P16, P18, charakterystyczne dla nich są tzw. powtórzenia ankyrinowe. Dwa
z tych genów; CDKN2A (MTS1) kodujący białko P16 i CDKN2B (MTS2) kodujący białko P15 leżą
w prążku 9p21.3. Niedawno odkryto, że locus CDNK2A koduje również białko P14 (odpowiednik biał-
ka P19
u myszy). Geny białek P16 i P14 są zlokalizowane w tym samym locus, chociaż każdy z nich
ma różny promotor i ekson 1 (odpowiednio: ekson 1α i 1β), inną ramkę odczytu i produkują zupełnie
inne rodzaje białek. Eksony 2 i 3 są jednakowe dla obydwu genów; w nowotworach człowieka często
spotyka się uszkodzenie drugiego eksonu. Lokalizację, budowę i kształtowanie transkryptu powyższych
genów przedstawia Rycina 1. Białka obu genów pełnią rolę inhibitorów podczas przejścia z fazy G1 do
fazy S; mimo że w cyklu komórkowym obierają zupełnie inną drogę oddziaływania- P16 działa jako
inhibitor cyklin D: kompleksów CDK4/6; natomiast P14 stabilizuje P53 poprzez inhibicję MDM-2 [4,
5]. Druga grupa CDKIs obejmuje geny P21, P27 i P57, jednak zmiany strukturalne tych genów w no-
wotworach człowieka opisywane są bardzo rzadko [2].
Gen P16
P16 jest pierwszym opisanym genem z rodziny INK4, bardzo często ulegającym mutacjom
i delecjom w różnych nowotworach, w tym w chorobach hematologicznych. Oficjalna nomenklatura
wg HUGO to CDKN2A, jednakże akronim P16 jest używany znacznie szerzej. Gen P16 ma wielkość
40 kb, jest zlokalizowany na krótkim ramieniu chromosomu 9, w prążku p21; koduje białko zbudowane
z 360 aminokwasów (16 kD) pełniące funkcje inhibitora kinaz cyklinozależnych CDK4 i CDK6 [6].
Białko P16 wykazuje wysoki poziom ekspresji w progenitorowych komórkach hematopoetycznych
CD34
+
, w późnym stadium ich rozwoju ekspresja ta obniża się, co sugeruje jego udział w ich różnico-
waniu [3].
Ogólna charakterystyka zaburzeń genu P16
Gen
P16 spełnia kryteria supresora nowotworów; jego inaktywację mogą powodować mutacje,
delecje lub mechanizmy (np. metylacja) zmieniające jego aktywność bądź strukturę. Nowotwory (płuc,
piersi, pęcherza, nerki, skóry oraz białaczki) wykazywały najczęściej homozygotyczne delecje genu
P16 (Fot. 1). W komórkach, które utraciły tylko jeden allel, w pozostałym allelu sekwencjonowanie
ujawniało mutacje typu nonsens, zmiany ramki odczytu lub rzadziej misssens; zlokalizowane w cen-
tralnej część białka (powtórzenia ankyrinowe II i III w domenie wiążącej CDK) [1, 6].
Unieczynnienie
genu
P16 jest możliwe również poprzez hipermetylację wysp CpG (utworzonych
z promotora i końca regionu 5’), które stanowi alternatywną do mutacji drogę inaktywacji genu [7].
Gen P16 koduje inhibitor kinazy zależnej od cykliny, który wchodzi w negatywną interakcję z kinazą
cyklinozależną, hamującą poprzez fosforylację działanie białka RB1. Z kolei białko RB1 działa jako
inhibitor cyklu komórkowego; tak więc mutacje powodujące wyłączenie funkcji genu P16 prowadzi do
utraty kontroli nad cyklem komórkowym, co może skutkować rozwojem nowotworu [8]. Homozygo-
tyczne unieczynnienie genu P16, tak jak innych genów opisanych w tzw. mechanizmie „dwóch tra-
fień”, może uczestniczyć w inicjacji nowotworu. Odnotowano również przypadki w których transfor-
macja nowotworowa związana była z unieczynnieniem tylko jednej z dwu kopii genu, dotyczy to min.
właśnie genu P16 [9].
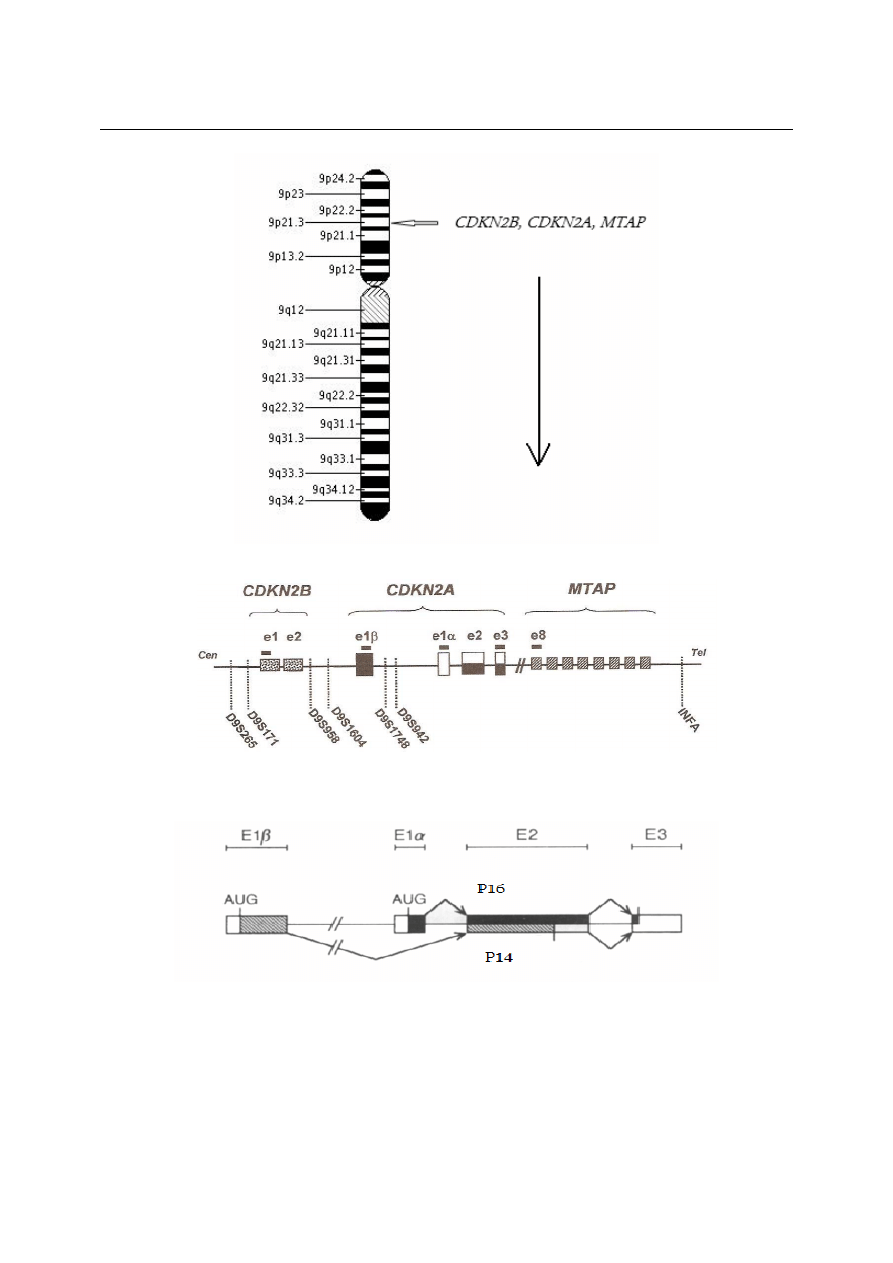
Współczesne standardy w diagnostyce i leczenie OBL
503
A.
B.
C.
Ryc. 1. A. Chromosom 9 z zaznaczonym locus 9p21.3 dla genów CDKN2B, CDKN2A, MTAP. B. Schemat przedstawiają-
cy eksony genów: CDKN2B, CDKN2A, MTAP (Bertin R. i wsp. Genes, Chromosomes Cancer 2003). C. Schemat przedstawia-
jący powstawanie dwóch różnych transkryptów genu CDKN2A: P16 i P14. (Quelle D.E. i wsp. Cell 1995)
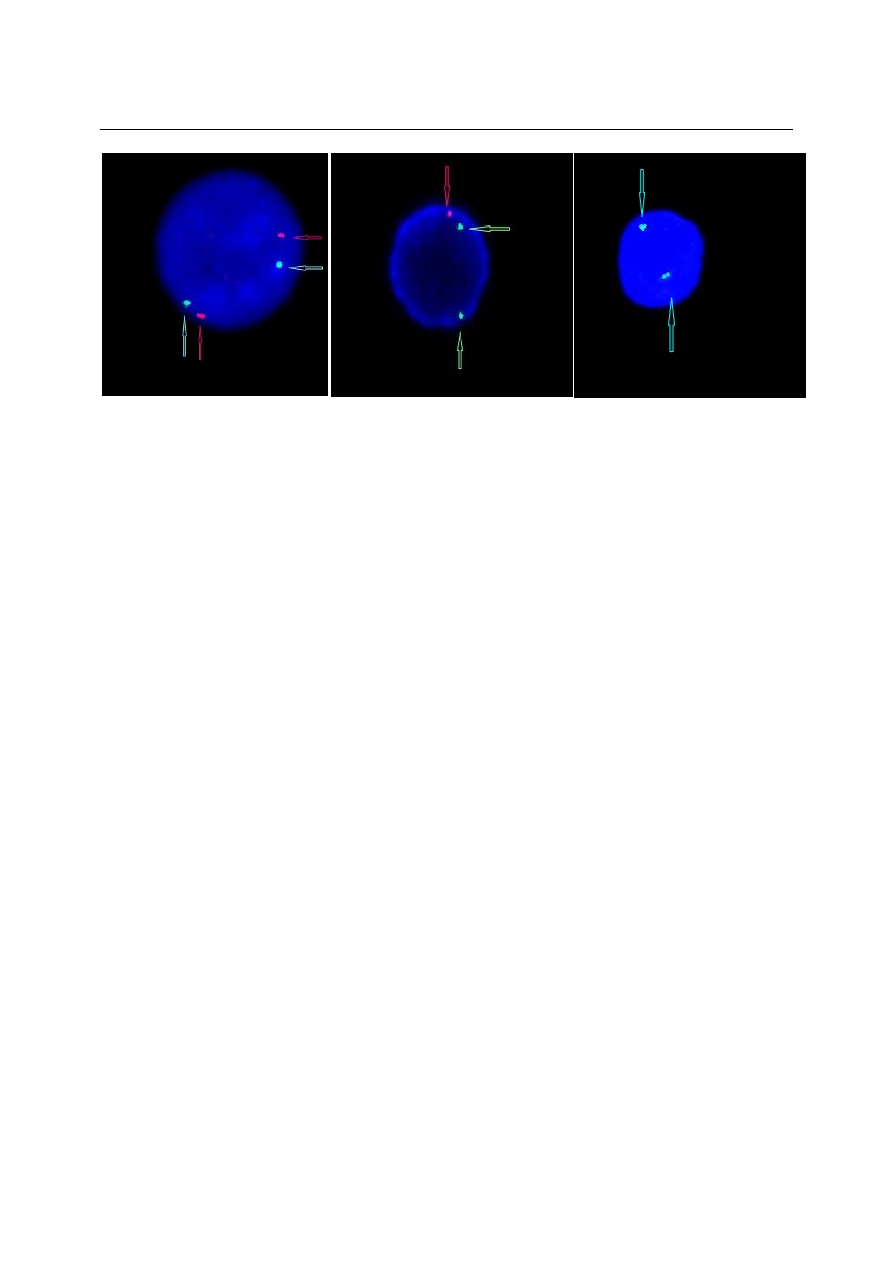
M. KRAWCZYK-KULIŚ, S. KYRCZ-KRZEMIEŃ
504
A.
B.
C.
Fot. 1. Hybrydyzacja in situ z użyciem sondy Vysis LSI P16(9p21)SpectrumOrange/CEP 9 SpectrumGreen Probe. A. Obec-
ność obu alleli genu P16 – widoczne dwa sygnały czerwone oraz dwa kontrolne sygnały zielone B. Delecja jednego allela
(hemizygotyczność genu P16) C. Delecja obu alleli (homozygotyczność genu P16).
Wiele
doniesień dotyczy zaburzeń w regionie 9p21 lub bezpośrednio samego genu jako czynników
wpływających również na przebieg kliniczny i rokowanie w białaczkach. W zależności od zastosowania
mniej lub bardziej dokładnej metody badania, zmiany te mogą być zdefiniowane poprzez aberracje
regionu 9p21 i obejmują wtedy zapewne większą liczbę genów, albo też poprzez molekularne zmiany
samego genu. Na kontrowersyjne opinie dotyczące tego problemu, może wpływać również duża hete-
rogenność jego nieprawidłowości, heterogenność grup badanych (dorośli, dzieci) i różne badane rodzaje
białaczek.
Aberracje chromosomowe i zmiany molekularne obszaru 9p21 w ALL
Aberracje i inne zaburzenia w obszarze 9p21 wśród białaczek najczęściej obserwuje się w ALL
(z ang. acute lymphoblastic leukemia); vice versa różne formy inaktywacji (delecja, uszkodzenie) sta-
nowią najczęstszą anomalię genetyczną stwierdzaną w przebiegu T-ALL [10]. Anomalie te bywają
zarówno aberracjami pierwotnymi jak i wtórnymi [11]. Zmiany te są badane przy użyciu wielu technik
przy różnym stopniu czułości i nadal jeszcze nie można z nich wyciągnąć sumarycznych końcowych
wniosków.
Po raz pierwszy zmiany cytogenetyczne w postaci hemizygotycznych delecji w tym regionie za-
uważyli Cimino i wsp. w 1979 roku u 2/19 chorych z ALL (12). W klasycznej ocenie kariotypu, delecje
9p obserwowali także Kowalczyk i wsp. u 7/90 pacjentów, Chilcote i wsp. u 6/8 oraz Pollack i wsp
u 20/148 analizowanych chorych [13, 14, 15]. Technikami cytogenetyki klasycznej w ostatnich latach
obecność delecji 9p w ALL u dzieci weryfikowali również Anderson i wsp, stwierdzając ją w 10%
u 152 badanych pacjentów. Autorzy ci zwracają uwagę na możliwość istnienia „ukrytej” delecji 9p,
którą można stwierdzić tylko techniką FISH (fluorescencyjna hybrydyzacja in situ) [16]. Odsetek pa-
cjentów wykazujących technikami klasycznej cytogenetyki delecje 9p można traktować w poszczegól-
nych badaniach jako zbliżony; wyjątkiem jest tylko publikacja Chilcote i wsp. którzy delecję tę stwier-
dzili u ok. 95% badanych pacjentów (6/8) co zapewne było związane z ogólną małą liczbą osób bada-
nych [14]. We wszystkich dotychczasowych obserwacjach zauważono, że w przeważającej liczbie pa-
cjentów wykryto białaczkę typu T, rzadziej typu null.
Wprowadzenie technik FISH do diagnostyki hematoonkologicznej w oczywisty sposób zwiększyło
wykrywanie delecji 9p, a zwłaszcza tzw. „delecji ukrytych”. Po raz pierwszy wykryto właśnie tymi

Współczesne standardy w diagnostyce i leczenie OBL
505
technikami nie tylko delecje hemizygotyczne w regionie 9p, ale również delecje homozygotyczne obu
homologicznych chromosomów. Jako homozygotyczne stwierdzono je u 8/65 pacjentów, natomiast
jako hemizygotyczne u 6/65; delecje te występowały nie tylko w T-ALL ale również w B-ALL [17].
Inni autorzy obserwowali znaczącą różnicę pomiędzy częstościami delecji homo- i hemizygotycznej
pomiędzy dziećmi i dorosłymi chorymi na białaczki; u dzieci pojawiają się one z częstością odpowied-
nio w 11% i 16%, natomiast u dorosłych odpowiednio w 30% i 20%. Zwraca zatem uwagę znacznie
częstsze manifestowanie się delecji u dorosłych przy jednoczesnym odwróceniu proporcji delecji homo
i hemizygotycznych [18]. Nieco odmienne częstości występowania delecji u dzieci i dorosłych o nie-
zdefiniowanym bliżej immunofenotypie obserwowali Lee i wsp. wykrywając je odpowiednio u 28%
i 35% [19]. Sulong i wsp. techniką FISH obserwowali delecję 9p u 21% dzieci z B-ALL, zwracając
uwagę na obecność delecji 9p w stosunkowo wysokim, bo aż w 50% odsetku ALL typu T [20]. W kon-
tekście badań FISH zwraca uwagę opublikowany niedawno przez Healey i wsp. pojedynczy przypadek
dziecka z T-ALL i hiperdiploidią w którego kariotypie we wszystkich trzech kopiach chromosomu 9,
stwierdzono hemizygotyczną delecję 9p21 [21]. Zbliżone wartości przedstawił Bungaro i wsp., którzy
homozygotyczne delecje obserwowali u 29% dzieci z ALL (7/24) i w 12% delecje hemizygotyczne
(3/24) [22]. Wdrożenie do diagnostyki hematoonkologicznej bardziej precyzyjnych technik: porów-
nawczej hybrydyzacji genomowej (CGH, z ang. Comparative Genomic Hybridization) i porównawczej
hybrydyzacji genomowej do mikromacierzy (a-CGH, z ang. array-CGH) pozwalało mieć nadzieję, że
metody te pozwolą dokładniej scharakteryzować zarówno częstość pojawiania się, jak i strukturę delecji
regionu 9p. Do chwili obecnej danych takich dostarcza Strefford i wsp. którzy badali techniką CGH 58
pacjentów (dorosłych i dzieci z ALL) stwierdzając delecję regionu 9p u 36% z nich. Istotną obserwacją
było stwierdzenie, że u poszczególnych pacjentów delecja ta charakteryzuje się różnym zasięgiem
i rozmiarami, zawsze jednak zawiera w sobie obszar genów CDKN2A i CDKN2B. Autorzy ci sugerują,
że udział innych genów z obszaru delecji może wpływać na przebieg kliniczny białaczki. Zwraca rów-
nież uwagę, że mimo użycia metody potencjalnie bardziej precyzyjnej aniżeli analizy FISH, odsetek
pacjentów wykazujących delecje w technice CGH nie był znacząco wyższy aniżeli w cytowanych
wcześniej badaniach z użyciem techniki FISH [23]. Delecje w obszarze 9p21 powodujące inaktywacje
genu P16, jak wspomniano wyżej, występują również w innych niż ALL nowotworach. Jednakże, jak
wykazali Kohno i Jokota w komórkach białaczkowych są one tworzone w nielicznych i zdefiniowanych
miejscach 9p, poprzez mechanizm „nieuprawnionego” działania kompleksu białkowego RAG uczestni-
czącego w reperacji pęknięć dwuniciowych DNA; podczas gdy takie same pęknięcia w guzach litych są
tworzone i naprawiane w przypadkowych miejscach 9p w sposób jeszcze nieznany, ale z pewnością
inny [24]. Z badań Schifamana i wsp. wynika jednoznacznie, że wzorzec molekularny delecji 9p jest
unikalny i odrębny w B-ALL w porównaniu do T-ALL, a delecje ograniczone wyłącznie do samego
genu CDKN2 bez utraty otaczających sekwencji pojawiają się niemal wyłącznie w T-ALL [25]. Nowe
znaczące informacje przyniosły badania grupy O. Zuffardi z 2009 r. Delecje 9p zaobserwowano jako
homo- i hemizygotyczne w 27% (18/65 pacjentów), główny jednak ciężar badań dotyczył poszukiwania
mechanizmów powstawania delecji i jej precyzyjnej diagnostyki molekularnej. Grupa ta, powstanie
delecji, śladem hipotez zaproponowanych przez Cauyela i wsp. w 1997 r., tłumaczy mechanizmem jej
flankowania przez sekwencje homologiczne do kodujących sygnałowych sekwencji rekombinacji hep-
tamerowych (heptamer RRS-s), które są odnajdywane przez regulujący rekombinacje V(D)J kompleks
RAG. Poprzednio Cayelo i wsp stwierdzili, że punkty złamań prowadzące do inaktywacji genu P16
występują w dwóch skupieniach: MTS1
bcrα-
i MTS2
bcrß.
. Odpowiadają one powtarzającemu się miejscu
rekombinacji mieszczących się naprzeciw siebie obszarów: 5’ egzonu 1 MTS2 oraz 5’ egzonu 1α
MTS1. Ich dokładniejsza lokalizacja odpowiada polimorficznym powtórzeniom (CA). W sąsiednich
rejonach częściej spotyka się dodatkowe krótkie delecje, losowe addycje nukleotydów bogate w GC
i klonalne naprzeciwległe sekwencje, co odpowiada obrazowi nieuprawnionej aktywności rekombinazy
V(D)J. Zatem proces, który jest niezbędny do prawidłowego różnicowania komórek T, odgrywa rów-
nież istotną rolę w patogenezie T-ALL. Jak sugeruje również grupa O. Zuffardi, powstanie delecji 9p21

M. KRAWCZYK-KULIŚ, S. KYRCZ-KRZEMIEŃ
506
jest najprawdopodobniej skutkiem nieprawidłowej, nieuprawnionej i niehomologicznej rekombinacji
w obszarze oddziaływania kompleksu RAG. Zastosowanie mikromacierzy pozwoliło na uzyskanie cha-
rakterystyki molekularnej aberracji 9p21 z rozdzielczością od 1 do 10 kb, zarówno w odniesieniu do
regionów zawierających pojedyncze kopie jak i sekwencje powtarzalne. Mimo że obserwacje te nie
zwiększyły, w stosunku do wcześniejszych oszacowań, częstości wykrywania delecji, pozwoliły jednak
na stwierdzenie w niej indywidualnie zróżnicowanych punktów złamań i sklonowanie poszczególnych
miejsc: złamanie–połączenie, a to z kolei umożliwiło dalsze scharakteryzowanie zarówno ich dokładnie
określonego miejsca lokalizacji jak i zidentyfikowanie sekwencji flankujących. Wszystkie badane dele-
cje obejmowały zawsze CDKN2A z dodatkiem delecji genu CDKN2B lub MTAP. Gdy delecje miały
charakter heterozygotyczny, pozostałe allele CDKN2A i/lub CDKN2B ulegały zawsze hipermetylacji
w wyspach powtórzeń CpG ich regionów promotorowych [10, 26]. Technikę hybrydyzacji do mikro-
macierzy zastosowali również cytowani wcześniej Sulong i wsp. stwierdzając, że średnia wielkość de-
lecji wynosiła 14,8 Mb, a w skład delecji biallelicznych wchodziła z reguły delecja duża (śr. 23.3 Mb)
i mała (1,4 Mb). Wśród badanych 86 pacjentów tylko dwie małe delecje były mniejsze niż rozdziel-
czość techniki FISH, i nie mogły być z jej pomocą wykryte, co potwierdza, że technika ta nadal może
być w sposób miarodajny stosowana. Częstość delecji różniła się w zależności od współwystępujących
innych uwarunkowań; niższy odsetek stwierdzanych delecji występował u chorych z wysoką hiperdi-
ploidią i rearanżacją ETV6/RUNX1 i 11q23/MLL (odpowiednio 11%, 15% i 13%). Natomiast u chorych
z t(9;22), t(1;19), rearanżacjami TELX3 lub TELX1 odsetek był znacząco wyższy (odpowiednio 61%,
42%, 78%, 89%). Mikromacierze SNIP pozwoliły wykazać utratę heterozygotyczności w miejscu dele-
cji, jednak nie zawsze przebiegającą z inaktywacją CDKN2A, co może sugerować obecność w tym re-
gionie innych genów które wnoszą swój wkład do patomechanizmu zaburzeń [20]. Otwartym proble-
mem jest, czy delecja 9p odzwierciedla jakieś zmiany preegzystujące w linii germinalnej czy też jest
zmianą o charakterze somatycznym. Za tą pierwszą możliwością przemawiają badania Morrisona
i wsp., którzy stwierdzili, że w 9/10 badanych przypadków delecja dotyczy allela pochodzenia matczy-
nego, sugerując że jakieś zdarzenia predysponujące do delecji pojawiają się już w linii germinalnej lub
nawet wcześniej [27]. Analizy molekularne genu P16 i jego obszaru w ALL były prowadzone od wielu
lat zarówno w ALL typu B, jak i ALL typu T. Stosowano w nich zarówno techniki o historycznym już
znaczeniu jak Southern blott, jak i nowoczesne metody RT-PCR (Reverse Transcription PCR) oraz
ilościowy PCR (Q-PCR/”real time” PCR). Porównywane wzajemnie wyniki wykazują bardzo znaczny
rozrzut zarówno, gdy rozpatruje się liczbę pacjentów wykazujących delecje, jak i rodzaj zastosowanych
technik molekularnych. I tak techniką Southern blott delecje wykrywano u 7,6%- 44,0% badanych pa-
cjentów z ALL [7, 9, 28, 29]. Techniką RT-PCR analogicznie wykrywano ją od 17,6% do 53% pacjen-
tów [28, 30, 31], a techniką Q-PCR od 31% do 72,7% [28, 32, 33, 34]. Wydaje się zatem, że spośród
stosowanych przez różnych badaczy technik molekularnych największą czułością w wykrywaniu dele-
cji P16 charakteryzowała się technika Q PCR. Jednakże ostateczne wnioski mogłyby być wyciągnięte
tylko wtedy, gdyby wszystkie te techniki były stosowane równocześnie w tej samej grupie pacjentów.
Odsetek ALL typu T i typu B również wahał się w obserwacjach poszczególnych badaczy: od (odpo-
wiednio typ T/B) w 29%/71%, co oznacza znaczne częstsze występowanie delecji w ALL typu B, po
wartości pośrednie ~50%/50%, aż po jednoznacznie częstsze występowanie delecji w ALL typu T:
67%/36% oraz 70%/30% [29, 33, 34, 33]. Warto w tym miejscu zauważyć, że wcześniej analizowane
dane uzyskane rożnymi technikami cytogenetycznymi jednoznacznie wskazywały na częstsze wystę-
powanie delecji P16 w ALL typu T. W żadnym z dostępnych opublikowanych opracowań nie przepro-
wadzono równoczesnego porównania częstości występowania delecji P16 w takiej samej ogólnej grupie
badanych dzieci i dorosłych. Poza jednym równoległym opracowaniem większych grup, częstość dele-
cji oceniano wyłącznie u dzieci. Tym samym nie można obecnie nawet oszacować czy częstość poja-
wiania się delecji P16 w ALL u dzieci i dorosłych jest taka sama czy różna [18]. Również nieliczni
tylko badacze technikami molekularnymi porównywali dane dotyczące wzajemnych częstości pojawia-
nia się pośród delecji, anomalii homo- i hemizygotycznych. Te dane wykazują wartości zbliżone, np.:

Współczesne standardy w diagnostyce i leczenie OBL
507
~17%-14% [32], ~50%-50% [31], ~35-30% [33]. Jedynie Kees i wsp. stwierdzili ponad dwukrotną
przewagę delecji hemizygotycznych (~33,3%) od homozygotycznych (~17%); podobnie Carter i wsp.
~25%-13% [9, 29].
Wiadomo,
że unieczynnienie genu jest nie tylko wynikiem jego delecji lub mutacji, ale również
może być skutkiem nieprawidłowej metylacji powtórzeń CpG promotora genu. Tego rodzaju zaburze-
nia metylacji genu P16 próbowano zatem identyfikować również w ALL, stwierdzając je odpowiednimi
technikami molekularnymi w rożnych odsetkach pacjentów, np. 6%, 15% i 44% [32, 35, 29]. Tylko
w jednej publikacji porównano dane dotyczące metylacji w ALL u dzieci i dorosłych stwierdzając za-
burzenia odpowiednio u 34% i 26% badanych [18]. W kontekście powyższych informacji zwraca uwa-
gę analiza wieloośrodkowych badań z obszaru Wielkiej Brytanii opracowana przez Sulong i wsp.,
w której częściowe zaburzenia metylacji zaobserwowano tylko u jednego z przebadanych w tym kie-
runku 99 pacjentów [20]. Kontrastuje to z analizą piśmiennictwa przeprowadzoną przez Kruga i wsp.,
którzy zaburzenia metylacji promotora odnotowali aż u 44% pacjentów z różnych badanych grup obej-
mujących zarówno T jak i B- ALL [3].
Ekspresja P15 i P16 w ALL
Oceniano
również przy użyciu różnych technik (immunocytochemia, RT-PCR, Q-PCR, microarray)
zmiany ekspresji genu P16 towarzyszące delecji, stwierdzając, jak można się było spodziewać, obniże-
nie ekspresji niekiedy do wartości niemal niewykrywalnych. Interesujące jest, że temu obniżeniu eks-
presji towarzyszy z reguły jej obniżenie również dla genów SMAD1 i JAG1 [22]. Stopień obniżenia
ekspresji był równoległy do charakteru delecji; i był widoczny w przypadku delecji hemizygotycznych,
a w delecjach homozygotycznych stwierdzano całkowity brak ekspresji [9]. Całkowity brak ekspresji
częściej występował w T – ALL [36].
Wartość prognostyczna zmian 9p21 w ALL
Wyniki
badań nie pozwalają obecnie na jednoznaczne interpretowanie rokownicze dele-
cji/inaktywacji w obszarze 9p21. Większość autorów uważa jednak, że jest ona jednym z możliwych
czynników złego rokowania [13, 37, 38]. Jednakże inni uważają, że zmiany te nie są odrębnym czynni-
kiem złego rokowania; w badanych grupach złe rokowanie wynika ich zdaniem raczej ze współistnienia
innych czynników wysokiego ryzyka [32, 33, 39]. Wydaje się jednak, że gorsze rokowanie występuje
częściej w przypadku homozygotycznej delecji i/lub całkowitym braku ekspresji białka P16. Również
w przytoczonych wcześniej danych Einsiedela i wsp. czas trwania remisji był znacząco krótszy w gru-
pie chorych z delecją niż w grupie chorych bez delecji [33]. Chorzy z delecją P16 wykazują w analizie
Kaplan-Meyer znacznie wyższe ryzyko wznowy; wskaźnik ryzyka wynosił 11,558 (P=.000539) dla
chorych z delecją homozygotyczną i 6,558 (P= 00687) dla chorych z delecją hemizygotyczną [29].
Według niektórych autorów, poziom hipermetylacji genów w obszarze 9p21 może być nowym marke-
rem pojawienia się kryzy i progresji u dorosłych chorych z T-ALL. Poziom hipermetylacji korespondu-
je również z krótszym czasem całkowitego przeżycia [35]. Inni autorzy uważają, że delecja ta wpływa
również na reakcje komórek białaczkowych na leki, zwłaszcza dotyczy to metotreksatu. Prawdopodob-
nie wynika to nie ze zmian samego genu P16, ale z współwystępujących jednoczesnych zmian sąsiadu-
jących genów P15 i MTAP [32]. Należy również wspomnieć, że niektórzy autorzy uważają, że istnienie
delecji/inaktywacji nie ma żadnego wpływu na rokowanie zarówno u dorosłych jak i u dzieci, ostrożnie
sugerując, że jedynym wyjątkiem może być sytuacja występowania delecji homozygotycznej u doro-
słych (ale nie u dzieci) pacjentów z B-ALL [18]. Powyższe dane sugerują, że znaczenie prognostyczne
delecji/unieczynnienia 9p u dzieci nie jest jednoznacznie określone; natomiast u dorosłych pacjentów
z ALL uważa się, że del(9p) jest czynnikiem lepszego rokowania zwłaszcza w porównaniu z pacjentami
Ph’ pozytywnymi lub pacjentami z kariotypem złożonym [11].

M. KRAWCZYK-KULIŚ, S. KYRCZ-KRZEMIEŃ
508
Aberracje 9p21 w AML
Delecja/unieczynnienie w opisywanym obszarze 9p była weryfikowana również u chorych z AML
(z ang. acute myeloid leukemia) zarówno przy użyciu metod cytogenetycznych i molekularnych; publi-
kacje na ten temat są jednak nieliczne. W porównaniu do wyżej cytowanych badań w ALL, w AML
zmiany w regionie 9p pojawiają się zarówno u dzieci jak i u dorosłych znacznie rzadziej. I tak np.: wy-
krywalną cytogenetycznie utratę materiału krótkiego ramienia chromosomu 9 znaleziono zaledwie
u 3/500 [15]. Inni po analizie 102 próbek szpiku kostnego różnych pacjentów, delecje wykazali u 2%
badanych [19]. Technikami molekularnymi LOH genu P16 nie znaleziono u żadnego z 14 dzieci
z AML [25]. Natomiast co najmniej tak samo częstym jak w ALL, mechanizmem unieczynnienia może
okazać się metylacja promotora; w jedynym jak dotąd opracowaniu pojawiała się ona u ~40% badanych
dzieci [7]. Wykazano również, że w AML zmniejszona jest ekspresja P16, u niektórych pacjentów jest
ona niewykrywalna. Poziom ekspresji może być zależny od wieku chorego, gdyż w tej samej grupie
badanej jest ona zmniejszona zwłaszcza u starszych pacjentów (średnia wieku 59 lat) [40, 19].
Aberracje 9p21 w MDS
W MDS (z ang. myelodysplastic syndromes), tylko jednostkowe opracowania dotyczą genu P16.
Papadhimitriou i wsp. nie odnaleźli jego delecji u żadnego z 34 badanych pacjentów, a Pollack i wsp.
utratę materiału 9p stwierdzili tylko u 1/120 [38, 15]. Zaskoczeniem może być wynik obserwacji Lee
i wsp. w porównaniu do poprzednio cytowanych badań, którzy oceniali ekspresję P16 w mieszanej
grupie pacjentów z rożnymi białaczkami, w tym u 5 z MDS. U 2 z nich stwierdzili oni podwyższenie
ekspresji P16, a u jednego współistniała ona z jednocześnie podwyższoną ekspresją P14; znikoma licz-
ba pacjentów nie pozwala jednak na wyciąganie wniosków [19].
Aberracje 9p21 w CML
Nieliczne badania utraty/inaktywacji regionu 9p21 u pacjentów z CML (z ang. chronic myeloid
leukemia) nie są jednoznaczne, wydają się jednak sugerować, że zjawisko to w tym typie białaczek
odgrywa raczej marginalną rolę. Utratę tego regionu znaleziono zaledwie u 1/200 pacjentów z CML
w kryzie blastycznej, a techniką mikromacierzy sond FISH nie znaleziono jej u żadnego z 47 badanych
[15, 19]. Inni stosując technikę RT-PCR wykazali delecję homozygotyczną u 6/21 pacjentów z przeło-
mu limfoblastycznego, podczas gdy w fazie przewlekłej i kryzie mieloblastycznej nie znaleziono delecji
w ogóle. Nie stwierdzono również hipermetylacji w 21 analizowanych przypadkach z przełomu limfo-
blastycznego. Zmiany genu P16 uważane są w kryzie blastycznej za rzadkie, nie wpływające na kli-
niczny obraz choroby i wyniki leczenia [41]. Stwierdzono jednak, że poziom ekspresji genu P16 mie-
rzony poziomem swoistego mRNA znacznie wzrasta u 4/5 chorych (4 w fazie przewlekłej, 1 w fazie
akceleracji) [40]. Odwrotne i niejednoznaczne wyniki otrzymali Cividin i wsp. u 109 pacjentów z CML,
u których poziom odpowiedniego mRNA był znacząco niski, natomiast znacząco wysoki u pacjentów
leczonych alfa interferonem bez korzystnej odpowiedzi na leczenie. Prawidłowy poziom ekspresji P16
obserwowano u pacjentów opornych na immatinib, być może, zatem poszczególne rodzaje lekooporno-
ści w CML korelują z poziomem ekspresji genu P16 [43].
Aberracje 9p21 w CLL
Podobnie jak w CML, pojedyncze dostępne opracowania pozwalają przypuszczać, że zmiany genu
P16 odgrywają marginalną rolę w CLL (z ang. chronic lymphocytic leukemia). Zmiany sekwencji
o charakterze polimorfizmu obserwowano u ~6% chorych, natomiast hipermetylację promotora genu
P16 u ~17,5%. Zwraca jednak uwagę mała liczba badań tego zagadnienia w CLL [44].

Współczesne standardy w diagnostyce i leczenie OBL
509
PODSUMOWANIE
Zmiany
genu
P16 w białaczkach, prowadzące do jego hemi- lub homozygotycznej inaktywacji,
mogą być identyfikowane poprzez stwierdzenie delecji większego przyległego do niego obszaru w ob-
rębie 9p, delecji tylko samego genu, wykrywane badaniem FISH, jak również zmiany w obrębie genu
wykrywane technikami molekularnymi o różnej rozdzielczości. Te ostatnie obejmują zarówno delecje
wewnątrzgenowe, mutacje punktowe, jak i zmiany metylacji unieczynniające gen. Porównując częstość
opisywanych mutacji punktowych do częstości delecji i zaburzeń metylacji, można stwierdzić, że muta-
cje punktowe są o wiele rzadsze, a wśród nich substytucje przeważają nad mutacjami typu stop bezpo-
średnio unieczynniającymi gen. Dotychczasowe badania powyższych procesów inaktywacji genu P16,
niezależnie od dokładności zastosowanej techniki i badanego typu białaczki, dają rozbieżne wyniki.
Zaburzenia w obrębie genu P16 wydają się dotyczyć głównie ALL, i mogą mieć znaczenie zarówno
w wyjaśnieniu patomechanizmu jak i w ocenie prognozy tego typu białaczki. W innych białaczkach jak
AML, CML i CLL oraz w zespołach mielodysplastycznych, zaburzenia takie odnotowuje się rzadko
i wydają się one mieć marginalne znaczenie.
PIŚMIENNICTWO
1. Epstein RJ. Kontrola cyklu komórkowego, apoptozy oraz procesu starzenia. Biologia molekularna człowieka. Red.
Lewiński A., Liberski P.P. Wyd. Czelej, Lublin 2006; 371-399.
2. Hatta Y, Koeffler HP. Role of tumor suppressor genes in development of adult T-cell leukemia/lymphoma (ATLL).
Leukemia 2002; 16:1069-1085.
3. Krug U, Ganser A, Koeffler HP. Tumor suppressor genes in normal and malignant hematopoesis. Oncogene 2002; 21:
3475-3495.
4. Bertin R, Acquaviva C, Mirebeau D, Guidal-Giroux C, Vilmer E, Cavé H. CDKN2A, CDKN2B and MTAP gene dosage
permits precise characterization of mono-and bi-allelic 9p21 deletions acute lymphoblastic leukemia. Genes,
Chromosomes Cancer 2003; 37: 44-57.
5. Quelle DE, Zindy F, Ashmun RA, Sherr C.J. Alternative reading frames of the INK4a tumor suppressor gene encode two
unrelated proteins capable of inducing cell cycle arrest. Cell 1995; 83: 993-100.
6. Okuda T, Shurtleff SA, Valentine MB, Raimondi SC, Head DR, Behm F. Frequent deletion of P16
INK4A
/MTS1,
P15
INK4B
/MTS2 in pediatric acute lymphoblastic leukemia. Blood 1995; 85: 2321-2330.
7. Guo SX, Taki T, Ohnishi H, Piao HY, Tabuchi K, Bessho F. Hypermethylation of p16 and p15 genes and RB protein
expression in acute leukemia. Leuk Res 2000; 24: 39-46.
8. Jorde LB, Carey JC, Bamshad MJ, White R.L. Genetyka nowotworów. Genetyka Medyczna. Red. Wojcierowski J. Wyd.
Czelej Lublin 2000; 260-282.
9. Kees U, Terry P. Ford J, Everett J, Murch A, Klerk N. Detection of hemizygous deletions in genomic DNA from leukemia
specimens for the diagnosis of patients. Leuk Res 2005; 29: 165-171.
10. Cayuela JM, Gardie B, Sigaux F. Disruption of the multiple tumor suppressor gene MTS1/p16(INK4a)/CDKN2 by
illegitimate V(D)J recombinase activity in T-cell acute lymphoblastic leukemias. Blood 1997; 90:3720–3726.
11. Moorman AV, Harrison J, Buck G, Richards SM, Secker-Walker LM, Martineu M. Karyotype is independent prognostic
factor in adult acute lymphoblastic leukemia (ALL): analysis of cytogenetic data from treated on the Medical Research
Council (MRC) OKALLXII/Eastern Cooperative oncology Group (ECOG) 2993 trials. Blood 2007; 109:3189-3197.
12. Cimino M, Rowley J, Kinnealey A, Variakojis D, Golomb H. Banding studies of chromosomal abnormalities in patient
with acute lymphocytic leukemia. Cancer Res 1979; 39: 227-238.
13. Kowalczyk J, Sandberg A. A possible subgroup of ALL with 9p-. Cancer Genet Cytogenet 1983; 9: 383-385.
14. Chilcote R, Brown E, Rowley J. Lymphoblastic leukemia with lymphomatous features associated with abnormalities of
the short arm of chromosome 9. N Engl J Med 1985; 313: 286-291.
15. Pollak Ch, Hagemeijer A. Abnormalities of the short arm of chromosome 9 with partial loss of material in hematological
disorders. Leukemia 1987; 1: 541-548.
16. Andreasson P, Höglund M, Békássy AN, Garwicz S, Heldrup J, Mitelman F. Cytogenetic and FISH studies of a single
center consecutive series of 152 childhood acute lymphoblastic leukemias. Eur J Haematol 2000; 65: 40-51.
17. Woo H, Kim D, Park H, Seong K, Koo H, Kim S. Molecular cytogenetic analysis of gene rearrangements in childhood
acute lymphoblastic leukemia. J Korean Med Sci 2005; 20: 36-41.

M. KRAWCZYK-KULIŚ, S. KYRCZ-KRZEMIEŃ
510
18. Kim M, Yim S, Cho N, Kang S, Ko D, Oh B. Homozygous deletion of CDKN2A (p16, p14) and CDKN2B (p15) genes is
a poor prognostic factor in adult but not in childhood B-lineage acute lymphoblastic leukemia: a comparative deletion and
hypermethylation study. Cancer Genet Cytogenet 2009; 195: 59-65.
19. Lee SD, Lee JH, Min CH, Kim TY, Oh BR, Kim HY. Application of high throughput cell array technology to FISH:
Investigation of the role of deletion of P16 gene in leukemia's. J Biotechnol 2007; 127: 355-360.
20. Sulong S, Moorman AV, Irving J, Strefford JC, Konn ZJ, Case MC. A comprehensive analyses of the CDKN2A gene in
childhood acute lymphoblastic leukemia reveals genomic deletion, copy number neutral loss of heterozygosity, and
association with specyfic cytogenetic subgroups. Blood 2009; 113: 100-107.
21. Healey K, Gray S.L, Halligan G.E, McKenzie A.S, Chadarevian J.P, Morrissette J.D. Hyperdiploidy with trisomy 9 and
deletion of the CDKN2A locus in T-cell acute lymphoblastic leukemia. Cancer Genet Cytogenet 2009; 190: 121-124.
22. Bungaro S, Dell'Orto MC, Zangrando A, Basso D, Gorletta T, Nigro LL. Integration of genomic and gene expression data
of childhood ALL without known aberrations identifies subgroups with specific genetic hallmarks. Genes Chromosomes
Cancer 2009; 48: 22-38.
23. Strefford JC, Worley
H, Barber K, Wright S, Stewart AR, Robinson H.M. Genome complexity in acute lymphoblastic
leukemia is revealed by array-based comparative genomic hybridization. Oncogene 2007; 26: 4306-4318.
24. Kohno
T
, Yokota J.
Molecular processes of chromosome 9p21 deletions causing inactivation of the p16 tumor suppressor
gene in human cancer: Deduction from structural analysis of breakpoints for deletions. DNA Repair 2006; 5:1273-1281.
25. Schiffman JD, Wang Y, McPherson LA, Welch K, Zhang N, Davis R. Molecular inversion probes reveal patterns of 9p21
deletion and copy number aberrations in childhood leukemia. Cancer Genet Cytogenet 2009; 193: 9-18.
26. Novara F, Beri S, Bernardo M, Bellazzi R, Malovini A, Ciccone R. Different molecular mechanisms causing 9p21
deletions in acute lymphoblastic leukemia of childhood. Hum Genet 2009; 126: 511-520.
27. Morison I, Ellis L, Teague L, Reeve A. Preferential loss of maternal 9p alleles in childhood acute lymphoblastic leukemia.
Blood 2002; 99: 375-376.
28. Hatta Y, Hirama T, Miller C, Yamada Y, Tomonaga M, Koeffler P. Homozygous deletion of the p15 (MTS2) and p16
(CDKN2/MTS1) genes in adult T-cell leukemia. Blood 1995; 85:2699-2704.
29. Carter TL, Watt PM, Kumar R, Burton PR, Reaman G. H, Sather H.N. Hemizygous p16
INK4A
deletion in pediatric acute
lymphoblastic leukemia predicts independent risk of relapse. Blood 2001; 97: 572-574.
30. Carter T, Terry P, Gottardo N, Baker D, Kees U, Watt P. Deletion of one copy the p16
INK4A
tumor suppressor gene is
implicated as a predisposing factor in pediatric leukemia. Biochem Biophys Res Commun 2004; 318:852-855.
31. Kustanovich A, Savitskaja T, Bydanov O, Belevtsev M, Potapnev M. Aberrant expression of tumor suppressor genes and
their association with chimeric oncogenes in pediatric acute lymphoblastic leukemia. Leuk Res 2005; 29:1271-1276.
32. Mirebeau D, Acquaviva C, Suciu S, Bertin R, Dastugue N, Robert A. The prognostic significance of CDKN2A, CDKN2B
and MTAP inactivation in B-lineage acute lymphoblastic leukemia of childhood. Result of the EORTC studies 58881 and
58951. Hematol J 2006; 91: 881-885.
33. Einsiedel H, Taube T, Hartmann R, Wellmann S, Seifert G, Henze G. Deletion analysis of p16
INKa
and p15
INKb
in relapsed
childhood acute lymphoblastic leukemia. Blood 2002; 99:4629-4631.
34. Zuna J, Muzikova K, Hrusak O, Stary J, Trka J. Significance of real-time quantitative polymerase chain reaction detection
of p16 gene deletion in childhood acute lymphoblastic leukemia. Haematologica 2002; 87:668-669.
35. Sato H, Oka T, Shinnou Y, Kondo T, Washio K, Takano M. Multi-step aberrant CpG island hyper-methylation is
associated with the progression of adult T-cell leukemia/lymphoma. Am J Pathol 2010;1:402-415.
36. Dalle J, Fournier M, Nelken B, Mazingue F, Laï J, Bauters F. p16INK4a immunocytochemical analysis is an independent
prognostic factor in childhood acute lymphoblastic leukemia. Blood 2002; 99:2620-2623.
37. Tutor O, Diaz M, Ramirez M, Algara P, Madero L, Martinez P. Loss of heterozygosity of p16 correlates with minimal
residual disease at the end of the induction therapy in non-high risk childhood B-cell precursor acute lymphoblastic
leukemia. Leukemia Res 2002; 26: 817-820.
38. Papadhimitriou S, Ploychronopoulou S, Tsakiridou A, Androutsos G, Paterakis G, Athanassiadou F. p16 inactivation
associated with aggressive clinical course and fatal outcome in TEL/AML1-positive acute lymphoblastic leukemia. J
Pediatr Hematol Oncol 2005; 27: 675-677.
39. Lemos J, Defavery R, Scrideli C, Tone L. Analysis of P16 gene mutations and deletions in childhood acute lymphoblastic
leukemia. Med J 2003; 12: 58-62.
40. Jonge H, Bont E, Valk P, Schuringa J, Kies M, Woolthuis C. AML at older age: age-related gene expression profiles
reveal a paradoxical down-regulation of P16 mRNA with prognostic significance. Blood 2009; 114:2869-2877.
41. Hernándes-Boluda J, Cervantes F, Colomer D, Vela M, Costa D, Fe-Paz M. Genomic p16 abnormalities is the progression
of chronic myleoid leukemia into blast crisis: A sequential study in 42 patients. Exp Hematol 2003; 31: 204-210.
42. Lee Y, Park J, Kang H, Cho H. Overexpression of p16
INK4A
and p14
ARF
in haematological malignancies. Clin Lab.
Haematol 2003; 25: 233-237.

Współczesne standardy w diagnostyce i leczenie OBL
511
43. Cividin M, Ayrault O, Sorel N, Séité P, Brizard F. Expression of the cycle regulators p14
ARF
and p16
INK4a
in chronic
myeloid leukemia. Leuk Res 2006; 30: 1273-1278.
44. Tsirigotis P, Pappa V, Labropoulus S, Papageorgiou S, Kontsioti F, Darvenoulas J. Mutational and methylation analysis of
the cyclin - dependent kinase 4 inhibitor (P16
INK4A
) gene in chronic lymphoblastic leukemia. Eur J Haematol 2006; 76:
230-236.
Praca wpłynęła do Redakcji 11.08.2010 r. i została zakwalifikowana do druku 21.10.2010 r.
Adres do korespondencji:
Ewa Studniak
Samodzielna Pracownia Cytogenetyki
Ul. Połabska 4
70-215 Szczecin
Tel. 91 466-15-45
e-mail: ewa.studniak@gmail.com
Document Outline
Wyszukiwarka
Podobne podstrony:
7 Wykl 7 str 4 tab 1 N 5 id 612 Nieznany (2)
Hurra I Lekcja 3 Str 24 pub id Nieznany
07 05 2013 odwiert (1)id 6788 Nieznany
Hurra I Lekcja 3 Str 27 pub id Nieznany
501 id 41999 Nieznany (2)
321[07] 01 122 Karta pracy egza Nieznany (2)
ntw 07 2005 str 62 63
07 popyt podaz www przeklej pli Nieznany (2)
07 woj dolnoslaskie uzdr zid 70 Nieznany
Aronson czlowiek istota spole str 501 516 id 69178 (2)
Hurra I Lekcja 3 Str 25 pub id Nieznany
Hurra I Lekcja 3 Str 28 pub id Nieznany
B 07 52 0264 Instr obslugi US 9 Nieznany
7 Wykl 7 str 4 tab 1 N 5 id 612 Nieznany (2)
Hurra I Lekcja 3 Str 24 pub id Nieznany
Das Volk und der Horn [w] Süddeutsche Zeitung Nr 169 2017 07 25, str 3
ntw 07 2005 str 62 63
więcej podobnych podstron