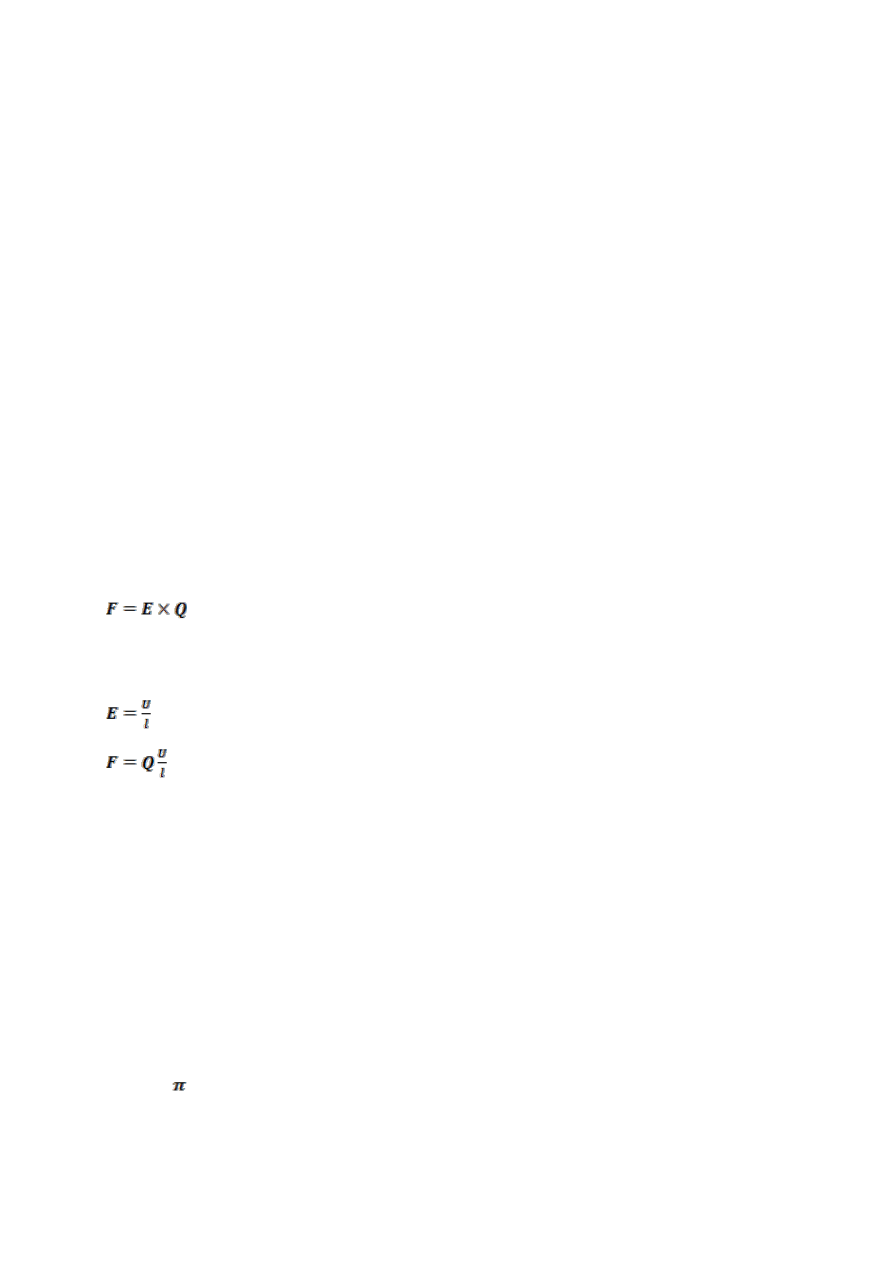
1
opracowała dr A. Żylicz-Stachula
I. ELEKTROFOREZA - PODSTAWY TEORETYCZNE.
Elektroforeza -
ruch cząstek obdarzonych ładunkiem w roztworze wodnym w polu
elektrycznym.
Naładowane cząsteczki poruszają się w kierunku elektrody o przeciwnym ładunku.
Różnorodność ich ładunków i mas, zdolność do dysocjacji lub tworzenia kompleksów
z obdarzonymi ładunkiem składnikami środowiska sprawia, iż w rezultacie różne
cząstki mieszaniny poruszają się z różną prędkością.
Jeżeli w trakcie doświadczenia zachowane zostaną stałe parametry:
różnica potencjałów
natężenie prądu
pH środowiska
temperatura roztworu
przewodność roztworu
lepkość roztworu
to dana cząstka będzie poruszać się ze stałą prędkością.
Siła F działająca na cząstkę w polu elektrycznym jest proporcjonalna natężenia pola
E
[V/cm]
i ładunku cząstki Q [C].
(1)
Natężenie pola elektrycznego E, definiowane jest jako iloraz napięcia elektrycznego
U
(różnicy potencjałów) oraz odległości pomiędzy potencjałami l.
(2) skąd
(3)
Im większe napięcie U - tym większa siła działa na cząsteczkę i szybsza jej migracja,
i odwrotnie im większa odległość l pomiędzy potencjałami (elektrodami), tym
mniejsze natężenie pola E i wolniejsza migracja cząsteczek.
Dlatego aby zapewnić optymalne natężenie pola elektrycznego E w aparatach
większych, o większej odległości pomiędzy elektrodami zwykle prowadzi się
elektroforezę przy wyższym napięciu niż w aparatach małych – o małej odległości
pomiędzy elektrodami.
Im większy jest wypadkowy ładunek elektryczny cząsteczki i natężenie pola
elektrycznego
– tym większa jest siła działająca na cząsteczkę. Cząstka osiąga
prędkość graniczną, która jest wypadkową oporu środowiska i pola elektrycznego.
Dla cząsteczki o kształcie kulistym (zgodnie z prawem Stokesa):
E·Q = 6
η r ν (4)
gdzie:
η- lepkość środowiska

2
r -
promień cząstki
ν- prędkość ruchu cząstki
Ważnym parametrem naładowanych cząstek jest ruchliwość elektroforetyczna, która
zależy od wartości pK naładowanych grup oraz od wielkości cząstek.
Ruchliwość elektroforetyczna (U): jest prędkością ν mierzoną w warunkach
standardowych
, wyrażoną na jednostkę pola E.
U =
(5)
Czynniki wpływające na ruchliwość elektroforetyczną:
rodzaj buforu
stężenie buforu
pH buforu
temperatura
natężenie pola elektrycznego
właściwości i rodzaj rozdzielanego materiału
Zazwyczaj określa się względną ruchliwość elektroforetyczną R
f
, czyli stosunek
odległości migracyjnej badanej substancji od odległości migracyjnej odnośnika.
Podstawę rozdzielenia cząstek za pomocą elektroforezy stanowi różnica prędkości
migracji cząstek w elektrolicie.
Rozdziały elektroforetyczne mogą być prowadzone:
w roztworach swobodnych np. elektroforeza kapilarna, elektroforeza
konwekcyjna, elektroforeza swobodna według Tiseliusa
w nośnikach np. bibuła, płytki cienkowarstwowe, żele
Metody rozdziału elektroforetycznego:
Prosta elektroforeza:
podczas której rozdział cząstek odbywa się na podstawie ich ruchliwości w danym
stałym pH i w stałym polu elektrycznym.
Izotachoforeza:
Jest metoda służącą głównie do analizy jakościowej. Podczas izotachoforezy
rozdział prowadzony jest w nieciągłym systemie buforów. Zjonizowana próbka
wędruje pomiędzy „szybkim’ elektrolitem prowadzącym a „wolnym” elektrolitem
kończącym. Cząstki o najwyższej ruchliwości elektroforetycznej poruszają się tuż za
prowadzącymi jonami, a te o najmniejszej ruchliwości elektroforetycznej wędrują
bezpośrednio przed elektrolitem kończącym. Tworzą się pasma zależne od stężenia
rozdzielanych substancji.
Ogniskowanie izoelektryczne:
Jest to metoda stosowana do rozdzielania substancji amfoterycznych np.
białek i
peptydów. Rozdział prowadzony jest w gradiencie pH. Cząstki przesuwają się w
kierunku anody lub katody do momentu osiągnięcia położenia w gradiencie pH, w
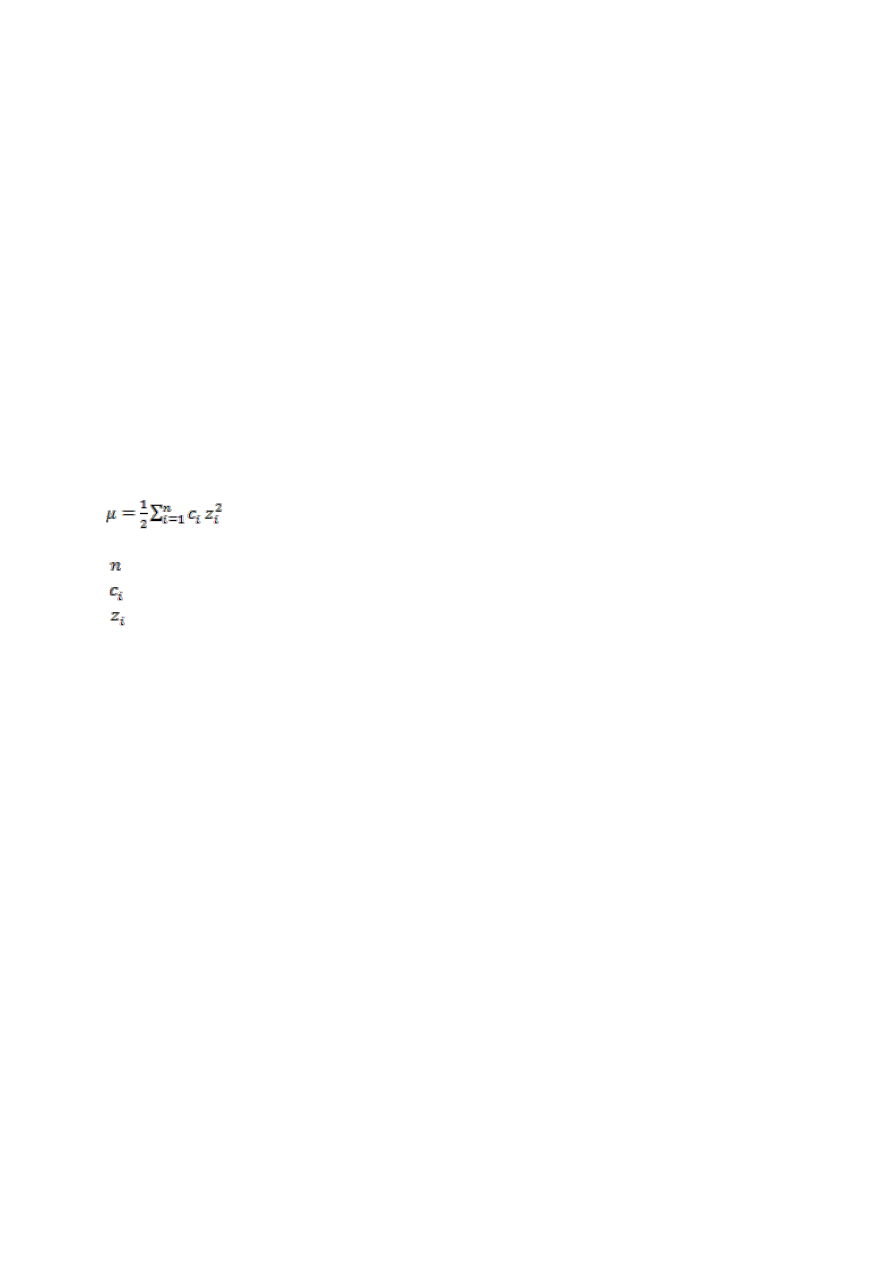
3
którym ich sumaryczny ładunek będzie wynosił zero. Ta wartość pH to tzw. punkt
izoelektryczny.
Istotnym kryterium wyboru metody elektroforetycznej jest rodzaj i charakter
analizowanej substancji. Makrocząsteczki takie jak białka są często wrażliwe na
pew
ne wartości pH i bufory. Źle dobrane warunki mogą powodować zmiany
konformacji, denaturację, tworzenie kompleksów i interakcje wewnątrzcząsteczkowe.
Należy również unikać efektu przeładowania (zbyt wysokiego stężenia rozdzielanej
substancji w roztworze). O
dpowiednio dobrane stężenie odgrywa ważną rolę w
momencie przejścia substancji z roztworu w żel. Do elektroforezy w warunkach
denaturujących (z solą sodową siarczanu dodecylu (SDS)) próbki muszą być
denaturowane, cz
yli przekształcone w micelle cząsteczka-detergent. Rodzaj
stabilizującego środowiska, np. żelu zależy od rozmiaru cząstki, która ma być
analizowana.
Istotnym czynnikiem jest również dobór odpowiedniego buforu. Elektroforetyczny
rozdział prowadzi się w buforze o ściśle określonym pH i stałej sile jonowej.
Siła jonowa roztworu µ dla mocnych elektrolitów wyraża się równaniem:
(6)
gdzie:
– liczba rodzajów jonów
– stężenie i-tego jonu w mol∙dcm
-3
– wartościowość i-tego jonu
Siła jonowa powinna być jak najniższa. Im wyższa jest siła jonowa, tym więcej prądu
przenoszą jony buforu i ruchliwość rozdzielanych cząstek maleje. Duże
rozcieńczenie buforu może powodować wypadanie niektórych białek z roztworu.
Konsekwencją przepływu prądu przez roztwór jest wydzielanie ciepła Joul’a, które
powoduje powstawanie prądów konwekcyjnych, utrudniających należyty rozdział.
Jeśli natomiast roztwór jest stabilizowany (np. bibułą czy żelem), to wywiązujące się
ciepło powoduje zagęszczenie elektrolitu na pasku, czego konsekwencją są znaczne
zmiany natężenia pola elektrycznego zachodzące wzdłuż paska w czasie trwania
elektroforezy.
Dlatego lepiej stosować w zasilaczu stałe natężenie (mA) niż stałe
napięcie prądu (V).
Do elektroforezy powinno się stosować bufory o minimalnej pojemności, żeby pH
analizowanej próbki nie miało wpływu na cały system. Ważny jest też rozmiar
naczynia do elektroforezy. Odpowiednio dobrany umożliwi zachowanie stałego i
stałej siły jonowej. W systemach kapilarnych i pionowych wartość pH buforu ustala
się w ten sposób, aby wszystkie cząsteczki badanej próbki były naładowane albo
ujemnie albo dodatnio i wędrowały w polu elektrycznym w określonym kierunku.
Ciepło Joul’a:
Podczas elektroforezy prąd wykonuje w żelu pracę, która powoduje wzrost
temp
eratury żelu. Efekt ten zwiększa dyfuzję polijonów podczas elektroforezy i
dlatego limituje użycie bardzo wysokich napięć podczas elektroforezy.
Ciepło Joul’a w – definiujemy jako moc wydzielaną w objętości podczas elektroforezy
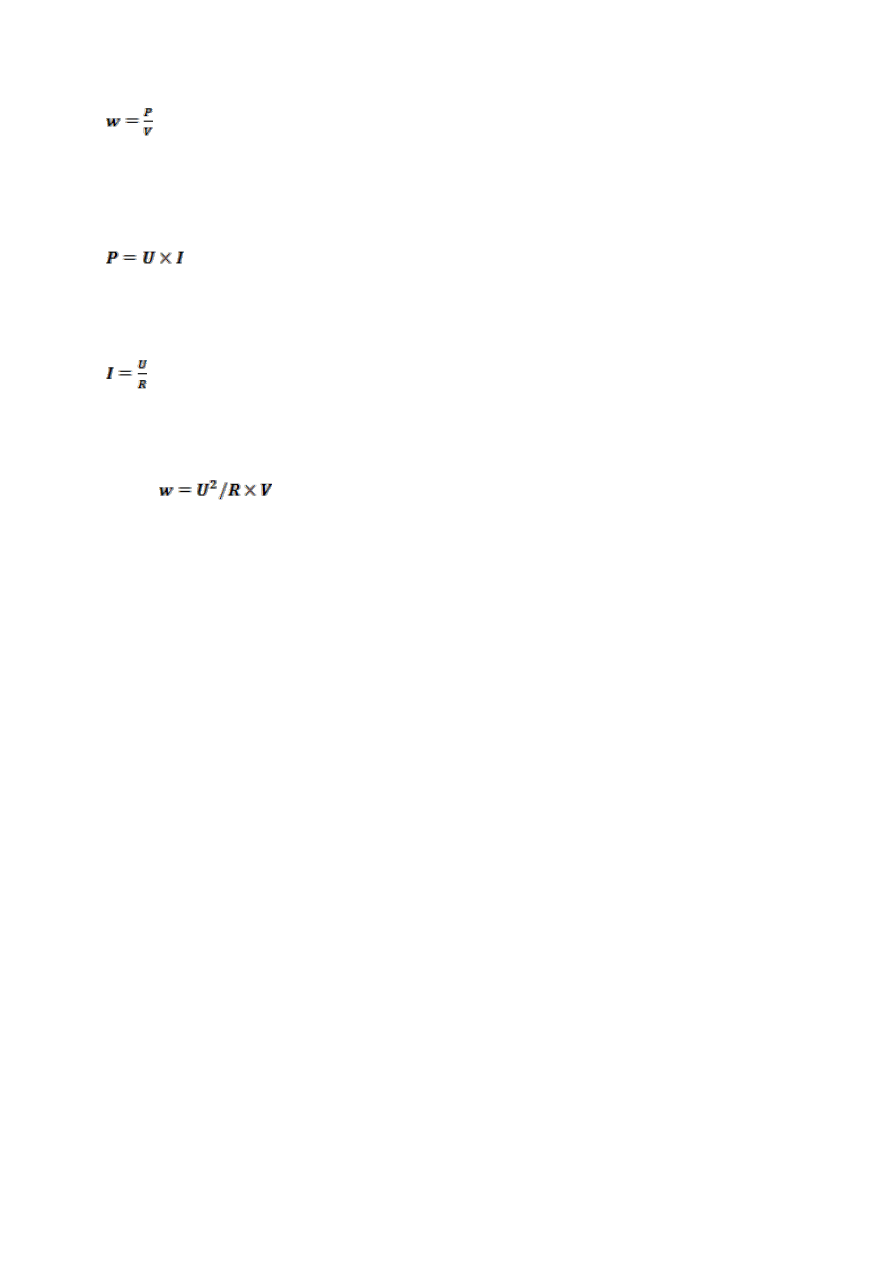
4
(7)
gdzie w-
ciepło Joul’a P - moc prądu [W], V- objętość w której wydzielana jest moc
[m
3
]
Ponieważ moc to iloczyn napięcia i natężenia prądu
(8)
gdzie U -
napięcie [V], I - natężenie prądu [A],
a z prawa Ohma wynika:
(9)
gdzie I
natężenie prądu [A], U napięcie prądu [V] , R – opór żelu [Ω],
Z równań (7), (8) i (9) otrzymujemy
(10)
Ciepło Joul’a jest więc proporcjonalne do kwadratu napięcia prądu.
Aby nie przegrzać żelu i nie doprowadzić do denaturacji lub nadmiernej dyfuzji
biomolekuł (wzrastającej wraz z temperaturą) – rozmyte prążki - stosuje się podczas
elektroforezy ograniczone napięcia.
Dla elektroforezy agarozowej DNA/RNA napięcie nie powinno przekraczać 5V/cm.
Dla elektroforezy w żelach poliakrylamidowych moc P wydzielana podczas
elektroforezy nie powinna przekraczać 5 [W]. Prowadzenie elektroforezy przy
wyższej mocy w aparatach nie posiadających efektywnego systemu odbierania
ciepła może powodować dużą dyfuzję bierną cząsteczek (nieostre prążki),
ni
erównomierną migrację związaną z przegrzewaniem się środka żelu (efekt
uśmiechu), utratę różnic konformacyjnych (SSCP) lub w przypadku natywnej
elektroforezy białek – denaturację białek.
Prowadzenie elektroforezy przy niskim napięciu redukuje moc wydzielaną w żelu – a
więc obniża ciepło Joul’a i wpływa pozytywnie na jakość rozdziału
elektroforetycznego. W niższej temperaturze dyfuzja bierna biomolekuł podczas
elektroforezy jest mniejsza
– prążki są bardziej ostre. Prowadzenie elektroforezy przy
niskim napi
ęciu (małej mocy) wydłuża jednak proporcjonalnie czas elektroforezy,
dlatego dużą zaletą jest możliwość schłodzenia żelu podczas elektroforezy, którą
dają aparaty z chłodzeniem.
Rozdział elektroforetyczny ze stałą mocą.
Rozwiązaniem problemu niekontrolowanego przegrzewania się żeli jest prowadzenie
rozdziału elektroforetycznego ze stałą mocą (W=const.).
Zasilacze
umożliwiające
utrzymywanie
stałej
mocy
podczas
rozdziału
elektroforetycznego
automatycznie dobierają napięcie U w czasie trwania
elektroforezy, ab
y utrzymać moc W na stałym zaprogramowanym poziomie.
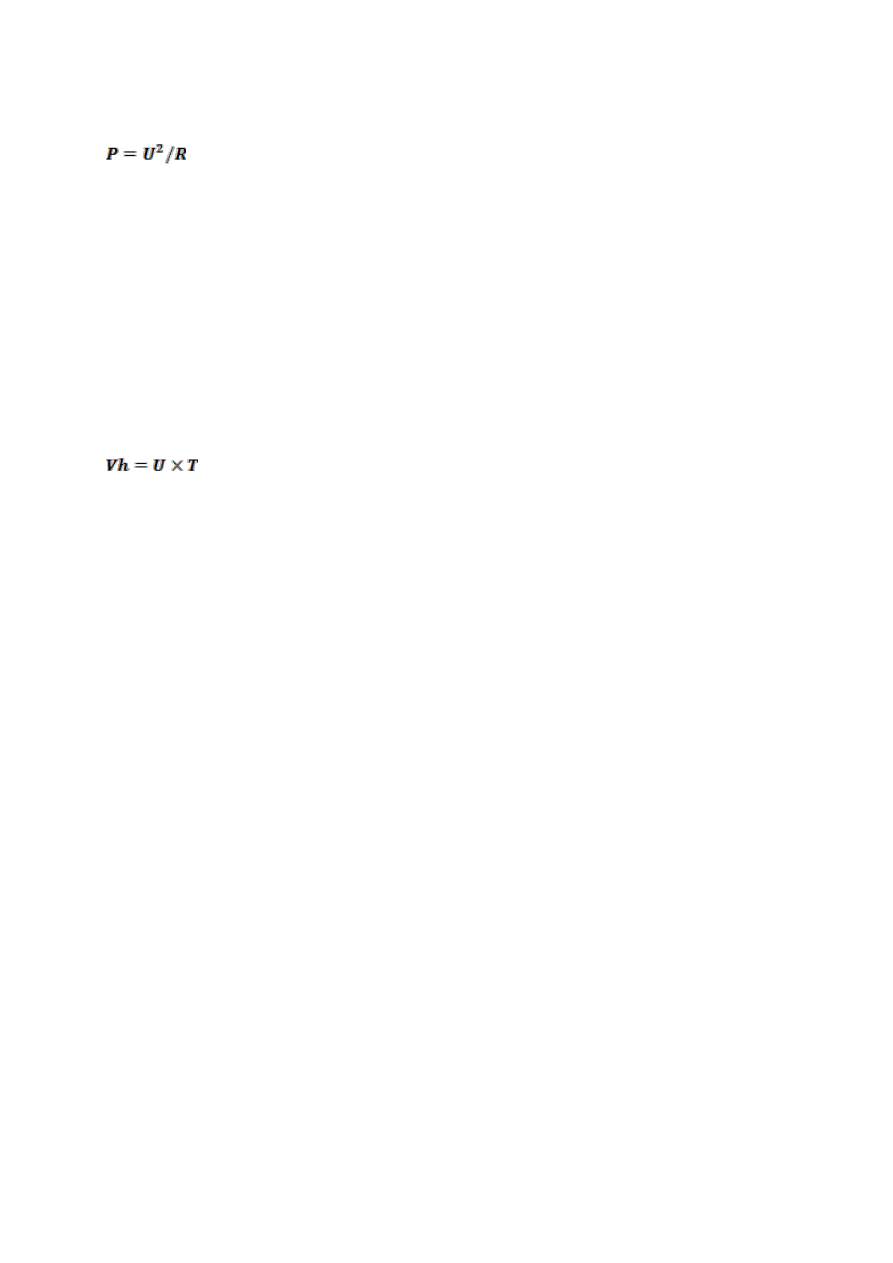
5
Z równań (8) i (9) wynika:
(11) gdzie P- moc [W], U-
napięcie [V], R- opór [Ω]
Ponieważ opór żelu R zazwyczaj rośnie w trakcie elektroforezy, prowadzenie
elektroforezy przy s
tałym napięciu prowadzi do niekontrolowanych zmian mocy
wydzielanej w postaci ciepła Joul’a, zaś prowadzenie elektroforezy przy stałej mocy
wymaga korekt napięcia.
Czas elektroforezy:
Podczas prowadzenia rozdziału elektroforetycznego ze stałą mocą długość
elektroforezy mierzona w czasie (np. w minutach) nie jest miarodajną wielkością
określającą przebieg elektroforezy ze względu na zmiany napięcia U. Parametrem
pozwalającym na w sposób powtarzalny określić długość elektroforezy są
woltogodziny. Jest to iloc
zyn napięcia U oraz czasu mierzony przez zasilacze
posiadające integrator Vh.
(11)
gdzie Vh- woltogodziny [V*min], U-
napięcie [V], T- czas [min]
Prowadzenie
elektroforezy przy stałej mocy oraz pomiar długości jej trwania przy
pomocy wolt
ogodzin, umożliwia prowadzenie rozdziałów elektroforetycznych
charakteryzujących się wysoką jakością i dużą powtarzalnością.
Zastosowanie elektroforezy:
preparatywne i ilościowe wydzielenie z badanych mieszanin czystych frakcji
określenie czystości i jednorodności badanych substancji
oznaczanie punktu izoelektrycznego badanych substancji
oznaczenie masy cząsteczkowej badanych substancji
zastosowanie do celów diagnostycznych i badawczych
zakres zastosowania rozciąga się od całych komórek i cząstek przez kwasy
nukleinowe, białka, peptydy, aminokwasy, organiczne kwasy i zasady do
narkotyków, pestycydów, nieorganicznych kationów i anionów (czyli
wszystkiego, co może nosić ładunek)
II.
ELEKTROFOREZA POLIAKRYLAMIDOWA BIAŁEK W WARUNKACH
NATYWNYCH.
Elektroforeza natywna:
Pozwala na rozdział elektroforetyczny białek ze względu na ich gęstość ładunku.
Bufory użyte podczas elektroforezy natywnej pozwalają na utrzymanie białek w
stanie niedenaturowanym. E
nzymy po elektroforezie zachowują swoją aktywność.
Elektroforeza nat
ywna jest używana głównie do określenia czystości białek – np.
oznaczenia czystości preparatyki enzymów. W elektroforezie natywnej białek używa
się nieciągłego systemu buforowego. Stosowane są dwa żele – zagęszczający o
niższym stężeniu akrylamidów i niższym pH, oraz rozdzielający o mniejszych porach
(wyższe stężenie akrylamidów) i wyższe pH. Rozwiązanie to, stosowane także w
żelach denaturujących, umożliwia uzyskanie zagęszczenia preparatu na styku żeli i
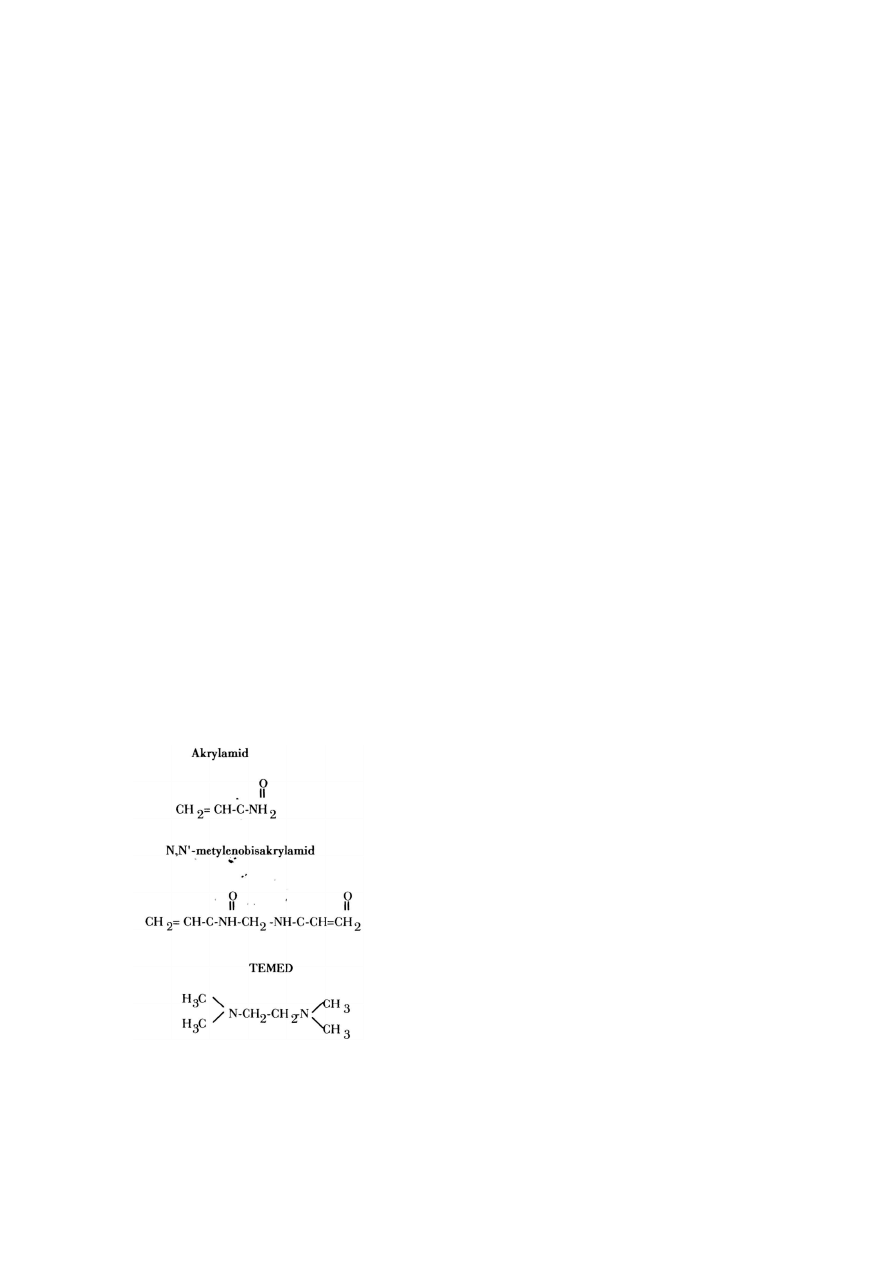
6
migracji białek w żelu rozdzielającym w postaci ostrych prążków. Dodatkowym
zabiegiem pozwalającym na poprawienie jakości rozdziałów – uzyskanie ostrych
prążków jest naniesienie na górną krawędź żelu rozdzielającego wody nasączonej n-
butanolem. Na górną krawędź żelu rozdzielającego, przed wylaniem żelu
zagęszczającego, należy ostrożnie pipetą (tak aby nie spowodować zmieszania
niespolimeryzowanego akrylamidu) na
kropić niewielką 0,1-0,5 ml ilość wody
nasączonej n-butanolem. Odetnie to dostęp powietrza do górnej krawędzi żelu
rozdzielającego, umożliwi jego pełna polimeryzację i zapewni ostry gradient pH. Ten
prosty zabieg wpływa w znacznym stopniu na poprawienie rozdzielczości rozdziałów
elektroforetycznych białek.
III.
ELEKTROFOREZA POLIAKRYLAMIDOWA BIAŁEK W WARUNKACH
DENATURUJĄCYCH (SDS-PAGE).
Elektroforeza w żelu poliakrylamidowym jest użyteczną i powszechnie stosowaną
metodą rozdziału i charakterystyki białek. Jest to stosunkowo prosta i szybka
technika. Na jednym żelu można przeprowadzić jednocześnie analizę wielu próbek, a
rozdzielane białka można skutecznie wykrywać w żelu np. metodami barwienia,
immunoprecypitacji czy autoradiografii. Można więc stosować tę technikę na przykład
przy analizie białek we frakcjach zbieranych w trakcie rozdziału na kolumnie
chromatograficznej.
Żel poliakrylamidowy powstaje w wyniku polimeryzacji monomerów akrylamidowych
w długie łańcuchy, które następnie łączone są kowalencyjnie przez N,N’-metyleno-
bis-akrylamid (Rys. 2,3)
. Gęstość żelu zależy od całkowitego stężenia obu
składników, natomiast stopień usieciowania zależy od stężenia bis-akrylamidu.
Reakcja polimeryzacji jest inicjowana przez wolne rodniki dostarczone przez
N,N,N’,N’-czterometylo-etylenodwuaminę
(TEMED)
i
stabilizowane
przez
nadsiarczan amonu (Rys. 1). Polimeryzacja zachodzi bez dostępu tlenu, który jest
inhibitorem tej reakcji. Możliwa jest także fotopolimeryzacja, czynnikiem inicjującym
reakcję polimeryzacji jest wówczas UV.
Rys. 1
Chemiczne struktury związków stosowanych do przygotowania żeli poliakrylamidowych.

7
Rys.2
Polimeryzacja bez udziału bisakrylamidu.
Rys. 3
Polimeryzacja z
udziałem bisakrylamidu.
Utworzony żel służy jako nośnik do rozdziału elektroforetycznego, funkcjonując
jednocześnie jak sito molekularne, co wpływa na skuteczny rozdział
makrocząsteczek.
Parametrami charakteryzującymi mieszankę akrylamidów są:
s
tężenie akrylamidu oznaczane jako T – jest to oznaczona % (w/v) ilość
akrylamidu i bisakry
lamidu w objętości roztworu.
stężenie cross-linker'ów oznaczane jako C - jest to % (w/w) wyrażony
stosunek ilości cross-linkerów do ilości akrylamidu i cross-linkerów. Np.
oznaczenie 20% T, 5 % C oznacza że roztwór zawiera 20% (w/v) akrylamidów
w stosunku akrylamidu do bisakrylamidu jak 19:1.
Wielkość porów tego sita uwarunkowana jest zarówno przez stężenie akrylamidu (ze
wzrostem stężenia wielkość porów maleje), jak i poprzez stopień usieciowania.
Wielkość ta musi być właściwie dobrana do wielkości rozdzielanych cząsteczek.
Najczęściej używa się żeli o stężeniu akrylamidu 7.5% - 10%, lecz przy rozdziale

8
cząsteczek bardzo dużych (rzędu 100 kDa), stosuje się żele o bardzo niskim
stężeniu akrylamidu, nawet 2.5%. Do rozdziału bardzo małych białek (m.cz. rzędu
2000 Da) używa się żeli zawierających 20% a nawet 30% akrylamidu. Ze wzrostem
czynnika sieciującego wielkość porów również maleje; stężenie to decyduje także o
własnościach mechanicznych żelu, jego elastyczności i przejrzystości.
Żel poliakrylamidowy może być formowany w rurkach lub jako żel płytowy o grubości
najczęściej 0.75-1.5 mm. Ze względu na możliwość naniesienia jednocześnie na
jeden żel wielu próbek, obecnie preferowana jest forma płytowa. Najczęściej stosuje
się elektroforezę w tzw. żelu nieciągłym, w którym próbkę nanosi się na żel
zagęszczający (o dużych porach) spolimeryzowany na wierzchu żelu rozdzielającego
(o małych porach). W tej technice bufor używany jako bufor elektrodowy oraz bufory
wchodzące w skład obu żeli różnią się składem i pH. Zaletą tego systemu jest
koncentracja białek podczas wędrówki przez żel zagęszczający, co powoduje, że
niezależnie od objętości próbki białka wchodzą w żel rozdzielający w postaci
wąskiego, zatężonego pasma.
Użycie anionowego detergentu soli sodowej kwasu dodecylosiarkowego (SDS – ang.
Sodium Dodecyl Sulfate)
pozwala na rozdział elektroforetytczny białek zgodnie z ich
masą cząsteczkową. Przed i w trakcie elektroforezy białka ulegają dysocjacji i
denaturacji w obecności SDS, który łączy się z białkami specyficznie w stosunku
masowym 4,1:1. Zapewnia to przejęcie przez białko ujemnego ładunku elektrycznego
netto o stałej gęstości bez względu na jego długość. Dla całkowitego rozwinięcia
polipeptydu, zapewnienia mu pierwszorzędowej struktury, niezbędne jest dodatkowo
zniszczenie mostków dwusiarczkowych (β-merkaptoetanol lub ditiotretiol).
Opłaszczenie białka przez SDS oraz redukcja mostków dwusiarczkowych pozwala
na separację białek w żelu poliakrylamidowym ze względu na ich wielkość, a zatem
pośrednio masę cząsteczkową. Szybkość migracji w SDS-PAGE nie jest
determinowana przez ładunek elektryczny białka zależny od pH i jego konformację
ale przez masę białka.
Dzięki możliwości rozdziału białek na podstawie ich wielkości możemy oznaczyć
masę cząsteczkową nieznanego białka, porównując jego położenie w żelu w
stosunku do innych białek (tzw. białek wzorcowych) po zakończonej migracji i
wybarwieniu.
Próbki nanosi się na żel po 5 minutowym ogrzaniu w 100
o
C (we wrzącej łaźni wodnej
lub w bloku grzejnym) w buforze denaturującym. Wchodząca w skład buforu
denaturującego sacharoza obciąża próbki, ułatwiając ich umieszczenie w studzience.
W obecności SDS i czynnika redukującego wiązania dwusiarczkowe (β-
merkaptoetanolu) białka ulegają denaturacji (rozdzielają się na podjednostki) a ich
wiązania niekowalencyjne ulegają rozkładowi. Początkowo rozdział prowadzi się przy
małym napięciu (60 V), aby białka jak najlepiej weszły w pory żelu. Następnie
zwiększa się napięcie (do 120 V), przyspieszając przepływ białek. Rozdział prowadzi
się w buforze glicynowym. Odpowiednie napięcie wraz z użyciem żelu o mniejszym
stężeniu akrylamidów w stosunku do żelu dolnego pozwala na zagęszczenie próbek.
Efekt taki uzyskujemy dzięki glicynie i jonom chlorkowym zawartym w buforze
elektrodowym. Glicyna w
pH 6.8 górnego żelu (wyższym od jej punktu
izoelektrycznego pI 6.3) przyjmuje ładunek lekko ujemny. W porównaniu z silnym
ładunkiem ujemnym jonów chlorkowych i białka (dzięki obecności SDS) jest to
ładunek znikomy. Glicyna jako cząsteczka o najmniejszym ładunku wypadkowym

9
ujemnym wędruje więc najwolniej ku anodzie. Przed nią przemieszczają się białka
poprzedzone przez jony chlorkowe,
charakteryzujące się dużą ruchliwością
elektroforetyczną. Takie rozmieszczenie w żelu migrujących cząsteczek powoduje
zagęszczenie próbki. Dzięki temu naniesione białka wchodzą równocześnie w żel
dolny. Przejście białek z żelu górnego do dolnego rozpoczyna proces ich właściwego
rozdziału. Żel dolny, poza stężeniem akrylamidów, a co się z tym wiąże wielkością
porów, różni się od żelu górnego również pH, które wynosi 8.8. Takie pH jest
znacznie wyższe od pI glicyny, co powoduje, że wypadkowy ładunek aminokwasu
staje się bardziej ujemny (wraz ze zmniejszeniem liczby jonów H
+
zwiększa się ilość
cząsteczek glicyny ze zjonizowanymi grupami karboksylowymi). Zmiana ładunku
glicyny jest przyczyną zmiany opisanej wcześniej kolejności migracji. Białka wędrują
za jonami chlorkowymi i glicyną, ulegając rozdziałowi pod względem masy
cząsteczkowej.
Białka w żelu można wybarwić następującymi metodami:
1. Barwienie w roztworze Coomassie Brilliant Blue R-250.
2.
Barwienie srebrem (np. według metody: Nesterenko i wsp., 1994).
3. Barwienie roztworem czerni amidowej.
Pojawia
się wówczas seria prążków (Rys.4). Po wybarwieniu błękitem Coomassiego
już 0.1 g (około 2 pmol) białka daje wyraźny prążek, a jeszcze mniejszą ilość (około
0.02 g) można uwidocznić wybarwiając srebrem. Tą metodą można na ogół
rozdzielić białka różniące się masą cząsteczkową o około 2% (np. 40 i 41 kDa, co
odpowiada różnicy około 10 reszt aminokwasowych). Znakowane białko można
zlokalizować metodą autoradiografii, umieszczając na żelu błonę rentgenowską.
Małe białka poruszają się w żelu szybko, natomiast duże zatrzymują się na górze,
blisko miejsca nałożenia mieszaniny na żel. W tych warunkach ruchliwość większości
łańcuchów polipeptydowych jest liniowo proporcjonalna do logarytmu ich masy
cząsteczkowej. Jednak do tej empirycznej zależności stosują się nie wszystkie
białka: np. niektóre białka bogate w reszty węglowodanowe i białka błonowe migrują
nietypowo.
Przykładowy rozdział białek w żelu poliakrylamidowym przedstawiono na Rys. 4. Żel
barwiono Coomassie Brilliant Blue R-250.
Elektroforezę typu SDS-PAGE możemy wykorzystać do sprawdzenia wydajności
stosowanej procedury izolacji, poddając rozdziałowi każdą frakcję z poszczególnych
etapów oczyszczania. Frakcje z pierwszych etapów rozdzielą się na dziesiątki, a
nawet setki białek, a w miarę postępu procesu oczyszczania ich liczba będzie się
zmniejszała, aż do pojawienia się jednego z nich, najbardziej intensywnego,
odpowiadającemu białku, które oczyszczamy (Rys. 4).
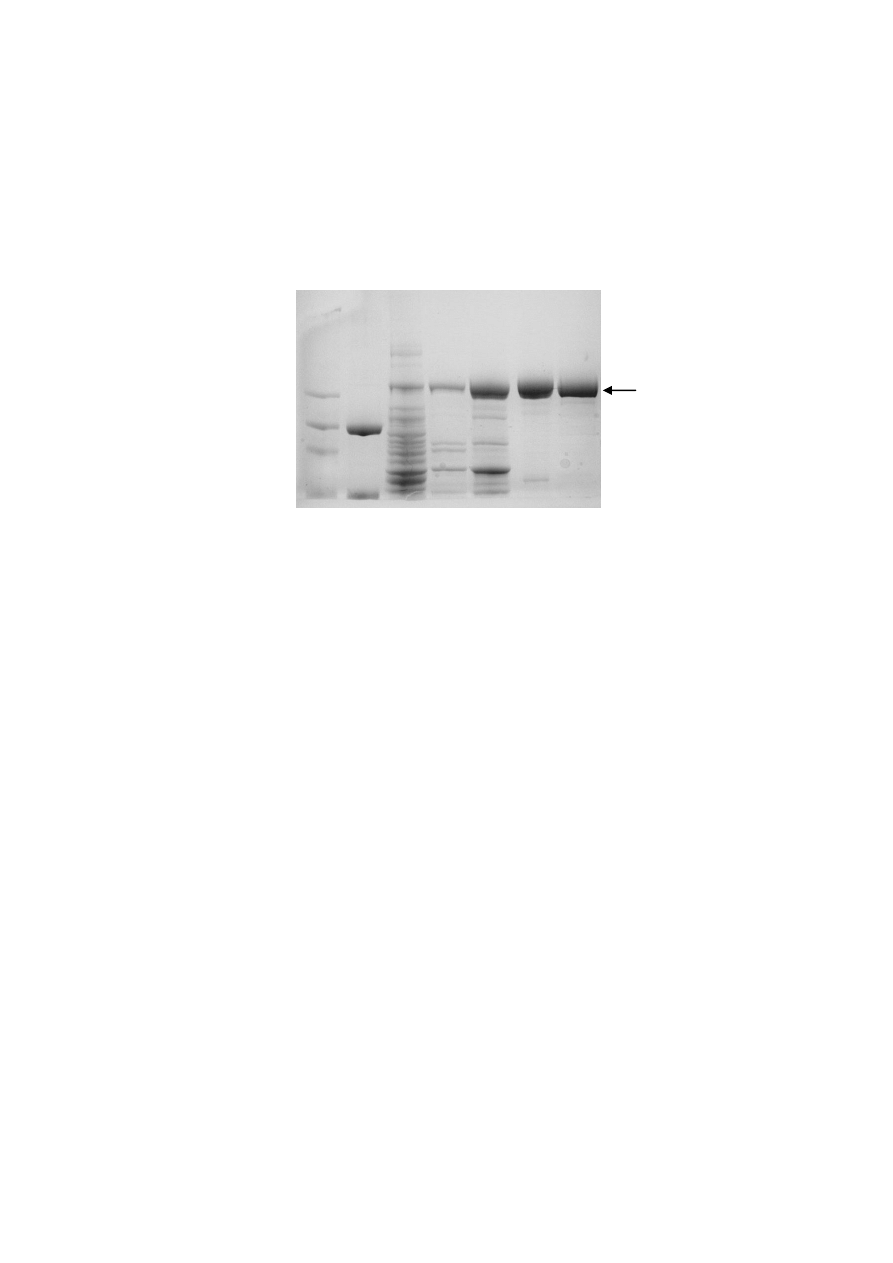
10
1 2 3 4 5 6 7
Rys. 4
Rozdział elektroforetyczny w żelu SDS-poliakrylamidowym (8%) białek uzyskanych z
poszczególnych etapów izolacji rekombinowanej endonukleazy restrykcyjnej TspGWI.
ścieżka 1: wzorzec masowy (Sigma)
ścieżka 2: wzorzec masowy (Pharmacia)
ścieżka 3: lizat białkowy
ścieżka 4: preparat po wytrąceniu białek siarczanem amonu w przedziale 35-65%
ścieżka 5: preparat po chromatografii na złożu Fosfocelulozy (P11)
ścieżka 6: preparat po zagęszczaniu i dializie do buforu do przechowywania
ścieżka 7: preparat natywnej REazy TspGWI
PIŚMIENNICTWO:
1.
Berg J.M, Tymoczko J.L, Stryer L. (2005) Biochemia. (według V wyd.
amerykańskiego) Wydawnictwo Naukowe PWN, Warszawa.
2.
Jóźwiak Z., Bartosz G. Biofizyka. Wybrane zagadnienia wraz z ćwiczeniami.
PWN (2007)
3.
Kur J. (1994) Podstawy inżynierii genetycznej. Wydawnictwo Politechniki
Gdańskiej.
4. http://www.kucharczyk.com.pl/ (Przepisy i porady)
5. Maniatis T., Fritsch E.F., Sambrook J. (1982) Molecular Cloning. A Laboratory
Manual. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY.
6. Nesterenko M.V., Tilley M., Upton S.J. (1994) A simple modification of Blum's
silver stain method allows for 30 minute detection of proteins in polyacrylamide
gels. J.Biochem.Biophys.Methods 28: 239-242.
nr ścieżki
116 kDa
84 kDa
67 kDa
TspGWI
Wyszukiwarka
Podobne podstrony:
KOROZJA PODSTAWY TEORETYCZNE I SPOSOBY ZAPOBIEGANIA
KOROZJA PODSTAWY TEORETYCZNE I SPOSOBY ZAPOBIEGANIA
Podstawy teoretyczne
lista 4a, Elektrotechnika, PODSTAWY ELEKTROTECHNIKI, ćwiczenia
zadania na egzaminie czerwcowym 2009, Elektrotechnika, PODSTAWY ELEKTROTECHNIKI, pytania
Zagad NE09, Politechnika Wrocławska, PWR - W10- Automatyka i Robotyka, Sem3, Elektro, Podstawy elekt
sc5 druk, Politechnika Wrocławska, PWR - W10- Automatyka i Robotyka, Sem3, Elektro, Podstawy elektro
zadania egzaminacyjne dzienne (PTM), elektro, 1, Podstawy Techniki Mikroprocesorowej
80C51 pytania i odpowiedzi, elektro, 1, Podstawy Techniki Mikroprocesorowej
FIG-02D, Elektrotechnika, PODSTAWY ELEKTROTECHNIKI, wyklad
terapia dzieci z trudnosciami w czytaniu i pisaniu - podstawy teoretyczne, Studia - pedagogika
Diody1, 1. TECHNIKA, Elektryka - Elektronika, Elektrotechnika, Podstawy elektotechniki i elektroniki
Diody1, 1. TECHNIKA, Elektryka - Elektronika, Elektrotechnika, Podstawy elektotechniki i elektroniki
SC3, Politechnika Wrocławska, PWR - W10- Automatyka i Robotyka, Sem3, Elektro, Podstawy elektrotechn
zadania egzaminacyjne zaoczne 2006 07 (PTM), elektro, 1, Podstawy Techniki Mikroprocesorowej
lista 3a, Elektrotechnika, PODSTAWY ELEKTROTECHNIKI, ćwiczenia
Egzamin z PTC podst kombinacyjne, elektro, 1, Podstawy Techniki Mikroprocesorowej
linie napowietrzne, Elektrotechnika, PODSTAWY ELEKTROTECHNIKI, sciagi
więcej podobnych podstron