
Mechanisms of Ageing and Development
123 (2002) 207 – 213
Protein turnover plays a key role in aging
Alexey G. Ryazanov *, Bradley S. Nefsky
Department of Pharmacology, Uni
6ersity of Medicine and Dentistry of New Jersey, Robert Wood Johnson Medical School,
675
Hoes Lane, Piscataway, NJ
08854
, USA
Abstract
Although the molecular mechanism of aging is unknown, a progressive increase with age in the concentration of
damaged macromolecules, especially proteins, is likely to play a central role in senescent decline. In this paper, we
discuss evidence that the progressive decrease in protein synthesis and turnover can be the primary cause of the
increase in the concentration of damaged proteins with age. Conversely, protein damage itself is likely to be the cause
of the decrease in protein turnover. This could establish a positive feedback loop where the increase in protein damage
decreases the protein turnover rate, leading to a further increase in the concentration of damaged proteins. The
establishment of such a feedback loop should result in an exponential increase in the amount of protein damage — a
protein damage catastrophe — that could be the basis of the general deterioration observed in senescent organisms.
© 2002 Published by Elsevier Science Ireland Ltd.
Keywords
:
Senescent decline; Protein damage; Protein turnover; Aging; Protein oxidation; Protein damage catastrophe
www.elsevier.com/locate/mechagedev
1. Increase in the concentration of damaged
proteins during aging
Various genes have been identified in worms,
flies and mammals whose mutation can drastically
affect life span. For example, mutations in the
insulin signaling pathway, such as age-
1
and daf-
2
, can double life span in Caenorhabditis elegans
(Friedman and Johnson, 1988; Kenyon et al.,
1993), the Methuselah mutation can significantly
increase life span in Drosophila (Lin et al., 1998),
and mutation of p66
shc
can increase life span in
mice (Migliaccio et al., 1999). However, the
molecular mechanism by which any of these genes
affects life span remains unknown. Similarly, the
molecular mechanism of aging in general remains
a mystery. It is also unclear whether there is a
general, or ‘public’, as George Martin called it,
mechanism of aging that is common to various
organisms, or if each type of organism ages in its
own way (Martin et al., 1996).
There is extensive evidence, however, that dam-
age to macromolecules, especially oxidative dam-
age, plays an important role in aging (Martin et
al., 1996). Increase in the concentration of dam-
aged proteins seems particularly important be-
cause it would lead to the malfunction of virtually
all biological processes. An increase in the concen-
tration of damaged intracellular proteins as well
as an increase in the concentration of inactive or
* Corresponding author. Tel.: + 1-732-235-5526; fax: + 1-
732-235-4073.
E-mail address
:
ryazanag@umdnj.edu (A.G. Ryazanov).
0047-6374/02/$ - see front matter © 2002 Published by Elsevier Science Ireland Ltd.
PII: S 0 0 4 7 - 6 3 7 4 ( 0 1 ) 0 0 3 3 7 - 2

A.G. Ryazano
6, B.S. Nefsky
/
Mechanisms of Ageing and De
6elopment
123 (2002) 207 – 213
208
partially active forms of various enzymes in aging
organisms is well-documented (reviewed in Stadt-
man, 1988, 1992; Rothstein, 1979, 1989; Rattan,
1996; Gershon, 1979; Rosenberger, 1991; Gafni,
1990). Extensive damage to extracellular proteins
such as collagen, elastin and proteoglycans is also
observed during aging (reviewed in Sell and Mon-
nier, 1995). The age-related increase in protein
damage is due to various post-translational mod-
ifications that include oxidation of amino acid
side chains, racemization of aspartyl and as-
paraginyl residues, deamidation of asparaginyl
and
glutaminyl
residues,
and
oxidation
of
sulfhydry groups (reviewed in Stadtman, 1988,
1992). Protein misfolding is also likely to con-
tribute to the increase in the concentration of
abnormal enzymes and proteins in senescent tis-
sues (Gafni, 1990).
Oxidation by free radicals is particularly impor-
tant in generating damaged proteins during aging
(Harman, 1956; Stadtman, 1992). There is exten-
sive evidence that the increased concentration of
oxidatively damaged proteins, can be a major
contributing factor in aging (Sohal et al., 1993;
Forster et al., 1996; Oliver et al. 1987; Carney et
al., 1991; Smith et al., 1991; Dubey et al., 1996;
Youngman et al., 1992).
The extent of protein damage observed during
aging is quite remarkable. Roughly 20 – 30% of all
cellular proteins are carbonylated in aged individ-
uals (Starke-Reed and Oliver, 1989; Stadtman,
1992). If all other forms of oxidative damage are
included, this number probably increases to 40 –
50% (Stadtman, 1992). If we consider other forms
of damage, it seems likely that most protein
molecules in senescent tissues contain some form
of damage.
What causes the increase in the concentration
of damaged proteins during aging? There are two
possible mechanisms: an increase in the rate at
which damage is generated or a decrease in the
rate at which damage is removed. Both mecha-
nisms appear to be involved in aging. For exam-
ple, an increase in the amount of oxidatively
damaged proteins during aging may be due to an
increase in the production of free radicals (re-
viewed in Sohal and Weindruch, 1996). On the
other hand, increased accumulation of damaged
proteins can result from an age-dependent de-
crease in the removal of protein damage by either
repair or replacement of the damaged protein.
The cells ability to repair protein damage, how-
ever, seems rather limited and most forms of
protein damage appear to be irreversible. The
only mechanism cells have to deal with irre-
versibly damaged proteins is to replace them
through protein turnover. The decrease in protein
turnover with age (see below) could therefore be a
major cause of the increased concentration of
damaged proteins.
2. Protein turnover in aging
The rate of protein turnover is determined by
the combined rates of protein synthesis and degra-
dation. Once growth is complete, organisms reach
a steady state in which the rate of protein synthe-
sis equals the rate of protein degradation. Most
studies of protein metabolism in aging were fo-
cused on protein synthesis, and there is over-
whelming evidence that the rate of protein
synthesis declines with age. This decrease in
protein synthesis appears to be a universal phe-
nomenon (reviewed in Makrides, 1983; Richard-
son and Cheung, 1982; Van Remmen et al., 1995;
Rattan, 1996). Since there is no pronounced de-
crease in protein mass with age, the age-depen-
dent decrease in protein synthesis must be
counterbalanced by a corresponding decrease in
protein degradation. In fact, there is experimental
evidence that certain proteolytic activities associ-
ated with both the lysosome and proteasome de-
crease with age (reviewed in Van Remmen et al.,
1995; Cuervo and Dice, 2000; Grune, 2000;
Friguet et al., 2000; Keller et al., 2000). The
age-dependent decrease in the rate of protein
turnover is quite significant and leads to a drastic
increase in protein half-life (see e.g. Sharma et al.,
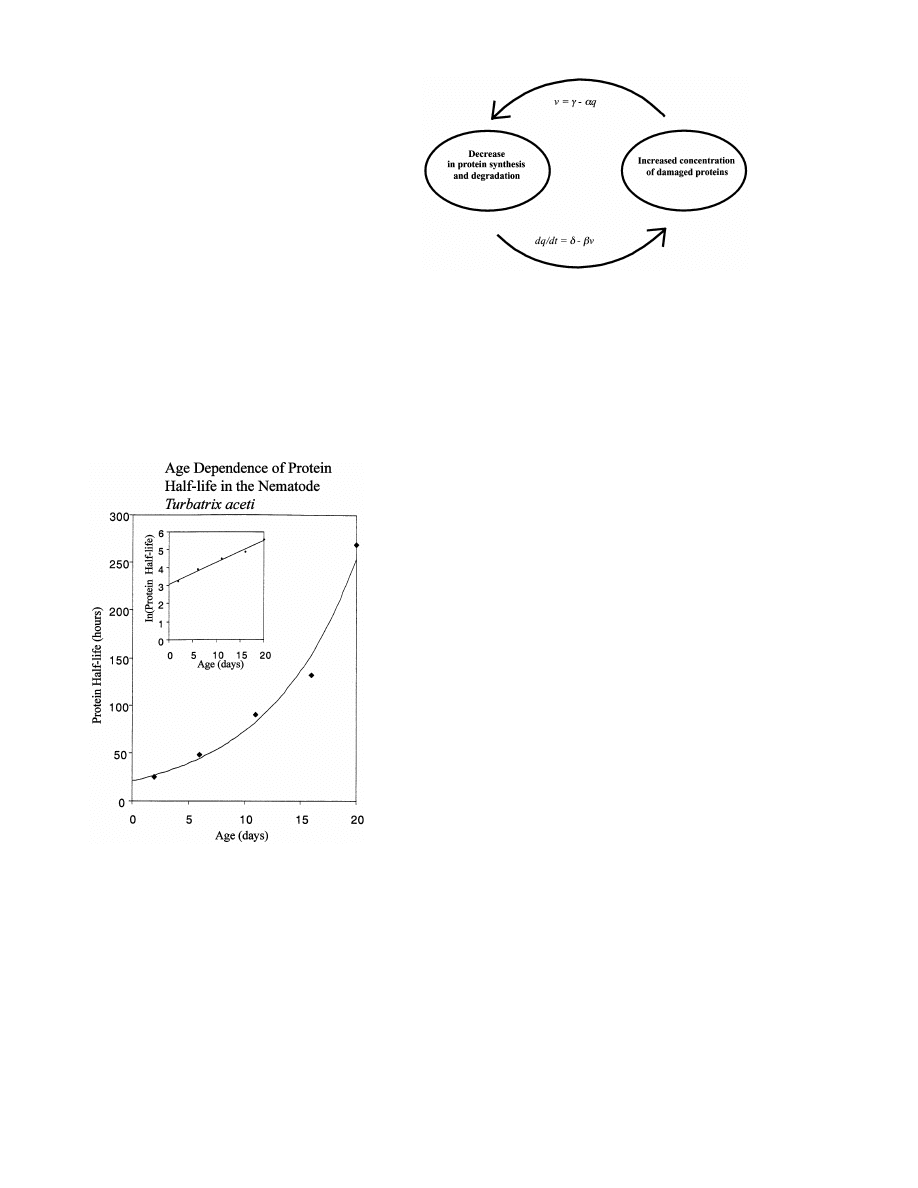
1979; Prasanna and Lane, 1979). As is shown in
Fig. 1, the half-life of an average protein increases
exponentially with age in the nematode Turbatrix
aceti, and is increased 10-fold in old nematodes in
comparison to young worms (Prasanna and Lane,
1979). The drastic decrease in protein synthesis
with age in C. elegans (Johnson and McCaffrey,
A.G. Ryazano
6, B.S. Nefsky
/
Mechanisms of Ageing and De
6elopment
123 (2002) 207 – 213
209
1985) suggests a similar increase in protein half-
life occurs during aging in this organism. Whole
body protein turnover is also significantly de-
creased during aging in rats (Lewis et al., 1985)
and humans (Young et al., 1975). It is striking
that despite a very significant decrease in protein
turnover during aging, there are no dramatic
changes in the spectrum of proteins synthesized in
C. elegans (Vanfleteren and DeVreese, 1994; John-
son and McCaffrey, 1985) or in the spectrum of
mRNA expressed in mammalian tissues (Goyns et
al., 1998; Lee et al., 1999). Therefore, the several-
fold decrease in overall protein turnover implies
that the rate of turnover of most proteins are
decreased several-fold.
As was suggested by Richardson and Cheung
(1982), an increase in protein half-life can signifi-
cantly decrease the rate at which protein expres-
sion is induced. This can significantly increase the
Fig. 2. Model for the positive feedback loop between the
increase in the concentration of damaged proteins and de-
creased protein turnover leading to a protein damage catastro-
phe.
time needed for cells to respond to external stim-
uli, and can explain diminished stress resistance
observed with age.
Decrease in protein turnover can also con-
tribute to the development of neurodegenerative
disorders that are associated with the deposition
of protein aggregates such as Parkinson’s disease
and Alzheimer’s disease (Alves-Rodriguez et al.,
1998; Andersen, 2000).
3. Protein damage catastrophe
Although the age-dependent decrease in protein
turnover is well-documented, the mechanism re-
sponsible for this decrease is unclear. Analysis of
the literature suggests that the activities of various
components of both the protein synthesis and
degradation machinery decline with age (reviewed
in Van Remmen et al., 1995; Cuervo and Dice,
2000; Grune, 2000; Friguet et al., 2000; Keller et
al., 2000). A possible cause of such age-dependent
decline in protein turnover can be the cumulative
effect of non-specific damage to various compo-
nents involved in protein synthesis and degrada-
tion. This could establish a positive feedback loop
where increase in protein damage decreases
protein synthesis and degradation rates, and this
decrease in protein turnover leads to a further
increase in the concentration of damaged proteins
(Fig. 2).
Fig. 1. Exponential increase of average protein half-life with
age in the nematode Turbatrix aceti. The graph was plotted
using data from Prasanna and Lane, 1979. In the inset, the ln
of protein half-life is plotted against age demonstrating a
linear relationship.

A.G. Ryazano
6, B.S. Nefsky
/
Mechanisms of Ageing and De
6elopment
123 (2002) 207 – 213
210
Increase in protein damage in this model can be
described by the following equations. Let
w be the
rate of protein turnover and q be the concentra-
tion of damaged proteins.
Then,
w=g−aq
dq/dt =
d−bn
Where
a, b, d and g are constants. From these
two equations, the rate of the increase in the
concentration of damaged proteins can be de-
scribed as:
dq/dt =
d−b(g−aq)=d−bg+baq
or, since
d−bg is a constant, (C):
dq/dt =
baq+C;
One can see that when q is small, dq/dt is
constant, while if q
\C/ab, then q#e
abt
and,
therefore, the concentration of damaged proteins
increases exponentially.
Therefore, the establishment of such a feedback
loop could result in an exponential increase in the
amount of protein damage, leading to a protein
damage catastrophe. An exponential increase in
the amount of damaged proteins with age was in
fact observed in several studies (Oliver et al.,
1987; Starke-Reed and Oliver, 1989; Stadtman,
1992). A decrease in the rate of protein turnover
with age also appears to be exponential (see Fig.
1), which is consistent with the idea that it is
caused by an exponential increase in damage to
the components of the protein synthesis and
degradation machinery. We suggest that this posi-
tive feedback loop between increasing protein
damage and decreasing protein turnover leading
to a protein damage catastrophe may be the ma-
jor cause of senescence.
Our model is reminiscent of Orgel’s error
catastrophe hypothesis that suggested aging is due
to an accumulation of abnormal proteins arising
from transcriptional and translational errors
(Orgel, 1963, 1970). He suggested that the accu-
mulation of errors in the transcriptional and
translational machinery would decrease their
fidelity, establishing a positive feedback loop,
which would further increase the accumulation of
errors in proteins leading to an ‘error catastro-
phe’. This hypothesis was widely discussed during
the 1970s and 1980s, and several mathematical
models of ‘error catastrophe’ have been devel-
oped. Although a number of studies (particularly
in tissue culture cells) found no evidence of an
‘error catastrophe’ during aging, in the absence of
a direct test we cannot determine the extent to
which transcriptional and translational errors
contribute to senescence. Our ‘protein damage
catastrophe’ model proposes a similar feedback
loop in which damage to the protein synthesis and
degradation machinery accelerates the accumula-
tion of abnormal proteins resulting in senescence.
Unlike the ‘error catastrophe’ model, however, we
propose that protein damage (covalent modifica-
tions and misfolding) instead of translational er-
rors gives rise to the abnormal proteins and that it
is the resulting decrease in protein turnover,
rather than a decrease in translational fidelity,
that leads to the increase in the concentration of
abnormal proteins.
Although our model assumes that the decrease
of protein turnover rate with age is the result of
cumulative damage to the various components of
the protein synthesis and degradation machinery,
it is possible that damage to some rate-limiting
components is particularly important in the over-
all decline in protein turnover. For example, there
is a consensus in the literature that the elongation
stage of protein synthesis is predominantly af-
fected by aging (reviewed in Van Remmen et al.,
1995). In addition, there is evidence that elonga-
tion factor-2 is particularly sensitive to oxidative
damage, and is one of the major carbonylated
proteins observed during oxidative stress in rat
liver as well as in yeast (Cabiscol et al., 2000;
Parrado et al., 1999; Ayala et al., 1996). There-
fore, it is possible that the overall exponential
decrease in protein turnover with age is due pre-
dominantly to damage to a rate-limiting compo-
nent such as an elongation factor, while damage
to other components that are not rate-limiting do
not affect the overall decline. If this is the case,
and there is a particular component whose decline
in activity is responsible for the overall decrease in
protein turnover, then this would suggest an obvi-
ous strategy for intervention that can increase
protein turnover and extend life span.

A.G. Ryazano
6, B.S. Nefsky
/
Mechanisms of Ageing and De
6elopment
123 (2002) 207 – 213
211
Is it possible that mutations or interventions
that extend life span act through an increase in
protein turnover? Dietary restriction is a well
established intervention which can extend the life
span of various organisms (Sohal and Weindruch,
1996). It was demonstrated in rats that an in-
crease in protein synthesis and degradation are
among the most prominent effects of dietary re-
striction (Lewis et al., 1985; Holehan and Merry,
1986; D’Costa et al., 1993). In their comprehen-
sive review on the mechanism of the anti-aging
effect of dietary restriction, Holehan and Merry
(1986) concluded that ‘‘protein turnover, both
directly and through amplification by adjustments
in endocrine feedback, is the primary effect of
underfeeding’’.
A
detailed
study
of
protein
turnover in rat liver revealed that protein turnover
is elevated throughout most of the life span of
dietary restricted versus ad libidum fed animals
(Ward, 1988a,b). This data, together with the data
that
an
increase
in
protein
degradation
is
observed shortly after the onset of dietary
restriction (Ishigami and Goto, 1990), argues that
an increase in protein turnover can play a
causative role in the anti-aging effect of dietary
restriction.
It was recently found that dwarf mice have a
significantly increased life span (Brown-Borg et
al., 1996; Bartke et al., 2001; Flurkey et al., 2001),
and it was argued that small size in animals was
associated
with
longevity
(Miller,
1999;
Bartke, 2000; Miller et al., 2000). Intriguingly, it
was reported that mice selected for small size
have
increased
protein
turnover
in
various
organs
(Priestley
and
Robertson,
1973).
It
will be interesting in the future to analyze whether
various
long-lived
mutants
of
C.
elegans,
Drosophila and mice have increased protein
turnover rates.
Overall, we suggest that the exponential in-
crease in the concentration of damaged proteins
with age in conjunction with the decrease in
protein turnover — protein damage catastrophe —
is the major mechanism underlying senescent de-
cline
during
aging,
and
increasing
protein
turnover can be a plausible strategy to retard
aging and extend life span.
References
Alves-Rodriguez, A., Gregori, L., Figueiredo-Pereira, M.E.,
1998. Ubiquitin, cellular inclusions and their role in neu-
rodegeneration. Trends Neurosci. 21, 516 – 520.
Andersen, J.K., 2000. What causes the build-up of ubiquitin-
containing inclusions in Parkinson’s disease? Mech. Age-
ing. Dev. 118, 15 – 22.
Ayala, A., Parrado, J., Bougria, M, Machado, A., 1996. Effect
of oxidative stress, produced by cumene hydroperoxide, on
the various steps of protein synthesis. Modifications of
elongation factor-2. J. Biol. Chem. 271, 23 105 – 23 110.
Bartke, A., 2000. Delayed aging in Ames dwarf mice. Rela-
tionships to endocrine function and body size. Results
Probl. Cell Differ. 29, 181 – 202.
Bartke, A., Brown-Borg, H., Mattison, J., Kinney, B., Hauck,
S., Wright, C., 2001. Prolonged longevity of hypopituitary
dwarf mice. Exp. Gerontol. 36, 21 – 28.
Brown-Borg, H.M., Borg, K.E., Meliska, C.J., Bartke, A.,
1996. Dwarf mice and the ageing process. Nature 384, 33.
Cabiscol, E., Piulats, E., Echave, P., Herrero, B., Ros, J.,
2000. Oxidative stress promotes specific protein damage in
Saccharomyces cere
6isiae. J. Biol. Chem. 275, 27 393–
27 398.
Carney, J.M., Starke-Reed, P.E., Oliver, C.N., Landum, R.W.,
Cheng, M.S., Wu, J.F., Floyd, R.A., 1991. Reversal of
age-related increase in brain protein oxidation, decrease in
enzyme activity, and loss in temporal and spatial memory
by chronic administration of the spin-trapping compound
N-tert-butyl-alpha-phenylnitrone. Proc. Natl. Acad. Sci.
USA 88, 3633 – 3636.
Cuervo, A.M., Dice, J.F., 2000. When lysosomes get old. Exp.
Gerontol. 35, 119 – 131.
D’Costa, A.P., Lenham, J.E., Ingram, R.L., Sonntag, W.E.,
1993. Moderate caloric restriction increases type 1 IGF
receptors and protein synthesis in aging rats. Mech. Ageing
Dev. 71, 59 – 71.
Dubey, A., Forster, M.J., Lal, H., Sohal, R.S., 1996. Effect of
age and caloric intake on protein oxidation in different
brain regions and on behavioral functions of the mouse.
Arch. Biochem. Biophys. 333, 189 – 197.
Flurkey, K., Papaconstantinou, J., Miller, R.A., Harrison,
D.E., 2001. Lifespan extension and delayed immune and
collagen aging in mutant mice with defects in growth
hormone production. Proc. Natl. Acad. Sci. USA 98,
6736 – 6741.
Forster, M.J., Dubey, A., Dawson, K.M., Stutts, W.A., Lal,
H., Sohal, R.S., 1996. Age-related losses of cognitive func-
tion and motor skills in mice are associated with oxidative
damage in the brain. Proc. Natl. Acad. Sci. USA 93,
4765 – 4769.
Friedman, D.B., Johnson, T.E., 1988. A mutation in the age-
1
gene in Caenorhabditis elegans lengthens life and reduces
hermaphrodite fertility. Genetics 118, 75 – 86.
Friguet, B., Bulteau, A.L., Chondrogianni, N., Conconi, M.,
Petropoulos, I., 2000. Protein degradation by the protea-
some and its implications in aging. Ann. N.Y. Acad. Sci.
908, 143 – 154.

A.G. Ryazano
6, B.S. Nefsky
/
Mechanisms of Ageing and De
6elopment
123 (2002) 207 – 213
212
Gafni, A., 1990. Altered protein metabolism in aging. Ann.
Rev. Gerontol. Geriatr. 10, 117 – 131.
Gershon, D., 1979. Current status of age altered enzymes:
alternative mechanisms. Mech. Ageing Dev. 9, 189 – 196.
Goyns, M.H., Charlton, M.A., Dunford, J.E., Lavery, W.L.,
Merry, B.J., Salehi, M., Simoes, D.C., 1998. Differential
display analysis of gene expression indicates that age-re-
lated changes are restricted to a small cohort of genes.
Mech. Ageing Dev. 101, 73 – 90.
Grune, T., 2000. Oxidative stress, aging and the proteasomal
system. Biogerontol. 1, 31 – 40.
Harman, D, 1956. Ageing: a theory based on free radical and
radiation chemistry. J. Gerontol. 11, 298 – 300.
Holehan, A.M., Merry, B.J., 1986. The experimental manipu-
lation of aging by diet. Biol. Rev. 61, 329 – 368.
Ishigami, A., Goto, S., 1990. Effect of dietary restriction on
the degradation of proteins in senescent mouse liver par-
enchymal cells in culture. Arch. Biochem. Biophys. 283,
362 – 366.
Johnson, T.E., McCaffrey, G., 1985. Programmed aging or
error catastrophe? An examination by two-dimensional
polyacrylamide gel electrophoresis. Mech. Ageing Dev. 30,
285 – 297.
Keller, J.N., Hanni, K.B., Markesbery, W.R., 2000. Possible
involvement of proteasome inhibition in aging: implication
for oxidative stress. Mech. Ageing Dev. 113, 61 – 70.
Kenyon, C., Chang, J., Gensch, E., Rudner, A., Tabtiang, R.,
1993. A C. elegans mutant that lives twice as long as wild
type. Nature 366, 461 – 464.
Lee, C.K., Klopp, R.G, Weindruch, R., Prolla, T.A., 1999.
Gene expression profile of aging and its retardation by
caloric restriction. Science 285, 1390 – 1393.
Lewis, S.E.M., Goldspink, D.F., Phillips, J.G., Merry, B.J.,
Holehan, A.M., 1985. The effects of aging and chronic
dietary restriction on whole body growth and protein
turnover in the rat. Exp. Gerontol. 20, 253 – 263.
Lin, Y.J., Seroude, L., Benzer, S., 1998. Extended life-span
and stress resistance in the Drosophila mutant methuselah.
Science 282, 943 – 946.
Makrides, S.C., 1983. Protein synthesis and degradation dur-
ing aging and senescence. Biol. Rev. 58, 343 – 422.
Martin, G.M., Austad, S.N., Johnson, T.E., 1996. Genetic
analysis of ageing: role of oxidative damage and environ-
mental stresses. Nature Genetics 13, 25 – 34.
Migliaccio, E., Giorgio, M., Mele, S., Pelicci, G., Reboldi, P.,
Pandolfi, P.P., Lanfrancone, L., Pelicci, P.G., 1999. The
p66
shc
adaptor protein controls oxidative stress response
and life span in mammals. Nature 402, 309 – 313.
Miller, R.A., 1999. Kleemeier award lecture: are there genes
for aging? J. Gerontol. 54, B297 – B307.
Miller, R.A., Chrisp, C., Atchley, W., 2000. Differential
longevity in mouse stocks selected for early life growth
trajectory. J. Gerontol. 55A, B455 – B461.
Oliver, C.N., Ahn, B.-W., Moerman, E.J., Goldstein, S.,
Stadtman, E.R., 1987. Age-related changes in oxidized
proteins. J. Biol. Chem. 262, 5488 – 5491.
Orgel, L.E., 1963. The maintenance of the accuracy of protein
synthesis and its relevance to ageing. Proc. Natl. Acad. Sci.
USA 49, 517 – 521.
Orgel, L.E., 1970. The maintenance of the accuracy of protein
synthesis and its relevance to ageing: a correction. Proc.
Natl. Acad. Sci. USA 67, 1476.
Parrado, J., Bougria, M., Ayala, A., Castano, A., Machado,
A., 1999. Effects of aging on the various steps of protein
synthesis: fragmentation of elongation factor 2. Free
Radic. Biol. Med. 26, 362 – 370.
Prasanna, H.R., Lane, R.S., 1979. Protein degradation in aged
nematodes (Turbatrix aceti ). Biochem. Biophys. Res.
Commun. 86, 552 – 559.
Priestley, G.C., Robertson, M.S.M., 1973. Protein and nucleic
acid metabolism in organs from mice selected for larger
and smaller body size. Genet. Res. 22, 255 – 278.
Rattan, S.I., 1996. Synthesis, modification, and turnover of
proteins during aging. Exp. Gerontol. 31, 33 – 47.
Richardson, A., Cheung, H.T., 1982. Current concepts: I. The
relationship between age-related changes in gene expres-
sion, protein turnover, and the responsiveness of an organ-
ism to stimuli. Life Sci. 31, 605 – 613.
Rosenberger, R.F., 1991. Senescence and the accumulation of
abnormal proteins. Mut. Res. 56, 255 – 262.
Rothstein, M., 1979. The formation of altered enzymes in
aging animals. Mech. Ageing Dev. 9, 197 – 202.
Rothstein, M., 1989. An overview of age-related changes in
proteins. Prog. Clin. Biol. Res. 287, 259 – 267.
Sell, D.R., Monnier, V.M., 1995. Aging of long-lived proteins:
extracellular matrix (collagens, elastins, proteoglycans) and
lens crystallins. In: Masoro, E.J. (Ed.), Handbook of Phys-
iology-Section 11: Aging. Oxford University Press, New
York, pp. 235 – 305.
Sharma, H.K., Prasanna, H.R., Lane, R.S., Rothstein, M.,
1979. The effect of age on enolase turnover in the free-liv-
ing nematode Turbatrix aceti. Arch. Biochem. Biophys.
194, 275 – 282.
Sohal, R.S., Agarwal, S., Dubey, A., Orr, W.C., 1993. Protein
oxidative damage is associated with life expectancy of
houseflies. Proc. Nati. Acad. Sci. USA 90, 7255 – 7259.
Sohal, R.S., Weindruch, R., 1996. Oxidative stress, caloric
restriction, and aging. Science 273, 59 – 63.
Smith, C.D., Carney, J.M., Starke-Reed, P.E., Oliver, C.N.,
Stadtman, E.R., Floyd, R.A., Markesbery, W.R., 1991.
Excess brain protein oxidation and enzyme dysfunction in
normal aging and in Alzheimer disease. Proc. Natl. Acad.
Sci. USA 88, 10 540 – 10 543.
Starke-Reed, P.B., Oliver, C.N., 1989. Protein oxidation and
proteolysis during aging and oxidative stress. Arch.
Biochem. Biophys. 275, 559 – 567.
Stadtman, E.R., 1988. Protein modification in aging. J. Geron-
tol. 43, B112 – B120.
Stadtman, E.R., 1992. Protein oxidation and aging. Science
257, 1220 – 1222.
Vanfleteren, J.R., DeVreese, A., 1994. Analysis of the proteins
of aging Caenorhabditis elegans by high resolution two-di-
mensional gel electrophoresis. Electrophoresis 15, 289 – 296.

A.G. Ryazano
6, B.S. Nefsky
/
Mechanisms of Ageing and De
6elopment
123 (2002) 207 – 213
213
Van Remmen, H., Ward, W.F., Sabia, R.V., Richardson, A.,
1995. Gene expression and protein degradation. In: Ma-
soro, E.J. (Ed.), Handbook of Physiology, Section 11:
Aging. Oxford University Press, New York, pp. 171 – 234.
Ward, W.F., 1988a. Food restriction enhances the proteolytic
capacity of the aging rat liver. J. Gerontol. 43, B121 – B124.
Ward, W.F., 1988b. Enhancement by food restriction of liver
protein synthesis in the aging Fischer 344 rat. J. Gerontol.
43, B50 – B53.
Young, V.R., Steffee, W.P., Pencharz, P.B., Winterer, J.C.,
Scrimshaw, N.S., 1975. Total human body protein synthe-
sis in relation to protein requirements at various ages.
Nature 253, 192 – 194.
Youngman, L.D., Park, J.-Y. K., Ames, B.N., 1992. Protein
oxidation associated with aging is reduced by dietary re-
striction of protein or calories. Proc. Natl. Acad. Sci. USA
89, 9112 – 9116.
Wyszukiwarka
Podobne podstrony:
[62]Protein Oxidation in Aging
Key Concepts in Language and Linguistics
Angielski tematy Performance appraisal and its role in business 1
Central Bank and its Role in Fi Nieznany
[WAŻNE] Minister Falah Bakir's letter to Wall Street Journal 'Don't forget Kurds' role in Iraq' (05
Societys Problems and my role in helping it
Key characteristics in 07
Key Concepts in Language and Linguistics
081121 NR 633 Australians take over ANA mentoring role in Uruzgan doc
Bacterial spore structures and their protective role in biocide resistance
KAK, S 2000 Astronomy and its role in vedic culture
The Army Special Operations Forces Role in Force Projection
0415226767 Routledge Fifty Key Figures in Twentieth Century British Politics Sep 2002
Brian Bond The Unquiet Western Front, Britain s Role in Literature and History (2002)
więcej podobnych podstron