
NARKOTYCZNE LEKI PRZECIWBÓLOWE I NIESTERYDOWE LEKI
PRZECIWZAPALNE
Kryteria stosowania leków p/bólowych
Przed przystąpieniem do terapii należy rozeznać się w rodzaju bólu, czasu jego
trwania i objawami towarzyszącymi.
― Opioidowe leki p/zapalne skuteczne w terapii bólów pourazowych,
pooperacyjnych i nowotworowych
― Nieopioidowe leki p/bólowe skuteczne w leczeniu
patofizjologicznego bólu pochodzenia zapalnego
― Trójcykliczne leki p/depresyjne i leki p/padaczkowe stosowane są w
leczeniu bólu neuropatycznego
― Triptany stosuje się w leczeniu migrenowych bólów głowy
Profilaktyka bólu jest lepsza niż leczenie bólu- np. przed operacja podaje się leki
p/bólowe w odpowiednich dawkach, by pacjent na nie nie cierpiał po zabiegu.
Schemat stopniowej farmakoterapii bólów nowotworowych wg WHO

Rozróżnia się dwie główne grupy leków przeciwbólowych:
leki przeciwbólowe opioidowe (analgetyki opioidowe, opioidy, opiaty,
narkotyczne środki przeciwbólowe = narkoanalgetyki, hipnoanalgetyki, silnie
działające analgetyki) – głownie z ośrodkowym, lecz również obwodowym
działaniem
nieopioidowe leki przeciwbólowe (słabe/małe analgetyki) – charakteryzujące
się działaniem zarówno obwodowym, jak i ośrodkowym, a ponadto
posiadające właściwości przeciwgorączkowe i przeciwzapalne
Opioidowe leki p/bólowe
(opioidy, opiaty, narkotyczne leki przeciwbolowe,
hipnoanalgetyki, silnie działające leki przeciwbolowe)
―
Działają p/bólowo dzięki łączeniu się w sposób agonistyczny z
jednym lub kilkoma typami rec opioidowych
―
Profil działania wszystkich leków z tej grupy jest podobny, nie
identyczny, ze względu na wspólne miejsce wychwytu
opioidowych leków p/bólowych w obrębie receptora
opioidowego
―
Ich działanie jest różnorodne, gdyż rec. opioidowe występują w
licznych organach i tkankach
Efekty ośrodkowe:
―
Analgezja (działanie przeciwbólowe):
∙ hamują na poziomie rdzenia kręgowego impulsy bólowe
∙ aktywują endogenny układ przeciwbólowy
∙ zmieniają odczuwanie bólu w obrębie układu limbicznego
(bole nie są odczuwane już jako bardzo nieprzyjemnie i
zagrażające)
―
Sedacja (nadmierne uspokojenie): opioidy zmniejszają uwagę i
zdolność koncentracji, jednak nie wywołują żadnej amnezji
―
Anksjoliza (redukcja/zmniejszenie lęku): opioidy wykazują działanie
przeciwlękowe;
―
Euforia lub dysforia: w zależności od (wyjściowego) nastroju opioidy
prowadzą do podwyższenia nastroju (euforii) lub drażliwości
(dysforii)
―
Depresja oddechowa: opioidy hamują działanie ośrodka
oddechowego
―
Działanie przeciwkaszlowe: opioidy blokują ośrodek kaszlu
―
Sztywność mięśni
―
Wymioty: opioidy pobudzają początkowo ośrodek wymiotny,
później jednak prowadzą do jego hamowania – a tym samym
ujawniają działanie przeciwwymiotne
―
Zwężenie źrenic: opioidy prowadzą do zwężenia źrenic – poprzez
pobudzenie przywspółczulnej części jądra nerwu okoruchowego
―
Działanie antydiuretyczne: opioidy zwiększają wydzielanie hormonu
antydiuretycznego
Efekty obwodowe:
―
Analgezja: opioidy ujawniają działanie przeciwbólowe również
przez działanie na obwodowe receptory opioidowe
―
Opóźnione opróżnianie się żołądka
―
Zaparcie: opioidy osłabiają motorykę i zwiększają napięcie mięśni
gładkich przewodu pokarmowego
―
Skurcz zwieraczy w obrębie dróg żółciowych
―
Zwiększenie napięcia mięśniówki pęcherza moczowego oraz
zwieraczy pęcherza moczowego (zatrzymanie moczu)
―
Zaburzenia ortostatyczne: opioidy obniżają napięcie mięśniówki
naczyń krwionośnych (spadek ciśnienia) z zagrożeniem reakcjami
ortostatycznymi
―
Uwalnianie histaminy: opioidy mogą – poprzez wyrzut histaminy –
prowadzić do zaczerwienienia skóry, powstania pęcherzy i
ujawnienia się świądu skory, jak również – szczególnie u pacjentów z
astmą oskrzelową – mogą wywołać skurcz oskrzeli
W dalszej kolejności istnieje ryzyko rozwinięcia się psychicznego i fizycznego
uzależnienia oraz ujawnienia się zjawiska tolerancji. Jednak w przypadku
zgodnego z zaleceniami oraz prawidłowego stosowania leków – ryzyko
uzależnienia jest stosunkowo niewielkie.
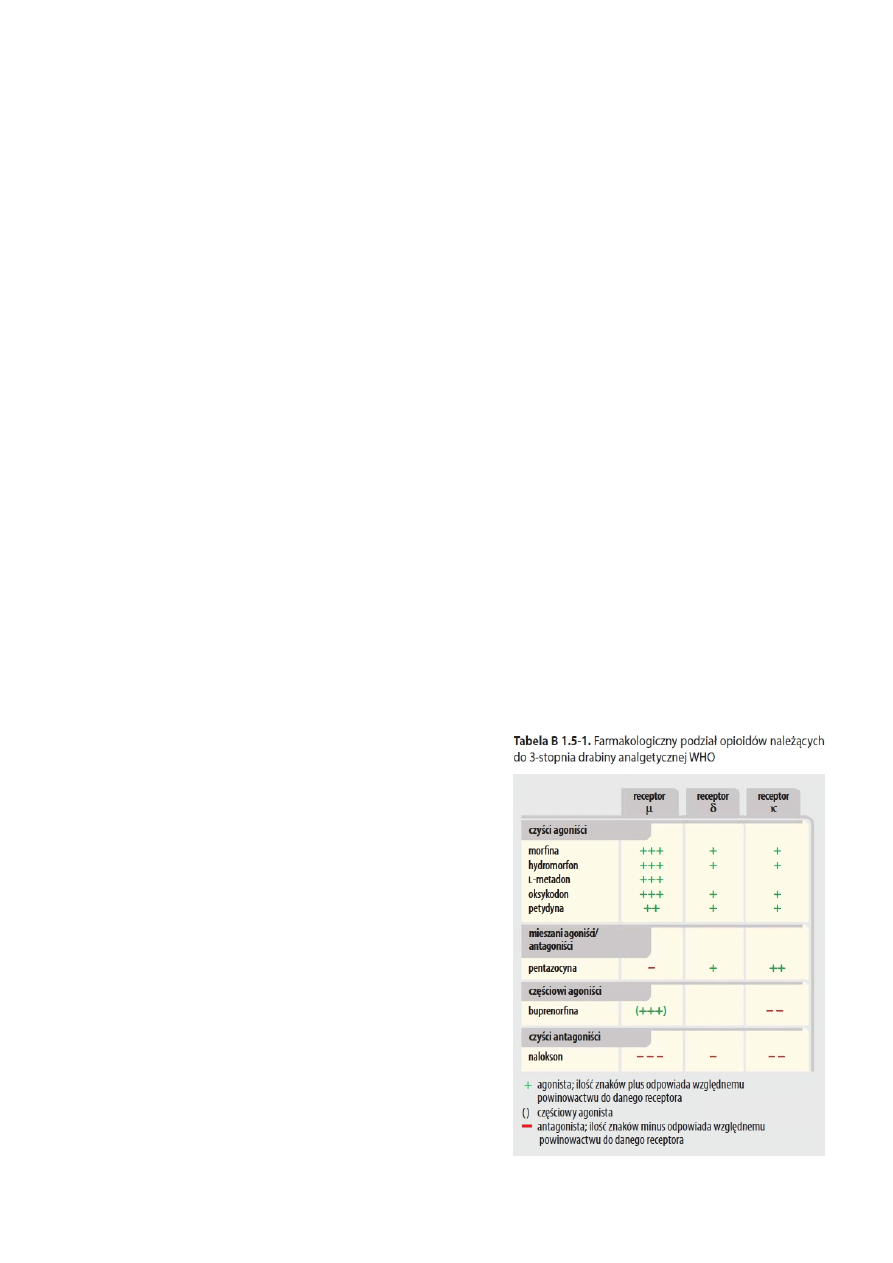
Farmakologiczny podział opioidowych leków p/bólowych
Opioidy różnią się między sobą w zakresie
powinowactwa do receptora, siły
działania, czasu działania
W zależności od powinowactwa do
rożnych typów receptorow opioidowych
oraz ich wewnętrznej aktywności– opioidy
dzieli się:
o czyści (pełni) agoniści (np. morfi
na)
o mieszani agoniści i antagoniści
(np. pentazocyna)
o częściowi agoniści (np.
buprenorfina)
o czyści antagoniści (np. nalokson)
klasyczne OLp/bólowe działają, jako
czyści agoniści przede wszystkim na rec. µ

czyści antagoniści (aktywność wewnętrzna=0) np. nalokson znoszą działanie
opioidów, ponieważ wiążą się mocniej z receptorami opioidowymi, lecz nie
wywołują żadnego działania. Substancje tego typu stosowane są m.in. w
opanowywaniu zatruć opioidami
związki o mieszanym mechanizmie działania np. pentazocyna (wykazują
działanie agonistyczne w stosunku do rec. Κ i równocześnie antagonistyczne
działanie w stosunku do rec. μ) zostały stworzone z założeniem, że ich
potencjał uzależniający mógłby być niższy niż czystych agonistów receptorów
μ
Podobny cel przyświecał badaniom nad rozwojem częściowych agonistow,
np. buprenorfiny. Podobnie jak morfina – buprenorfina działa przeciwbólowo
poprzez receptory opioidowe μ, jednakże z mniejszą aktywnością
wewnętrzną, tzn. buprenorfina nie jest w stanie osiągnąć maksymalnej
skuteczności przeciwbólowej morfiny (efekt pułapowy)
Efekt pułapowy dotyczy również objawów niepożądanych, np. depresji
oddechowej.
Jeżeli – głownie po długim stosowaniu dużych dawek czystych agonistów rec.
μ – dojdzie do zmiany na lek o mieszanym mechanizmie działaniu w obrębie
tych rec, mogą ujawnić się w określonych warunkach objawy zespołu
odstawionego.
Jeszcze nie stworzono silnie działających opioidów bez niebezpieczeństwa
uzależnienia
Stosowanie preparatow o opoźnionym/przedłużonym działaniu (typu retard)
zmniejsza niebezpieczeństwo uzależnienia i nadużywania leków opioidowych,
bo subst. czynna z preparatów retard uwalnia się wolniej i ze względu na
utrzymywanie się stężenia leku w przybliżeniu na podobnym poziomie
zapobiega powstawaniu pików stężenia w osoczu (z działaniem
euforyzującym!).Powolne zmniejszanie się poziomu substancji w osoczu
zmniejsza ryzyko ujawnienia się objawów odstawiennych.
Wskazania:
―
Lek z wyboru w terapii silnych bolow (np. środ- i pooperacyjnych,
nowotworowych)
―
W przypadku zawału serca( równoczesne działanie
psychosedatywne)
―
W przypadku ostrego obrzęku płuc (powodują przerwanie
błędnego koła prowadzącego do duszności, lęku, pogorszenia się
ekonomiki pracy serca – związanego ze stymulacją układu
wspołczulnego – i nasilenia się obrzęku płuc)
―
w przypadku bólów występujących w chorobach układu ruchu (w
zmianach zwyrodnieniowych stawów, reumatoidalnym zapaleniu
stawów)gdy nie można zastosować NLPZ
―
bóle neuropatyczne
―
w terapii substytucyjnej u osob uzależnionych od narkotykow
(metadon, buprenorfi na; heroina w modelowym projekcie)
―
w biegunce (np. loperamid)
―
jako leki przeciwkaszlowe
Ze względu na różnorodność postaci opioidów, różne są drogi podania: drogą
doustną, parenteralnie lub w pobliże rdzenia kręgowego
(podpajęczynówkowo – intratekalnie, nadtwardówkowo- zewnątrzoponowo –
epiduralnie). Wszystkie te możliwości podania leku mają szczególne znaczenie
wobec rozmaitych postaci bólów i ich intensywności.
Dawkowanie:
―
jako leków p/bólowych dawkowanie ma charakter indywidualny,
zależy od intensywności bólu, czasu trwania i charakteru
―
Ze względu na rozwój tolerancji konieczne jest z czasem zwiększenie
dawki
―
W przypadku przewlekłego stosowania opioidow – leczenie
powinno być prowadzone według określonego planu, a sam
pacjent powinien prowadzić „kalendarzyk” bólu
―
Opioidy przedostają się przez łożysko i do mleka matki
Przeciętne dawki ogólnie stosowanych opioidów:
I.
Pochodne morfiny (dawki dzienne w mg)
Morfina
Kodeina
Dimorfina= heroina
Lek p/ bólowy
p/ kaszlowy, p/bólowy
substytucja- w badaniach
10-60 parenteralnie
60-120 p.o. lub
doodbytniczo
60-120 jako fosforan kodeiny
-----------------------------------
II.
Pochodne dihydromorfiny
Dihydrokodeina
Hydromorfon
Oksykodon
Hydrokodon
p/ kaszlowy, p/ bólowy
p/ kaszlowy
p/ bólowy
p/kaszlowy
20-60 (winian dihydrokodeiny)
2 (parenteralnie)
8-32 (kapsułki typu retard)
20-40
10-15
III.
Grupa petydyny
Petydyna
lek p/ bólowy
25-50
IV.
Grupa metadonu
Lewometadon
piritramid
Lek p/bólowy
Lek p/ bólowy
2,5- 7,5
15- 60

V.
Grupa fenantylu
VI.
Mieszani/ częściowi agoniści
Pentazocyna
Buprenorfina
Tilidyna( w skojarzeniu z
naloksonem)
Tramadol
p/ bólowy
p/ bólowy
p/ bólowy
p/bólowy
30-90
0,3-0,9- parenteralnie
0,2-0,8- podjęzykowo
0,8-1,6 przezskórnie
100-300
100-300
Działania niepożądane:
―
Są wynikiem pobudzenia receptorów opioidowych
―
nudności i wymioty (szczegolnie na początku terapii)
―
sedacja (nadmierne uspokojenie;na początku terapii)
―
depresja oddechu/oddechowa (tłumienie ośrodka
oddechowego): objaw ten jest wyrażony znacznie słabiej u
pacjentów z dolegliwościami bólowymi niż u pacjentów bez bólów,
gdyż ból stymuluje ośrodek oddechowy. Szczególna ostrożność u
pacjentów z obturacyjną chorobą płuc oraz rozedmą. Niemowlęta
i dzieci bardzo wrażliwe na opioidowe leki przeciwbólowe. W takim
przypadku zalecane jest stosowanie częściowych agonistów, np.
jak buprenorfiny, gdyż w przypadku hamowania oddechu
wykazuje ona efekt pułapowy
―
spadek ciśnienia krwi (hipotensja): należy zwracać szczególną
uwagę w przypadku hipowolemii lub u pacjentów biorących
równocześnie leki obniżające ciśnienie
―
zaparcia(spastyczne): występuje w przypadku przewlekłej terapii
opioidami (np. w terapii bólów nowotworowych) i musi być prawie
zawsze opanowywany za pomocą leków przeczyszczających
―
działanie polegające na zatrzymaniu moczu- okresowo
kontrolować stan wypełnienia pęcherza moczowego, gdyż może
dojść do jego przepełnienia, czego pacjent może nie wyczuwać
m.in. w związku z efektem przeciwbólowym
―
zwężenie źrenic (mioza)
―
świąd
―
(nadmierne) pocenie się
―
uzależnienie
: psychiczne i fizyczne uzależnienie przy kontrolowanym i
prawidłowym stosowaniu nie stanowi zwykle poważnego problemu. Prawdziwym
problemem zastosowania opioidowa nie jest powstanie uzależnienia w trakcie
terapii przeciwbólowej, lecz ich wykorzystanie, jako narkotyków. Osoby stają się
uzależnione z powodu euforyzującego działania opioidowa oraz ekstremalnie
nieprzyjemnych objawów abstynencyjnych po ich odstawieniu. Osoby uzależnione
od opioidowa (morfiniści) – charakteryzujące się labilnym nastrojem i bladożółtym
wyglądem – błyskawicznie przyzwyczajają się do dużych dawek morfiny, które
mogą sięgać 1 grama (!) morfiny dziennie (rozwój tolerancji). W zaawansowanym
stadium występują bezsenność, wychudzenie, impotencja, drżenie mięśniowe,

zaburzenia koordynacji oraz zaburzenia psychiczne: pacjent degraduje się
fizycznie i psychicznie. Jeżeli osobie uzależnionej odstawi się nagle opioid – w
ciągu godzin ujawniają się objawy abstynencyjne, takie jak niepokój, depresja,
stany lękowe, poczucie marznięcia lub nadmierne pocenie się i nasilone łzawienie.
Po okresie 24–48 godzin zespół abstynencyjny, który może być wyjaśniony zarówno
odhamowaniem neuronów noradrenergicznych w miejscu sinawym, jak i
warunkowanym efektem z odbicia po odstawieniu opioidow zwiększeniem
aktywności cyklazy adenylanowej, osiąga swój punkt maksymalnego nasilenia.
Zespół ten charakteryzuje się nudnościami, wymiotami, biegunkami, nasileniem
oddychania, zwiększenie częstotliwości pracy serca i skurczowego ciśnienia krwi,
zwiększeniem temperatury i odwodnieniem – jako objawami wegetatywnej
dysfunkcji. W dalszej kolejności występują drgawki mięśniowe i skurcze jelitowe.
Opisane objawy abstynencyjne mogą wystąpić u osób uzależnionych również po
podaniu antagonistów opioidowych. W sumie trwa to około tygodnia, zanim
objawy abstynencyjne całkowicie ustąpią. Nasilenie symptomatyki odstawiennej
udaje się złagodzić za pomocą α2-adrenomimetyku– klonidyny. Leczenie
odwykowe jest możliwe do przeprowadzenia wyłącznie w warunkach
stacjonarnych. Oprócz odstawienia środka uzależniającego niezbędne jest
wdrożenie metod psychoterapeutycznych i postępowania resocjalizacyjnego. W
przypadku tzw. programów substytucyjnych, usiłuje się za pomocą pozostającego
pod państwową kontrolą stosowania lewometadonu, racematu metadonu,
buprenorfiny, a ostatnio nawet także heroiny– zmniejszyć związaną z
nadużywaniem środków odurzających kryminogenność oraz zwiększyć
skuteczność terapii odwykowej polegającej na długotrwałym podawaniu
stopniowo zmniejszanych dawek opioidu. Po przeprowadzonej detoksykacji osób
uzależnionych od opioidów w terapii odwykowej opartej na oddziaływaniach
psychoterapeutycznych skuteczną farmakologiczną pomocą może być
włączenie naltreksonu. Podobnie jak nalokson jest on antagonistą receptorów
opioidowych i zapobiega łączeniu się innych agonistów, takich jak heroina, z
receptorem. Jego biodostępność przy doustnym podaniu wynosi 10–40% i jest
znamiennie większa od biodostępności naloksonu (ok. 2%), tak, więc naltrekson
może być stosowany doustnie
―
tolerancja:
∙ w trakcie długotrwałego stosowania może nastąpić jej rozwój-
konieczne jest zwiększanie dawki żeby uzyskać ten sam efekt
p/bólowy
∙ dotyczy również działań niepożądanych: nudności, sedacji i
depresji oddechowej– co powoduje, że występują one
głownie na początku terapii i z czasem zmniejsza się ich
nasilenie.
∙ nie rozwija się jednak w stosunku do takich objawów, jak
zaparcia czy zwężenie źrenic, które z tym samym nasileniem
ujawniają się nawet po długotrwałej ekspozycji na opioidy
∙ rozwój tolerancji wiąże się z mechanizmami adaptacyjnymi
receptorów opioidowych, w których ważną funkcję zdają się
pełnić receptory NMDA. U niektórych pacjentów możliwe jest
osłabienie tolerancji opioidowej za pomocą ketaminy –
antagonisty NMDA
Przeciwwskazania:
―
Stany chorobowe, w których należy unikać tłumienia ośrodka
oddechowego
―
Ostra porfiria wątrobowa
―
Niedoczynność tarczycy – względnie
―
Wrzodziejące zapalenie jelita grubego

―
Zapalenie trzustki
―
W przebiegu mocznicy zwiększa się wrażliwość na opioidy
Interakcje:
Równoczesne spożywanie środków o ośrodkowym działaniu hamującym oraz
alkoholu nasila określone działania niepożądane opioidów.
Ostre zatrucie opioidami:
―
Ostre zatrucie charakteryzuje się głęboką śpiączką z płytkim, a
niekiedy prawie niewyczuwalnym oddechem oraz maksymalnym
zwężeniem źrenic ( typowa triada objawów: utrata przytomności,
depresja oddechu i zwężenie źrenic). Pojawia się również sinica,
zimna skóra i hipotermia.
―
Zgon następuje w wyniku porażenia ośrodka oddechowego.
―
Dawka śmiertelna morfiny u osoby dorosłej nieuzależnionej od leku
wynosi < 0,1 g przy podaniu pozajelitowym oraz 0,3–1,5 g przy
podaniu doustnym (dla noworodka podanie już 2–3 kropli nalewki
opiumowej może być śmiertelne)
―
Terapia zatruć morfiną- najważniejsze jest opanowanie braku tlenu-
niedotlenienia
―
Zastosować sztuczne oddychanie i podać antagonistę
opioidowego- nalokson
―
Dawkowanie naloksonu- początkowo 0,4-2 mg dożylnie,
domięśniowo lub podskórnie, następnie, jeśli to niezbędne 0,4-2mg,
co 2 do 3 minuty
―
Z powodu zagrożenia wystąpieniem niebezpiecznego dla życia
zespołu ostrej abstynencji u osób uzależnionych należy stosować
mniejsze dawki przy jednocześnie krótszych odstępach między
kolejnymi podaniami
Opium
―
Zawiera ponad 20 alkaloidów, których proporcje mogą być bardzo
zmienne
―
Gł. alkaloidem jest morfina; pozostałe ważniejsze alkaloidy
towarzyszące: noskapina (dawniej nazywaną narkotyną), kodeina,
papaweryna, tebaina i narceina
―
Opium bywa jeszcze b. rzadko stosowane pod postacią nalewki
opiumowej w celu uspokojenia motoryki jelitowej w biegunkach
―
Ze względu na zawartość innych alkaloidów, zwłaszcza
papaweryny, podawanie opium prowadzi do zaparcia
atonicznego (w przeciwieństwie do zaparcia spastycznego –
wywoływanego przez morfinę)
Silnie działające leki opioidowe stopnia 3 wg WHO

1. Morfina
― Standardowa substancja opioidowa
― Drogi podania: pozajelitowa, doustna, doodbytnicza
― Biodostępność- 25%, związków retard- 40%
― Wolne wchłanianie z przewodu pokarmowego
― Silny efekt pierwszego przejścia
― Podczas pierwszego przejścia przez wątrobę ulega przekształceniu
w 6-glukuronian morfiny, który ma podobne działanie agonistyczne
na receptory µ
― Główny metabolit to nieczynny 3-glukuronian morfiny
― T
1/2
= 2-3 godz.
― U chorych z niewydolnością nerek należy zmniejszyć dawkę lub
podać inny lek opioidowy (bo kumuluje się 6-glukuronian morfiny)
― Dawki
∙ dożylnie- 5-10 mg
∙ podskórnie- 10-30 mg
∙ doustnie- 30-60 mg
2. Diamorfina (heroina, diacetylomorfina)
― Ulega szybkiej hydrolizie do morfiny
― Szybciej niż morfina pokonuje barierę krew- mózg (bo jest lipofilna)
― Przez łatwą penetrację do OUN- szybko wywołuje uzależnienie
― Jedyny kraj, który legalnie stosuje ją jako środek przeciwbólowy to
Anglia
― Obecnie wykorzystywana tylko w specjalnych programach
odwykowych
3. Hydromorfon
― 7,5x silniejszy od morfiny
― T
1/2
= 3 godz.
― Biodostępność- 40%
― Główny metabolit- 3-glukuronian hydromorfonu nie działa
p/bólowo
4. Lewometadon
― 4x silniejszy od morfiny
― Ma dłuższy okres działania niż morfina
― Ma słabsze działania niepożądane niż morfina
― Objawy abstynencyjne rozwijają się wolniej i są mniej nasilone
― Prawie całkowita biodostępność i dobra wchłanialność
― T
1/2
= 20-60 godz. (indywidualnie zmienny)
5. Oksykodon

― Nieco silniejszy niż morfina
― Biodostępność większa od morfiny
― T
1/2
= 3-56 godz.
― Stosowany obecnie w preparatach z naloksonem
∙ 10mg oksykodonu i 5mg naloksonu, lub 20mg o. i 10mg n.
∙ Nalokson łączy się z receptorami opioidowymi po
wewnętrznej stronie ściany jelit- zapobieganie zaparciom
indukowanym przez opioidy
∙ Nalokson ulega całkowitemu rozkładowi przy pierwszym
przejściu przez wątrobę, dlatego nie znosi działania
p/bólowego opioidów
6. Petydyna
― Działa 5x słabiej od morfiny
― T
1/2
= 2-6 godz.
― Biodostępność- 50%
― Metabolit to norpetydyna- ma właściwości drgawkorodne
― Nie nadaje się do długotrwałej terapii, bo norpetydyna się kumuluje
7. Piritramid
― Ma podobną siłę działania do morfiny
― Dobra sterowalność przy podaniu dożylnym
― T
1/2
= 4-10 godz.
8. Buprenorfina
― Działanie:
∙ Częściowy agonista receptorów opioidowych µ- duże
powinowactwo do nich
∙ Agonistyczne z małym powinowactwem do receptorów
N/OFQ
∙ Antagonistyczne na receptory opioidowe κ
― 40x silniejsze działanie niż morfina
― Niższa aktywność wewnętrzna- nigdy nie osiąga maksymalnego
efekty analgetycznego (bo częściowy agonista)
― Taki sam potencjał uzależniający jak inne opioidy
― Poza terapią p/bólowa stosowana w leczeniu uzależnień
(wypieranie lub uniemożliwianie wiązania heroiny z receptorami)
― T
1/2
= 12-16 godz.
― Wysoka biodostępność przy podaniu podjęzykowym, a niska przy
podaniu doustnym
― Wolno oddziela się od receptora- duże dawki czystych
antagonistów opioidowych przy przedawkowaniu
― Może być stosowana w przezskórnych systemach terapeutycznych

9. Pentazocyna
― Działanie:
∙ Pełny agonista receptorów opioidowych κ
∙ Antagonista receptorów opioidowych µ
∙ Wykazuje efekt pułapowy (
po przekroczeniu określonej dawki
skuteczność przeciwbólowa nie poprawia się, natomiast znacznie zwiększa się
ryzyko wystąpienia objawów niepożądanych)
― 1/3 siły działania morfiny
― Podwyższa ciśnienie tętnicze i przyspiesza czynność serca
― T
1/2
= 2-4 godz.
10. Fentanyl
― Może być stosowany w przezskórnych systemach terapeutycznych
11. Alfentanyl
12. Sufentanyl
13. Remifentanyl
Słabo działające leki opioidowe stopnia 2 wg WHO
1. Kodeina
― Stosowana w terapii skojarzonej z nieopioidowymi lekami
p/bólowymi
― Demetylacja kodeiny do morfiny- to wywołuje efekt p/bólowy
― Zależnie od metabolizmu CYP2D6 9enzym odpowiadające za
demetylację) może dojść do zatrucia morfiną, lub
niewystarczającego efektu terapeutycznego
― T
1/2
= 2-3 godz.
2. Dihydrokodeina
― 3x silniejsza od kodeiny
― W wątrobie ulega przemianie do dihydromorfiny (DHC ma duży
potencjał uzależniający)
― DHC ma nasilony efekt pierwszego przejścia
― T
1/2
= 3-5 godz.
3. Tilidyna
― Słabe działanie agonistyczne
― Substancja właściwie działająca to nortilidyna
― Stosowana w preparatach łączonych z naloksonem (50mg t.+4mg
n.)
― Terapia skojarzona nie jest stosowana u chorych z niewydolnością
wątroby, ale polecana u osób z niewydolnością nerek
― Terpaia skojarzona nie wywołuje uzależnień

4. Tramadol
― Najczęściej stosowany opioid
― Działanie:
∙ Słaby agonista receptorów opioidowych µ (1/6000 morfiny)
∙ Hamowanie wychwytu zwrotnego monoamin (NA,
serotonina)
∙ (+)-tramadol hamuje serotoninę, a (-)-tramadol hamuje NA
∙ (+)-tramadol dużo silniejszy niż (-)-tramadol
― Siła działania słabsza niż morfiny
― Słabszy wpływ depresyjny na ośrodek oddechowy i słabsze
działanie uzależniające
― Działania niepożądane:
∙ W dużych dawkach wywołuje nudności i wymioty
― T
1/2
= 5 godz.
Dronabinol
― Stereoizomer 9-tetrahydrokanabinolu (THC)
― Podawanie jest dyskusyjne
― Tylko postępowanie uzupełniające, wspomagające w terapii bólu
(gł. stwardnienie rozsiane, lekooporny świąd)
― Max dawka: 500 mg na 30 dni
― B. słaba dostępność doustnie
― Przechodzi przez barierę krew-mózg, łożysko i do mleka matki
― Gł. metabolit: 11-hydroksy-9-THC jest związkiem czynnym
― Potencjał uzależniający względnie słaby, ale objawy
abstynencyjne po odstawieniu dawek leczniczych, możliwe
uzależnienie fizyczne
― Działanie:
∙ Nieselektywne wiązanie z receptorami kannabinoidowymi
CB
1
i CB
2
― Działania niepożądane:
∙ Tachykardia
∙ Suchość w jamie ustnej
∙ Działania ośrodkowe: osłabienie sprawności psychofizycznej,
pamięci
Leki przeciwkaszlowe
Tłumienie odruchu kaszlu w wyniku hamowania ośrodka kaszlu i/lub blokowania
receptorów kaszlowych w drzewie oskrzelowym.
Wskazane tylko w suchym kaszlu.

1. Kodeina
― Pochodna morfiny
― Działanie p-bólowe osłabione przez eteryfikację gr. hydroksylowej
pierścienia fenolowego morfiny
― Wykazuje pełne działanie przeciwkaszlowe morfiny
― Może być stosowana p-bólowo
2. Dihydrokodeina
― Jeden z najczęstszych leków
― Może być stosowana p-bólowo
3. Hydrokodon
― Stosowany w ciężkich przypadkach
― Może prowadzić do uzależnienia
4. Noskapina
― Alkaloid maku lekarskiego
― Działanie p-kaszlowe porównywalne z kodeiną, brak działania p-
bólowego
― Rzadkie działanie niepożądane: zespół Stevensa-Johnsona
5. Dekstrometorfan
― Prawoskrętny enencjomer metyloeteru leworfanolu
― Oddziałuje na receptory opioidowe dopiero po przedawkowaniu
― Antagonistyczne działanie na receptor NMDA
― Coraz częściej stosowany w neuropatiach
6. Klobutinol
― Strukturalnie podobny do metadonu
― Brak działania p-bólowego i depresyjnego na ośrodek oddechowy
― Wydłuża odstęp QT
C
(wycofany z rynku)
7. Pentoksyweryna
― Zasadowy ester
― Brak działania nasennego i depresyjnego na ośrodek oddechowy
Nieopioidowe leki p/bólowe stopnia 1 wg WHO
― Określane jako małe lub obwodowe (błędnie) analgetyki
― Działanie p-bólowe i p-gorączkowe

― Brak działania psychotropowego i uspakajającego (sedatywnego)
― Zakres działania bardzo duży (wyj: NEFOPAM, FLUPIRTYNA)
― W bólach wywołanych stanami zapalnymi ich działanie jest lepsze
od opioidów
Patofizjologia gorączki i zapalenia
― Termoregulacja i gorączka
∙ Ośrodek termoregulacji: w przedniej części podwzgórza
∙ Przegrzanie: wzmożone pocenie i nasilenie przepływu krwi
przez skórę
∙ Przechłodzenie: skurcz naczyń obwodowych
― Gorączka: termoregulacja z przestawieniem wartości należnej
temperatury na wyższy poziom. (temp. 37
0
odczuwalna jest jako
wychłodzenie)
∙ Związki powodujące gorączkę: egzogenne pirogeny ( części
patogennych mikroorganizmów, wirusy)
∙ Powstawanie odczynu gorączkowego: współdziałanie
układu immunologicznego, endokrynnego i OUN
∙ Krążeniowe pirogeny: cytokiny IL-1, IL-2, czynnik martwicy
nowotworu α (TNF-α)
∙ IL-1- gł. w miejscu uszkodzenia tkanki
∙ W mózgu cytokiny indukują COX-2 -> synteza prostaglandyn,
gł. PGE
2
∙ PGE
2
– końcowy przekaźnik odczynu gorączkowego
(pobudzenie receptorów prostaglandynowych, zwiększenie
zależnej od cAMP przemiany komórkowej neuronów ośrodka
termoregulacyjnego podwzgórza, wzrost ciepłoty ciała)
― Potwierdzające badania na zwierzętach:
∙ antagoniści IL-1 i leki hamujące COX-2 (wybiórcze i
nieselektyne) działają przeciwgorączkowo
∙ myszy pozbawione COX-2 (znokautowane genetycznie) po
podaniu egzogennego pirogenu nie reagują odczynem
gorączkowym
― Faza gorączki:
∙ Skurcz skórnych naczyń krwionośnych, dreszcze i
subiektywne odczucie zimna
∙ (temp. 37
0
odczuwalna jest jako wychłodzenie)
― Faza odgorączkowania:
∙ Panująca temperatura ciała odczuwalna jest jako zbyt
wysoka
∙ Obfite pocenie się, rozszerzenie naczyń skórnych,
subiektywne uczucie gorąca
Zapalenie
― Odczyn zapalny na skutek działania fizycznych i chemicznych
czynników szkodliwych (noksów)
― Ścisłe powiązanie reakcji naczyniowych i komórkowych oraz
odczynów obronnych zarówno specyficznych, jak i
niespecyficznych antygenowo.
― Objawy podstawowe ostrej reakcji zapalnej Aulusa Corneliusza
Celsusa:
∙ zaczerwienienie (rubor)
∙ obrzęk (tumor)
∙ miejscowe podwyższenie ciepłoty (calor)
∙ ból (dolor)
∙ upośledzenie czynności (functio laesa) –piąty objaw
dodatkowy Galena
następstwa zaburzeń w obrębie mikrokrążenia z
ucieczką składników osocza do przestrzenie
międzykomórkowej (wysięk)
pobudzenie i uwrażliwienie nocyceptorów przez
mediatory zapalenia
może dołączać się migracja elementów
komórkowych krwi (granulocyty, monocyty), rozplem
histiocytów i fibroblastów, co może mieć wpływ
szkodliwy np. w przewlekłych zapaleniach
reumatycznych
Właściwości farmakologiczne nieopioidowych leków p/bólowych
Podział:
ANELGETYKI NIEOPIOIDOWE
NLPZ (NSAID)
inne
Pochodne kwasowe (prócz
niektórych inhibitorów COX-2)
Mają wł. Lipo- i hydrofilne
Działanie:
1. Przeciwbólowe
2. Przeciwgorączkowe
3. PRZECIWZAPALNE
Bardzo silnie wiążą się z białkami
osocza (99%)
Pochodne obojętne lub lekko
zasadowe
Brak wł. Lipo- i hydrofilnych
Działanie:
1. Przeciwbólowe
2. Przeciwgorączkowe
Znacznie słabiej wiążą się z
białkami osocza
Wł. przeciwzapalne NLPZ-ów jest sprawą wątpliwą:

― NLPZy gromadzą się w tkankach o kwaśnym pH (stan zapalny)
― Docierają tam z białkami osocza (wysięk zapalny osocza)
― Hipoteza opiera się na tym, że w tej tkance jest odpowiednia ilość
frakcji niezwiązanej z białkami
― Przeciwko w/w hipotezie: inhibitory COX niekwasowe mają takie
samo działanie jak NLPZy
NIEOPIOIDOWE ANALGETYKI
Poch. Kwasowe - NLPZ
Poch. Niekwasowe - inne
NP.
Salicylany
Poch. Kw. Octowego
Poch. Kw. Propionowego
Oksamy
Fenamaty i in.
NP.
Paracetamol
Fenazon
Propyfenazon poch.
pirazolowe
Metamizol
fenylobutazon
Mechanizm działania NLPZów:
― Oddziaływują na biosyntezę PG
― W dawkach leczniczych
hamują COXy (zamiana kw.
Arachidonowego
cykliczne nadtlenki, np. PG H2, która jest
prekursorem dla PG, THA2, prostacyklin)
― Blokują syntezę PG (działanie):
∙ Przeciwbólowo
∙ Przeciwgorączkowo
∙ Przeciwzapalnie
― PG syntetyzowana prawie we wszystkich komórkach i tkankach
COX-1
COX-2
Enzym KONSTYTUTYWNY
(stale występujący)
Występowanie: żołądek,
płytki krwi, nerki
COX-1 ujawnia DN w/w
narządach
Enzym SZYBKO
INDUKOWANY (gen dla
COX-2 – gen
natychmiastowej wczesnej
odpowiedzi) przez różne
czynniki, np. cytokiny
COX-2 w czasie:
- zapalenia
- reakcji bólowej
- inne uszkodzenia tkanek
COX-2 odp. Za działanie
NLPZów, tj. przeciwbólowo,
gorączkowo i zapalnie
Biorą udział w powstawaniu: bólu,
gorączki i stanu zapalnego

NLPZ w dawkach leczniczych
hamują oba COXy (izoformy)
wyjątek KOKSYBY selektywne na COX-2
COX-2 jako enzym konstytutywny w:
- rdzeń kręgowy
- nerki
- macica
- śródbłonek naczyń
Jego duże wytwarzanie (COX-2):
- w miejscu gojenia się ran lub wrzodu
- w macicy w procesie zagnieżdżania się jaja
- komórki śródbłonka naczyń
Farmakokinetyka:
― Szybkie i dobre wchłanianie
Wskazania:
― Różne stany bólowe (np. bóle głowy i zębów)
― Bóle w przebiegu choroby zwyrodnieniowej stawów
― Migrena
― Gorączka
― Patofizjologiczne (warunkowane procesem zapalnym bóle
nocyceptywne, włącznie z występującymi w związku z chorobą
reumatyczną
― Bóle związane z przerzutami nowotworowymi do kości
― Inhibitory syntezy prostaglandyn -> zamknięcie przetrwałego
przewodu tętniczego Botalla
Działania niepożądane:
― Zaburzenia żołądkowo-jelitowe, nadżerki, owrzodzenia, krwawienia,
perforacje
― Reakcje skórne
― Zaburzenia czynności nerek – retencja sodu i wody -> tworzenie się
obrzęków
― Zahamowanie agregacji płytek krwi
― Objawy ze strony OUN
― Zmniejszenie aktywności motorycznej macicy
― Powikłania sercowo naczyniowe
― Wyzwolenie napadu astmy (związane z zahamowanie
cyklooksygenaz i podaż substratu dla lipooksygenaz)
Zwróć uwagę

― COX-2 mają działania podobne do nieselektywnych NLPZ poza:
∙ brakiem hamowania COX-1
∙ hamowania agregacji płytek
Przeciwwskazania:
― Choroba wrzodowa żołądka i XII-cy
― Astma
― Skaza krwotoczna
― Ostatnie tygodnie ciąży (bo przedwcześnie zamknie się przewód
Botalla)
― Uszkodzenie wątroby i nerek -> stosujemy z dużą ostrożnością
Interakcje:
― GKS zwiększają niebezpieczeństwo powikłań żołądkowo- jelitowych
― Zmniejsza się urykozouryczne działanie probenecidu
― Zmniejszają moczopędne działanie saluretyków
― Nasilają działanie hipoglikemiczne doustnych leków
p/cukrzycowych
― Spowalniają eliminację metotreksatu
― Zmniejszają wydalanie Li+
― Osłabiają działanie p/zakrzepowe pochodnych kumaryny
― Zmniejszają efekt antyhipertensyjny leków p/nadciśnieniowych,
szczególnie inhibitorów ACE
Kwas acetylosalicylowy (ASA)
― Działanie
∙ P/gorączkowe i p/zapalne w szczególności zahamowanie
agregacji płytek krwi
∙ Inaktywuje cyklooksygenazy przez nieodwracalną
acetylację reszty serynowe
∙ Płytki krwi -> nie mają jąder -> nie mogą się regenerować ->
krótki czas działania ASA ok 15 min utrzymuje się przez kilka
dni zanim kolejna pula trombocytów dojrzeje
― Szybkie wchłanianie
― T1/2= 15 min w osoczu, kwas salicylowy 2-3h
― Wydalanie -> nerki
― Dawkowanie:
∙ W stanach bólowych i gorączkowych 1,5-3g/dobę
∙ Ch. Reumatyczne 4-6g/dobę
∙ Profilaktyka zawałów 100mg/dobę
― Specyficzne działania niepożądane (częściej niż w innych NLPZ):
∙ Zgaga, dolegliwości żołądkowe
∙ Mikrokrwawienia śluzówki żołądka
― Ciężkie działania niepożądane:

∙ Szum w uszach, upośledzenie słuchu, zawroty głowy,
nudności wymioty, krwawienia z przew pokarmowego,
owrzodzenia
∙ Obniżenie protrombiny
∙ Zespół Reye’a u dzieci– dlatego raczej nie stosuje się u
dzieci!
∙ Występują w wysokich dawkach po dłuższym stosowaniu
― Interakcje z NLPZ:
∙ ASA i ibuprofen – ibuprofen może związać się z katalitycznym
centrum COX-1 przed ASA -> dlatego możliwe jest zniesienie
działania kardioprotekcyjnego -> pacjenci przy profilaktyce
zawału mogą zażywać nie więcej niż 1 dawkę ibuprofenu 2h
po przyjęciu ASA
∙ Równoczesne przyjmowanie paracetamolu, diklofenaku,
celekoksybu nie wpływa na działanie ASA
― Zatrucia:
∙ OSTRE:
- hiperwentylacja (nadmierne wydychanie CO
2
->
oddechowa zasadowica)
- poty, pobudzenie
- porażenie czynności oddechowej
- utrata przytomności
- hipertermię
- odwodnienie
- postępujące zatrucie prowadzi do kwasicy
∙ LECZENIE:
- zahamowanie dalszego wchłaniania ASA -> wlew
wodorowęglanu sodu
- w przypadku zatruć zagrażających życiu wykonuje się
hemodializę
Pochodne kwasu octowego
1. Indometacyna
― bardzo silny inhibitor cyklooksygenaz (mocniej na COX-1)
― przeważające działanie:
∙ przeciwzapalne
∙ przeciwreumatyczne
― dawka 50-150 (-200) mg
― działania niepożądane:
∙ zaburzenia żółądkowo-jelitowe (częściej niż w innych NLPZ
∙ zaburzenia czucia
∙ bóle głowy (okol. czołowa)
∙ zaburzenia widzenia
∙ wymioty
― prolek dla indometacyny – acematacyna

2. Diklofenak
― dawka 12,5-25 mg, terapia przeciwreumatyczna 50-100 mg
― w celu uzyskania środków lepiej znoszonych niż indometacyna
zsyntetyzowano aromatyczne lub heteroaromatyczne pochodne
kwasu octowego, np. diklofenak
― bardzo silny inhibitor COX (niewielka preferencja do COX-2)
― działania niepożądane:
∙ zaburzenia żołądkowo-jelitowe (mniej niz indometacyna)
∙ prowadzi do zwiększenia się poziomu enzymów
wątrobowych
∙ przy podaniu pozajelitowym – szok anafilaktyczny
∙ owrzodzenia przewodu pokarmowego – podajemy razem z
0,2 mg mizoprostolu
3. Pochodne kwasu arylopropionowego
― posiadają asymetryczny atom węgla
― enancjomery typu S hamują cyklooksygenazy 100-1000 razy silniej
niż R
― występują jako racematy (mieszanki S i R) albo czyste S (np.
naproksen)
― enancjomery S nie wywołują mniej działań niepożądanych
― niektóre enancjomery R mogą in vivo przechodzic w S
Ibuprofen:
― względnie słaby, nieselektywny inhibitor cyklooksygenaz
― najmniejsze ryzyko ciężkich żołądkowo-jelitowych działań
niepożądanych
― sól D,L-lizynowa dobrze rozpuszcza się w obrębie przewodu
pokarmowego, dlatego potrzebuje mniej czasu, żeby osiągnąć
skuteczny poziom w osoczu
― deksibuprofen - S-ibuprofen (działa silniej niz R-ibuprofen, mimo to
stosowany w połowie dawki powoduje podobne działania
niepożądane)
― dawka: pojedyncza 200-400 mg (przeciwbólowe)
400-800 mg (przeciwreumatyczne)
maksymalnie 1200-2400 mg
Naproksen: dawka 200 mg, max 600 mg
4. Oksykamy
― Piroksykam – nieslektywny inhibitor cykooksygenazy
∙ Dawka dzienna w leczeniu przeciwreumatycznym: 20 mg
― Meloksykam – hamuje COX-2> COX-1
∙ Dawka dzienna w leczeniu przeciwreumatycznym:7,5 –
15mg
― Lornoksykam - nieslektywny inhibitor cykooksygenazy

∙ Dawka dzienna w leczeniu przeciwreumatycznym:12-16mg
Niesteroidowe leki przeciwzapalne selektywne względem COX-2 (koksyby)
Wszystkie koksyby powinny być stosowane w możliwie najmniejszych dawkach, przez
możliwie jak najkrótszy czas gdyż ryzyko powikłań sercowo naczyniowych wzrasta
prawopodobnie z dawką i czasem stosowania.
― Działanie: hamują tylko COX-2
― Wskazania :terapia rodzinnej polipowatośći gruczalakowatej jelita
grubego – COX-2 hamują znacznie wzrost różnych komórek
nowotworowychi polilów jelita grubego.
― Działania niepożądane: mniej niż w tradycyjnie stosowanych NLPZ
∙ infekcje górnych dróg oddechowych
∙ niestrawność
∙ biegunki
∙ uczucie ucisku w nadbrzuszu
∙ bóle głowy
∙ obwodowe obrzęki~Jak w tradycyjnych NLPZ
∙ podwyższenie ciśnienia krwi~jak w tradycyjnych NLPZzawał
serca i udar mózgu (rzadko)
― Przeciwwskazania: ciąża
1. Celekoksyb
― Dawka:
∙ W zmianach zwyrodnieniowych stawów (jednorazowo) 200
mg
∙ W reumatoidalnym zapaleniu stawów (dwa razy) 100 – 200
mg
∙ W FAP – rodzinnej polipowatości gruczolakowatej jelita
grubego (dwa razy) 400 mg
― Farmakokinetyka
∙ Biodostępność 50 – 70%
∙ Maksymalne stężenie w osoczu po 3h
∙ Metabolizm: przez CYP2A9 do nieczynnych metabolitów
∙ Okres półtrwania: 6 – 12h
― Działania niepożądane: celekoksyb jest pochodną sulfonamidu
dlatego może dojść do alergii
2. Etorikoksyb
― Dawka:
∙ w zmianach zwyrodnieniowych stawów (jednorazowo) 60
mg
∙ w reumatoidalnym zapaleniu stawów (jednorazowo) 90 mg
∙ w ostrym zapaleniu stawów (jednorazowo) 120 mg (do 8 dni)

― Przeciwwskazania: zespoły bólowe z nadciśnieniem
3. Lumirakoksyb (wycofany – ze wzgl. Na ddoniesenia o ciężkich reakcjach
ze str. Wątroby.)
― Dawka: 100 mg/doba
― Skuteczność : taka jak innych koksybów
― Działania niepożądane: jako pochodna diklofenaku podnosi
poziom enzymów wątrobowych
4. Parekoksyb (prolek waldekoksybu)
― Podawany pozajelitowo
― Dawka: 40 mg i.v., maksymalnie 80 mg/d i.v.
― Zastosowanie: krótkotrwała terapia bólów pooperacyjnych\
5. Rofekoksyb (wycofany)
― Wycofany ze względu na podwyższone ryzyko wystąpienia
powikłań sercowo naczyniowych
Inne niesteroidowe leki p/zapalne
1. Nabumeton – prolek
― Dawka: 500 mg 2x dziennie
― Nie posiada działania drażniącego
― Działania niepożądane – takie jak inne NLPZ
2. Nimesulid
― Dawka: 100 mg 2x
3. Aceklofenak
― Dawka: 200 mg dziennie – w leczeniu zwyrodnienieniowych zmian
kostno– stawowych lub RZS
― Działanie:
∙ słaby inhibitor COX
∙ hamuje syntezę interleukiny-1 prostaglandyny zapalne
― Zastosowanie:
∙ reumatoidalne zapalenie stawów
∙ zwyrodnienia kostno-stawowe
4. Oksaceprol [N-acetylohydroksyprolina]
― Dawka: 200 – 400 mg 3x dziennie
― Zastosowanie: choroby degeneracyjne i zapalne stawów
― Działania niepożądane:
∙ zaburzenia żołądkowo-jelitowe (bóle brzucha, utrata
łaknienia, biegunki)
∙ alergie skórne
∙ ulcerogenność i nefrotox – rzadko

Leki p/gorączkowe i p/bólowe niebędące kwasami
Mechanizm działania:
― Przenikają barierę krew-mózg i hamują syntezę prostaglandyn
wywołaną bodźcami bólowymi (na poziomie rdzenia kręgowego i
mózgowia, dlatego tez nie hamują syntezy prostaglandyn na
obwodzie i m-cu zapalenia, poza tym nie wykazują typowych
działań niepożądanych)
― Przykład : paracetamol ---> ulega deacetylacji do 4-aminofenolu,a
następnie przez enzym FAAH do arachidonoilofenolaminy (AM-
404), która jest nieselektywnym inhibitorem cyklooksygenaz
― Ale tylko w OUN jest wystarczające stez FAAH
Pochodne aniliny
1. Paracetamol
― Dobre działanie p/gorączkowe, słabe p/bólowe i bardzo słabe
p/zapalne (nie działa w ognisku, bo brak FAAH)
― Per os wchłania się szybko i dobrze
― Okres półtrwania 2-3h
― Wiazanie z bialkami 30%
― Eleminacja przez biotrans. do glukuronianu i siarczanu
― Dawki :500-1000 mg/24h dorośli, 50mg/kg m.c. dzieci
― Zatrucia
∙ Wykazuje głownie działanie hepatotoksyczne
∙ Dawki powyżej 10g prowadza do śmiertelnej martwicy
hepatocytow (leczenie środkami z grupami -SH : metionina ,
cysteamine, acetylocysteine)
Pochodne pirazolu
1. Metamizol:
― Dobrze zwalcza bol (dobrze rozpuszczalny w wodzie--> i.v., ale
istnieje ryzyko wstrząsu ! dlatego powolne podawanie np. jako
krótkotrwały wlew)
― Działa spazmolitycznie (kolki)
― Może być podawany doustnie (substancja czynna jest wtedy 4-
metylofenazon)
― Okres połowicznego rozpadu 2-5h po per os
― Wydalanie przez nerki (czasem czerwone zabarwienie moczu-kw.
rubazonowy)
― Dawka : per os 0,5-1 g , i.v. 1-2,5g
― Działania niepożądane:
∙ agranulocytoza

∙ zmiany na bl. śluzowych i skórze
∙ przy zatruciach- drgawki
∙
2. Fenazon i propyfenazon:
― Działania niepożądane i skuteczność jak metamizol
3. Fenylobutazon:
― Per os dobrze wchłania się
― Dobre wiązanie z białkami (98%)
― T1/2 ok.70h
― Metabolity również działają p/zapalnie, poza tym może być i.v.
(jako sól)
― Zastosowanie:
∙ Ostre napady dny,
∙ Ch. Bechterewa (zesztywniajace zap. stawow kregoslupa)
― Dawkowanie:
∙ Dna-> 400-800mg/24 przez 3 doby
∙ Ch.Bechterewa 200-400mg/24 do 7 dni
― Działania niepożądane: o wiele częściej (30% pacjentów), niż w
metamizolu
― Interakcje:
∙ Obniża wiązanie leków p/cukrzycowych i p/zakrzepowych
typu dikumarolu z bielakami osocza
Flupirtyna
― Mechanizm działania:
∙ Właściwości p/bólowe + ośrodkowe działanie
miorelaksacyjne (brak działania p/gorączkowego i
p/zapalnego)
∙ Zwiększa przepuszczalność kanałów przez jony potasu
(słabsze pobudzanie przez bodźce nycoreceptywne)
― Wchłaniana w 90%, wydalana przez nerki
― Dawka:
∙ per os 100mg, max.600mg (niewydolność nerek max. 300mg)
― Działania niepożądane:
∙ Zaburzenia OUN
∙ Zaburzenia żołądkowo-jelitowe
∙ Suchość w ustach
∙ Nadmierne pocenie się
∙ Reakcje skórne
∙ Zaburzenia widzenia
― Przeciwwskazania:
∙ encefalopatia wątrobowa
∙ zastój żółci
∙ miastenia

― Nasila działanie : etanolu, l. uspakajających, zwiotczających
mięsnie, p/zakrzepowych
Zikonotyna
― Pochodna trucizny ślimaka morskiego Conus magnus
― Blokuje na poziomie rdzenia kręgowego presynaptyczne kanały
wapniowe typu N (hamuje uwalnianie mediatorów bólowych)
― Jest do podawania podpajęczynówkowego
― Nie jest metabolizowana w płynie mózgowo-rdzeniowym
― T1/2=4-5h
― Podawanie we wlewie za pomocą cewnika dokanałowego
(dawka początkowa 2,4 mikra/gram/dzień), zalecany odstęp 48 h
miedzy kolejnymi dawkami, max dawka = 21,6 mikrogram
― Działania niepożądane:
∙ oczopląs
∙ bole głowy
∙ zawroty
∙ zaburzenia chodu
∙ zab. Pamięci
∙ astenia
∙ senność
∙ działania niepożądane sercowe, nerkowe, płucne
Złożone leki p/bólowe
― Znaczna część preparatów w połączniu z kofeina, środkami
nasennymi, lekami rozkurczowymi
― Istnieje ryzyko uszkodzenia nerek
Wyszukiwarka
Podobne podstrony:
do 3 id 137583 Nieznany
odpowiedzi do testu id 332437 Nieznany
Montaz zamka do drzwi id 307578 Nieznany
mnozenie do 25 2[1] id 304290 Nieznany
DO Gimn 1 id 137870 Nieznany
dodawanie do 10 4 id 138940 Nieznany
F Zadania do kol 1 id 167111 Nieznany
Droga Polski do NATO id 142564 Nieznany
mnozenie do 25 11 id 304283 Nieznany
mechanika do poprawki id 290847 Nieznany
DO P gimn 4 id 137938 Nieznany
od bollanda do deminga id 33072 Nieznany
karteczki do wydruku id 232392 Nieznany
ZestawY do Dziekonskiej id 5891 Nieznany
więcej podobnych podstron