
Vol. 9
CHARACTERIZATION OF POLYMERS
159
CHARACTERIZATION
OF POLYMERS
Introduction
Over the last 50 years, systematic investigation has established a number of
structure–property relationships for polymeric materials, which allow prediction
of their physical properties from a knowledge of the chemical structure of the re-
peat units and molar mass of the material. All polymeric materials can be divided
into a series of subclasses, reflecting either their method of synthesis or some
particular characteristic of the material. Using this classification, it is possible to
quickly identify how the material will respond to external factors such as change
of temperature, pressure, stress, impact, etc.
The term polymer (poly–many, and monomer–low molar mass unit) reflects
the simplicity of the high molecular weight macromolecular structure. Natural
polymers, although they may have complex sequences of repeat units, can exhibit
physical properties that obey the same physical laws as their equivalent synthetic
polymers. The increased use of polymers in everyday life has come with the discov-
ery of simple synthetic routes to the raw materials and the ability to tailor their
physical properties to particular applications. Despite the wide range of methods
available for their synthesis, the physical characteristics of many polymers can
be predicted by recognizing certain generic features in their structure.
Many polymers are created from the reaction of monomers that contain an
unsaturated carbon (ene/vinyl) bond. For example, polyethylene [ (CH
2
CH
2
)
n
],
where n is the degree of polymerization and indicates the number of the repeat
units in the chain. Synthesis of the polymer can be achieved using either free-
radical processes, ionic (anionic or cationic initiation), or using a coordination
polymerization route that uses an addition reaction. These synthetic routes pro-
duce macromolecular chains that have similar chain lengths and well-defined
physical properties. Polymers with heteroatoms in the polymer backbone are fre-
quently produced by a step-growth method that may either involve ring opening or
elimination of a low molecular weight molecular species; eg, poly(ethylene oxide)
produced by ring opening of ethylene oxide and poly(ethylene adipate) produced
by the condensation of ethylene diol and adipic acid with the elimination of a
molecule of water. Step-growth methods produce materials with a broad spread
of lengths, and consequently a range of physical properties. The distribution of
chain lengths is reflected in a range in the physical properties.
Knowledge of the chemical structure of the monomer and the distribution of
chain lengths allows prediction of the method used for the production of that poly-
mer. If the monomer is effectively difunctional, then the polymer will have a linear
backbone structure and is a thermoplastic. Such polymers can be heated to an ele-
vated temperature and shaped. If the monomer has functionality greater than two,
then a cross-linked three-dimensional matrix can be created and the material is a
thermoset, which cannot be reshaped by heating to an elevated temperature. Ther-
moplastics and thermosets (qv) can be created using similar chemistry, the only
difference being the number of functions associated with the primary monomer
unit.
Encyclopedia of Polymer Science and Technology. Copyright John Wiley & Sons, Inc. All rights reserved.
160
CHARACTERIZATION OF POLYMERS
Vol. 9
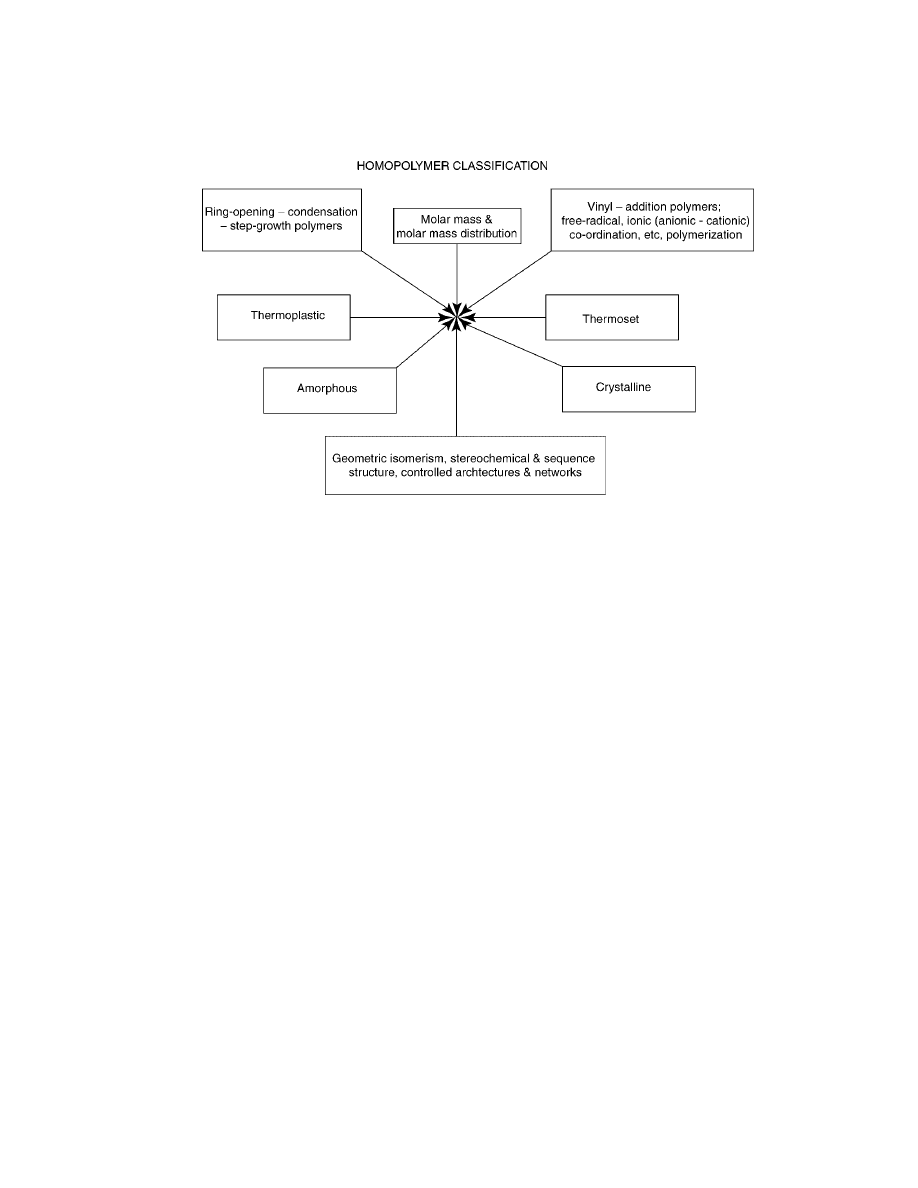
Fig. 1.
Classification of homopolymer types.
The chemical structure of the repeat unit has a dominant effect on the ability
of the polymer chains to pack together to form an ordered structure. If there is ei-
ther a high level of symmetry or relative simplicity of the polymer backbone, then
packing is encouraged and a crystalline structure generated (see S
EMICRYSTALLINE
P
OLYMERS
). If the monomer structure is asymmetric and/or contains bulky pen-
dant groups, packing may be inhibited and the solid structure is disordered and
amorphous (see A
MORPHOUS
P
OLYMERS
). Being able to differentiate between these
different types of organization can help with prediction of the physical properties
of the solid.
A useful classification of the polymers is presented in Figure 1.
Blending together two different types of compatible polymer material will
create physical properties that are different from those of the individual materials
(see P
OLYMER
B
LENDS
). Polymeric materials may be formed using more than one
repeat unit, and these are classified as copolymers. In real polymeric materials,
there may be present a range of additives (qv): plasticizers (qv), antioxidants (qv),
processing aids, fillers (qv), reinforcement (qv), antistatic modifiers, pigments,
dyes, etc. These additives will influence the physical properties of the material,
and understanding their influence is critical to understanding the properties of
the material.
The questions that need to be asked in order to fully characterize a polymeric
material include the following:
(1) What is the chemical structure of the repeat units from which the poly-
mer is constructed? Does the polymer have a specific geometrical isomeric
structure, particular sequence or branched-chain structures?

Vol. 9
CHARACTERIZATION OF POLYMERS
161
(2) Is the sample a thermoplastic or a thermoset?
(3) What is the average length of the polymer chains present? Are these chains
of the same length or is there a broad distribution of lengths present?
(4) Is the solid a crystalline or an amorphous material?
In addition, knowledge of a number of physical properties is required to
determine whether the polymer is suitable for a particular application.
Sample Preparation
All methods used by organic chemists for the determination of molecular structure
are appropriate for the characterization of polymers. However, the simple analysis
may not be able to differentiate between a homopolymer, a copolymer, or a blend
of polymers. Even with a homopolymer, such as a commercial polypropylene, the
solid may contain varying amounts of polymers with different molar masses and
stereochemical and block sequence structure. Therefore it is desirable to be able
to dissolve the polymer in selective solvents and extract species on the basis of
solubility.
To obtain an unambiguous characterization of a particular material, it is
often essential to fractionate a material (1–3). Synthetic polymers are rarely
homogeneous chemical species, but have multivariate distributions in molecu-
lar weight, chemical composition, chain architecture, and functionality (4). For a
precise characterization of a synthetic polymer, all the distributions need to be
determined, which is a difficult, if not virtually impossible, task. Traditionally,
fractionation has allowed separation of polymers on the basis of molecular mass
or chemical composition (2). With proper techniques it is often possible to separate
and characterize complex homo- and copolymer species on the basis of chemical
heterogeneity and molar mass.
Separation by Solubility.
Fractionation (qv) of a polymer has tradition-
ally been based on solubility using precipitation, fractional dissolution (extrac-
tion), coacervate extraction, thin-layer chromatography, counter current distribu-
tion, and turbidimetry (5).
Two methods have been developed that are in common usage and aid the
fractionation of polymer materials. These are temperature rising elution fraction-
ation (TREF) (6–8) and Field Flow Fractionation (FFF) (9).
TREF was developed to separate semicrystalline polymers according to dif-
ferences in molecular structure or composition that influence the crystallinity
and solubility. TREF can be divided into crystallization and elution stages. In the
crystallization stage, the polymer is dissolved in a good solvent, and then allowed
to crystallize under controlled conditions by slowly decreasing the temperature.
Crystallization may take place on an inert support or the support may be added
later. In the elution step, solvent is pumped through a column packed with the
polymer-support mixture, while the temperature is increased. Polymer elutes in
the reverse order that it was crystallized, with less crystalline material eluting at
lower temperatures followed by more crystalline polymer at higher temperatures
and has been used for polyalkenes (6,8). In preparative TREF (P-TREF) polymer

162
CHARACTERIZATION OF POLYMERS
Vol. 9
fractions are collected at predetermined but different elution temperature inter-
vals from those used in the crystallization (8). Critical, isobaric temperature rising
elution fractionation (CITREF) is carried out using supercritical fluids, such as
propylene and propane, in a column containing a high surface area stainless steel
mesh packing in a manner analogous to P-TREF (10,11).
FFF is based on the simultaneous action of the effect of field forces and the
carrier liquid flow on the fractionated species inside a belt-shaped fractionation
channel (9,12). FFF is used to fractionate polymers or mixtures. Separation is
based on the use of intensive properties, such as chemical composition and struc-
ture of the polymer chains, electrical charge, density, etc. Separation is influenced
by application of a temperature gradient (ThFFF) (13) or an electrical field (EFFF)
(14), and separation is aided by flow. In ThFFF, a temperature difference between
the walls of the channel produces a flux, known as the Soret effect, usually towards
the cold wall. In the EFFF, an electric potential across the channel generates a
transverse flux of charged macromolecules or particles (14). In preparative FFF,
a lower dilution of the separated species during the elution occurs as compared
with flow FFF, due to partial depletion of the liquid due to the cross-flow. Two-
dimensional thermal field-flow fractionation (2D-ThFFF) is devised for continuous
fractionation of macromolecules (15). The sample introduced into a disc-shaped
channel undergoes radial and tangential flow. Random copolymers of styrene and
acrylonitrile (SAN) in toluene have been fractionated. (16). Polymer Brushes (qv)
with poly(methyl methacrylate) (PMMA) backbone and polystyrene side chains
have been separated using continuous counter-current extraction (17). FFF is ap-
plicable for the separation of polymers with molar mass in the range 10
3
–10
8
and
variants include sedimentation as an applied field (18–20). For small-scale analy-
sis, components are sometimes quickly and conveniently separated by thin-layer
chromatography (TLC), (21).
Molecular Size.
Separation of soluble macromolecules can be achieved in
size-exclusion chromatography (SEC) or gel-permeation chromatography (GPC)
as a consequence of the differential permeation of the molecules into the porous
solid column packing (see C
HROMATOGRAPHY
, S
IZE
E
XCLUSION
). Small molecules will
penetrate more effectively and are eluted last from the column; the large species
are eluted first. The separation is based on the size of the polymer molecule in
solution—hydrodynamic volume that is directly related to molar mass of the poly-
mer molecule. Since the hydrodynamic volume takes into account the polymer–
solvent interactions, it is possible to create a universal calibration that takes into
account variations in size due to change in polymer–solvent interaction. Care must
be taken to ensure that size exclusion is the only separation mechanism operating
in the column and to avoid errors arising because of Adsorption (qv) and aggrega-
tion (22). The SEC/GPC technique is used for the determination of the molar mass
relative to some calibration standards, but can also be used for the separation of
the mixtures into narrow molar mass fractions.
Molecular Structure Characterization
The first step in any analysis is determination of the elemental composition of
the polymer. Combustion analysis can establish the presence of carbon, hydrogen,

Vol. 9
CHARACTERIZATION OF POLYMERS
163
halogen, sulfur, and the Kjeldahl oxidation for nitrogen and, occasionally, phospho-
rus. The structure of the polymer will then usually be determined by a combination
of spectroscopic and mass spectroscopic analysis of degradation products.
Infrared and Raman Spectroscopy.
Identification of the type of func-
tional groups present in a polymer is effectively achieved by infrared and Raman
analysis (23–28). The spectroscopic selection rules for infrared and Raman activ-
ity are respectively a dipole moment or polarizability change during interaction
of electromagnetic radiation with the atomic grouping (see V
IBRATIONAL
S
PEC
-
TROSCOPY
). A vibration that gives a strong infrared signature may be weak in the
Raman spectrum and vice versa. It is relatively easy to identify the occurrence
of carbonyl, ethers, aromatic functions, hydroxyl groups, epoxy rings, carbon–
halogen, and carbon–hydrogen bonds. The techniques are applicable to the study
of solids (usually as very thin films), powders, and solutions. Raman spectroscopy
is particularly useful when studying aqueous solutions, wet samples, or where dif-
ferentiation between polymers with similar structures is necessary. Using laser
sources, depolarization data associated with the Raman spectra can be obtained
and conformational differences in molecular structure determined. At low fre-
quencies, ca 100 cm
− 1
, the longitudinal acoustic mode (LAM) is observed in the
Raman and is characteristic of the all trans structure of the polymer backbone and
used to study the lamellar structure in crystalline polymers, such as polyethylene
(29–32). Shifts in the Raman spectra have been used to indicate how local stresses
are distributed within a sample (29,33).
Application of stress can induce changes in the infrared spectrum (34,35).
Fourier transform infrared (FTIR) spectroscopy, in which rapid multiple scanning
of the sample is possible (22,29), has allowed real-time observation of changes
in the spectra. Studies have been reported of materials subjected to an applied
external stress or used to follow the cure reaction of a thermoset. Computer li-
braries allow assignment of the spectra and identification of various functional
groups within the polymer. Attenuated total reflection (ATR) attachments allow
the surface of films to be explored (36). The ATR technique uses a glancing an-
gle and total internal reflection of the infrared beam to collect information on
the groups that lie close to the surface of the film (5–10
µm). Data manipulation
techniques such as difference spectroscopy (spectral subtraction), factor analysis,
spectral deconvolution, and least squares fitting of calibration plots allow quanti-
tative determination of the species present.
Nuclear Magnetic Resonance Analysis.
Nuclear Magnetic Resonance
(qv) (NMR) provides both qualitative and quantitative analysis with respect to
monomer composition and the average configuration of the polymer chain (22,37,
38). Both solid-state and conventional NMR techniques provide information on
molecular structure, dynamics of the chain, crystallinity, network formation, and
chain entanglement (39–41). Many types of nuclei may be observed; but proton
(
1
H) and carbon-13 (
13
C) are usually investigated. Other useful nuclei studied
include silicon (
29
Si), nitrogen (
15
N), fluorine (
19
F), and phosphorus (
31
P). The use
of FTNMR with superconducting magnets and wide bore magnets has allowed a
wide range of materials to be studied.
Proton (
1
H) NMR, initially, was used for structural characterization, and
use of the Karplus equation has allowed conformational and configurational anal-
ysis to be carried out on a wide range of polymers in solution (42). The advent of

164
CHARACTERIZATION OF POLYMERS
Vol. 9
various pulse sequences has enhanced the signal sensitivity and consequently the
popularity of carbon-13 NMR spectroscopy. The natural abundance of
13
C is 1.1%
and although there are problems of sensitivity, the spectra are simplified because
carbon–carbon coupling is rarely observed in naturally-occurring materials.
1
H
decoupling leads to spectra that consist of a series of single lines indicative of
the types of chemical environment present. The inherent spectral separation of
carbon chemical shifts is generally greater than for protons, ie, 0–220 ppm for
13
C
compared with 0–10 ppm for
1
H, making assignment relatively easy. The
1
H de-
coupled line width is typically about 2–10 Hz for solution spectra. Grant and Paul
(43) established that since the shifts are dependent on the s-orbital density, the
observed shifts can be used to characterize the sequence structure of the polymer
backbone. (22,44–50). Many molecular modeling packages contain software which
will predict the
13
C spectrum. Subtle differences in both configuration and confor-
mation of the polymer chain and changes in the dynamics of the chain backbone
as a consequence of substitution can be observed (22,45).
Use of various pulse sequences has allowed solid-state NMR to be used
to explore morphological features in solids. Studies of diblock copolymers of
poly(styrene)–poly(methyl methacrylate) indicate domain sizes from 1 to 100 nm
(51). Studies of the morphology and dynamics in poly(ethylene terephthalate)
(52) and polyethylene (53) indicate the presence of a number of different phases.
Motional heterogeneities within the phases of core shell particles composed of
poly(n-butyl acrylate) (PBuA) and poly(methyl methacrylate) (PMMA) have also
been demonstrated (54). The scope of the technique is continuing to develop and
provide insight into the way in which chain–chain interactions influence the poly-
mer morphology (55).
Detailed analysis of
1
H and
13
C NMR coupling constants obtained from
solution spectra has allowed characterization of the chemical composition and
conformation of racemic and meso dyads in glassy polystyrenes (56), and the
stereoregularity in poly(methyl methacrylates) (57). With sufficient resolution,
detection of tacticity at a pentad and higher level is possible, allowing validation
of Bernouillian, Markov, or other statistical models for the description of polymer-
ization processes and the tendency for block formation in copolymers. Geometric
isomerism is readily detected in polybutadiene or polyisoprene, which form cis
and trans double bonds in the polymer backbone as well as undergoing 1,4 or 1,2
addition. Imaging has allowed the mapping of the diffusion of labeled species in
solids (58,59).
Pyrolysis.
Spectroscopic methods can provide an identification of the
monomeric species present; however, pyrolytic degradation plays an important
role in the final assignment of the structure. Pyrolysis-gas chromatography is used
extensively for the analysis of synthetic (60–63) and natural polymers (64). Appli-
cation of heat to a sample leads to degradation (qv) and release of low molecular
mass fragments. The composition and relative abundance of the pyrolysis prod-
ucts are characteristic of a given polymer. The technique is used in conjunction
with other analytical methods, such as mass spectroscopy, infrared, and combina-
tions such as pyrolysis-gas chromatography-mass spectroscopy. Pyrolysis can be
carried out either in a continuous or a pulse mode; the latter has the advantage
of minimizing the effects of secondary reactions on the fragmentation products.
Small samples, in the microgram to milligram range, are rapidly heated either

Vol. 9
CHARACTERIZATION OF POLYMERS
165
directly in the inlet of a gas chromatograph or separately in an attached unit.
For pyrolysis-mass spectroscopy, the pyrolysis is carried out in vacuum in the
mass spectrometer. In a pulse mode pyrolyzer, the sample is frequently heated by
resistive heating of a filament or by radio frequency inductive heating of a ferro-
magnetic metal (Curie-point pyrolyzer). The sample may be deposited as a thin
film obtained by drying a solution or a small sample placed on the filament.
Pyrolysis of poly(methyl methacrylate) at low temperature produces
monomer, whereas other acrylics fragment with loss of side chains, scission of the
chain backbone, elimination or rearrangement of the products. Knowledge of the
degradation pathways for particular polymer sequences is required to interpret
the fragmentation patterns obtained from pyrolysis (65–70).
UV Spectroscopy.
UV spectroscopy is appropriate for characterization of
aromatic or conjugated systems (22). The high sensitivity allows study of species
at high dilution and is useful for intrinsically conducting polymers and light-
emitting species (71,72). The absorption is characteristic of the
π–π
∗
transition;
fluorescence and phosphorescence lifetimes are influenced by the state of aggre-
gation and the matrix structure in which the absorbing species is localized. Many
pigments and dyes have characteristic absorption and emission spectra. Lumines-
cence techniques are valuable for the study of degradation and aging in polymers.
Rotation and local motion of groups in dilute solution on the order of 10
− 10
–10
− 8
s in amorphous polymers and orientated films may be studied by fluorescence
depolarization. Doped polymer films excited with polarized light will exhibit flu-
orescence depolarization, which is characteristic of the motion in the system.
Luminescence may be suppressed by the presence of antioxidants, light sta-
bilizers, and pigments, which are able to transfer energy form the excited state
to other nonfluorescing electronic states. Quenching of excited states also occurs
when either oxygen or moisture is present.
Mass Spectrometry.
Direct observation of polymers by means of mass
spectrometry (qv) has traditionally been limited to molecules with molar mass
of about 5000 or less. However, the advent of matrix-assisted laser desorp-
tion/ionization (MALDI) (73) has allowed accurate analysis of high molecular mass
species to bed one (22). A high powered pulsed laser is used to transfer into the
vapor phase the polymeric species which has been dispersed in a low molar mass
matrix. The matrix, present in large excess (molar ratio 1:2000), is chosen for its
ability to absorb the laser energy and protect the analyte, transferring energy to
it in a way that allows desorption and ionization of molecules without significant
fragmentation. Surprisingly, this simple change in handling technique has allowed
species of up to 10
6
to be studied. Delayed extraction techniques enhance resolu-
tion (74,75). Calibration is necessary to quantify the intensities, lighter molecules
are preferentially desorbed and ionized with respect to those having higher mass,
and correction to the data is necessary. The resolution achievable has been en-
hanced by the use of time-of-flight mass spectroscopy to give TOF-MALDI (76,77).
TOF-MALDI has also been applied successfully to biopolymer systems with pre-
cise molecular weight determination being possible up to 400,000 Da (78).
Electrospray mass spectroscopy (ESMS) is used for the study of biological
macromolar species (79,80). Spraying produces droplets of the polymer solution
which are ionized in an applied electric field and the charged species measured
using a mass spectrometer.

166
CHARACTERIZATION OF POLYMERS
Vol. 9
Molar Mass and Molar Mass Distribution Determination
The GPC/SEC approach can be used for the determination of the molar mass or
the distribution of molar masses. Traditionally, refractive index detection has been
used and allows concentration of polymer to be measured as a function of elution
volume. Using this approach, and calibration with narrow molar mass distribution
samples, it is possible to determine the relative molar mass. Use of more complex
detector arrays allows internal calibration of the system to be achieved and ab-
solute molar mass determination (22,81). The usual triple detector combination
uses refractive index, capillary viscometry, and light scattering (82–88). The lat-
ter provides an absolute calibration of the instrument; the capillary viscometry
data gives through the intrinsic viscosity and hydrodynamic volume, information
on the polymer–solvent interactions. Using this approach, studies have been car-
ried out on multibranched star-shaped polyethers having poly(ethylene oxide)s
(PEO) arms (82) which illustrate how the method can provide information on the
architecture as well as molar mass. Due attention must be given to validating
the accuracy of the theory for the particular type of polymer being studied (82–
88). The multidetector systems are able to provide information on the size of the
polymer molecule in solution (89).
Average Molar Mass.
The molar mass of a polymer is usually described
by an average molar mass. If the chains are counted by number it is the number-
average molar mass ¯
M
n
, whereas if it is counted by weight it is the weight-average
¯
M
w
; higher averages,
¯
M
z
, etc, can also be calculated. The parameter M
w
/M
n
,
known as the molar mass distribution, is characteristic of the method of synthesis.
Ionic and coordination polymerization will have an M
w
/M
n
approaching 1.05,
whereas a polymer produced using a radical initiated reaction has values between
1.5 and 4.5. Insertion polymerization, eg Ziegler–Natta olefin polymerization,
yields values between 5 and 20. With condensation and ring opening step growth
polymers, the value of M
w
/M
n
will be between 3 and 20. The distribution pa-
rameter can indicate whether the material is a blend of polymers or is a single
material.
Three types of polymer distribution are typically observed in samples ob-
tained from polymerization: logarithmic normal, Poisson, and Schulz–Flory dis-
tributions. The Poisson distribution can be very narrow and occurs when a con-
stant number of polymer chains grow simultaneously and addition of the next
monomeric unit is independent of previous units and is found in anionic polymer-
ization (qv). The Schulz–Flory distribution, typical of radical polymerization (qv),
arises when a constant number of chains growing ends exist and when termina-
tion and chain initiation processes are also active. This is in contrast to the Poisson
distribution. A logarithmic-normal distribution is found for the polymerization of
polyethylene and polypropylene (90,91).
For simple quality assurance, GPC/SEC with a simple refractive index de-
tector is used, as the shape of the trace indicates directly the distribution of mo-
lar masses. Comparison of the distribution that is obtained from GPC/SEC with
MALDI-TOF has helped clarify the validity of both techniques, especially in the
case of low molar mass calibration samples. Simpler traditional methods are of-
ten more appropriate where a specific average is required to correlate variations
in a polymer material with a physical property. Solution viscosity measurements

Vol. 9
CHARACTERIZATION OF POLYMERS
167
Table 1. Methods for Determination of Average Molar Mass
Absolute
Average
Molar mass
Method
or relative measured
range, g/mol
Characteristics
Ebulliometry
A
M
n
Up to 10
4
Low sensitivity
Cryoscopy
A
M
n
Up to 10
4
Small samples size,
fast
Membrane osmometry
A
M
n
5
× 10
3
–10
6
Suitable membranes
required
Vapor phase osmometry
R
M
n
<3 × 10
4
Suitable standards for
accurate calibration
required
End group analysis
A
M
n
10
2
∼ 3 × 10
4
Low sensitivity at
higher molar masses
Light scattering
A
M
w
3
× 10
4
∼ 10
7
Low sensitivity at low
molar mass
Ultracentrifugation
A
M
n
/M
w
2
× 10
2
∼ 10
7
Small samples,
time-consuming,
difficult technique
Dilute solution viscometry
R
M
v
10
2
∼ 10
7
Fast, low cost small
samples
SEC/GPC
R
M
n
/M
w
10
2
∼ 10
7
Popular, small
samples, requires
calibration samples
SEC/GPC–triple detection
A
M
n
/M
w
10
2
∼ 10
7
Requires skill and care
to obtain reliable
results, expensive
MALDI-TOF
A
M
n
10
∼ 10
7
Sensitivity varies with
molar mass
provide a method of monitoring the effects of degradation (qv) caused by polymer
processing and the assessment of physical aging (qv) when polymers are exposed
to extreme environments.
Some of the traditional methods are listed in Table 1.
In order to obtain accurate data it is essential that care be taken in the sam-
ple preparation. Because M
n
is more sensitive to low molar mass components,
removal of residual solvents, monomers, salts, catalysts, etc, from a sample be-
fore measurement is essential. M
w
is more sensitive to the higher molar mass
components, as are the higher averages M
z
. Incomplete dissolution of a sample
often leads to meaningless results. Similarly, polymer degradation during solution
preparation or in the process of molar mass measurement must be prevented. Use
of ultrasound to aid production of a solution is strongly discouraged. The issues
associated with the measurements are discussed in detail elsewhere (22,92–94).
Analysis of complex structures, branched chains, dendritic molecules, and regular
copolymers has been successfully undertaken by combining data from a series of
different methods (95–100). MALDI-TOF, in conjunction with other approaches,
is allowing considerable detail to be obtained on copolymer composition changes

168
CHARACTERIZATION OF POLYMERS
Vol. 9
during reaction, probability of branching reactions, sequence structure within
polymers with more than one reaction mechanistic possibility, etc (101,102).
The importance of knowing the molar mass of a polymer and its distribution
of molar masses cannot be overemphasized. Commercially, many polymers, such
as polystyrene, are available in different grades. Often the difference between
these grades is their molar mass and/or molar mass distribution. One grade will
be ideal for injection molding, another for compression molding. Incorrect selection
of the grade of polymer would lead to selection of a material that is unsuitable for
the application for which it is intended.
Solution Characteristics of Polymers.
The molar mass characteriza-
tion techniques often provide additional information on the nature of the polymer
species in solution. In most cases, the solutions used are sufficiently dilute so that
the properties being measured are those of the “isolated” molecule. At infinite
dilution the size of the polymer coil is dictated by both inter- and intramolecular
interactions, the nature of the solvent used, and the temperature. In a viscosity or
GPC/SEC experiment the properties being observed can be related to the hydro-
dynamic volume of the polymer coil. This hydrodynamic volume is the “effective
volume” which the polymer occupies in solution. The hydrodynamic volume is im-
plicit in the Mark-Houwink relationship, which describes the value of the limiting
infinitely dilute increment to the viscosity—the intrinsic viscosity [
η] to the molar
mass:
[
η] = K. ¯
M
α
η
where K and
α are constants valid over a limited molar mass range and specific to a
particular temperature–solvent combination. Generally, the value of
α is between
0.5 and 1.0, and depends on the conformation of the polymer in solution. For
random coil conformations, obtained at theta conditions,
α is around 0.5, and
extended conformations resulting in rod-like molecules give values approaching
1.0. Because
α usually falls somewhere between 0.6 and 0.8, the viscosity average
molar mass ¯
M
η
ν
is always somewhere between ¯
M
n
and ¯
M
w
. The value of ¯
M
η
ν
is
often closer to ¯
M
w
than ¯
M
n
. The value of K is typically 1
× 10
− 4
dL/g, and values
of the constant are to be found in the Polymer Handbook (103). Because the values
of
α and K reflect the hydrodynamic volume and are sensitive to changes in the
polymer architecture, long-chain branching will lower the values in comparison
to an equivalent linear polymer.
The concentration dependence of many physical properties indicates the
quality of the polymer–solvent interactions, through the second virial coefficients.
Study of the concentration and temperature dependence of these parameters pro-
vides information on the theta temperature, radius of gyration, and the mean
square end-to-end distance for a polymer coil. The radius of gyration is defined as
the root mean square distance from the center of mass of a polymer chain to a given
mass element. The characteristic ratio is a measure of the ratio of the square of the
average random-flight end-to-end distance to the product of a number of backbone
units times the interactions along the polymer chain. These parameters provide
useful information on the validity of a particular theoretical model and its ability
to describe the architecture and conformation of a polymer (104).

Vol. 9
CHARACTERIZATION OF POLYMERS
169
Molecular Organization and Dynamic Behavior of a Polymer
The ability to produce fractions of polymers has allowed their true physical prop-
erties to be determined. Polymer chain interactions are determined by the local
interactions and longer range effects that scale with the molar mass of the species.
The former will dictate whether it is possible to achieve sufficient favourable in-
teractions for crystallization to occur or whether local interactions are sufficiently
strong to inhibit ordering and an amorphous material is generated. Relatively
small changes in the size and distribution of these ordered regions can be re-
sponsible for dramatic differences in the physical and mechanical properties of
ostensibly the same polymer. The degree of ordering depends on the local organi-
zation; hence it is not uncommon to find that an isotactic form of a polymer may
be very different to that observed in a syndiotactic molecule. In polypropylene the
isotactic form of the polymer has a helical structure that crystallizes, whereas
the syndio or atatic forms are unable to pack and have rubbery characteristics
(105,106).
The ability of a material to absorb energy reflects the mobility of the chemical
entities it contains and the types of interaction that are broken as the temperature
is increased. In crystallizable polymers, the interactions between chains depend
on the chemical structure and range from the relatively weak van der Waals in-
teractions found in polyethylene to the strong dipole and hydrogen bonding inter-
actions found in nylon and polyurethanes. In between these extremes lie a range
of materials with interactions of a dipole–dipole nature and associated with ester,
amide, nitro, chloro, and ether groups which may or may not form crystalline
phases, depending on the ability of the chains to form a regular packed structure.
Hydrogen bonding is the strongest of the interactions and has a dominant effect
on the structures formed. In the case of aromatic containing polymers, significant
contributions can arise from dipole–induced dipole and higher order interactions
associated with the delocalized electronic structure of the molecules. Typically
the organized regions, even for the most crystalline of polymers, will be disrupted
by conformational (gauche) defects. The clustering of these defects will allow the
chains to loop or deviate from the lower energy (trans) extended structure that is
incorporated into the ordered regions. As a consequence of the defect structure,
the size of the crystalline domains will be typically of the order of a few tens of
nanometers. Higher order structures are created by organization of these primary
lamellae structures. Because of the possibility of chains folding back on one an-
other, the number of chains bridging from one crystallite to another will be fewer
than those reentering the structure from which they originate. These bridging
chains form a disordered structure that is amorphous. The geometry (spherulitic
or row-nucleated), size, and distribution of crystalline regions are complex and
affect physical properties in a complex way.
In other polymer systems, such as polystyrene, the steric constraints associ-
ated with the group pendant to the chain backbone inhibit packing and create a
disordered, amorphous, structure. Chain–chain interactions, however, can achieve
a level of packing density that leads to motion of the backbone being inhibited.
In this state, the polymer has properties of a glass. Increasing the temperature
leads to expansion, increased chain separation, and a decrease in the strength
of the interactions, and the solid exhibits flexible-rubbery characteristics. This

170
CHARACTERIZATION OF POLYMERS
Vol. 9
transformation is associated with the inclusion of volume for the chains to move
and is known as the glass–rubber transition T
g
(see G
LASS
T
RANSITION
). The rela-
tionship between supermolecular order/disorder and physical or mechanical prop-
erties depends on the thermal and mechanical history of a polymer (107).
Thermal Methods.
Heating a polymer sample allows observation of the
changes in physical properties, associated with changes in the degree of packing
and chain–chain interaction. In thermal analysis, the properties are measured
as a function of temperature or time at constant temperature. Methods include
differential thermal analysis (DTA), differential scanning calorimetry (DSC), ther-
mogravimetric analysis (TGA), evolved gas analysis (EGA), adiabatic calorimetry
(AC), and a recent variant of DSC: modulated DSC (MDSC) (108–121). Use of so-
phisticated mathematical techniques and adaptation of the microscanning tech-
niques have allowed mapping of thermal properties at the micron level. These
techniques are not yet widely available, but will make a significant impact on our
understanding of the way in which one aspect of morphology influences another
in phase-separated materials and apparently homogeneous materials.
Amorphous Materials.
The characteristic feature of amorphous polymers
(qv) is the observation of a glass–rubber transition temperature (T
g
). The glass
transition (qv) temperature is associated with cooperative motion of the backbone
chain of a polymer and involves the collective motion of between six and 12 chem-
ical bonds on average. (122). The important difference between the T
g
and other
transitions that are observed below this temperature is that the T
g
process re-
quires free volume in the region into which the chains are going to move. The free
volume is produced by the expansion of the solid as a consequence of the increase
in chain–chain separation. As a consequence, the T
g
is sensitive to pressure; in-
creasing the pressure to which a material is subjected leads to an increase in Tg.
In contrast, the motion of groups attached to the backbone via a flexible link-
age exhibits simple Arrhenius/thermal activation behavior, the rate of the motion
varying linearly with temperature. The rate of chain motion associated with the
T
g
does not vary linearly with temperature, usually showing a marked tempera-
ture dependence that reflects the limitations imposed by the free volume. The T
g
process reflects a dramatic change in physical properties of the material, changing
from being a rigid glass to a flexible rubber. The T
g
is usually reported as a single
value; however, the nature of the processes and its kinetic nature dictates that it
occurs over a broad temperature range. The process is complex, and hence differ-
ent methods of observation lead to a slightly different value being reported for the
same material (22,122). In addition, the length of the polymer chains can have an
influence on the value observed. The chain ends will require less free volume than
the motion of the center of a long chain. As a consequence, it is observed that for
many amorphous polymers, the value of T
g
varies with molar mass according to
the relationship
T
g
(M)
= T
∞
g
−
K
¯
M
n
where T
g
(M) is the value for that material, K is a constant characteristic of that
material, T
g
∞
is the limiting value observed for a very high molar mass poly-
mer, and M
n
is the molar mass of the sample being studied. Recognition of the

Vol. 9
CHARACTERIZATION OF POLYMERS
171
sensitivity to the molar mass is very important in understanding how commercial
polymers with the same chemical structure have apparently different physical
properties. Low molar mass fractions will lower the glass transition temperature
and soften the material.
Sub-T
g
processes are very important in defining the physical properties of
a polymer. The side chain motions can occur below the T
g
and absorb energy,
helping to dissipate the energy associated with impact and allowing the mate-
rial to demonstrate ductile fracture behavior. The ability of a polymer to resist
impact can be designed into the chemical structure by changing the nature of
the side groups, their ability to move, and the efficiency with which they absorb
energy. Backbone elements, such as aromatic entities, rotate about the backbone
axis without being involved in the collective T
g
process and impart high impact
resistance to the material, eg, polycarbonate. Backbone and side-chain motions
can become closely coupled on certain time–temperature scales, eg, poly(methyl
methacrylate). The value of T
g
is sensitive to changes in the chemical structure of
the backbone and varies between
∼160 to ∼570 K. Because it is a kinetic process,
the T
g
is sensitive to the thermal history of the sample. The fabrication process
will usually involve the freezing in of a level of entropy reflecting the processing
temperature. Heating the sample up close to the T
g
will allow slow reorganization
of the chain conformations, leading to changes in the bulk physical properties. The
time dependence of the physical properties has been extensively studied and is
called physical aging (107). The side-chain relaxation, designated T
β
, often oc-
curs at a temperature of about 0.75T
g
. This correlation reflects the connectivity of
the nature of the potential functions responsible for the processes that are taking
place. Cross-linking of the polymer structure leads to an increase in the T
g
and the
introduction of certain fillers can also increase the value of T
g
. The presence of low
molar mass additives will have the reverse effect and swell the matrix lowering
the value of the T
g
.
Crystalline Materials.
Polymers do not form perfect crystalline structures;
however, many polymers, eg polyethylene, exhibit the characteristics of materials
in which large elements of the material are regularly packed (see S
EMICRYSTALLINE
P
OLYMERS
). As in the case of low molecular weight materials, it is possible to ob-
serve alignment of the polymer chains that lead to optical characteristics of typical
nematic or smectic liquid crystal phases. The extreme degree of organization is the
crystalline phase. Only in solution-grown, low molar mass polymers are nearly
perfect crystalline phases observed. In the typical polymer sample, there will be
frozen in, at the freezing point, a certain amount of disorder. Order in the crys-
talline phase is usually associated with the all trans conformation of the backbone,
whereas disorder is associated with the gauche conformation. A coincidence of
gauche conformations will loop the chain back in its original direction and develop
a folded crystalline structure. The loops form a disordered-amorphous interface
between the crystalline regions, which are packed into lamellae. The lamellae will
in turn organize themselves into spherulite structures. The morphology of crys-
talline materials is varied and reflects the nature of the nucleation process and the
thermal history. As in the case of amorphous polymers, post-thermal treatment
can lead to changes in the size of the crystalline content with subsequent vari-
ation in the physical properties. The mechanical properties of polyethylene are
thus dictated by the ratio of the crystalline to amorphous content. Just below and

172
CHARACTERIZATION OF POLYMERS
Vol. 9
above the melting point T
m
, the polymer chains can undergo extensive transla-
tional movement or flow. The melting of semicrystalline polymers (qv) as observed
by DSC indicates that the melting process often occurs over a range of temper-
atures. The breadth of the distribution is a reflection of the distribution of sizes
of the crystallites in the material. The value of T
m
is a function of a number
of properties that include molar mass, molar mass distribution, percent crys-
tallinity, and dimensions of the polymer crystallites.
An empirical rule which has been observed for many polymers is that T
g
∼
2/3T
m
, where the temperatures are in Kelvin. This rule is applicable to most
vinyl, vinylidene, and condensation polymers. This ratio is lower for polymers
with unsubstituted backbones and higher for poly(
α-olefins) with long alkyl side
groups. T
αc
occurs in crystalline and semicrystalline polymers and is ascribed to
an oscillation about the chain axes within the chain-folded crystals or a translation
along the axis. T
αc
is related to T
m
by
T
αc
T
m
= 0.85 +
25
T
m
where all the temperatures are in Kelvin (123–125). The introduction of branches
and bulky side chains into linear polymers can inhibit crystallization and in the
case of polytetrafluorethylene aids the processing of the polymer by introducing
amorphous structure.
The usual approach to the study of the transitions is by the use of ther-
mal methods (see T
HERMAL
A
NALYSIS
). The instruments for DTA and DSC are
different in construction. In DTA, the temperature difference between a sample
and a reference chamber is measured as a function of time or temperature as
heat is supplied to both chambers. In DSC, the heat flow into or from a sample
chamber compared to a reference chamber is measured as a function of time or
temperatures. Modern instrumentation is capable of measuring the heat input
or output of samples in the 0.2–30-mg range with high accuracy. The information
obtained from DSC and DTA with proper and suitable calibration is in many cases
similar, if not identical. Modulated DSC (113–116) is an adaptation of the DSC
method in which the heating, instead of being carried out in a linear manner, is
applied in an increasing sigmoidal fashion. Fourier transformation of the heat
flux against the heat flow allows separation of real and imaginary components.
The real part of the transform function is a direct measure of the heat capacity,
whereas the imaginary component is a measure of nonreversible heat flow. The
latter can be associated with crystallization, cure in the case of a not completely
cured thermoset resin, polymer degradation, and other similar types of process.
The curves can be used to directly measure the energy associated with the T
m
, or
the entropy associated with the T
g
or parameters characterizing other transitions.
The melting behavior can strongly influence the physical properties, and DSC
and DTA studies allow quantitative investigation of phase structure in a polymer
sample.
Plasticizer efficiency may be evaluated by studying the effects of plasticizers
on T
g
using thermal methods. The effects of the addition of nucleating agents on
the rate and extent of crystallite formation can similarly be quantified. Current

Vol. 9
CHARACTERIZATION OF POLYMERS
173
instrumentation can approach the temperatures which materials approach during
processing and allow modeling of the processes that occur at a microscopic level.
Thermal stability can be determined in DSC by measuring the temperature
of reaction or degradation under isothermal or scanned temperature conditions.
Other conditions include an inert or an oxidative atmosphere at ambient or ele-
vated temperature. It is also possible to study the effects of increased pressure
using pressurized DSC cells. By varying the conditions, thermooxidative processes
can be separated from thermodegradative processes. The induction time before re-
action begins is taken as a measure of the stability. The effect of antioxidants and
stabilizers on this time may be readily studied.
Mechanical Properties.
The measurement of mechanical properties is
concerned with load–deformation or stress–strain relationships. Forces may be
applied as tension, shear, torsion, and compression and bending. Stress is the
force divided by the cross-sectional area of the sample. Strain is the change in a
physical dimension of the sample divided by the original dimension. The ratio of
the stress to strain is referred to the modulus. Stress may be applied continuously
or periodically at varying rates for different tests. The characteristic stress–strain
curve, stress relaxation, or impact behavior is very important in determining the
applications and limitations of a polymer.
The condition, size, and history of a sample are of great importance in per-
forming mechanical measurements. Samples should be uniform and free of im-
perfections that can act as stress concentrators. Mechanical properties depend on
production techniques. For example, an injection-molded test specimen may be
strongly influenced by the molecular orientation resulting from flow in the mold.
Sample size should be controlled and is often set for a given test. The thermal
history of a sample is very important because aging often produces change; eg,
crystallinity may develop in a supercooled polymer over time, or oxidation may
produce an alteration of the polymer structure. It is usual with mechanical tests
to measure a number of samples and average the results. Even taking the aver-
age of a set of data must be carried out intelligently as sometimes there are good
reasons; eg, differences in morphology within a sample which are reflected in the
measurements. Samples may be subjected to equilibration prior to measurement
at some set humidity and temperature; however, this process in itself may be in-
ducing changes in the structure. A myriad of mechanical tests are available, some
standardized (126) and some not. A comprehensive list of standards is available
on the Web site www.astm.org. It is important to select a test which is applicable to
the conditions in the material is going to be used (see M
ECHANICAL
P
ERFORMANCE
).
Stress–Strain Tests.
The steady application of a stress (force per unit area)
to a material will lead to a measured strain (change in length). The initial slope
for many materials is linear, and with reduction in the stress there is also a re-
duction in the strain. The material is behaving as a simple linear Hookian solid.
However, application of higher levels of stress will ultimately lead to the material
breaking, and for a hard material this occurs rapidly at a limiting value of the
strain. The failure of such a material is designated as being brittle. Some poly-
mers may exhibit curvature in their stress–strain plots at high stresses and this
nonlinear behavior is often indicative of ductile behavior (see Y
IELD AND
C
RAZ
-
ING
). A further characteristic of a ductile material is the observation of uniform
extension or a yield point where the stress remains constant or even decreases

174
CHARACTERIZATION OF POLYMERS
Vol. 9
as the strain (elongation) increases. This type of behavior is typical of a necking-
down or reduction of sample cross-section at the yield point due to cold drawing
of the specimen. A levelling of stress may often occur after the yield point until
the break point is reached. As the material takes on a more rubbery characteris-
tic, the length of the region over which the stress remains constant is increased.
The initial slope of the stress–strain curve allows measurement of the modulus
of the material. The area under the curve indicates the energy required to frac-
ture the sample and is directly related to the toughness of the material. A good
elastomer is capable of exhibiting ultimate elongations before failure approaching
1000%.
As the temperature of a sample is raised, the polymer will be transformed
from exhibiting brittle to ductile and eventually rubbery characteristics, provided
the material is of sufficiently high molar mass. A material that shows tough ductile
behavior when stress is applied slowly often behaves as if it were brittle when a
large stress is applied rapidly. This observation reflects the ease with which the
polymer chains are able to reorganize to redistribute the stress. Similarly, when
the temperature is lowered, the tough ductile specimen often becomes brittle even
if the stress is applied slowly. The mechanical properties are a direct reflection
of the mobility of the chains and can be described by the Williams–Landel–Ferry
(WLF) equation. The WLF equation relates through a shift factor a
T
, the ratio of
the time constant
τ of a particular response at a temperature T to a corresponding
time constant
τ
0
at a reference temperature T
0
. Because of the relevance of T
g
to defining large-scale molecular motion, this is often selected as the reference
temperature. The WLF equation is
a
T
=
− A(T − T
g
)
B
+ (T − T
g
)
where A and B are constants specific for a given polymer. The constants obtained
are typically 17.4 for A and 51.6 for B (127). In practice, slight variations in the
values of the constants are observed which reflect the differences in the processes
that are responsible for the shift factors.
Dynamic Mechanical Thermal Analysis.
Dynamic mechanical thermal
analysis (DMTA) subjects the sample to a low amplitude cyclic stress and observes
the corresponding strain induced (see D
YNAMIC
M
ECHANICAL
A
NALYSIS
). By com-
parison of the amplitude and the phase of the response, it is possible with suitable
calibration to deduce the dynamic modulus, storage modulus (energy dissipated
per cyclic of the stress), and tan
δ, the ratio of storage to the dynamic modulus for
a particular form of deformation. In the typical apparatus it is possible to subject
samples to bending, shear, or torsion. Mapping these values of the physical param-
eters obtained as a function of the temperature allows identification of the major
transitions and quantification of the modulus of the glass and rubbery states.
The technique is ideally suited for the detection of phase separation in polymers,
where two values may be observed for the T
g
. These tests are highly sensitive to
crystallinity, orientation, morphology, dependence on molar mass in thermoplas-
tics, degree of cure in thermosets, and miscibility of multiphase systems including
composites and polymer blends (22,110).

Vol. 9
CHARACTERIZATION OF POLYMERS
175
Creep Tests.
Creep tests measure deformation upon the application of con-
stant stress such as that obtained by hanging a weight on a sample and noting
the change in elongation as a function of time. Creep tests may be performed by
applying stress in tension, compression, flexural bending, shear, or torsion (twist-
ing) modes in such a way that the deformation increases with time. Dimensional
stability under static loads can be estimated in this manner and this information
is very useful to designers, fabricators, and engineers using plastics. Creep results
are often reported as the inverse of modulus or compliance (strain divided by the
applied stress), which for the reasons outlined above will be a time-dependent
quantity. If the level of stress is small it is possible to observe recovery of the
original dimensions, creep recovery.
Stress Relaxation Tests.
Stress relaxation involves measurement of the
stress required to hold a given deformation as a function of time. The deformation
is initially applied as rapidly as possible to strain the sample to a certain degree.
The modulus calculated from such an experiment decreases with increasing time,
reflecting the polymer structure ability to redistribute the stress.
Both creep and stress relaxation is modeled using computer simulation soft-
ware based on simple spring (elastic deformation) and dashpot (viscous flow) mod-
els. Many polymers, when they approach the T
g
, will exhibit viscoelastic behavior
in which the physical characteristics are best described by considering the mate-
rial as having both solid- and liquid-like properties. Viscoelasticity is an important
property to be found in polymeric materials (see V
ISCOELASTICITY
).
Impact Testing.
Although not a fundamental materials property, the im-
pact strength is a very valuable parameter when attempting to determine the
suitability of a material for a particular application (128). Impact tests usually in-
volve striking a sample of a specified geometry with an object that is travelling at
high velocity. The most common tests are the Izod, Charpy, and falling weight. The
Izod and Charpy tests both employ a weighted pendulum. In the Izod test, a bar
of the polymer is suitably notched to act as a stress concentrator. The notched bar
is held vertical while clamped at one end. Charpy test has the specimen clamped
at both ends. The falling weight test is usually conducted with a weighted dart or
ball striking either a sheet or plate of material (see I
MPACT
R
ESISTANCE
).
The underlying theme that emerges from the mechanical testing is the re-
quirement to characterize the molecular organization at various levels (129). This
organization–morphology is a function of the thermal history and various chemi-
cal and molar mass parameters implicit in the material being studied.
Visualization of the Morphology of Polymer Materials
Amorphous materials are defined by a lack of organized structure, but crystalline
materials exhibit a range of levels of organization (22,130–133). Visualization of
the structure of the polymer is therefore an important component of the charac-
terization of any material that is capable of showing order.
X-Ray Diffraction.
Improvements in diffraction methods have allowed the
rapid characterization of the order within polymer solids. Synchrotron Radiation
(qv) has allowed real-time examination of the creation of order as polymers are

176
CHARACTERIZATION OF POLYMERS
Vol. 9
cooled from the melt (134). The X-ray diffraction techniques can be subdivided, de-
pending on the range over which the scattered electrons are collected. The major
structural types that are of interest are single crystals, polycrystalline aggre-
gates with crystals, randomly orientated “powder samples,” and polycrystalline
aggregates in which the crystals show a preferred orientation. The typical dis-
tances that are responsible for diffraction can be anywhere between 1 and 20
nm, which requires measurement of very small angles (5
◦
–15
◦
) close to the inci-
dent beam. With synchrotron sources it is possible to measure such small angles,
whereas with conventional sources this is difficult. The technique is referred to
as small-angle X-ray scattering (SAXS). Studies of aromatic polyesters indicate
that even with these relatively complex molecules, regions of crystalline order
can exist and have a profound effect on their physical properties. The extension
of the technique, which scans a broader range of angles (
∼10
◦
–40
◦
), is designated
wide-angle X-ray scattering (WAXS) and provides information on spacing between
0.1 and 1 nm. This technique can provide valuable information on the crystalline
packing within the crystallites. Whereas in single crystals diffraction spots can be
observed, in many polymers halos are observed and these may have bright spots
which reflect the preferred orientation of the crystallites within the sample (see
X-
RAY
M
ICROSCOPY
).
Neutron Diffraction and Inelastic Scattering.
Use of neutron scatter-
ing (qv) can, in principle, allow observation of the location of hydrogen atoms in
a lattice (135). The contrast between the scattering cross-section of hydrogen and
deuterium allows the distribution of a single labeled chain within a low scatter-
ing matrix. Although in principle deuteration leaves the structure of the polymer
essentially unchanged, there are distinct thermodynamic differences between the
protonated and deuterated polymer raising the question whether the structure
may have been changed by the insertion of the labeled polymer (136). Although
publications have been produced on neutron diffraction from polymeric single
crystals, most of the research has been carried out on inelastic scattering mea-
surements. The latter are concerned with energy transfer processes between the
neutron and the matrix and are ideal for the study of dynamic processes, but are
less easily interpreted in terms of the structure of the polymer. The one application
where neutron scattering has proved to be useful is for the determination of the
size of a labeled polymer coil in a nonlabeled matrix. It has been shown that the
dimensions of an amorphous polymer in the solid state approximate to those of the
ideal random coil dimensions that would be predicted to occur in near-ideal inter-
action conditions. Low energy or long-wavelength neutrons (0.4–2.0 nm), unlike
X-rays, produce little damage and can penetrate samples of up to 1-cm thickness,
which is thicker than those used for X-ray analysis.
Electron Scattering and Electron Microscopy Techniques.
High en-
ergy electrons can be scattered by the molecule in the polymer structure and the
diffraction patterns will resemble those obtained from X-rays. Electron diffraction
is only really possible on very highly crystalline materials. If the sample is suf-
ficiently thin, then the analogue of the optical microscope can be used in which
the electron beam is passed through the sample and imaged on a sensitive pho-
tographic, or now an image, plane. The resolution of such a system can be at the
0.1-nm level and provides useful information that is below the resolution limit of
the conventional optical microscope. A problem with this method is that the high

Vol. 9
CHARACTERIZATION OF POLYMERS
177
energy electron beam (100–200 kV) can produce degradation of the sample, and
resolution that is usually associated with high acceleration voltages has to be bal-
anced against the time required to produce an satisfactory image. Transmission
electron microscopy (TEM) method has allowed visualization of the chain packing
in polyethylene-type of polymer (137). The TEM method requires that the sam-
ple be thinned to dimensions which allow the electron beam to pass through the
material which is typically below 100 nm. This requirement usually necessitates
that the section be prepared using a cold working microtome which generates
wafers that are of the order of a few tens of nanometers thick. The TEM method
can be used to provide electron diffraction data that can be used to calculate the
molecular spacing in crystalline phases in the polymer material. This type of in-
formation is very complementary to that obtained from X-ray diffraction, and with
the use of computer models of the order structure it is impossible to obtain quite
a precise definition of the chain-packing dimensions and orientation (138). The
alternative and slightly lower resolution approach to electron microscopy is scan-
ning electron microscopy (SEM) (139). In SEM, the back-scattering of electrons
used to provide information on the structure of polymers has resolution limits and
cannot normally provide molecular resolution data. The back-scattered electron
has an energy which is characteristic of the atoms that produce the scattering,
and imaging selective on a particular atomic distribution is possible (140). How-
ever, for polymers this is, not usually used as the resolution of light atoms such as
carbon and oxygen is very difficult and the method is only useful for heavy atoms.
It is, however, possible to identify the segregation of metal catalyst residues in
polyolefins. Electron microscopic techniques usually require that the sample be
coated with conducting media to avoid loss of resolution as a consequence of the
buildup of trapped charge, and hence sample preparation can be problematic.
Supermolecular Organization.
Both in biological and synthetic polymer
systems, the local order at the nanometer scale very rarely extends to the micro
level (140). Interruption of the chain packing occurs by either chain folding or
conformational defects leading to major disruption in the perfection of the chain
packing. A variety of models have been formulated to assist visualization of these
structures, an example of which is the so-called fringe micelle model. Much of our
knowledge of the organization at this lengthy scale is obtained from either TEM or
SEM studies combined with X-ray diffraction measurements. The SEM technique
is ideally suited for the study of polymers allowing the magnification to be changed
from relatively low values, where the object can be imaged at optical resolution,
to length scales which are submicron to those where the resolution approaches
the subnanometer scale. The organization of the polymer chains observed at this
scale reflects the packing of smaller scale crystallites and allows visualization of
liquid crystalline order within extended domains of the polymer. The discussion
of this level of organization is often referred to under the general heading of tex-
tured polymer and may, in the case of a polymer such as cellulose, be constrained
to the microfibrils. In polyethylene, the organization of the crystalline lamellar
structures within domains leads to the creation of microcrystallites that can have
micron-size dimensions or larger (130,131) but from the point of view of their
mechanical properties are distinct entities. The dimensions of the range of the
structure at this level are defined by the extent to which the entity can be consid-
ered to have identifiable physical properties. In polyethylene, the cool deformation

178
CHARACTERIZATION OF POLYMERS
Vol. 9
of the substrate can lead to significant alignment of the crystallites and enhance-
ment of the modulus in the direction of the applied stress. The alignment of the
crystallites, however, has a negative effect: enhancement of the modulus in one
direction leads to a reduction in the strength in the direction transverse to the
draw direction (130). The development of anisotropy in a crystalline solid can be
visualized from examination of the angular dependence of the X-ray scattering
plots. The “pole” diagrams (131), which is a plot of scattering intensity as a func-
tion of the direction of the draw axis to the X-ray beam axis, allow the extent of
the anisotropy to be determined.
Optical Microscopy.
Traditionally, light microscopy has played an im-
portant role in the visualization of ordered polymers, and a range of different ap-
proaches can be adopted to enhance the contrast, phase contrast, polarized light
microscopy, orientation birefringence, strain birefringence, modulated contrast,
interference microscopy, etc (132,141–144). Light microscopy rarely provides res-
olution better than several micrometers but can give a quick and easy assessment
of the extent of order in the polymer material.
Scanning Atomic Force Microscopy and Related Methods.
Whereas
the characterization of the structure of the bulk solid is of relevance to the me-
chanical properties, a knowledge of the surface structure may be important in
understanding adsorption (qv), tribology, friction, adhesion, and other surface-
dominated phenomena (145,146) (see S
URFACE
P
ROPERTIES
). The development of
atomic force microscopy (qv) (AFM) has allowed imaging at better than a nanome-
ter resolution of phase-separated and similar structure (147) (see also S
CANNING
F
ORCE
, M
ICROSCOPY
). The surface analytical techniques will not be included in
this review but do provide a very important part of the characterization of atomic
composition and alignment and segregation of the polymer chains in the surfaces
(22) (see S
URFACE
A
NALYSIS
).
Positron Annihilation Lifetime Spectroscopy (PALS).
A positive
electron of very high energy can penetrate matter and through inelastic collisions
be slowed down until it has thermal energies. The inelastic collisions will
often produce damage resulting in the emission of electrons. The combination
of a positron (
+ve electron) with an electron forms positronium. Positron-
ium is only stable if thermalized within a void of molecular dimensions. The
para-positronium can undergo spin allowed annihilate with the generation of
energy. The ortho-positronoium does not have the correct spin and will usually
annihilate through spin exchange with the electrons that form the walls of the
cavity in which it resides. The process allows determination of the size of the
molecular cavity through the lifetime of the ortho-positronium annihilation and
the intensity is directly proportional to the number density of the events. The
voids measured by this method are essentially those associated with the free
volume in an amorphous polymer. Study of PALS can indicate the mechanisms of
aging, plasticization, gas diffusion, and other processes that are morphologically
controlled in a disordered material (148).
Molecular Dynamics of Polymer Systems
Physical properties are a reflection of the degree and extent of the freedom of the
polymer molecule to undergo spatial rearrangement as a consequence of thermal

Vol. 9
CHARACTERIZATION OF POLYMERS
179
or an applied perturbation. A variety of methods exist which allow the direct
probing of these molecular motions.
Nuclear Magnetic Resonance—Relaxation Time Measurements.
The NMR method is usually used in solution for the determination of the struc-
ture of a polymer; however, when used in the Fourier transform model it can
also probe the rate at which nuclei reorientate within the applied magnetic field
(22,122). The relaxation of a particular nucleus can be used to characterize the
motion of a specific group.
1
H NMR signals are usually not sufficiently decoupled
from one another to allow the motion of individual groups to be characterized, and
it is usual to limit the discussion to gross differences as between aliphatic and aro-
matic protons. The low natural abundance of the
13
C nucleus allows separation
of the resonance of individual groups, and hence the relative rates of the motion
between individual atoms can be probe (122). In practice, this level of detailed
information can only be obtained for polymer molecules dispersed in solution;
the equivalent solid-state spectrum reflects a more highly coupled state in which
motions are not resolved. Solid state is sensitive to short-range interactions and
has been used to study crystallinity, rotational transitions, relative orientations of
polymer chains, solid glassy polymers, compatibility of solid polymer blends, and
cross-linking and entanglement (149,155).
Dielectric Relaxation Spectroscopy.
Many polymers contain polar
groups and the internal motions of these groups lead to fluctuations in the lo-
cal dielectric permittivity. Measurement of the dielectric relaxation (qv) spectrum
over a broad frequency range (10
− 3
–10
10
Hz) can provide valuable information
on the dynamic nature of a polymer at any given temperature (156–162). As with
other relaxation measurements, it is conventional to combine temperature and
frequency to allow calculation of activation energies and map the dynamics of the
polymer system. Because of the breadth of most relaxation processes, it is desir-
able to explore the influence of factors such as molar mass and morphology on the
distribution parameter describing the motion of the dipoles. Various approaches
have been used to describe the distribution parameter; however, the most adapt-
able is that of Havriliak and Negami (163,164). The parameters obtained have
no molecular significance, but provide a method of characterizing the influence of
various factors on the process being investigated. In amorphous polymers, DRS
has been extensively used for the study of sub-T
g
processes and, in particular, the
effects of physical aging on the nature of these relaxation processes. In the case of
thermosets, DRS can be used to monitor the cure process (165,166). In solution,
DRS has the capability of probing the backbone local dynamics, and studies as a
function of molar mass allow an understanding of the development of cooperative
motion as the chain length is increased.
Electron Spin Resonance Spectroscopy.
Through the use of stable
free-radical species, it has been possible to follow the relaxation processes of these
labels and infer the nature of the motion of the elements of the polymer chain
to which they are attached. This technique is limited, in that it does not see the
actual motion of the linkage to which the probe is attached to the polymer but can
provide important information on the nature of the effects of the matrix on the
motion of the probe (167–175) (see E
LECTRON
S
PIN
R
ESONANCE
).
Luminescence.
Many polymer groups, eg naphthalene, are able to ex-
hibit fluorescence, or even phosphorescence, when exposed to light of a suitable

180
CHARACTERIZATION OF POLYMERS
Vol. 9
excitation frequency. If the excitation is polarized, it is possible to follow the sub-
sequent motion of the probe during the fluorescence or phosphorescence lifetime.
The technique has been successfully applied to the study of a range of polymer
systems.
Combining the data obtained from a range of dynamic observations of poly-
mers, it is possible to develop an understanding of the factors that influence the
physical properties of these materials. It is clear that short-range forces can con-
trol both side-chain and local motion of the polymer backbone. However, in an
amorphous solid, provided the motions of the backbone are thermally labile, it is
free volume which dictates the temperature at which the T
g
occurs (176–180).
Rheology and Melt Flow Behavior.
In many applications the ability of
the polymer to undergo flow under the influence of an applied force is critical in
understanding whether or not that material can be processed using a particular
technology. A variety of different types of equipment exist for the measurement
of flow under particular constraints: constant shear stress, constant strain, elon-
gation flow, normal force measurements, oscillatory stress, capillary flow, etc. A
typical polymer when subjected to oscillatory shear will exhibit a viscosity that
reflects the ease with which it can move. Low molar mass polymers exhibit flow
characteristics which are similar to simple liquids and are said to be Newtonian.
However, as the chain length increases, cooperative motions of the chain become
possible leading to the so-called normal mode relaxation processes. These collec-
tive motions of a flexible polymer chain are a function of the length of the chain
and molar mass. These motions will contribute to the flow resistance at low rates
of shear but, as the frequency is increased, cease to be able to contribute and lead
to the observation of a lower effective viscosity-shear thinning. This phenomenon
is observed in many polymer systems and is molar mass dependent. If the chain
is sufficiently long, then entanglement is possible and additional constraints are
imposed on the flow, and a marked increase in viscosity is observed. For low molar
masses the viscosity (
η) varies as η α M
n
whereas for polymers above a certain criti-
cal molar mass M
c
, the viscosity varies as
η α M
n
3
.5
. Many applications of polymers
in the melt phase require knowledge of M
c
that typically has a value of 1.5
×
10
4
to 6
× 10
4
for most materials (181–184) (see R
HEOLOGICAL
M
EASUREMENTS
).
Thermosetting Polymer Systems
An important group of polymers are classified as being thermosetting materials
(165) (see T
HERMOSETS
). The chemistry associated with their formation is often
the same as that used for thermoplastic materials, except that in the case of these
polymers the monomers used have more than two functions per monomer unit. It
is therefore possible to have two materials that ostensibly look chemically similar,
but one is a thermoplastic and the other a thermoset. This situation is to be found
in Polyurethanes (qv) where, depending on the functionality of the isocyanate or
soft block used, the resulting material may be a thermoplastic or a thermoset. In
most cases it is desirable to monitor the cure process to avoid either too slow a for-
mation of the three-dimensional network, and hence poor production efficiency, or
too quick a cure with the possibility of excessive exotherms and possible degrada-
tion of the material. The formation of a network for a material that has a T
g
above

Vol. 9
CHARACTERIZATION OF POLYMERS
181
the cure temperature can also lead to trapped stresses, and these will usually
have to be relieved by post-thermal treatment. A number of rheological, thermal,
dielectric relaxation, and spectroscopic methods exist for the monitoring of the
cure processes. The techniques used for the study of thermoplastics are generally
appropriate for the study of thermosets, provided flow is not required to make the
observation.
Other Important Characteristics of Polymers
In terms of application, polymers are used in a variety of different ways that lead
to the requirement to measure specific physical properties.
Electrical Characteristics.
Although polymers have been traditionally
used as insulators, in recent years the ability to achieve intrinsic conductivity
has been demonstrated and is associated with systems in which the backbone has
developed an extended delocalized electronic structure, eg polyacetylene. Other
materials such as poly(vinyl chloride) or polyethylene are used in electric insula-
tion applications. In the latter it is their ability to inhibit electron migration that is
the important characteristic. A range of tests exists to help measure such features
as the electric breakdown resistance. Other specific types of property observed in
polymers include semiconductivity, photoconductivity, piezoelectricity, pyroelec-
tricity, and static electrical charging (triboelectricity). Each of these features is a
direct consequence of a combination of chemical and specific morphological fea-
ture in the polymer or composite material. These properties have been used to
specific effect in microphones, load speakers, photocopying, display technology,
etc (185–188) (see E
LECTRICALLY
A
CTIVE
P
OLYMERS
; P
IEZOELECTRIC
P
OLYMERS
).
Stability.
Polymers are used in a wide range of environments and their
ability to resist environmental factors is often a determining factor in their ap-
plication. The factors which will lead to degradation include mechanical stresses
due to handling, radiation, light, X-rays, and gamma rays; exposure to various
gases including oxygen, ozone, sulfur dioxide, nitrogen dioxide and to solvents
and corrosive chemicals; and strong acids and strong bases. A variety of different
specific tests exist for the study of these features and relate to specific problems.
The most commonly encountered problem is thermal degradation associated with
injection molding and similar methods and directly related to the relative stability
of particular bonds forming the backbone and side chains (189) (see D
EGRADATION
;
S
TABILIZATION
).
Permeability.
Many polymers are used in packaging and, in particular,
for food. In this latter case the permeability to gases and vapors is of prime im-
portance. The permeation or transmission of a gas or vapor is a function of the
solubility of a gas or vapor in the polymer and the rate of diffusion through the
matrix. The permeability coefficient, diffusion constant, and solubility coefficients
can all be measured and are influenced by the chemical structure and morphol-
ogy. In order to achieve the required permeability characteristics it is common
to co-extrude a series of polymers to form a laminated structure. Such materials
allow selective permeation of a specific species and enhance the life of the product
(190,191) (see T
RANSPORT
P
ROPERTIES
).

182
CHARACTERIZATION OF POLYMERS
Vol. 9
Flammability.
The ability to withstand exposure to fire is often a critically
important feature when selecting a specific plastic for a particular application. The
oxygen index, defined as the percentage of oxygen in an oxygen/nitrogen mixture
required to sustain combustion, is often used to rank plastics. The higher the
value the better the material’s ability to withstand exposure to heat. TGA data is
often used to assess the degradation characteristics of a polymer; however, specific
simulation tests are required to determine how factors such as heat flow, thermal
conductivity, drip, etc, in reality influence the burning characteristics of a material
(192) (see F
LAMMABILITY
; F
IRE
R
ETARDANTS
).
Solubility.
Solubility of a polymer will reflect the compatibility with the
solvent. For a polymer to be dissolved there is a requirement for the forces between
the solvent to match those between the elements of the polymer itself. If the
match is close, then it is possible to create a situation in which the dissolved
polymer is in a collapsed state corresponding to the thermodynamic requirements
of the theta condition. At this point the forces between the segments balance the
forces between the segments and the solvent. If the strength of the interaction
is increased, the quality of the solvent is improved and the effective size of the
polymer increases; it swells. Thermoplastic polymers can be completely dissolved
in a good solvent, whereas thermoset materials are only able to swell to a size that
reflects the cross-link density of the matrix. It is possible for two nonsolvents to
combine to form a solvating mixture, and this phenomenon is termed co-solvency
(see M
ISCIBILITY
).
Density.
The ability to achieve a particular level of packing is critical in de-
termining the density of a material and is directly related to the morphology of the
polymer. The density may be measured directly or alternatively by determination
of the composition of a liquid in which the material is just buoyant (193).
A comprehensive survey of test methods is to be found in the Handbook of
Polymer Testing (194).
BIBLIOGRAPHY
“Characterization of Polymers” in EPST 1st ed., Vol. 3, pp. 611–631, by W. R. Sorenson,
Continental Oil Co.; in EPSE 2nd ed., Vol. 3, pp. 290–327, by W. J. Freeman, Hercules, Inc.
1. R. Koningsveld, Adv. Polym. Sci. 7, 1 (1970).
2. L. H. Tung, ed., Fractionation of Synthetic Polymers: Principles and Practices, Marcel
Dekker, Inc., New York, 1977.
3. L. H. Tung, ed., J. Macromol. Sci. Rev. Makromol. Chem. 6(1), 58 (1971).
4. T. Y. Chang, Adv. Polym. Sci. 163, 1 (2003).
5. G. Reiss and P. Callot, in L. H. Tung, ed., Fractionation of Synthetic Polymers: Prin-
ciples and Practices, Marcel Dekker, Inc., New York, 1977.
6. C. A. Fonseca and I. R. Harrison, in R. A. Pethrick and J. V. Dawkins, eds., Modern
Techniques for Polymer Characterization, John Wiley & Sons, Inc., New York, 2003,
pp. 1–13.
7. P. Vastamaki, M. Jussila, and M. L. Riekkola, Analyst 128, 1243 (2003).
8. J. B. P. Soares and A. E. Hamielec, in R. A. Pethrick and J. V. Dawkins, eds., Modern
Techniques for Polymer Characterization, John Wiley & Sons, Inc., New York, 2003,
pp. 1–13.

Vol. 9
CHARACTERIZATION OF POLYMERS
183
9. J. Janca, in R. A. Pethrick and J. V. Dawkins, eds., Modern Techniques for Polymer
Characterization, John Wiley & Sons, Inc., New York, 2003. pp. 1–13,
10. B. Folie, M. Keltchtermans, J. R. Shutt, H. Schoneman, and V. J. Krukonis, J. Appl.
Polym. Sci. 64, 2015 (1997).
11. J. J. Watkins, V. J. Krukonis, P. D. Condo, D. Pradham, and P. Ehrlich, J. Supercrit.
Fluids 4, 24 (1991).
12. J. C. Giddings, Sep. Sci. 1, 123 (1966).
13. G. H. Thompson, M. N. Myers, J. C. Giddings, Sep. Sci. 2, 797 (1967).
14. K. D. Caldwell, L. K. Kesner, M. N. Myer, and J. C. Giddings, Science 176, 296 (1972).
15. S. Loske, A. Schneider, and B. A. Wolf, Macromolecules 36, 5008 (2003).
16. G. E. Kassalainen and S. K. R. Williams, J. Chromatogr. A 988, 285 (2003).
17. N. Hugenberg, S. Loske, A. H. E. Muller, W. Schartl, M. Schmidt, P. F. W. Simon,
A. Strack, and B. A. Wolf, J. Non-Cryst. Solids 307, 765 (2002).
18. J. C. Giddings, J. Chem. Educ. 50, 667 (1973).
19. J. C. Giddings, M. N. Meyers, F. J. F. Yang, and J. K. Smith, in M. Kerker, ed., Colloid
and Interfacial Science, Vol. 4, Academic Press, Inc., New York, 1976, pp. 381–398.
20. J. C. Giddings, K. A. Graff, K. D. Caldwell, and M. N. Meyers, Adv. Chem. Ser. 203,
257 (1983).
21. H. Inagaki, Adv. Polymer Sci. 24, 189 (1977).
22. D. Campbell, R. A. Pethrick, and J. R. White, Polymer Characterization: Physical
Techniques, Stanley Thornes (Publishers) Ltd., Cheltenham, U.K., 2000, p. 38.
23. P. C. Painter, M. M. Coleman, and J. L. Koenig, The Theory of Vibrational Spectroscopy
and Its Application to Polymeric Materials, John Wiley & Sons, Inc., New York,
1982.
24. J. Haslam, H. A. Willis, and D. C. M. Squirell, Identification and Analysis of Plastics,
2nd ed., Butterworth Publishers Ltd., London, 1972.
25. P. J. Hendra, Adv. Polym. Sci. 6, 151 (1969).
26. B. Jasse, in J. V. Dawkins, ed., Developments in Polymer Characterization, Vol. 4,
Elsevier Applied Science Publishers, Ltd., Barking, U.K., 1983.
27. D. J. Cutler, P. J. Hendra, and G. Fraser, Laser Raman Spectroscopy of Synthetic
Polymers, Vol. 2, Elsevier Applied Science Publishers, Ltd., Barking, U.K., 1980,
p. 121.
28. R. S. McDonald, Anal. Chem. 54, 1250 (1982).
29. W. Y. Yeh and R. J. Young, J. Macromol. Sci., Phys. B 37, 83–118 (1998).
30. V. Galitsyn, S. Khizhnyak, P. Pakhomov, and A. Tshmel, J. Macromol. Sci., Phys. B
42, 1085–1095 (2003).
31. S. Webster and D. I. Bower, Polymer 36, 4351–4353 (1995).
32. T. Achibat, A. Boukenter, E. Duval, A. Mermet, M. Aboulfaraj, S. Etienne, and
C. Gsell, Polymer 36, 251–257 (1995).
33. J. L. Koenig, Spectroscopy of Polymers, American Chemical Society, Washington, D.C.,
1992. ACS Professional Reference Book.
34. J. M. Chalmers, M. W. Mackenzie, H. A. Willis, H. G. M. Edwards, J. S. Lees, and
D. A. Long, Spectrochim. Acta, Part A: Mol. Biomol. Spectrsc. 47, 1677–1683 (1991).
35. R. P. Wool, Bull. Am. Phys. Soc. 26, 327 (1981).
36. R. J. Jakobsen, in J. R. Ferraro and L. J. Basile, eds., Fourier Transform Infrared
Spectroscopy: Applications to Chemical Systems, Vol. 1, Academic Press, Inc., New
York, 1978, pp. 61–97.
37. F. A. Bovey, High Resolution NMR of Macromolecules, Academic Press, Inc., New
York, 1972.
38. K. J. Ivin, Pure Appl. Chem. 55, 1529 (1983).
39. V. J. McBrierty and D. C. Douglass, J. Polym. Sci., Macromol. Rev. 16, 195 (1981).
40. B. C. Gerstein, Anal. Chem. 55, 781A, 899A (1983).

184
CHARACTERIZATION OF POLYMERS
Vol. 9
41. J. R. Havens and J. L. Koenig, Applications of High Resolution Carbon -13 NMR
Method, Academic Press, Inc., New York, 1977.
42. K. Wuthrich, NMR of Proteins and Nucleic Acids, John Wiley & Sons, Inc., New York,
1986.
43. D. M. Grant and E. G. Paul, J. Am. Chem. Soc. 86, 2984 (1964).
44. C. Delides, R. A. Pethrick, A. V. Cunliffe, and P. G. Klein, Polymer 22, 1205 (1981).
45. A. V. Cunliffe and R. A. Pethrick, Polymer 21, 1026 (1980).
46. J. C. Randall, Polymer Sequence Determination, Carbon-13 NMR Method, Academic
Press, Inc., New York, 1977.
47. R. A. Pethrick and B. Thomson, Br. Polym. J. 18(3), 171–180 (1986).
48. R. A. Pethrick and B. Thomson, Br. Polym. J. 18, 380–386 (1986).
49. J. B. Stothers, Carbon-13 NMR Spectroscopy, Academic Press, Inc., New York, 1972.
50. R. J. Abraham, J. Fisher, and P. Loftus, Introduction to NMR Spectroscopy, John Wiley
& Sons, Inc., Chichester, U.K., 1988.
51. J. Clauss, K. Schmidtrohr, and H. W. Spiess, Acta Polym. 44(1), 1–17 (1993).
52. J. B. Miller, J. Therm. Anal. 49, 521–524 (1997).
53. F. Horii, H. Kaji, H. Ishida, K. Kuwabara, K. Masuda, and T. Tai, J. Mol. Struct. 441,
303–311 (1998).
54. K. Landfester and H. W. Spiess, Acta Polym. 49, 451–464 (1998).
55. H. N. Cheng and A. D. English, ACS Symp. Series 834, 3–20 (2003).
56. P. Robyr, Z. Gan, and U. W. Suter, Macromolecules 31, 8918–8923 (1998).
57. E. Klesper, in D. O. Huummel, ed., Polymer Spectroscopy, Verlag Chemie, Weinheim,
Germany, 1974, Chapt. 1
58. S. Hafner, D. E. Demco, and R. Kimmich, Solid State Nucl. Magn. Reson. 6, 275–293
(1996).
59. B. Blumich and P. Blumler, Makromol. Chem., Macromol. Chem. Phys. 194, 2133–
2161 (1993).
60. W. J. Irwin, Analytical Pyrolysis: A Comprehensive Guide, Vol. 22, Chromatographic
Science Series, Marcel Dekker, Inc., New York, 1982.
61. C. J. Wolf, M. A. Grayson, and D. L. Fanter, Anal. Chem. 52, 348A (1980).
62. S. A. Liebman and E. J. Levy, Adv. Chem. Ser. 203, 617 (1983).
63. E. Reiner and T. F. Mason, Adv. Chem. Ser. 203, 703 (1983).
64. F. L. Bayer, Adv. Chem. Ser. 203, 693 (1983).
65. C. G. Smith, J. Anal. Appl. Pyrolysis 15, 209–216 (1989).
66. J. K. Haken and P. I. Iddamalgoda, J. Chromatogr. A 756, 1–20 (1996).
67. J. K. Haken, J. Chromatogr. A 825, 171–187 (1998).
68. F. C. Y. Wang, J. Chromatogr. A 843, 413–423 (1999).
69. J. W. Lyons, D. Poche, F. C. Y. Wang, and P. B. Smith, Adv. Mater. 12, 1847–1854
(2000).
70. S. S. Choi, J. Anal. Appl. Pyrolysis 57, 249–259 (2001).
71. N. N. Barashkov, and O. A. S. Gunder, Fluorescent Polymers, Polymer Science and
Technology, Ellis Horwood Series, Chichester, U.K., 1994.
72. M. Freemantle, Chem. Eng. News 81(8), 6 (2003).
73. E. Scamporrino and D. Vitalini, in R. A. Pethrick and J. V. Dawkins, eds., Modern
Techniques for Polymer Characterization, John Wiley & Sons, Inc., New York, 2003,
p. 233.
74. R. J. Cotter, Anal. Chem. 64, 1027A (1992).
75. R. S. Brown and J. J. Lennon, Anal. Chem. 67, 1998 (1995).
76. G. Montaudo, Polym. Prepr. 290 (1996).
77. E. Scamporrrino, D. Vitalini, and P. Mineo, Macromolecules 29, 5520 (1996).
78. M. Karas, U. Bahr, A. Deppe, B. Stahl, F. Hillenkamp, and U. Giessmann, Makromol.
Chem. Macromol. Symp. 61, 397–404 (1992).

Vol. 9
CHARACTERIZATION OF POLYMERS
185
79. S. D. Hanton, Chem. Rev. 101, 527–569 (2001).
80. L. Q. Muang, A. Paiva, R. Bhat, and M. Wong, J. Am. Soc. Mass Spectrosc. 7, 1219
(1996).
81. N. Aust, I. Beytollahi-Amtmann, and K. Lederer, Int. J. Polym. Anal. Charact. 1,
245–258 (1995).
82. G. Lapienis and S. Penczek, Macromol. Symp. 195, 317 (2003).
83. Y. Brun, J. Liq. Chromatogr. Relat. Technol. 21, 1979–2015 (1998).
84. W. S. Bahary, M. P. Hogan, M. Jilani, and M. P. Aronson, Adv. Chem. Ser. 247, 151–166
(1995).
85. M. Netopilik, J. Chromatogr. A 915, 15–24 (2001).
86. M. Netopilik, Polymer 35, 4799–4803 (1994).
87. B. Trathnigg, S. Feichtenhofer, and M. Kollroser, J. Chromatogr. A 786, 75–84
(1997).
88. M. Helmstedt and J. Stejskal, Int. J. Polym. Anal. Charact. 4, 219–230 (1998).
89. M. Andersson, B. Wittgren, and K. G. Wahlund, Anal. Chem. 75, 4279–4291
(2003).
90. U. Zucchi and G. Cecchin, Adv. Polym. Sci. 51, 108 (1983).
91. H. G. Ellias, Macromolecules, Vols. 1 and 2, Plenum Press, New York, 1977.
92. T. C. Ward, J. Chem Educ. 58, 867 (1981).
93. W. Burchard, Adv. Polym. Sci. 48, 1 (1983).
94. P. Kratochvil, Pure Appl. Chem. 54, 379 (1982).
95. H. Pasch, Macromol. Symp. 178, 25–37 (2002).
96. H. Pasch, Macromol. Symp. 174, 403–412 (2001).
97. H. Pasch, Adv. Polym. Sci. 150, 1–66 (2000).
98. H. Pasch, Phys. Chem. Chem. Phys. 1, 3879–3890 (1999).
99. W. Burchard, Adv. Polym. Sci. 143, 113–194 (1999).
100. H. Pasch, Adv. Polym. Sci. 128, 1–45 (1997).
101. A. Jacobs and O. Dahlman, Nord. Pulp Paper Res. J. 15, 120–127 (2000).
102. J. Spickermann, H. J. Rader, K. Mullen, B. Muller, M. Gerle, K. Fischer, and
M. Schmidt, Macromol. Rapid Commun. 17, 885–896 (1996).
103. J. Brandrup, E. H. Immergut, E. A. Grulke, and D. Bloch, eds., Polymer Handbook,
4th ed., John Wiley & Sons, Inc., New York, 1999.
104. W. L. Mattice and U. W. Suter, Conformational Theory of Large Molecules: The Ro-
tational Isomeric State Model in Macromolecular Systems, John Wiley & Sons, Inc.,
New York, 1994.
105. P. Blumler and B. Blumich, Rubber Chem. Technol. 70, 468–518 (1997).
106. P. B. Smith, A. J. Pasztor, M. L. McKelvy, D. M. Meunier, S. W. Froelicher, and F. C.
Y. Wang, Anal. Chem. R95–R121 (1997).
107. L. C. E. Struick, Internal Stresses, Dimensional Instabilities and Molecular Orienta-
tion in Plastics, John Wiley & Sons, Inc., New York, 1990.
108. R. H. Still, Br. Polym. J. 11(3), 101 (1979).
109. J. R. Freid, in J. V. Dawkins, ed., Developments in Polymer Characterization, Vol. 4,
1983, pp. 39–90.
110. J. N. Hay, in L. S. Bark and N. S. Allan, eds., Analysis of Polymer Systems, Applied
Science Publishers, London, 1982, pp. 103–154.
111. M. J. Richardson, in J. V. Dawkins, ed., Developments in Polymer Characterization,
Vol. 1, 1978, pp. 205–244.
112. D. M. Price, M. Reading, T. J. Lever, A. Hammiche, and H. M. Pollock, Thermochim.
Acta 376, 95–97, (2001).
113. M. Reading, D. M. Price, D. B. Grandy, R. M. Smith, L. Bozec, M. Conroy, A. Ham-
miche, and H. M. Pollock, Macromol. Symp. 167, 45–62 (2001).
114. M. Reading, J. Therm. Anal. Calorim. 64(1), 7–14 (2001).

186
CHARACTERIZATION OF POLYMERS
Vol. 9
115. F. Oulevey, N. A. Burnham, G. Gremaud, A. J. Kulik, H. M. Pollock, A. Hammiche,
M. Reading, M. Song, and D. J. Hourston, Polymer 41, 3087–3092 (2000).
116. D. M. Price, M. Reading, and T. J. Lever, J. Therm. Anal. 56, 673–679 (1999).
117. X. Ramis, A. Cadenato, J. M. Morancho, and J. M. Salla, Polymer 44, 2067–2079
(2003).
118. I. R. Harrison, Thermochim. Acta 367, 85–92 (2001).
119. J. Rieger, Polym. Test. 20(2), 199–204 (2001).
120. J. Wolfrum, G. W. Ehrenstein, and M. A. Avondet, J. Compos. Mater. 34, 1788–1807
(2000).
121. I. Lacik, I. Krupa, M. Stach, A. Kucma, J. Jurciova, and I. Chodak, Polym. Test. 19,
755–771 (2000).
122. R. T. Bailey, A. M. North, and R. A. Pethrick, Molecular Motion in High Polymers,
Oxford University Press, New York, 1981.
123. R. G. Beaman, J. Polym. Sci. 9, 470 (1952).
124. R. F. Boyer, J. Appl. Phys. 25, 825 (1954).
125. R. F. Boyer, Polym. Yearbook 2, 233–344 (1985).
126. ASTM Standards, American Society for Testing and Materials, Philadelphia Pa.,
1984. Web site: www.astm.org.
127. M. L. Williams, R. F. Landel, and J. D. Ferry, J. Am. Chem. Soc. 77, 3701 (1955).
128. A. J. Kinloch and R. J. Young, Fracture Behavior of Polymers, Applied Sciences Pub-
lishers, New York, 1983, pp. 182–225.
129. R. Brown, Handbook of Polymer Testing, Marcel Dekker, Inc., New York, 1999.
130. I. M. Ward, in I. M. Ward, ed., Structure and Properties of Orientated Polymers, Applied
Science Publishers, London, 1975.
131. B. W. Cherry, Polymer Surfaces, Cambridge Solid State Science Series, Cambridge
University Press, London, 1981, p. 31.
132. R. A. Pethrick, in R. A. Pethrick and C. Viney, eds., Techniques for Polymer organi-
zation and Morphology Characterization, John Wiley & Sons, Inc., Chichester, U.K.,
2003, pp. 1–30.
133. M. E. Vickers, in R. A. Pethrick and C. Viney, eds., Techniques for Polymer Organi-
zation and Morphology Characterization, John Wiley & Sons, Inc., Chichester, U.K.,
2003, pp. 35–69.
134. A. J. Ryan, J. Stanford, W. Bras, and T. M. W. Nye, Polymer 38, 759 (1997).
135. D. M. Sadler, in I. H. Hall, ed., Structure of Crystalline Polymers, Elsevier Applied
Science Publishers, New York, 1984, Chapt. 4, p. 125.
136. V. Arrighi, D. C. Bucknall, and A. Triolo, in R. A. Pethrick, and C. Viney, eds., Tech-
niques for Polymer Organization and Morphology Characterization, John Wiley &
Sons, Inc., Chichester, U.K., 2003, pp. 169–197.
137. E. L. Thomas, in I. H. Hall, ed., Structure of Crystalline Polymers, Elsevier Applied
Science Publishers, New York, 1984, Chapt. 3, p. 79.
138. R. Vialluzzi, in R. A. Pethrick and C. Viney, eds., Techniques for Polymer Organization
and Morphology Characterization, John Wiley & Sons, Inc., Chichester, U.K., 2003,
pp. 145–163.
139. H. Assender, in R. A. Pethrick and C. Viney, eds., Techniques for Polymer Organization
and Morphology Characterization, John Wiley & Sons, Inc., Chichester, U.K., 2003,
pp. 201–218.
140. D. C. Bassett, Principles of Polymer Morphology, Cambridge Solid State Science Series,
Cambridge University Press, London, 1981, p. 19.
141. A. D. Curson, in D. A. Hemsley, ed., Applied Polymer Light Microscopy, Elsevier
Applied Science, Barking, U.K., 1989, p. 19.
142. B. P. Saville, in D. A. Hemsley, ed., Applied Polymer Light Microscopy, Elsevier Applied
Science, Barking, U.K., 1989, p. 73.

Vol. 9
CHARACTERIZATION OF POLYMERS
187
143. R. Hoffman, in D. A. Hemsley, ed., Applied Polymer Light Microscopy, Elsevier Applied
Science, Barking, U.K., 1989, p. 151.
144. D. A. Hemsley, in D. A. Hemsley, ed., Applied Polymer Light Microscopy, Elsevier
Applied Science, Barking, U.K., 1989, p. 151.
145. R. A. L. Jones and R. W. Richards, Polymers at Surfaces and Interfaces, Cambridge
University Press, U.K., 1999, p. 94.
146. D. Sarid, Scanning Force Microscopy, Oxford University Press, Oxford, 1994.
147. S. Affrossman, G. Henn, S. A. O’Neil, R. A. Pethrick, and M. Stamm, Macromolecules
29, 5010–5016 (1996).
148. R. A. Pethrick, Prog. Polym. Sci. 22, 1–47 (1997).
149. W. P. Slichter and D. W. McCall, J. Polym. Sci. 25, 230 (1957).
150. M. J. Richardson, Br. Polym. J. 1(3), 132 (1969).
151. J. Schafer, E. O. Stejskal, and R. Buchdal, Macromolecules 10, 384 (1977).
152. K. R. Doolan, J. Polym. Sci., Part B: Polym. Phys. 40, 572–584 (2002).
153. B. C. Min and J. W. Lee, J. Mol. Liq. 80(1), 33–51 (1999).
154. M. Laviolette, M. Auger, and S. Desilets, Macromolecules 32, 1602–1610
(1999).
155. M. Geppi, R. K. Harris, A. M. Kenwright, and B. J. Say, Solid State Nucl. Magn. Reson.
12(1), 15–20 (1999).
156. J. F. Mano, J. Macromol. Sci., Phys. B 42, 1169–1182 (2003).
157. J. F. Mano and J. L. G. Ribelles, Macromolecules 36, 2816–2824 (2003).
158. J. P. Eloundou, J. F. Gerard, J. P. Pascault, and D. Kranbuehl, Macromol. Chem. Phys.
203, 1974–1982 (2002).
159. J. Mijovic and J. W. Sy, Macromolecules 35, 6370–6376 (2002).
160. J. Mijovic, M. Y. Sun, and Y. F. Han, Macromolecules 35, 6417–6425 (2002).
161. G. Williams, I. K. Smith, G. A. Aldridge, P. A. Holmes, and S. Varma, Macromolecules
34, 7197–7209 (2001).
162. F. Kremer and A. Schonhals, Broadband Dielectric Spectroscopy, Springer, Berlin,
2003.
163. S. Havriliak and J. Havriliak, Dielectric and Mechanical Relaxation in Materials,
Hanser Publishers, Munich, 1997.
164. C. C. Ku, and R. Liepins, Electrical Properties of Polymers-Chemical Principles,
Hanser Publishers, Munich, 1987.
165. R. A. Pethrick, in A. K. Kulshreshtha and C. Vasile, eds., Handbook of Polymer
Blends and Composites, Vol. 1, RAPRA Technology, Shrewsbury, U.K., 2002, Chapt. 10,
p. 393.
166. R. A. Pethrick and D. Hayward, Prog. Polym. Sci. 27, 1983–2017 (2002).
167. F. Lembicz, Polimery 48, 557–560 (2003).
168. Y. Miwa, T. Tanase, K. Yamamoto, M. Sakaguchi, M. Sakai, and S. Shimada, Macro-
molecules 36, 3235–3239 (2003).
169. K. Kruczala, B. Varghese, B. G. Bokria, and S. Schlick, Macromolecules 36, 1899–1908
(2003).
170. M. Lucarini, G. F. Pedulli, and M. V. Motyakin, and S. Schlick, Prog. Polym. Sci. 28,
331–340 (2003).
171. Z. Veksli, M. Andreis, and B. Rakvin, Prog. Polym. Sci. 25, 949–986 (2000).
172. E. J. Harbron, V. P. McCaffrey, R. X. Xu, and M. D. E. Forbes, J. Am. Chem. Soc. 122,
9182–9188 (2000).
173. G. G. Cameron, M. Y. Qureshi, and S. C. Tavern, Eur. Polym. J. 32, 587–591 (1996).
174. G. G. Cameron, D. Stewart, R. Buscall, and J. Nemcek, Polymer 35, 3384–3388
(1994).
175. G. G. Cameron, I. S. Miles, and A. T. Bullock, Br. Polym. J. 19(2), 129–134 (1987).
176. M. Danko, P. Hrdlovic, and E. Borsig, Chem. Listy 97, 1052–1060 (2003).

188
CHARACTERIZATION OF POLYMERS
Vol. 9
177. S. Kimata, D. L. Jiang, and T. Aida, J. Polym. Sci., Polym. Chem. 41, 3524–3530 (2003).
178. J. Duhamel, S. Kanagalingam, T. J. O’Brien, and M. W. Ingratta, J. Am. Chem. Soc.
125, 12810–12822 (2003).
179. R. A. L. Vallee, M. Cotlet, J. Hotkens, and F. C. De Schryver, Macromolecules 36,
7752–7758 (2003).
180. R. A. L. Vallee, N. Tomczak, L. Kuipers, G. J. Vancso, and N. F. van Hulst, Phys. Rev.
Lett. 91, 38301 (2003).
181. M. Doi and S. F. Edwards, The Theory of Polymer Dynamics, Oxford Science Publica-
tions, Oxford, 1986.
182. J. D. Ferry, Viscoelastic Properties of Polymers, John Wiley & Sons, Inc., New York,
1970.
183. R. Byron Bird, R. C. Armstrong, and O. Hassager, Dynamic of Polymeric Liquids,
Vol. 1; with C. F. Curtis, Vol. 2, John Wiley & Sons, Inc., New York, 1987.
184. A. A. Collyer, Techniques in Rheological Measurement, Chapman and Hall, London,
1993.
185. J. I. Kroschwitz, Electrical and Electronic Properties of Polymers: A State of the Art
Compendium, John Wiley & Sons, Inc., New York, 1988.
186. H. Kuzmany, M. Mehring, and S. Roth, Electronic Properties of Conjugated Polymers
Solid State Sciences, Vol. 76, Springer-Verlag, Berlin, 1987.
187. H. Kuzmany, M. Mehring, and S. Roth, Electronic Properties of Conjugated Polymers
Solid State Sciences, Vol. 63, Springer-Verlag, Berlin, 1985.
188. G. Zerbi, Polyconjugated Materials, European Materials Research Society, Amster-
dam, 1992.
189. D. Kockott, in R. Brown, ed., Polymer Testing, Marcel Dekker, Inc., New York, 1999,
p. 697.
190. D. Hands, in R. Brown, ed., Polymer Testing, Marcel Dekker, Inc., New York, 1999,
p. 747.
191. J. Crank, The Mathematics of Diffusion, Oxford Science Publications, Oxford, 1985.
192. K. Paul, in R. Brown, ed., Handbook of Polymer Testing, Marcel Dekker, Inc., New
York, 1999, p. 659.
193. R. Brown, in R. Brown, ed., Handbook of Polymer Testing, Marcel Dekker, Inc., New
York, 1999, p. 157.
194. R. Brown, ed., Handbook of Polymer Testing, Marcel Dekker, Inc., New York, 1999.
R
ICHARD
A. P
ETHRICK
University of Strathclyde
CHITIN AND CHITOSAN.
See Volume 1.
Wyszukiwarka
Podobne podstrony:
Sience Direct Characterization of adsorption properties of extracellular polymeric substances (EPS)
Fundamentals of Polymer Chemist Nieznany
Electrochemical properties for Journal of Polymer Science
Modeling of Polymer Processing and Properties
Huang et al 2009 Journal of Polymer Science Part A Polymer Chemistry
Drying, shrinkage and rehydration characteristics of kiwifruits during hot air and microwave drying
Improved Characterization of Nitromethane, Nitromethane Mixtures, and Shaped Charge Jet
Production and Characterisation of extracts
1 3 16 Comparison of Different Characteristics of Modern Hot Work Tool Steels
Characteristics of young Ls
In silico characterization of the family of PARP like
characters of cindersmella
Practical Analysis Techniques of Polymer Fillers by Fourier Transform Infrared Spectroscopy (FTIR)
Characterization of Nucleotide Nieznany
Characterization of Particle Size Distribution
Genetic Methods of Polymer Synthesis
Fatigue Behavior of Polymers
Fundamentals of Polymer Chemist Nieznany
więcej podobnych podstron