
MASS SPECTROMETRY
Introduction
For many years, mass spectroscopists have searched for ways to place charged
polymeric molecules in the gas phase without degrading the polymer molecule.
With charged polymer molecules in the gas phase, the many methods of mass
spectrometry (ms) used for small molecules can be applied to large molecules. In
the same way, placing unfragmented polymer molecules into the gas phase offers
polymer science one of the most powerful absolute techniques that can be ap-
plied to single-chain characterization. The ms techniques offer the possibility to
obtain polymer single-chain structural information as a function of its molecular
mass, eg, repeat units, end groups, copolymer composition distributions, and poly-
mer architecture, as well as the overall molecular mass distribution (MMD). One
can also envision studying gas-phase chemical reactions involving polymer chains
to further study chain structure, ie, copolymer sequencing, or chain branching.
This hope has been realized in the last 10 years with the advent of soft ioniza-
tion methods in mass spectrometry, matrix-assisted laser desorption/ionization
(MALDI), and electrospray ionization (esi) methods. These methods place unfrag-
mented high molecular weight polymer molecules in the gas phase. Using MALDI
methods, one can examine polymers with molecular mass from about 500 to about
1,500,000 (1). Using esi methods, polymers with molecular masses up to 5,000,000
have been measured in the gas phase (2).
In this article, focus is on the soft ionization technique MALDI, which is
mainly in use for synthetic polymers. ESI is briefly discussed, although the
electrospray method has been generally confined to polar polymers, which are
water-soluble. Other ionization methods that have been used on synthetic poly-
mers, mainly laser desorption (LD) and field desorption (FD), are confined to low
160
Encyclopedia of Polymer Science and Technology. Copyright John Wiley & Sons, Inc. All rights reserved.

Vol. 3
MASS SPECTROMETRY
161
molecular mass polymers. LD and FD methods are discussed in some detail in
other references on ms (3,4).
Readers should note: A search of the current literature will reveal that esi
and MALDI methods are most commonly used on biopolymers. Any simple search
using the names of these methods will give many more articles on biopolymer uses
of these methods than articles on synthetic polymers.
In the last edition of this encyclopedia, a detailed discussion of traditional
small molecule ms was given in connection with studies on the degradation of
polymers and studies on additives in polymers. These topics will not be considered
in this article. The reader is referred to the 1987 Encyclopedia article or other more
current reviews (5,6).
This review begins with an abbreviated history of the work done to success-
fully place a high molecular mass polymer into the gas phase. Then there is a
description of the two most common methods used to place charged high mass
molecules into the gas phase, esi, and MALDI. Because of its dominant use with
synthetic polymers, the MALDI method is described in detail, including sample
preparation, data analysis, and the problems this method poses. The time-of-flight
mass spectrometry (tof-ms) system is also described in detail since it is the most
common in use today for synthetic polymers. References to other ms and detection
systems are also provided for the interested reader. Additionally, issues of quan-
tification will be reviewed because to determine MMD or molecular composition
distribution (MCD) for copolymers one must be able to quantify the ms data. A
variety of applications of MALDI to synthetic polymers are described.
History of MALDI and Electrospray
Electrospray ionization of polymers was introduced in 1968 (7) in an effort to study
polystyrene by ms. In 1984, the technique was further developed for biopolymers
(8). Spectra of poly(ethylene glycols) (PEG) up to molecular masses of 17,500 were
also obtained using esi (9). In 1992, esi techniques were used to obtain ms on
a poly(ethylene oxide) (PEO) of about 5,000,000 (2). Since esi techniques mul-
tiply charge macromolecules, both these studies were able to get spectra on a
quadrupole ms with an m/z of less than 2000, where z is the number of charges.
However, with many different n-mers in a normal narrow MMD of a synthetic
polymer, the effect of z often as high as 40 means that one needs to obtain resolu-
tion in m/z of a small fraction of a mass unit to see the entire MMD.
In 1988, a laser-based desorption method was developed (10) which pro-
duced ions of both high molecular mass synthetic polymers and biopolymers. In
that same year, the laser desorption technique using an organic molecule matrix
now commonly called MALDI was developed (11). The technique was originally
described for biopolymers. In 1992, it was shown that the MALDI technique could
be applied to synthetic polymers (12–15).
The Analysis Process
Mass spectrometry may be viewed as comprised of three distinct processes: (1)
The production of charged gas-phase species from the original analyte. This step
162
MASS SPECTROMETRY
Vol. 3
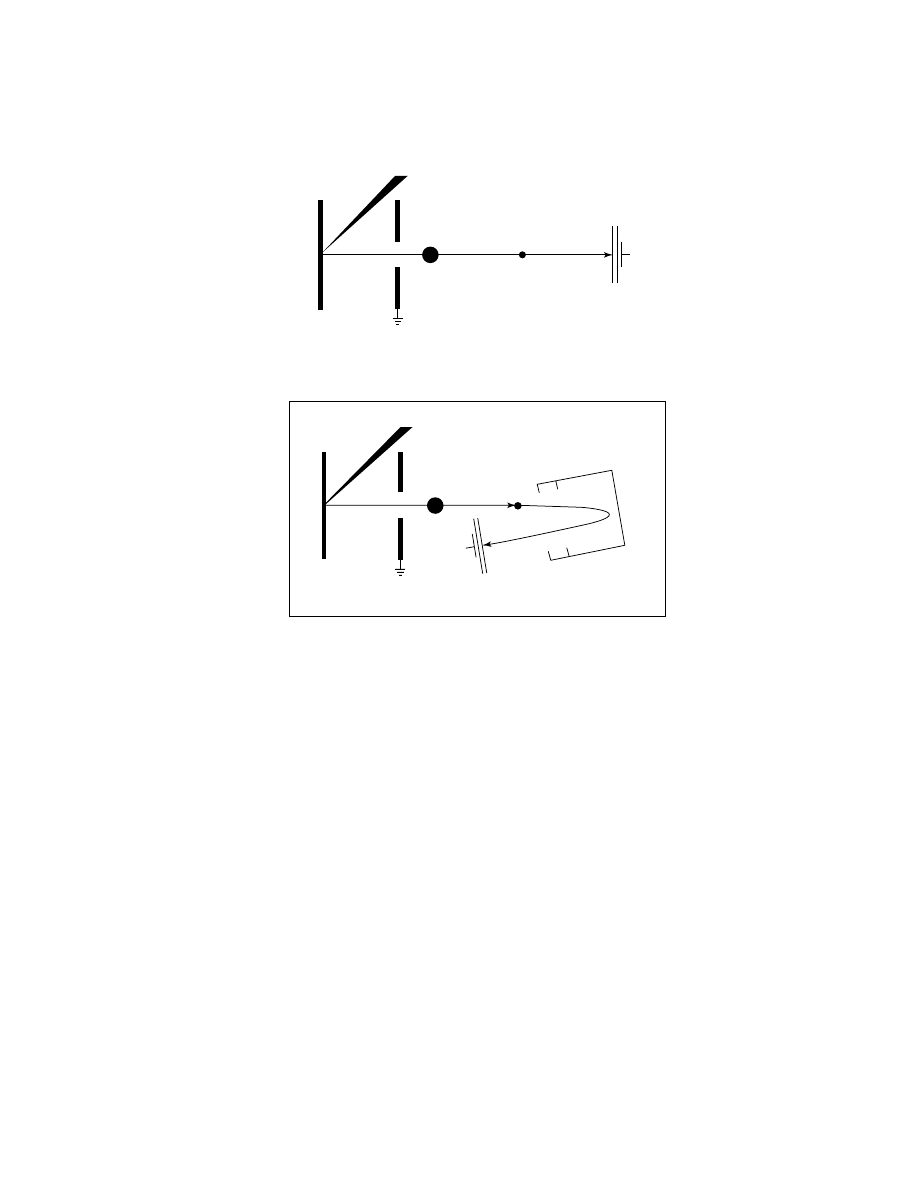
Detector
Drift region
Laser
Ion source
M
+
m
+
Fig. 1.
Schematic of linear tof-MALDI instrument.
Reflector
Detector
Ion source
Laser
m
+
M
+
Fig. 2.
Schematic of reflectron tof-MALDI instrument.
involves a way to get the analyte into the gas phase and a way to ionize it. For both
MALDI and esi, these events occur in the same process; for other ms techniques
used on lower mass molecules, this is not necessarily the same process; (2) The
separation of the analytes by mass or, more correctly, by m/z; and (3) The detection
of the ions.
Herein will be considered the MALDI-tof-ms with a microchannel plate
(MCP) detector (see Fig. 1 for the schematic of a linear MALDI-tof-ms and Fig. 2
for the schematic of a reflectron MALDI-tof-ms). This is currently the instrument
most commonly used to analyze synthetic polymers.
Introduction of the Polymer Molecule into the Gas Phase and Ion
Formation.
Since the advent of lasers, it has been hoped that with very short
laser pulses of an appropriately chosen wavelength, energy could be transferred
into the translational modes of the polymer molecule in such a rapid way that the
molecule would go into the gas phase before it broke apart (fragmented). Although
there had been numerous tries (7), only small polymers with masses less than 2000
or so could be gotten into the gas phase without significant fragmentation (16,17).
These methods of laser desorption or field desorption are described in more detail
in the previous encyclopedia article and recent books (3,4,18). Since these methods
are limited to low molecular mass polymers they will not be considered here.

Vol. 3
MASS SPECTROMETRY
163
The MALDI Process.
The MALDI process is the ablation of the analyte
cocrystallized with an organic small molecule matrix, most commonly an organic
acid. The process (11) initially used and also in common use today is as follows:
A polymer (biological or synthetic) is cocrystallized or co-mixed with the matrix
molecule in the solid phase. A short uv or ir laser pulse is used to ablate the
matrix and the analyte mixture creating a plume of matrix and analyte species.
The ablation process involves the uv or ir absorption by the matrix molecule. The
laser energy excites the matrix molecule, causing it to vaporize and decompose
into what is now considered a supersonic phase transformation (19). This process
excites many modes of the matrix and transfers the energy from electronic or
vibrational modes into translational modes in the matrix. For MALDI-tof-ms, the
most commonly used laser is the 337-nm wavelength uv nitrogen laser. This laser
has a pulse width of about 3 ns and generally performs well in the ablation of the
matrices commonly used.
The laser energy per unit area is a variable that one can adjust with a vari-
able absorbing neutral density filter. The ablation energy per unit area is usually
adjusted to the lowest level that will produce polymer ions. This keeps the ini-
tial energy of the plume lower. Lower initial plume energy results in lower initial
velocity distributions of the molecular ions, minimizing broadening and reducing
the fragmentation of the polymer.
The matrix is selected to absorb most of the energy; few (if any) electronic
states of the analyte (the polymer) are excited directly. However, since the analyte
is intimately mixed with the matrix, the analyte is carried into the gas phase as
a consequence of the phase transformation in the matrix. The analyte, the matrix
molecules, clusters of matrix molecules, cations and cation clusters, as well as
various combinations of each of these species have been detected in the plume
from this laser ablation process (21–23).
For biopolymers, it is thought that the biopolymer is protonated by the many
protons released in the plume and thus charged as a positive ion (4). For synthetic
polymers, only those which have labile protons are easily charged in the same
way (like polyacrylic acid and polystyrene sulfonate). Most synthetic polymers
are charged by the addition of a metal ion. In some cases, adventitious metal ions
are present in the matrix at concentrations sufficient to allow for their addition to
the polymer in the gas phase. (Careful analysis of polymers of known end groups
allows unambiguous identification of the attached metal cation.) In other cases,
the ions have to be added to the matrix–polymer mixture to give enough of the
proper ion to attach to the polymer. Generally, polar polymers are charged by
alkali metal ions. These are either added to the solutions used to create the solid
matrix analyte mix or often exist as impurities, either from the glassware or
solvent used in the sample preparation or in the matrix material. For nonpolar
polymers containing double bonds, transition-metal salts with Ag or Cu cations
are added to the solution containing the matrix analyte mixture, and these ions
charge the polymer in the gas phase. For reasons not completely understood at
this time, regardless of the normal stable charged state of the metal ion ie,
+1,+2,
or
+3, the MALDI process generally favors only singly charged n-mers in the
gas phase (24). This generalization does not hold for very large molecular mass
where multiply charged synthetic polymers can often be seen (1). Once charged,
the metal polymer species is then accelerated into the separation region of the ms.

164
MASS SPECTROMETRY
Vol. 3
Other Soft Ionization Processes for Large Molecules.
Electrospray ion-
ization creates ions by a potential difference between a metal capillary tube and
the inlet to the mass spectrometer. The electric field generates charged droplets in
the form of a fine mist (see Ref. 25). Application of a drying gas or heat evaporates
the solvent, reducing the droplet size and eventually resulting in the formation of
molecular ions.
Truly monodispersed polymers like large biomolecules may be examined by
esi-ms. These samples typically show a distribution of multiply charged molecules
and no evidence of fragmentation. The multiple charging process has the effect of
lowering the m/z values to a range easily measured by quadrupole ms, molecular
mass typically in the range 500–2500. The molecular mass of biopolymers can be
calculated since more than one charged state is observed. However, the multiple
charging of esi is a drawback for synthetic polymers. Here, there are many n-mers
even with a synthetic polymer having a narrow MMD. Each n-mer in the MMD
forms many multiply charged species. These multiply charged species for each
n-mer together with the distribution of n-mers results in an m/z spectrum that
requires a resolution often not available in many mass spectrometers to separate
all the species present. Because esi-ms uses solutions of the polymer as the sample,
esi-ms is easily connected to liquid chromatography systems, such as hplc or sec.
The coupling of esi with chromatographic process is often used to deconvolute the
complex spectra described above (26).
The Separation Process.
Once the analyte is in the gas phase as an
ion, analytes must be separated on the basis of mass to charge (m/z). A variety
of separation techniques are available. The most common form of ms separation
is the quadrupole mass filter (3,4). Because this method is limited to m/z less
than 3000 or so, it is not generally used with MALDI. However, a quadrupole is
commonly used with esi where multiply charged analytes allow one to examine
high masses for m/z less than 3000.
Fourier transform mass spectrometry (ftms) is an ion cyclotron resonance
method (3,4). This method is currently limited to masses less than 20,000. Thus,
it works well for lower molecular mass polymers. Often it is used with MALDI.
However, the MALDI process is so energetic that the initial velocity of the analytes
can cause some intensity difficulties that make the method difficult to quantify.
Because of the expense of the magnet required to obtain quality data with this
method, only a few laboratories use this method.
For MALDI, the most common mass separation technique is tof-ms. In its
simplest form, the ions in the MALDI plume are accelerated by a high voltage
(often as high as 25 kV) for a distance of a few millimeters during which the ions
obtain a velocity v. The accelerated ions drift in an evacuated tube, typically about
a meter long, at this velocity. (Some instruments have flight tubes as long as 6 m).
The equation describing this simple process is
zeV
= 1/2mv
2
(1)
where V is the electric potential applied to accelerate an ion of charge ze. Once
in the drift tube the translational energy of the ion is given by the r.h.s. of the
equation. There is a correction for the initial velocity of the particle in equation
(1) but if the field is large enough, this is a small correction (18,27). If the drift

Vol. 3
MASS SPECTROMETRY
165
tube is long compared to the acceleration region, then v
= L/(t − t
0
) where L is the
length of the drift tube and t
− t
0
is the time from some arbitrary zero time t
0
set
by the arrival of the ablating laser pulse. Thus, we have
m
/z = 2eV(t − t
0
)
2
/L
2
(2)
or
m
/z = a(t − t
0
)
2
(3)
Equation (3) is the equation relating mass and charge to time, which is used as
the general calibration for a tof-ms instrument. Often some small corrections are
added to equation (3) (18,28) for the final tof calibration equation.
In current instruments, two modifications from a simple tof instrument are
included. One is the reflectron, a set of ion mirrors which slow the ions down,
reverses their direction, and by increasing the actual length of the flight path,
increases the resolution. See Figure 2 for a schematic of an instrument with a
reflectron. By appropriate choice of reflectron voltages and shape of the voltage
fields, one can improve resolution even more. This is described in detail in other
references (18). The overall effect of the reflectron is to make little change in
equation (2) for use as a calibration equation.
Although the reflectron improves the resolution of the instrument, there can
be loss of signal because of ion optics as well as late fragmentation. Fragmen-
tation that occurs while the molecule is in the linear flight tube is considered
late fragmentation. (Early fragmentation is fragmentation in the initial plume.)
Molecules that fragment in the late fragmentation process will have the proper
kinetic energy and arrive at the correct time at the detector in the linear region.
Once in the reflectron, these ions are reacclerated with the mass of the charged
fragment. Thus, they arrive at the reflectron detector at the “wrong” time.
The second modification is called delayed extraction. In earlier instruments,
a single high voltage accelerating electric field was continuously applied while the
plume was formed by laser ablation. Instruments with delayed extraction have
a second field plate located a few millimeters away from the original field plate.
The second field plate is energized for a brief time, keeping the plume trapped for
a short time (hundreds of nanoseconds), between the two field plates. The second
field is then suddenly dropped to zero potential, accelerating the ions from the
contained plume. This brief containment of the analyte plume allows it to come to
some modest equilibration, decreasing the range of initial translational velocities
and thus improving the resolution of the final spectra (27,28).
Detectors.
Several detectors are available for tof-ms instruments. An op-
timum detector should show no ion velocity dependence. The detector should not
saturate or should have a sufficiently rapid recovery rate on the time scale of the
smallest collection time interval. No current detector satisfies these requirements.
Many of the most common detectors have been discussed (3).
The MCP detector is the most commonly used on instruments employed for
the analysis of synthetic polymers. The MCP detector is made up of an assembly
of lead glass capillaries coated on the inside with electron-emissive materials and
fused together. The capillaries are biased by a high voltage. Ions strike the inside

166
MASS SPECTROMETRY
Vol. 3
wall, creating secondary electrons which amplify each ion impact signal with a
gain of 10
3
or 10
4
. Higher gains are obtained with two MCP detectors in series.
In a study (29) of differences in signals from two detectors, using the same tof
instrument, equimass blends of narrow PMMA standards were used to simulate
a wide polydispersity polymer and it was shown that different detection systems
produced different MMD for the polymer blend using the same sample preparation
method and the same analyzer conditions. The differences arose from detection
mechanisms, saturation effects in the detector, and signal to noise problems.
Matrices for the MALDI Process.
The matrix is crucial to the MALDI
process. It is generally believed that how the polymer is situated in the matrix has
a significant influence on the success of the MALDI process. Liquid matrices have
been used in a number of applications for biopolymers but only a few applications
for synthetic polymers. Solid matrices are most common for both biopolymers and
synthetic polymer work. The earliest experiments (11) that described the MALDI
technique employed solid matrices.
Liquid Matrices.
Liquid matrices have the advantage that they present a
homogeneous material for the polymer to reside in (in contrast to the crystal of a
solid matrix which is discussed in a later section) and thus show less variation in
signal as the laser probe is moved across the sample compared to use of a solid
matrix with hand spotting (30). Furthermore, with a liquid matrix it is more likely
that one can detect precipitation of the polymer sample in the sample preparation.
Such an observation cannot be made during the solvent evaporation step of the
solid matrix polymer preparation.
The major disadvantage of the liquid matrix is high vapor pressure of the
liquid, often too high for the vacuum systems of many current tof instruments. The
use of atmospheric pressure ms in MALDI analyses (31) may lead to the increased
use of liquid matrices. Furthermore, the liquid matrices seem to have poorer mass
resolution than the same polymer in solid matrices. Finally, many instruments are
designed with vertical sample holders, which are not impossible to work with but
certainly this sample alignment does not favor the use of these matrices. Little
work has been done on this promising method of sample introduction.
m-Nitrobenzyl alcohol (m-NBA) and 2-nitrophenyl octyl ether (o-NPOE) are
commonly used MALDI liquid matrices. These substances were originally used as
fast ion bombardment (FAB) ms (3,4) liquid matrices. Liquid systems with liquid
mediators and uv absorbers, some of which were normal solid matrices, have been
developed (30). These gave excellent sodium cationized PEO spectra. A glycerol
mediator with sodium and potassium ferrocyanide as the matrix absorber has
been used (32,33).
Solid Matrices.
Most work on synthetic polymers use the solid matrices de-
veloped for the biopolymer analysis. To use these matrices, solutions of polymer,
matrix, and cationizing salt are mixed. The solvent is then allowed to evaporate
from these solutions deposited onto a sample surface. The mass proportion ratios
of the matrix:polymer:salt in the final solid mixture cover the range of 5:1:2 to
2000:1:1. These proportions are often dependent on molecular mass of the poly-
mer (1). The choice of matrix compounds for synthetic polymers with respect to
the polarity of the polymer have been discussed (34). As a general rule, matrix
polarity should be matched with the polarity of the polymer so that both are solu-
ble in a common solvent. Since the MALDI sample preparation requires intimate

Vol. 3
MASS SPECTROMETRY
167
mixing of matrix, analyte, and salt, this choice will assure the best co-mixing of
the final solid mixture of the analyte and the matrix. The following table for the
appropriate choice of matrix and polymer in terms of solvent strength may be
helpful. The arrangement in the table is more hydrophilic at the top proceeding
to more hydrophilic.
Matrix
Polymer
Thiourea
Poly(ethylene glycol)
Dihydrobenzoic acid
Poly(propylene oxide)
Cyanohydrobenzoic acid
Poly(vinyl acetate)
Ferulic acid
Poly(tetramethylene glycol)
Indol acrylic acid
Poly(methyl methacrylate)
Dithranol
Polystyrene
Retinoic acid
Polybutadiene
Diphenyl butadiene
Polydimethylsiloxane
Although this table provides a good first rule for the choice of various matrices
for each polymers, its usefulness is often overridden by the sample preparation
methods described later in this review.
A recent review (20) of MALDI of synthetic polymers offers an extensive
reference list of polymers and the appropriate matrix to use with them.
Sample Preparation Methods for Solid Matrices. Sample preparation is crit-
ical to MALDI using solid matrices. The presumption is that the polymer and the
salt must be well dispersed in the final matrix mixture to achieve a one-to-one
representation of the polymer MMD in the solution to the polymer MMD in the
gas phase. Yet, the matrix is commonly crystalline and the polymer may be either
semicrystalline, like PEO, or glassy, like atactic polystyrene. Kinetic processes
occurring during the loss of solvent from the solution of the mixture of matrix,
salt, and polymer must occur either to co-crystallize the polymer with the matrix
and salt or to embed the polymer in the defect structure of the organic matrix. To
obtain the correct representation of the MMD in the ms, each n-mer in the MMD
must occur in the ms in proportion to its appearance in the original MMD.
The intimate dispersion of the salt in the matrix is also important (35,36);
if the salt is not well dispersed, there may be a loss of signal. The matrix is often
an organic acid. By making the salt of the matrix acid and mixing this with the
parent matrix acid, increased polymer signal is obtained. Too much salt, however,
leads to no polymer signal since matrix salt alone is often not a good matrix for
the polymer.
Generally, there are two common approaches to sample deposition onto the
target surface, hand-spotting and electrospraying. In one approach, the solutions
described above are hand-spotted from a microliter pipette onto a target plate;
0.5–2
µL of solution are used to deposit micrograms of polymer, matrix, and salt
mixtures onto the plate. The solvent is allowed to evaporate rapidly (often with
help from a fan or heating or by drawing the pipette tip across the plate, spread-
ing the solvent out). One usually obtains crystals of the matrix. This is called
“hand-spotting” or the “air-dried droplet technique.” The advantage to this method
is that it requires little additional equipment. However, the samples have large

168
MASS SPECTROMETRY
Vol. 3
signal variations across the target plate; one finds areas of large polymer signal,
“a sweet spot,” and other regions where no polymer signal is found. This inconsis-
tency across the sample is reduced somewhat by a variety of modifications of the
hand-spotting method. In a procedure used by various workers (37,38), the matrix
crystals are crushed with a spatula. This leads to additional sample homogeneity.
Others have examined the different modifications of the hand-spotting technique
(39).
However, in general, hand-spotting leads to a relative standard deviation of
40% in the total signal intensity variation across the target surface. The electro-
spraying method of sample preparation, described below, can produce targets that
exhibit less than 5% relative standard deviation of total signal intensity across
the target surface (40).
The same solutions that are used for hand-spotting are used in the electro-
spray technique. These solutions, after mixing, are then drawn into a microliter
syringe that is placed into a syringe pump. The needle of the syringe is held at a
potential between 3 kV and 7 kV against the sample target as ground. When the
solution is delivered at 2–20
µL/min, a fine spray of charged droplets is delivered
out of the needle. The sprayed solvent evaporates from the droplets, and the poly-
mer:salt:matrix mixture is deposited on the sample plate nearly dry (40,41). This
procedure keeps the crystals of the matrix small (ca 2–5
µm diameter) (35,40) and
the polymer matrix and salt in an intimate mixture.
The signal from electrospray sample deposition shows little shot-to-shot vari-
ability as long as the sample depth is thicker than the amount of sample that can
be ablated by a few laser shots at the same location of the target (42). This method-
ology also shows good shot-to-shot repeatability for biopolymers (41).
Choice of Salts. Synthetic polymers generally obtain a charge by a different
mechanism than biopolymers. Most biopolymers obtain a charge by addition or
loss of a proton from the parent molecule. This is because biopolymers have acid
groups available to them. For synthetic polymers like polystyrene sulfonic acid,
this mode is also available. However, most synthetic polymers do not contain labile
protons. Those synthetic polymers that are polar or contain double bonds can be
cationized, however. The polar polymer molecules require a metal ion, often an
alkali like Na
+
or K
+
, to attach to the polymer n-mers to obtain a charge. The
Na and K metal ions are usually found in the matrix itself or introduced through
the glassware used in sample preparation. For example, with PEO or PMMA, the
common matrices used for these polymers have enough Na and K so that one
commonly sees ions with both Na and K attachment without the addition of any
Na or K salt. One can also add salts to the sample preparation solutions causing
these salts to attach to the polymer n-mers. For polymers with double bonds,
polybutadiene or polystyrene for example, one normally has to add Ag or Cu salts.
These metal salts are found to work best, although other salts also give spectra
(43). In fact, some matrices give spectra for these polymers with Na or K salts but
the quality of the spectra is poor compared to spectra obtained with transition-
metal salts. Polymers without polarity or double bond, for example polyethylene or
polypropylene, have difficulty being cationized. Only a few papers report MALDI
spectra on these saturated hydrocarbon polymers (44,45).
Commonly, salts that are soluble in the same solvent as the polymer and
the matrix are added to the polymer matrix solvent mix. Nitrate, chlorides, etc

Vol. 3
MASS SPECTROMETRY
169
are commonly tried, but these do not easily dissolve in the organic solvents com-
monly used (tetrahydrofuran, acetone, chloroform). One often uses trifluoroacetic
acid (TFA) salts; for example, Ag TFA or CuTFA are commonly used to cationize
polystyrene. Only one paper has reported any effect of the anion on the cationiza-
tion process (46).
Other materials have been used to cationize the polymers. Metallocenes (fer-
rocene, nickelocene, and cobaltocene) have been used as cationizing agents for
polystyrene and PEO (47). These cations have the advantage that they are solu-
ble in many organic solvents and also do not have the isotopic splitting seen with
Ag, which often complicates the interpretation of the MALDI spectra.
Quantification of MALDI for MMD and Moments of MMD
Mass Axis Quantification.
Mass axis quantification is most easily done.
Calibration of most tof instruments is usually done with biopolymers of known
molecular masses. These biolpolymer are selected because they typically provide
a single major peak whose mass is known accurately; thus, mass axis quantifi-
cation is quite straightforward. Calibration usually can be done using three of
these biopolymers. Collecting data with 2-ns time intervals, one can get better
than single-unit resolution on an instrument with a 1.5-m flight tube in reflec-
tron mode at about 7000. Good mass resolution and calibration is required for
end group determination, copolymer composition determination, and for polymer
architecture studies.
Signal Axis Quantification.
For synthetic polymers, signal axis quan-
tification is of utmost importance to obtain a good representation of the MMD or
MCD or to compute the moments of the MMD to compare with classical polymer
characterization methods like light scattering or membrane osmometry.
The signal axis may be in error due to a variety of reasons. Many of these
issues have been discussed (48). Most of the issues related to uncertainties arising
from sample preparation have been discussed in some detail in the earlier sections
on matrices and sample preparation.
Uncertainties Arising from Ablation, Ionization, and Drift Regions.
Little
is known about the desorption/ionization process in MALDI. Work on the same
type of polymer of different molecular masses suggests that higher ablation ener-
gies are required for polymers of higher molecular masses. Thus, since the normal
procedure is to adjust the laser ablation energy to just above the threshold needed
to obtain the polymer spectra in MALDI, the desorption process of MALDI could
also cause the moments determined by mass spectrometry to be lower than the
true moment. It is not known whether low mass molecules desorb better than
higher mass molecules, but, if preferential desorption of low mass molecules oc-
curs, it could account for the low MALDI values often reported when the moments
from MALDI are compared to those obtained by classical methods (49–53).
For homopolymers, the dominant polymer repeat units commonly show high
signal intensity. Also, less intense intermediate peaks may be seen adjacent to
the major high intensity repeat units. These intermediate peaks more often than
not have the same repeat unit as the major peaks. These intermediate peaks may
arise from a variety of sources. It is most obvious that end groups or cations may
170
MASS SPECTROMETRY
Vol. 3
differ. One can have two different sets of end groups on linear chains or many
different types of end groups on branched chains. This gives rise to a main group
series of peaks arising from the major pair of end groups and a second series of
peaks from the second set of end groups.
One may see additional peaks from different metal cations attaching to the
polymer. As noted before for polar polymers like PEO and PMMA, one does not
need to add salts to the matrix. Impurities in the matrix in the form of Na or
K seem to be in sufficient concentration to cause the addition of Na or K to the
polymers. For example, with PMMA it is not uncommon to see peaks separated by
16 u, the difference between the molecular mass of Na and K. This peak intensity
will change by the addition of one or the other metal ion. All the above peaks will
appear in both the linear and reflectron mode.
Some intermediate peaks will often appear differently in linear and reflec-
tron modes for reasons other than sensitivity differences between the two modes.
If the reflectron spectra is different from the linear spectra, this may be due to
fragmentation during the flight down the linear flight tube. Of course, fragmen-
tation occurring in the ablation region (the source region) cannot be detected this
way. Ablation region fragmentation may be detected by the appearance of more
n-mers in the low mass regions of the MMD as the laser energy is increased.
Finally, the intermediate peaks may arise from matrix adducts to the poly-
mer. Matrix adducts of polystyrene polymers of molecular mass 3900 and 7000
of the form MatrixAgPolymer and MatrixAgAgPolymer have been found (21).
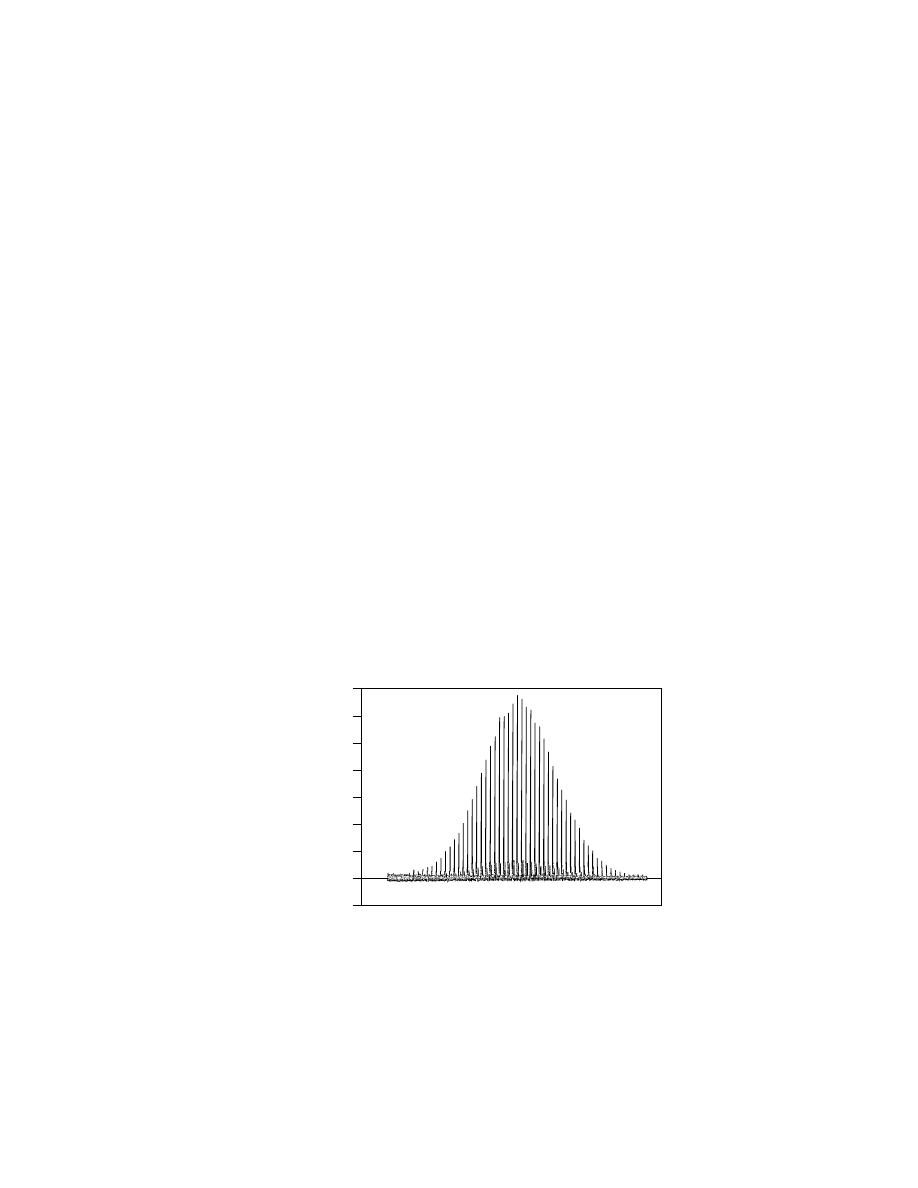
Figure 3 shows the entire spectrum, and Figure 4 shows these adduct peaks in
a polystyrene of molecular mass of about 7000 used in the NIST Interlaboratory
Comparison (49,54).
Uncertainties Arising from the Detector.
The detector is another influence
on the MMD that may impact the moments calculated by MALDI-tof-ms. The
3000
2000
−2000
4000
6000
8000
10000
12000
14000
0
5000
7000
9000
m/z
Arbitr
ar
y intensity
Fig. 3.
MALDI-tof-ms of polystyrene around 7000 molecular mass. This MALDI spectrum
was generated using dithranol as the matrix and AgTFA as the salt. The polystyrene,
dithranol, and salt were dissolved in tetrahydrofuran. The solution was then electrosprayed
onto the sample plate. The polymer analyzed in reflectron mode with an extraction voltage
of about 25 keV.
Vol. 3
MASS SPECTROMETRY
171
6100
5900
5700
6300
6500
1400
1000
600
200
−200
m/z
Arbitr
ar
y intensity
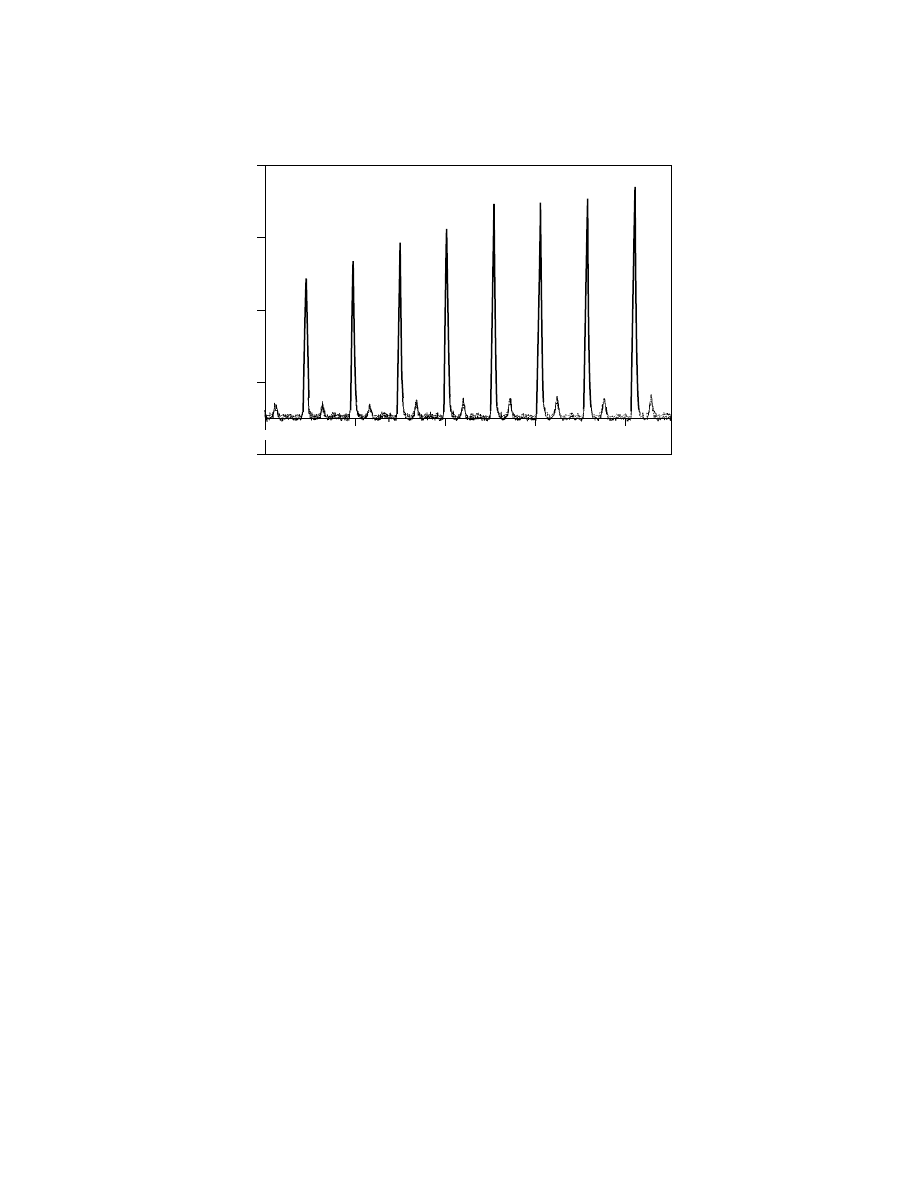
Fig. 4.
Detail of MALDI-tof-ms of polystyrene polymer shown in Figure 3. The smaller
peaks between the main peaks have been shown (21) to be the result of matrix adducts. This
is an example where these intermediate peaks are not from polymer molecules terminated
with other end groups or from n-mers charged with different salts.
detector in most of the instruments is an MCP detector. The smaller molecular
mass molecules hit the detector first, and the larger molecules follow later. If
there is saturation occurring in the detector, then there would be a discrimination
against the high mass molecules. The issue of detector saturation has been dis-
cussed by many authors (51,55,56). Since the matrix molecules or low molecular
mass polystyrene molecules could saturate the detector before the higher molec-
ular mass molecules arrive at the detector, this would cause the computation of a
lower M
n
and M
w
than the true values.
The detector may also be less sensitive to larger mass polymer molecules
because of the way the ions are detected. MCP detectors count the number of
ions by an ion-to-electron conversion when the ionized polymer collides with the
detector plate. A bias occurs against the high mass species when the ion-to-electron
conversion is diminished due to a decrease in the impact velocity of the larger
ionic species (51,57). If a discrimination against the high mass polymer molecules
exists, this would cause the estimated moments of the distribution to be less than
the true value of the moment.
Uncertainties from Data Analysis.
Baselines drawn in MALDI-tof-ms are
similar to that of a spectroscopic or chromatographic system where a many-point
baseline must be drawn. Baseline corrections may be a cause of the disagreement
between the moments determined by classical methods and those determined by
MALDI. There is much more noise in the low mass end of the spectra obtained by
MALDI-ms. Some of this noise may arise from fragments of the polymer, clusters
of matrix and salt, or metal clusters. If the baseline correction does not account for
this difference in baseline noise, then these data may overweight the lower part

172
MASS SPECTROMETRY
Vol. 3
of the MMD and thus the calculated M
n
and M
w
would be expected to be lower
than the true moments.
Assuming a correct baseline is drawn, the question arises as to how to com-
pare the MMD estimated from a tof mass spectrometer to that obtained from
size exclusion chromatography (sec) using a UV or differential refractive index
(DRI) detector. A sec is the instrument most commonly used to obtain the MMD
for synthetic polymers. The most obvious difference between the two techniques
is that the tof detector counts numbers of ions of n-mers while the DRI or UV
detectors on sec instruments measures mass concentration of the polymer. The
mass concentration of the polymer is proportional to the product of the molecular
mass of the n-mer and the number of molecules of that n-mer per unit volume.
Thus, in the ms we obtain the number MMD while in the sec we normally obtain
the mass MMD. Furthermore, the raw signal of each must be corrected for the
transformation from time to mass, which is different for each instrument. This
transformation has been discussed in detail (58).
Finally, there is a problem of the detection for higher n-mer masses in
MALDI. Since the detector is an ion detector, a counter of the number of n-mers,
peaks disappear into the noise before peaks would disappear into the noise on
a normal sec detector, which detects the total mass associated with each n-mer.
The consequences of this signal-to-noise problem on the polydispersity (M
w
/M
n
)
studied by MALDI-tof-ms has been discussed (59).
The above discussion assumes that the polymer is a linear homopolymer. For
branched polymer there is not a simple direct relationship of elution volume in
sec to molecular mass. For a copolymer, the signal differs in both the DRI or the
uv detector for different groups in the copolymer. Further, it is usually assumed
that there is no effect of molecular mass on the DRI or uv response. Although this
is the case for high molecular mass species, as we go to very short chains the DRI
and uv are affected by the presence of end groups. This is discussed in more detail
in the section on narrow MMD.
The effect of instrumental broadening on the sec data has not been discussed.
For a polymer with little polydispersity the chromatographic broadening is impor-
tant in sec and may lead to prediction of larger polydispersities than those found
by MALDI. Some measurement condition differences which cause effects greater
than those between the mass and number MMDs have been studied (60).
Applications of MALDI-tof-ms
Narrow MMD Homopolymers.
Generally, most laboratories obtained
reasonable agreement for narrow MMD polymers between moments of MMD ob-
tained from MALDI and those obtained by classical methods, eg light scattering
to obtain M
w
and end group analysis or osmometry to obtain M
n
or sec. Good
agreement between classical methods and MALDI for polymers with M
w
/M
n
<
1.2 has been observed (61,62). A study (63) of PMMAs by sec and MALDI support
this view. A careful study of polystyrene of molecular mass 5050, 7000, and 11,600
and blends of the three has been done (48). Using the polystyrene 5050 and 11,700
and studying changes in the MMD of the polymer of 7000 by blends, showed no
systematic uncertainties within 0.5% in the MMD or the moments of the MMD.

Vol. 3
MASS SPECTROMETRY
173
A MALDI-tof-ms interlaboratory comparison was conducted by NIST among
23 laboratories using MALDI-tof-ms on a well-characterized polystyrene to assess
the robustness of the method to determine the MMD (49). An example of a MALDI-
tof-ms spectrum from this polymer is given in Figure 3. The polystyrene was
synthesized to have a tertiary butyl end group at one end and a proton at the other.
An nmr characterization of M
n
was found to give 7050
± 400. The M
w
was found
to be 7300
± 600 by light scattering. The expanded uncertainty values following
the “
±” represent both type A uncertainties (statistical uncertainty) and type B
uncertainties (systematic uncertainties) (64). An ftir spectroscopy confirmed the
presence of a single pair of end groups and no other end groups in measurable
amounts.
Each participating laboratory was asked to perform MALDI-ms using two
distinct protocols. Each laboratory was asked to do three repeats of each protocol
to check for intralaboratory variability.
By compiling all the returned data using all protocols into one preliminary
analysis, it was found that MALDI-ms returned an M
n
of 6600
± 100 and an
M
w
of 6700
± 90. The uncertainty values following the “±” represent only type A
standard uncertainties (statistical uncertainty). These M
w
and M
n
obtained from
MALDI were below the M
w
and M
n
of the classical methods but still within the
overlapping uncertainty ranges. The statistical uncertainty in the ms measure-
ments was very small, indicating that from laboratory to laboratory, reproducibil-
ity was extremely good.
In a few reported cases, the agreement for narrow distribution polymers has
not been good (52). This can generally be attributed to the failure of the classical
method at low molecular mass. For example, calibration of sec at low molecular
mass is at best difficult. Furthermore, the effect of end groups on the detector
response is often not taken into account nor is the effect of broadening on the
computed moments properly taken into account at the lower molecular masses.
For example, the DRI detector requires the dn/dc be constant, independent of
molecular mass for the calculation performed by most current software programs.
But dn/dc is known to be affected by end groups (65).
dn
/dc = a + b/M
(4)
For most polymers, the second term on the r.h.s. has a contribution below
100 repeat units or molecular mass less than 10,000 for polystyrene.
Generally, the effect of not correcting for broadening is to increase the appar-
ent values of sec values of M
n
and M
w
. This effect may be seen by comparing the
moments calculated from sec and from MALDI in the data (63) on narrow MMD
polymers.
Broad MMD Homopolymers.
Early in the studies on MALDI of synthetic
polymers, it was hoped that MALDI could be used successfully for both narrow
and broad MMD. Soon it was discovered that for a variety of reasons the correct
MMD of broad MMD polymers could not be obtained in one step by MALDI-tof-ms.
Good agreement between MALDI and gpc for MMD moments for polymers with
polydispersities less than M
w
/M
n
= 1.1–1.2 was found (50). Polymers with higher
polydispersities regularly showed incorrect moments of the MMD (50,66).
A number of authors used the procedure of simulating a broad MMD by
preparing a multicomponent blend of a number of narrow fractions to mimic a

174
MASS SPECTROMETRY
Vol. 3
broad MMD. This way of controlling the amount of each part of the MMD allowed
them to show that they do not get the correct representation of the expected
MMD from the mixed systems. This procedure also provides a method to examine
the various causes of this problem, including instrumental effects (55), sample
preparation (67), desorption/ionization issues (51), and laser power (68).
None of these studies have lead to significant improvements in doing the
MALDI-ms of polymers with high polydispersities. Generally, this problem has
been overcome by coupling MALDI-tof-ms to various chromatographic methods to
separate polymers.
MALDI as a Chromatography Detector.
The sec is the most commonly
used chromatography technique for analysis of polymers. Two common approaches
have been used to obtain a MMD from the combination of MALDI and sec. In one
case, one fractionates the polymer using sec, collecting each fraction and obtaining
the MALDI on each fraction. One then must combine the data from each MALDI
spectra to create the MMD on the polymer. This is done by summing each MALDI
chromatogram used in obtaining the moments to obtain M
n
and M
w
and the poly-
dispersity (69). This technique is thought to suffer some of the discrimination
issues brought out in the discussion in the broad MMD section (20).
In another method, fractions are taken from the sec at well-defined elution
volumes. A MALDI analysis of these fractions is made and the peak of the mass
MMD M
p
is computed from the MALDI data on the fraction. The M
p
so obtained
is then used to calibrate the sec. The calibrated sec is then run to get the exact
MMD of the polymer (70). A number of authors have used this technique. Two
limitations of this technique should be mentioned. The first problem arises be-
cause one must have relatively narrow mass fractions in each sec fraction used
for this to work. Thus, linear or lightly branched homopolymers or random or
alternating copolymers meet this criterion. Branched polymers or polymers with
rings or closed loops may show broad MMD fractions for narrow sec fractions.
This is because branched polymers or polymers with rings and loops can have the
same hydrodyamic volumes for molecules with widely varying masses (71–74).
Block copolymers in a solvent such that one block is extended and the other is col-
lapsed (with very different hydrodynamic volumes) may result in mass broadening
too. This effect will make it difficult to assign a mass to the broadened fraction by
MALDI and thus to get a good calibration for the sec columns. The second problem
comes from the assignment of the total mass of the material in each sec fraction.
For a copolymer, for example, a single detector may not be enough to assign the
total mass of different groups at a given molecular mass. Two detectors at least
may be required for copolymers.
In one case (71) a different phenomenon is observed: Poly(bisphenol A car-
bonate) terminated with hydroxyl groups undergoes self-association by hydrogen
bonding in solution and these molecular aggregates remain in the sec column. In
the application of the sec MALDI method, self-aggregation is suppressed in the
sample preparation for the MALDI analysis and thus a very broad molecular mass
for each MALDI appears. This is an extreme case where sec alone cannot provide
a narrow MMD for MALDI.
A number of papers discuss placing a MALDI-ms instrument in line with a
continuous chromatographic process (75,76). For example, a method called Aerosol
MALDI has been developed. In this method, elutant from an sec is mixed with

Vol. 3
MASS SPECTROMETRY
175
matrix prior to pneumatic nebulization, which sprays directly into a mass spec-
trometer. MALDI is performed on aerosol particles using a 355-nm laser. The
solvent is evaporated from the aerosol particles by passing the aerosol through a
heated tube. This technique has been used successfully with PEG of mass 1000 u
and polypropylene glycol also of mass 1000 u. Mass resolution with this method
is less than with normal MALDI. In addition, the problem with data analysis is
overwhelming. Baseline determinations, identification of peaks, and quantifica-
tion are all problems.
The sec method separates polymers on the basis of the polymer size alone. In
sec, only the interaction of a polymer which excludes it from the wall is operative.
An attractive interaction between the polymer in solution and the solid portion of
the porous separation medium can substantially affect molecular separation. The
attractive interaction counterbalances the exclusion effect until a compensation
point, “the adsorption theta point,” is obtained (77,78). Selective molecular sepa-
ration is lost at this compensation point. For an even greater attractive polymer
porous media interaction, the polymer becomes adsorbed onto the surface of the
porous media so that the higher molecular weight polymers exhibit an inhibited
flow through the porous media. The change from an excluding interaction to an at-
tractive interaction with the porous media can be affected by changing the solvent
strength by use of mixed solvents.
For block copolymers, one can choose a solvent system that will have one block
at the polymer compensation point and another block in the exclusion regime.
This chromatographic method has been used to examine the polyethylene
oxide-co-polymethylene (PM) block system (79). The solvent is chosen so that the
PEO is at its critical condition, ie, there is no separation with respect to the molec-
ular mass of the PEO, but there is separation with respect to PM. MALDI-tof-ms
was used as a detector on this system. Each separate PM peak was continuously
transferred onto a MALDI target. The fractionation was made into separate PM
peaks. Within each PM peak there is a MMD of PEO. This allowed the authors to
estimate both the MMD and MCD for the whole polymer.
End Groups.
Polymer end groups play an important role in determining
polymer properties. This is increasingly important as the molecular mass of the
polymer decreases. Low molecular mass polymers with well-defined end group
functionality are used as prepolymers for many important final polymer products,
ie, polyurethanes, epoxies, uv cure adhesives, and various other thermosets (qv).
For example, polyurethanes are commonly made by a three-step processing in
which the activity of the end groups is important in determining the final product
(see P
OLYURETHANES
).
Bulk functionality of prepolymers can be determined by nmr, uv–vis spec-
troscopy, or titration methods. The sec data from multiple detectors can sometimes
be used to obtain estimates of functionality, but this is often not available due to
the lack of difference of detector signal between the end group compared to the
central groups. Even if this is possible, sec does not allow one to separate mono-
from multifunctional end groups. Further, the calibration of sec at low molecu-
lar masses is, at best, difficult. MALDI-tof-ms offers a unique probe of the end
group functionality as a function of the molecular mass of the polymer itself. For
polymers of molecular masses below 10,000 or so, current commercially available
MALDI-tof-ms instruments can differentiate end groups differing by a few mass

176
MASS SPECTROMETRY
Vol. 3
units. With the sensitivity of ftms, one can achieve single mass unit end group
identification up to 20,000 (3).
PMMA with a variety of end groups was analyzed and it was shown that
the various end groups can be identified (80). A variety of PMMAs of industrial
relevance have also been examined and it was possible to distinguish many end
groups using “time-lag-focusing” (delayed extraction) for polymers with molecular
masses of 16,000 (81,82).
MALDI has been applied to the analysis of poly(alkylthiophenes) (PAT), a
rigid rod-type polymer (83), and it was possible to determine the end-group com-
positions of the PAT samples; H/H, H/Br, and Br/Br end groups were found. The
samples showed large variations in the presence of each end-group pair as a func-
tion of preparation conditions. Such end-group monitoring is necessary for the
development of new materials from these polymers. Further, for the polymers
with Br end groups, some end-group loss as a function of laser power was found,
with more fragmentation at higher laser powers.
Finally, the end groups in poly(bisphenol A carbonate) have been examined
(84). It was possible to distinguish among a variety of end groups for polymers with
masses up to 16,000. This work was done with the MALDI acting as an sec detector.
Copolymers.
MALDI-tof-ms of copolymers offers the opportunity to ob-
tain an MMD and MCD. However, to obtain an MCD the ms can only be done on
low mass copolymers. This is generally difficult because the number of n-mers sep-
arated by only a few mass units quickly becomes very high for copolymer composi-
tion near 50-50. If the mole faction of one of the copolymers is small, it is possible
to obtain single or nearly single n-mer resolution for the MCD of the copolymer.
As simple example, MALDI-tof-ms has been used to study the number of
α-
methyl styrene (
α-MeSty) repeat units in SRM 1487, a narrow MMD PMMA NIST
standard reference material of about 6300 g/mol (62). Here, the major copolymer
component is MMA and
α-MeSty is the minor component. The α-MeSty is, in fact,
part of the initiator for this polymer and the material is either a “MMA styrene
MMA” triblock copolymer or a “styrene MMA” diblock copolymer, with the
α-MeSty
block length containing 0–6
α-MeSty.
In a more comprehensive study, the copolymer systems polystyrene-block-
poly(
α-MeSty) and poly(α-MeSty)-block-poly(4-vinyl pyridine) were examined
(85–87). These copolymers have a much more complex MCD as a function of molec-
ular mass than the one just discussed (62), and the resulting MALDI-tof-ms spec-
tra reflect this complexity. In the work (87) on poly(
α-MeSty)-block-poly(4-vinyl
pyridine), polymers with different mole ratios of
α-MeSty and 4-vinyl pyridine
were studied. The composition of the maximum peak was used to start the anal-
ysis. The compositional assignment of this maximum peak was made from nmr
data and/or the initial chemical composition of the polymer reaction mixture. The
rest of the peaks were assigned by a procedure that references the composition of
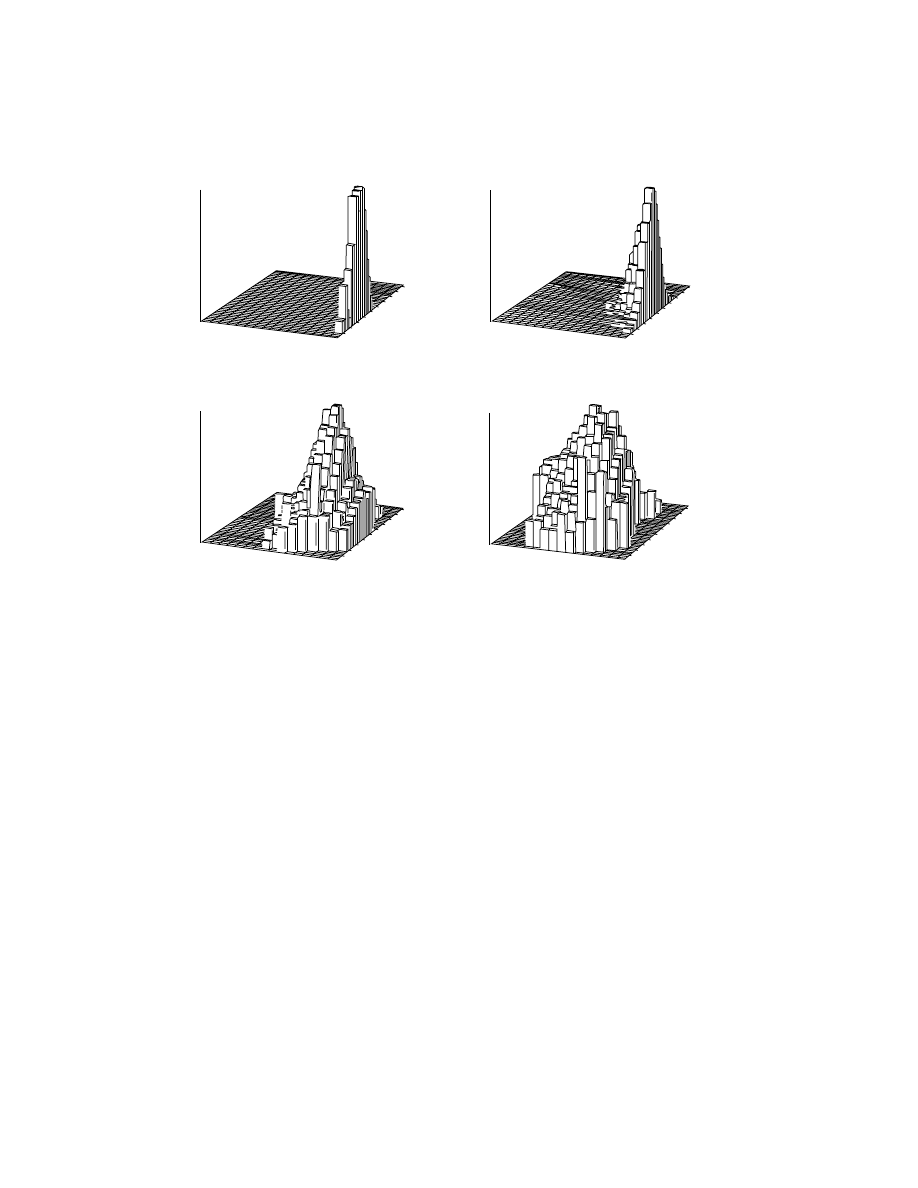
the main peak. From this analysis the two dimensional distribution of the di-block
copolymer was obtained as shown in Figure 5. Such use of collateral data shows
that ms alone may not be enough to obtain such a complex copolymer distribution.
However, with the copolymer composition distribution, a richer understanding of
the copolymer chemistry can be obtained.
In a study (85) on the copolymer system polystyrene-block-poly(
α-MeSty), it
was possible to confirm that the copolymer composition agrees with the predicted
Vol. 3
MASS SPECTROMETRY
177
(a) Sample PVP 1
(b) Sample PVP 2
(c) Sample PVP 3
(d) Sample PVP 4
relativ
e intensity
0.02
relativ
e intensity
0.005
1
3
5
7
9
11
13
15
0.04
relativ
e intensity
0.08
0.04
relativ
e intensity
0.010
0.008
1
3
5
7
9
11
13
15
1
3
5
7
9
11
13
15
1
3
5
7
9
11
13
15
A – 4 – vin
yl pyridine
A – 4 – vin
yl pyridine
A – 4 – vin
yl pyridine
A – 4 – vin
yl pyridine
4
B – meth
yl styrene
8 12
16
20
24
28
32
36
40
4
B – meth
yl styrene
8 12
16
20
24
28
32
36
40
4
B – meth
yl styrene
8 12
16
20
24
28
32
36
40
4
B – meth
yl styrene
8 12
16
20
24
28
32
36
40
Fig. 5.
MMD for one homopolymer and three copolymers of block copolymer of poly(
α-
MeSty)-block-poly(4-vinyl pyridine) as a function of mole fraction of poly(
α-MeSty). (a)
PVP1 is the MMD for poly(
α-MeSty) homopolymer; (b) PVP2 is the MMD and MCD of the
diblock copolymer poly(
α-MeSty)-block-poly(4-vinyl pyridine) with 0.9 ± 0.1 mole fraction
poly(
α-MeSty); (c) PVP3 is the MMD and MCD of the diblock copolymer poly(α-MeSty)-
block-poly(4-vinyl pyridine) with 0.63
± 0.1 mole fraction poly(α-MeSty); (d) PVP2 is the
MMD and MCD of the diblock copolymer poly(
α-MeSty)-block-poly(4-vinyl pyridine) with
0.45
± 1 mole fraction poly(α-MeSty). Reproduced from Ref. 87.
Zimm–Schulz distribution (88,89). Using an analysis method similar to that used
previously on the poly(
α-MeSty)-block-poly(4-vinyl pyridine) system, the MMD
of both parts of the copolymer were determined. The data analysis method was
claimed to verify the random coupling hypotheses. The hypothesis (90) that the
polydispersity of individual blocks is higher than the polydispersity of the whole
polymer was confirmed (85). That is, block copolymers with narrow MMD have
broad complex chemical composition distribution.
The composition and sequence distribution of poly(butyleneadipate-co-
butyleneterephthalate) have been studied (91). The intensity of the ms peaks
was compared with theoretical intensities obtained using a Bernoulli distribution
of the sequences. This result is in excellent agreement with nmr analysis of the
bulk polymer.
Many other authors have shown MALDI-tof-ms of copolymers, but have
not offered analysis to obtain a composition distribution as have the previous

178
MASS SPECTROMETRY
Vol. 3
authors. For example, the alternating copolymers of polycarbonate and two ran-
dom copolyethers used as nonlinear optical materials were examined (92). Poly(p-
phenylene ethynylene)-block-poly(ethylene oxide) has been studied (93). Also,
styrene-block-isoprene copolymers have been examined (94). Much of the current
work on copolymers has been reviewed (95).
MALDI to Elucidate Polymer Chemistry.
Perhaps one of the most prof-
itable future uses of MALDI will be for elucidation of polymer chemistry. Exam-
ining a reaction not yet complete or looking for polymer side products differing
only by an end group will yield important reaction kinetics information. Unlike
nmr, uv, or ftir, where one can perhaps find the presence of differing end groups
or internal species for the overall MMD, MALDI can identify these species and
how they can appear as a function of molecular mass. A caveat to all this work
is the matrix attachment studies (21). Without careful studies of the linear and
reflectron modes for each reaction examined and the effect of matrix and attached
ion on the resultant spectra, one has to consider many of the conclusions from
these studies tentative.
Some of the early examples of this use of MALDI to elucidate polymer chem-
istry are presented here. One can often elucidate polymer chemistry by examining
the polymer products during or at the end of a reaction. Much of what has been
discussed in the copolymer section relates to this. The work in References 85–87
describes the distribution from which the chemistry can be elucidated.
In the work on SRM 1487, a low molecular mass PMMA, the authors related
their findings to the polymer chemistry of the initiator (62). Based on the synthesis
information, SRM 1487 would have the following structure:
H-PMMA-
{α-MeSty}
m
-PMMA-H
(5)
where m is the number of
α-MeSty repeat units. The course of the anionic reaction
of PMMA initiated by
α-MeSty with a Na gegen-ion had been extensively studied
(96,97) and the conclusion was that dimer and tetramer
α-MeSty difunctional
anion initiators are the dominant species in the initiation reaction (m
= 2 or 4).
MALDI studies suggested that all values of m from m
= 0 to m = 7 were possible.
However, no chains with m
= 6 were found.
MALDI-tof-ms was used to examine the products from some anionic polymer-
izations with styrene initiated by sec-butyl-Li or t-C
4
H
9
O(CH
2
)
3
Li and extended
by isoprene (98). Several electrophilic reagents affected termination. The results
showed unusual end group rearrangements to yield some end groups not normally
expected. From this observation it was possible to suggest new mechanisms for
the termination reaction of the polymer.
MALDI-tof-ms has been used to study the preparation of linear polysilox-
anes with hydride end-capped and cyclic polysiloxanes from the ring-opening an-
ionic polymerization reaction with hexamethylcyclotrisiloxane and octamethylcy-
clotrisiloxane (99). The PDMS reaction was followed by MALDI and the incor-
poration of the hydride end cap and the creation of the cyclics was studied as a
function of time. Further studies (100) employed MALDI and sims; it was found
that MALDI provides more reproducible relative intensities of the n-mers than
does SIMS.
Vol. 3
MASS SPECTROMETRY
179
Polymer Architecture Elucidated by MALDI.
Although ms is a mass
determining device, ms studies in combination with an understanding of polymer
chemistry allows one to make deduction on the architecture of polymer molecules.
In this section, a study of trifunctional polymerizations similar to those studied by
Flory in the early 1940s (see, for example, Ref. 101 is described). Here, however,
instead of a carbon-based backbone a silicon-based backbone was studied.
The prepolymers of the polysilsesquioxanes were studied by MALDI-tof-ms
(102,103). Polysilsesquioxanes are three-dimensional polymers with a repeat unit
of the form [RSiO
3
/2
] where each silicon is coordinated with three oxygen atoms.
After a low temperature sol–gel synthesis and processing, silsesquioxanes are
then fully cross-linked to from a fourfold coordinate structure. One important
unknown in the processing of silsesquioxanes is the “degree-of-condensation” of
the prepolymer silsesquioxanes before it is converted into the final product. That
is, how many of the silicon atoms are threefold coordinated with bridging oxygen
atoms and how many have terminal silanol (SiOH) groups?
In the polymerization process to create the prepolymer, a condensation re-
action of two silanol groups to create one Si O Si bridge with the elimination of
water occurs. In the prepolymer, these terminal silanol groups control the reac-
tivity of the polymer in subsequent processing steps that result in a cross-linked
product. Before the use of MALDI-ms only the average number of terminal silanol
groups in the whole sample could be measured by ir spectroscopy. Condensation
of SiOH groups out of the growing prepolymer silsesquioxane molecule can lead
to the formation of intramolecular Si O Si bridges with the loss of water. This
elimination reaction is easily identified via high resolution tof-ms. Figure 6 shows
the overall spectrum while Figure 7 shows the detail of a single low mass oligomer
Mass
Relativ
e intensity
1600
2600
3600
4600
5600
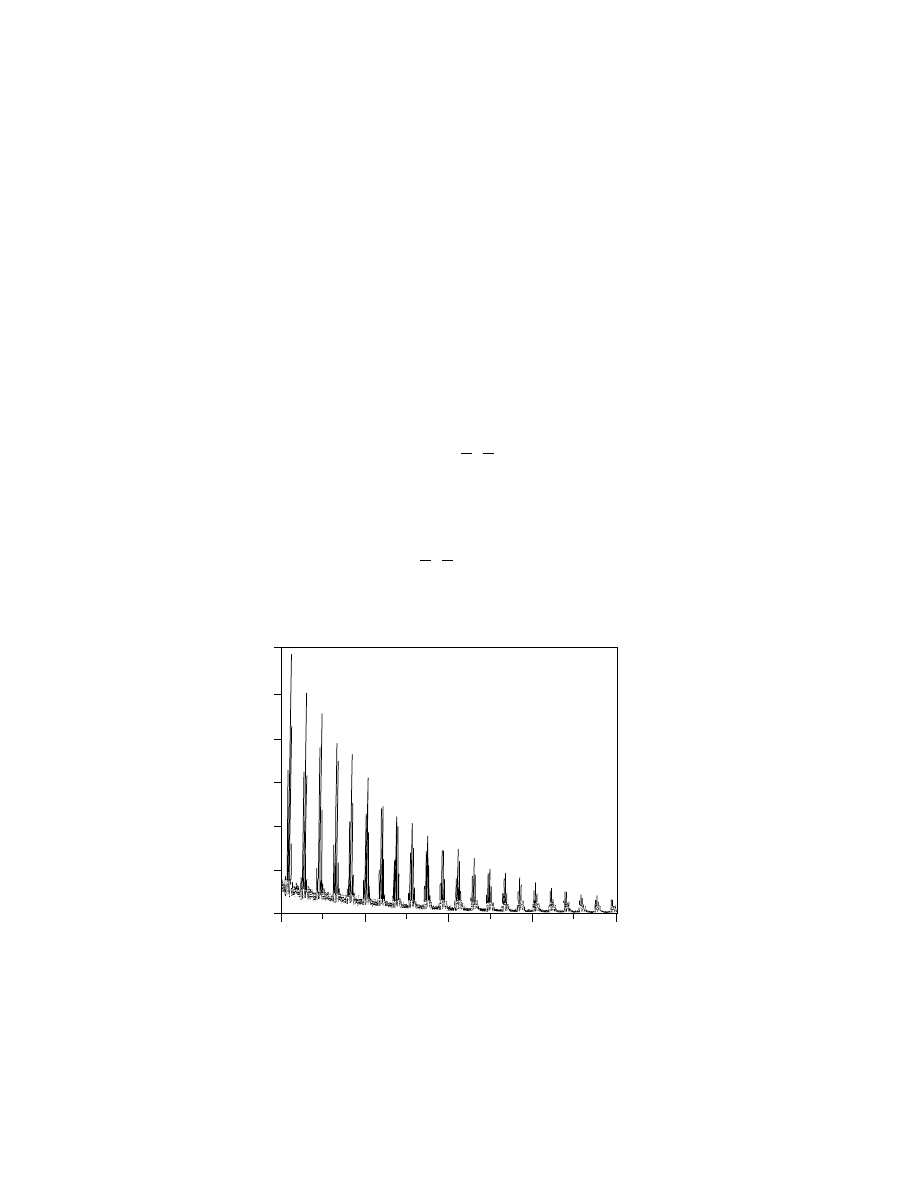
Fig. 6.
The central portion of the full mass spectrum of a polysilsesquioxane prepolymer
showing the characteristic shape of a condensation polymer. The major repeats show the
changing number of RSiO
3
/2
with a repeat of 188, 12 u.
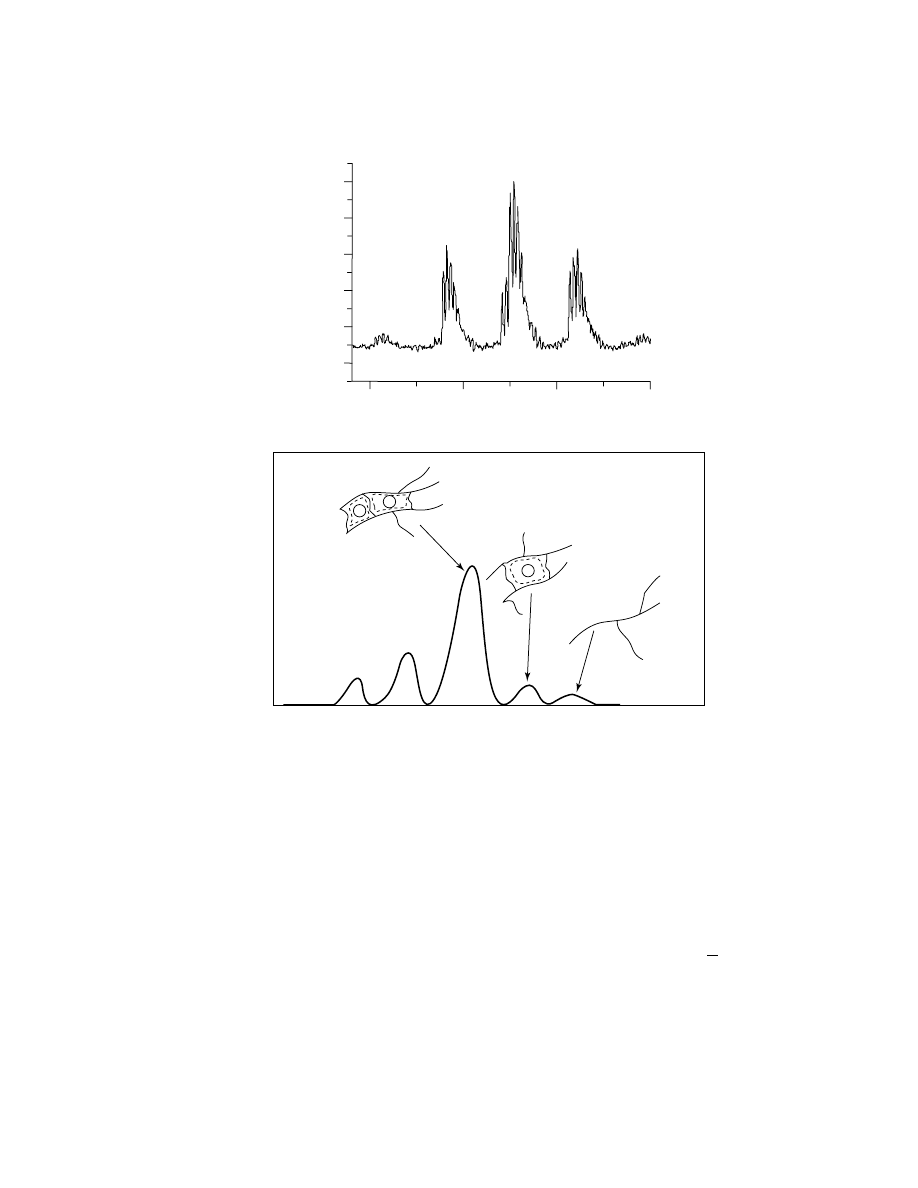
180
MASS SPECTROMETRY
Vol. 3
Mass
Detector Response
1825
1850
1875
1900
0
500
1000
1500
2000
2500
(a)
Mass/charge
Detector response
2
1
1
2 Closed Loops
1 Closed Loop
1
0
2
3
4
Branched
Linear
(b)
Fig. 7.
(a) A single-repeat unit detail of the full mass spectrum shown in Figure 6. The
distance between peaks in a single major cluster is 18 u, indicative of the intermolecular
loss of water; (b) The schematic of the polymer ms shows the relation of each peak of the ms
to the degree of intermolecular polymer condensation, the important polymer architecture
change in this system.
from the overall spectrum. The maximum possible mass of an oligomer with n re-
peat units occurs when every silicon atom has one silanol group in addition to one
R-group and two bridging oxygen atoms. However, the highest intensity peak gen-
erally does not occur at the maximum possible mass. Instead, lower mass peaks
are more intense. These peaks correspond to the loss of water as a pair of
SiOH
groups react. This indicates that intramolecular reactions are occurring.
Each loss of 18 u closes a loop in the molecule moving it away from a highly
branched linear structure toward a closed polyhedron. In the limit of the loss of

Vol. 3
MASS SPECTROMETRY
181
all silanol groups, a fully condensed polyhedral structure results. For example,
for n
=8 the fully condensed structure is a cube with Si atoms at each corner and
Si O Si linkages for the cube edges. Thus, the structure and chemical architec-
ture of each n-mer can be deduced from MALDI-tof-ms.
In another determination of architecture by MALDI (84), the appearance of
cyclics and linears in the poly(bisphenol A carbonate) has been investigated. It
was possible to distinguish between the linear and cyclic species. From MALDI
studies, a separate sec calibration line was obtained for the cyclic and the linear
species. This work was done with the MALDI acting as an sec detector.
BIBLIOGRAPHY
“Mass Spectrometry” in EPST 1st ed., Vol. 8, pp. 412–419, by G. P. Shulman, Jet Propulsion
Laboratory; “Mass Spectromety” in EPSE 2nd ed., Vol. 9, pp. 319–356, by K. D. Cook,
University of Tennessee.
1. D. C. Schriemer and L. Li, Anal. Chem. 68, 2721–2725 (1996).
2. T. Nohmi and J. B. Fenn, J. Am. Chem. Soc. 114, 3241–3246 (1992).
3. J. L. Watson, Introduction to Mass Spectrometry, 3rd ed., Lippencott-Raven, New
York, 1997.
4. G. Siuzdak, Mass Spectrometry for Biotechnology, Acedemic Press, San Diego,
1996.
5. P. B. Smith and co-workers, Anal. Chem. 69, 95R–121R (1997).
6. P. B. Smith and co-workers, Anal. Chem. 71, 61R–80R (1999).
7. M. Dole and co-workers, J. Chem. Phys. 49, 2240–2249 (1968).
8. M. Ymashita and J. B. Fenn, J. Phys. Chem. 88, 4451–4459 (1984).
9. S. F. Wong, C. K. Meng, and J. B. Fenn, J. Phys. Chem. 92, 546–550 (1988).
10. K. Tanaka and co-workers, Rapid Commun. Mass Spectrom. 2, 151 (1988).
11. M. Karas and F. Hillenkamp, Anal. Chem. 60, 2299 (1988).
12. U. Bahr and co-workers, Anal. Chem. 64, 2866 (1992).
13. P. O. Danis and co-workers, Org. Mass Spec. 27, 843 (1992).
14. P. O. Danis and D. E. Karr, Org. Mass Spectrom. 28, 923–925 (1993).
15. R. Abate and co-workers, Rapid Commun. Mass Spectrom. 7, 1033–1036 (1993).
16. R. J. Cotter and co-workers, Macromolecules 19, 2996–3001 (1986).
17. S. J. Pastor and C. L. Wilkins, J. Amer. Soc. Mass Spectrom. 8, 225–233 (1997).
18. R. J. Cotter, Time-of-Flight Mass Spectrometry, American Chemical Society, Washing-
ton, D.C., 1997. ACS Professional Reference Books.
19. A. Vertes, G. Irinyi, and R. Gijbels, Anal. Chem. 65, 2389–2393 (1993).
20. M. W. F. Nielen, Mass Spectrom. Rev. 18, 309–344 (1999).
21. R. J. Goldschmidt and C. M. Guttman, J. Amer. Soc. Mass Spectrom. 11, 1095–1106
(2000).
22. H. Rashidezadeh and B. C. Guo, J. Amer. Soc. Mass Spectrom. 9, 724–730
(1998).
23. R. Knochenmuss, E. Lehmann, and R. Zenobi, Eur. Mass Spectrom. 4, 421–426
(1998).
24. M. Karas, M. Gluckmann, and J. Schaefer, J. Mass Spectrom. 35, 1–12 (2000).
25. A. G. Bailey, Electrostatic Spraying of Liquids, John Wiley & Sons, Inc., New York,
1988.
26. W. J. Simonsick and L. Prokai, Adv. Chem. Ser. 247, 41–56 (1995).
27. M. L. Vestal, P. Juhasz, and S. A. Martin, Rapid Commun. Mass Spectrom. 9, 1044–
1050 (1995).

182
MASS SPECTROMETRY
Vol. 3
28. P. Juhasz, M. L. Vestal, and S. A. Martin, J. Amer. Soc. Mass Spectrom. 8, 209–217
(1997).
29. H. C. M. Byrd and C. N. Mcewen, Anal. Chem. 72, 4568–4576 (2000).
30. J. B. Williams, A. I. Gusev, and D. M. Hercules, Macromolecules 29, 8144–8150
(1996).
31. V. V. Laiko, M. A. Baldwin, and A. L. Burlingame, Anal. Chem. 72, 652–657
(2000).
32. P. Zollner, E. R. Schmid, and G. Allmaier, Rapid Commun. Mass Spectrom. 10, 1278–
1282 (1996).
33. P. Zollner and co-workers, Int. J. Mass Spectrom. Ion Processes 169, 99–109
(1997).
34. S. D. Hanton and K. G. Owens, in Proceedings of the 46th ASMS Conference on Mass
Spectrometry and Allied Topics, Orlando, Fla., 1998, Vol. 46, p. 1185.
35. S. D. Hanton, P. A. C. Clark, and K. G. Owens, J. Amer. Soc. Mass Spectrom. 10,
104–111 (1999).
36. R. C. King and K. G. Owens, in Proceedings of the 42nd ASMS Conference on Mass
Spectrometry, Chicago, Ill., 1994, Vol. 42, p. 977.
37. F. M. L. Amado and co-workers, Rapid Commun. Mass Spectrom. 11, 1347–1352
(1997).
38. D. A. Allwood and co-workers, Appl. Surf. Sci. 103, 231–244 (1996).
39. O. Vorm, P. Roepstorff, and M. Mann, Anal. Chem. 66, 3281–3287 (1994).
40. W. R. Blair, C. M. Guttman, and W. E. Wallace, in Proceedings of the 46th ASMS
Conference on Mass Spectrometry and Allied Topics, Orlando, Fla., 1998, Vol. 46, p.
1059.
41. R. R. Hensel, R. C. King, and K. G. Owens, Rapid Commun. Mass Spectrom. 11,
1785–1793 (1997).
42. J. Axelsson and co-workers, Rapid Commun. Mass Spectrom. 11, 209–213 (1997).
43. A. M. Belu and co-workers, J. Amer. Soc. Mass Spectrom. 7, 11–24 (1996).
44. R. Chen and L. Li, in Proceedings of the 48th ASMS Conference on Mass Spectrometry,
Long Beach, Calif., 2000, Vol. 48, p. 1211.
45. S. Weidner, A. Hollander, and G. Kuhn, Rapid Commun. Mass Spectrom. 11, 447–450
(1997).
46. A. M. Hoberg and co-workers, Eur. Mass Spectrom. 4, 435–440 (1998).
47. S. K. Poehlein and co-workers, Rapid Commun. Mass Spectrom. 13, 1349–1353
(1999).
48. H. H. Zhu, T. Yalcin, and L. Li, J. Amer. Soc. Mass Spectrom. 9, 275–281
(1998).
49. C. M. Guttman and co-workers, Polym. Prepr. 41, 678–679 (1999).
50. G. Montaudo and co-workers, Rapid Commun. Mass Spectrom. 9, 453–460 (1995).
51. D. C. Schriemer and L. A. Li, Anal. Chem. 69, 4169–4175 (1997).
52. R. S. Lehrle and D. S. Sarson, Rapid Commun. Mass Spectrom. 9, 91–92
(1995).
53. B. S. Larsen, W. J. Simonsick, and C. N. Mcewen, J. Amer. Soc. Mass Spectrom. 7,
287–292 (1996).
54. C. M. Guttman and co-workers, Anal. Chem. 73, 1252–1262 (2001).
55. C. N. McEwen, C. Jackson, and B. S. Larsen, Int. J. Mass Spectrom. Ion Processes 160,
387–394 (1997).
56. A. Westman, G. Brinkmalm, and D. F. Barofsky, Int. J. Mass Spectrom. Ion Processes
169/170, 79–87 (1997).
57. P. W. Geno and R. D. Macfarlane, Int. J. Mass Spectrom. Ion Processes 92, 195–210
(1989).
58. C. M. Guttman, Polym. Prepr. 37, 837–838 (1996).

Vol. 3
MASS SPECTROMETRY
183
59. D. A. Saucy and L. Zhu, in Proceedings of the 47th ASMS Conference on Mass Spec-
trometry, Dallas, Tex., 1999, Vol. 47, p. 3021.
60. C. Jackson, B. Larsen, and C. Mcewen, Anal. Chem. 68, 1303–1308 (1996).
61. G. Montaudo and co-workers, Macromolecules 28, 7983–7989 (1995).
62. C. M. Guttman, W. R. Blair, and P. O. Danis, J. Polym. Sci., Part B: Polym. Phys. 35,
2409–2419 (1997).
63. P. M. Lloyd and co-workers, Eur. Mass Spectrom. 1, 293–300 (1995).
64. S. F. Wong, C. K. Meng, and J. B. Fenn, J. Phys. Chem. 92, 546–550 (1988).
65. U. Bahr and co-workers, Anal. Chem. 64, 2866–2869. (1992).
66. G. Montaudo and co-workers, Macromolecules 28, 4562–4569 (1995).
67. B. C. Guo and co-workers, Rapid Commun. Mass Spectrom. 11, 781–785 (1997).
68. K. Martin and co-workers, Rapid Commun. Mass Spectrom. 10, 1471–1474 (1996).
69. C. E. Kassis and co-workers, Rapid Commun. Mass Spectrom. 11, 1134–1138 (1997).
70. M. W. F. Nielen, Anal. Chem. 70, 1563–1568 (1998).
71. C. Puglisi and co-workers, Rapid Commun. Mass Spectrom. 13, 2260–2267
(1999).
72. P. C. Hiemetz, Polymer Chemistry, Marcel Dekker, Inc., New York, 1984.
73. C. Wu, Handbook of Size Exclusion Chromatography, Marcel Dekker, Inc., New York,
1995.
74. W. W. Yau, J. J. Kirkland, and D. D. Bly, Modern Size Exclusion Liquid Chromatog-
raphy, John Wiley & Sons, Inc., New York, 1979.
75. K. K. Murray and D. H. Russell, J. Amer. Soc. Mass Spectrom. 5, 1–9 (1994).
76. A. I. Gusev, Fresenius, J. Anal. Chem. 366, 691–700 (2000).
77. C. M. Guttman, E. A. Dimarzio, and J. F. Douglas, Macromolecules 29, 5723–5733
(1996).
78. S. G. Entelis, V. V. Evreinov, and A. V. Gorshkov, Adv. Polym. Sci. 76, 129 (1986).
79. J. Falkenhagen and co-workers, Int. J. Polym. Anal. Char. 5, 549–562 (2000).
80. D. R. Maloney and co-workers, J. Chem. Soc. Chem. Commun. 561–562 (1995).
81. A. T. Jackson and co-workers, J. Amer. Soc. Mass Spectrom. 8, 132–139 (1997).
82. A. T. Jackson and co-workers, Rapid Commun. Mass Spectrom. 11, 520–526
(1997).
83. J. S. Liu, R. S. Loewe, and R. D. McCullough, Macromolecules 32, 5777–5785
(1999).
84. C. Puglisi and co-workers, Rapid Commun. Mass Spectrom. 13, 2260–2267
(1999).
85. G. Wilczek-Vera, P. O. Danis, and A. Eisenberg, Macromolecules 29, 4036–4044
(1996).
86. Wilczek-Vera and co-workers, Macromolecules 32, 2180–2187 (1999).
87. G. Wilczek-Vera and co-workers, Rapid Commun. Mass Spectrom. 13, 764–777
(1999).
88. G. V. Schulz, Z. Phys. Chem. B 43, 25 (1939).
89. B. H. Zimm, J. Chem. Phys. 16, 1093, 1099 (1948).
90. D. Freyss, P. Rempp, and H. Beniot, J. Polym. Sci (Phys.) B2, 217 (1969).
91. M. S. Montaudo and F. Samperi, Eur. Mass Spectrom. 4, 459–465 (1998).
92. D. Vitalini and co-workers, Macromolecules 29, 4478–4485 (1996).
93. V. Francke and co-workers, Macromol. Rapid Commun. 19, 275–281 (1998).
94. V. Schadler and co-workers, Macromolecules 29, 4865–4870 (1996).
95. M. S. Montaudo, Macromol. Symp. 141, 95–101 (1999).
96. V. Warzelhan, H. Hocker, and G. V. Schulz, Makromol. Chem. 181, 149 (1978).
97. V. Warzelhan and G. V. Schulz, Makromol. Chem. 177, 2185 (1976).
98. C. Wesdemiotis and co-workers, Polym. Prepr. 41, 629–630 (2000).
99. S. K. Pollack and A. M. Morgan, Polym. Prepr. 41, 631 (2000).

184
MASS SPECTROMETRY
Vol. 3
100. A. M. G. J. A. Hackridge, Polym. Prepr. 41, 635 (2000).
101. P. C. Painter and M. M. Coleman, Fundamentals of Polymer Science, Technomic Pub-
lishing Corp., Lancaster, Pa., 1994.
102. W. E. Wallace, C. M. Guttman, and J. M. Antonucci, J. Amer. Soc. Mass Spectrom. 10,
224–230 (1999).
103. W. E. Wallace, C. M. Guttman, and J. M. Antonucci, Polymer 41, 2219–2226 (2000).
C
HARLES
M. G
UTTMAN
NIST Polymers Division
MECHANICAL TESTING.
See M
ECHANICAL
P
ERFORMANCE
.
MELAMINE RESINS.
See A
MINO
R
ESINS AND
P
LASTICS
.
Wyszukiwarka
Podobne podstrony:
Electron ionization time of flight mass spectrometry historical review and current applications
Mass spectroscopy overview
Mass spectroscopy in food aplication
2B 4 Mass Spectroscopy; Summary
Szewczyk, Rafał i inni Rapid method for Mycobacterium tuberculosis identification using electrospra
019 Masowe środki przekazu mass media
Critical Mass UM
Mass Index, giełda(3)
SPEKTOMETRIA MASS W POŁĄCZENIU Z CHROMATOGRAFIĄ GAZOWĄ
In vivo MR spectroscopy in diagnosis and research of
EMC Spectrum Analyzer v2
Testy mass, Ekonomia, ekonomia
spectral tworzywa sztuczne, Elektra boczki
Wpływ mass mediów na dzieci, media w edukacji
więcej podobnych podstron