Nowotwory
dziedziczne

Przykłady nowotworów
(zespołów) dziedzicznych
Nowotwór/zespół
gen
lokalizacja Nowotwory
towarzyszące
dziedziczny rak sutka
BRCA1
BRCA2
17q21
13q12-13
rak jajnika, rak
trzonu macicy
rak jajnika
dziedziczny
niepolipowaty rak jelita
grubego (zespół Lynch)
MSH2
MLH1
2p15-22
3p21
rak trzonu macicy,
rak jajnika
rodzinna polipowatość
jelit
APC
5q21
polipy jelita
cienkiego, żołądka,
desmoid
nerwiekowłókniakowat
ość
NF1
17q11-22
nerwiakowłókniaki,
glejaki, mięsaki
czerniak złośliwy
(postać rodzinna)
CDKN9
9p21
glejaki, nerwiaki
Guz Wilmsa
WT1
11p13
guz nerki u dzieci

Przykłady nowotworów
(zespołów) dziedzicznych
cd.
Nowotwór/zespół
gen
lokalizacj
a
Nowotwory
towarzyszące
zespół
gruczolakowatości
wewnątrzwydzielniczej
typu 1 (zespół MEN1)
MENIN
11q13
gruczolaki przysadki,
przytarczyc, wyspiaki
trzustki, rakowiaki
zespół
gruczolakowatości
wewnątrzwydzielniczej
typu 2 (zespół MEN2)
RET
10q12
rak rdzeniasty tarczyc,
gruczolaki przytarczyc,
pheochromocytoma
siatkówczak płodowy
(retinoblastoma)
RB
13q14
mięsak kostny,
białaczki
zespół Li-Fraumeni
P53
17p13
rak sutka, białaczki,
mięsaki, guzy mózgu
zespół Von Hippel-
Lindau
VHL
3p25
rak nerki
Rak sutka i jajnika

5-10% wszystkich nowotworów złośliwych (w
tym tzw. pospolitych, jak rak sutka, jajnika czy
jelita grubego) powstaje w wyniku
predyspozycji wykazującej rodowe cechy
dziedziczenia autosomalnego dominującego.
Rak sutka (piersi) jest w Polsce najczęstszym
nowotworem złośliwym u kobiet (około 8 tys.
nowych zachorowań rocznie). Najwięcej
zachorowań przypada u kobiet >50 roku życia
(x = 61 lat).

Najistotniejsze czynniki
ryzyka:
rak sutka w rodzinie
wiek powyżej 50 lat
bezdzietność
późna pierwsza donoszona ciąża (>34 roku życia)
wczesna pierwsza i późna ostatnia miesiączka
otyłość
nadmiar tłuszczów zwierzęcych w diecie
długotrwałe stosowanie hormonów płciowych
(antykoncepcja)
promieniowanie jonizujące

Rak sutka i jajnika –
postać dziedziczna
8-10% raków sutka i jajnika ma podłoże
dziedziczne.
Około połowa wszystkich raków dziedzicznych
sutka wywołana jest mutacją genu BRCA1.
Dziedziczenie autosomalne dominujące.
Nowotwór często występuje przed menopauzą.
Wcześniejszy wiek występowania w
porównaniu do postaci sporadycznej.
Mutacja genu BRCA1 nadaje określony fenotyp
raka sutka (rak rdzeniasty).

Gen BRCA1
17q21
wielkość około 100 kb
22 eksony
mRNA o długości 7,8 kb
białko – 1863 aa
Najczęstsze mutacje:
nt. 5382insC (50%); nt. C61G (21%) (PL)
nt. 185del AG; nt. 5382insC (Żydzi
aszkenazyjscy)

Ryzyko rozwinięcia się raka sutka w
ciągu całego życia kobiety z mutacją w
genie BRCA1 wynosi 56-87%.
ryzyko 50% - przed 50 rokiem życia
ryzyko 80-95% - do 70 roku życia
W około 50% przypadków raka sutka
rozpoznaje się do 41 roku życia.

Ryzyko rozwinięcia się raka jajnika z
mutacją genu BRCA1 w ciągu całego
życia kobiety wynosi 16-44%.
ryzyko 23% - przed 50 rokiem życia
ryzyko 63% - przed 70 rokiem życia

Gen BRCA2
13q12-13
wielkość 70 kb
27 eskonów
mRNA o długości 11-12 kb
białko – 3418 aa
dziedziczenie autosomalne dominujące
Częste mutacje:
nt. 6174delT (Islandia, Żydzi aszkenazyjscy)
U kobiet z mutacją genu BRCA2 ryzyko
rozwinięcia się raka sutka w ciągu całego
życia jest podobne jak w przypadku mutacji w
genie BRCA1.
Około 35% dziedzicznych raków sutka
związana jest z mutacją genu BRCA2.
Fenotyp raka sutka z mutacją genu BRCA2 –
rak cewkowo-zrazikowy.

Ryzyko jest mniejsze dla wystąpienia
raka jajnika.
Mutacje genu BRCA2 zwiększają ryzyko
wystąpienia raka sutka u mężczyzn (5%
do 70 roku życia).
Rodowodowo-kliniczne
kryteria rozpoznawania
rodzin z wysokim ryzykiem
dziedzicznych raków piersi i
jajnika

Jeden przypadek raka
piersi lub jajnika w
rodzinie
rak piersi w wieku poniżej 35 lat
rak piersi rdzeniasty lub atypowy rdzeniasty
rak piersi u mężczyzny
rak piersi i jajnika u tej samej osoby
niezależnie od wieku
rak piersi obustronny, jeden z nich
rozpoznany przed 50 rokiem życia
co najmniej jedno z powyższych kryteriów musi
być spełnione

Dwa przypadki raka piersi
lub jajnika w rodzinie
dwa raki piersi lub jajnika wśród krewnych
I°; co najmniej jeden rak piersi rozpoznany
przed 50 rokiem życia
raki jajnika rozpoznane w jakimkolwiek
wieku
jeden rak piersi rozpoznany przed 50
rokiem życia i jeden rak jajnika rozpoznany
w dowolnym wieku wśród krewnych I°
co najmniej jedno z powyższych kryteriów musi
być spełnione

Trzy przypadki raka piersi
lub jajnika w rodzinie
co najmniej 3 chore z rakiem piersi lub
jajnika rozpoznanym w dowolnym
wieku, jedną z tych chorych jest krewną
I°
Lubiński i wsp., Współ. onkologia 2000, 4: 186-189
Program badań kontrolnych w
rodzinach z wysokim ryzykiem
dziedzicznego raka piersi i jajnika
Lubiński i wsp., 2000 Współ. onkologia
narząd
badanie
początek
częstość
pierś
palpacyjne przez
pacjentkę
18-21 lat
co 1 mies.
palpacyjne przez
lekarza
20-30 lat
co 6 mies.
mammografia
(MG)
30-35 lat
co 12 mies.
USG
25 lat
co 12 mies.
(6 mies. po MG)
jajnik
USG dopochwowe
(Doppler)
30-35 lat
co 12 mies.
antygen CA-125
białko HER
30-35 lat
30-35 lat
co 12 mies.
(6 mies. po USG)
co 12 mies.
Dziedziczny niepolipowaty
rak jelita grubego

Geny biorące udział w naprawie
postreplikacyjnej (Mismatch
repair; MMR)
gen
chromosom
HNPCC*
hMLH1
3p21
33%
hMSH2
2p11-22
31%
hPMS2
7q22
4%
hPMS1
2q31-33
2%
* Hereditary nonpolyposis colorectal cancer

Funkcje genów
mutatorowych
Produkty białkowe genów tworzą kompleks o
właściwościach naprawczych błędnie
sparowanych zasad.
Kompleks ten rozpoznaje niewłaściwe zasady
poprzez porównanie stopnia metylacji adeniny w
sekwencjach GATC obu nici DNA.
Mechanizm takiej naprawy został wykryty m. in.
u bakterii Escherichia coli, u drożdży
Saccharomyces cerevisiae, u których
zaobserwowano niestabilność
mikrosatelitarnych, powtarzających się
sekwencji DNA (tandemy CA).

Charakterystyczne cechy
kliniczne HNPCC
autosomalny dominujący tor dziedziczenia
wczesny wiek raka (duży rozrzut wieku
występowania; wiek zachorowania obniża się z
pokolenia na pokolenie)
częsta lokalizacja prawostronna (kątnica,
poprzecznica, zagięcie wątrobowe i
śledzionowe)
częste współistnienie raków pozaokrężnicowych
(rak narządu rodnego, rak jajnika, rak pęcherza
moczowego, rak żołądka, rak dróg żółciowych,
rak jelita cienkiego) – zespół Lynch 2.

Kryteria diagnostyczne
HNPCC
U co najmniej 3 członków danej rodziny
wykryto zweryfikowanego
histopatologicznie RJG, przy czym jeden z
pacjentów jest krewnym I° dla pozostałych
dwóch; wykluczono polipowatość rodzinną.
Co najmniej 2 z tych osób to krewni I° w 2
różnych pokoleniach.
Przynajmniej u 1 spośród tych osób
zdiagnozowano RJG przed 50 rokiem życia.
Rodzinna polipowatość
gruczolakowata jelita
grubego (Familial
adenomatous polyposis)

Rodzinna polipowatość
gruczolakowata jelita grubego
(FAP)
dziedziczenie autosomalne dominujące
podłoże genetyczne – mutacje
germinalne genu APC
objawy – liczne (setki lub tysiące) polipy
w śluzówce jelita grubego
występowanie jednego lub kilku
objawów pozajelitowych

Gen APC
Adenomatous polyposis
coli
gen supresorowy
5q21-22
21 eksonów
białko – 2843 aa
białko APC bierze udział w regulacji wielu
procesów w komórce, jak: podział,
migracja, adhezja, różnicowanie komórki;
wchodzi w interakcje z wieloma białkami
np. β-kateniną, białkiem mikrotubul,
białkiem p34

Mutacje genu APC
region o podwyższonej częstości
występowania mutacji MCR (mutation
cluster region) – 1055-1309
23% wszystkich mutacji germinalnych –
region 5’ eksonu 15
delecje lub insercje kilku par zasad – 68%
substytucje – 30%
98% mutacji prowadzi do skrócenia produktu
białkowego
Mutacje somatyczne występują najczęściej
między kodonami 1286 i 1513
Zespoły mnogiej
gruczolakowatości
wewnątrzwydzielniczej
(multiple endocrine neoplasia,
MEN)

Zespoły mnogiej
gruczolakowatości
wewnątrzwydzielniczej
Typu 1 (MEN1) – zespół Wermera
Typu 2 (MEN2)
MEN 2A – zespół Sipple’a
MEN 2B – zespół Gorlina
Dziedziczenie autosomalne dominujące
Częstość występowania zespołów MEN
1/30.000 – 1/50.000

Gen MENIN
„kandydat” dla zespołu
MEN1
region 11q13 (locus D11427-D11460)
wielkość 9kb
10 eksonów
obecność transkryptu genu (2,8 kb
mRNA) wykryto w wielu komórkach, m.in.
neuroendokrynnych, trzustce, tarczycy,
jądrach, korze nadnerczy, leukocytach
krwi
produkt genu – białko jądrowe (610 aa)
Stopień penetracji
zmutowanego genu
MENIN
wiek
objawy kliniczne
zespołu MEN1*
<20 lat
43% pacjentów
20-35 lat
85% pacjentów
50 lat
94% pacjentów
* Nowotwory związane z MEN1 pojawiają się synchronicznie
lub metachronicznie.

MEN1
Typ guza
Czynność
hormonalna
Względna
częstość
Nowotwory
przytarczyc
parathormon
90-100%
Hormonalnie czynne
guzy wysp
trzustkowych:
90-100%
Gastrinoma
gastryna
50-70%
Insulinoma
insulina
20-40%
Glucagonoma
glukagon
≤10%

MEN1 cd.
Typ guza
Czynność
hormonalna
Względna
częstość
VIPoma
wazoaktywne
peptydy jelitowe
≤5%
PPoma
polipeptydy
trzustkowe
≤5%
Somatostatinom
a
somatostatyna
≤5%
Rakowiak
substancje
wazoaktywne
≤5%

MEN1 cd.
Typ guza
Czynność
hormonalna
Względna
częstość
Gruczolaki
przysadki (przedni
płat)
20-40%
Prolactinoma
prolaktyna
20-40%
Somatotropinoma hormon
wzrostu
10-20%
Corticotropinoma
ACTH
5-28%
Gruczolaki
nieczynne
hormonalnie
20%

Wskazania do badań
genetycznych w kierunku
zespołu MEN1
wszyscy pacjenci z hormonalnie czynnymi
guzami trzustki
wszyscy pacjenci z rakowiakiem oskrzeli/płuc
u pacjentów < 30 roku życia z guzem(ami)
związanymi z zespołem MEN1
u pacjentów > 50 lat z pierwotną
nadczynnością przytarczyc
u pacjentów z co najmniej dwoma lub więcej
guzami związanymi z zespołem MEN1

Genetyczne badania
przesiewowe rodziny z
MEN1
dokładna analiza mutacji genu MENIN jest
zalecana u krewnych I° nosicieli mutacji (zarówno
z objawami MEN1, jak i bez)
zalecany wiek przeprowadzenia analizy mutacji –
10-12 rok życia
poznanie miejsca i rodzaju mutacji w genie MENIN
u pacjenta z MEN1 ułatwia rodzinne badania
przesiewowe w kierunku MEN1
kliniczne badania kontrolne wymagane są u
nosicieli mutacji
konieczny jest podpis formularza świadomej
zgody

Kliniczne badania kontrolne u
pacjentów z MEN1 i
bezobjawowych nosicieli genu
MENIN
Każdego roku (obowiązkowo)
szczegółowy wywiad i badanie fizykalne
wapń w surowicy, PTH, prolaktyna, gastryna, GH
USG jamy brzusznej
Każdego roku (opcjonalnie, zależnie od
wskazań klinicznych)
dodatkowe procedury diagnostyczne, np.
biochemiczne

Kliniczne badania kontrolne u
pacjentów z MEN1 i
bezobjawowych nosicieli genu
MENIN cd.
Co 3-5 lat (obowiązkowo)
badanie MRI przedniego płata przysadki
badanie obrazowe trzustki; jeśli USG jest
niewystarczające, to np. ultrasonografia
endoskopowa, TK, MRI

MEN2
Typ zespołu Rodzaj guza
Koincydencj
a
MEN2A
Rak rdzeniasty tarczycy
>90%
Nowotwory przytarczyc
50%
Pheochromocytoma
20%
MEN2B
Rak rdzeniasty tarczycy
80%
Pheochromocytoma
60%
Nerwiako-włókniakowatość
skóry i błon śluzowych
>90%
Inne zaburzenia, np.
marfanoidalna budowa ciała,
megacolon

Rak rdzeniasty tarczycy
postać sporadyczna
75%
postać dziedziczna
składowa zespołu MEN2A
15%
składowa zespołu MEN2B
4%
postać rodzinna (FMTC)
6%
FMTC – familial medullary thyroid carcinoma
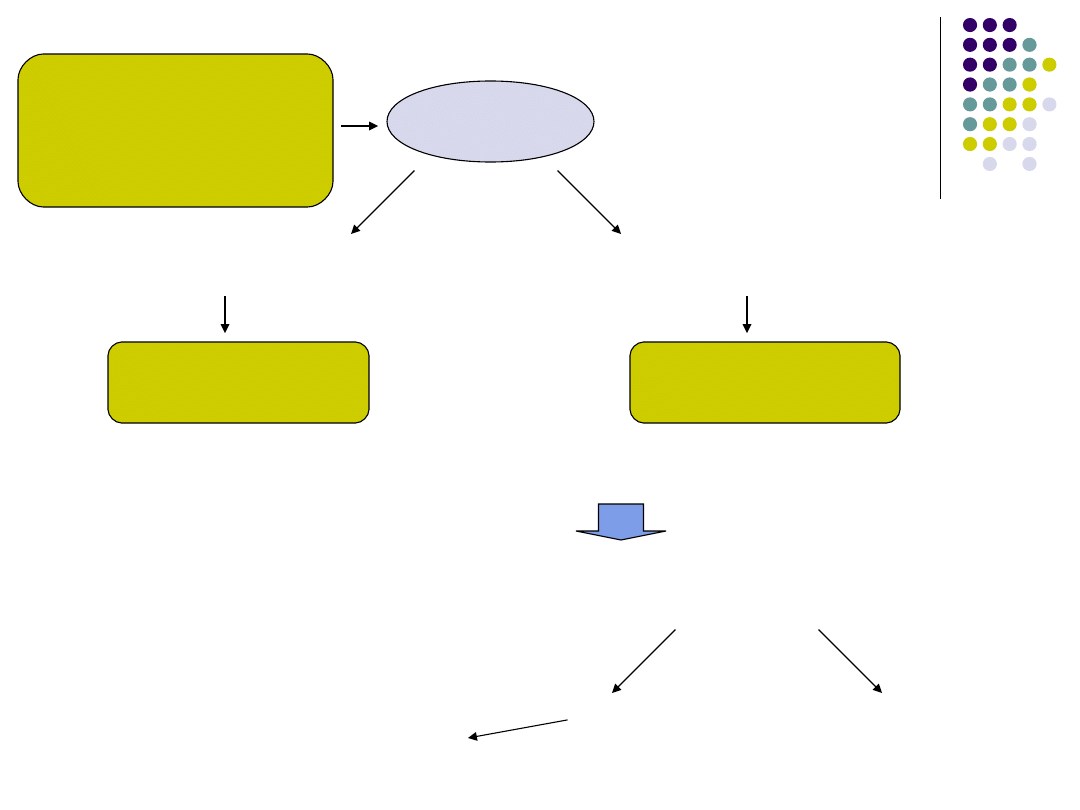
rak rdzeniasty tarczycy
(zdiagnozowany)
BAC
badanie histopatologiczne
DNA genomowy
rak rdzeniaty tarczycy
postać sporadyczna
rak rdzeniaty tarczycy
postać dziedziczna
DNA z limfocytów krwi obwodowej (-)
DNA z tkanki guza (+)
DNA z tkanki guza (+)
DNA z limfocytów krwi obwodowej (+)
RET
RET
rodziny zwiększonego ryzyka
(I, II stopień pokrewieństwa)
1. przesiewowe badania biochemiczne
(stężenie kalcytoniny, test
pentagastrynowy)
2. badanie DNA z
limfocytów
krwi obwodowej
i
RET
RET
(+)
(-)
badania biochemiczne
(test pentagastrynowy – regularnie,
okres wieloletni)

Czerniak rodzinny
dziedziczenie autosomalne dominujące
gen CMM1 locus 1p36
gen CDKN2 locus 9p21
mutacje typu nonsens, missens, insercje i inne
gen CDK4 locus 12q14
mutacje germinalne
częstość występowania: 5-7% pacjentów z
czerniakiem należy do rodzin z grupy
wysokiego ryzyka
wzrost występowania czerniaków, raka
trzustki, niekiedy astrocytoma

DIAGNOZOWANIE
10-100 barwnych znamion na tułowiu i
kończynach w rozmiarze 5-15 mm
co najmniej 2 krewnych I-stopnia z
czerniakiem
W rodzinach obciążonych mutacjami liczne
pierwotne czerniaki pojawiają się w młodym
wieku.

Retinoblastoma
dziedziczenie autosomalne dominujące
gen Rb locus 13q14
mutacje punktowe, delecje w obrębie całego
genu
dochodzi do inaktywacji obu alleli genu Rb;
pierwsza mutacja często germinalna lub na
wczesnym etapie rozwoju embrionalnego,
druga – somatyczna
w 20-30% przypadkach z mutacją germinalną
dochodzi do drugiej mutacji germinalnej

częstość występowania
~ 60% - siatkówczak jednostronny, niedziedziczny
~ 15% - siatkówczak jednostronny, dziedziczny
~ 25% - siatkówczak obustronny, dziedziczny
~ 90% przypadków jest diagnozowanych do 3 r.ż.
u ~ 49% dzieci rodziców z siatkówczakiem
obustronnym rozwija się ten nowotwór
kolejne nowotwory nie dotyczące oczu to:
kostniakomięsaki (wzrost ryzyka 500-krotny),
włókniakomięsaki, złośliwe raki epitelialne, mięsaki
Ewinga, białaczki, chłoniaki, czerniaki, nowotwory
mózgu, łagodne nowotwory siatkówki, tłuszczaki

Syndrom Li-Fraumeni
dziedziczenie autosomalne dominujące
gen p53 locus 17p13.1
częstość występowania: 2-4/100 000;
400 zarejestrowanych rodzin na świecie
najczęściej dochodzi do mutacji punktowych w
eksonach 5-8
ryzyko wystąpienia nowotworu przed 30 r.ż. wynosi
50% (w populacji ogólnej 1%); przed 70 r.ż. – 90%
zwiększone wystąpienie u jednego chorego różnych
nowotworów, jak: rak piersi, mięsaki, kostniaki,
białaczki, nowotwory mózgu, rak nadnerczy
zanotowano także przypadki czerniaków, raka płuc,
trzustki, żołądka i prostaty.
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
- Slide 23
- Slide 24
- Slide 25
- Slide 26
- Slide 27
- Slide 28
- Slide 29
- Slide 30
- Slide 31
- Slide 32
- Slide 33
- Slide 34
- Slide 35
- Slide 36
- Slide 37
- Slide 38
- Slide 39
- Slide 40
- Slide 41
- Slide 42
- Slide 43
- Slide 44
- Slide 45
- Slide 46
Wyszukiwarka
Podobne podstrony:
Nowotwory dziedziczne
GENETYKA KLINICZNA V rok seminarium Nowotwory dziedziczne wprowadzenie Nowotwory jelita grubeg
Nowotwory dziedziczne 2
więcej podobnych podstron