
Wykłady do końca semestru
Grupy poniedziałkowe 2.1 - 2.6:
05.12 (pon.)
12.12 (pon.)
19.12 (pon.)
02.01 (pon.)
09.01 (pon)
II kolokwium
23.01 (pon.)
poprawa I kolokwium
Grupy piątkowe 2.7 - 2.12
09.12 (pt)
16.12 (pt)
13.01 (pt)
16.01 (pn)
20.01 (pt)
II kolokwium
27.01 (pt)
poprawa I kolokwium
Termin poprawy II kolokwium
zostanie ustalony później
i będzie to prawdopodobnie po 27 stycznia
Istnieje możliwość poprawy
kolokwium I i II
dla niewielkiej liczby osób.
Terminy:
16.01, 20.01 i 27.01
Godz 8.00 i 9.00
Sala 3.3
Konieczne jest wcześniejsze
Zgłoszenie na adres:
ziemk@ch.pw.edu.pl
(najpóźniej poprzedniego dnia
Do godz 18 przed kolokwium)
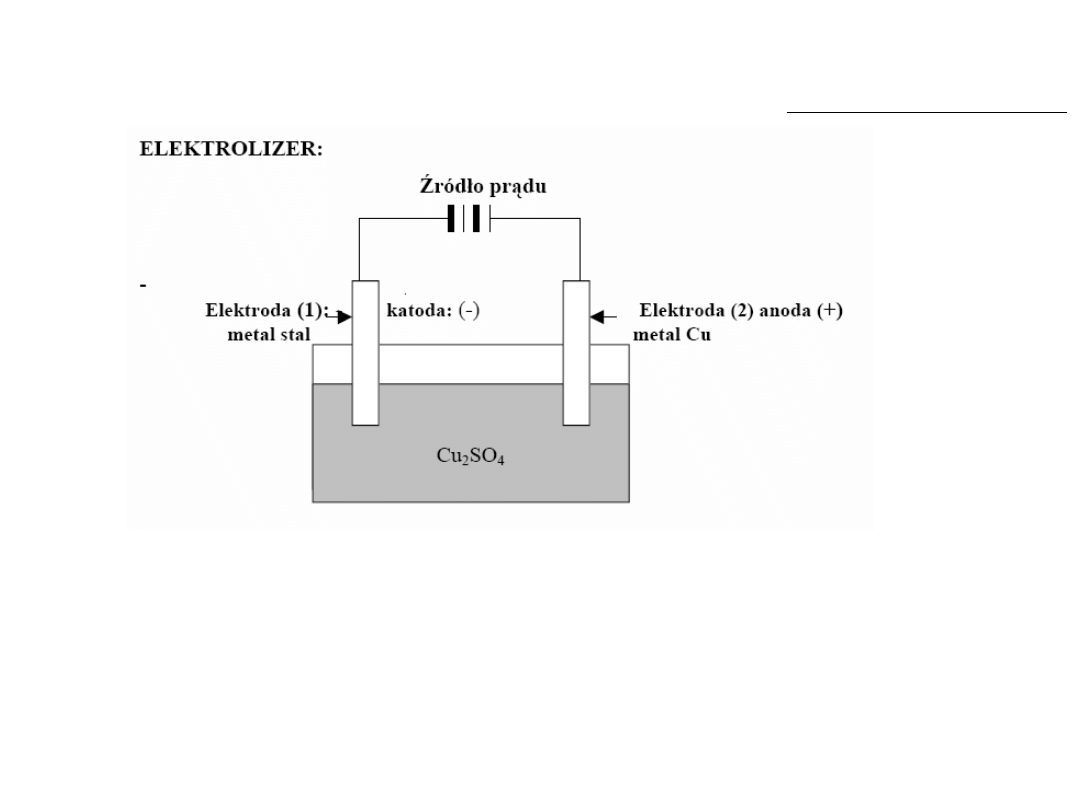
Elektroliza
Uporządkowany ruch jonów pod wpływem prądu
Przemiana energii elektrycznej na energię chemiczną, proces wymuszony
W trakcie elektrolizy prąd podłącza się tak aby na katodzie był ładunek ujemny
a na anodzie dodatni.
Kation
(jon dodatni) podąża do
katody
(elektroda ujemna)
Anion
(jon ujemny) podąża do
anody
(elektroda dodatnia)

Urządzenia:
elektrolizery
. Elektrody znajdują się w jednym naczyniu z roztworem
1 elektrolitu.
Elektrolit
: stopione sole, wodne roztwory kwasów, zasad i soli
Elektroliza roztworu chlorku miedzi CuCl
2
Proces stosowany do wydzielania miedzi metalicznej
Utlenianie: anoda
(+) 2Cl
–
Cl
2
+ 2e
Redukcja: katoda
(-) Cu
2+
+ 2e Cu
Sumarycznie: Cu
2+
+ 2 Cl
–
Cu + Cl
2
Elektroliza stopionego chlorku sodu NaCl
Metoda przemysłowego otrzymywania sodu metalicznego
Utlenianie: anoda
(+) 2Cl
–
Cl
2
+ 2e
Redukcja: katoda
(-) 2Na
+
+ 2e 2Na
Sumarycznie: 2Na
+
+ 2Cl
–
2Na + Cl
2

Elektroliza stężonego wodnego roztworu chlorku sodu NaCl:
Przemysłowy proces otrzymywania chloru i wodorotlenku sodu
Utlenianie: anoda(+) 2Cl
–
Cl
2
+ 2e
Redukcja: katoda(-) 2Na
+
+ 2e 2Na
Metaliczny sód natychmiast reaguje z wodą 2Na + H
2
O 2
NaOH
+
H
2
W wyniku elektrolizy na anodzie wydziela się chlor, na katodzie wodór
i otrzymuje się bardzo stężony roztwór NaOH.
Elektroliza rozcieńczonego roztworu NaCl
Rozcieńczony wodny roztwór NaCl daje w wyniku elektrolizy
wodór
i
tlen
.
W procesach elektrodowych rozcieńczonych roztworów elektrolitów cząsteczki
wody ulegają elektrolizie.
Utlenianie: anoda(+) 2H
2
O O
2
+ 4H
+
+ 4e
-
wydziela się gazowy tlen
Redukcja: katoda(-) 4H
+
+ 4OH
-
+ 4e
-
2H
2
+ 2OH
-
wydziela się gazowy wodór

Elektroliza wodnych roztworów kwasów:
Roztwory kwasów tlenowych (H
2
SO
4
, H
3
PO
4
, HNO
3
, H
2
CO
3
) dają zawsze tlen i wodór.
Katoda(-) 2H
+
+ 2e
-
H
2
Anoda (+) 2H
2
O
O
2
+ 4H
+
+ 4e
-
lub 2OH
–
O
2
+ 2H
+
+ 4e
-
W przypadku kwasów beztlenowych utlenieniu ulegają aniony reszt kwasowych
Utlenianie: anoda(+) 2Cl
–
Cl
2
+ 2e
Redukcja Katoda(-) 2H
+
+ 2e
-
H
2
Elektroliza wodnych roztworów zasad:
Jeżeli poddaje się elektrolizie bardzo rozcieńczony roztwór zasady, a do elektrod
nie przyłoży się z zewnętrznego źródła prądu zbyt dużego napięcia,
to
rozkładowi ulega głównie woda
;
katoda(-) 2H
2
O + 2e
-
2OH
-
+
H
2
anoda(+) H
2
O 2H
+
+
1/2O
2
+ 2e
-
W przypadku roztworów stężonych:
katoda(-) 2H
2
O + 2e
-
2OH
-
+
H
2
(tak samo jak dla roztworów rozc.)
Anoda (+) 2 OH
–
- 2e H
2
O +
½ O
2
Wniosek:
Prawie wszystkie reakcje elektrodowe zachodzące podczas przepływu prądu
elektrycznego przez bardzo rozcieńczone roztwory wodne prowadzą do wydzielenia
wodoru i tlenu.
Roztwór wokół anody staje się kwaśny w wyniku tworzenia się jonów wodorowych
H
+
a roztwór wokół katody zasadowy w wyniku tworzenia się jonów
wodorotlenowych OH
–
.
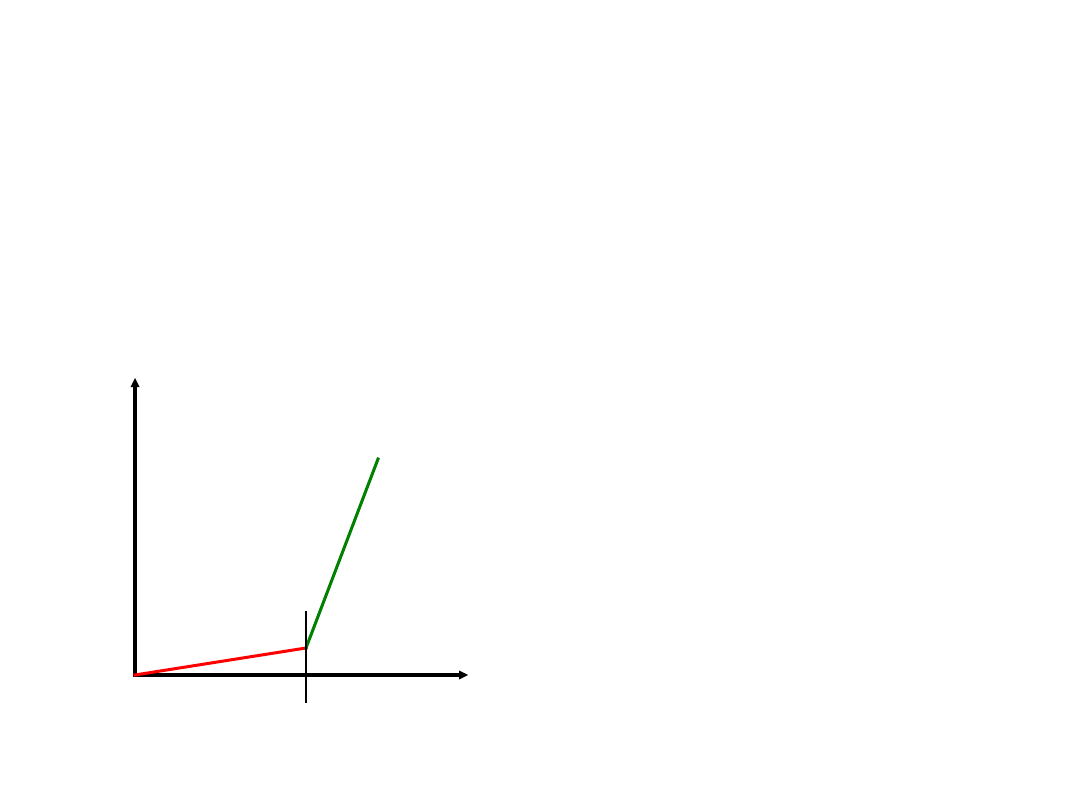
Napięcie rozkładowe
– najmniejsze napięcie konieczne do wywołania elektrolizy.
W praktyce wartość napięcia rozkładowego jest powiększana o
nadnapięcie
.
U rozkł.
I
U
Wykres zmian natężenia prądu w funkcji napięcia
Wielkość napięcia rozkładowego niezbędnego
do przeprowadzenia jonów w obojętne atomy
zależy od położenia pierwiastka w szeregu
napięciowym.
Najłatwiej
redukują się kationy
metali z największym potencjałem dodatnim
np.
Au, Ag
,
najtrudniej
metale o najmniejszych
potencjałach normalnych np.
Mg, Be
.
Ta właściwość jest wykorzystywana do
elektrolitycznego rozdzielenia metali.

Związek pomiędzy ilością produktu a ilością elektryczności zużytej
podczas elektrolizy (prawo Faradaya)
Przykład:
na redukcję na katodzie 1 mola jonów srebra trzeba zużyć 1 mol elektronów
Ag
+
+ 1e
-
Ag
Ilość moli elektronów możemy zamienić na ładunek, wykorzystując fakt, że
Stała Faradaya – stała fizyczna, która oznacza ładunek elektryczny przypadający
na 1 mol elektronów.
F = stała Avogadra N
A
• ładunek elektronu e
N
A
= 6,023•10
23
,
ładunek elementarny e = 1,602•10
-19
C (Kulomba),
1C(1 Kulomb) = 1A(amper)•1s(sekunda)
F = ok. 96500 C
= ładunek jednego mola elektronów
Aby otrzymać 1 mol srebra (108g) należy przez roztwór soli srebra przepuszczać
prąd o natężeniu 1A w ciągu 96500 s (26 godz 48 min 20 s)

Zadanie 1: Jak długo należy prowadzić elektrolizę prądem o natężeniu 1A
aby otrzymać 6,35 g miedzi z jonów Cu
2+
.
Zadanie 2: Przez roztwór soli złota zawierającego kationy Au
3+
przepuszczano
prąd o natężeniu 1 A w ciągu 13 godz. 24 min. 10s. Ile g złota otrzymano.
M
au
= 197 g/mol

Korozja metali
Degradacja środowiskowa materiałów –
degradacja
mikrostruktury i właściwości materiałów w
wyniku działania naprężeń, agresywnych
chemicznie środowisk, temperatury i czasu.
Korozja metali
– niszczenie metali pod wpływem
chemicznego lub elektrochemicznego działania
środowiska.
Korozja chemiczna –
korozja w suchych gazach i
nieelektrolitach
Korozja elektrochemiczna -
korozja w wilgotnych
gazach i elektrolitach

Rdza
– głównym składnikiem jest uwodniony tlenek żelaza 2Fe
2
O
3
•3H
2
O.
Inne składniki rdzy:
woda, tlenki i wodorotlenki żelaza o innym składzie, węglany żelaza.
Żelazo nie rdzewieje w obecności wody pozbawionej tlenu i w suchym powietrzu.
Korozja chemiczna
– polega na bezpośrednim ataku czynnika korozyjnego
(np. gazu w podwyższonej temperaturze) na metal lub stop metalu.
Przez metal podczas korozji nie przepływa prąd elektryczny a zachodzi tylko
utlenianie. Żelazo w atmosferze utleniającej powleka się warstewką tlenków
żelaza Fe
3
O
4
, Fe
2
O
3
, FeO (
pasywacja
) wskutek bezpośredniej reakcji z tlenem.

Korozja elektrochemiczna
– polega na tworzeniu lokalnych mikroogniw
na powierzchni metalu.
Elektrolit
w mikroogniwach powstaje przez rozpuszczenie w wodzie tlenu,
dwutlenku węgla, dwutlenku siarki, tlenków azotu.
Elektrody
w ogniwie korozyjnym powstają wskutek:
1. różnicy naprężeń w samym metalu, powstałe w czasie chłodzenia stopionego
metalu, po stłuczce, na powierzchniach poddanych obróbce skrawaniem
(
korozja naprężeniowa
).
Powierzchnia zdefektowana = anoda(-) (utlenianie) Fe
s
Fe
aq
2+
+ 2e
żelazo rozpuszcza się
obszar katodowy – redukcja jonów Fe
aq
2+
nie zachodzi, ale zachodzą inne reakcje:
2 H
+
+ 2e H
2
4H
+
+ O
2
+ 4e 2H
2
O
2. obecności w samym metalu wtrąceń innego metalu, grafitu w przypadku
stali węglowych lub też elementów konstrukcyjnych wykonanych z metalu
bardziej szlachetnego (
korozja jako wynik wtrąceń innych substancji
),

Wtrąceniami mogą być inne metale, związki chemiczne, niemetale, grafit.
One są katodą. Anodą jest żelazo.
anoda(-) (utlenianie) Fe
s
Fe
aq
2+
+ 2e żelazo rozpuszcza się
Katoda(+) (redukcja) 2H
2
O + O
2
+ 4e 4OH
–
Następnie jony Fe
2+
reagują z OH
–
tworząc rdzę
3. różnicy stężeń w roztworach soli lub tlenu, mającymi kontakt z powierzchnią
metalu (
korozja wynikająca z różnicy stężeń
).
Mikroogniwa korozyjne mogą powstawać jako ogniwa stężeniowe przez różne
napowietrzenie elektrolitu. Rozpuszczony w elektrolicie tlen tworzy elektrodę
dodatnią (katodę).
Obszary o mniejszym stężeniu tlenu stanowią anodę (-), na której rozpuszcza się
żelazo.

Skutki procesów korozyjnych
:
Korozja równomierna zmniejsza przekrój poprzeczny przedmiotu.
Korozja miejscowa: plamy, punkty i wżery – zmniejszenie wytrzymałości
materiału.
Korozja międzykrystaliczna – postępuje w głąb materiału na granicach ziaren,
bardzo niebezpieczna.
Ochrona przed korozją
:
Malowanie, galwaniczne pokrywanie metalu cienką warstwą cynku.
Cynk jest bardziej elektroujemny niż żelazo, dlatego będzie anodą.
Zn Zn
aq
2+
+ 2e
Żelazo nie będzie ulegało rozpuszczeniu.
Ochrona katodowa
– podłączenie zewnętrznej anody, inaczej płyty zwanej
protektorem.

Termodynamika – nauka o czynnikach energetycznych
reakcji chemicznych
Tematyka:
przepływ ciepła pomiędzy ciałami i ich układami.
skąd pojawia się ciepło, jakie są jego postaci i jakim ulega przemianom.
Reakcja egzotermiczna – przepływ energii z układu do otoczenia
Reakcja endotermiczna – przepływ energii z otoczenia do układu
3 zasady termodynamiki:
I zasada:
zasada zachowania energii
Jest ona związana z entalpią H.
Pojawieniu się ciepła musi towarzyszyć dostarczenie energii, znikaniu ciepła
musi towarzyszyć znikanie energii.
Energia wewnętrzna
układu izolowanego
(oddzielonego zarówno mechanicznie
jak i termicznie od otoczenia) jest stała:
U = const ΔU = 0
3 ważne pojęcia: entalpia, entropia i entalpia swobodna

Dowodem na tę właściwość układu izolowanego jest to, że nie udało się
zbudować poruszającej się ciągle maszyny, która nie pobierałaby energii
z zewnętrznego źródła, (czyli
perpetum mobile
pierwszego rodzaju)
Zmiana energii wewnętrznej
układu zamkniętego
(czyli takiego, w którym
masa nie może przepływać przez granicę układu z otoczeniem) jest równa energii,
która przepływa przez jego granice na sposób ciepła lub pracy.
ΔU = W + Q
W>0 - energia przepływa do układu na sposób
pracy
Q>0 - energia przepływa do układu na sposób
ciepła
W<0 - układ traci energię na sposób
pracy
Q<0 - układ traci energię na sposób
ciepła
Entalpia H
– suma energii wewnętrznej i pracy objętościowej
Fizycznie można zmierzyć jedynie ΔH, czyli ilość energii wymienionej między
układem zamkniętym a otoczeniem.

Pierwsza zasada termodynamiki - podsumowanie:
Jest to zasada zachowania energii. Ma ona fundamentalne znaczenie dla chemii,
gdyż większości reakcji chemicznych towarzyszy wydzielanie lub pobranie energii.
Prawo to uświadamia nam, że w przyrodzie dokonuje się jedynie przemiana
jednej formy energii w inną, a nie można jej ani wytworzyć z niczego ani zniszczyć
bez śladu.

II Zasada
: Każdy układ dąży do wzrostu
entropii
, czyli do możliwie
największej różnorodności podziału energii między składniki układu, a więc
do największego stopnia nieuporządkowania
.
Entropia S
jest miarą stopnia nieuporządkowania układu
S = k
B
lnW
S – entropia, k
B
– stała Boltzmana,
W –liczba sposobów podziału energii w układzie
Energia w układzie jest dzielona między różne drobiny na wiele sposobów – energia
translacyjna, rotacyjna, oscylacyjna.
W układach chemicznych entropia rośnie:
•W miarę rozpadania się wiązań
•Ze wzrostem temperatury
•Przy obniżaniu ciśnienia
Jeżeli nieuporządkowanie układu rośnie, rośnie też jego entropia.

W przemianach samorzutnych entropia układu izolowanego rośnie.
ΔS > 0
Proces samorzutny – proces, który aby zaszedł nie wymaga wykonania żadnej pracy.
Przykłady procesów samorzutnych:
-Gaz rozpręża się aby wypełnić całą objętość naczynia
-Gorący przedmiot ochładza się do temperatury otoczenia
Przemianom samorzutnym towarzyszy rozproszenie
energii w postać
bardziej nieuporządkowaną. Przykład: gorący przedmiot chłodzony w atmosferze
gazu przekazuje ciepło (rozprasza energię) cząsteczkom gazu, konsekwencją czego
jest szybszy ruch termiczny tych cząsteczek, czyli wzrost nieporządku.
W układzie termodynamicznie izolowanym
entropia nigdy nie maleje
,
czyli rośnie lub pozostaje stała.
Entropia wszechświata ma tendencję do zwiększania.
II zasada termodynamiki wyjaśnia dlaczego pewne procesy biegną samorzutnie
(np. dyfuzja gazów) a inne nie (np. rozdział mieszaniny).

Hipoteza „śmierci cieplnej Wszechświata”
Śmierć ta miałaby polegać na tym, że po jakimś czasie, wszechświat jako całość
dojdzie do
stanu równowagi termodynamicznej
. Będzie miał wtedy jednakową
temperaturę w każdym punkcie i wymiana energii termicznej całkowicie zaniknie.
To pociąga za sobą zaniknięcie wszelkich innych rodzajów wymiany energii,
które są zawsze związane ze zmianą temperatury.
Taka interpretacja II zasady termodynamiki w stosunku do kosmologii zakłada,
że Wszechświat jako całość jest układem izolowanym.
Jest to nieprawdą, ponieważ rozszerzający się
Wszechświat jest układem otwartym
.

III Zasada:
W warunkach stałej temperatury i ciśnienia zamknięty układ reagentów
dąży do osiągnięcia minimalnej wartości
entalpii swobodnej G
(inaczej funkcji Gibbsa).
Na układ działają dwa czynniki: oprócz dążenia do
wzrostu entropii
jest też
dążenie do
zmniejszenia energii.
Te dwa czynniki konkurują ze sobą.
Wypadkowy efekt tych dwóch czynników dla układów zamkniętych i dla stałej
temperatury i ciśnienia, określa
entalpia swobodna G.
G
=
H
–
TS
H – entalpia, T – temperatura, S – entropia
H - czynnik energetyczny
,
TS - czynnik entropowy
Kierunek reakcji w układzie reagentów jest też zdeterminowany dwoma czynnikami:
dążeniem do obniżenia energii i dążeniem do wzrostu entropii.
W przemianach endotermicznych czynnik entropowy przewyższa
czynnik energetyczny i dlatego reakcja zachodzi mimo, iż układ powiększa energię.
Układ, w którym
funkcja G
osiągnęła minimum znajduje się w stanie równowagi
termodynamicznej czyli nie działa już na niego czynnik energetyczny i entropowy.
Z makroskopowego punktu widzenia, w stanie równowagi termodynamicznej,
parametry stanu nie ulegają zmianie.
W rzeczywistości dla układu reagentów, stan równowagi termodynamicznej
jest stanem równowagi chemicznej, czyli równowagi dynamicznej.
Polega ona na równoczesnym przebiegu dwóch reakcji lecz z tą samą szybkością:
1. Przekształcania substratów w produkty
2. Przekształcania produktów w substraty

Termodynamiczny warunek równowagi chemicznej można ująć w następującej
postaci:
Zamknięty układ reagentów osiąga stan równowagi chemicznej wówczas,
gdy entalpia swobodna układu osiąga wartość minimalną.
W warunkach stałej temperatury i ciśnienia zamknięty układ reagentów dąży
do osiągnięcia minimalnej wartości entalpii swobodnej G.
Żadna reakcja chemiczna nie przebiega od mieszaniny samych substratów
do mieszaniny samych produktów lecz zawsze od mieszaniny wyjściowej
do mieszaniny równowagowej.
Tzn., że nawet reakcje biegnące praktycznie do końca do wyczerpania substratów
w rzeczywistości biegną do mieszaniny, w której oprócz produktów są minimalne
ilości substratów.

Kinetyka chemiczna
dział chemii fizycznej zajmujący się badaniem reakcji chemicznej w czasie.
Kinetyka zajmuje się:
•badaniem szybkości reakcji
•wpływem rozmaitych czynników na szybkość reakcji
•zmianami stężeń i produktów w czasie
Równanie chemiczne:
aA + bB + ... mM + nN + ...
Równanie kinetyczne
- równanie opisujące zmiany stężenia substratów
w czasie trwania reakcji
ν = kc
A
α
•c
B
β
• ...
ν– szybkość reakcji, k – stała szybkości reakcji, c – stężenia substratów
Wykładniki potęgowe α i β nie wynikają z równania chemicznego ale zależą
od mechanizmu reakcji.

Szybkość reakcji zależy od:
•Temperatury
•Ciśnienia
•Rodzaju rozpuszczalnika
•Rodzaju i stężenia katalizatora
•Wyjściowych proporcji substratów
Na podstawie powyższych danych wyznacza się stałe
szybkości reakcji
i mechanizm reakcji
.
Badanie kinetyki reakcji nie jest potrzebne przy syntezie związków na skalę
laboratoryjną, natomiast jest niezbędne w procesach przemysłowych.
Reakcje chemiczne przebiegają z różną szybkością:
•momentalne (reakcje jonowe),
•powolne (reakcje polimeryzacji),
•bardzo powolne (niektóre reakcje rozpadu promieniotwórczego).
Szybkość reakcji zależy od liczby zderzeń reagujących cząstek liczonych na jednostkę
czasu.
O szybkości decydują tylko zderzenia skuteczne prowadzące do wytworzenia produktu.
Do skutecznego zderzenia i przereagowania cząstek potrzebna jest odpowiednio
duża energia.

Rząd reakcji:
Suma wszystkich wykładników potęg w równaniu kinetycznym
Jeżeli ν = kc
A
m
c
B
n
c
c
p
to rząd reakcji wynosi: m + n + p
Szybkość reakcji wyznacza się eksperymentalnie
Dla reakcji jednocząsteczkowej przebiegającej według równania:
A = B + C + ........
szybkość reakcji wyraża się wzorem:
dc
ν =
–
— = k • c
A
dt
dc
Δc
—
= ubytek stężenia substratu w jednostce czasu. Można go zapisać jako
——
dt
Δt
C
A
= stężenie molowe substancji A
k
= stała szybkości reakcji

Szybkość reakcji mierzona w dwóch różnych stanach postępu reakcji, gdy stężenie
substancji zmalało z c
1
do c
2
w odstępie czasu t
2
-t
1
wyrazi się ułamkiem:
C
2
– C
1
ν = ———
t
2
– t
1
dc
jest
wartością ujemną
i z tego względu przed wyrażeniem — stawia się
znak ujemny
dt
aby szybkość była wartością dodatnią.
Reakcja zerowego rzędu:
Szybkość nie zależy od stężenia substratów
. Wykładnik potęgi =
0
.
dc
ν = - — = kc
A
0
= k
dt
Przykład: reakcje fotochemiczne. Szybkość reakcji jest zależna od strumienia
dostarczanej energii w postaci fotonów a nie od stężenia substratu A ulegającego
przemianie chemicznej.

Reakcje pierwszego rzędu:
Szybkość zależy od stężenia substratu
. Wykładnik potęgi =
1
ν = kc
A
1
= kc
A
Przykład: reakcje jednocząsteczkowe, które nie są wynikiem zderzeń
cząsteczek różnych reagentów np. reakcje rozpadu promieniotwórczego
lub termicznego.
Reakcje drugiego rzędu (reakcje dwucząsteczkowe):
Szybkość zależy od stężenia substratu
. Wykładnik potęgi =
2
ν = kc
A
2
lub ν = kc
A
c
B
Wpływ temperatury na szybkość reakcji:
Podwyższenie temperatury o 10° podwaja w przybliżeniu szybkość reakcji.
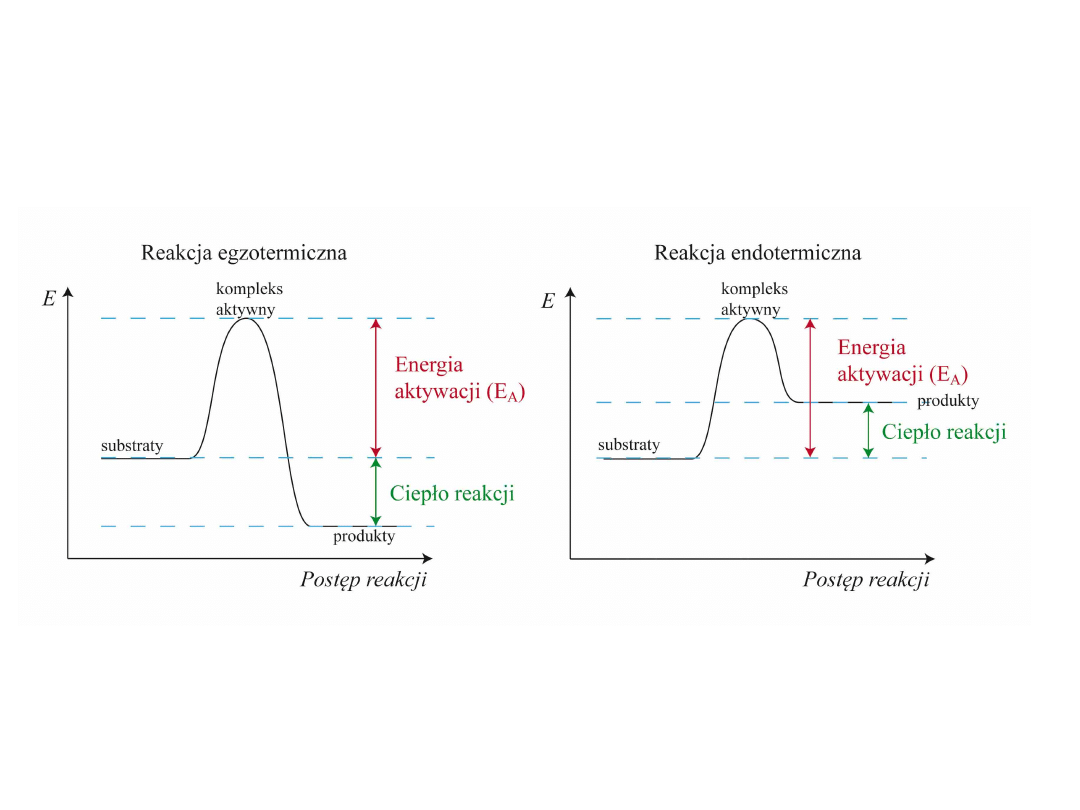
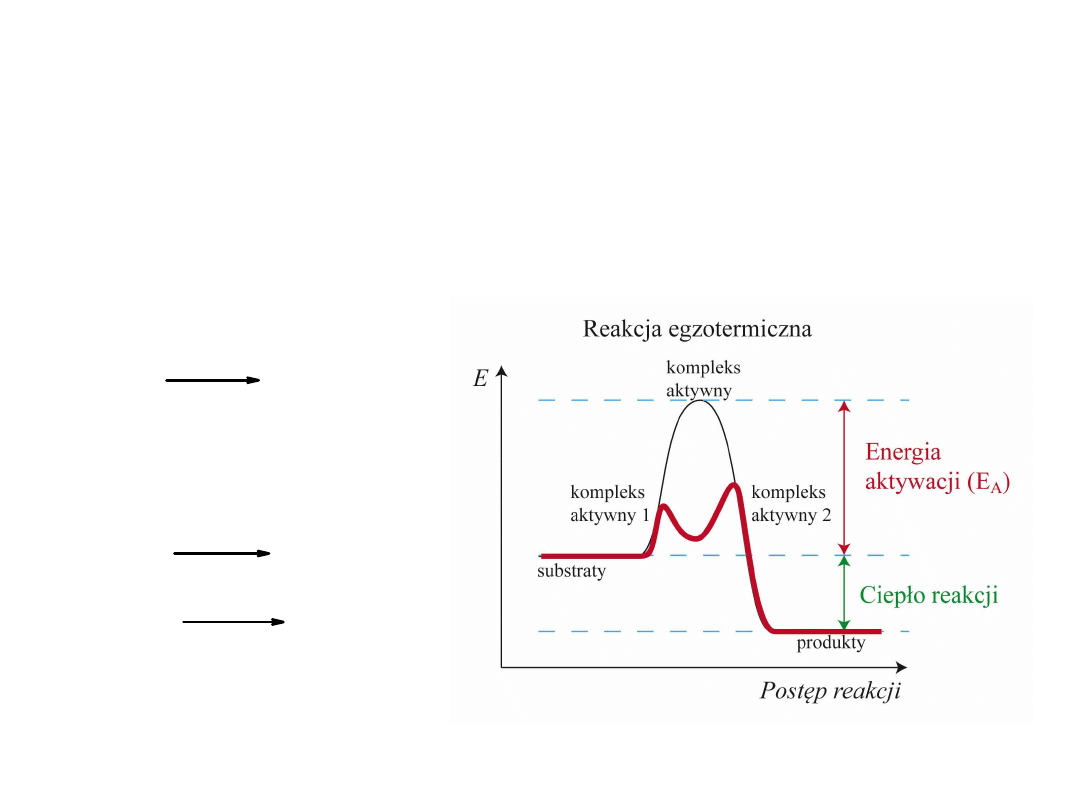
Energia aktywacji
: najmniejsza energia jaką muszą posiadać cząsteczki
substratów aby mogła zajść reakcja chemiczna.
Katalizator:
substancja, która dodana do układu reagującego zwiększa
szybkość reakcji nie ulegając przy tym wypadkowej zmianie.
Rola katalizatora polega na obniżeniu
energii aktywacji E
A
poprzez utworzenie
aktywnego kompleksu
lub (lub kilku kompleksów).
Efektem działania katalizatora w danej temperaturze jest zwiększenie
szybkości reakcji w tej samej temperaturze.
A + B
powoli
C
Reakcja bez katalizatora
A + K
AK
szybko
szybko
AK + B
C + K
Reakcja z katalizatorem
AK – kompleks aktywny
substratu z katalizatorem

Energia aktywacji
: (podawana w przeliczeniu na 1 mol substancji) jest to
wielkość bariery energetycznej, którą musi pokonać układ reagujących
indywiduów chemicznych, aby doszło do reakcji chemicznej.
Przykład:
Tlen zmieszany z wodorem
w temperaturze pokojowej nie reaguje:
chociaż bilans energetyczny reakcji jest korzystny, aby reakcja zaszła
cząsteczki muszą zderzyć się z odpowiednią energią, co pozwoli pokonać im
barierę potencjału. Jeżeli takich cząsteczek jest zbyt mało, wówczas energia
uzyskana w tych bardzo rzadkich zderzeniach efektywnych czyli tych, w których
doszło do reakcji (decyduje o tym nie tylko energia, ale również np. kąty
zderzenia i to, czy są to zderzenia centralne itd.) - jest rozpraszana
w zderzeniach z pozostałymi zimnymi (niskoenergetycznymi) cząsteczkami
układu.
Reakcja będzie zachodzić, gdy:
•proporcja O
2
:H
2
będzie w odpowiednim zakresie (nadmiar/niedomiar jednego
•ze składników zmniejsza prawdopodobieństwo efektywnych zderzeń)
•układowi zostanie lokalnie dostarczona
dodatkowa energia
, dostatecznie duża,
aby nie rozproszyła się w zderzeniach z zimnymi cząsteczkami układu
(iskra elektryczna, płomień)
•zostanie wykorzystany odpowiedni
katalizator
zmniejszający energię aktywacji
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
- Slide 23
- Slide 24
- Slide 25
- Slide 26
- Slide 27
- Slide 28
- Slide 29
- Slide 30
Wyszukiwarka
Podobne podstrony:
Plan wykładu 3 ogarnijtemat com
wyklad 6 ogarnijtemat com
Plan wykładu 4 ogarnijtemat com
Plan wykładu 5 ogarnijtemat com
Wykład 4 OgarnijTemat com
wykład 5 OgarnijTemat com
Analiza Wykład 6 (16 11 10) ogarnijtemat com
Plan wykładu 3aaaa ogarnijtemat.com, Chemia
więcej podobnych podstron