
Miastenia

Stadia kliniczne miastenii (wg
Ossermana)
I
miastenia oczna
IIa
postać łagodna uogólniona
IIb
postać średnio ciężka uogólniona
w II stadium nie są zajęte mm. oddechowe
III
postać ostra i szybko postępująca.
Nagły początek i progresja choroby z zajęciem mm.
oddechowych w
ciągu 6 miesięcy
IV
postać przewlekła i ciężka
Progresja od stadium I lub II po dwóch latach
ustabilizowanego
przebiegu
W stadium III i IV częsty jest grasiczak

Leczenie miastenii
- inhibitory cholinoesterazy
- tymektomia
- kortykosteroidy i inne środki immunosupresyjne
- terapia immunologiczna o krótkotrwałym działaniu
(plazmafereza,
immunoglobuliny)
Przełom miasteniczny
- osłabienie mięśni
- niewydolność oddechowa
- niepokój, lęk

Przełom cholinergiczny
- osłabienie mięśni,
- niewydolność oddechowa,
- objawy nikotynowe (drżenie, fascykulacje, bolesne kurcze mięśni),
- objawy muskarynowe (poty, nudności, kurczowe bóle brzucha,
nasilona motoryka jelit,
zwiększona sekrecja w tchawicy i
oskrzelach),
- niepokój, lęk.

Przełom cholinergiczny
- osłabienie mięśni,
- niewydolność oddechowa,
- objawy nikotynowe (drżenie, fascykulacje, bolesne kurcze mięśni),
- objawy muskarynowe (poty, nudności, kurczliwe bóle brzucha,
nasilona motoryka jelit,
zwiększona sekrecja w tchawicy i
oskrzelach),
- niepokój, lęk.
Zespół miasteniczny Lamberta – Eatona
- w 80% wiązany z rakiem drobno komórkowym oskrzela
- może występować z chorobą Addisona – Biermera, niedoczynnością
tarczycy,
nadczynnością tarczycy, cukrzycą t. I
- osłabienie mm. kończyn i tułowia, u 70% lekkie przejściowe objawy
oczne, Odruchy
osłabione, lub zniesione, zaburzenia
wegetatywne,
- EMG – po stymulacji bodźcami elektrycznymi. Pierwsze potencjały
czynnościowe mięśnia są niskie, ale wraz ze zwiększającą się częstością
bodźców ich amplituda rośnie

Zespół miasteniczny Lamberta – Eatona
- w 80% wiązany z rakiem drobnokomórkowym
oskrzela
- może występować z chorobą Addisona –
Biermera, niedoczynnością tarczycy,
nadczynnością tarczycy, cukrzycą t.I
- osłabienie mm. kończyn i tułowia, u 70%
lekkie przejściowe objawy oczne, odruchy
osłabione, lub zniesione, zaburzenia wegetatywne,
- EMG – po stymulacji bodźcami elektrycznymi,
pierwsze potencjały czynnościowe mięśnia są
niskie, ale wraz ze zwiększającą się częstością
bodźców ich amplituda rośnie

Charakterystyczne objawy miastenii:
- narastająca nużliwość mięśni w czasie ich aktywności, z
największym nasileniem objawów wieczorem,
- poprawa po spoczynku,
- często początek choroby dotyczy mięśni ocznych,
podniebienia miękkiego i
gardła,
- objawy mają zmienne nasilenie
- proces obejmuje mięśnia unerwione przez różne nerwy
obwodowe
- nie ma zaburzeń czucia, ani bólów,
- poprawa lub ustąpienie objawów po podaniu inhibitora
acetylocholinesterazy
- w EMG spadek amplitudy potencjałów przy powtarzanym
drażnieniu bodźcami
elektrycznymi
- w surowicy autoprzeciwciała przeciw receptorom
acetylocholinowym
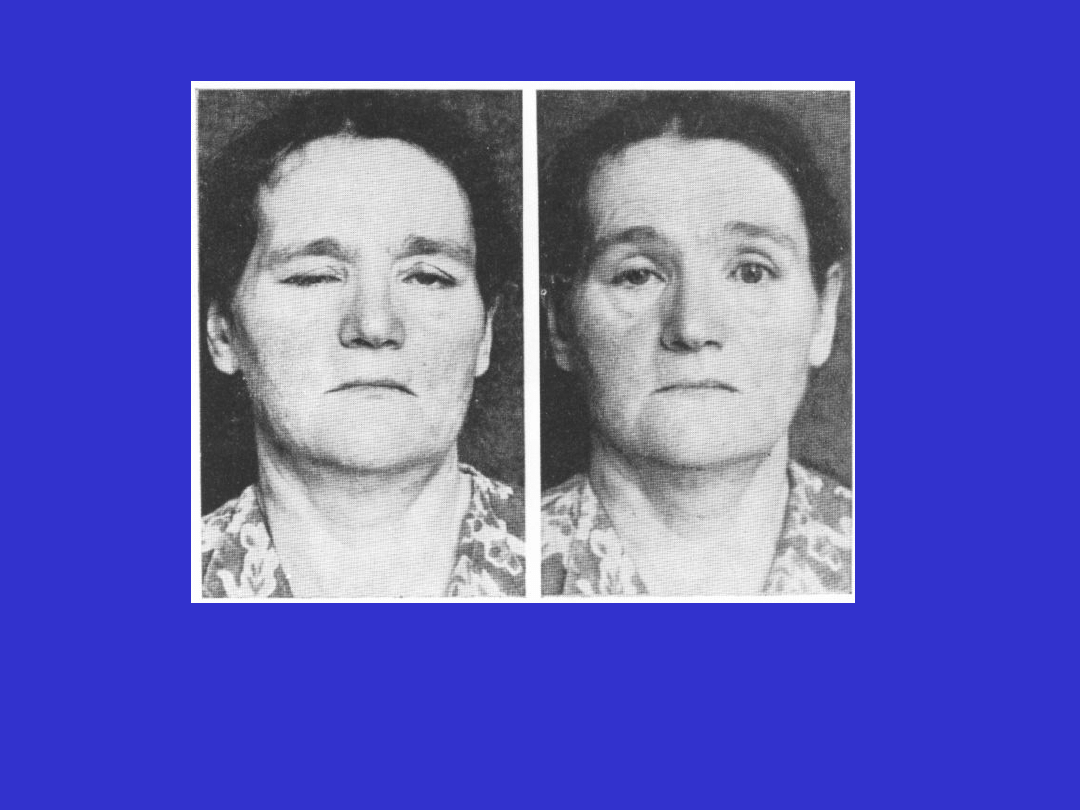
Miastenia

Współistnienie z miastenią innych
schorzeń
- grasiczak
10,0%
- nadczynność tarczycy
5,7%
- niedoczynność tarczycy
5,3%
- zapalenie stawów
3,6%
- niedokrwistość Addison-Biermera
0,9%
- toczeń rumieniowaty układowy
0,2%
- sarkoidoza
0,2%

Leczenie miastenii
- inhibitory cholinoesterazy
- tymektomia
- kortykosteroidy i inne środki immunosupresyjne
- terapia immunologiczna o krótkotrwałym działaniu
(plazmafereza,
immunoglobuliny)
Przełom miasteniczny
- osłabienie mięśni
- niewydolność oddechowa
- niepokój, lęk

Choroby mięśni

Objawy chorób mięśni:
- osłabienie mięśni z osłabieniem lub zniesieniem odruchów
- zanik mięśni,
- męczliwość mięśni,
- ból mięśni
- sztywność mięśni,
- kurcze mięśni,
- mioglobinuria,
- inne objawy (miotonia, fascykulacje, miokimie, przerost mięśni,
przykurcze mięśni)
-
Badania dodatkowe w miopatiach
- elektromiografa i elektroneurografa
- enzymy mięśniowe w surowicy (kinaza kreatynowa, Alat, Aspat,
dehydrogeneza
mleczanowa, aldolaza),
- badania immunologiczne surowicy (zapalenie mięśni),
- badania przeciwciał (miastenia),
- elektrolity w surowicy (porażenie okresowe, endokrynopatie,
zaburzenia metaboliczne,
nowotwory),
- biopsja mięśnia,
TK i MR mięśni (zapalenie mięśni – obraz jednorodny, wzrost
zawartości wody),
dystrofe – obraz heterogenny, czasem obecność
tłuszczu),

Systematyka
miopatii

- Dystrofie mięśniowe:
o dystrofa mięśniowa postępująca (Duchenne’a – Beckera),
o dystrofa kończynowo-obręczowa,
o dystrofa łopatkowo-strzałkowa,
o dystrofa twarzowo-łopatkowo-ramienna
o dystrofa odsiebna
o dystrofa miotoniczna Steinerta
- Miotonie
o Miotonia wrodzona Thomsena,
o Miotonia wrodzona Eulenburga,
- Porażenie okresowe
o porażenie okresowe hipokaliemiczne,
o porażenie okresowe hiperkaliemiczne
o porażenie okresowe normokaliemiczne,
- Miopatie metaboliczne
o Zobacz przemiany węglowodanów, miopatie w lipidozach,

- Miopatie mitochondrialne i encefalomiopatie
o postępująca oftalamoplegia zewnętrzna,
o zespół Kearnsa i Sayre’a,
o zespół MERFF,
o zespół MELAS,
- Miopatie wrodzone
o miopatia central core,
o miopatia nemalinowa (nitkowata),
- Miopatie zapalne
o zapalenie wielomięśniowe,
o zapalenie skórno-mięśniowe
o zapalenie wielomięśniowe i skórno-mięśniowe w nawrotach
złośliwych,
o zapalenie wielomięśniowe w kolagenozach, sarkoidoza
o zapalenie mięśni w zakażeniach

- Miopatie w endokrynopatiach
o niedoczynność tarczycy,
o nadczynność tarczycy,
o choroba Cushinga
o akromegalia
o niedoczynność przytarczyc
o nadczynność przytarczyc
o miopatia posteroidowa
- Objawy mięśniowe z zaburzeniach elektrolitycznych
o hipo i hiperkaliemia,
o hiponatremia,
o hipo i hiperkalcemia ,
o hipomagnezemia,
o hipofosfatemia,
- Miopatie toksyczne i indukowane przez leki
o miopatia alkoholowa
o kokaina, heroina
o kolchicyna, chlorochina, winkrystyna
o leki antylipemiczne
o penicylamina, cymetydyna
o niedobór witaminy E

Miopatae mitochondrialne
- Zespół Kearnsa – Sayre’a
niski wzrost, obustronna oftalmoplegia
zewnętrzna, zaburzenia
móżdżkowe,
niedorozwój umysłowy
- zespół MELAS (mitochondrialne
encefalomiopatia z kwasicą mleczanową i
epizodami udarowymi)
otępienie, napady drgawek, głuchota
- zespół MERRF (mitochondrialna
encefalomiopatia z
padaczką miokloniczną i
włóknami szmatowatymi w mięśniach) ataksje,
zanik nerwu wzrokowego, głuchota.

Polineuropatie

Objawy kliniczne polineuropatii
- parestezje i zaburzenia czucia
pojawiają się zwykle symetrycznie, dotyczą głównie dystalnych
części kończyn, pojawiają się najpierw w kończynach dolnych
rozmieszczenie przypomina skarpety i rękawiczki
zanik czucia wibracji w odsiebnych odcinkach kończyn lub
epikrytyczne zaburzenia czucia na czubkach palców.
- ruchowe objawy ubytkowe
pojawiają się nieco później. Są zazwyczaj symetryczne
rozpoczynają się w kończynach dolnych zwykle dotyczą
grzbietowych prostowników stopy i palców.
W późniejszym stadium mogą być objęte ręce
- zniesienie odruchów
najbardziej charakterystyczny objaw polineuropatii
przede wszystkim obustronny brak odruchów skokowych
nieco później zniesienie innych odruchów

- zaburzenia troficzne
zaniki mięśniowe
zmniejszenie wydzielania potu, czasem wzmożenie
skóra sucha i gładka
owrzodzenie trofczne, zwykle na podeszwowej powierzchni
stopy
zmiany dystrofczne policzków stopy
- bolesność uciskowa nerwów
np. ból uciskowy łydki
- ataksja
u chorych z ciężkimi zaburzeniami czucia – polineuropatia
pseudotabetyczna

Badania dodatkowe w polineuropatiach
- elektromiografie
dla rozpoznania różnicowego porażeń neurogennych i miogennych
(potencjały
fbrylacyjne, znamiona reinerwacji przy maksymalnym
wysiłku, zapis interferencyjny)
- elektroneurografie
dla potwierdzenia polineuropatii oraz odróżnienia polineuropatii
aksonalnej od demielinizacyjnej
wczesny zanik przewodnictwa czuciowego znaczne zwolnienie
przewodnictwa
ruchowego, ale dopiero wtedy, gdy doszło do
znacznego uszkodzenia osłonki
mielinowej
- biopsja mięśni
w celu odróżnienia porażeń neurogennych od miopatycznych
wykazanie
ewentualnego zapalenia naczyń
- biopsja nerwów
najczęściej biopsja nerwu łydkowego pozwala na odróżnienie
polineuropatii zapalnej od demielinizacyjnej
wykazuje obecność zapalenia naczyń
wykazuje obecność złogów (np. amyloidu)
wykazuje np. struktury cebulaste, komórki Schwanna (w
dziedzicznej polineuropatii
czuciowo-ruchowej

- nakłucie lędźwiowe
często wynik prawidłowy rozszczepienie białkowo-komórkowe w
zespole Guillaina-
Barre’go
Podwyższenie zawartości białka (polineuropatie demielinizacyjne,
choroba Refsuma,
nerwiaki korzeniowe)
Pleocytoza w zapaleniu oponowo-korzeniowo-nerwowym
Komórki nowotworowe
- OB.
Różnicowanie kolagenoz, zapalnych dys- i paraproteinemii, oraz
choroby nowotworowej
- morfologia
stany zapalne, zatrucia ołowiem (zasadochłonne nakrapiania),
białaczki, policytemie
- glikemia
cukrzyca
- kreatynina, mocznik
mocznica
- próby wątrobowe
zaburzenia krzepliwości,
neuropatia alkoholowa
- badania czynności tarczycy
niedoczynność tarczycy
- stężenie w surowicy witaminy B
12
, kwasu foliowego,
tiaminy (witamina B
1
)
witaminy E
upośledzenie wchłaniania, niedobory pokarmowe

Serologiczne próby reumatyczne, przeciwciała
przeciwjądrowe, krążące kompleksy immunologiczne
reumatyczne zapalenie wielostawowe
kolagenozy
- elektroforeza białek surowicy krwi
kolagenozy, dys- i paraproteinowe
- badania bakteriologiczne, serologiczne
neuropatia zakaźna i okołozakaźna
- uro- i koproforfryny w moczu
porfrie
- stężenie w surowicy kwasu fitanowego
choroba Refsuma
- chromatografia gazowa – badanie w kierunku
trucizn i metali ciężkich
neuropatie egzotoksyczne

- badania szpiku kostnego
białaczki, szpiczak
- badania radiologiczne
stany paranowotworowe (rtg klatki piersiowej, pasaż
żołądkowo-
dwunastniczy, badania jelita grubego)
szpiczak (rtg czaszki i kręgosłupa)
zatrucia ołowiem (rtg kości długich)
- badania endoskopowe
zespoły paranowotworowe
- badania scyntygraficzne
przerzuty nowotworowe do kości, szpiczak

Najczęściej występujące
polineuropatie
- Polineuropatie uwarunkowane genetycznie
dziedziczne polineuropatie czuciowe i ruchowe
o polineuropatie ze skłonnością do porażeń z ucisku
o w porfrii
o skrobiawicy pierwotnej
- Polineuropatie spowodowane zaburzeniami
metabolicznymi
o cukrzycowa (symetryczna dystalna, asymetryczna
proksymalna,
mononeuropatia, zanik mięśnia lub
mielopatia)
o uremiczna
o w marskości wątroby
o w dnie moczanowej
o w niedoczynności tarczycy

- Polineuropatie w wyniku niedoborowego lub
wadliwego żywienia
- Polineuropatie spowodowane wadliwym
wchłanianiem witaminy B
12
- Polineuropatie w dysproteinemii i paraproteinemii
- Polineuropatie w chorobach zakaźnych
Trąd, zapalenie przyusznic, mononukleoza, dur brzuszny i
paradury, dur
plamisty, zakażenie wirusem HIV, błonica,
botulizm, po ukąszeniu kleszcza
- Polineuropatie w sprue lub innych zaburzeniach
wchłaniania
- Polineuropatie w zatruciach egzogennych
Rodniki etylowe, ołów, arsen, tal rozpuszczalniki organiczne,
leki
(izoniazyd, talidomid, nitrofurantoina)
- Inne polineuropatie
Serogenne (posurowicze), w sarkoidozie, nowotworowe

Dziedziczne neuropatie ruchowo-czuciowe
Typ I (choroba Charcota-Marie’a Tootha)
- dziedziczenie autosomalne dominujące
- początek w 2 –4 dekadzie życia
- zaniki mm dystalnych odcinków kończyn
- deformaje stóp
- nieznaczne zaburzenia czucia
- wyraxne zwolnienie szybkości przewodnictwa nerwowego
- pogrubienie i zwiększenie konsystencji nerwów obwodowych
- w biopsji nerwu łydkowego – zwyrodnienie osiowe, demielinizacja z
cechami remienizacji,
cebulowato zmienione komórki Schwanna
-
Typ II (neuronalny, strzałkowy zanik mięśni)
- dziedziczenie autosomalne dominujące
- początek w 2 –4 dekadzie życia
- dystalne zaniki mięśniowe głównie na stopach i podudziach
- stopa wydrążona
- nieznaczne zaburenia czucia
- szybkość przewodzenia nieznacznie zwolniona lub prawidłowa
- nerwy obwodowe niepogrubiałe
- w biopsji nerwu łydkowego – zwyrodnienie osiowe z nieznaczną
segmentową demielinizacją,
komórki Schwanna niezmienione

Typ III (neuropatia przerostowa Dejerine’a - Sottasa)
- dziedziczenie autosomalne recesywne
- początek w 1 dekadzie życia
- opóźnienie rozwoju motorycznego szybki postęp choroby, wyraźne
niedowłady
- wyraźne zaburzenia czucia
- znaczące zwolnienie szybkości przewodzenia (większe niż w typie I)
- pogrubiałe nerwy obwodowe, o miękkiej konsystencji
- w biopsji nerwu łydkowego – hypomielinizacja, de- i remielinizacja,
zmienione cebulowato komórki Schwanna, śródnerwie wyraźnie poszerzone
Typ IV (neuropatia przerostowa w chorobie Refsuma)
- dziedziczenie autosomalne recesywne
- początek w 1-3 dekadzie życia
- retinitis pigmentosa, neuropatia czuciowo-ruchowa zaburzenia słuchu,
objawy sercowe i skórne
deformacje szkieletowe
- wyraźne zwolnienie przewodnictwa
- w biopsji nerwu łydkowego – zwyrodnienie osiowe, segmentowa de- i
remielinizacja, komórki cebulowate, złogi lizosomalne w komórkach
Schwanna
- podwyższone stężenie kwasu ftanowego w surowicy, gromadzenie
kwasu ftanowego w
różnych tkankach

Typ V (ze spastyczną paraparezą)
- dziedziczenie autosomalne dominujące
- początek w 2 dekadzie życia lub później
- przebieg wolno postępujący z towarzyszącym niedowładem
spastycznym
- brak subiektywnych i obiektywnych zaburzeń czucia
- przewodnictwo prawidłowe lub nieznacznie poniżej normy
- w biopsji nerwu łydkowego – wyraźne zmniejszenie włókien osiowych u
niektórych chorych
Typ VI (z zanikiem nerwu wzroku)
- dziedziczenie autosomalne dominujące
- początek w różnym wieku
- postępująca ślepota
- dystalne zaniki mięśni
- zaburzenia neurofzjologiczne – nieznane
- w pojedynczych przypadkach hipertrofczne zmiany w nerwach

Typ VII (z barwnikowym zwyrodnieniem siatkówki)
- dziedziczenie prawdopodobnie autosomalne recesywne
- początek w różnym wieku
- osłabienie mięśni dystalnych z zanikami
- nieznaczne dystalne zaburzenia czucia
- zwolnienie przewodnictwa

Uszkodzenie
obwodowego
układu
nerwowego w
cukrzycy

Czuciowo-ruchowa neuropatia cukrzycowa
- symetryczna, przeważnie dystalna
- parestezje i piekące bóle głównie kończyn dolnych
- zniesienie odruchów (głównie OA)
- zaburzenie czucia wibracji
- nadwrażliwość na ból
- odsiebne ubytki czucia czasem prowadzące do ataksji
- kurcze mięśni, zwłaszcza łydek
- osłabienie ruchowe prostowników grzbietowych stóp
- owrzodzenia, zniszczenie proksymalnych stawów palców stopy
Proksymalna, asymetryczna polineuropatia cukrzycowa
- rzadsze, niż postać dystalna
- jednostronne zajęcie kilku korzeni nerwowych lub zwoju
nerwowego
- nagły początek
- intensywne bóle narastające w nocy zlokalizowane w dosiebnych
odcinkach przeważnie kończyn dolnych
- osłabienie i zaniki mięśni (zginacze biodra, mięsień czworogłowy)
trudności w
chodzeniu po schodach lub wstawaniu z krzesła
- zniesienie odruchu kolanowego
- ubytki czucia w obrębie pola unerwienia udowego, lub brak
zaburzeń czucia

Mononeuropatia cukrzycowa
- szczególnie okoruchowe, bolesne, mięśnie oczne wewnętrzne
niedotknięte
- nerw łokciowy, promieniowy, kulszowy, strzałkowy, piszczelowy,
skórny uda boczny
mogą być następstwem działania czynników
mechanicznych, lub niedokrwienia nerwu
już uprzednio
uszkodzonego z powodu zaburzeń metabolicznych
Ostra zapalna polineuropatia demielinizacyjna i
poliradikuloneuropatia (zespól Guillaina – Barre,go
- u ¾ poprzedzona infekcją górnych dróg oddechowych lub
zaburzeniami żołądkowo -
jelitowymi (też szczepienia, operacje,
ciąża, nowotwory)
- parestezje (początkowo tylko w kończynach dolnych) i bóle
- osłabienie mięśni rozpoczynające się od stóp i postępujące
dosiebnie
- może dojść do tetraplegii z porażeniem mięśni twarzy,
zaburzeniami połykania i
oddychania
- rozszczepienie białkowo komórkowe w płynie m-r (w pierwszych 3
dniach płyn może
być prawidłowy
- zwyrodnienie szybkości przewodzenia, wydłużenie fali F
- u większości objawy stopniowo cofają się w odwrotnej kolejności
do ich pojawienia

Przewlekła, zapalna nawracająca poliradikuloneuropatia
- przebieg przewlekły, lub progresywny z rzutami
- zajęcie również mięśni odsiebnych i wczesny zanik
- rokowania mniej pomyślne jak w zespole Guillaina – Barre’go

Bólowe zespoły
korzeniowe

Ogólna symptomatologia
uszkodzeń korzeni rdzeniowych
- bóle, w większości przypadków z typowym
promieniowaniem
korzeniowym
- ubytki czucia odpowiadające dermatomom, nie zawsze
łatwe do
sprecyzowania
- niedowład odpowiadający korzeniowemu unerwieniu
pojedynczych mięśni
(nigdy całkowite porażenie
mięśnia)
- zanik mięśni mniej nasilony, niż w uszkodzeniu nerwów
obwodowych i
widoczny przeważnie dopiero po 3
tygodniach
- osłabienie odruchów (rzadko całkowity brak)
- drżenie pęczkowe (wyjątkowo, w przewlekłym
uszkodzeniu)
- objawy ze strony kręgosłupa, bóle, nieprawidłowe
ustawienie ograniczenie
ruchomości

Przepukliny krążków międzykręgowych i zmiany
zwyrodnieniowe w odcinku szyjnym kręgosłupa - rwa
ramienna
bóle karku promieniujące do ramienia, wzdłuż kończyny
górnej do palców,
niekiedy promieniowanie do łopatki i
przedniej części klatki piersiowej
napięcie i przykurcz mięśni karku
bólowe ograniczenie ruchów szyi
bolesność w dole nadobojczykowym
wyzwalanie bólu przez pociąganie do tyłu w stawie
barkowym
wyprostowanego ramienia
osłabienie odruchów
zaburzenia czucia
najczęstsze zespoły to C
7
, C
6
i
C8
potwierdzenie rozpoznania: rezonans magnetyczny
ewentualnie TK, zdjęcie
skośne kręgosłupa szyjnego

Typowy wywiad w przypadkach przepukliny jądra
miażdżystego krążka międzykręgowego w odcinku
lędźwiowym - rwa kulszowa
prawie zawsze „postrzały” w wywiadzie
„uraz” doznany podczas dźwigania ciężkich przedmiotów
wysiłek w pozycji
schylonej lub skręconej
objawy początkowe w obrębie pleców z upośledzeniem
ruchów
promieniowanie bólu do kończyny dolnej lub stopy
stałe umiejscowienie bólu z nasileniem przy kaszlu,
kichaniu, parciu
subiektywne odczuwanie zaburzeń czucia
utrudnienie chodzenia na palcach i piętach
zmienność strony występowania rwy kulszowej (duża
przepuklina)
zaburzenia oddawania moczu (ucisk na ogon koński)

Typowe objawy stwierdzone w badaniu przedmiotowym u
chorych z lędźwiową przepukliną jądra miażdżystego
spłycenie lordozy lędźwiowej
ewentualne skrzywienie kręgosłupa
ograniczenie ruchomości kręgosłupa
bóle przy ruchach kręgosłupa
bolesność uciskowa i opukowa wyrostków kolczastych
wzmożenie napięcia mięśni przykręgosłupowych, często z
bolesnością uciskową
ewentualna bolesność przy ucisku długie osi kręgosłupa
(objaw szczytowy)
dodatni objaw Laseque’a
ewentualnie dodatni, skrzyżowany objaw Laseque’a (przy
dużej przepuklinie lub
wypadnięciu jądra miażdżystego)
bolesność uciskowa w punktach Valleixa
dodatni objaw Neriego (zgięcie w stawach kolanowych przy
zginaniu karku do
przodu)
osłabienie odruchów głębokich (często brak OK i OA)
niedowład mięśni( pośladkowy wielki, prostowniki stawu
kolanowego, dźwigacze
stopy, prostownik palucha, zginacz
stopy i palców)
zanik mięśni (pomiar obwodu uda i łydki)
zaburzenia czucia - przednia powierzchnia uda i podudzia,
grzbiet i boczny brzeg
stopy

Rwa udowa
promieniowanie bólu wzdłuż przedniej powierzchni
uda
dodatni objaw Mackiewicza
zaburzenia czucia na przedniej powierzchni uda
osłabienie lub zniesienie odruchu kolanowego
czasem niedowład i zanik mięśnia czworogłowego
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
- Slide 23
- Slide 24
- Slide 25
- Slide 26
- Slide 27
- Slide 28
- Slide 29
- Slide 30
- Slide 31
- Slide 32
- Slide 33
- Slide 34
- Slide 35
- Slide 36
- Slide 37
- Slide 38
- Slide 39
- Slide 40
Wyszukiwarka
Podobne podstrony:
6 tydzień, VI Wielki Poniedziałek
6 tydzień, VI Wielki Czwartek (Msza z poświęceniem krzyżma)
II tydzie, VI rok, VI rok, Pediatria, Pediatria, PEDIATRIA OLA
6 tydzień, VI Wielki Czwartek (Msza Wieczerzy Pańskiej)
6 tydzień, VI Wielki Wtorek
6 tydzień, VI Wielka Środa
6 tydzień, VI Wielki Poniedziałek
6 tydzień Wielkanocy, VI Niedziela Wielkanocna A
6 tydzień Wielkanocy, VI środa
6 tydzień Wielkanocy, VI czwartek
6 tydzień Wielkanocy, VI Niedziela Wielkanocna B
6 tydzień Wielkanocy, VI sobota
6 tydzień Wielkanocy, VI poniedziałek
VI tydzień Menstruacja, Geografia Intymna
6 tydzień Wielkanocy, VI wtorek
6 tydzień Wielkanocy, VI piątek
6 tydzień Wielkanocy, VI Niedziela Wielkanocna C
6 tydzień Wielkanocy, VI Niedziela Wielkanocna A
więcej podobnych podstron