
The Journal of Nutrition
Proceedings of the Fourth International Scientific Symposium on Tea and Human Health
Targeting Multiple Neurodegenerative Diseases
Etiologies with Multimodal-Acting Green
Tea Catechins
1,2
Silvia A. Mandel,* Tamar Amit, Limor Kalfon, Lydia Reznichenko, and Moussa B. H. Youdim
Eve Topf Center for Neurodegenerative Diseases Research and Department of Pharmacology, Faculty of Medicine, Technion,
Haifa, Israel
Abstract
Green tea is currently considered a source of dietary constituents endowed with biological and pharmacological activities
relevant to human health. Human epidemiological and new animal data suggest that the pharmacological benefits of tea
drinking may help to protect the brain as we age. Indeed, tea consumption is inversely correlated with the incidence
of dementia and Alzheimer’s and Parkinson’s diseases. In particular, its main catechin polyphenol constituent (2)-
epigallocatechin-3-gallate has been shown to exert neuroprotective/neurorescue activities in a wide array of cellular and
animal models of neurological disorders. The intense efforts dedicated in recent years to shed light on the molecular
mechanisms participating in the brain protective action of green tea indicate that in addition to the known antioxidant
activity of catechins, the modulation of signal transduction pathways, cell survival/death genes, and mitochondrial function
all contribute significantly to the induction of neuron viability. Because of the multietiological character of neurodegen-
erative disease pathology, these natural compounds are receiving significant attention as therapeutic cytoprotective
agents that simultaneously manipulate multiple desired targets in the central nervous system. This article elaborates on
the multimodal activities of green tea polyphenols with emphasis on their recently described neurorescue/neuro-
regenerative and mitochondrial stabilization actions.
J. Nutr. 138: 1578S–1583S, 2008.
Introduction
Despite the lack of well-controlled clinical trials with tea
polyphenols in neurodegenerative diseases, human epidemiolog-
ical and new animal data suggest that the pharmacological
benefits of tea drinking may help protect the brain as we age.
Indeed, tea consumption is inversely correlated with the incidence
of dementia, Alzheimer’s disease (AD),
3
and Parkinson’s disease
(PD), which may explain why there are significantly lower rates of
age-related neurological disorders among Asians than in Euro-
peans or Americans (1). In a cross-sectional study conducted in
Japan aimed at investigating the association between consump-
tion of green tea and cognitive function in elderly Japanese
subjects, it was found that consumption of 2 or more cups/d (100
mL/cup) of green tea is associated with lower prevalence of
cognitive impairment (2). In a case-control study in the United
States, it was found that people who consumed 2 cups/d or more
of tea presented a decreased risk of PD (3). In support of this
finding, a recent prospective cohort study of nearly 30,000
Finnish adults aged 25–74 y followed for 13 y found that drinking
3 or more cups (200 mL/cup) of tea is associated with a reduced
risk of PD (4). These findings emphasize the importance of well-
designed controlled studies to assess risk reduction for PD and AD
in consumers of green and black tea. The Michael J. Fox
Foundation has awarded a grant to the team of Piu Chan from
Xuanwu Hospital, Beijing, China, to carry out the first-ever
3
Abbreviations used: Ab, amyloid b-peptide; AD, Alzheimer’s disease; APP,
amyloid precursor protein; COMT, catechol-O-methyl transferase; DA, dopa-
mine; DAT, dopamine transporter; EGCG, (2)-epigallocatechin-3-gallate; ERK1/2,
extracellular signal-regulated kinase; MAPK, mitogen-activated protein kinase;
MPTP, N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; 6-OHDA, 6-hydroxydopa-
mine; OS, oxidation stress; PD, Parkinson’s disease; PKC, protein kinase C;
sAPPa, soluble amyloid precursor protein-a; SOD, superoxide dismutase.
1
Published in a supplement to The Journal of Nutrition. Presented at the
conference ‘‘Fourth International Scientific Symposium on Tea and Human
Health,’’ held in Washington, DC at the U.S. Department of Agriculture on
September 18, 2007. The conference was organized by the Tea Council of the
U.S.A. and was cosponsored by the American Cancer Society, the American
College of Nutrition, the American Medical Women’s Association, the American
Society for Nutrition, and the Linus Pauling Institute. Its contents are solely the
responsibility of the authors and do not necessarily represent the official views of
the Tea Council of the U.S.A. or the cosponsoring organizations. Supplement
coordinators for the supplement publication were Lenore Arab, University of
California, Los Angeles, CA and Jeffrey Blumberg, Tufts University, Boston, MA.
Supplement coordinator disclosure: L. Arab and J. Blumberg received honorar-
ium and travel support from the Tea Council of the U.S.A. for cochairing the
Fourth International Scientific Symposium on Tea and Human Health and for
editorial services provided for this supplement publication; they also serve as
members of the Scientific Advisory Panel of the Tea Council of the U.S.A.
2
Author disclosures: S. A. Mandel received travel support from the Tea Council
of the U.S.A. for speaking at the Fourth International Scientific Symposium on
Tea and Human Health and for preparing this manuscript for publication; T. Amit,
L. Kalfon, L. Reznichenko, and M. B. H. Youdim, no conflicts of interest.
* To whom correspondence should be addressed. E-mail: mandel@tx.technion.
ac.il.
1578S
0022-3166/08 $8.00
ª 2008 American Society for Nutrition.
jn.nutrition.org
Downloaded from

multicenter, double-blind, randomized, placebo-controlled study
to investigate the safety, tolerability, and potential neuroprotec-
tive effects of green tea polyphenols in patients with early PD.
Intense efforts have been dedicated during the past 5 y to shed
light on the molecular mechanisms and cell-signaling pathways
participating in the neuroprotective/neuroregenerative action
of green tea. The emerging data indicate that the antioxidant/
metal-chelating attributes of the catechin polyphenols are un-
likely to serve as the sole explanation for their neuroprotective
and neurorescue capacity. This article presents the state of the
art in the molecular mechanisms and cell-signaling pathways
implicated in the neuroprotective action of green tea catechins,
with emphasis on their recently described neurorescue effect and
mitochondrial stabilization potency.
Etiopathology of neurodegenerative diseases
Neurodegenerative disorders are progressive diseases of the
nervous system involving damage or loss of neurons in the brain
and/or spinal cord, which can occur at any time of life.
Neurodegeneration in PD or AD or other neurodegenerative
diseases, such as Huntington disease and amyotrophic lateral
sclerosis, appears to be multifactorial, where a complex set of
toxic reactions lead to the demise of neurons (5,6). Common
features involve impairment of protein handling and aggregation
associated with dysfunction of the ubiquitin-proteasome system,
depletion of endogenous antioxidants, reduced expression of
trophic factors, inflammation, glutamatergic excitotoxicity, ex-
pression of proapoptotic proteins, and increases of iron and
nitric oxide leading to oxidative-stress (OS) damage (7–9). An
unresolved question, however, is to determine which of these
factors constitute the primary event, the sequence in which they
act, and where the point of convergence is or the final pathway
by which the predisposed neuronal cell types die in the affected
brain areas. Because of the multietiological character of the
pathology, novel therapeutic neuroprotective strategies support
the idea that simultaneous manipulation of multiple desired
targets in the central nervous system will exert higher therapeu-
tic effectiveness (10,11). Thus, it is not surprising, that green tea
catechins have attracted increasing interest as therapeutic
cytoprotective agents for the treatment of neurological disorders
because of their broad spectrum of biological/pharmacological
activities, including cardiovascular, antiinflammatory, and anti-
carcinogenesis effects (12–14) and, more recently recognized,
antidiabetic (15,16), antiobesity (17), and neuroprotective/
neurorestorative properties (18).
Neuroprotection/neurorescue by green
tea polyphenols
There is a growing recognition that polyphenolic catechins exert
a protective role in neurodegeneration. The neuroprotective
effect has been long established in animal models of neurological
disorders: (2)-epigallocatechin-3-gallate (EGCG), the major
polyphenol component of green tea, has been shown to improve
age-related cognitive decline and to protect against cerebral
ischemia/reperfusion injuries (19,20) and brain inflammation
and neuronal damage in experimental autoimmune encephalo-
myelitis (21). Furthermore, the treatment of EGCG significantly
prolonged the symptom onset and life span and attenuated death
signals in a mouse amyotrophic lateral sclerosis model with the
human G93A-mutated Cu/Zn-superoxide dismutase (SOD)
gene (SOD1) (22). Similarly, a green tea polyphenol extract or
isolated EGCG prevented striatal dopamine (DA) depletion and
substantia nigra dopaminergic neuron loss when given chron-
ically to mice treated with the parkinsonism-inducing neuro-
toxin, N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)
(23). More recently, long-term administration of a preparate
of green tea catechins (polyphenol E) or EGCG has been dem-
onstrated to improve spatial cognition and learning ability in
rats (24) and to reduce cerebral amyloidosis in Alzheimer’s
transgenic mice, respectively (25).
In line with the in vivo findings, cell culture studies have
demonstrated that green tea catechins prevented neuronal cell
death caused by the neurotoxins 6-hydroxydopamine (6-OHDA),
1-methyl-4-phenylpyridinium and amyloid b-peptide (Ab) (26–
29). More recently, EGCG was reported to exert a neurorescue
activity in long-term serum-deprived rat pheochromocytoma
(PC12) cells and to promote neurite outgrowth (30). It remains
to be established whether there is any mechanistic relation
between survival and differentiation induced by EGCG and
also to what extent the in vitro findings could be replicated in
vivo. To test this assumption, we have examined the possible
neurorescue/neurorestorative activity in a post-MPTP-induced
nigrostriatal DA neurodegeneration model of PD in mice. MPTP
(20 mg/kg, i.p., per d) was administered for 4 d, followed by a
further 4-d resting period, and, at d 8, EGCG (5 mg/kg or water)
was administered orally, over a total treatment period of 22 d.
MPTP caused a significant reduction in viability of tyrosine-
hydroxylase-positive cells, whereas oral EGCG administration
post-MPTP resulted in a substantial recovery of the neurons.
Current experiments are in progress to determine effective doses
and duration of treatment. Thus, the neurorescue action of
EGCG may suggest a potential disease-modifying effect for the
drug, similar to what has been recently described for the novel
anti-Parkinson drug rasagiline (N-propargyl-1(R)-aminoindan),
a second-generation selective inhibitor of monoamine oxidase-B
(31).
Molecular mechanisms of neuroprotective/
neurorescue action of EGCG
The protein kinase C pathway.
Emerging evidence suggests
that the biological actions of green tea catechins relate not simply
to their antioxidant/radical-scavenging potential but also to the
modulation of various protein kinase signaling pathways. Our
recent in vitro cell-signaling studies on the neuroprotective
mechanistic action of EGCG revealed a specific involvement of
protein kinase C (PKC) (26,32), a family of serine/threonine
kinases consisting of 11 isoforms, which are divided into 3
subclasses: conventional (a, b
I
, b
II
, g), novel (d, e, u, h, m), and
atypical [i (mouse)/l (human), z] (33). PKC has a fundamental
role in the regulation of cell survival, programmed cell death,
long-term potentiation (34), and consolidation of different types
of memory (35,36). Indeed, it has been suggested that phar-
macological interventions aimed at modulating specific PKC
isozymes or PKC-mediated signal transduction pathways may
constitute a potential therapeutic tool for senescence or age-
related pathologies that are responsible for memory disturbances
(37). The induction of PKC activity in neurons is thought to be a
prerequisite for neuroprotection against several exogenous
insults. Indeed, PKCe activation after ischemic preconditioning
or pharmacological preconditioning (with either PKCe, NMDA,
or A1AR agonists) was shown to be essential for neuroprotection
against oxygen/glucose deprivation in organotypic slice cultures
(38). In accordance, activation of PKC by estrogen or by the grape
flavonoid resveratrol protected rat cortical or hippocampal
neurons against Ab toxicity, respectively (39,40).
PKC activation by EGCG prevents apoptosis and mito-
chondrial membrane potential collapse.
A rapid phosphor-
ylative activation of PKC by EGCG is thought to be the main
Targeting neuroprotection with green tea catechins
1579S
jn.nutrition.org
Downloaded from

mechanism accounting for its neuroprotective activity against
several neurotoxins such as Ab (28), serum withdrawal (30,41),
and 6-OHDA (26) and for its neurorescue effect against long-
term growth factor withdrawal (30). In addition, EGCG induced
a rapid translocation of the isoform PKCa to the membrane
compartment in response to EGCG in human astroglioma or rat
PC12 cells (30,42). This isozyme is particularly important in
neuronal growth and differentiation in the brain. These findings
are supported by animal studies showing that a 2-wk oral
consumption of EGCG prevented the extensive depletion of
PKCa isoform and counteracted the robust increase of Bax
protein in the striatum and the dopaminergic neurons of the
substantia nigra pars compacta of mice intoxicated with MPTP,
respectively (43).
Recently, we identified a novel pathway in the neuroprotective
mechanism of action of EGCG that involves a rapid PKC-
mediated degradation of the Bad protein by the ubiquitin-
proteasome system and a more pronounced reduction after 24 h
in cell culture (32). Bad may directly contribute to the opening of
the mitochondrial megachannel permeability transition pore by
its heterodimerization with the mitochondrial death suppressor
proteins Bcl-2 and/or BclxL, thus neutralizing their antiapoptotic
function (44). Indeed, we have recently found that the adminis-
tration of EGCG for 30 min prevented the dissipation of the
mitochondrial membrane potential, DCm, induced by short-term
(4 h) exposure to 6-OHDA (data not presented). This appears to
involve activation of the PKC signaling pathway because
pretreatment with the pharmacological general PKC inhibitor
GF109203X blunted the protective effect of EGCG on DCm.
PKC activation by EGCG is beneficial for AD and PD.
Neuronal amyloidosis in AD is characterized by extracellular
deposition of Ab peptide, derived from proteolytic cleavage of
amyloid precursor protein (APP), a type I integral membrane
protein. APP can be processed via alternative pathways: a
nonamyloidogenic secretory pathway includes cleavage of APP
to sAPPa by a putative a-secretase within the sequence of the
amyloidogenic Ab peptide, thus precluding the formation of Ab,
whereas the formation of Ab is regulated by the sequential
action of b- and g-secretases (45). Our pioneer studies have
demonstrated that either short- or long-term incubation with
EGCG promotes the generation of the nontoxic sAPPa via PKC-
dependent activation of a-secretase (28,46). New supportive
data came from a study conducted in an Alzheimer’s transgenic
mice model, showing that EGCG promotes sAPPa generation
through activation of a-secretase cleavage (25). This was ac-
companied by a significant reduction in cerebral Ab levels and
b
-amyloid plaques.
Another potential beneficial effect of PKC activation in AD is
related to the recent finding that neuronal overexpression of
PKCe in transgenic mice expressing familial AD mutant forms of
the human APP decreases Ab levels and plaque burden, and this
is accompanied by increased activity of endothelin-converting
enzyme, which degrades Ab (47). Because EGCG has been
shown to increase the levels of PKC isoforms a and e in mouse
hippocampus and striatum (28,43), it can be hypothesized that
in AD pathology, EGCG may reduce Ab levels, both via
concomitant stimulation of sAPPa secretion and promotion of
Ab clearance through increased endothelin-converting enzyme
activity.
In PD, a possible beneficial effect of green tea polyphenols
maybe related to the increased internalization of the DA
presynaptic transporters (DAT) by EGCG, eventually resulting
in a rise in the synaptic DA level. This effect was mimicked by
phorbol 12-myristate 13-acetate, a potent activator of PKC, and
abolished by blockade of the PKC pathway (48), suggestive of a
potential therapeutic target of PKC in the brain as a result of
green tea intake. This observation, together with the finding that
EGCG inhibited catechol-O-methyltransferase (COMT) activity
at a low IC50 concentration (0.2 mmol/L) in rat liver cytosol
homogenates (49), may be of particular significance for PD
patients given that DA and related catecholamines are physio-
logical substrates of COMT. Indeed, COMT inhibitors entaca-
pone and tolcapone, clinically prescribed to PD-affected
individuals, dose-dependently inhibit the formation of the major
metabolite of levodopa, 3-O-methyldopa, thereby improving its
bioavailability in the brain (50).
Other signaling pathways.
In addition to PKC, other cell-
signaling pathways have been implicated in the action of green
tea catechins, such as the mitogen-activated protein kinases
(MAPK), phosphatidylinositide 3#-OH kinase/AKT and protein
kinase A signaling cascades, and cell calcium influx regulation
[for review see Mandel et al. (18)]. These cascades have been
shown to play central functions in neuronal protection against a
variety of extracellular insults and to be essential for neuronal
differentiation and survival (51,52). In general, flavonoids can
activate MAPK signaling cascades in both neuronal and ex-
traneuronal tissues and neutralize the decline in the mitogen
and growth factor-induced extracellular signal-regulated kinase
(ERK1/2) activity caused by exogenous OS-inducing agents
(26,53). Low doses of (2)-epicatechin were recently shown to
stimulate phosphorylation of the cAMP-response element bind-
ing protein, a regulator of neuronal viability and synaptic
plasticity activity through both ERK1/2 and AKT in primary
cortical neurons (54). Using the same cell culture conditions, this
group of researchers demonstrated that activation/phosphorylation
of both kinases was also involved in the antiapoptotic action of
submicromolar concentrations of the flavanone hesperetin and
its metabolite, 5-nitro-hesperetin (55). A number of flavonoids
and phenolic antioxidants, at their respective low protective con-
centrations, were demonstrated to activate the expression of some
stress-response genes, such as the phase II drug-metabolizing
enzymes glutathione-S-transferase and heme-oxygenase-1, likely
via activation of the MAPK pathway (56). Although EGCG had
no effect on ERK1/2 phosphorylative levels in the absence of any
exogenous damage, it was able to counteract the decline in
ERK1/2 induced by 6-OHDA in neuroblastoma cells (26).
Antioxidant and iron chelating activity of green tea
polyphenols.
Tea catechins are powerful hydrogen-donating
antioxidants and free radical scavengers of reactive oxygen and
nitrogen species in in vitro systems (57–59). The neuroprotective
effect of green tea polyphenols may also involve the regulation of
antioxidant protective enzymes such as SOD and catalase in
mouse striatum (23). In peripheral tissue, it has been shown
that a number of flavonoids and phenolic antioxidants activate
the expression of some stress-response genes such as the phase II
drug metabolizing enzymes, glutathione-S-transferase and heme-
oxygenase-1 in correlation with an increase in the activity and
nuclear binding of the transcription factors Nrf1 and Nrf2 to the
antioxidant regulatory element sequences contained in their
promoters (60).
It is well established that iron progressively accumulates in
the brain with age, as well as in those brain areas affected by
neurodegenerative diseases, and is considered to be a major
contributor to OS (7,61). Transcranial sonography has detected
increased iron and decreased neuromelanin levels at the sub-
stantia nigra, even before the clinical manifestation of PD (62).
1580S
Supplement
jn.nutrition.org
Downloaded from
Similarly, analysis of AD brains indicates iron accumulation
within specific brain regions displaying selective vulnerability
to neurodegeneration, such as the hippocampus and cerebral
cortex (63,64), in particular in association with neurofibrillary
tangles and Alzheimer’s Ab-containing senile plaques, both
considered central pathological hallmarks of AD.
These observations have formed the basis for the implementation
of iron-complexing molecules that can cross the blood-brain
barrier and possess neuroprotective/neurorestorative activities
as a new therapeutic approach in neurologic disorders. Exam-
ples include the novel nontoxic lipophilic, brain-permeable
multifunctional iron chelators HLA20 and M30, in which the
N-propargylamine neuroprotective moiety of the antiparkinso-
nian drug rasagiline was incorporated into the skeleton of the
prototype iron chelator 8-hydroxyquinoline derivative VK28
(Varinel, West Chester, PA) [for review see Youdim and Buccafusco
(65)]. Recent lines of research reported that several metal-binding
natural antioxidants, including polyphenols of wine (e.g., resver-
atrol, myricetin, quercetin, kaemferol), curcumin, (1)-catechin,
(2)-epicatechin, nordihydroguaiaretic acid, and rosmarinic acid
inhibit formation of nascent Ab and a-synuclein fibrils, elongation
of the fibrils, and destabilization of the formed assemblies (66,67),
suggesting a promising therapeutic approach of naturally occur-
ring polyphenols for the treatment of neurodegenerative diseases.
In light of the multietiological character of neurodegenerative
disease pathology, novel pharmacological approaches suggest
the use of antioxidant metal-chelating molecules possessing 2 or
more active neuroprotective moieties that simultaneously ma-
nipulate multiple desired targets. A wealth of new data suggests
that green tea catechins may well fulfill the requirements for a
putative neuroprotective drug displaying diverse pharmacolog-
ical activities. Originally viewed as simple radical scavengers,
green tea catechin polyphenols are considered at present to be
compounds that invoke a spectrum of cellular mechanisms of
action related to their neuroprotection/neurorescue activities.
These mechanisms may include activation of signaling pathways
(e.g., PKC, MAPK, AKT); promotion of neurite outgrowth;
antioxidant action (direct radical scavenging and induction of
endogenous defenses such as SOD, catalase, and phase II de-
toxifying enzymes); antiapoptotic action (induction/reduction of
survival/death genes, respectively); bioenergetic action (mito-
chondrial stabilization); increase of synaptic DA (by promoting
DAT internalization and inhibition of COMT activity); prefer-
ential processing of APP by a-secretase to engender the
nonamyloidogenic sAPPa; reduction of Ab and a-synuclein
generation/fibrillization and plaque burden (a direct action on
formation of nascent or destabilization of assembled fibrils); and
reduction of membrane-associated APP hippocampal levels,
presumably via the iron-chelating effect on APP mRNA trans-
lation. Thus, EGCG may influence Ab levels, either via trans-
lational inhibition of APP or by stimulation of sAPPa secretion.
It cannot be ruled out that some of the biological effects de-
scribed above may share a common signaling pathway. For exam-
ple, activation of the PKC signaling pathway by EGCG (26,30)
might be responsible for the acute decrease in Bad protein (32)
as well as regulation of DA transporters (48) and elevation of
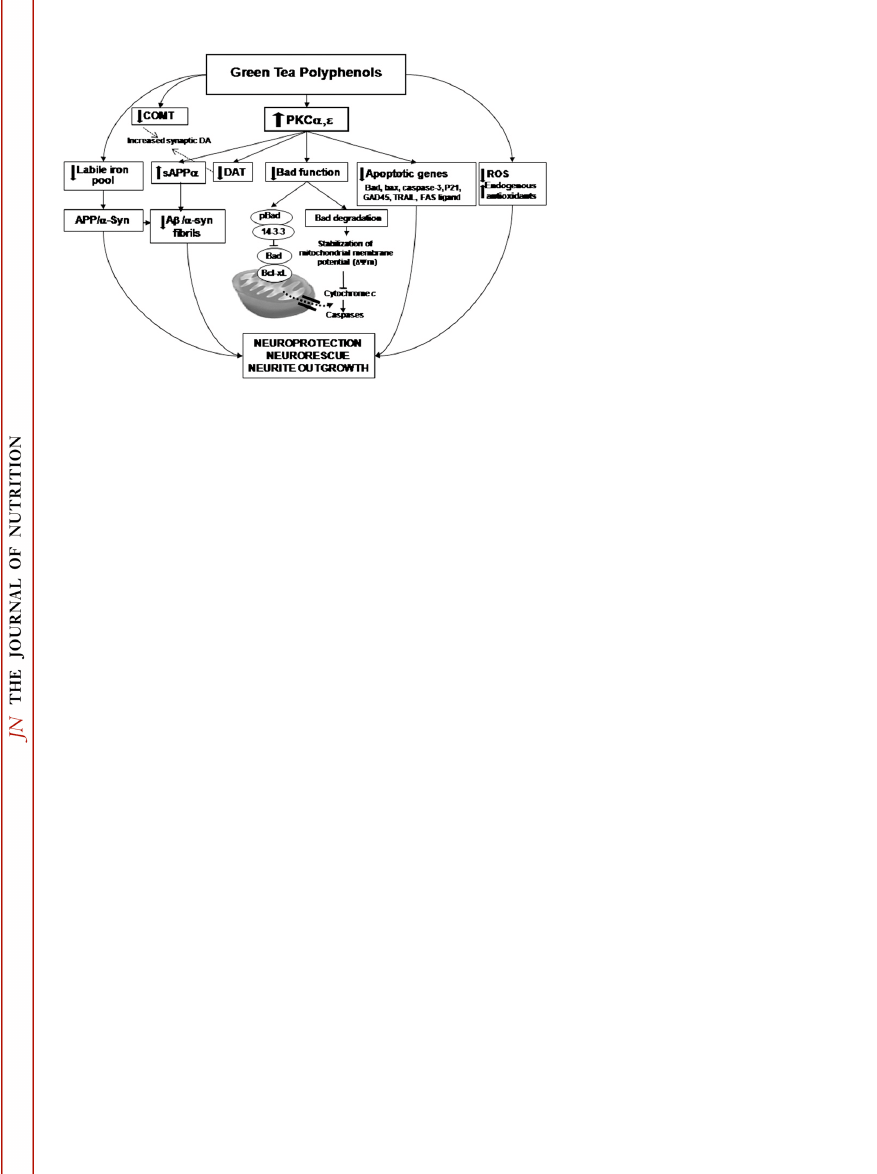
sAPPa secretion (28). A proposed schematic model for the neuro-
protective/neurorestorative effect by EGCG is illustrated in Fig. 1.
Collectively, the accumulated data support and extend the
emerging view that green tea catechins are multimodal-acting,
brain-permeable natural iron chelators-antioxidants endowed with
polypharmacological activities and acting at multiple brain targets
to prevent or delay neuronal death in the degenerating brain. The
intervention with these compounds is assumed to have a profound
impact on neuron preservation and disease progression. The
presently described neurorescue/neurorestorative action of EGCG
may suggest a potential disease-modifying effect for this polyphenol.
Other articles in this supplement include references (68–77).
Literature Cited
1.
Ritchie K, Lovestone S. The dementias. Lancet. 2002;360:1759–66.
2.
Kuriyama S, Hozawa A, Ohmori K, Shimazu T, Matsui T, Ebihara S,
Awata S, Nagatomi R, Arai H, Tsuji I. Green tea consumption and
cognitive function: a cross-sectional study from the Tsurugaya Project 1.
Am J Clin Nutr. 2006;83:355–61.
3.
Checkoway H, Powers K, Smith-Weller T, Franklin GM, Longstreth
WT Jr, Swanson PD. Parkinson’s disease risks associated with cigarette
smoking, alcohol consumption, and caffeine intake. Am J Epidemiol.
2002;155:732–8.
4.
Hu G, Bidel S, Jousilahti P, Antikainen R, Tuomilehto J. Coffee and tea
consumption and the risk of Parkinson’s disease. Mov Disord. 2007;22:
2242–8.
5.
Dauer W, Przedborski S. Parkinson’s disease: mechanisms and models.
Neuron. 2003;39:889–909.
FIGURE 1
Proposed schematic model for
EGCG neuroprotective/neurorescue action.
For explanation see text. Abbreviations: Ab,
amyloid b-peptide; a-syn, a-synuclein;
COMT, catechol-O-methyl transferase; DAT,
dopamine transporter; PKC, protein kinase C;
sAPPa, soluble amyloid precursor protein-a.
Targeting neuroprotection with green tea catechins
1581S
jn.nutrition.org
Downloaded from

6.
Mandel S, Grunblatt E, Riederer P, Gerlach M, Levites Y, Youdim
MBH. Neuroprotective strategies in Parkinson’s disease: an update on
progress. CNS Drugs. 2003;17:729–62.
7.
Riederer P, Sofic E, Rausch WD, Schmidt B, Reynolds GP, Jellinger K,
Youdim MBH. Transition metals, ferritin, glutathione, and ascorbic
acid in parkinsonian brains. J Neurochem. 1989;52:515–20.
8.
Fahn S, Cohen G. The oxidant stress hypothesis in Parkinson’s disease:
evidence supporting it. Ann Neurol. 1992;32:804–12.
9.
Berg D, Gerlach M, Youdim MBH, Double KL, Zecca L, Riederer P,
Becker G. Brain iron pathways and their relevance to Parkinson’s
disease. J Neurochem. 2001;79:225–36.
10. Mandel S, Amit T, Bar-Am O, Youdim MB. Iron dysregulation in
Alzheimer’s disease: multimodal brain permeable iron chelating drugs,
possessing neuroprotective-neurorescue and amyloid precursor protein-
processing regulatory activities as therapeutic agents. Prog Neurobiol.
2007;82:348–60.
11. Van der Schyf CJ, Gal S, Geldenhuys WJ, Youdim MB. Multifunctional
neuroprotective drugs targeting monoamine oxidase inhibition, iron
chelation, adenosine receptors, and cholinergic and glutamatergic
action for neurodegenerative diseases. Expert Opin Investig Drugs.
2006;15:873–86.
12. Higdon JV, Frei B. Tea catechins and polyphenols: health effects,
metabolism, and antioxidant functions. Crit Rev Food Sci Nutr. 2003;
43:89–143.
13. Khan N, Mukhtar H. Tea polyphenols for health promotion. Life Sci.
2007;81:519–33.
14. Kuriyama S, Shimazu T, Ohmori K, Kikuchi N, Nakaya N, Nishino Y,
Tsubono Y, Tsuji I. Green tea consumption and mortality due to
cardiovascular disease, cancer, and all causes in Japan: the Ohsaki study.
JAMA. 2006;296:1255–65.
15. Li C, Allen A, Kwagh J, Doliba NM, Qin W, Najafi H, Collins HW,
Matschinsky FM, Stanley CA, Smith TJ. Green tea polyphenols
modulate insulin secretion by inhibiting glutamate dehydrogenase.
J Biol Chem. 2006;281:10214–21.
16. Anderson RA, Polansky MM. Tea enhances insulin activity. J Agric
Food Chem. 2002;50:7182–6.
17. Wolfram S, Wang Y, Thielecke F. Anti-obesity effects of green tea: from
bedside to bench. Mol Nutr Food Res. 2006;50:176–87.
18. Mandel SA, Avramovich-Tirosh Y, Reznichenko L, Zheng H, Weinreb
O, Amit T, Youdim MB. Multifunctional activities of green tea
catechins in neuroprotection. Neurosignals. 2005;14:46–60.
19. Lee S, Suh S, Kim S. Protective effects of the green tea polyphenol (2)-
epigallocatechin gallate against hippocampal neuronal damage after
transient global ischemia in gerbils. Neurosci Lett. 2000;287:191–4.
20. Sutherland BA, Shaw OM, Clarkson AN, Jackson DN, Sammut IA,
Appleton I. Neuroprotective effects of (2)-epigallocatechin gallate
following hypoxia-ischemia-induced brain damage: novel mechanisms
of action. FASEB J. 2005;19:258–60.
21. Aktas O, Prozorovski T, Smorodchenko A, Savaskan NE, Lauster R,
Kloetzel PM, Infante-Duarte C, Brocke S, Zipp F. Green tea
epigallocatechin-3-gallate mediates T cellular NF-kappa B inhibition and
exerts neuroprotection in autoimmune encephalomyelitis. J Immunol.
2004;173:5794–800.
22. Koh SH, Lee SM, Kim HY, Lee KY, Lee YJ, Kim HT, Kim J, Kim MH,
Hwang MS, et al. The effect of epigallocatechin gallate on suppressing
disease progression of ALS model mice. Neurosci Lett. 2006;395:
103–7.
23. Levites Y, Weinreb O, Maor G, Youdim MBH, Mandel S. Green tea
polyphenol (2)- epigallocatechin-3-gallate prevents N-methyl-4-phenyl-
1,2,3,6-tetrahydropyridine-induced dopaminergic neurodegeneration.
J Neurochem. 2001;78:1073–82.
24. Haque AM, Hashimoto M, Katakura M, Tanabe Y, Hara Y, Shido O.
Long-term administration of green tea catechins improves spatial
cognition learning ability in rats. J Nutr. 2006;136:1043–7.
25. Rezai-Zadeh K, Shytle D, Sun N, Mori T, Hou H, Jeanniton D, Ehrhart
J, Townsend K, Zeng J, et al. Green tea epigallocatechin-3-gallate
(EGCG) modulates amyloid precursor protein cleavage and reduces
cerebral amyloidosis in Alzheimer transgenic mice. J Neurosci. 2005;25:
8807–14.
26. Levites Y, Amit T, Youdim MBH, Mandel S. Involvement of protein
kinase C activation and cell survival/cell cycle genes in green tea
polyphenol (2)-epigallocatechin-3-gallate neuroprotective action. J Biol
Chem. 2002;277:30574–80.
27. Choi YT, Jung CH, Lee SR, Bae JH, Baek WK, Suh MH, Park J, Park
CW, Suh SI. The green tea polyphenol (2)-epigallocatechin gallate
attenuates beta-amyloid-induced neurotoxicity in cultured hippocampal
neurons. Life Sci. 2001;70:603–14.
28. Levites Y, Amit T, Mandel S, Youdim MBH. Neuroprotection and
neurorescue against amyloid beta toxicity and PKC-dependent release
of non-amyloidogenic soluble precusor protein by green tea polyphenol
(2)-epigallocatechin-3-gallate. FASEB J. 2003;17:952–4.
29. Ban JY, Jeon SY, Bae K, Song KS, Seong YH. Catechin and epicatechin
from Smilacis chinae rhizome protect cultured rat cortical neurons
against amyloid beta protein (25–35)-induced neurotoxicity through
inhibition of cytosolic calcium elevation. Life Sci. 2006;79:2251–9.
30. Reznichenko L, Amit T, Youdim MB, Mandel S. Green tea polyphenol
(2)-epigallocatechin-3-gallate induces neurorescue of long-term serum-
deprived PC12 cells and promotes neurite outgrowth. J Neurochem.
2005;93:1157–67.
31. Sagi Y, Mandel S, Amit T, Youdim MB. Activation of tyrosine kinase
receptor signaling pathway by rasagiline facilitates neurorescue and
restoration of nigrostriatal dopamine neurons in post-MPTP-induced
parkinsonism. Neurobiol Dis. 2007;25:35–44.
32. Kalfon L, Youdim MB, Mandel SA. Green tea polyphenol (2)-
epigallocatechin-3-gallate promotes the rapid protein kinase C- and
proteasome-mediated degradation of Bad: implications for neuro-
protection. J Neurochem. 2007;100:992–1002.
33. Liu WS, Heckman CA. The sevenfold way of PKC regulation. Cell
Signal. 1998;10:529–42.
34. Berra E, Municio MM, Sanz L, Frutos S, Diaz-Meco MT, Moscat J.
Positioning atypical protein kinase C isoforms in the UV-induced
apoptotic signaling cascade. Mol Cell Biol. 1997;17:4346–54.
35. Durkin JP, Tremblay R, Chakravarthy B, Mealing G, Morley P, Small D,
Song D. Evidence that the early loss of membrane protein kinase C is a
necessary step in the excitatory amino acid-induced death of primary
cortical neurons. J Neurochem. 1997;68:1400–12.
36. Vianna MR, Barros DM, Silva T, Choi H, Madche C, Rodrigues C,
Medina JH, Izquierdo I. Pharmacological demonstration of the differ-
ential involvement of protein kinase C isoforms in short- and long-term
memory formation and retrieval of one-trial avoidance in rats.
Psychopharmacology (Berl). 2000;150:77–84.
37. Pascale A, Amadio M, Govoni S, Battaini F. The aging brain, a key
target for the future: the protein kinase C involvement. Pharmacol Res.
2007;55:560–9.
38. Lange-Asschenfeldt C, Raval AP, Dave KR, Mochly-Rosen D, Sick TJ,
Perez-Pinzon MA. Epsilon protein kinase C mediated ischemic tolerance
requires activation of the extracellular regulated kinase pathway in the
organotypic hippocampal slice. J Cereb Blood Flow Metab. 2004;24:
636–45.
39. Cordey M, Gundimeda U, Gopalakrishna R, Pike CJ. Estrogen activates
protein kinase C in neurons: role in neuroprotection. J Neurochem.
2003;84:1340–8.
40. Han YS, Zheng WH, Bastianetto S, Chabot JG, Quirion R. Neuro-
protective effects of resveratrol against beta-amyloid-induced neuro-
toxicity in rat hippocampal neurons: involvement of protein kinase C.
Br J Pharmacol. 2004;141:997–1005.
41. Mandel S, Reznichenko L, Amit T, Youdim MB. Green tea polyphenol
(2)-epigallocatechin-3-gallate protects rat PC12 cells from apoptosis
induced by serum withdrawal independent of P13-Akt pathway.
Neurotox Res. 2003;5:419–24.
42. Kim SY, Ahn BH, Kim J, Bae YS, Kwak JY, Min G, Kwon TK, Chang JS,
Lee YH, et al. Phospholipase C, protein kinase C, Ca
21
/calmodulin-
dependent protein kinase II, and redox state are involved in
epigallocatechin gallate-induced phospholipase D activation in human
astroglioma cells. Eur J Biochem. 2004;271:3470–80.
43. Mandel S, Maor G, Youdim MB. Iron and alpha-synuclein in the
substantia nigra of MPTP-treated mice: effect of neuroprotective
drugs R-apomorphine and green tea polyphenol (2)-epigallocatechin-
3-gallate. J Mol Neurosci. 2004;24:401–16.
44. Shimizu S, Narita M, Tsujimoto Y. Bcl-2 family proteins regulate the
release of apoptogenic cytochrome c by the mitochondrial channel
VDAC. Nature. 1999;399:483–7.
45. Selkoe DJ. The molecular pathology of Alzheimer’s disease. Neuron.
1991;6:487–98.
46. Reznichenko L, Amit T, Zheng H, Avramovich-Tirosh Y, Youdim MB,
Weinreb O, Mandel S. Reduction of iron-regulated amyloid precursor
1582S
Supplement
jn.nutrition.org
Downloaded from

protein and beta-amyloid peptide by (2)-epigallocatechin-3-gallate in
cell cultures: implications for iron chelation in Alzheimer’s disease.
J Neurochem. 2006;97:527–36.
47. Choi DS, Wang D, Yu GQ, Zhu G, Kharazia VN, Paredes JP, Chang WS,
Deitchman JK, Mucke L, Messing RO. PKCepsilon increases endothelin
converting enzyme activity and reduces amyloid plaque pathology in
transgenic mice. Proc Natl Acad Sci USA. 2006;103:8215–20.
48. Li R, Peng N, Li XP, Le WD. (2)-Epigallocatechin gallate regulates
dopamine transporter internalization via protein kinase C-dependent
pathway. Brain Res. 2006;1097:85–9.
49. Lu H, Meng X, Yang CS. Enzymology of methylation of tea catechins
and inhibition of catechol-O-methyltransferase by (2)-epigallocatechin
gallate. Drug Metab Dispos. 2003;31:572–9.
50. Deleu D, Northway MG, Hanssens Y. Clinical pharmacokinetic and
pharmacodynamic properties of drugs used in the treatment of
Parkinson’s disease. Clin Pharmacokinet. 2002;41:261–309.
51. Gary DS, Milhavet O, Camandola S, Mattson MP. Essential role for
integrin linked kinase in Akt-mediated integrin survival signaling in
hippocampal neurons. J Neurochem. 2003;84:878–90.
52. Singer CA, Figueroa-Masot XA, Batchelor RH, Dorsa DM. The mitogen-
activated protein kinase pathway mediates estrogen neuroprotection
after glutamate toxicity in primary cortical neurons. J Neurosci. 1999;19:
2455–63.
53. Schroeter H, Spencer JP, Rice-Evans C, Williams RJ. Flavonoids protect
neurons from oxidized low-density-lipoprotein- induced apoptosis
involving c-Jun N-terminal kinase (JNK), c-Jun and caspase-3. Biochem
J. 2001;358:547–57.
54. Schroeter H, Bahia P, Spencer JP, Sheppard O, Rattray M, Cadenas E,
Rice-Evans C, Williams RJ. (2)Epicatechin stimulates ERK-dependent
cyclic AMP response element activity and up-regulates GluR2 in
cortical neurons. J Neurochem. 2007;101:1596–606.
55. Vauzour D, Vafeiadou K, Rice-Evans C, Williams RJ, Spencer JP. Acti-
vation of pro-survival Akt and ERK1/2 signalling pathways underlie the
anti-apoptotic effects of flavanones in cortical neurons. J Neurochem.
2007;103:1355–67.
56. Chen C, Yu R, Owuor ED, Kong AN. Activation of antioxidant-
response element (ARE), mitogen-activated protein kinases (MAPKs)
and caspases by major green tea polyphenol components during cell
survival and death. Arch Pharm Res. 2000;23:605–12.
57. Nanjo F, Goto K, Seto R, Suzuki M, Sakai M, Hara Y. Scavenging
effects of tea catechins and their derivatives on 1,1-diphenyl-2-
picrylhydrazyl radical. Free Radic Biol Med. 1996;21:895–902.
58. Salah N, Miller NJ, Paganga G, Tijburg L, Bolwell GP, Rice-Evans C.
Polyphenolic flavanols as scavengers of aqueous phase radicals and as
chain-breaking antioxidants. Arch Biochem Biophys. 1995;322:339–46.
59. Guo Q, Zhao B, Li M, Shen S, Xin W. Studies on protective mechanisms
of four components of green tea polyphenols against lipid peroxidation
in synaptosomes. Biochim Biophys Acta. 1996;1304:210–22.
60. Owuor ED, Kong AN. Antioxidants and oxidants regulated signal
transduction pathways. Biochem Pharmacol. 2002;64:765–70.
61. Zecca L, Youdim MB, Riederer P, Connor JR, Crichton RR. Iron, brain
ageing and neurodegenerative disorders. Nat Rev Neurosci. 2004;5:
863–73.
62. Berg D. In vivo detection of iron and neuromelanin by transcranial
sonography–a new approach for early detection of substantia nigra
damage. J Neural Transm. 2006;113:775–80.
63. Lovell MA, Robertson JD, Teesdale WJ, Campbell JL, Markesbery WR.
Copper, iron and zinc in Alzheimer’s disease senile plaques. J Neurol Sci.
1998;158:47–52.
64. Pinero DJ, Hu J, Connor JR. Alterations in the interaction between iron
regulatory proteins and their iron responsive element in normal and
Alzheimer’s diseased brains. Cell Mol Biol. 2000;46:761–76.
65. Youdim MBH, Buccafusco JJ. Multi-functional drugs for various CNS
targets in the treatment of neurodegenerative disorders. Trends
Pharmacol Sci. 2005;26:27–35.
66. Ono K, Yoshiike Y, Takashima A, Hasegawa K, Naiki H, Yamada M.
Potent anti-amyloidogenic and fibril-destabilizing effects of polyphenols
in vitro: implications for the prevention and therapeutics of Alzheimer’s
disease. J Neurochem. 2003;87:172–81.
67. Ono K, Yamada M. Antioxidant compounds have potent anti-
fibrillogenic and fibril-destabilizing effects for alpha-synuclein fibrils
in vitro. J Neurochem. 2006;97:105–15.
68. Arab L, Blumberg JB. Introduction to the Proceedings of the Fourth
International Scientific Symposium on Tea and Human Health. J Nutr.
2008;138:1526S–8S.
69. Henning SM, Choo JJ, Heber D. Nongallated compared with gallated
flavan-3-ols in green and black tea are more bioavailable. J Nutr.
2008;138:1529S–34S.
70. Auger C, Mullen W, Hara Y, Crozier A. Bioavailability of polyphenon E
flavan-3-ols in humans with an ileostomy. J Nutr. 2008;138:1535S–42S.
71. Song WO, Chun OK. Tea is the major source of flavan-3-ol and flavonol
in the U.S. diet. J Nutr. 2008;138:1543S–7S.
72. Kuriyama S. The relation between green tea consumption and cardio-
vascular disease as evidenced by epidemiological studies. J Nutr. 2008;
138:1548S–53S.
73. Grassi D, Aggio A, Onori L, Croce G, Tiberti S, Ferri C, Ferri L, Desideri
G. Tea, flavonoids, and NO-mediated vascular reactivity. J Nutr. 2008;
138:1554S–60S.
74. Arts ICW. A review of the epidemiological evidence on tea, flavonoids,
and lung cancer. J Nutr. 2008;138:1561S–6S.
75. Hakim IA, Chow HHS, Harris RB. Green tea consumption is associated
with decreased DNA damage among GSTM1 positive smokers regard-
less of their hOGG1 genotype. J Nutr. 2008;138:1567S–71S.
76. Kelly SP, Gomez-Ramirez M, Montesi JL, Foxe JJ. L-Theanine and
caffeine in combination affect human cognition as evidenced by oscilla-
tory alpha-band activity and attention task performance. J Nutr. 2008;
138:1572S–7S.
77. Stote KS, Baer DJ. Tea consumption may improve biomarkers of insulin
sensitivity and risk factors for diabetes. J Nutr. 2008;138:1584S–8S.
Targeting neuroprotection with green tea catechins
1583S
jn.nutrition.org
Downloaded from
Wyszukiwarka
Podobne podstrony:
Green tea catechins as brain permeable, natural iron chelators antioxidants for the treatment of neu
Maternal diseases associated with pregnancy
Targeting Profitable Entry & Exit Points with Alan Farley
MOISTURIZING BODY SCRUB CUBES WITH GREEN TEA AND GINGER
Osteochondritis dissecans in association with legg calve perthes disease
Dietary Patterns Associated with Alzheimer’s Disease
Intermediate Dialogues with Multiple Choice Quesitions A Cookie
Neurodegeneration in multiple scerosis novel treatment strategies
The TRUE Coldwar Our?ttle with Diseases
Dungeons and Dragons 3 5 Accessory Color Character Sheets with Multiple Spell Lists
Legg Calvé Perthes disease multipositional power Doppler sonography of the proximal femoral vascular
Applying Principles of Neurodevelopment to Clinical Work with Maltreated and Traumatized Children
Femoral head vascularisation in Legg Calvé Perthes disease comparison of dynamic gadolinium enhanced
Osteochondritis dissecans in association with legg calve perthes disease
Dietary Patterns Associated with Alzheimer’s Disease
Applying Principles of Neurodevelopment to Clinical Work with Maltreated and Traumatized Children
Beginning Dialogues with Multiple Choice Questions How are You
więcej podobnych podstron