
The Incorporation of Carboxylate Groups into
Temperature-Responsive Poly(
N-isopropylacrylamide)-Based
Hydrogels Promotes Rapid Gel Shrinking
MITSUHIRO EBARA,
1
TAKAO AOYAGI,
2
KIYOTAKA SAKAI,
1
TERUO OKANO
2
1
Department of Applied Chemistry, Waseda University, 3-4-1 Ohkubo, Shinjuku-ku, Tokyo 169-8555, Japan
2
Institute of Biomedical Engineering, Tokyo Women’s Medical University, 8-1 Kawada-cho, Shinjuku-ku,
Tokyo 162-8666, Japan
Received 4 October 2000; accepted 16 November 2000
ABSTRACT:
Aqueous gel deswelling rates for copolymer hydrogels comprising N-isopro-
pylacrylamide (IPAAm) and 2-carboxyisopropylacrylamide (CIPAAm) in response to
increasing temperatures were investigated. Compared with pure IPAAm-based gels,
IPAAm–CIPAAm gels shrink very rapidly in response to small temperature increases
across their lower critical solution temperature (their volume is reduced by five-sixths
within 60 s). Shrinking rates for these hydrogels increase with increasing CIPAAm
content. In contrast, structurally analogous IPAAm–acrylic acid (AAc) copolymer gels
lose their temperature sensitivity with the introduction of only a few mole percent of
AAc. Additionally, deswelling rates of IPAAm–AAc gels decrease with increasing AAc
content. These results indicate that IPAAm–CIPAAm copolymer gels behave distinctly
from IPAAm–AAc systems even if both comonomers, CIPAAm and AAc, possess car-
boxylic acid groups. Thus, we propose that the sensitive deswelling behavior for
IPAAm–CIPAAm gels results from strong hydrophobic chain aggregation maintained
between network polymer chains due to the similar chemical structures of CIPAAm and
IPAAm. This structural homology facilitates aggregation of chain isopropylamide
groups for both IPAAm and CIPAAm sequences with increasing temperature. The
incorporation of AAc, however, shows no structural homology to IPAAm, inhibiting
chain aggregation and limiting collapse. A functionalized temperature-sensitive
poly(N-isopropylacrylamide) hydrogel containing carboxylic acid groups is possible with
CIPAAm, producing rapid and large volume changes in response to smaller tempera-
ture changes.
© 2000 John Wiley & Sons, Inc. J Polym Sci A: Polym Chem 39: 335–342, 2001
Keywords:
poly(N-isopropylacrylamide);
2-carboxyisopropylacrylamide;
tempera-
ture-responsive hydrogels; volume phase transition; deswelling kinetics; functional
groups; anionic gel
INTRODUCTION
Stimuli-responsive hydrogels that exhibit sub-
stantial property changes in response to temper-
ature,
1,2
pH,
3,4
light,
5
and electric fields
6 – 8
have
been investigated in the context of new devices for
drug delivery systems and biomaterials. Poly(N-
isopropylacrylamide) (PIPAAm) gels are known
for their reversible swelling– deswelling behavior
in response to temperature changes across a
lower critical solution temperature (LCST).
9
PIPAAm gels have been applied to control drug
Correspondence
to:
T.
Aoyagi
(E-mail:
taoyagi@lab.
twmu.ac.jp)
Journal of Polymer Science: Part A: Polymer Chemistry, Vol. 39, 335–342 (2001)
© 2000 John Wiley & Sons, Inc.
335

release rates in temperature-modulated drug de-
livery systems.
1,2,10
We already reported ther-
mally induced on– off drug release by using the
formation of a rate-controlling collapsed polymer-
gel-skin layer impermeable to drug molecules
and water.
11,12
Polymer-gel-skin-layer formation
leads to very slow deswelling of the bulk polymer
network. To avoid rate-limiting skin formation
and facilitate rapid water– drug efflux, we pre-
pared comb-type PIPAAm gels that contained a
large amount of freely mobile PIPAAm graft
chains.
13,14
These comb-type PIPAAm gels shrink
rapidly in response to temperature changes or
solvation changes near the LCST because mobile
PIPAAm graft chains are able to undergo rapid
dehydration in response to small temperature in-
creases.
Another method to accelerate deswelling rates
in PIPAAm-based gels is the introduction of hy-
drophilic groups into PIPAAm gels. Kim et al.
15
reported deswelling kinetics studies of random
copolymer
PIPAAm
gels
containing
small
amounts of acrylic acid (AAc). N-Isopropylacryl-
amide (IPAAm)–AAc gels showed rapid deswell-
ing with increasing temperature because a small
amount of ionized AAc suppressed collapsed poly-
mer-skin-layer formation on the gel surface,
whereas the deswelling volumes for IPAAm–AAc
gel decreased with an increasing AAc content. We
propose that the small content of AAc in the gel
decreases PIPAAm chain hydrophobic aggrega-
tion forces due to both charge– charge repulsion
and excess hydration by AAc proximal to the
IPAAm polymer backbone. In our recent study,
hydrophilic poly(ethylene oxide) (PEO)-grafted
chains were introduced into PIPAAm gel net-
works.
14,16
The deswelling rate of this PEO-
grafted PIPAAm gel was also accelerated over
pure IPAAm gel because hydrophilic PEO chains
plausibly form channels for water through the
IPAAm collapsed skin layer. In contrast to col-
lapsible IPAAm–AAc gels, high PIPAAm thermo-
sensitivity in this case was maintained in the
presence of significant hydrophilic PEO grafting
in the PIPAAm network. This might be due to
significant association of IPAAm chain units in-
dependent of PEO-grafted chains because of the
gel architecture. On the basis of these previous
reports, sensitive gel temperature response might
result from the readily reversible association of
critical lengths of isopropylamide groups as in an
IPAAm homopolymer.
To establish molecular design criteria for func-
tionalized hydrogels with high thermosensitivity,
we newly designed the functional monomer 2-car-
boxyisopropylacrylamide
(CIPAAm).
CIPAAm
shares all the structural features of IPAAm ex-
cept for the terminal carboxyl group.
17,18
Hence,
random copolymers of IPAAm with CIPAAm were
proposed to maintain continuous lengths of asso-
ciating polymer isopropylamide groups similar to
IPAAm homopolymers with similar gel thermo-
sensitivity in the presence of significant amount
of hydrated, electrostatically repulsive carboxy-
late groups in the PIPAAm network. We recently
reported that IPAAm–CIPAAm soluble linear co-
polymers containing less than 20 mol % CIPAAm
exhibit very sensitive phase transitions in re-
sponse to temperature changes with phase-tran-
sition temperatures nearly the same as those of
the IPAAm homopolymer and its gel over a wide
pH range.
17,18
In contrast, the phase-transition
temperature and collapse sensitivity for IPAAm–
AAc analogous copolymers and gels were influ-
enced considerably by small amounts of AAc, so-
lution pH, and ionic strength.
With this study, we sought to clarify the effects
of both gel carboxyl groups and polymer isopro-
pylamide aggregation in IPAAm–CIPAAm gels on
their deswelling kinetics in response to tempera-
ture changes and temperature cycles across the
LCST. From these results, the deswelling mech-
anism for IPAAm–CIPAAm gels versus IPAAm–
AAc gels was contrasted. Preserving the associa-
tion of critical lengths of gel network chain iso-
propylamide
groups
is
shown
to
play
an
important role in the resulting strong hydropho-
bic aggregation that produces thermosensitivity
in the respective gel networks.
EXPERIMENTAL
Materials
IPAAm was kindly provided by Kojin Co. (Tokyo,
Japan) and was purified by recrystallization from
n-hexane. AAc was distilled under reduced pres-
sure. N,N,N
⬘,N⬘-Tetramethylethylenediamine (TE-
MED), ammonium persulfate (APS), and N,N
⬘-
methylenebisacrylamide (MBAAm) were purchased
from Kanto Chemical Co., Ltd. (Tokyo) and were
used as received.
Synthesis of CIPAAm
CIPAAm was synthesized with the same methods
reported in a previous article.
17
In brief,
D
,
L
-3-
336
EBARA ET AL.

aminobutyric acid was esterified with benzyl al-
cohol, and acryloyl chloride was reacted with
D
,
L
-3-aminobutyric acid benzyl ester in the pres-
ence of triethylamine. CIPAAm was obtained by
the hydrolysis of benzyl ester with a sodium hy-
droxide aqueous solution. Protonation of result-
ing carboxyl groups was carried out with an ex-
cess amount of hydrochloric acid.
Gel Synthesis
For each gel composition, appropriate amounts of
IPAAm, CIPAAm, MBAAm, and TEMED were
dissolved in distilled water, and the solution was
transferred to a test tube and bubbled with dry
nitrogen gas for 15 min to remove oxygen. APS
was added to the solution, and then glass capil-
laries (300-
m inner diameter) were set into the
solution. The test tube was kept at 4 °C for 1 day
for polymerization and spontaneous gelation. The
poly(IPAAm-co-CIPAAm) gels were abbreviated
as IPAAm–CIPAAm (X) gel, where X is the molar
percentage of CIPAAm in the feed. Poly(IPAAm-
co-AAc) gels and PIPAAm homopolymer gel were
synthesized with the same methods and were ab-
breviated as IPAAm–AAc (X) gel and PIPAAm
gel, respectively. After the gelation was complete,
the 300-
m diameter gels were removed from the
glass capillaries, cut into small pieces, and im-
mersed in pure cold water to remove unreacted
compounds for 1 day. The gels were set in glass
capillaries 1.34 mm in diameter filled with phos-
phate-buffered saline (pH 7.4, 0.15 M), and the
glass capillaries were sealed.
18
Measurements of Gel Deswelling Kinetics
Gel deswelling kinetics from the equilibrium
swollen state to equilibrium deswollen conditions
were continuously monitored with a charge-cou-
pled device camera connected to a microscope.
The swelling ratio of the gel, (d
t
/d
o
)
3
, during the
deswelling change was calculated from the ratio
of the gel diameter at a certain time to the origi-
nal diameter (300
m).
RESULTS AND DISCUSSION
Molecular Design of the Hydrogels
Random copolymerization of IPAAm with func-
tional monomers such as AAc or other ionizable
comonomers is very conventional, readily intro-
ducing ionizing groups into temperature-respon-
sive PIPAAm gels.
19
In IPAAm–AAc random co-
polymer gels, however, small amounts of AAc
significantly affect the temperature-responsive
behavior. Specifically, gel volume changes are re-
duced, and their transition temperatures shift to
higher temperatures. We hypothesized that reg-
ular aggregations of critical lengths of isopropy-
lacrylamide groups along PIPAAm chains are re-
quired to produce their sensitive dehydration
changes that result in rapid shrinking of gels. Our
inclusion of the new monomer CIPAAm, despite
its content of carboxylic acid functional groups,
was intended to maintain IPAAm chain aggrega-
tion behavior characterized in previous IPAAm–
CIPAAm copolymers
17
and gels.
18
This hypothe-
sis was confirmed: phase-transition temperatures
and transition sensitivity for IPAAm–CIPAAm
linear polymers and its hydrogels were nearly
independent of solution pH and carboxyl group
content.
18
These results are distinctly different
from those of IPAAm–AAc and its gels.
17,18
Spe-
cific evidence supporting this hypothesis is pro-
vided later.
Hydrogel Deswelling Kinetics
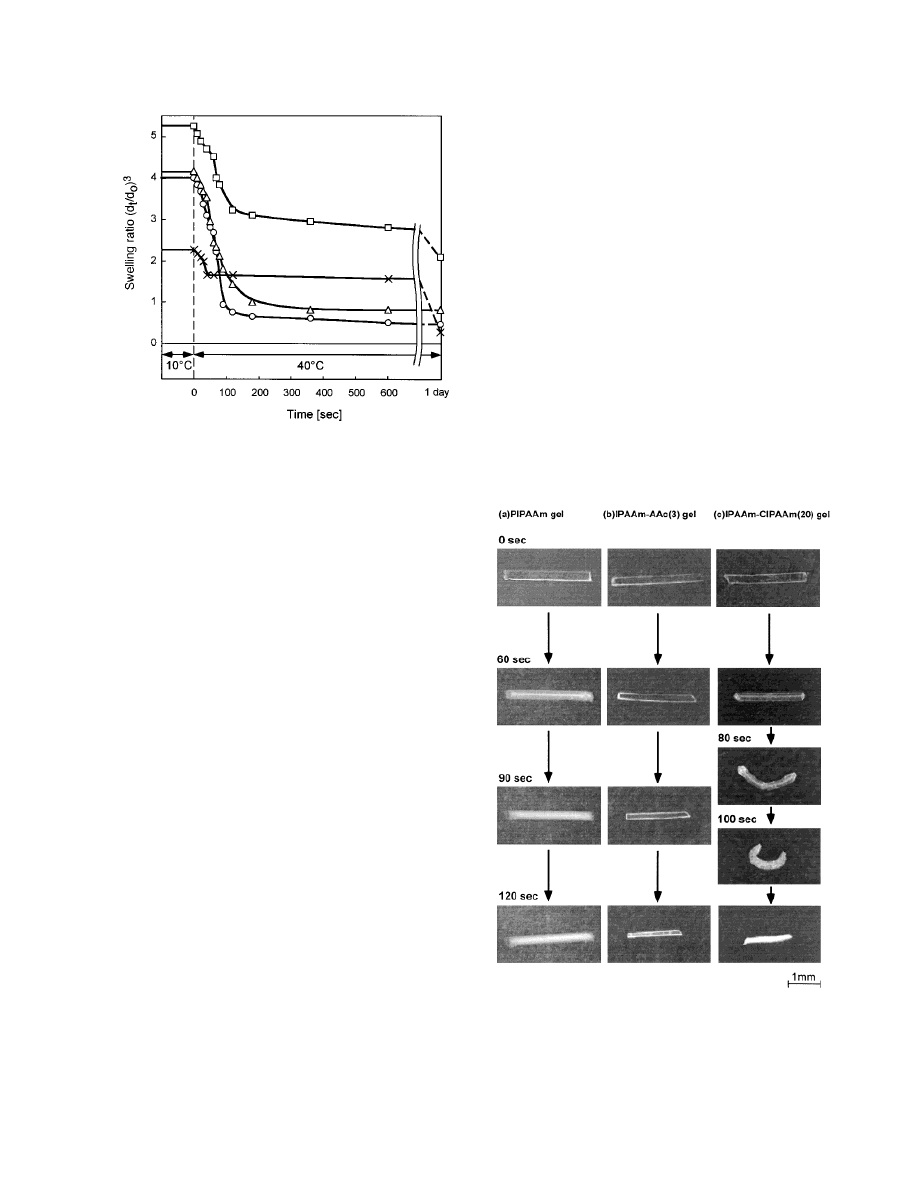
Figure 1 compares the deswelling kinetics data
from PIPAAm and IPAAm–AAc copolymer gels
exposed to stepwise temperature changes from 10
°C under equilibrium swelling conditions to 40 °C
in PBS (pH 7.4). Pure PIPAAm gels behaved as
previously shown.
11,12,19,20
Rapid initial shrink-
ing was observed just after the temperature
change
was
induced.
Deswelling,
however,
abruptly stopped, and volume change rates be-
came very slow for long periods. This phenome-
non, that is, instantaneous shrinking followed by
a slow volume change, has been attributed to a
surface collapsing polymer layer, the so-called
polymer-skin layer.
11,12,20
The gel shrinking be-
havior is demonstrated in Figure 2. The PIPAAm
gel changed from transparent to opaque within
60 s after a temperature change was induced,
suggesting the formation of a heterogeneous gel
structure. Then, this gel shrank gradually from
its surface inward, mediated by cooperative diffu-
sion of the collapsing polymer networks and the
gradual permeating release of entrapped water
from within the gel. As explained previo-
usly,
11,12,20
the shrinking rate of PIPAAm gel is
controlled by hindered water permeation from the
inner gel because of the collapsed polymer skin
POLY(N-ISOPROPYLACRYLAMIDE)-BASED HYDROGELS
337
and the onset of internal hydrostatic pressure
from collapse.
As shown in Figure 2(b), the IPAAm–AAc (3)
gel shrinks rapidly, maintaining a gel transpar-
ence. In contrast to the PIPAAm gel, IPAAm–AAc
gel with a small AAc content shrinks rapidly
without polymer-skin-layer formation because
hydrophilic AAc suppresses the formation of the
collapsed hydrophobic skin layer.
15,19
However,
the gel deswelling magnitudes decrease with in-
creasing AAc content in the series of IPAAm–AAc
copolymer gels. Hydrophobic polymer aggregation
forces in the phase transition are apparently
weakened because hydrated charged AAc units
disrupt regular aggregation of isopropylamide
groups in polymer chains. As a result, water per-
meation from inside these gels increases with in-
creasing AAc content.
16
In particular, volume
changes for the IPAAm–AAc (5) gel were reduced
over time: the collapsed size of IPAAm–AAc (5)
gel at 40 °C was only half that of the equilibrium
swelling state at 10 °C, whereas lower AAc con-
tent gels showed higher swollen/collapsed ratios.
Figure 3 shows deswelling kinetics for the
IPAAm–CIPAAm gels exposed to stepwise tem-
perature changes from 10 (equilibrium swelling
state) to 40 °C (above the LCST), enough to
readily prompt gel collapse in PBS (pH 7.4).
IPAAm–CIPAAm (5) gel shrank rapidly after ini-
tial temperature changes. However, its volume
change stopped after skin-layer formation was
observed on the gel surface after 80 s of temper-
ature change. This indicates that despite its
charged, albeit small, monomer content, IPAAm–
CIPAAm gel maintains strong hydrophobic aggre-
gability, an effect distinct from the analogous
IPAAm–AAc
gels
attributed
to
the
unique
CIPAAm structure. This gel deswelling rate and
collapse magnitude gradually increases with in-
creasing CIPAAm content. Figure 2(c) shows pho-
tographs of the shrinking process for the IPAAm–
CIPAAm (20) gel. The initially transparent
IPAAm–CIPAAm (20) gel became opaque within
80 s after being heated from 10 to 40 °C. The
formation of a heterogeneous polymer network
structure due to polymer chain surface aggrega-
tion was observed. However, the deswelling rate
of IPAAm–CIPAAm (20) gel was not hindered,
Figure 2.
Photographs for deswelling changes ob-
served for (a) PIPAAm gels, (b) IPAAm–AAc (3), and (c)
IPAAm–CIPAAm (20) in response to a temperature
increase from 10 to 40 °C in PBS (initial gel dimen-
sions: 0.3-mm diameter and 2.7-mm length).
Figure 1.
Shrinking kinetics for PIPAAm and
IPAAm–AAc gels at 40 °C under equilibrium swelling
conditions at 10 °C (initial gel dimensions: 0.3-mm
diameter and 5-mm length).
⫻ ⫽ PIPAAm gel; E ⫽
IPAAm–AAc (1) gel; ‚
⫽ IPAAm–AAc (3) gel; 䊐 ⫽
IPAAm–AAc (5) gel.
338
EBARA ET AL.
regardless of the skin-layer formation, because of
sufficient ionic monomer content. These results
indicate that the uniquely structured carboxyl
groups of CIPAAm influence the gel shrinking
process. More specifically, the density of the col-
lapsed polymer-skin layer of IPAAm–CIPAAm
(20) gel must be lower than that of PIPAAm gel to
permit squeezing water efflux and avoid hydro-
static pressure increases, whereas the chain ag-
gregation forces between polymer chains produc-
ing collapse are maintained in comparison with
that in IPAAm–AAc gels. Therefore, water within
the collapsing gel readily permeates out through
its porous skin layer.
These results emphasize the balance of two
factors important for producing rapid shrinking of
IPAAm copolymer gels: (1) maintenance of effec-
tive hydrophobic aggregation forces between col-
lapsing polymer chains and (2) facilitation of wa-
ter channel formation in the gels to readily re-
lease water through the collapsing gel-skin layer.
If the chain aggregation force is too strong and
the gel-skin-layer density is very high, water in-
side the gel cannot permeate, and the gel deswell-
ing change is halted by the resulting hydrostatic
pressure as seen in PIPAAm gels. On the con-
trary, if aggregation forces are weakened by the
introduction of hydrophilic comonomer moieties,
deswelling rates become inordinately slow and
diffuse, and the collapse magnitude becomes
small, as seen in IPAAm–AAc gels. Deswelling
rates and magnitudes in the PIPAAm copolymer
gels are readily controlled with CIPAAm because
the collapsed skin-layer density is altered by
changes in the CIPAAm content. This result is
completely different from that of conventional
IPAAm–AAc hydrogels, where the introduction of
only 1 mol % AAc prevents gel-skin-layer forma-
tion. Moreover, increasing the AAc content re-
duces hydrophobic aggregation forces consider-
ably, and gel deswelling rates are subsequently
reduced.
Gel Deswelling Changes in Response to Small
Temperature Changes
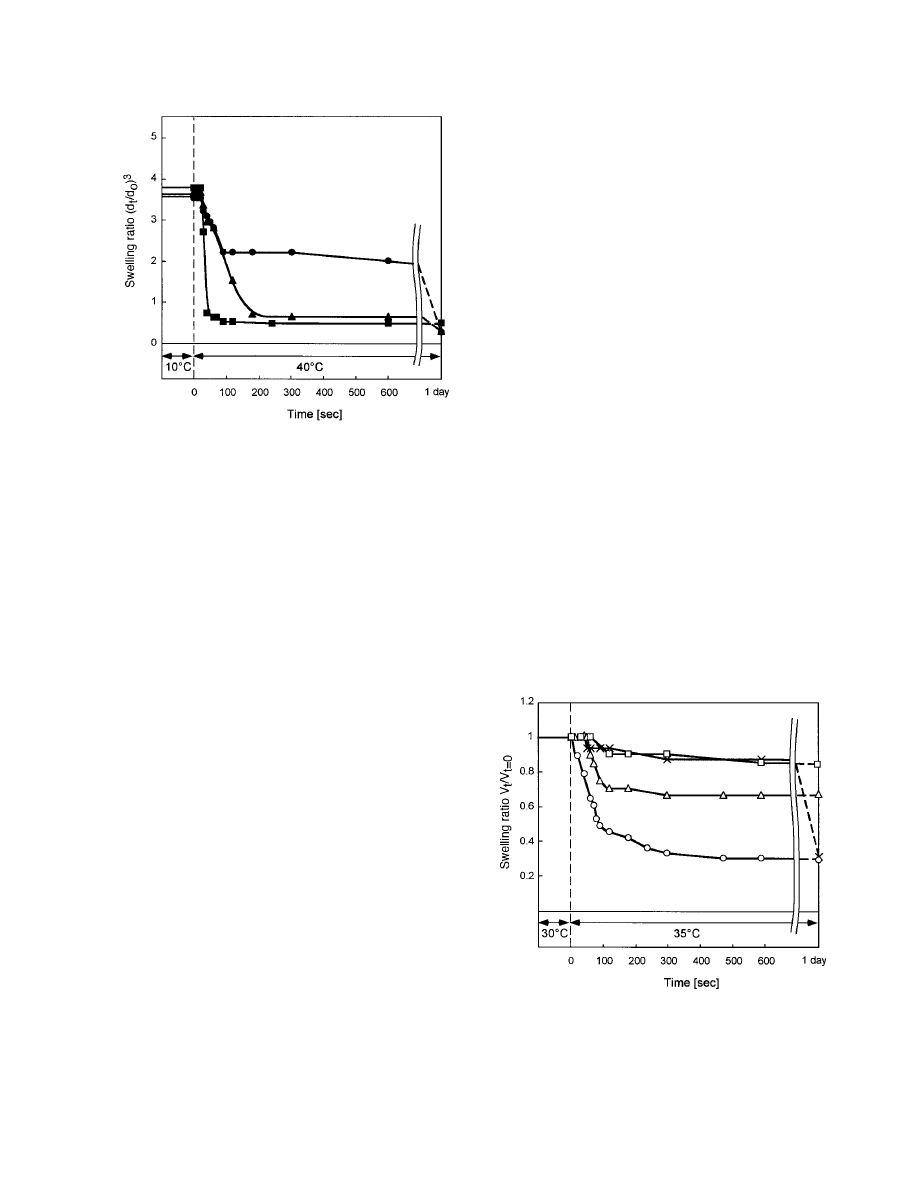
Figure 4 shows deswelling kinetics for pure
PIPAAm gel and IPAAm–AAc gels at 35 °C from
equilibrium swelling states at 30 °C in PBS (pH
7.4). Swelling ratios during the continuous de-
swelling changes are converted to normalized
swelling ratios, V
t
/V
t
⫽0
, calculated from the ratio
of the gel volume at a certain time (V
t
) to the
initial volume at 30 °C (V
t
⫽0
). As seen in Figure 4,
changes in the PIPAAm gel volume after the tem-
perature increased to 35 °C were not observed
over this timescale because of rapid skin-layer
formation. Equilibrium in this case takes weeks
or months.
11,12,19,20
In contrast, the IPAAm–AAc
Figure 4.
Shrinking kinetics for PIPAAm and
IPAAm–AAc gels at 35 °C under equilibrium conditions
at 30 °C (initial gel dimensions: 0.3-mm diameter and
5-mm length).
⫻ ⫽ PIPAAm gel; E ⫽ IPAAm–AAc (1)
gel; ‚
⫽ IPAAm–AAc (3) gel; 䊐 ⫽ IPAAm–AAc (5) gel.
Figure 3.
Shrinking kinetics for IPAAm–CIPAAm
gels at 40 °C under equilibrium swelling conditions at
10 °C (initial gel dimensions: 0.3-mm diameter and
5-mm length). F
⫽ IPAAm–CIPAAm (5) gel; Œ ⫽
IPAAm–CIPAAm (10) gel; ■
⫽ IPAAm–CIPAAm (20)
gel.
POLY(N-ISOPROPYLACRYLAMIDE)-BASED HYDROGELS
339
(1) gel shrinks rapidly because hydrophilic AAc
moieties inhibit the formation of a gel-skin layer
and readily release water from inside to outside,
relieving opposing hydrostatic pressure and facil-
itating collapse. The IPAAm–AAc (3) gel also
shrinks rapidly. However, deswelling changes are
not so large because of repulsive ionic comonomer
content. Additionally, deswelling changes in
those IPAAm–AAc gels become smaller with in-
creasing AAc content. These phenomena are due
to the introduction of hydrophilic AAc into
PIPAAm chains; increased hydrated, charged hy-
drophilic sites, and a decreased tendency to ag-
gregate critical lengths of isopropylacrylamide
chain units. Figure 5 shows deswelling kinetics
for IPAAm–CIPAAm gels at 35 °C from equilib-
rium swelling states at 30 °C in PBS (pH 7.4). The
deswelling behaviors of IPAAm–CIPAAm gels
correspond very well to deswelling kinetics from
heating modes from 10 to 40 °C, as seen in Figure
3. The IPAAm–CIPAAm (10) and IPAAm–
CIPAAm (20) gels exhibit large volume changes
in response to small temperature changes, and
gel shrinking is nearly complete within 1 min
after the temperature change is induced. These
results indicate that the IPAAm–CIPAAm gel
maintains strong hydrophobic chain aggregation
capabilities despite relatively large amounts of
carboxyl groups introduced into the PIPAAm net-
work via CIPAAm.
Reversibility of the Gel Swelling–Deswelling
Kinetics
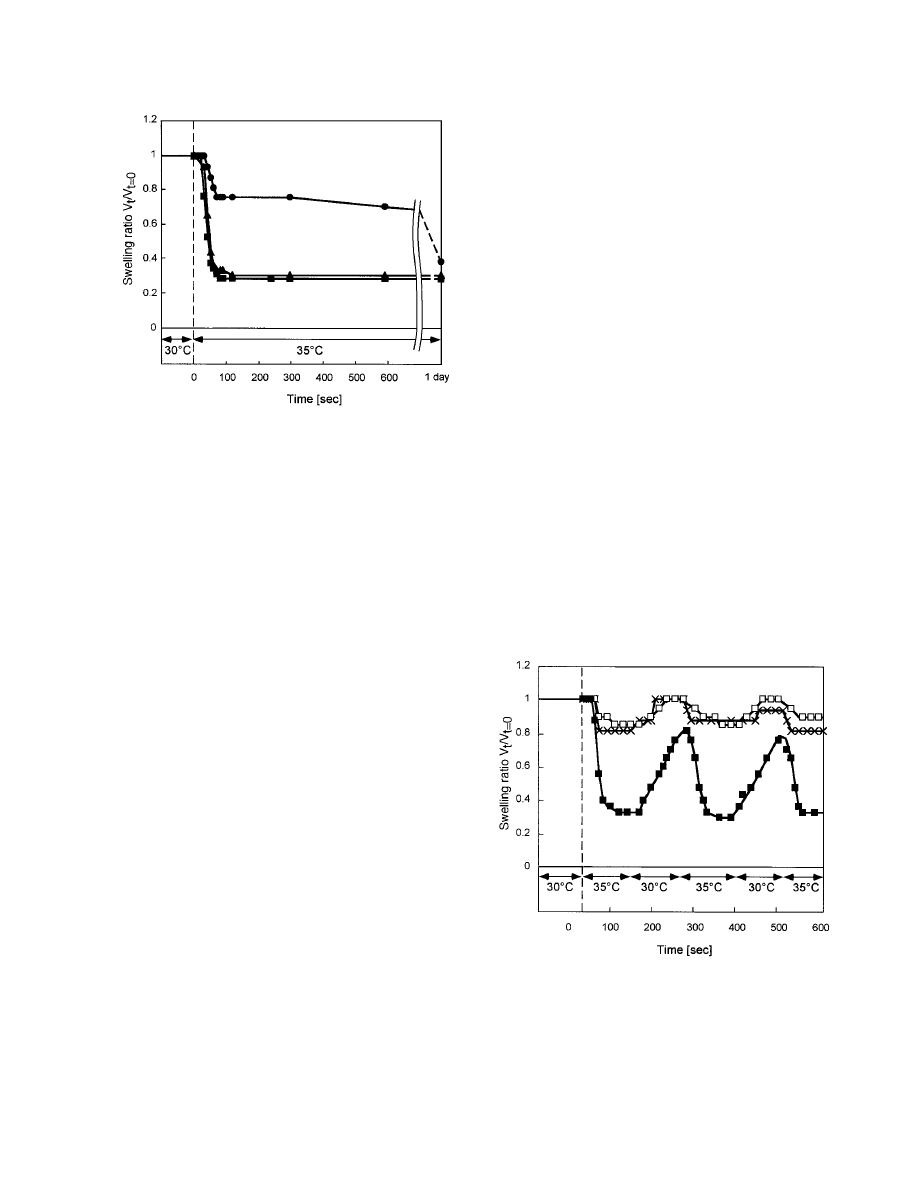
Figure 6 demonstrates the swelling– deswelling ki-
netics for PIPAAm, IPAAm–AAc (5), and IPAAm–
CIPAAm (20) gels over 2-min temperature cycles
between 30 and 35 °C in PBS (pH 7.4). Normalized
swelling ratios calculated from the ratio of the gel
volume at a certain time (V
t
) to the initial volume at
30 °C (V
t
⫽0
) were used to compare the swelling–
deswelling behavior. A small swelling– deswelling
change of the PIPAAm gel was observed.
11,12,19,20
PIPAAm gel showed completely repeatable and re-
producible oscillations between 0.8 and 1.0 swelling
ratios. The PIPAAm gel became opaque with tem-
perature increases from 30 to 35 °C, supporting
heterogeneous collapse. When the temperature was
decreased to 30 °C from 35 °C, PIPAAm gels imme-
diately became transparent again. In contrast, a
large swelling– deswelling change was observed for
the IPAAm–CIPAAm (20) gel, with swelling ratios
oscillating between 0.3 and 0.8. The size of the
IPAAm–CIPAAm (20) gel at 30 °C was more than
twice that of the deswollen state at 35 °C, whereas
those for the PIPAAm and IPAAm–AAc (5) gels
were about 1.2 times those of their deswollen states
at 35 °C. In addition, shrinking behavior differences
between the IPAAm–CIPAAm (20) and IPAAm–
Figure 6.
Swelling– deswelling kinetics for PIPAAm,
IPAAm–AAc (5), and IPAAm–CIPAAm (20) gels in re-
sponse to stepwise temperature changes between 30
and 35 °C for 2-min cycles (initial gel dimensions:
0.3-mm diameter and 5-mm length).
⫻ ⫽ PIPAAm gel;
䊐 ⫽ IPAAm–AAc (5) gel; ■ ⫽ IPAAm–CIPAAm (20)
gel.
Figure 5.
Shrinking kinetics for IPAAm–CIPAAm
gels at 35 °C under equilibrium conditions at 30 °C. F
⫽ IPAAm–CIPAAm (5); Œ ⫽ IPAAm–CIPAAm (10); ■
⫽ IPAAm–CIPAAm (20).
340
EBARA ET AL.

AAc (5) gels are observed. The deswelling rate of the
IPAAm–AAc (5) gel from 30 to 35 °C is nearly iden-
tical to that from 35 to 30 °C because both phenom-
ena are governed by simple diffusion of polymer
chains in water (no skin-layer hindrance). However,
the deswelling rate of IPAAm–CIPAAm (20) gel
from 30 to 35 °C was 2.5 times faster than its swell-
ing rate from 35 to 30 °C. These results suggest that
the shrinking mechanism for the IPAAm–CIPAAm
(20) gel is based on strong chain aggregation medi-
ated by dehydration and hydrophobic interaction,
as well as retention of water pores throughout the
aggregated collapsed polymer chain network. In
other words, the IPAAm–CIPAAm (20) gel main-
tains strong chain aggregation despite a 20 mol %
content of carboxyl groups. The IPAAm–AAc ran-
dom copolymer gel exhibits a lower limit of carboxyl
group content (5 mol %) sufficient to maintain such
a rapid shrinking behavior response to temperature
changes, but it loses its swelling– deswelling mag-
nitude with this content. In contrast, the IPAAm–
CIPAAm gel exhibits a much greater magnitude of
swelling changes in response to small temperature
cycles between 30 and 35 °C with substantial (20
mol %) carboxyl group content.
CONCLUSION
Deswelling kinetics for temperature-responsive
IPAAm copolymer gels containing new functional
CIPAAm monomer were compared to known tem-
perature-sensitive gels. Deswelling kinetics for
IPAAm–CIPAAm gels at 40 °C (above their LCST)
from equilibrium swelling states at 10 °C (below
their LCST) in PBS (pH 7.4) were compared to
those of pure PIPAAm and IPAAm–AAc ionic gels.
The conventional PIPAAm gel becomes opaque just
after this temperature increase, with limited de-
swelling changes for long time periods because of
collapsed skin-layer formation. In contrast, known
IPAAm–AAc gels shrink rapidly without opposing
internal hydrostatic pressure buildup because hy-
drophilic AAc suppresses the formation of the hy-
drophobic collapsed polymer-skin layer.
19
However,
gel deswelling rates and volume changes decrease
with increasing AAc content because network chain
aggregation forces are weakened by AAc ionomer
incorporation. In contrast to IPAAm–AAc gels, the
deswelling rates for ionized IPAAm–CIPAAm gels
gradually increase with increasing CIPAAm con-
tent. Although deswelling changes for the IPAAm–
CIPAAm (5) gel (5 mol % CIPAAm) are limited over
longer periods, IPAAm–CIPAAm (20) gels shrink
rapidly at ionic contents where IPAAm–AAc gels
have lost their phase transition.
19
Despite the large
amount of carboxyl groups, this gel maintains col-
lective IPAAm chain aggregation forces under ther-
mally induced collapse in comparison with IPAAm–
AAc gels. Swelling– deswelling kinetics for gels
compared over 2-min temperature cycles between
30 and 35 °C in PBS (pH 7.4) show that the
PIPAAm and IPAAm–AAc (5) gels exhibit small
swelling– deswelling changes accompanied by a
slight and gradual decrease in gel diameter over
repeated temperature cycles. In contrast, a rapid
and large volume swelling– deswelling change was
observed for the IPAAm–CIPAAm (20) gel. The size
of IPAAm–CIPAAm (20) gel at 30 °C was nearly
three times that of its deswelling gel state at 35 °C
and significantly greater than any IPAAm–AAc
ionic gels under comparable conditions. These be-
haviors are consistent with the proposed hypothesis
that chain aggregation forces in phase transitions
(near the LCST) can be maintained in ionic gels if
chain– chain association is facilitated by careful
monomer design.
The authors are grateful to Professor David Grainger
(Colorado State University) for valuable comments.
This work was supported in part by a Grant-in-Aid for
Scientific Research on Priority Areas (A, No. 1167276)
from the Ministry of Education, Science, Sport and
Culture (Japan) and the Japan Society for the Promo-
tion of Science Research for the Future Program (JSPS-
RFTF96I00201).
REFERENCES AND NOTES
1. Feil, H.; Bae, Y. H.; Heijan, J.; Kim, S. W. Macro-
molecules 1993, 26, 2496 –2500.
2. Yoshida, R.; Kaneko, Y.; Sakai, K.; Okano, T.;
Sakurai, Y.; Bae, Y. H.; Kim, S. W. J Controlled
Release 1994, 32, 97–102.
3. Dong, L. C.; Hoffman, A. S. J Controlled Release
1991, 15, 141–152.
4. Park, S. Y.; Bae, Y. H. Macromol Rapid Commun
1999, 20, 269 –273.
5. Suzuki, A.; Tanaka, T. Nature 1990, 346, 345–347.
6. Takeoka, Y.; Aoki, T.; Sanui, K.; Ogata, N.;
Yokoyama, M.; Okano, T.; Sakurai, Y.; Watanabe,
M. J Controlled Release 1995, 33, 79 – 87.
7. Kwon, I. C.; Bae, Y. H.; Okano, T.; Berner, B.; Kim,
S. W. Makromol Chem Macromol Symp 1990, 33,
265–277.
8. Kwon, I. C.; Bae, Y. H.; Kim, S. W. Nature 1991,
354, 291–293.
9. Bae, Y. H.; Okano, T.; Kim, S. W. J Polym Sci Part
B: Polym Phys 1990, 28, 923–936.
POLY(N-ISOPROPYLACRYLAMIDE)-BASED HYDROGELS
341

10. Yoshida, R.; Kaneko, Y.; Sakai, K.; Okano, T.;
Sakurai, Y.; Bae, Y. H.; Kim, S. W. J Controlled
Release Short Commun 1994, 32, 97–102.
11. Yoshida, R.; Sakai, K.; Okano, T.; Sakurai, Y.; Bae,
Y. H.; Kim, S. W. J Biomater Sci Polym Ed 1991, 3,
155–162.
12. Yoshida, R.; Okano, T.; Sakurai, Y.; Kaneko, Y.;
Sakai, K. Drug Delivery Syst 1995, 10, 31–35.
13. Yoshida, R.; Uchida, K.; Kaneko, Y.; Sakai, K.;
Kikuchi, A.; Sakurai, Y.; Okano, T. Nature 1995,
374, 240 –242.
14. Kaneko, Y.; Nakamura, S.; Sakai, K.; Aoyagi, T.;
Kikuchi, A.; Sakurai, Y.; Okano, T. Polym Gels
Networks 1998, 6, 333–345.
15. Gutowska, A.; Bae, Y. H.; Feijen, J.; Kim, S. W. J
Controlled Release 1992, 22, 95–104.
16. Kaneko, Y.; Nakamura, S.; Sakai, K.; Aoyagi, T.;
Kikuchi, A.; Sakurai, Y.; Okano, T. Macromole-
cules 1998, 31, 6099 – 6105.
17. Aoyagi, T.; Ebara, M.; Sakai, K.; Sakurai, Y.;
Okano, T. J Biomater Sci Polym Ed 2000, 11, 101–
110.
18. Ebara, M.; Aoyagi, T.; Sakai, K.; Sakurai, Y.;
Okano, T. Macromolecules, 2000, 33, 8312– 8316.
19. Yu, H.; Grainger, D. W. J Appl Polym Sci 1993, 49,
1553–1563.
20. Kaneko, Y.; Yoshida, R.; Sakai, K.; Sakurai, Y.;
Okano, T. J Membr Sci 1995, 101, 13–22.
342
EBARA ET AL.
Document Outline
Wyszukiwarka
Podobne podstrony:
The role of BRCA1 in DNA damage response
Trithemius The Arte Of Drawing Spirits Into Crystals
Study of temperature responsibility on the surfaces of a thermo responsive polymer modified stationa
J Slowacki Into the midst of riotous squabblers
Kradin Placebo Response and the Power of Unconscious Healing (Routledge, 2008)
Student Roles and Responsibilities for the Masters of Counsel
A Bite Into the History of the Autopsy
Forstchen, William R Into the Sea of Stars
The effect of temperature on the nucleation of corrosion pit
Bondeson; Aristotle on Responsibility for Ones Character and the Possibility of Character Change
Borovik Mirrors And Reflections The Geometry Of Finite Reflection Groups (2000) [sharethefiles com
Phillip G Zimbardo A Situationist Perspective On The Psychology Of Evil Understanding How Good Peo
social networks and the performance of individualns and groups
S D Houston Into the Minds of Ancients Advances in Maya Glyph Studies
Modification of Intestinal Microbiota and Its Consequences for Innate Immune Response in the Pathoge
William R Forstchen Into the Sea of Stars
British Patent 16,709 Improvements relating to the Conversion of Alternating into Direct Electric Cu
Latin in Legal Writing An Inquiry into the Use of Latin in the M
więcej podobnych podstron