
Multiplex PCR and minisequencing of SNPs—
a model with 35 Y chromosome SNPs
Juan J. Sanchez
, Claus Børsting
, Charlotte Hallenberg
, Anders Buchard
Alexis Hernandez
, Niels Morling
a
Department of Forensic Genetics, Institute of Forensic Medicine, University of Copenhagen, 11 Frederik V’s Vej,
DK-2100 Copenhagen, Denmark
b
Departamento de Canarias, Instituto Nacional de Toxicologı´a, Campus de Ciencias de la Salud, 38320 La Laguna, Tenerife, Spain
Received 22 January 2003; received in revised form 2 July 2003; accepted 7 July 2003
Abstract
We have developed a robust single nucleotide polymorphism (SNPs) typing assay with co-amplification of 25 DNA-fragments
and the detection of 35 human Y chromosome SNPs. The sizes of the PCR products ranged from 79 to 186 base pairs. PCR
primers were designed to have a theoretical T
m
of 60
5 8C at a salt concentration of 180 mM. The sizes of the primers ranged
from 19 to 34 nucleotides. The concentration of amplification primers was adjusted to obtain balanced amounts of PCR products
in 8 mM MgCl
2
. For routine purposes, 1 ng of genomic DNA was amplified and the lower limit was approximately 100 pg DNA.
The minisequencing reactions were performed simultaneously for all 35 SNPs with fluorescently labelled dideoxynucleotides.
The size of the minisequencing primers ranged from 19 to 106 nucleotides. The minisequencing reactions were analysed by
capillary electrophoresis and multicolour fluorescence detection. Female DNA did not influence the results of Y chromosome
SNP typing when added in concentrations more than 300 times the concentrations of male DNA. The frequencies of the 35 SNPs
were determined in 194 male Danes. The gene diversity of the SNPs ranged from 0.01 to 0.5.
# 2003 Elsevier Ireland Ltd. All rights reserved.
Keywords: Y chromosome; Single nucleotide polymorphism; Multiplex PCR; Minisequencing; Genotyping
1. Introduction
A large number of single nucleotide polymorphisms
(SNPs) have been identified
. Investigations of SNPs
on the Y chromosome in various populations have given
us important information on the history of the human male
populations (e.g.
). Due to the low mutation rates of
SNPs, the information relates to longer periods of time
compared to the information obtained with e.g. short tandem
repeat (STR)
and minisatellite markers as, for exam-
ple MSY1
.
Presently, typing of selected short tandem repeat (STR)
systems is the state of the art in forensic routine casework. It
is, however, anticipated that SNP typing will be used for
parentage testing and forensic casework in the future. The
advantage of SNPs in forensic casework is that small DNA
fragments of 40–50 bps from e.g. heavily degraded DNA can
be SNP typed. Furthermore, the SNP technology has a high
potential for automation. Although the genetic information
obtained by a SNP, in average, is much lower than that
obtained by an STR system, typing of 50–100 selected SNPs
would be sufficient for forensic casework
. The low
mutation rate of SNPs
makes these markers an
attractive tool for parentage testing.
Genetic markers on the Y chromosome are valuable tools
in forensic casework in special situations, e.g. in cases with
mixtures of DNA with a dominant amount of female DNA
and a very small amount of male DNA. In such cases, the
DNA profile of the autosomes of the male cannot be
obtained, but the Y chromosome markers can usually be
typed, even in situations with a very large relative amount of
female DNA
. In special cases of parentage testing, e.g.
Forensic Science International 137 (2003) 74–84
*
Corresponding author. Tel.:
þ45-35-32-62-25;
fax:
þ45-35-32-61-20.
E-mail address: juan.sanchez@forensic.ku.dk (J.J. Sanchez).
0379-0738/$ – see front matter # 2003 Elsevier Ireland Ltd. All rights reserved.
doi:10.1016/S0379-0738(03)00299-8

if the alleged father is unavailable for testing while close
male relatives are available, investigation of genetic markers
on the Y chromosome are valuable.
If SNP typing is going to be used in forensic casework, it
is essential that the investigations can be performed on small
amounts of DNA, if possible, <1 ng DNA. If the polymerase
chain reaction (PCR) is used, the amplifications of all DNA
fragments to be investigated must be done in one or very few
amplification reactions.
We decided to explore a SNP typing method that is based on
multiplex PCR and multiplex minisequencing. We chose SNP
markers on the Y chromosome because these markers, in
forensic genetics, offer additional information to the informa-
tion obtained by STR typing. Furthermore, the Y chromosome
SNPs are useful tools for the study of genetics of populations.
In the last years, a number of multiplex PCR Y chromo-
some SNP analyses have been reported. Most of them
included a limited amount of SNPs (often 3–10 SNPs) in
each PCR (e.g.
) although larger multiplexes have
been reported
We selected Y chromosome SNPs that were reported to be
polymorphic in European and other populations
However, the main purpose of the study was to explore the
technical issues related to multiplexing a larger number of
DNA fragments and simultaneous detection of a large
number of SNPs. The intention was not to make a final
panel for typing of major Y chromosome haplogroups. In
order to assess the technical performance of the SNP typing
system, we included four pairs of SNPs each of which pair
was expected to give concordant results (e.g. M40 and M96).
Here, we describe a method for typing 35 SNPs on the Y
chromosome. The typing was performed by (1) multiplex
PCR amplification of 25 Y chromosome DNA fragments, (2)
multiplex primer extension reactions of 35 SNPs with
fluorescence labelled nucleotides, and (3) detection of the
35 SNPs by capillary electrophoresis and multicolour fluor-
escence detection.
2. Materials and methods
2.1. Donors and DNA preparations
A total of 194 unrelated males and 15 unrelated female
Danes donated blood samples or buccal cells. DNA was
isolated from 200 ml of peripheral blood using QIAamp
DNA Blood Mini Kit according to the manufacturer’s pro-
tocol (Qiagen, Hagen, Germany). Alternatively, 1.2 mm
(diameter) FTA
1
paper (Whatman International, Cam-
bridge, UK) soaked with blood or buccal cells was used.
Mixtures of DNA from males and females were prepared in
checker board with three concentrations of male DNA (0.16,
0.8 and 1.6 ng) and female DNA ranging from 0 to 60 ng.
Fluorometric measurement of DNA concentration was
done by SYBR Green I and analysed in a LightCycler
instrument (Roche Diagnostics GmbH, Germany) and
Hoechst 33258 (Molecular Probes Inc., Eugene, OR) using
a Hoefer DyNA Quant 200 instrument (Molecular Vision).
Calibration reference curves were established using a calf
thymus DNA standard (Sigma–Aldrich, Missouri, USA).
2.2. Selection of PCR amplification primers
The Y chromosome SNPs selected (
) included
those used by Semino et al.
for a study of the distribu-
tion of Y chromosome SNPs in European populations. In
addition, we included SNPs that were reported to be poly-
morphic in other ethnic groups.
DNA segments including the SNPs selected were identi-
fied and complementary primers were designed so that the
lengths of the amplified genomic Y chromosome DNA
fragments would range from 79 to 186 nucleotides. Some
SNPs were situated very closely to each other and it was
decided to include a number of amplification targets with
two or three SNPs (
The sequence of each locus was obtained from GenBank
1
(
) using a nucleotide basic local
alignment search tool (BLAST). Published PCR primers
were initially used as the reference sequence for each Y
SNP locus, but all of them needed to be redesigned.
The primers for the genomic segments spanning one or more
Y chromosome markers were designed with the Primer
3.0 program v. 0.2 (
http://www-genome.wi.mit.edu/cgi-bin/
). All primers were selected to have
theoretical melting temperatures of 60
5 8C at a salt con-
centration of 180 mM and a purine:pyrimidine content close
to 1:1, when possible. The lengths of the primers ranged
between 19 and 34 nt. Primers with four or more bases at the
3
0
end complementary to another part of the primer were
discarded or redesigned to avoid artefacts due to hairpin
formation. Each primer pair was tested for primer–primer
interactions, and the primer sequences were checked to avoid
similarities with repetitive sequences or with other loci in the
genome. The primers were checked for homology to other
amplicons in the pool of 25 primer pairs.
shows the
sequences of the amplification primers selected.
2.3. PCR conditions
HPLC purified primers for amplification were purchased
from TAG A/S (Copenhagen, Denmark). A primer stock
solution was prepared by dissolving the lyophilized primers
in Tris/EDTA buffer (10 mM Tris, 100 mM EDTA, pH 7.5;
Sigma–Aldrich) to a final DNA concentration of 100 pmol/ml.
Each primer pair was tested in singleplex PCR. Ten ng
template was amplified by PCR in a 25 ml reaction volume
containing 1
PCR buffer, 1.5 mM MgCl
2
, 200 mM of each
dNTP, 0.4 mM of each primer, and 0.6 units of AmpliTaq
Gold DNA polymerase at 94 8C for 5 min followed by 30
cycles of 30 s at 95 8C, 30 s at 60 8C, 30 s at 72 8C, and a
final extension for 5 min at 72 8C. The products were
analysed by electrophoresis in 11% polyacrylamide gels.
J.J. Sanchez et al. / Forensic Science International 137 (2003) 74–84
75

TBE (1
) (89 mmol/l Tris base, 89 mmol/l boric acid,
2 mmol/l EDTA, pH 8.3) was used as electrophoresis buffer.
The gels were stained with 0.5 mg/ml ethidium bromide. The
10 bp ladder from invitrogen (Groningen, The Netherlands)
was used to assign the sizes of the fragments.
The final setup of the PCR amplification included 1 ng
DNA in a 50 ml reaction volume containing 1
PCR buffer,
8 mM MgCl
2
, 400 mM of each dNTP, 0.01–0.42 mM of each
primer, and 2.5 units of AmpliTaq Gold DNA polymerase
(AB, Foster City, CA).
All DNA amplifications were performed in a GeneAmp
9600 thermal cycler (Perkin-Elmer, Wellesley, USA) using
the following programme: denaturation at 94 8C for 5 min
followed by 33 cycles for 30 s at 95 8C, 30 s at 60 8C, and 30 s
at 65 8C, followed by a final extension for 7 min at 65 8C.
The concentrations of the primers in the multiplex reac-
tion were adjusted in order to obtain equal amount of each
PCR product. The primer concentrations ranged from 0.01 to
0.42 mM (
The PCR products were analysed on 11% polyacrylamide
gels as described later (
In order to eliminate the excess of primers and dNTPs, the
PCR products was purified on a MinElute PCR purification
spin column (Qiagen, Hagen, Germany) following the man-
ufacturer’s protocol. The DNA was eluted in 30 ml of Milli-Q
water.
E. coli exonuclease I (Exo I) and shrimp alkaline phos-
phatase (SAP) was also used to remove primers and unin-
corporated dNTPs (USB Corporation, Cleveland, USA). Six
microliters ExoSAP-IT kit or 5 units of SAP and 2 units of
Table 1
Y chromosome SNPs and primer sequences for PCR amplification of 25 Y chromosome DNA fragments with SNPs
Locus
GenBank or
dbSNPs accesion
Mutation
PCR primers (5
0
! 3
0
)
mM
Amplicon
size (bp)
Forward primer
Reverse primer
M2/sY81
Rs3893
A/G
acggaaggagttctaaaattcagg
aaaatacagctccccctttatcct
0.15
128
Rs3900
C/G
aggaccctgaaatacagaactg
aaatatttcaacatttcacaaaggaa
0.36
186
Rs3908
4G/3G
cctggtcataacactggaaatc
agctgaccacaaactgatgtaga
0.09
170
Rs3909
2 bp insertion
cctggtcataacactggaaatc
agctgaccacaaactgatgtaga
0.09
170
M19
Rs3010
T/A
cctggtcataacactggaaatc
agctgaccacaaactgatgtaga
0.09
170
M32
AC009977
T/C
tgaccgtcataggctgagaca
ttgaagcccccaagagagac
0.07
160
M33
AC009977
A/C
tgaccgtcataggctgagaca
ttgaagcccccaagagagac
0.07
160
M35
Rs1179188
G/C
agggcatggtccctttctat
tccatgcagactttcggagt
0.42
96
M40/SRY
4064
AC006040
G/A
tggtctcaatctcttcaccctgt
catttcagtaaatgccacacaaga
0.18
119
Rs2032631
G/A
gagagaggatatcaaaaattggcagt
tgacagtggcaccaaaggtc
0.03
138
M46/Tat
AC002531
T/C
tatatggactctgagtgtagacttgtga
ggtgccgtaaaagtgtgaaataatc
0.46
115
M52
AC009977
A/C
cctcaacttcccagagtgttg
gacgaagcaaacatttcaagagag
0.03
152
AC010889
C/T
tgcattactccgtatgttcgac
tggaagcttaccatctttttatga
0.08
132
Rs2032640
C/T
catctcttaacaaaagaggtaaattttgtcc
cattgtgttacatggcctataatattcagt
0.24
179
M89
Rs2032652
C/T
tggattcagctctcttcctaaggttat
ctgctcaggtacacacagagtatca
0.03
135
M96
AC010889
G/C
tgccctctcacagagcactt
ccacccactttgttgctttg
0.27
143
M123
AC010889
G/A
gttgcccaggaatttgcat
cacagagcaagtgactctcaaag
0.02
88
AC010137
5G/4G
ccccgaaagttttattttattcca
ttctcagacaccaatggtcctatc
0.06
113
AC010889
G/A
catctcttaacaaaagaggtaaattttgtcc
cattgtgttacatggcctataatattcagt
0.24
179
AC010137
T/A
ccccgaaagttttattttattcca
ttctcagacaccaatggtcctatc
0.06
113
AC010889
T/C
catctcttaacaaaagaggtaaattttgtcc
cattgtgttacatggcctataatattcagt
0.24
179
AC010889
A/C
gagagaggatatcaaaaattggcagt
tgacagtggcaccaaaggtc
0.03
138
AC009977
A/C
aggaccctgaaatacagaactg
aaatatttcaacatttcacaaaggaa
0.36
186
M167/SRY
2627
AC006040
C/T
cggaaccactaccagcttca
agttaaggccccacgcagt
0.03
113
M170
Rs2032597
A/C
cagctcttattaagttatgttttcatattctgtg gtcctcattttacagtgagacacaac
0.07
119
M172
Rs2032604
T/G
tgagccctctccatcagaag
gccaggtacagagaaagtttgg
0.16
179
M173
Rs2032624
A/C
ttttcttacaattcaagggcatttag
ctgaaaacaaaacactggcttatca
0.10
81
M175
Rs2032678
5 bp
gatttaaactctctgaatcaggcacat
ttctactgatacctttgtttctgttcattc
0.02
79
Rs2032664
C/A
ccatataaaaacgcagcattctgtt
tggagagaacttgagaaaaagtagagaa
0.12
176
Rs2032665
T/C
ccatataaaaacgcagcattctgtt
tggagagaacttgagaaaaagtagagaa
0.12
176
AC010889
T/C
tgcattactccgtatgttcgac
tggaagcttaccatctttttatga
0.08
132
SRY
10831
/SRY
1532
Rs2534636
A/G
tcatccagtccttagcaaccatta
ccacataggtgaaccttgaaaatg
0.06
150
12f2
AC005820
Present/absent
cactgactgatcaaaatgcttacagat
ggatcccttccttacaccttataca
0.06
90
92R7
Rs2535813
GA/A
ttaaatccctcctatttgtgctaacc
aatgcatgaacacaaaagacgtaga
0.04
89
P25
Rs150173
C/CA
tggaccatcacctgggtaaagt
ggcagtataaggttgtcacatcacat
0.01
109
a
SNP markers on the same DNA fragment: (M9 and M163), (M17, M18 and M19), (M32 and M33), (M45 and M157), (M78 and M224),
(M81, M151 and M154), (M139 and M153), (M212 and M213). All primers were redesigned compared to previously published primers.
76
J.J. Sanchez et al. / Forensic Science International 137 (2003) 74–84

Exo I were added to 15 ml of PCR product, mixed, and
incubated at 37 8C for 1 h. The enzymes were inactivated at
75 8C for 15 min.
2.4. Design of PCR minisequencing primers
shows the genotyping primers designed for each
SNP. Primers for detection of deletions and insertions were
designed with the 3
0
, base corresponding to the last base
before the possible deletion or insertion. For each SNP
system investigated in the present study, the following base
would identify the polymorphism. The sequences of the
primers were checked for the possibility of primer–dimer
and hairpin formation and investigated in PCR without
template (‘self-extension reaction’). In order to distinguish
between the sizes of the detection primers, the primers
were synthesized with lengths between 19 and 106 nucleo-
tides with intervals of four nucleotides for the great major-
ity of the primers (
). The lengths of the template
specific parts of the primers ranged from 16 to 29 nucleo-
tides. The desired length of a primer was adjusted at the
5
0
end by addition of a piece of a ‘neutral’ sequence
and, if necessary, a poly-C tail. The neutral sequence,
5
0
-AACTGACTAAACTAGGTGCCACGTCGTGAAAGT-
CTGACAA-3
0
, is a random sequence that did not match
with any human sequence in the NCBI non-redundant
database
.
For each 4 bp DNA fragment size interval of the detection
primers, two SNP loci were detected. This was done by
selecting two SNP loci with different nucleotide polymorph-
ism. One SNP could be, e.g. an A/T SNP and the other a C/G
SNP. Thus, the minisequencing primers for the two SNPs
could have the same length and the two polymorphisms
would still be detectable. Primers for minisequencing were
HPLC purified (DNA-Technology A/S, Aarhus, Denmark
and Proligo France SAS, Paris, France).
2.5. Minisequencing reaction and capillary
electrophoresis
Multiplex PCR minisequencing was performed in 8 ml
reactions with 0.2 ml purified PCR product (6–10 ng equiva-
lent to 5–8 fmol of each fragment), 4 ml of SNaPshot
TM
reaction mix and 0.01–0.5 mM of the primers (
). The
thermal cycling was performed with a rapid thermal ramp to
96 8C for 10 s, 50 8C for 5 s, and 60 8C for 30 s for 25 cycles.
Fig. 1. Multiplex PCR products of 25 Y chromosome DNA fragments. Ethidium bromide stained polyacrylamide gel with
PCR products obtained from various sources of blood. A negative control with DNA from a female was included. (L) 10 bp ladder
from invitrogene.
J.J. Sanchez et al. / Forensic Science International 137 (2003) 74–84
77

A positive control (provided with the kit) and negative
control (sterile water or PCR product from a female), was
performed for each batch of 44 samples.
The homogeneity of each primer was checked in single-
plex minisequencing. The occurrence of extra peaks one or
more nucleotides smaller than the expected size indicated
heterogeneity of the minisequencing primer.
After the minisequencing reaction, 1 Unit of SAP was
added and the tube was incubated at 37 8C for 1 h in order to
remove the 5
0
phosphoryl groups of the unincorporated
[F]ddNTPs. SAP was inactivated by incubation at 75 8C
for 15 min.
One ml of the purified minisequencing PCR product was
analysed on an AB Prism 3100 Genetic Analyser with a
36 cm capillary array, POP-4 polymer and 10 s at 3000 V
injections. GeneScan-120 LIZ
TM
was used as internal size
standard. The data were analysed using GeneScan Analysis
software v. 3.7 (Applied Biosystems). After background
substraction and colour separation, peaks were sorted
into bins according to sizes by comparison to the internal
size standard. Peaks above 400 relative fluorescence units
were considered positive signals and a SNP type was
assigned.
2.6. Reproducibility studies
DNA samples from 194 unrelated male Danes were typed
twice with the minisequencing technique and assigned SNP
types for the 35 SNP systems. The assignments of SNP types
of the duplicate testing were compared.
Table 2
Minisequencing primer sequences for typing of 35 Y chromosome SNP markers
Locus
Poly
(dC)
Neutral Sequence
(5
0
! 3
0
)
Target specific sequence
(5
0
! 3
0
)
Orientation
mM
Primer
size (nt)
M170
None
None
caacccacactgaaaaaaa
Reverse
0.02
19
M45
None
caa
ctcagaaggagctttttgc
Reverse
0.02
22
M139
None
aa
taatctgacttggaaagggg
Forward
0.01
22
M2/sY81
None
gacaa
ctttatcctccacagatctca
Reverse
0.28
26
M46/Tat
None
None
gctctgaaatattaaattaaaacaac
Reverse
0.25
26
M167/SRY
2627
None
tgaaagtctgacaa
aagccccacagggtgc
Forward
0.35
30
M213
None
tgacaa
tcagaacttaaaacatctcgttac
Reverse
0.02
30
M52
None
tctgacaa
aatatcaagaaacctatcaaacatcc
Reverse
0.02
34
P25
None
tcgtgaaagtctgacaa
tgcctgaaacctgcctg
Forward
0.04
34
M78
None
gaaagtctgacaa
cttattttgaaatatttggaagggc
Reverse
0.02
38
92R7
None
gtgaaagtctgacaa
catgaacacaaaagacgtagaag
Reverse
0.01
38
M89
None
cacgtcgtgaaagtctgacaa
aactcaggcaaagtgagagat
Reverse
0.09
42
M123
None
acgtcgtgaaagtctgacaa
atttctaggtattcaggcgatg
Reverse
0.03
42
M35
None
ggtgccacgtcgtgaaagtctgacaa
tcggagtctctgcctgtgtc
Reverse
0.25
46
M153
None
ggtgccacgtcgtgaaagtctgacaa
gctcaaagggtatgtgaaca
Forward
0.02
46
M40/SRY
4064
None
aaactaggtgccacgtcgtgaaagtctgacaa
tccaccctgtgatccgct
Reverse
0.08
50
M154
None
gccacgtcgtgaaagtctgacaa
gttacatggcctataatattcagtaca
Reverse
0.03
50
M32
None
taggtgccacgtcgtgaaagtctgacaa
agacaagatctgttcagtttatctca
Forward
0.50
54
M151
None
aggtgccacgtcgtgaaagtctgacaa
caatctactacatacctacgctatatg
Forward
0.02
54
M17
None
actaaactaggtgccacgtcgtgaaagtctgacaa
ccaaaattcacttaaaaaaaccc
Reverse
0.02
58
M96
None
aactgactaaactaggtgccacgtcgtgaaagtctgacaa
ggaaaacaggtctctcataata
Forward
0.15
62
M172
7
aactgactaaactaggtgccacgtcgtgaaagtctgacaa
caaacccattttgatgctt
Forward
0.10
66
M173
3
aactgactaaactaggtgccacgtcgtgaaagtctgacaa
tacaattcaagggcatttagaac
Forward
0.03
66
M19
4
aactgactaaactaggtgccacgtcgtgaaagtctgacaa
aaactatttttgtgaagactgttgta
Forward
0.10
70
M224
7
aactgactaaactaggtgccacgtcgtgaaagtctgacaa
aattgatacacttaacaaagatacttc
Forward
0.13
74
SRY
10831
/SRY
1532
10
aactgactaaactaggtgccacgtcgtgaaagtctgacaa
ttgtatctgactttttcacacagt
Forward
0.03
74
M18
17
aactgactaaactaggtgccacgtcgtgaaagtctgacaa
gtttgtggttgctggttgtta
Forward
0.05
78
M157
18
aactgactaaactaggtgccacgtcgtgaaagtctgacaa
caccaaaggtcatttgtggt
Reverse
0.20
78
M81
14
aactgactaaactaggtgccacgtcgtgaaagtctgacaa
cttggtttgtgtgagtatactctatgac
Reverse
0.03
82
M163
25
aactgactaaactaggtgccacgtcgtgaaagtctgacaa
cacaaaggaattttttttgag
Reverse
0.51
86
M212
20
aactgactaaactaggtgccacgtcgtgaaagtctgacaa
gcattctgttaatataaaacacaaaa
Forward
0.20
86
M9
22
aactgactaaactaggtgccacgtcgtgaaagtctgacaa
catgtctaaattaaagaaaaataaagag
Reverse
0.40
90
12f2
29
aactgactaaactaggtgccacgtcgtgaaagtctgacaa
aacatgtaagtctttaatccatctc
Forward
0.02
94
M33
29
aactgactaaactaggtgccacgtcgtgaaagtctgacaa
cagttacaaaagtataatatgtctgagat
Reverse
0.18
98
M175
46
aactgactaaactaggtgccacgtcgtgaaagtctgacaa
cacatgccttctcacttctc
Forward
0.28
106
a
The detection orientation has been probed relative to the YCC information reported in
.
78
J.J. Sanchez et al. / Forensic Science International 137 (2003) 74–84

2.7. Statistical methods
Gene diversities and standard errors were calculated
according to the methods of Nei
3. Results
3.1. DNA purification methods
DNA purified with Qiagen columns and DNA from FTA
1
paper with bloodstains in all cases gave satisfactory results
(
). DNA from buccal cells on FTA
1
paper gave
variable intensities of the results of samples.
3.2. Design of primers
When no band or only a very weak band was observed,
suggesting that the affinities of the primers were suboptimal,
the primers were redesigned. In one case, the PCR amplifica-
tion was very weak and four different sets of primers were
tried before an acceptable yield was obtained. It was not
possible to understand the reason since the primer set best
suited from a theoretical point gave the lowest yield. In three
cases with unsatisfactory yields, the primers were redesigned
with ‘GC’ at the 3
0
end with successful results. Twenty-one of
the 25 primer pairs worked satisfactorily at the first design.
3.3. PCR buffer and efficiency of multiplex PCR
amplification
We found that the best results of amplification of all 25
DNA targets were obtained by increasing the concentration
to 8 mM MgCl
2
. Higher concentrations inhibited the ampli-
fication (data not shown).
3.4. Quality of DNA primers for template PCR
amplification
Unpurified primers could be combined into multiplexes
up to seven systems while HPLC purified primers could be
combined to amplify at least 25 templates in one reaction.
3.5. Titration of primer concentrations in PCR
amplification
It was necessary to titrate primer concentrations to obtain
a balanced PCR multimix for minisequencing. The final
concentrations of primers ranged from 0.11 to 0.46 mM.
Fig. 2. Electropherogramme with 35 Y chromosome SNP profiles from a male donor. GeneScan analysis of SNaPshot
TM
minisequencing of
the Y chromosome SNP multiplex.
J.J. Sanchez et al. / Forensic Science International 137 (2003) 74–84
79
3.6. Sensitivity of the target multiplex PCR amplification
In our hands, the lower limit for reproducible results was
approximately 100 pg DNA with a range up to approxi-
mately 10 ng and an optimum at 1–2 ng (
Figs. 2 and 3
).
3.7. Purification of the PCR template amplification
product
Both spin column and enzymatic purified PCR amplifica-
tion products gave satisfactory minisequencing typing reac-
tions. The recovery with the Exo I-SAP was 100% while the
column purification had a recovery of approximately 80%
(data not shown).
3.8. Design of DNA primers for minisequencing
None of the 35 detection primers had to be redesigned.
3.9. Quality of DNA primers for minisequencing
Clear, homogeneous peaks were obtained only if the
purity of the primers was higher than approximately 90%.
If the purity was less, the signal from degenerated primers
(n
1, n 2, etc.) would decrease the discrimination.
3.10. Annealing temperature of minisequencing
primers
Annealing temperatures from 50 to 60 8C gave almost the
same overall results in the 35 SNP multiplex when judged by
inspection of the peak areas.
3.11. Y chromosome SNP typing results
shows a representative electropherogramme of
typing of 35 Y chromosome SNPs in an individual. In
one of the 194 males, typing could no reaction was obtained
in M81. The same lack of reaction in M81 was found in the
son of the investigated man. The remaining 34 Y chromo-
some SNPs were detected in the man and his child. All other
male samples gave a full 35-Y-SNP profile.
3.12. Reproducibility of Y chromosome SNP typing
with minisequencing
SNP typing was performed twice in all 194 male Danes
and the duplicate types were consistent. In each minisequen-
cing experiments, at least one sample with known types for
all 35 SNPs was included, and concordant assignments of
SNP types were obtained in all cases.
Four samples were typed for the 11 SNPs SRY2627,
M213, M35, M153, SRY
4064
, M17, M18, M9, SRY
10831
,
92R7, and P25 as part of an interlaboratory exercise of the
European DNA Profiling Group, and correct results were
obtained.
50
100
250
500
1000 2000 4000 8000
0
5
10
15
20
25
DNA (pg)
Relative Fluorescence Units
(%)
Fig. 3. Sensitivity of the 35 Y chromosome SNP typing assay.
For each DNA concentration, the relative fluorescence units
(RFUs from GeneScan) of investigations of four SNPs detected
with each of the four dyes: blue, green, yellow and red were
collated from typing of two individuals. For each DNA
concentration, the median RFU value of the two individuals
was calculated for each dye, and for each concentration the
median RFUs were normalized as a percentage of the total RFUs
of all the RFUs for the dye in question. Finally, for each DNA
concentration, the median of the normalized RFU values for all
four dyes was calculated as a percentage of the sum of all
normalized median RFU values of all concentrations. Thus, the
sum of RFUs in the figure sum up to 100%. The error bars
indicate the standard error of the mean (S.E.M.).
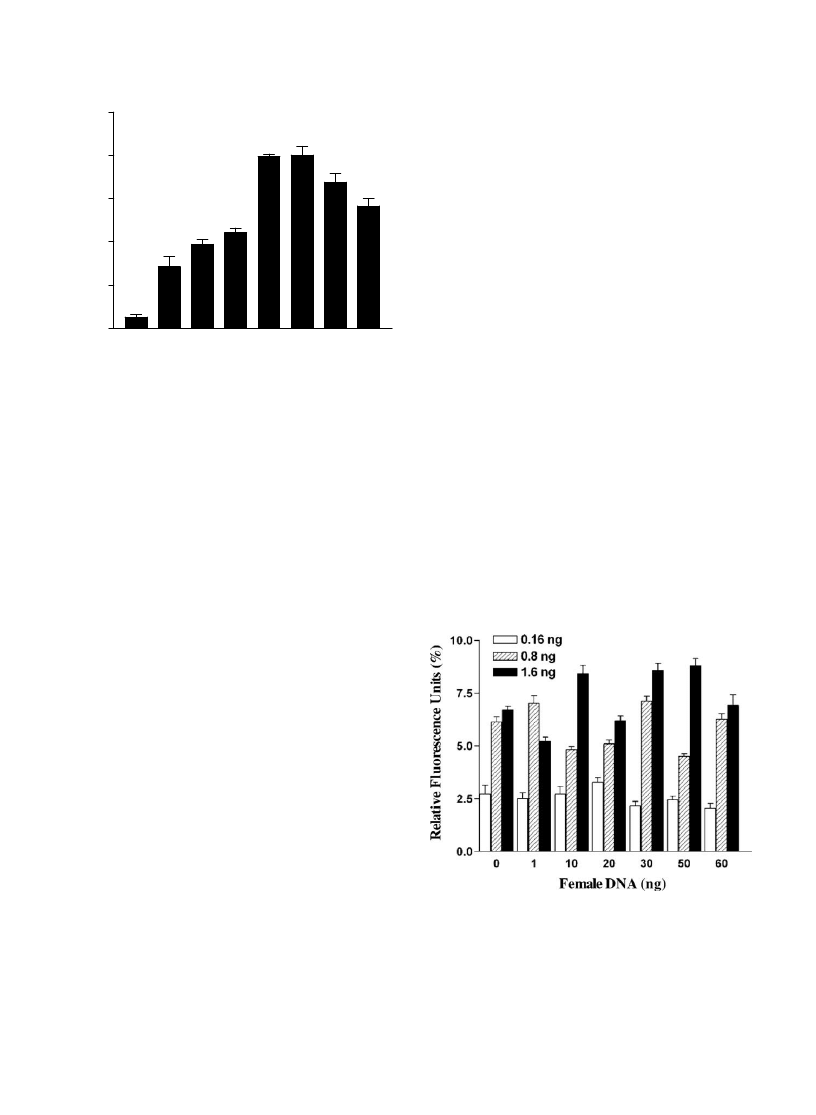
Fig. 4. Effect of excess DNA from females on the 35 Y
chromosome SNP typing assay. The relative fluorescence units
(RFUs from GeneScan) of mixtures of male DNA and female
DNA in great excess. The RFUs were calculated as indicated in
. In general, there was a dose response relation between the
concentration of male DNA and the RFU signal strength, while
female DNA had practically no influence on the RFU signal in
the concentration range investigated.
80
J.J. Sanchez et al. / Forensic Science International 137 (2003) 74–84

3.13. Male–female mixtures of DNA
Female DNA did not influence the results of Y chromo-
some SNP typing when added in concentrations more than
300 times the concentrations of male DNA (
3.14. Y chromosome SNP population data in Danes
shows the frequency distribution of the 35 SNPs
investigated in 194 male Danes. No SNP signal was obtained
in 15 female Danes. A total of 19 SNPs showed variation
while 16 SNPs were monomorphic in the male Danes
studied.
Two signals were obtained in P25 and 92R7 in some
individuals (cf discussion). DNA from individuals with two
signals in theses systems was investigated with STR-tech-
nique. Only one STR-profile was obtained in each individual
demonstrating that contamination of DNA was not the
reason for the two signals in P25 and 92R7.
4. Discussion
We have developed a PCR multiplex-based system for
typing of a large number of SNPs using Y chromosome
SNPs as an example. An important part of the work was to
explore the various aspects of the multiplex PCR methods.
The 35 Y chromosome SNPs presented here are not our final
set of Y chromosome SNPs for population studies or forensic
genetic applications.
Table 3
Frequencies of 35 Y chromosome SNP markers in male Danes
Locus
Fragment number
Polymorphism
Frequency (number)
Frequency (%)
M2/sY81
1
A/G
194/0
100.0/0.0
M9
2
C/G
85/109
43.8/56.2
M17
3
4G/3G
162/32
83.5/16.5
M18
3
No ins./2 bp ins.
194/0
100.0/0.0
M19
3
T/A
194/0
100.0/0.0
M32
4
T/C
194/0
100.0/0.0
M33
4
A/C
194/0
100.0/0.0
M35
5
G/C
190/4
97.9/2.1
M40/SRY
4064
6
G/A
190/4
97.9/2.1
M45
7
G/A
86/108
44.3/55.7
M46/Tat
8
T/C
193/1
99.5/0.5
M52
9
A/C
194/0
100.0/0.0
M78
10
C/T
192/2
99.0/1.0
11
C/T
193/0
100.0/0.0
M89
12
C/T
4/190
2.1/97.9
M96
13
G/C
190/4
97.9/2.1
M123
14
G/A
193/1
99.5/0.5
M139
15
5G/4G
0/194
0.0/100.0
M151
11
G/A
194/0
100.0/0.0
M153
15
T/A
194/0
100.0/0.0
M154
11
T/C
194/0
100.0/0.0
M157
7
A/C
194/0
100.0/0.0
M163
2
A/C
194/0
100.0/0.0
M167/SRY
2627
16
C/T
194/0
100.0/0.0
M170
17
A/C
119/75
61.3/38.7
M172
18
T/G
189/5
97.4/2.6
M173
19
A/C
89/105
45.9/54.1
M175
20
No del./5 bp del.
194/0
100.0/0.0
M212
21
C/A
194/0
100.0/0.0
M213
21
T/C
4/190
2.1/97.9
M224
10
T/C
194/0
100.0/0.0
SRY
10831
/SRY
1532
22
A/G
32/162
16.5/83.5
12f2
23
Present/absent
189/5
97.4/2.6
92R7
24
GA/A
86/108
44.3/55.7
P25
25
C/CA
124/70
63.9/36.1
a
Some PCR products contain more than one SNP in the same fragment.
b
Following the Y chromosome consortium nomenclature system
c
One male gave no reaction in minisequencing of M81.
d
Two signals were detected in some individuals
J.J. Sanchez et al. / Forensic Science International 137 (2003) 74–84
81

Successful PCR multiplexing depends on a number of
factors. Below, we present some of our considerations
concerning the selection of the SNPs and the generation
of the multiplex PCRs for amplification and minisequen-
cing.
At an early stage, it was decided to use the multicolour
fluorescence electrophoresis technique combined with PCR
multiplexing at approximately 60 8C in high concentrations
of MgCl
2
. The spacing between minisequencing primers
was decided to be four nucleotides because we wanted to
obtain reliable separation in the electrophoresis.
We attempted to avoid SNPs situated in regions reported
to be replicated. Two exceptions were the P25 and 92R7
SNPs that are situated in a region that most probably is part
of a duplication
. Both SNPs seem to discriminate
between European and other populations
Multiplex PCR amplification primers between 19 and 34
bases pairs long were selected because it was anticipated that
such long primers would work well under multiplex condi-
tions
Qiagen purified DNA from blood samples and blood
stains on FTA
1
paper worked equally well in the assay.
Chelex treated blood samples worked as well (data not
shown). Optimal multiplex SNP typing results were obtained
with 1 ng DNA (range 0.1
20 ng DNA). Thus, quantifica-
tion of DNA is not mandatory for the SNP assay. It should,
however, be noticed that the balance of the amounts of
amplification products of the DNA fragments is changed
with increasing amounts of templates. With increasing
concentrations of PCR amplified fragments, small, fluores-
cent adenosinnucleotide peaks with sizes of PCR amplified
fragments plus one nucleotide were seen, most likely do to
non-template addition of a single adenosin molecules to the
3
0
end of some PCR amplified fragments. At low amounts of
template DNA, loss of signal will occur due to stochastic
phenomena
Commonly used PCR buffers include only KCl, Tris and
MgCl
2
. It has been reported that many primer pairs produ-
cing short amplification products (<200 bp) work better at
higher salt concentration (KCl) in multiplex systems
.
Increasing the concentration of KCl in the PCR buffer 1.6
and 2-fold in our 35-plex did not increase the yield of PCR
product significantly and had no effect on the synthesis of
fragments >150 bp. Increase of MgCl
2
concentration from 2
to 8 mM increased the yield of amplicons; higher MgCl
2
concentration inhibited the amplification (data not shown).
We used AmpliTaq Gold DNA polymerase (Applied
Biosystems) because this enzyme minimizes primer dimer
formation. Even with a 4-base 3
0
overlap between two
primers we obtained homogeneous PCR products (data
not shown). The most efficient enzyme concentration
seemed to be around 2.5 U/50 ml reaction volume.
In our hands, primer concentrations below 0.01 mM were
insufficient and concentrations above 0.5 mM seemed to
inhibit multiplex PCRS probably by inducing dimer–dimer
formation. Primer concentrations were adjusted to be
approximately 10
3
times more than the concentration of
the template.
We stored dNTPs in small aliquots at
20 8C for up to 8
months. However, we observed that dNTPs were sensitive to
repeated freezing and thawing. As a rule of thumb, the
multiplex PCR would fail if the dNTPs have been frozen
and thawed more than four times. The amount of time in
freezer was less important as it has been reported by others
authors
The enzymatic purification method is obviously easy, has
an almost 100% recovery and a very limited risk of con-
tamination.
We chose to adjust the length of the minisequencing
primers by means of (1) a part of a neutral sequence of
up to 40 nt and for the longer primers (2) an additional poly-
C part. The neutral sequence was selected in order to obtain a
more balanced base composition. We chose poly-C for the
tail because, in theory, poly-G would give a higher molecular
mass, poly-A would have a risk of depurination during
synthesis, and poly-T tails may interfere with the addition
of 3
0
ddA in the minisequencing reaction (SNaPshot
TM
protocol recommendation, Applied Biosystems).
The quality of minisequencing primers is important
because
primer
batches
with
heterogeneous
primer
sequences consisting of the intended DNA sequence of
‘n’ nucleotides plus a spectrum of shorter nucleotides
(n
1, n 2, n 3, etc.) in many cases will destroy the
minisequencing reaction. In addition, we observed amplifi-
cation failure due to a heterogeneous primer batch in the
PCR multiplex with seven systems even though each of the
seven works in singleplex reactions. Therefore, we recom-
mend that each primer batch is tested before the multiplex
PCR and subsequent analyses, e.g. by minisequencing or
mass spectrometry. Purification of the primers with e.g.
HPLC or gel purification techniques can to some extent
solve these problems.
The minisequencing system was rather insensitive to the
annealing temperature. It was necessary to adjust primer
concentrations from 0.01 to 0.50 mM in the minisequencing
multimix.
The longer extension products had electrophoretic mobi-
lities corresponding to those predicted by the number of
bases. The mobility of shorter extension products with the
same number of bases varied to some extent. This is most
probably due to the fact that differences in the masses of the
various fluorochromes used and in the exact composition of
purines and pyrimidines have a relatively high influence on
the mobility of short DNA molecules.
The SNP-typing results were highly reproducible. A total
of 194 males were SNP typed in duplicate and no discre-
pancies were observed. Furthermore, five of the most poly-
morphic SNPs were analysed by a DNA hybridisation assay
using the Nanogen technology
. Concordant results were
obtained for all 194 individuals (data not shown).
In one father-child combination, no allele of M81 was
detectable. An amplified fragment was present in the first
82
J.J. Sanchez et al. / Forensic Science International 137 (2003) 74–84

PCR because two other SNPs (M151 and M154) on the
fragment were detected, but no reaction of M81 was detected
in the minisequencing reaction. Work is in progress in order
to determine the nature of the variant.
A total of 19 of 29 SNPs reported to be polymorphic in
Europeans in a previous study
and 9 of 10 SNPs reported
in another study
turned out to be polymorphic in the
male Danes studied. The gene diversity for the loci showing
polymorphism ranged from 0.01 to 0.5 (
). M173,
M45, 92R7 and M9 were the most polymorphic markers in
Danes. The data were described as frequencies of individual
SNPs and not as Y chromosome haplogroups because the
study was a technical study and the Y chromosome multi-
plex is not ideal for typing of Y chromosome haplogroups. A
larger study of Y chromosome haplogroups in Danes and
other populations will be published elsewhere.
P25 and 92R7 were previously reported as SNPs
However, the P25 and 92R7 minisequencing primers were
extended with two different dideoxynucleotides during the
minisequencing reaction of numerous samples. This indi-
cates that at least two different, almost identical fragments
were amplified during the PCR reaction. Hurles et al.
previously observed that SNP typing of 92R7 gave two
results in some individuals. Further studies have confirmed
that P25 and 92R7 are paralogous sequence variants and that
at least one of the sequence variants in each group of loci is
polymorphic
.
The multiplex PCR SNP typing format presented here
seems to be useful for forensic casework because small
amounts of DNA (100 pg DNA) can be reliably typed.
The multiplex presented is not our final package for Y
chromosome SNPs for forensic purposes. The way forward
would go either through (1) the development of SNP
packages optimised for an initial screening plus further
packages optimised for the major populations or (2) the
development of a large multiplex package that include Y
chromosome SNPs that can discriminate between individual
lineages in all populations.
Acknowledgements
We thank Dr. Rebecca Reynolds, Roche Molecular Sys-
tems, for advice concerning the design of the multiplex PCR
for template generation in the initial phase of the project. We
thank Ms. AnneMette Holbo Birk for technical assistance.
The work was supported by grants to Juan Sanchez from
Ellen and Aage Andersen’s Foundation and Manuel Morales
Foundation.
References
[1] R. Sachidanandam, D. Weissman, S.C. Schmidt, J.M. Kakol,
L.D. Stein, G. Marth, S. Sherry, J.C. Mullikin, B.J.
Mortimore, D.L. Willey, S.E. Hunt, C.G. Cole, P.C. Coggill,
C.M. Rice, Z. Ning, J. Rogers, D.R. Bentley, P.Y. Kwok, E.R.
Mardis, R.T. Yeh, B. Schultz, L. Cook, R. Davenport, M.
Dante, L. Fulton, L. Hillier, R.H. Waterston, J.D. McPher-
son, B. Gilman, S. Schaffner, W.J. Van Etten, D. Reich, J.
Higgins, M.J. Daly, B. Blumenstiel, J. Baldwin, N. Stange-
Thomann, M.C. Zody, L. Linton, E.S. Lander, D. Altshuler,
A map of human genome sequence variation containing 1.42
million single nucleotide polymorphisms, Nature 409 (2001)
928–933.
[2] S. Paracchini, B. Arredi, R. Chalk, C. Tyler-Smith,
Hierarchical high-throughput SNP genotyping of the human
Y chromosome using MALDI-TOF mass spectrometry,
Nucleic Acids Res. 30 (2002) e27.
[3] M. Raitio, K. Lindroos, M. Laukkanen, T. Pastinen, P.
Sistonen, A. Sajantila, A.C. Syvanen, Y-chromosomal SNPs
in Finno-Ugric-speaking populations analyzed by minise-
quencing on microarrays, Genome Res. 11 (2001) 471–482.
[4] P.A. Underhill, G. Passarino, A.A. Lin, P. Shen, M. Mirazon
Lahr, R.A. Foley, P.J. Oefner, L.L. Cavalli-Sforza, The
phylogeography of Y chromosome binary haplotypes and
the origins of modern human populations, Ann. Hum. Genet.
65 (2001) 43–62.
[5] M.A. Jobling, C. Tyler-Smith, Fathers and sons: the Y
chromosome and human evolution, Trends Genet. 11 (1995)
449–456.
[6] H. Oota, W. Settheetham-Ishida, D. Tiwawech, T. Ishida, M.
Stoneking, Human mtDNA and Y chromosome variation is
correlated with matrilocal versus patrilocal residence, Nat.
Genet. 29 (2001) 20–21.
[7] M. Seielstad, Asymmetries in the maternal and paternal
genetic histories of Colombian populations, Am. J. Hum.
Genet. 67 (2000) 1062–1066.
[8] G. Passarino, G.L. Cavalleri, A.A. Lin, L.L. Cavalli-Sforza,
A.L. Borresen-Dale, P.A. Underhill, Different genetic com-
ponents in the Norwegian population revealed by the analysis
of mtDNA and Y chromosome polymorphisms, Eur. J. Hum.
Genet. 10 (2002) 521–529.
[9] P. de Knijff, Messages through bottlenecks: on the combined
use of slow and fast evolving polymorphic markers on
the human Y chromosome, Am. J. Hum. Genet. 67 (2000)
1055–1061.
[10] M.A. Jobling, Y-chromosomal SNP haplotype diversity in
forensic analysis, Forensic Sci. Int. 118 (2001) 158–162.
[11] J.L. Mountain, A. Knight, M. Jobin, C. Gignoux, A. Miller,
A.A. Lin, P.A. Underhill, SNPSTRs: empirically derived,
rapidly typed, autosomal haplotypes for inference of popula-
tion history and mutational processes, Genome Res. 12 (2002)
1766–1772.
[12] M.A. Jobling, E. Heyer, P. Dieltjes, P. de Knijff, Y
chromosome-specific microsatellite mutation rates re-exam-
ined using a minisatellite, MSY1, Hum. Mol. Genet. 8 (1999)
2117–2120.
[13] M. Brion, R. Cao, A. Salas, M.V. Lareu, A. Carracedo, New
method to measure minisatellite variant repeat variation
in population genetic studies, Am. J. Hum. Biol. 14 (2002)
421–428.
[14] P. Gill, An assessment of the utility of single nucleotide
polymorphisms (SNPs) for forensic purposes, Int. J. Legal
Med. 114 (2001) 204–210.
[15] D.E. Reich, S.F. Schaffner, M.J. Daly, G. McVean, J.C.
Mullikin, J.M. Higgins, D.J. Richter, E.S. Lander, D.
J.J. Sanchez et al. / Forensic Science International 137 (2003) 74–84
83

Altshuler, Human genome sequence variation and the
influence of gene history, mutation and recombination, Nat.
Genet. 32 (2002) 135–142.
[16] R. Thomson, J.K. Pritchard, P. Shen, P.J. Oefner, M.W.
Feldman, Recent common ancestry of human Y chromo-
somes: evidence from DNA sequence data, Proc. Natl. Acad.
Sci. U.S.A. 97 (2000) 7360–7365.
[17] M.A. Jobling, A. Pandya, C. Tyler-Smith, The Y chromo-
some in forensic analysis and paternity testing, Int. J. Legal
Med. 110 (1997) 118–124.
[18] N.M. Makridakis, J.K. Reichardt, Multiplex automated
primer extension analysis: simultaneous genotyping of several
polymorphisms, Biotechniques 31 (2001) 1374–1380.
[19] K. Lindblad-Toh, E. Winchester, M.J. Daly, D.G. Wang, J.N.
Hirschhorn, J.P. Laviolette, K. Ardlie, D.E. Reich, E.
Robinson, P. Sklar, N. Shah, D. Thomas, J.B. Fan, T.
Gingeras, J. Warrington, N. Patil, T.J. Hudson, E.S. Lander,
Large-scale discovery and genotyping of single nucleotide
polymorphisms in the mouse, Nat. Genet. 24 (2000) 381–386.
[20] R. Reynolds, K. Walker, L. Steiner, SNP genotyping using
megaplex PCR amplification and linear probe assays, in:
Proceedings of the 19th International Congress ISFG,
Mu¨nster, Germany (P. Gill), FSS, 2002, WS2.
[21] O. Semino, G. Passarino, P.J. Oefner, A.A. Lin, S. Arbuzova,
L.E. Beckman, G. De Benedictis, P. Francalacci, A. Kouvatsi,
S. Limborska, M. Marcikiae, A. Mika, B. Mika, D. Primorac,
A.S. Santachiara-Benerecetti, L.L. Cavalli-Sforza, P.A. Un-
derhill, The genetic legacy of Paleolithic Homo sapiens
sapiens in extant Europeans: a Y chromosome perspective,
Science 290 (2000) 1155–1159.
[22] P. Malaspina, F. Cruciani, B.M. Ciminelli, L. Terrenato, P.
Santolamazza, A. Alonso, J. Banyko, R. Brdicka, O. Garcia,
C. Gaudiano, G. Guanti, K.K. Kidd, J. Lavinha, M. Avila, P.
Mandich, P. Moral, R. Qamar, S.Q. Mehdi, A. Ragusa, G.
Stefanescu, M. Caraghin, C. Tyler-Smith, R. Scozzari, A.
Novelletto, Network analyses of Y-chromosomal types in
Europe, northern Africa, and western Asia reveal specific
patterns of geographic distribution, Am. J. Hum. Genet. 63
(1998) 847–860.
[23] M. Nei, Molecular Evolutionary Genetics, Columbia Uni-
versity Press, New York, 1982, p. 512.
[24] C. Bøsting, J.J. Sanchez, N. Morling, The two Y chromosome
loci, P25 and 92R7, are polymorphic paralogous sequence
variants, Forensic Sci. Int. (2003), submitted for publication.
[25] M.F. Hammer, T.M. Karafet, A.J. Redd, H. Jarjanazi, S.
Santachiara-Benerecetti, H. Soodyall, S.L. Zegura, Hierarch-
ical patterns of global human Y chromosome diversity, Mol.
Biol. Evol. 18 (2001) 1189–1203.
[26] O. Henegariu, N.A. Heerema, S.R. Dlouhy, G.H. Vance, P.H.
Vogt, Multiplex PCR: critical parameters and step-by-step
protocol, Biotechniques 23 (1997) 504–511.
[27] B.E. Krenke, A. Tereba, S.J. Anderson, F. Buel, S. Culhane, C.J.
Finis, C.S. Tomsey, J.M. Zachetti, A. Masibay, D.R. Rabbach,
E.A. Amiott, C.J. Sprecher, Validation of a 16-locus fluorescent
multiplex system, J. Forensic Sci. 47 (2002) 773–785.
[28] P. Markoulatos, N. Siafakas, M. Moncany, Multiplex
polymerase chain reaction: a practical approach, J. Clin.
Lab. Anal. 16 (2002) 47–51.
[29] P.N. Gilles, D.J. Wu, C.B. Foster, P.J. Dillon, S.J. Chanock,
Single nucleotide polymorphic discrimination by an electro-
nic dot blot assay on semiconductor microchips, Nat. Biotech.
17 (1999) 365–370.
[30] M.F. Hammer, A.J. Redd, E.T. Wood, M.R. Bonner, H.
Jarjanazi, T. Karafet, S. Santachiara-Benerecetti, A. Oppen-
heim, M.A. Jobling, T. Jenkins, H. Ostrer, B. Bonne´-Tamir,
Jewish and middel eastern non-jewish populations share a
common pool of Y-chromosome biallelic haplotypes, Proc.
Natl. Acad. Sci. U.S.A. 97 (2000) 6769–6774.
[31] N. Mathias, M. Baye´s, C. Tyler-Smith, Highly informative
compound haplotypes for the human Y chromosome, Hum.
Mol. Genet. 3 (1994) 115–123.
[32] The Y Chromosome Consortium, A nomenclature system for
the tree of human Y chromosomal binary haplogroups,
Genome Res. 12 (2002) 339–3480.
[33] M.E. Hurles, R. Veitia, F. Arroyo, M. Armenteros, J.
Bertranpetit, A. Perez-Lezaun, E. Bosch, M. Shlumukova,
A. Cambon-Thomsen, K. McElreavey, A. Lopez De Munain,
A. Rohl, I.J. Wilson, L. Singh, A. Pandya, F.R. Santos, C.
Tyler-Smith, M.A. Jobling, Recent male-mediated gene flow
over a linguistic barrier in Iberia, suggested by analysis of a
Y-chromosomal DNA polymorphism, Am. J. Hum. Genet. 65
(1999) 1437–1448.
84
J.J. Sanchez et al. / Forensic Science International 137 (2003) 74–84
Document Outline
- Multiplex PCR and minisequencing of SNPs-a model with 35 Y chromosome SNPs
- Introduction
- Materials and methods
- Results
- DNA purification methods
- Design of primers
- PCR buffer and efficiency of multiplex PCR amplification
- Quality of DNA primers for template PCR amplification
- Titration of primer concentrations in PCR amplification
- Sensitivity of the target multiplex PCR amplification
- Purification of the PCR template amplification product
- Design of DNA primers for minisequencing
- Quality of DNA primers for minisequencing
- Annealing temperature of minisequencing primers
- Y chromosome SNP typing results
- Reproducibility of Y chromosome SNP typing with minisequencing
- Male-female mixtures of DNA
- Y chromosome SNP population data in Danes
- Discussion
- Acknowledgements
- References
Wyszukiwarka
Podobne podstrony:
Dornbusch Fischer Samuelson Comparative Advantage, Trade, and Payments in a Ricardian Model with a
Review and critique of reading Zeliger with Granovetter
An Assessment of the Efficacy and Safety of CROSS Technique with 100% TCA in the Management of Ice P
Capote In Cold Blood A True?count of a Multiple Murder and Its Consequences
Aerobic granules with inhibitory strains and role of extracellular polymeric substances
0622 Removal and installation of control unit for airbag seat belt tensioner Model 126 (from 09 87)
0620 Removal and installation of control unit for airbag seat belt tensioner Model 126 (to 08 87)
Engle And Lange Predicting Vnet A Model Of The Dynamics Of Market Depth
Dialectic Beahvioral Therapy Has an Impact on Self Concept Clarity and Facets of Self Esteem in Wome
Preparation of garlic powder with high allicin content by using combined microwave–vacuum and vacuum
A rapid and efficient method for mutagenesis with OE PCR
Development and Evaluation of a Team Building Intervention with a U S Collegiate Rugby Team
Identification and fault diagnosis of a simulated model of an industrial gas turbine I6C
Interaction between ascorbic acid and gallic acid in a model of
Differences between the gut microflora of children with autistic spectrum disorders and that of heal
Removal and installation of interior temperature sensor Heating, ventilation Model 126 A To 06 81,
Computer Modelling with CATT Acoustic Theory and Practice of Diffusion Reflection and Array Modelin
Dietary intake and biochemical, hematologic, and immune status of vegans compared with nonvegetarian
więcej podobnych podstron