TEORIE
Najogólniejszy model katalitycznej reakcji chemicznej zachodzącej pomiędzy cząstkami można zbudować w oparciu o dwie teorie:
teorię zderzeń aktywnych i
teorię stanu przejściowego zwaną również teorią kompleksu aktywnego.
Reakcja katalityczna może zachodzić w roztworze gazowym, ciekłym, na powierzchni, i w środowiskach które mogą być mikroskopową fazą, taką jak komory o wymiarach cząsteczkowych. Reakcje katalityczne zachodzące w roztworze są najlepiej poznane i dlatego rozważania na temat katalizy zgodnie z logiką powinny rozpoczynać się od nich. Fakt, że reakcje te są najlepiej rozpoznane, wynika z jednorodności cząsteczek katalizatora i jednorodności środowiska otaczającego reagujące cząsteczki. Struktura reagujących cząsteczek, z dużym prawdopodobieństwem zawierających związki pośrednie może być określona metodami spektroskopowymi (IR, UV, NMR), a zależności szybkości reakcji od stężenia reagentów i katalizatorów mogą być względnie łatwo określone.
Dla zrozumienia działania katalizatorów pomocnym jest rozważenie zachowania cząsteczek w roztworze. Cieczami o najprostszej budowie są gazy. W gazach o niskiej gęstości, oddziaływania pomiędzy cząsteczkami można zaniedbać z wyjątkiem ich przypadkowych zderzeń. Zderzenie pomiędzy cząsteczkami może powodować, co najwyżej transfer energii pomiędzy nimi, czasami może także prowadzić do utworzenia lub zerwania wiązań chemicznych.
Częstotliwość zderzeń można oszacować na podstawie kinetycznej teorii gazów, która daje podstawy teorii zderzeń aktywnych; podstawowym założeniem tej teorii jest konieczność zderzenia reagujących cząsteczek
A + B X +.........
Reakcja chemiczna zajdzie wtedy i tylko wtedy, gdy zderzające się cząsteczki będą miały dostatecznie dużą energię, o pewnej dla danego układu określonej wartości, zwaną energią aktywacji Eakt. Oznacza to, że ze wszystkich chaotycznych zderzeń zachodzących w układzie tylko część będzie zderzeniami aktywnymi. Ilość cząsteczek o energii ≥ E wynosi
aE = a • exp (-E/RT)
gdzie a - całkowita liczba cząsteczek w układzie
aE -liczba cząsteczek o energii ≥ E
Dla danego układu zakłada się, że stała szybkości reakcji jest równa
kr = A • Tw • exp (-Eakt/RT)
W równaniu zawarty jest współczynnik (Arheniusa) wyrażający zależność szybkości reakcji od bezwzględnej temperatury reakcji. Wielkość ta wskazuje, że tylko część zderzeń pomiędzy cząsteczkami pozwala na zajście reakcji, są to zderzenia pomiędzy cząsteczkami o odpowiedniej energii. Wielkość Eakt to wielkość energii aktywacji, energii wymaganej dla pokonania bariery energetycznej charakterystycznej dla danej reakcji.
W teorii zderzeń aktywnych wielkość A = PZ, w której
- P to współczynnik steryczny obrazujący wymagania przestrzenne, które muszą być spełnione dla przebiegu reakcji; a
- Z to liczba zderzeń pomiędzy cząsteczkami w jednostce czasu, mówi że ze zderzeń aktywnych nawet wtedy gdy cząsteczki posiadają odpowiednią energię to tylko te prowadzą do zajścia reakcji w których biorące udział cząsteczki mają odpowiednią konfigurację przestrzenną.
Teoria zderzeń aktywnych ma ograniczony zakres, najlepiej opisuje reakcje zachodzące w fazie gazowej, zastosowanie jej do opisu reakcji w fazie ciekłej czy w obecności stałych katalizatorów jest bardzo ograniczone.
Teorię stanu przejściowego zwaną również teorią kompleksu aktywnego czy teorią absolutnej szybkości reakcji sformułowali Eyring, Polanyi i Evans w 1935r.
W teorii tej zakłada się, że reagenty tworzą nietrwałe, krótko żyjące połączenia określane jako stan przejściowy, kompleks aktywny, lub kompleks pośredni. Połączenia te rozpadają się dając produkty, a zatem jest to stan pośredni pomiędzy substratami a produktami. Schemat reakcji można przedstawić równaniem
X + Y <===> [HY]* Z
gdzie [XY]* oznacza kompleks aktywny. Na krzywej zmian energii potencjalnej położenie kompleksu aktywnego odpowiada maksimum energii układu, jest to stan niemożliwy do wyizolowania, czas jego życia jest rzędu 10-13s, reagujące cząsteczki znajdujące się w tym stanie mają niezbędną energię do przejścia w produkty reakcji.
A zatem żeby mogła zajść reakcja, cząsteczki substratów muszą przekroczyć barierę energetyczną równą różnicy energii kompleksu aktywnego i energii substratów, a zatem muszą osiągnąć energię aktywacji.
Zgodnie z teorią stanu przejściowego kompleks aktywny jest w stanie równowagi z cząsteczkami substratów. Stałą szybkości reakcji powstawania kompleksu aktywnego wyraża zależność
kr* = ![]()
![]()
gdzie k - stała Boltzmana (1,38065 10-23J/K)
h - stała Plancka (6,62607 10-34J/s)
T - temperatura bezwzględna, a
![]()
= K* stała równowagi tworzenia kompleksu aktywnego, może być przedstawiona również jako funkcja sumy stanów poszczególnych cząstek biorących udział w reakcji czyli jest funkcją energii (1) translacji, (2) oscylacji, (3) rotacji i (4) różnego rodzaju ruchu elektronów reagujących cząsteczek
K* = ![]()
= 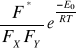
Osiągnięcie konfiguracji kompleksu aktywnego o najwyższej w układzie, nadmiarowej energii, umożliwia przegrupowanie elektronów i atomów prowadzące do powstania nowych wiązań chemicznych co w końcowym efekcie daje zawsze przejście kompleksu w produkty reakcji. A zatem szybkość reakcji chemicznej jest bezpośrednio zależna od szybkości powstawania kompleksu aktywnego i jest wyrażana równaniem
r = 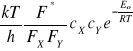
w równaniu tym wyrażenie 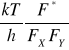
określa częstość rozpadu kompleksu aktywnego do produktów, jest wielkością charakterystyczną dla danego układu reakcyjnego i nosi nazwę współczynnika częstości. Współczynnik ten jest również zależny od rodzaju i budowy cząsteczek tworzących kompleks aktywny.
W przypadku reakcji odwracalnych kompleks aktywny dla reakcji odwrotnej ma tę samą konfigurację co kompleks dla reakcji wprost, ale przeciwny kierunek ruchu wzdłuż współrzędnej reakcji; a zatem nie jest słuszne twierdzenie, że kompleks aktywny jest w stanie równowagi z substratami i produktami.
Zmianę energii potencjalnej układu w czasie trwania reakcji obrazuje nam poniższy wykres:
W obecności katalizatora reakcja może przebiegać w jednym etapie
X + Y + K <===> [XKY]* Z + K
lub w kilku etapach z utworzeniem produktu przejściowego
X + Y + K <===> [XK]* XK
XK + Y <===> [XKY]* Z + K
gdzie XK produkt przejściowy.
W jednym i drugim przypadku energia utworzenia kompleksu aktywnego w skład którego wchodzą substraty/substrat i katalizator jest niższa od energii utworzenia kompleksu aktywnego zawierającego tylko substraty.
Pojęcia produkt przejściowy substrat-katalizator i kompleks aktywny substrat/substraty-katalizator nie są synonimami.
W każdej reakcji katalitycznej zarówno homogenicznej jak i heterogenicznej powstające kompleksy aktywne/przejściowe mogą być uznane jako pewna forma związków kompleksowych. Bardzo często centra aktywne katalizatorów heterogenicznych i powstałe przy ich udziale kompleksy przejściowe reakcji rozpatruje się jako cząstki wbudowane w matrycę/ nośnik, który wymusza strukturę fizyczną tych cząstek, wpływa na ich strukturę elektronową oraz jest dostawcą uprzednio zaadsorbowanych reagentów. Powstające kompleksy aktywne mają najczęściej strukturę przestrzenną oktaedryczną o liczbie koordynacji 6, lub strukturę tetragonalną lub płaskiego prostokąta, odpowiadające liczbie koordynacji 4.
W katalizie heterogenicznej rozważa się centrum aktywne jako molekułę wbudowaną w matrycę (rodzaj nośnika), którą również może stanowić część substancji aktywnej zlokalizowanej poza centrum aktywnym (angielskie określenie takiego układu Embeded Surface Molecule - ESM).
W podobny sposób w katalizie heterogenicznej rozpatruje się kompleks powierzchniowy.
Powstawanie określonych rodzajów wiązań obecnych w kompleksach aktywnych może być wyjaśnione w oparciu o teorię wiązania walencyjnego, proste oddziaływania elektrostatyczne, teorię pola kryształu czy teorię orbitali molekularnych.
Teoria wiązania walencyjnego
(Linus Pauling - Nobel 1954; wyjaśnił rolę pary elektronowej w wiązaniach związków kompleksowych)
wiązanie metal - ligand jest wynikiem reakcji zasady Lewisa (ligand np.: NH3, H2O, CH3OH) z kwasem Lewisa (metal; wolne orbitale d, s lub p).
Należy przypomnieć, że:
kwas Lewisa to substancja, której cząsteczka lub jon może utworzyć wiązanie koordynacyjne przez pobranie pary elektronów od innej cząsteczki, natomiast
kwas Broenstedta (Lowry) jest to substancja dostarczająca proton, na przykład kation wodorowy.
utworzenie wiązania metal - ligand wymaga obecności wolnej pary elektronów w ligandzie i wolnego orbitalu metalu
atom metalu wykorzystuje zwykle orbitale zhybrydyzowane, co ułatwia kierunkowe właściwości orbitalu i daje w rezultacie lepsze nakładanie się orbitali metalu i ligandu, i bardziej stabilne wiązanie
geometria oktaedryczna (hybrydyzacja d2sp3,), kompleks sześcioskoordynowany; geometria tetraedralna (sp3) lub kwadroplanarna (dsp2), cztero skoordynowane kompleksy;
doświadczalne rozróżnienie hybrydyzacji sp3 i dsp2 przez pomiar właściwości magnetycznych. Struktura płaska jest diamagnetyczna, natomiast czworościanu paramagnetyczna.
Teoria wiązania walencyjnego jest użyteczna, aczkolwiek nie jest kompletna, pozwala na interpretację faktów lecz nie wyjaśnia widm związków kompleksowych.
Teoria prostych oddziaływań elektrostatycznych
Oddziaływanie zachodzi między dodatnim ładunkiem jonu lub atomu metalu (dodatni ładunek jądra) i ujemnym ładunkiem ligandu (halidki, lub koniec dipolu).
Teoria ta nie wyjaśnia dużej stabilności karbonylków metali (atom neutralny i prawie niepolarny O). Poza tym nie zawsze zachowana jest zależność siły wiązania metal - ligand dla tego samego ligandu i serii jonów metali przejściowych.
Teoria pola kryształu
W tej teorii atom metalu i ligand rozpatrywane są jako ładunki punktowe. Uwzględnia ona jednak odpychanie elektronów liganda i walencyjnej powłoki metalu. Powoduje to, że elektrony metalu przechodzą w miarę możliwości do orbitali nie leżących na drodze zbliżania się liganda. Orbitale d metalu nie są zatem zdegenerowane. Rozszczepienie na dwa lub więcej poziomów energetycznych zależy od ich zorientowania w stosunku do wiązania metal - ligand M-L.
Model wyjaśnia właściwości magnetyczne kompleksów metali przejściowych, obserwowane tendencje stabilności i jakościowo tłumaczy widma absorpcyjne tych kompleksów.
Jednakże obliczenia oparte o tę teorię nie dają zgodności z widmami przejścia elektronów mierzonych dla metali przejściowych. Teoria ta nie podobnie jak teoria prostych oddziaływań elektrostatycznych nie tłumaczy silnych wiązań metal-ligand wtedy gdy atom centralny metalu jest na zerowym poziomie utlenienia a ligand jest niepolarny.
Teoria orbitali molekularnych
Jej konieczność wynika z istnienia kompleksów metali na zerowym lub ujemnym poziomie utlenienia z niepolarnymi ligandmi, zwystępowania wspólntych orbitali M-L w zdecydowanie jonowych kompleksach (np. CoF63-).
Niestety dokładna jej postać jest zbyt skomplikowana w przypadku związków koordynacyjnych, a tym bardziej kompleksów z katalizatorami heterogenicznymi.
Teoria musi być uproszczona przez rozwiązanie dla orbitali tylko warstwy walencyjnej w metalu oraz nisko energetycznych, nieobsadzonych orbitali atomowych lub molekularnych dla ligandu.
Rozważania symetrii dalej upraszczają problem. Wykorzystanie symetrii daje w wyniku jakościowo takie same rozszczepienie orbitali d jak teoria pola kryształu.
Uproszczona teoria orbitali daje wyniki spójne z doświadczeniem. Zastosowanie jej do kompleksów metalu na niskim stopniu wartościowości i niepolarnymi ligandami dało wprowadzenie zasady oddawania elektronu metalu do orbitalu drugiego liganda. Takie oddziaływanie wpływa na aktywność liganda i duże znaczenie dla przebiegu reakcji katalitycznych.
Jakkolwiek teoria ta jest dużo lepsza niż podano poprzednio, jej wykorzystanie wymaga nie tylko wysokich kwalifikacji i doświadczenia ale również dużych, szybkoliczących komputerów.
Jeżeli pamiętamy o ograniczeniach to w prostszych przypadkach można stosować teorie podane wcześniej.
P
Energia układu
Postęp reakcji
Energia aktywacji reakcji termicznej (Ert)
Erc reakcji katalitycznej
ΔHa
S
P1
P2
Ed
ΔHd
Ea
ΔHr
Wyszukiwarka
Podobne podstrony:
spektro6, Technologia chemiczna pw, 2rok, spektra
spektro2, Technologia chemiczna pw, 2rok, spektra
spektroX, Technologia chemiczna pw, 2rok, spektra
spektro3, Technologia chemiczna pw, 2rok, spektra
spektro4, Technologia chemiczna pw, 2rok, spektra
spektroY, Technologia chemiczna pw, 2rok, spektra
spektro10, Technologia chemiczna pw, 2rok, spektra
spektro1, Technologia chemiczna pw, 2rok, spektra
spektro16, Technologia chemiczna pw, 2rok, spektra
spektro12, Technologia chemiczna pw, 2rok, spektra
Zestawy egzaminacyjne, Technologia chemiczna pw, 2rok, aparatura
spektro17, Technologia chemiczna pw, 2rok, spektra
spektro13, Technologia chemiczna pw, 2rok, spektra
spektroB, Technologia chemiczna pw, 2rok, spektra
spektro14, Technologia chemiczna pw, 2rok, spektra
spektro5, Technologia chemiczna pw, 2rok, spektra
spektro15, Technologia chemiczna pw, 2rok, spektra
spektro7, Technologia chemiczna pw, 2rok, spektra
więcej podobnych podstron