Spis treści
Tkanka mięśniowa, białka kurczliwe mechanizm skurczu.
Trawienie i wchłanianie
Równowaga kwasowo zasadowa - zaburzenia metaboliczne .diagnostyka
Kwasy nukleinowe i biosynteza białek
Tkanka mięśniowa, białka kurczliwe mechanizm skurczu.
Mięsień jest wysoce wyspecjalizowaną tkanką zdolną do przetwarzania energii chemicznej w energię mechaniczną skurczu
Odróżniamy 3 rodzaje mięśni:
• Mięśnie gładkie - zbudowane z tkanki mięśni gładkich
• Mięśnie szkieletowe - zbudowane z włókien poprzecznie prążkowanych
• Mięsień sercowy
Jak zbudowana jest tkanka mięśniowa?
Tkanka mięśniowa zbudowana jest z komórek (miocytów) wydłużonych, otoczonych blaszką podstawną, która wspólnie z tkanką łączną tworzy zrąb. Pochodzi z mezodermy.
Jakiego rodzaju są mięśnie?
Wyróżnia się
1. tkankę mięśniową poprzecznie prążkowaną szkieletową,
2.tkankę mięśniową poprzecznie prążkowaną sercową
i3. tkankę mięśniową gładką.
Tkanka mięśniową poprzecznie prążkowaną - zbudowana jest z włókien wielojądrowych, wykazujących charakterystyczne prążkowania widoczne pod mikroskopem, zbudowane są z niej mięśnie szkieletowe,
Tkanka sercowa - zbudowana jest z wielojądrowych włókien prążkowanych, tworzących rozgałęzienia łączące się z sąsiednimi włóknami (mięsień sercowy, serce).
Tkanka mięśniowa gładka - zbudowana jest ze ściśle ułożonych, jednojądrowych komórek kształtu wrzecionowatego, występującą w narządach układu pokarmowego, oddechowego, moczowego, rozrodczego (macica, pochwa, jajowody) oraz w niektórych innych organach wewnętrznych (np.: mięśniach poruszających gałką oczną, naczyniach krwionośnych)
Jak zbudowane są mięśnie prążkowane?
Tkanka mięśniowa, tkanka zwierzęca zbudowana z wydłużonych cylindrycznych lub wrzecionowatych komórek mięśniowych, zawierających kurczliwe włókienka mięśniowe zwane miofibrylami. Miofibryle zbudowane są z dwóch głównych białek - aktyny i miozyny, dzięki którym tkanka mięśniowa może kurczyć się i rozkurczać, umożliwiając wszelkie ruchy zwierząt.
Retikulum sarkoplazmatyczne jest systemem kanalików i cystern utworzonych przez błony elementarne. Jest to retikulum agranularne, czyli gładkie. Przylega do miofibryli i kontaktuje się z kanalikami T. W obszarze odcinków I leżą cysterny końcowe. W obszarze linii Z leżą kanaliki T, które wpuklają się do wnętrza miocytu prostopadle do powierzchni sarkolemmy. W odcinku A leżą kanaliki retikulum sarkoplazmatycznego, łączące zbiorniki (cysterny) końcowe. Układ złożony z 1 kanalika T i z dwóch cystern końcowych (przylegających) zwie się triadą. Należy pamiętać, że kanalik T jest wpukleniem sarkolemmy i służy do szybkiego przenoszenia pobudzenia w głąb komórki, do sarkomerów. Retikulum sarkoplazmatyczne gromadzi wapń, niezbędny do skurczu. W trakcie pobudzenia wapń przenika do cytoplazmy (cytozolu).
Budowa mięśni.
Przez jasne mięśnie prążków izotropowych przechodzą ciemniejsze linie o dużej gęstości optycznej - linie Z. Są to tzw. błony graniczne. Odcinek między dwiema liniami Z to sarkomer, będący jednostką czynnościową miofibryli. Miofibryle zbudowaną są z dwóch rodzajów filamentów:
- filamentów grubych o średnicy 100 A
- filamentów cienkich o średnicy 60 A
Prążki anizotropowe posiadają filamenty grube i częściowo zachodzące filamenty cienkie.
W prążkach izotropowych znajdują się filamenty cienkie. Filamenty zbudowane są z białek. Stosując metodę elektroforezy na żelu poliakrylamidowym w obecności siarczanu dodecylu, otrzymano całe spektrum białkowe miofibryli.
W skład grubych filamentów wchodzą:
Miozyna, białko linii M i białko C
Na cienkie filamenty składają się: aktyna, tropomiozyna i tropnnina
Cząsteczka miozyny to białko typu -kreatyny o c. cz. około 500 kDa ma kształt pałeczki i składa się z dwu metrycznych łańcuchów o długości około 1400 A. Każdy z łańcuchów ma przestrzeń 1300 A i ma strukturę spiralną i oba łańcuchy związane są w superspiralę. W końcowym odcinku o długości 100 A łańcuchy nie są skręcone, tu znajdują się tzw dwie głowy o średnicy 30 A. W tej części łańcucha zawarte jest cała aktywność biologiczna miozyny tzw. aktywność ATP-azowa i miejsce przyłączenia się aktyny
Pod wpływem enzymów proteolitycznych cząsteczka miozyny rozpada się na fragmenty, co pozwala lepiej poznać strukturę tego białka. W wyniku krótkotrwałego działania trypsyny uwalniane są dwie charakterystyczne podjednostki
lekka meromiozyna (LMM) typowe białko włókienkowe
ciężka meromiozyna (HMM) - zawiera dwie głowy - białko globularne
Dłuższe trawienie trypsyną prowadzi do rozdzielenia ciężkiej meromiozyny (HMM) na subfragment S2 i subfragment S1 - czyli pojedyncze głowy. Subfragment S1 można otrzymać bezpośrednio z miozyny w wyniku trawienia papaniną.
Ta fragmentacja cząsteczki miozyny pozwoliła ustalić, że aktywność ATP-azowa miozyny zawarta jest wyłącznie w jej „głowie” i że na cząsteczkę miozyny przypadają dwa miejsca katalityczne - po jednym w każdym z dwóch fragmentów S1. Ustalono także, że miozyna wiąże aktynę również w tych dwóch miejscach tworząc aktomiozynę i że w wiązaniu tym biorą udział grupy tiolowe miozyny.Cząsteczki miozyny in vivo agregują zarówno końcami jak i wzdłuż pałeczkowatych fragmentów. Powstają grube filamenty, których trzon stanowią wiązki pałeczkowatych odcinków miozyny, głowy zaś wystają na boki tworząc tzw. mostki poprzeczne. W ostatnich latach wykryto w miozynie niskocząsteczkowe fragmenty tzw. lekkie łańcuchy, które łatwo oddysocjowują od reszty cząsteczki nie powodując rozwinięcia superspirali całej cząsteczki.
Przypuszcza się, że obserwowane różnice we właściwościach enzymatycznych miozyny różnego pochodzenia mogą wynikać z różnic w składzie i budowie lekkich łańcuchów.
Środkowy odcinek grubego filamentu zajmuje białko linii M. Jest to białko globularne o c. cz. 88 kDa daltonów. Powoduje ono agregację miozyny. Wydaje się, że funkcje tego białka polega na łączeniu pojedynczych cząsteczek miozyny w walec tworzący gruby filament. Białko C wykryte niedawno w grubych filamentach posiada ciężar cząsteczkowy 130 kDa daltonów. Zawiera ono jeden łańcuch peptydowy i silne się wiąże z częścią pałeczkowatą miozyny. Funkcja tego białka polega na spinaniu filamentu niczym klamrami.Główną masę cienkich filamentów stanowi aktyna. Białko to można otrzymać z mięśni w postaci monomeru tzw. G-aktyny. Jest to białko globularne o c. cz. około 45 kDa daltonów, posiadające silne związaną cząsteczkę ATP oraz wapń. Jeżeli z G-aktyny usuniemy Ca 2+ i ATP, spowodujemy nieodwracalną denaturację białka. Gdy G-aktyna znajduje się w warunkach o tej samej sile jonowej jaka występuje w komórce mięśnia i w obecności Mg2+ polimeryzuje tworząc nić F-aktyny. F-aktyna tworzy cienkie filamenty zbudowane z dwóch spiralnie wokół siebie skręconych sznurów cząsteczek G-aktywnych. Podczas polimeryzacji ATP silnie związany z G-aktywną ulega hydrolizie, z uwolnieniem nieorganicznego fosforanu do środowiska W wyniku tego F-aktyna zawiera ADP jako silne związany nukleotyd
n ( G-aktyna-ATP) KCl,Mg 2+ F-aktyna-ADP +Pi
In vivo aktyna występuje w formie spolimeryzowanej. Jej biologiczna rola polega na aktywacji miozyny.Oprócz miozyny cienkie filamenty wiążą tzw. białka regulujące tropomiozynę i troponinę
1 cz.aktyny + 3 cz. miozyny Ca2+ Tropomiozyna, troponina, aktomiozyna
Cząsteczka tropomiozyny zbudowana jest z dwu łańcuchów prawoskrętnej spirali, skręconych w superspiralę. Posiada c. cz. 70 kDa i około 400 A i znajduje się w warunkach spirali F-aktyny. Wpływa na interakcję aktyny z miozyną.
Troponina jest białkiem globuralnym, składającym się z trzech komponent o średnich c. cz. 40 kDa ; 23 kDa; 18 kDa. Białko o c. cz. 23 500 jest inhibitorem interakcji aktyny z miozyną. Niezależnie od stężenia jonów Ca 2+ hamuje aktywność ATP-azy aktomio-zynowej i nazywane jest czynnikiem inhibującym lub TN-I.Białko o c.cz. 18 kDa jest akceptorem jonów wapnia(razem z tropomiozyną hamuje aktywność ATP-azy w nieobecność jonów Ca 2+ . Nosi nazwę czynnika uczulającego na Ca +2 lub TN-C. Trzeci składnik troponiny o c. cz. 40,2 kDa ma duże powinowactwo do tropomiozyny i F-aktyny. Wydaje się, że stanowi on miejsce przez które cała troponina wiąże się z pozostałymi składnikami cienkich filamentów. Nosi ona nazwę składnika wiążącego lub TN- B.
W cienkim filamencie jedna cząsteczka troponiny przypada na jedną cząsteczkę tropomiozyny i na siedem monomerów aktyny.Wyróżnić można trzy stany mięśnia: stan rozkurczu, stan skurczu odwracalnego i stan skurczu nieodwracalnego - w stężeniu pośmiertnym - rigor mortis. Skurcz mięśnia - teoria przesuwania filamentów względem siebie. Wzajemne zachodzenie obu rodzajów filamentów stwarza możliwość bezpośredniego kontaktu miozyny z aktyną.
Połączenie to następuje za pomocą mostków poprzecznych. W czasie spoczynku łączą się one pod kątem prostym. W czasie skurczu wykonują ruch zmieniający kąt nachylenia o 45°, powoduje to przesunięcie filamentu aktynowego wzdłuż miozynowego Sygnałem do rozpoczęcia skurczu mięśnia jest uwolnienie jonów Ca+2 z błon sarkoplazmatycznego reticulum.Uwalniany wapń wiąże się z tropiną C (TN-C) i umożliwia wystąpienie oddziaływania między aktyną i miozyną a w konsekwencji następuje skurcz.
Do skurczu mięśnia niezbędna jest energia chemiczna, która powstaje w samym mięśniu. Mięśnie szkieletowe posiadają znaczne ilości enzymów zarówno do przemian węglowodanów jak i tłuszczów które biorą udział w beztlenowej glikolizie po rozkładzie glikogenu.W mięśniach zachodzi także przemiana tlenowa dzięki mioglobinie, która odwracalnie wiąże cząsteczki tlenu. Mioglobina jest homoproteiną podobną do hemoglobiny. Jej krzywa dysocjacji ma kształt hiperboli i jest przesunięta w lewo w stosunku do krzywej dysocjacji oksyhemoglobiny. Ma ona większe powinowactwo do tlenu niż hemoglobina, od której pobiera tlen w niskich wartościach PO2. Mioglobina wiąże 1 cząsteczkę tlenu a nie jak hemoglobiny 4.
W bardzo aktywnych mięśniach czerwonych, zawierających dużą ilość mioglobiny i cytochromów energia powstaje głównie w wyniku fosforylacji oksydacyjnej. W mięśniach białych głównym źródłem energii jest glikoliza .
U ssaków mięśnie w stanie spoczynku wykorzystują energię głównie przez wykorzystanie kwasów tłuszczowych i acetylooctanu (z wątroby). Podczas maksymalnej pracy mięśniowej głównie wykorzystywana jest glukoza.
Bezpośrednim źródłem energii niezbędnej do skurczu mięśnia jest hydroliza ATP do ADP. Zawartość ATP w mięśniach w spoczynku wynosi około 2,5 mmola/100gsuchej tkanki Rozpad 1 cząsteczki ATP prowadzi do uwolnienia 7.3 kcal Zapas energii zgromadzonej w postaci ATP jest więc niewielki i może wystarczyć zaledwie na kilka sekund pracy mięśnia. Dlatego w mięśniach zgromadzony jest wysokoenergetyczny fosforan w postaci fosfokreatyny. Związek ten źródłem wysokoenergetycznego fosforanu dla szybkiej resyntezy ATP i służy do utrzymania odpowiedniej ilości ATP. W stanie spoczynki mięśnie ssaków zwierają 4-6 krotnie więcej fosfokreatyny niż ATP. Przemieszczanie wysokoenergetycznego fosforanu fosforanu z fosfokreatyny na ADP katalizowane jest przez kinazę kreatynową. Inną możliwością powstania ATP w mięśniach jest przenoszenie wysokoenergetycznego fosforanu z jednej cząsteczki ADP na drugą. Reakcję tą katalizuje kinaza adenylowa (miokinaza)Nie jest to proces korzystny dla organizmu, gdyż w wyniku tej reakcji powstają znaczne ilości AMP z którego pod wpływem swoistej dezaminazy odczepiony zostaje amoniak. Powstający IMP podlega kondensacji z asparaginianem. Produktem tej reakcji jest kwas adenylobursztynowy, który rozpada się odtwarzając AMP i fumaran. Jest to tzw. cykl purynowy. W jego wyniku powstaje wolny amoniak, sprawnie wiązany w mięśniach przez syntetazę glutaminy.
Uważa się, że interakcja dwóch strukturalnych białek mięśniowych - aktyny i miozyny w obecności ATP stanowi podstawowy mechanizm skurczu mięśniowego.
Aktomiozyna wiąże ATP, który powoduje jej rozpad. W rezultacie powstaje kompleks ATP-miozyna. Hydroliza ATP następuje pod wpływem miozyny. Jest to proces bardzo szybki. Znacznie wolniejsze jest uwalnianie produktów ADP i Pi. Etap ten zachodzi pod wpływem aktyny. Ważną cechą tego cyklu jest fakt, że aktyna ma duże powinowactwo do miozyny i do jej kompleksu ADP i Pi natomiast małe powinowactwo do kompleksu ATP-miozyna. Wszystkie te rozwiązania obejmują interakcję miozyny z aktyną w czasie skurczu mięśnia.
ŹRÓDŁA ENERGII DLA MIĘŚNI - ZASOBY ENERGETYCZNE W MIĘŚNIACH
Źródłem energii dla mięśni są: glikogen, zw. wysokoenergetyczne ATP oraz fosfokreatyna. Fosfokreatyna + ADP = kreatyna + ATP. W czasie wysiłku w mięśniach wytwarza się kwas mlekowy, który wnika do krwi jako glukoza, następnie glukoza wędruje żyłą wrotną do wątroby gdzie przekształcona zostaje w glikogen , z wątroby (przemieniany w glukozę) przemieszczany jest do mięśni dostarczając im energię. W wysiłkach krótkotrwałych o dużej mocy, energia jest czerpana prawie wyłącznie z beztlenowych procesów metabolicznych. Dużą rolę odgrywa tu ATP i fosfokreatyna. Jeśli wysiłek trwa dłużej jako źródło energetyczne wykorzystywany jest glikogen mięśniowy. W wysiłkach dłuższych o dużej mocy głównym źródłem energetycznym są węglowodany. Wykonywanie takiej pracy uzależnione jest w dłuższym stopniu od wyjściowego poziomu glikogenu w mięśniach i sprawności mechanizmów umożliwiających oszczędzanie węglowodanów. W miarę przedłużania czasu pracy i zmniejszania intensywności wysiłku zwiększa się udział procesów tlenowych. Powtarzane wysiłki fizyczne o charakterze treningu pobudzają procesy adaptacyjne przejawiające się m.in. w zwiększaniu zdolności gromadzenia glikogenu przez mięśnie. Kreatyna - związek azotowy występujący głównie w mięśniach. Fosfokreatyna - połączenie kratyny z kwasem fosforanowym.
CHEMIZM MIĘŚNI - SKURCZE I ROZKURCZE
Podczas skurczu mięśnia filamenty cienkie wsuwają się między filamenty grube. Proces ten wymaga ATP jako detonatora energii oraz aktywności układu enzymatycznego. Poza ATP istotnymi czynnikami w procesie skurczu mięśnia są jony Ca oraz układ białek rozkurczowych: tropomizyna i tropanina. Proces kurczowo rozkurczowy ma charakter cykliczny. Bodziec nerwowy pobudzający skurcz mięśnia powoduje zmiany w potencjale retikulum i przejście z niego jonów Ca do białek (tropomizyny i troponiny). Następuje aktywacja ATP-razy aktomiozynowej i skurcz mięśnia po ustaniu działania bodźca rozpoczyna się odbieranie wapnia z troponiny przez pompę wapniową retikulum, co stwarza warunki blokujące czynności ATP- razy i rozkurcz mięśnia
Struktura mięśnia szkieletowego wg.Harpera PZWL 1994 str.795
Schematyczna prezentacja cienkiego filamentu pokazująca przestrzenną. konfigurację 3 głównych sktadników białkowych: aktyny, tropomiozyny i troponiny. wg.Harpera PZWL 1994 str.797
Ułożenie filamentów w mięśniu poprzecznie prążkowanym. A — w rozkurczu, B — w skurczu. wg.Harpera PZWL 1994 str.788
Schematyczna prezentacja cienkiego filamentu pokazująca przestrzenna. konfigurację 3 głównych składników biatkowych: aktyny, tropomiozyny i troponiny. - wg.Harpera PZWL 1994 str.797
Diagram cząsteczki miozyny pokazujący 2 zwinęte ze sobą. heliksy (część nitkowata), główkę (G), lekkie łańcuchy (L) i efekt trawienia przez trypsyne i papainę. HMM — ciężka meromiozyna; LMM — lekka meromiozyna; S-1 — podjednostka 1; S-2 — podjednostka 2. wg.Harpera PZWL 1994 str.798
Trawienie i wchłanianie
Układ pokarmowy
Przewód pokarmowy rozpoczyna się jama ustną, w której pokarm ulega rozdrobnieniu i zostaje zapoczątkowane trawienie, następnie pokarm zmieszany ze śliną dostaje się gardłem i przełykiem do żołądka, gdzie zachodzą właściwe procesy trawienne. Następnym odcinkiem przewodu pokarmowego jest jelito cienkie (dzielące się na dwunastnicę, jelito czcze i kręte), do którego uchodzą enzymy trzustki i żółć produkowana przez wątrobę - tutaj pokarm zostaje rozłożony do składników prostych wchłanianych przez kosmki jelitowe. Natomiast niestrawione resztki pokarmowe dostają się do jelita grubego (dzielącego się na jelito ślepe, okrężnicę i odbytnicę), w którym zachodzi wchłanianie wody i formowanie kału. Transport treści pokarmowej w przewodzie pokarmowym zachodzi dzięki pracy jego mięśni (perystaltyka).
Składniki pokarmowe
Składniki pokarmowe, to składniki odżywcze które po uwolnieniu w procesie trawienia mogą być wchłonięte w przewodzie pokarmowym i wykorzystane przez organizm. Składniki pokarmowe pełnią w organizmie funkcje budulcową, energetyczną i regulującą. Do składników budulcowych należą: woda, białko i związki mineralne. Do składników energetycznych: węglowodany, tłuszcze i częściowo białko. Do składników regulujących: witaminy, enzymy i niektóre związki mineralne.
Żółć
Żółć, wydzielina komórek wątrobowych, W jej skład wchodzą: sole kwasów żółciowych, cholesterol, barwniki żółciowe, będące produktami degradacji hemoglobiny, substancje śluzowe, lipidy, kwasy tłuszczowe i kwasy nieorganiczne .Żółćw pęcherzyku żółciowym , gdzie zostaje zagęszczona . W momencie pojawienia się pokarmu w jelicie żółć zostaje dostaje siaz pęcherzyka żółciowego do jelita, gdzie bierze udział w trawieniu. Sole żółciowe, które są głównym składnikiem czynnym żółci, powodują emulgację tłuszczów, zwiększając powierzchnię kropelek tłuszczu i zapoczątkowują działanie lipazy triacyloglicerolowej.
Enzymy trawienne
Sok trzustkowy, zawiera
Enzymy rozkładające węglowodany- amylaza trzustkowa,
Enzymy rozkładające tłuszcze na glicerol i kwas tłuszczowy lipaza
Enzymy rozkładające rozkładającą białka na aminokwasy - proteaza
Wątroba zwana fabryką chemiczną spełnia następujące funkcje:
wytwarza żółć
magazynuje nadmiar cukru w postaci glikogenu
przechowuje witaminy
odtruwa organizm
rozkłada nadmiar aminokwasów
Do najważniejszych soków trawiennych w przewodzie pokarmowym należą: ślina, sok żołądkowy, sok trzustkowy (trzustka), żółć i sok jelitowy. Ilość wydzielanych soków trawiennych wynosi ok. 8 - 10 l. na dobę.
Enzymy trawienne,to enzymy a właściwie cały ich zespół działajacych w przewodzie pokarmowym, które są odpowiedzialne za trawienie treści pokarmowej.
Do enzymów trawiennych .należą:
1) Amylaza, maltaza, laktaza, sacharaza. -enzymy trawiące węglowodany tj.
2)Chymotrypsyna trypsyna,,peptydazy: aminopeptydazy, karboksypeptydazy i dwupeptydazy -enzymy trawiące białka: pepsyna,.
3) Lipazy, fosfolipazy. - enzymy trawiące tłuszcze tj.
4) Nukleazy - enzymy trawiące kwasy nukleinowe -
.
Pepsyna
Enzymem wytwarzanym przez gruczoły zlokalizowane w błonie śluzowej żołądka jest pepsyna, składnik soku żołądkowego,. Pepsyna wydzielana jest najpierw w postaci nieczynnej tzw.zymogenu o nazwie pepsynogen, Jest to proenzymu, który staje się aktywny dopiero w kwaśnym środowisku soku żołądkowego. Pepsyna rozkłada białka do polipeptydów.
Gastryna
Gastryna jest hormonem polipeptydowym wydzielanym do krwi przez błonę śluzową okolicy odźwiernika żołądka. Gastryna pobudza wydzielanie pęcherzyka żółciowego, trzustki i błony śluzowej jelita cienkiego.
Trypsyna
Trypsyna jest enzymem należącym do grupy peptydaz, rozrywający wiązania peptydowe w łańcuchach białkowych. Trypsyna podobnie jak pepsyna wydzielana jest w postaci nieczynnego trypsynogenu do dwunastnicy, .Synteza trypsynogenu zachodzi w trzustce. W dwunastnicy pod wpływem czynnej trypsyny i enzymu enterokinazy trypsynogen zostaje przekształcony w trypsynę. Optimum dla działania trypsyny wynosi ok. pH 8.
Chymotrypsyna
Chymotrypsyna, jest enzymem należącym do grupy peptydaz serynowych, Chymotrypsyna rozkłada białka i peptydy. Chymotrypsyna rozszczepia łańcuchy peptydowe w miejscu wiązań tworzonych przez grupy karboksylowe L-aminokwasów aromatycznych tj. tryptofan, tyrozyna, fenyloalanina., Reakcja trawienia zachodzi w jelicie cienkim . Chymotrypsyna powstaje z chymotrypsynogenu pod wpływem aktywacji przez trypsynę. W przeciwieństwie do trypsyny chymotrypsyna hydrolizuje kazeinę.
Erepsyna
Erepsyna, to zespół enzymów trawiennych, wytwarzanych przez trzustkę i jelito cienkie, Działanie enzymów wchodzących w skład erepsyny polega na rozkładzie polipeptydów do aminokwasów.. Do enzymów tej grupy należą peptydazy: karboksypeptydazy, aminopeptydazy i dwupeptydazy
Enterokinaza
Enterokinaza to enzym aktywujący trypsynogen (poprzez odszczepienie od niego heksapeptydu) i przekształcający go w formę aktywną - Enterokinaza, jest enzymem, proteolitycznym wytwarzanym przez gruczoły błony śluzowej jelita cienkiego,
Peptydazy
Peptydazy, sa enzymami należącymi do grupy hydrolaz, i rozkładającymi wiązania peptydowe w białkach
Rozróżnia się następujace peptydazy:
endopeptydazy, hydrolizujące wiązania wewnątrz łańcucha polipeptydowego (np. pepsyna, trypsyna, enterokinaza)
egzopeptydazy, hydrolizujące krańcowe wiązania w cząsteczce białka. . Końcowymi produktami działania peptydaz są, poza aminokwasami, również dwupeptydy,
dwupeptydazy. rozkładają dipeptydy do aminokwasów.
.
Lipazy
Lipazy to enzymy które występują w soku żołądkowym, trzustkowym, jelitowym i we krwi, jak również w wielu roślinach, głównie nasionach. Lipazy,należą do grupy esteraz, katalizujących rozkład lub syntezę lipidów
Nukleazy
Nukleazy występują w każdej komórce, jak i poza komórką. Nukleazy, jest to potoczna nazwa enzymów należących do grupy hydrolaz,.Zadaniem nukleaz jest rozkładanie kwasów nukleinowych. Nukleazy katalizują rozkład wiązań diestrowych utworzonych pomiędzy kwasem fosforowym a grupą hydroksylową cukru rybozy lub dezoksyrybozy.
W zależności od miejsca działania nukleazy dzielą się na
endonukleazy - hydrolizują wewnętrzne wiązania fosfodwuestrowe
egzonukleazy - powodujące odszczepianie końcowych nukleotydów.
W zależności od rodzaju rozkładanego kwasu rozróżnia się:
dezoksyrybonukleazy - działają na DNA
rybonukleazy - działają na RNA.
.
Hydrolazy glikozydowe
Enzymy rozrywające wiązanie glikozydowe węglowodanów należą do grupy Hydrolaz glikozydowych ,.Do glikozydaz ( popularna nazwa) należy m.in. maltaza (α-glukozydaza) występująca w ślinie i soku trzustkowym, czy też β-glukozydaza.
Wchłanianie
Wchłanianie to proces fizykochemiczny powodujący utylizacjie np. produktów trawienia. Wchłanianie może być bierne (na zasadzie dyfuzji) albo czynne (z wykorzystaniem energii Wchłanianie czyli resorpcja jest procesem pobierania przez komórki cząstek stałych i płynnych z przechodzeniem substancji do krwi lub limfy przez skórę, błony śluzowe przewodu pokarmowego i dróg oddechowych..
TRAWIENIE WĘGLOWODANÓW
Z węglowodanów spożywanych przez człowieka główną rolę odgrywa skrobia. W znacznie mniejszych ilościach spożywana jest sacharoza, laktoza mleka, glikogen czy cukry proste.
Proces trawienia węglowodanów praktycznie zaczyna się w dwunastnicy, gdzie w soku trzustkowym występuje amylaza hydrolizująca wiązania Błąd! Nie zdefiniowano zakładki.-1,4-glikozydowe polisacharydów. Enzym ten nie wykazuje swoistości substratowej i hydrolizuje każde wiązanie Błąd! Nie zdefiniowano zakładki.-1,4-glikozydowe znajdujące się wewnątrz cząsteczki. Nie rozkłada jednak wiązań skrajnych ani wiązań 1,4 sąsiadujących z wiązaniami 1,6-glikozydowymi. W wyniku działania amylazy powstają więc cząsteczki maltozy, maltotriozy oraz oligosacharydy zawierające wiązania 1,6-glikozydowe (dekstryny graniczne). Dalszy rozkład zachodzi w świetle jelita, w soku jelitowym. Tu występują przynajmniej 4 oligosacharydazy: Błąd! Nie zdefiniowano zakładki.-galaktozydazy (laktazy) rozkładając tylko laktozę, sacharazy - rozkładające sacharozę, glukoamylazy (maltazy) hydrolizujące wiązania Błąd! Nie zdefiniowano zakładki.-1,4-glikozydowe we wszystkich oligosacharydach, oraz dekstrynazy (izomaltazy) rozkładające oba typy wiązań glikozydowych.
TRANSPORT MONOSACHARYDÓW W JELICIE
Monosacharydy jako związki polarne nie podlegają biernej dyfuzji lecz są transportowane za pomocą nośnika, znajdującego się wewnątrz błony. Można je podzielić na dwie grupy: do jednej należą glukoza i galaktoza do drugiej fruktoza, mannoza i pentozy. Te drugie są wchłaniane zgodnie z gradientem stężeń, bez zużycia energii czyli na zasadzie ułatwionej dyfuzji. Glukoza i galaktoza wchłaniają się znacznie szybciej a także wbrew gradientowi stężeń lecz transport ten zależy od obecności jonów sodowych w środowisku i związany jest ze zużyciem energii. Jest to tzw transport wtórnie aktywny, energia jest zużywana na utrzymanie gradientu elektrochemicznego jonów sodowych. Transport monosacharydów w jelicie odbywa się w obrębie mikrokosmków. Na zewnętrznej powierzchni błony rozmieszczone są białka enzymatyczne (tzw ektoenzymy) hydrolizujące oligo- i disacharydy. W ich bezpośrednim sąsiedztwie znajdują się transportujące białka integralne posiadające odpowiednie receptory służące do połączenia się z odpowiednim monosacharydem.
TKANKOWY TRANSPORT GLUKOZY
Wchłonięte monosacharydy przedostają się do płynu pozakomórkowego i dopiero stąd dyfundują do naczyń włosowatych , a następnie poprzez układ żyły wrotnej dostają się do wątroby. Tutaj znaczna część glukozy zostaje zatrzymana. Wychwyt glukozy przez komorki odbywa się przy udziale integralnych białek błonowych na zasadzie ułatwionej dyfuzji. Białka transportujące glukozę, występujące w komórkach różnych tkanek, wykazują duże podobieństwo. Są to glikoproteiny o masie ok. 55000 i podobnej strukturze I rzędowej. Białka te nie wykazują swoistości w stosunku do glukozy,mogą transportować także inne monosacharydy.
Istnieją trzy główne typy transportera glukozy:
1. typ mózgowy występujący także w erytrocytach
2. typ wątrobowy występujący też w komórkach wysp trzustkowych a nawet w komórkach nadnerczy.
3. typ mięśniowy występujący też w tkance tłuszczowej.
Typ mózgowo erytrocytarny może występować także w innych tkankach. Charakteryzuje się dużym powinowactwem do glukozy i jest niemal całkowicie wysycony przy prawidłowym stężeniu glukozy we krwi. Komórki mózgu i erytrocyty zawierają w błonie dużą ilość cząsteczek transportera, charakteryzują się więc dużą pojemnością transportową.
Typ wątrobowy charakteryzuje się małym powinowactwem do glukozy i dużą maksymalną szybkością transportu. Każdy wzrost glikemii powoduje przyśpieszenie transportu. Transporter występujący w komórkach wątroby współdziała z glukokinazą - enzymem fosforylującym swoiście glukozę.
Typ mięśniowy dominuje w mięśniach szkieletowych, w mięśniu sercowym oraz w tkance tłuszczowej podskórnej. Szczególną właściwością tego transportera jest jego zależność od insuliny. Hormon ten pobudza transport glukozy do komórek mięśni i tkanki tłuszczowej, nawet 10-krotnie. Przy braku insuliny komórki tych tkanek pobierają bardzo mało glukozy ze środowiska. Insulina zwiększając szybkość maksymalną transportu nie zmienia powinowactwa transportera do glukozy. Zmiany stężenia glukozy we krwi nie mają wpływu na transport. Sądzi się, że insulina wpływa na translokację cząsteczek transportera. Przy małych stężeniach insuliny we krwi transporter glukozy, w tych komórkach znajduje się wewnątrz komórki, dopiero gdy stężenie insuliny wzrasta transporter pojawia się w błonie komórkowej.Transport przebiegający drogą ułatwionej dyfuzji odbywa się zgodnie z gradientem stężeń. W większości tkanek, stężenie wolnej glukozy w komórkach jest bliskie zeru i to warunkuje transport glukozy do wnętrza komórki. W komórkach mózgu i w erytrocytach stężenie glukozy jest bliskie stężeniu w płynie pozakomórkowym. Ciągłe jednak zużywanie glukozy w przemianach powoduje dokomórkowy jej transport. Inaczej jest w przypadku wątroby, która ma zdolność wewnątrzkomórkowego wytwarzania glukozy. Kierunek transportu zależy tutaj od kierunku działania gradientu - do wnętrza komórek lub poza ich obręb.
Transport acylo-CoA do matriks mitochondrium
Acylo-CoA znajduje się w stanie równowagi z inną makroergiczną pochodną, estrem karnityny (acylokarnityną).
Aktywowane kwasy tłuszczowe o dlugich łańcuchach są przenoszone przez wewnętrzna błonę mitochondrialną przy udziale karnityny w postaci acylokarnityny, po przeniesieniu grypy acylowej z atomu siarki CoA na grupę hydroksylową karnityny
Po matriksowej stronie błony grupa acylowa jest z powrotem przenoszona na CoA, co jest termodynamicznie możliwe, gdyż wiązanie O-acylowe jest wiązaniem wysokoenergetycznym.
Reakcje transacylacji katalizowane są przez acylotransferazę karnitynową.
Przeniesienie acylokarnityny przez błone mitochondrialna katalizuje palmitoilotransferaza karnitynowa I ,w mitochondrium działa palmitoilotransferaza karnitynowa II,która katalizuje odtworzenie Acetylo-CoA.
Równowaga kwasowo zasadowa - Zaburzenia metaboliczne .Diagnostyka
PŁYNY USTROJOWE - KREW
KREW
Skład i funkcje krwi
Krew stanowi ok. 8% masy ciała. Dzieli się na osocze, które stanowi 55% jej objętości i na elementy morfotyczne. Ponieważ elementy morfotyczne są cięższe niż osocze, więc możliwe jest ich rozdzielenie poprzez wirowanie
Krew - to tkanka płynna, jej substancja pozakomórkowa - Osocze - składa się prawie w stu procentach z wody. W osoczu zawieszone są elementy morfologiczne - krwinki. Krew krąży w układzie krwionośnym zamkniętym lub otwartym - wtedy nazywany jest hemolimfą.
Osocze - jest to płynna substancja pozakomórkowa Stanowi 55% objętości krwi W jego skład wchodzi woda - 92%
- białka - 7% - albuminy, globuliny, fibrynogen
- inne związki organiczne - glukoza, aminokwasy, lipidy
- składniki nieorganiczne o charakterze elektrolitów - zawierają jony sodowe, potasowe, chlorowe, wapniowe, magnezowe, węglowodanowe, fosforowe. sole mineralne oraz substancje transportowane (rozpuszczone gazy, substancje odżywcze, zbyteczne produkty przemiany metabolicznej, hormony). Osocze dzięki zawartości albumin i elektrolitów utrzymuje stałe ciśnienie osmotyczne płynów ciała oraz ich PH
KRWINKI - dzielimy na trzy grupy:
1. erytrocyty - czerwone krwinki
2. leukocyty - białe krwinki
3. trombocyty - płytki krwi
Krwinki czerwone (erytrocyty) transportują tlen. Nie zawierają jądra komórkowego (jest ono wypychane w trakcie rozwoju). Są elastyczne (mogą zmienić kształt, a potem wrócić do pierwotnej formy). Mają formę dwuwklęsłego krążka o średnicy 1-2 ၭm. W organizmie człowieka ok. 4,3 mln. (kobiety) i 4,8 mln. (mężczyźni) erytrocytów w 1ml krwi.W krwi znajduje się ich średnio ok. 30 bilionów (ok. 5 milionów/1 mm3). Wytwarzane są w czerwonym szpiku kości (kręgi, żebra, mostek, kości czaszki, kości długie). W trakcie rozwoju wytwarzają duże ilości hemoglobiny. Erytrocyty żyją ok. 120 dni. Gdy są zużyte lub stare wychwytywane są na drodze fagocytozy przez wyspecjalizowane komórki wątroby i śledziony. Następnie są niszczone, a niektóre ich składniki są odzyskiwane. W ciągu każdej sekundy zniszczeniu ulega ok. 2,4 miliona erytrocytów (tyle samo jest tworzonych). Niewystarczająca ilość erytrocytów lub zbyt mała zawartość hemoglobiny prowadzą do anemii (niedokrwistości).
Krwinki białe (leukocyty) bronią organizm przed drobnoustrojami. W zdrowym organizmie jest ich ok. 7000/1 mm3 (ok. 1/700 erytrocytów) . W razie infekcji bakteryjnych ich liczba może gwałtownie wzrosnąć (stąd ich dokładna ilość ma znaczenie diagnostyczne). Mogą się poruszać ruchem pełzakowatym („pod prąd” krwi, lub przez ściany naczyń). Białaczka to nowotwór powodujący zbyt duży przyrost leukocytów nie w pełni uformowanych wypierających trombocyty i erytrocyty (w szpiku), co prowadzi do zaburzenie krzepliwości krwi i śmierci najczęściej na skutek wylewu wewnętrznego (np. do mózgu).
Grupy krwi.
Pomimo, że prawidłowe erytrocyty są z wyglądu identyczne u wszystkich ludzi, to różnią się między sobą w znacznym stopniu. Są na nich zawarte pewne charakterystyczne białka, które określa się mianem antygenów grup krwi. Istnieje szereg układów grupowych krwi. Najważniejszymi jest układ grup głównych (AB0) i układ Rh. Ze względu na obecność lub brak substancji A i B na krwinkach czerwonych rozróżnia się cztery główne grupy krwi: grupa A (40% ludności w Polsce, występuje substancja A), grupa B (12%, występuje substancja B), grupa AB (8%, występuje substancja A i B) oraz grupa 0 (40%, brak substancji A i B na krwinkach). Każda z tych grup może posiadać substancję z układu grupowego Rh - antygen D, daną osobę określa się wtedy jako Rh-dodatnią. Przeciwnie, u osoby Rh-ujemnej, substancja D nie występuje. Oznaczenie grup krwi ma podstawowe znaczenie przy doborze krwi do przetoczeń wymaganych np. w trakcie wielu zabiegów operacyjnych lub podczas leczenia chorób krwi. W razie potrzeby podawać trzeba krew identyczną w zakresie przynajmniej tych dwóch układów, a więc osobie z grupą krwi A Rh+ należy podać krew A Rh+. Grupa krwi jest niezmienna w ciągu życia, jedynie sporadycznie, po przeprowadzeniu allogenicznego przeszczepu szpiku (od rodzeństwa lub dawcy niespokrewnionego), może dojść (choć nie musi) do zmiany grupy krwi u biorcy przeszczepu.
pH KRWI. Równowaga kwasowo-zasadowa, pojęcie biochemiczne informujące o wzajemnym stosunku ilościowym jonów o cechach zasadowych i jonów o cechach kwasowych we krwi i w tkankach.
Prawidłowy stan równowagi kwasowo-zasadowej cechuje się niewielkim nadmiarem jonów zasadowych - jego liczbowy odpowiednik określany jako pH (potentia hydrogenii, ujemny logarytm ze stężenia jonów wodorowych, wykładnik wodorowy) wynosi ok. 7,35-7,40.
Przesunięcie wartości pH powyżej 7,40 świadczy o zaburzeniu równowagi kwasowo-zasadowej i przesunięciu odczynu krwi w kierunku zasadowym (alkaloza, zasadowica) wskutek wzrostu stężenia jonów zasadowych lub spadku stężenia jonów kwaśnych.
Przesunięcie wartości pH poniżej 7,35 świadczy o zaburzeniu równowagi kwasowo-zasadowej i przesunięciu odczynu w kierunku kwaśnym (acydoza, kwasica) wskutek spadku stężenia jonów zasadowych lub wzrostu stężenia jonów kwaśnych. Im wartość liczbowa pH jest niższa, tym bardziej kwaśny jest odczyn krwi.
W celu określenia stanu równowagi kwasowo-zasadowej oznacza się pH w próbce krwi, a w celu interpretacji przyczyn, jakie wywołały zmiany pH krwi, oznacza się stężenie dwuwęglanów, prężność dwutlenku węgla, zasób zasad (gazometria, gospodarka gazowa).
pH, ujemny dziesiętny logarytm stężenia jonów wodorowych w roztworze (pH=-log[H+]). Dla roztworów wodnych wartość pH mieści się w przedziale 0-14; roztwory kwaśne: pH<7, roztwory zasadowe: pH>7, obojętne: pH=7. Pomiar pH roztworów wykonywany jest za pomocą półogniwa chinhydronowego lub elektrody szklanej.
Największą przydatność kliniczną ma pomiar pH krwi tętniczej lub arterializowanej krwi włośniczkowej. Prawidłowo pH krwi tętniczej waha się od 7,36 do 7,44. W warunkach chorobowych stwierdza się wartości 6,8-7,8. pH krwi niższe niż 7,35 wskazuje na istnienie kwasicy, a wyższe niż 7,45 na zasadowicę. Stwierdzenie prawidłowego pH nie wyklucza jednak istnienia poważnych zaburzeń metabolicznych lub oddechowych. Wynika to jasno z równania Hendersona-Hasselbalcha według którego przy równoleg-łym wzroście lub spadku stężenia ![]()
i ![]()
pH może nie ulec żadnej zmianie.
. Pomiar ciśnienia parcjalnego dwutlenku węgla we krwi tętniczej jest najlepszym wskaźnikiem zmian w równowadze kwasowo-zasadowej uwarunkowanych oddechowo. Znajomość samej tylko wartości ![]()
nie pozwala określić przyczyny stwierdzonej zmiany (może ona być przejawem zaburzonej wymiany gazowej w płucach lub też kompensacji zaburzeń metabolicznych) ani też przewidzieć aktualnego pH krwi, dlatego też ma ona ograniczoną wartość diagnostyczną.
Prawidłowe wartości ![]()
wahają się od 4,66 do 5,99 kPa (33-44 mmHg), wynosząc średnio 5,32 kPa (40 mmHg).
Stężenie wodorowęglanów. Przy znajomości pH krwi i ![]()
stężenie ![]()
można obliczyć z równania Hendersona- Hasselbalcha:
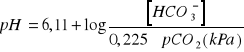
![]()
Stężenie wodorowęglanów odpowiada aktualnemu stężeniu ![]()
w osoczu jeśli ozna-czone zostało we krwi pobranej w warunkach bezpowietrznych. Wartość ta jest w istotny sposób zależna od ![]()
krwi. Prawidłowe stężenie ![]()
wynosi 25 mmol/dm (wahania od 22 do 28 mmol/l).
ZASADY BUFORUJĄCE
Zasady buforujące (buffer base-BB). Są sumą stężenia wodorowęglanów, białek osocza, fosforanów i hemoglobiny. Bardzo niskie stężenie fosforanów w osoczu sprawia, że wartość BB osocza jest faktycznie zdeterminowana sumą stężenia anionów wodorowęglanowych (25 mEq/l) i białczanowych osocza (17 mEq/l) i wynosi łącznie 42 mmol\l. Każde 0,6205 mmol/l (=1 g%) hemoglobiny podnosi wartość BB o 0,36 mEq/l. Przy prawidłowym stężeniu Hb (9,3 mmol/l = 15,0 g%) wartość BB dla krwi całkowitej wynosi 48 mEq/l.
KWASICE METABOLICZNE
Kwasicę metaboliczną określa się jako odbiegający od normy proces fizjologiczny prowadzący do zwiększenia w organizmie ilości nielotnych kwasów albo utraty wodoro-węglanów z płynu pozakomórkowego.
Przyczyną kwasicy metabolicznej może być:
A. Nadmierna produkcja endogennych kwasów
1. Kwasica ketonowa:
a) cukrzycowa
b) głodowa
c) u chorych z nadczynnością tarczycy
d) u chorych gorączkujących
2. Kwasica mleczanowa
3. Kwasice o innej etiologii:
a) kwasica polekowa po podaniu inhibitorów anhydrazy węglanowej lub chlorku wapniowego
b) kwasica po zatruciu alkoholem metylowym lub glikolem etylenowym
c) kwasica po wszczepieniu moczowodów do esicy lub prostnicy
B. Kwasice uwarunkowane utratą zasad:
1. Przez przewód pokarmowy:
a) w zespołach biegunkowych
b) przy dużym drenażu przewodu pokarmowego i przez przetoki
2. Przez nerki:
a) kwasica cewkowa dystalna (typ 1)
b) kwasica cewkowa proksymalna (typ 2)
C. Kwasice uwarunkowane zaburzona regeneracją zasad przez nerki:
1. Kwasica mocznicowa przewlekla
2. Kwasica mocznicowa ostra
D. Kwasice wywołane rozcieńczeniem zasad w przestrzeni wodnej pozakomórkowej
ZASADOWICE METABOLICZNE
Zasadowicę metaboliczną określa się jako odbiegający od normy proces fizjologiczny prowadzący do utraty kwasów lub wzrostu stężenia jonów wodorowęglanowych w płynie pozakomórkowym.
Zasadowicę metaboliczną powodują:
a) utrata kwaśnej treści żołądkowej (wymioty, płukanie żołądka)
b) utrata kwasów z moczem na tle hiperaldosteronizmu
c) przesunięcie jonów wodorowych z płynu pozakomórkowego do wnętrza komó-rek na tle niedoboru jonów potasowych
d) utrata jonów potasowych z kałem na tle uporczywych biegunek
e) nadmierna utrata chlorku sodowego po podaniu leków moczopędnych
f) nadmierna podaż zasad (zarówno doustnie jak i drogą pozajelitowa)
g) nadmierna podaż soli organicznych
ROLA PŁUC I NEREK W REGULACJI RÓWNOWAGI KWASOWO-ZASADOWEJ
Utrzymanie stałej równowagi kwasowo-zasadowej jest jednym z podstawowych warunków prawidłowego funkcjonowania organizmu. W wyniku stałej przemiany materii istnieje prawie nieustanna tendencja do zakwaszania organizmu z powodu tworzenia się kwaśnych produktów przemiany materii, z których najważniejszy jest dwutlenek węgla tworzący się pod-czas spalania niemal każdej substancji organicznej. Przesunięcie oddziaływania organizmu w stronę zasadową jest znacznie rzadsze, ponieważ zasadowe substancje tylko wyjątkowo tworzą się w przebiegu prawidłowej przemiany materii. Ze względu na możliwość przesunięcia równowagi kwasowo-zasadowej w kierunku kwaśnym lub zasadowym niezbędna jest stała i sprawna regulacja tego oddziaływania.
Regulacja płucna czyli wydalanie![]()
w płucach. Dynamikę tego procesu wyraża wzór:
![]()
Zgodnie z prawem działania mas zwiększenie stężenia jonów wodorowych przesuwa natychmiast reakcję w lewo-jon wodorowy łącząc się z ![]()
daje ![]()
,który dysocjuje na ![]()
i ![]()
, po czym ![]()
zostaje natychmiast wydalony przez płuca. Dzięki temu układ wodorowęglanowy przyjmuje pierwotną postać, a stężenie jonów wodorowych - mimo napły-wu ![]()
-pozostaje nie zmienione.
REGULACJA NERKOWA.
Rola nerek w utrzymywaniu równowagi kwasowo-zasadowej polega obok wydalania jonu wodorowego także na regulacji poziomu w osoczu i utrzymaniu zasobów ustrojowych jonu ![]()
wchodzącego w skład buforu wodorowęglanowego. Nerki współpracują tutaj z płucami regulującymi zasób drugiego składnika tego buforu-kwasu węglowego, poprzez wydalanie pozostającego z nim w równowadze ![]()
. Proces resorpcji ![]()
i sekrecji kanalikowej ![]()
są ze sobą ściśle powiązane, szczególnie w obrębie kanalika krętego bliższego.
Głównym układem buforowym stabilizującym pH płynu pozakomórkowego jest bufor wodorowęglanowy.
DIAGNOSTYKA MEDYCZNA
Homeostaza
Homeostaza (z greki homoíos - podobny, równy i stásis - trwanie) jest to zespół mechanizmów zapewniających stałość pewnego dynamicznego układu. Określenie to odnosi się także do stanu takiej dynamicznej równowagi.
W naukach biologicznych homeostaza określa stałość określonych parametrów środowiska wewnętrznego organizmu. Homeostaza jest niezbędnym warunkiem zdrowia (prawidłowego funkcjonowania) organizmu, a co za tym idzie wiele chorób u swego podłoża ma zaburzenie mechanizmów homeostazy.
Mechanizmy homeostazy działają ciągle, na wielu poziomach:
śródkomórkowym (w cytoplazmie danej komórki)
tkankowym (w danej tkance np. tkance nerwowej)
układów (w zakresie danego układu np. krwionośnego)
Do najważniejszych parametrów, które muszą być stale kontrolowane i regulowane przez mechanizmy homeostatyczne należą:
temperatura ciała (u organizmów stałocieplnych)
pH krwi i płynów ustrojowych
ciśnienie osmotyczne
objętość płynów ustrojowych (stan nawodnienia organizmu)
stężenie związków chemicznych w płynach ustrojowych (np. glukozy w osoczu)
ciśnienie tętnicze krwi
ciśnienie parcjalne tlenu i dwutlenku węgla we krwi
Mechanizmy homeostatyczne
• wegetatywny układ nerwowy
• układ hormonalny
• układy tkankowe
Normy badań laboratoryjnych
Najbardziej wiarygodnym źródłem norm laboratoryjnych jest zwykle personel laboratorium, które wykonywało badania. Wynika to przede wszystkim z korzystania z różnych metod analitycznych, mających różną dokładność.
Tabela
KREW
Składniki chemiczne krwi
Azot mocznikowy |
|
Składniki morfotyczne krwi |
OB (Odczyn Biernackiego) |
HEMATOKRYT (Ht, HCT) |
Stosunek objętościowy erytrocytów do osocza krwi, określany jako % objętości krwinek w pewnej objętości krwi |
HEMOGLOBINA (Hb, HGB) |
Ilość barwnika krwi wiążącego tlen i dwutlenek węgla. Określana w gramach na 100 ml [g%] lub w milimolach na litr [mmol/l] |
ERYTROCYTY (Krwinki czerwone - E, RBC) |
Erytrocyty dzięki swemu kształtowi i zawartości hemoglobiny służą wymianie gazowej (przenoszeniu tlenu i dwutlenku węgla). |
Liczba |
Średnia objętość w fl (MCV) |
Średnia masa Hb w pg (fmol) (MCH) |
Średnie stężenie Hb w % (mmol/l) (MCHC) |
Grubość w um |
Średnica w um |
TROMBOCYTY (Płytki krwi - T, PLT) |
Płytki krwi biorą udział w procesie hemostazy, krzepnięciu krwi, a także magazynują i transportują niektóre aminy (m. in. serotoninę, adrenalinę) |
LEUKOCYTY (Krwinki białe - L, WBC) |
Krwinki białe odgrywają bardzo ważną rolę w mechanizmach obronnych ustroju. |
We krwi obwodowej znajduje się kilka rodzajów krwinek białych. Ich liczbę określa się zwykle w procentach (%) lub w wartościach bezwzględnych (G/l). |
granulocyty obojętnochłonne |
granulocyty kwasochłonne ("kw") |
granulocyty zasadochłonne ("zas") |
limfocyty ("lim") |
monocyty ("mono") |
MOCZ |
Właściwości fizykochemiczne moczu |
Ilość dobowa moczu |
Gęstość względna moczu |
Zabarwienie moczu |
Odczyn moczu |
Składniki chemiczne moczu |
Białko |
Białkomocz to stan wydalania z moczem ponad 150 mg białka na dobę. |
Urobilinogen |
Cukier |
Związki ketonowe |
Składniki morfotyczne moczu |
Krwinki czerwone - Erytrocyty (E) |
Krwinkomocz - obecność krwinek czerwonych wykryta w badaniu osadu moczu. |
Krwinki białe - Leukocyty (L) |
Wałeczki |
Kryształy |
Bakterie |
|
Rodzaj elementów |
Przeciętna liczba (/mm3) lub udział |
Funkcje |
Patologia |
Erytrocyty |
M: 4,2-5,4 mln K: 3,6-5,0 mln |
Transport tlenu i CO2 |
Za mało: anemia Za dużo: czerwienica |
Trombocyty |
150 - 400 tyś |
Udział w procesach krzepnięcia krwi |
Zaburzenia krzepnięcia krwi; skaza krwotoczna; łatwość powstawania sińców |
Leukocyty |
5000 - 10000 |
||
Obojętno- chłonne |
60% |
Fagocytoza |
Liczba ich wzrasta w wyniku infekcji bakteryjnych, stanów zapalnych, białaczki mieloblastycznej |
Kwasochłonne |
1-3% |
Czynne w reakcjach alergicznych |
Liczba ich wzrasta w wyniku reakcji alergicznych oraz zakażeń pasożytami |
Zasadochłonne |
1% |
Odgrywają rolę w zapobieganiu krzepnięciu krwi w organizmie |
|
Limfocyty |
25-35% |
Wytwarzają przeciwciała, niszczą obce komórki |
Liczba ich wzrasta w wyniku białaczki limfatycznej oraz infekcji wirusowych; przy mononukleozie zakaźnej pojawiają się nietypowe limfocyty |
Monocyty |
6% |
Pełnią funkcję makrofagów |
Liczba ich wzrasta w białaczce monocytowej i grzybicach |
Kwasy nukleinowe i biosynteza białek
Kwasy nukleinowe
Kwasy nukleinowe są to związki występujące we wszystkich żywych komórkach, odgrywające zasadniczą rolę w przekazywaniu cech dziedzicznych i kierowaniu syntezą białek. Łańcuchy cząsteczek kwasów nukleinowych zbudowane są z elementów zwanych nukleotydami, zawierających 3 składniki: kwas fosforowy, cukier (pentozę) oraz purynową lub pirymidynową zasadą.Kwas dezoksyrybonukleinowy (DNA) zawiera cukier D-dezoksyrybozę, a jako zasady - tyminę, cytozynę, adeninę i guaninę. Kwas rybonukleinowy (RNA) zawiera cukier D-rybozę i zasady: uracyl, cytozynę, adeninę i guaninę. Kwas DNA występuje w chromosomach, chloroplastach i mitochondriach, a jego rolą jest przekazywanie informacji genetycznej (kod genetyczny).
Rozróżniamy następujące rodzaje kwasów nukleinowych:
RybosomalnY RNA - r RNA
Olbrzymia ilość rRNA jest wynikiem ogromnej ilości kopii kodujących ten specyficzny kwas rybonukleinowy
Przenośnikowy (transportujący) tRNA
Cząsteczka tRNA pełniąca rolę transportera musi w jakiś sposób rozpoznawać tryplet na mRNA. Nie czynią tego same aminokwasy (co więcej nie maja one wpływu na rozpoznawanie kodonu), ale ma to miejsce dzięki pętli antykodonu, w której 3 zasady stanowiące antykodon są komplementarne do trypletu - kodonu na mRNA. Przy czym kod genetyczny czyta się od końca 5'→3' a antykodon od 3'→5', są one więc antyrównoległe w swojej komplementarności
Tworzenie aminoacetylo-tRNA
Aby tRNA mógł pełnić swoja funkcje musi zostać połączony z aminokwasem. Aminokwas musi być uprzednio zaktywowany.Aktywację aminokwasów przeprowadzają enzymy syntetazy aminoacylo-tRNA (tzw. Enzymy aktywujące).
Kodon
Kodon to triplet, trójka nukleotydów w cząsteczce DNA (kwasy nukleinowe) kodująca określony aminokwas włączany w strukturę białka w trakcie biosyntezy białek w komórce.
Antykodon
Antykodon, w genetyce termin określający trzy kolejne nukleotydy RNA (kwasy nukleinowe) transportującego (tRNA), posiadające zdolność rozpoznawania kodonów w RNA matrycowym (mRNA) w procesie biosyntezy białka w komórce
REPLIKACJA
Replikacja to złożony proces prowadzący do podwojenia ilości DNA komórkowego. Zachodzi ona w fazie S cyklu komórkowego. Aby zachować trwałość organizmu replikacja musi być całkowita i wierna. Mechanizm replikacji poznano po odkryciu budowy DNA zgodnego z modelem dwuniciowej helisy zaproponowanej w 1953 r. przez Waotsona i Cricka.
Główne założenia tego modelu to: dwa helikalne łańcuchy polinukleotydowe zawijają się dookoła wspólnej osi, biegnąc w przeciwległych kierunkach ( jedna nić 3' do 5' druga 5' do 3')
zasady azotowe znajdują się w wewnątrz prostopadle do osi helisy, a fosforany i dezoksyrybozy na zewnątrz ( płaszczyzny deoksyrybozy sa prawie prostopadłe do płaszczyzny zasad)
średnica helisy wynosi 2nm, odległość między zasadami 0,34 nm na całkowity skręt przypada po 10 nukleotydów
łańcuchy wiążą się ze sobą wiązaniami wodorowymi, w ściśle określonych parach: adenina z tyminą ( 2 wiązania wodorowe) a guanina z cytozyną (3 wiązania wodorowe) Reguła Chargaffa
Kolejność zasad jest nie ograniczona. Ściśle określona sekwencja zasad w nici DNA niesie informację genetyczną.
W procesie replikacji możemy wyróżnić kilka etapów:
1. Przygotowanie nici DNA do replikacji
2. Inicjacja replikacji
3. Elongacja
NAPRAWA DNA
Dla przeżycia pojedynczego organizmu jak i przetrwania gatunku istotne znaczenie ma utrzymanie całości informacji w cząsteczkach DNA. Gatunki przetrwałe musiały rozwinąć mechanizmy naprawy szkód w DNA jako błędy replikacji lub oddziaływanie środowiska. Główną rolę odgrywa tu tautomeryczna zdolność łączenia się w pary puryn i pirymidyn. Nieprogramowana synteza DNA zwana również REPARACJĄ DNA zachodzi niezależnie od cyklu komórkowego. Czynniki uszkadzające wyeliminowują jedną nić DNA w wyniku czego powstają dimery między sąsiednimi pirymidynami. Rozleglejsze uszkodzenia obu nici bądź chromosomów są nieodwracalne.
Biosynteza białka
Biosynteza białka jest to zachodzący w żywych komórkach organizmu proces powstawania białka uwarunkowany przez zapisaną w DNA (kwasy nukleinowe) informację genetyczną (gen).Pierwszym etapem biosyntezy białek jest transkrypcja odpowiedniego odcinka DNA, która polega na syntezie RNA na matrycy określonego odcinka DNA przy udziale polimerazy RNA. RNA powstały w wyniku transkrypcji, zawierający informacje dla syntezy białek, zwany jest mRNA. mRNA przenosi transkrybowaną informację genetyczną z jądra do cytoplazmy. Tutaj dochodzi do modyfikacji mRNA, tzn. do wycinania, z udziałem odpowiednich enzymów, sekwencji niekodujących - intronów i pozostawiania sekwencji kodujących - egzonów. Tak zmodyfikowane i skrócone cząsteczki mRNA wnikają pomiędzy dwie podjednostki rybosomów, gdzie odbywa się właściwe odczytywanie kodu genetycznego i przepisywanie go na sekwencję aminokwasową białka w procesie zwanym translacją. Znajdujące się w cytoplazmie aminokwasy są przenoszone na rybosomy za pomocą tRNA. Cząsteczki tRNA z doczepionymi aminokwasami przedostają się do rybosomów i kolejno dopasowują się, na zasadzie komplementarności, swoimi antykodonami do odpowiednich kodonów mRNA. Translacja zaczyna się od kodonu startowego, zapewniającego dalsze odczytywanie mRNA we właściwej kolejności - najczęściej jest to kodon AUG lub GUG, a kończy się kodonem symbolizującym ostatni aminokwas (u prokariontów są to kodony nonsensowne - nie oznaczające żadnego aminokwasu). Po zakończeniu syntezy cząsteczki białka wędrują przez przestrzenie pomiędzy błonami reticulum endoplazmatycznego do aparatu Golgiego albo wydzielane są na zewnątrz komórki, lub pozostają przez jakiś czas związane z błonami ziarnistego (szorstkiego) reticulum endoplazmatycznego i wykorzystywane jako białka wewnątrzkomórkowe. Energia potrzebna do syntezowania wiązań peptydowych pochodzi z wysokoenergetycznych wiązań ATP.
STRUKTURA CHROMATYNY, CECHY DNA EUCARYOTA
Jądrowy DNA zbudowany z deoksyrybonukleotydów połączonych wiązaniami 5' - 3' jest związkiem o największej masie cząsteczkowej (3,5 x 106) w komórce. Metodami biochemicznymi można go rozdzielić na 2 frakcje.
80-90% stanowi frakcja główna (ułożona równomiernie w jądrze)
10% stanowi DNA frakcji satelitarnej - tworzy skupiska heterochromatyny w okolicach centromerów, w chromatynie organizującej jąderka oraz na końcu ramion chromosomów. Funkcjonalnie frakcja ta odpowiada za transkrypcję rRNA, tRNA, we frakcji tej występują liczne kopie powtarzające się.
Nieodłącznym składnikiem chromatyny są białka zasadowe - histony, białka niehistonowe - zasadowe i kwaśne.
Białka niehistonowe
Białka niehistonowe (m. cz. 20 kDa do 22,5 kDa ) można podzielić na 4 białka o m. Cz. 30 kDa - 40 kDa -aktyna i białka ochraniające hnRNA (heterogeny RNA), cztery białka o m. Cz. 50 kDa - tubuliny 4 białka o m. cz. 25 kDa to miozyna. Ponadto znajdują się w chromatynie białka kwaśne (m. cz. 100 kDa) łączące histony w oktamery nukleosomów (patrz niżej).
Powyższe białka stanowią około 50% białek niehistonowych, tzw. strukturalnych. Pozostała część stanowią białka niehistonowe mające aktywność enzymatyczną --polimerazy kwasów nukleinowych, proteazy, nuleazy, transferazy, a około 5% białek niestrukturalnych jest na stałe związana z DNA.
HISTONY
Histony są one pojedynczymi łańcuchami polipeptydowymi (masa od 11 kDa do 21 kDa). Wyróżniono 5 klas histonów na podstawie stosunku aminokwasów zasadowych do kwaśnych i stosunku lizyny do argininy.
Polimerazy DNA i RNA
Enzymami katalizującymi syntezę DNA i RNA. są polimerazy DNA i RNA,- nukleotydylotransferazy DNA i RNA, łównym zadaniem polimerazy DNA jest tworzenie łańcucha komplementarnego do istniejącego łańcucha DNA, który działa jako starter. Polimeraza RNA natomiast uczestniczy w syntezie łańcucha RNA, wymagając obecności DNA jako matrycy, z której czerpie instrukcje.
Intron
Z powstałego w wyniku transkrypcji pierwszego genu - tzw. pre mRNA - zostają wycięte odcinki będące odpowiednikami intronów w procesie zwanym składaniem genowym, natomiast zostają odcinki kodujące białko (egzony).Introny są więc odcinkami pozbawionymi informacji o strukturze białka (nie kodujące).
Egzony
W procesie modyfikacji genów na poziomie cytoplazmy zostają wycięte odcinki niekodujące (introny) a pozostają tylko sekwencje kodujące tworzące skrócony mRNA, wnikający pomiędzy dwie podjednostki rybosomu. Egzony są więc to odcinki zawierające informację o strukturze białka (kodujące białko) występujące w tzw. genach podzielonych występujących u organizmów eukaryotycznych.
Rybosomy
Rybosomy biorą udział w biosyntezie białek, dlatego szczególnie licznie występują w młodych, silnie rosnących komórkach. Nazwę swą zawdzięczają wysokiej zawartości kwasu rybonukleinowego Rybosomy to drobne ciałka o średnicy 0,01-0,025 µm występujące w komórkach roślin i zwierząt, zbudowane z białka i kwasu rybonukleinowego (RNA). Rybosomy rozmieszczone są w jądrze komórkowym (głównie w jąderku), mitochondriach,
.
Kod genetyczny
Kod genetyczny jest to przepisana informacja genetyczna z DNA na mRNA w konsekwencji jest to współzależność między sekwencją zasad w mRNA stanowiącym transkrypt DNA a sekwencją aminokwasów w białku.
W sekwencji nukleotydów w cząsteczce mRNA są zawarte słowa kodu (trójnukleotydowe) dla każdego aminokwasu, zespół tych słów to kod genetyczny.
Kod genetvczny jest:
Trójkowy - każde słowo kodu zwane kodonem, zbudowane jest z 3 kolejnych nukleotydów. Przy 4 różnych zasadach w mRNA, istnieją 64 triplety nukleotydów - kodujące 20 aminokwasów
Uniwersalny, tzn. że w całym świecie istot żywych tym samym nukleotydom (tripletom) odpowiadają te same aminokwasy w syntetyzowanych peptydach. Wszystkie organizmy używają do translacji swoich genów na swoiste białko tego samego kodu.
Niezachodzący - każda trójka nukleotydów decyduje o jednym aminkowasie, przy czym ostatni nukleotyd kodonu nie jest wspólny dla tripletu następnego. Odczyt prowadzony jest kolejno trójkami nukleotydów.
Bezprzecinkowy - jeżeli raz rozpocznie się odczytywanie w miejscu swoistego kodonu, to informacja jest odczytywana bez przerwy w sekwencji tripletów aż do momentu osiągnięcia kodonów nonsensowynch (UAG, UAA, UGA), które nie są odczytywane przez tRNA, ale przez specyficzne białka zwane czynnikami uwalniającymi (releasing factors). Nie ma więc tripletów przerywających ciągłość odczytu.
Jednoznaczny (niedwuznaczny) -oznacza to, że dany kodon (triplet) koduje tylko jeden aminokwas np. UAA oznacza tylko i wyłącznie triplet dla leucyny i inny aminokwas nie może zostać w to miejsce wbudowany. Rozpoznanie swoistych kodonów w mRNA przez cząsteczki t-RNA zależy od antykodonu i praw łączenia się zasad w komplementarne pary.
Zdegenerowany - wiele aminokwasów jest kodowanych przez więcej niż jeden triplet (kodonów sensownych jest 61, a aminokwasów 20). Tylko tryptofan i metionina kodowane są przez jeden triplet (odpowiednio UGG i AUG). Osiemnaście pozostałych kodowanych jest przez dwa lub więcej tripletów. aż po sześć kodonów odpowiada leucynie, argininie i serynie.
Kodony, które określają ten sam aminokwas nazywają się synonimami np. GUU, GUC, GUA, GUG są synonimami waliny. Ulokowanie aminokwasów nie jest przypadkowe, zajmują one z reguły jeden blok, którego cechą charakterystyczną jest to, że dwie pierwsze zasady są z reguły takie same, różnią się natomiast trzecią zasadą , która można by powiedzieć ma najmniejsze znaczenie. Jest to tzw. h i p o t e z a t o l e r a n c j i .Zdegenerowanie kodu zmniejsza szkodliwe skutki mutacji. Gdyby kod nie był zdegenerowany to 20 kodonów określałoby 20 aminokwasów, a 44 oznaczałyby terminację, wówczas prawdopodobieństwo mutacji, która powodowałaby terminację łańcucha byłoby dużo większe.
Transkrypcja
Proces transkrypcji zachodzi w komórkach pod wpływem enzymów polimeraz RNA. Przeprowadzają one syntezę RNA na nici DNA .Transkrypcja,to przepisywanie informacji genetycznej z DNA na mRNA (kwasy nukleinowe), zachodzące w jądrze komórkowym w drodze syntezy RNA na matrycy DNA, kodującej białko. Z kolei mRNA w procesie translacji przekazuje informację genetyczną, według której zostaje ustalona sekwencja aminokwasów w łańcuchu polipeptydowym. W procesie transkrypcji powstają: tzw. informacyjny, czyli matrycowy RNA - mRNA, rybosomowy RNA - rRNA i przenoszący RNA - tRNA
Translacja
Komórka rozporządza aparatem pozwalającym przekładać informację zakodowaną w sekwencji nukleotydów w mRNA na sekwencje aminokwasów. Proces ten nazywamy translacją.
MECHANIZM TRANSLACJI - BIOSYNTEZA BIAŁKA
Cząsteczkami pośredniczącymi są kwasy tRNA w połączeniu z aminokwasami (aminoacylo-tRNA), a dzieje się to na rybosomach (lub polirybosomach). Przypuszcza się,że asocjacja jednostek rybosomowych tworzy jakby tunel, w którym przesuwa się nić m RNA. Aktywnie syntetyzujące białko rybosomy są nanizane na nić m-RNA i tworzą polisomy - skupisko rybosomów powiazanych jedną nicią mRNAW obrębie polisomu odbywa się proces syntezy tego samego łańcucha peptydowego, (z tym, że na każdym rybosomie w danej chwili jest inny etap). Informacyjny RNA ulega translacji w kierunku 5'→ 3', w takim samym kierunku był on syntetyzowany.Tak więc białko syntetyzuje się od N końca do C - końca.
W translacji wyróżniamy następujące etapy:
I Inicjacji
a) powstawanie kompleksu preinicjującego
b) związanie kompleksu preinicjującego z mRNA → kompleks pośredni
c) powstanie aktywnego kompleksu inicjującego
d) odtwarzanie aktywnego czynnika IF-2-GTP
I Elongacja
W rybosomie są dwa sąsiadujące ze sobą miejsca:
akceptorowe (aminokwasowe) - A - wiążące wszystkie aminoacylotRNA- z wyjątkiem Met-tRNA
peptydowe ( donorowe) -P- wiążące Met-tRNA i peptydylo-tRNA
W elongacji możemy wyróżnić kilka etapów:
Przyłączanie Met-RNA w miejsce P.
Przyłączenie aminoacylo-tRNA do miejsca A
Wytwarzanie wiazania peptydowego - transpeptydylacja
III Terminacja
W miejscu A pojawia się jeden z kodonów nonsensownych ( UAA, UAG, UGA), którego nie odczytuje żaden z aminoacylo tRNA
Potranslacyjne modyfikacje białek
Po translacji mRNA wiele powstałych polipeptydów ulega modyfikacjom, które obejmować mogą:
Wycinanie kilku lub pojedynczych polipeptydów poprzez aminopeptydazy.
Przez utlenienie dwóch cystein mogą powstawać wiazania dwusiarczkowe. Dzieje się tak z insuliną, która jako proinsulina powstała po translacji.
Łańcuchy boczne reszt aminokwasowych mogą być modyfikowane.
Biosynteza białka - transkrypcja
Biosynteza białka - translacja
1
34
Plasma
Erytrocyt
Granulocyt
Lymfocyt
Monocyt
Trombocyt
Wyszukiwarka
Podobne podstrony:
Suplement z biochemii(1), biologia, biochemia
suplement biochemia 3
Kolo biochemia KOMPLET NOTATEK AMINOKWASY, Szkoła Rolnictwo studia, Szkoła, Materiały studia, bioche
zadania do opracowania(1), ^^Szkoła ^^, ^^Biochemia^^
zagadnienia do egzaminu z biochemi(1), ^^Szkoła ^^, ^^Biochemia^^
sciaga biochemia(1), ^^Szkoła ^^, ^^Biochemia^^
BIALKA CALE, Szkoła Rolnictwo studia, Szkoła, Materiały studia, biochemia cwiczenia
BIOCHEMIA EGZAMIN, Szkoła Rolnictwo studia, Szkoła, Materiały studia, biochemia cwiczenia, biochemia
Biochemia egzamin, Szkoła Rolnictwo studia, Szkoła, Materiały studia, biochemia cwiczenia, biochemia
4, Szkoła, Biochemia, Wykład
kolo 2 biocha sciaga22, Szkoła Rolnictwo studia, Szkoła, Materiały studia, biochemia cwiczenia
exam bio, Szkoła Rolnictwo studia, Szkoła, Materiały studia, biochemia cwiczenia
CUKRY!!!!!!!, Szkoła Rolnictwo studia, Szkoła, Materiały studia, biochemia cwiczenia
więcej podobnych podstron