INŻYNIERIA GENETYCZNA L
Przygotowanie komórek kompetentnych XL1-Blue
Komórki kompetentne - komórki zdolne przyjąć obce DNA.Ich przygotowanie - zamrażanie, szok cieplny.
Bufor A - jony Mg
Bufor B - jony Cl
Bufor C - jony Ca, glicerol (20%)
Inokulum- hodowla wstępna, dajemy do niej antybiotyk, aby zablokować rozwój nieporządanych mikroorganizmów, albo rozwijać tylko te mikroorganizmy, które są odporne na specyficzne antybiotyki.
Liczenie rozcienczenia:
Stezenie wyjściowe=12,5mg/ul
stezenie koncowe ma byc= 12,5ug/ul
R=Co/Ck= 12500/ 12,5= 1000
Vkoncowe= 70ml
Vpoczatkowe antybiotyku= Vk/R= 70/1000=70 ul
Kompetencja- zdolność komórek do pobrania materiału genetycznego z zewnątrz. Może być naturalne i prowadzona sztucznie.
Strefy adhezyjne u bakteri- pory, przez które może przeniknąć materiał DNA. Błonę należy zobojętnić, ponieważ jest naładowana ujemnie. Zobojętniamy ją np. jonami wapni, aby wprowadzić DNA do środka. Obniżamy temperaturę i błonę wtedy łątwiej opłaszczyć. W ukompetententnianiu używamy jonów magnezu i wapnia. Gdy następuje podwyższenie temperatury, usztywniona komórka się rozluźnia i wtedy może przyjąć DNA.
Czysta kultura- wszystkie kopie komórek pochodzą od hodolwi macierzystej
Glicerol zwiększa gęstość, żeby białka bakteryjne i oddziaływania między białkami były bardziej stabilne. Glicerol chroni komórki - zapobiega lizie.
Jony są potrzebne do neutralizacji ładunku błony.
Temperatura - jej obniżanie powoduje, że błona jest bardziej przepuszczalna i sztywna. Podniesienie temperatury powoduje rozciąganie się błony, dzięki czemu może wejść DNA.
Błona komórkowa - fosfolipidy (P - ładunek ujemny)
Transformacja DNA - proces, dzięki któremu DNA jest wprowadzane do bakterii.
OD600 - mówi o rozproszeniu światła. Jest to gęstość optyczna. (komórki kompetentne OD600= 0,4-0,5)
Wykres wzrostu bakterii:
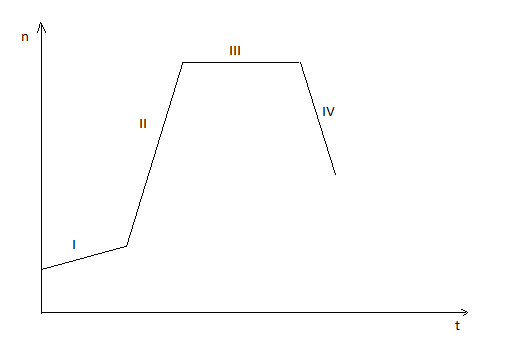
I - faza adaptacyjna (komórki nie dzielą się)
II - faza logarytmiczna (największy wzrost komórek)
III - faza stacjonarna (ilość komórek powstających i umierających jest taka sama)
IV - faza zamierania
X-Gal - pochodna laktozy. Β-galaktozydaza hydrolizuje X-Gal do galaktozy i glukozy.
Galaktoza daje kolor niebieski. Kolonie dobrze stransformowane widać na płytce jako kolonie niebieskie (rozkładają X-Gal)
transpozon - gen ruchomy
Transformacja- proces aktywnego pobierania materiału genetycznego z zewnątrz (prokariota)
transfekcja -- proces aktywnego pobierania materiału genetycznego z zewnątrz u organizmów eukartiotycznych
Transdukcja- przekazania materiału genetycznego z jednego prokariota do drugiego za pomocą wirusa
Metody wszczepiania genów do komórek eukariotycznych:
Elektroporacja - poddawanie komórek działaniu pola elektrycznego, dzięki czemu tworzą się odwracalne pory.
Mikroiniekcja - wszczepianie DNA, bombardowanie błony komórkowej w polu elektrycznym (błona komórkowa jest otoczona złotem lub wolframem).
Liposomy - pęcherzyki lipidowe, które mogą powstawać w fosfolipidów, są to wodne przestrzenie ograniczone dwuwarstwą lipidową, mogą służyć do badania przepuszczalności błon czy do dostarczania komórkom różnych związków chemicznych
Działanie dekstranem i fosforanem wapnia
Transdukcja - wprowadzenie DNA do komórki bakteryjnej za pomocą bakteriofagów
U roślin musimy zrobić protoplastyzację, żeby zniszczyć ścianę komórkową.
Plazmid - kolisty fragment DNA pełniący funkcje chromosomów pomocniczych; służą jako wektory do klonowania DNA w bakteriach.
Komórki bakteryjne zawierają DNA koliste, które łatwiej przenika i łatwiej się łączy. Komórka bakteryjna nie przyjęłaby DNA liniowego.
Enzymy restrykcyjne - białka pochodzące z bakterii, które wykorzystują te enzymy, by bronić się przed obcym DNA. Enzymy restrykcyjne nie tną DNA gospodarza ponieważ jest ono zmetylowane. Restryktazy generują lepkie końce.
BamHI - rozpoznaje sekwencje G▼CCGC, tnie DNA tworząc lepkie końce
EcoRI - tnie DNA tworząc lepkie końce, 5'-G▼AATTC-3', tnie tworząc 3 fragmenty
HindIII - tnie tworząc 2 fragmenty
Metylazy - metylują w rozpoznawanej sekwencji 1 resztę DNA, dzięki czemu DNA bakteryjne nie jest cięte przez enzymy restrykcyjne
METYLACJA
U prokariota
U eukariota
u eukariotów i archeonów - ATP
u bakterii - NAD+
endA1 - mutacja genu endonukleazy A (endonukleaza A tnie DNA) specyficznej dla DNA, zwiększająca wydajność i jakość izolowanych plazmidów (chodzi o to, żeby nasze plazmidy nie były rozcinane)
lacIq - zdolność do nadprodukcji lac represora hamującego transkrypjcę z promotorów zawierających lac operator (bo nie chcemy, żeby komórka sama transkrybowała laktozę)
recA1 - niezdolność do rekombinacji DNA; zwiększa stabilność niektórych insertów podczas propagacji wektorów (chodzi o to, żeby nasze plazmidy nie były rozcinane)
TcR - obecność genu tet, nadającego oporność na terracyklinę (tertacyklina jest markerem selekcyjnym, nietranformowane komórki nie wyrosną na płytce gdy jest ona obecna w podłożu)
czystości DNA - zanieczyszczenia DNA wpływające na aktywność enzymu: białka, fenol, chloroform, etanol, EDTA, SDS, wysokie stężenie soli
temperatura i czas inkubacji
dobór buforu
za duże stężenie glicerolu
zbyt wysokie pH
jeden kofaktor (Mn jest zastąpiony innym)
za długa inkubacja
zła temperatura
za duża ilość enzymu lub substratu
zbyt niska siła jonowa
elektroforeza
termicznie - 80°C, 30 minut ← podstawowy sposób
chemicznie (EDTA, fenol, chloroform, EtOH)
wysiewamy pożądane DNA na bakterie
wycinami DNA z żelu
agaroza = galaktoza D + galaktoza L |+| ← |-| (bo DNA ma ładunek|-|)
rozdział molekuł w polu elektrycznym następuje na podstawie ich wielkości, kształtu i ładunku. DNA przemieszcza się w polu elektrycznym od ujemnej elektrody katody (-) do dodatniej elektrody anody (+).
-liniowe -> nie zostaną powielone przez bakterie, gdyż one mają DNA koliste
- koliste =>
- koliste superskręcone => musimy je oczyścić, bo zostaną powielone przez bakterie
długość cząsteczki DNA - im dłuższa tym wolniej migruje V=1/[log(bp)]
konformacja
bromek etydyny - wchodzi między pary zasad (interkaluje), wiec spowalania czasteczki liniowe
napięcie - im większe tym szybciej migrują, ale spada rozdzielczość
stężenie agarozy - im bardziej stężona tylko wolniej migrują, ale jest lepsza rozdzielczość
bufor (siła jonowa)
12%- żel dolny, rozdzielający
4% - żel górny, zagęszczający
APS - nadsiarczam amonu (NH4)2S2O8, ryboflawina
TEMED - N,N,N',N'-tetrametyloetylenodiamina;
HCl → jony Cl- migrują najszybciej
glicyna - mogruje wolno
metanol - rozpuszcza barwnik i wytrąca białka
kwas octowy - wiąże się z białkiem i utrwala je w żelu
Rodzaj metylacji |
Zasada metylowana |
Rozpoznawane sekwencje |
dam |
A |
GATC |
dcm |
C |
CCAGG / CCTGG |
dam - nie pokrywa miejsca metylacji
dcm - może pokrywać miejsce metylacji
Rodzaj metylacji |
Zasada metylowana |
Rozpoznawane sekwencje |
CpG |
C |
CpG |
Metylacja ta służy do wybierania DNA, które ma być czynne restrykcyjnie.
DNA niezmetylowane - ulega ekspresji
DNA zmetylowane - nieaktywne
Ligaza DNA - łączy końce DNA w regionach dwuniciowych. Enzym ten katalizuje tworzenie się wiązania fosfodiestrowego między grupą 3'-OH jednego końca nici DNA i grupą 5'-fosforanową na końcu drugiej nici. Do przeprowadzanie tej reakcji potrzebne jest źródło energii:
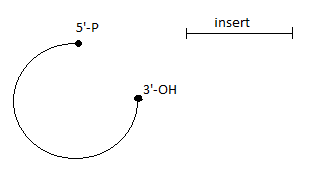
Znaczenie markerów genetycznych:
Operon laktozowy:
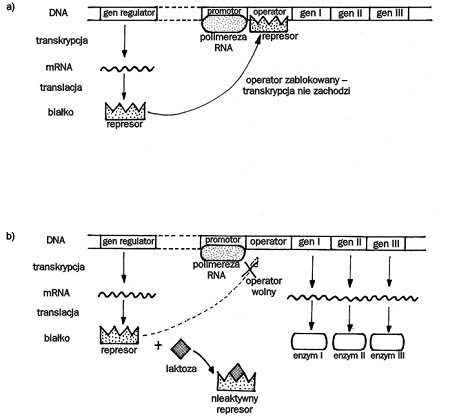
Enzym I - β-galaktozydaza
Enzym II - permeaza
Enzym III - acetylotransferaza
Aktywność enzymów restrykcyjnych zależy od:
* Mg2+ - niezbędne do aktywności enzymu
* Tris-HCl - utrzymanie odpowiedniego pH
*Kcl/NaCl - odpowiednia siła jonowa
*BSA - stabilizacja cząsteczek białkowych, zapobieganie adhezji enzymów do probówki
*Triton X-100 - detergent, stabilizacja białek
Aktywność gwiezdna - tzw. „rozluźniona” specyficzność enzymu będąca skutkiem nie optymalnych warunków, utrata specyficzności działania:
Metody inaktywacji enzymu:
Jednostka aktywności - taka ilość enzymu, która trawi całkowicie 1 μg charakterystycznego substratu testowego przez 1h w optymalnych warunkach podanych przez producenta.
Elektroforeza preparatywna - oczyszczenie pożądanego DNA z innych pasm DNA
Po ligacji:
1) DNA superskręcone → najbardziej niebezpieczne
2) wektor + insert
3) wektor bez insertu → niebezpieczne
4) DNA liniowe
Elektroforeza: DNA
FORMY DNA:
Elektroforeza jest techniką do rozdziału molekuł w polu elektrycznym na podstawie wielkości, kształtu i ładunku. DNA przemieszcza się od (-) katody do (+) anody.
Gdy mamy cząsteczki DNA podobnej długości- potrzebne większe stężenie agarozy.
Czynniki wpływające na ruchliwość DNA kolistego/liniowego w żelu agarozowym:
* koliste superskręcone - najszybciej
*koliste - średnia prędkość
* liniowe - najwolniej
* kolisty ujemnie superskręcony - bromek etydyny dodaje skręty dodatnie
7 (-) superskręconych → 7 (+) superskręconych prędkość nie zmienia się
1 (-) superskręconych → 3 (+) superskręconych prędkość zwiększa się
6 (-) superskręconych → 3 (+) superskręconych prędkość zmniejsza się
* liniowy
bromek etydyny wysłuża i usztywnia DNA, co zmniejsza prędkość
ELEKTROFOREZA SDS-PAGE → Elektroforeza w warunkach denaturujących (bo jest SDS)
SDS- Sodium Dodecyl Sulfate
żele:
MIX = akryloamid + bisakryloamid
Bufory: UPPER
LOWER
|
Górny [ml] |
Dolny [ml] |
MIX |
0,933 |
4,00 |
UPPER |
1,75 |
- |
LOWER |
- |
2,5 |
SDS 10% [μl] |
70 |
100 |
H2O |
4,205 |
3,340 |
Bufor Tris- pH=6,8
SDS- rozfałdowanie białek, nadanie im ładunku ujemnego (-)
β-merkaptoetanol - denaturuje wiązania disiarczkowe (dodaje się go, ponieważ po lizie komórek mogą powstać mostki disiarczkowe, które są niepożądane)
Glicerol - zagęszcza próbkę, stabilizuje białka
Błękit bromofenolowy - wiąże się z białkiem, zabarwia je (żeby było je widać)
Gdy nasze białko jest przed dodaniem SDS częściowo bardzo sfałdowane, denaturyzujemy je w 90°C.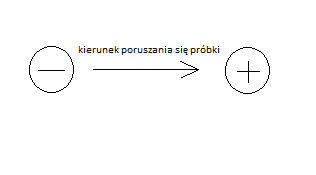
Polimeryzacja żelu poliakrylamidowego
T= ((masa akrylamidu [g] + masa bisakrylamidu [g]) / obj. Całkowita [ml]) * 100%
C= (masa bisakrylamidu / (masa akrylamidu [g] + masa bisakrylamidu [g]) ) * 100%
Układ nieciągły wg Laemlli'ego
Bufor do elektroforezy (przy elektrodach) pH=8,3, zawiera glizynę
Im mniejsza cząsteczka tym szybciej porusza się w żelu.
Skład żelu:
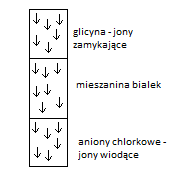
GRADIENT pH → pH rośnie, bo HCl wędruje w dół-glicyna zmienia stopień uprotonowania (glicyna z jonu obojnaczego przechodzi w jon glicynianowy), zaczyna migrować szybciej i „spycha” białka w dół „zbijając” je, żeby tworzyły cienką warstwę.
W żelu rozdzielającym glicyna zagęszcza, wyprzedza białka.
Wybarwianie żelu ( np. barwnikiem Coomassie)
Kolor białka zależy od jego pI (punktu izoelektrycznego) oraz od modyfikacji chemicznych np. glikozylacji czy fosforylacji.
EcRDBD - wiąze DNA, zawiera palce cynkowe, 8 cystein, brak wiązań disiarczkowych.
Enzymy restrykcyjne- Endonukleazy (enzymy należące do klasy hydrolaz, które działając na DNA i RNA doprowadzają do ich rozkładu do oligonukleotydów przez rozerwanie wiązań fosfodiestrowych wewnątrz łańcucha kwasu nukleinowego.) i Egzonukleazy (enzymy należące do klasy hydrolaz, które działając na jedno- lub dwuniciowe DNA i RNA powodują odłączenie nukleotydów od końców ich łańcuchów.)
3 literowy skrót od nazwy organizmu gospodarza, z którego go wyizolowano; oznaczenie szczepu lub typu, oraz numer enzymu (cyfra rzymska).
Enzymy restrykcyjne typu II:
- ponad 600 enzymów
-tną dsDNA wewnątrz rozpoznawalnej sekwencji lub określonej odległości od niej
-miejsce rozpoznawane 4-8 nukleotydow (4-6) PALIDRONOWOŚĆ
- 2 sposoby cięcia: - produkty o lepkich końcach (kohezyjność) i tępych (cięcie w osi symetrii)
IZOSCHIZOMERY- pochodzą z różnych organizmów, rozpoznają te same sekwencje, tną w obrębie sekwencji rozpoznawalnej lub w jej sąsiedztwie
NEOSCHIZOMERY - podgrupa izoschizomerów, rozpoznają te same sekwencje ale generują inne końce
IZOKAUDOMERY- enzymy rozpoznają różne sekwencje, ale generują te same końce
R7S- sól chaotropowa- związki, które zaburzają oddziaływania wodorowe, wchodzi w oddziaływania z krzemionką. Chlorowodorek guanidyny też tam jest- powoduje denaturację białek. Oczyszcza DNA.
PCR- reakcja łańcuchowa polimerazy. Metoda powielania łańcuchów DNA polegająca na łańcuchowej reakcji polimerazy DNA w wyniku wielokrotnego podgrzewania i oziębiania próbki, w warunkach laboratoryjnych. Do przeprowadzenia tej reakcji dodajemy:
1. parę starterów, które hybrydyzują z sekwencjami przylegającymi do sekwencji docelowych
2. trifosforany wszystkich czterech nukleozydów dNTP
3. termostabilną polimerazę DNA.
Trzy etapy PCR: - oddzielenie nici DNA (denaturacja) dwie nici oddzielają się przez ogrzewanie roztworu w temperaturze ok. 95stopni przez 15 sekund
- hybrydyzacja starterów. Roztwór jest gwałtownie chłodzony do temperatury 54stopni, aby umożliwić hybrydyzację starterów z nićmi DNA. Jeden starter hybrydyzuje z sekwencją przy końcu 3 sekwencji docelowej, drugi do końca 5 nici komplementarnej. Wyjściowa cząsteczka DNA nie ulega renaturacji do formy dwuniciowej, ponieważ startery są dodawane w nadmiarze.
- Synteza DNA (elongacja). Roztwór ogrzewa się do 72 stopni (temperatura optymalna dla polimerazy DNA Taq) Polimeraza wydłuża sekwencje od starterów w kierunku sekwencji docelowej, ponieważ synteza przebiega w kierunku 5->3. Synteza DNA zachodzi na obu niciach i rozciąga się poza sekwencję docelową.
Termostabilność polimerazy umożliwia przeprowadzenie reakcji w zamkniętych probówkach. Dupleksy są podgrzewane i rozpoczyna się drugi cykl reakcji, w wyniku którego powstają 4 cząsteczki dwuniciowe, po czym inicjowany jest trzeci cykl. Na końcu trzeciego cyklu pojawiają się dwie krótkie nici z sekwencjami docelowymi i starterowymi. Następne cykle prowadzą do powielenia cząsteczki wykładniczo, podczas gdy cząsteczki DNA są powielane liniowo. Sekwencja powielanego DNA nie musi być znana. Musimy znać tylko sekwencje przylegające do niego. Można powielać bardzo długie fragmenty. Wykrywa się całę rodziny genów, można izolować geny. Wykrywa się tym wirusy i bakterie, mutacje, monitoruje się przebiegi chemioterapii, porównuje DNA.
Polimeraza wykazuje 3 cechy aktywności:
-synteza DNA od końca 5->3
-egzonukleaza 5->3
-egzonukleaza 3->5
Polimeraza przeprowadza reakcje ataku nukleoiflowego.
5->3 nić zawierająca kodony (nić kodująca=sensowna)
3->5 nić niekodująca( antysensowna)
Do reakcji potrzebna jest:
-termostabilna polimeraza
-bufor dla polimerazy
- matryca (może być liniowe, koliste DNA)
- startery (zależy od nich ilość produktów, musza być w ilości równomolowej)
- dNTP (w nadmiarze)
Po 3 cyklu uzyskujemy pierwsze produkty o pożądanej długości. Temperatura hybrydyzacji nie może przekroczyć 72stopni, jak będzie za niska, to primer się nie przyłączy. Temperatura topnienia- tylko dla reszt parujących się z matrycą
TM=4*(G-C)+2*(A-T) Ta/n=Tm-3C- temp hybrydyzacji
Starter przedni paruje się z nicią matrycową i jest kompetentny do kodującej. Starter tylny paruje się z nicią matrycową i jest kompetentny do kodującej. Starter tylny paruje się z nicią kodującą i jest kompetentny do matrycy. Nie musimy używać 2 cyklów, gdy nie chcemy wprowadzać np. miejsc restrykcyjnych. „Gorący start”- powinien trwać 3-5minut. Liczymy temp. Hybrydyzacji i czas polimeryzacji(zależy od procesywności enzymu np. 1000bp/30s -> jeśli mamy do spolimeryzowania 1500bp zajmie nam to 45s+15% z tego czasu; z instrukcji Time6 jest suma Time 3 i Time 4).
Projektowanie starterów:
5' G/C |
m. rozpoznawane przez enzym restrykcyjny |
Dodatkowe nukleotydy uzgadniające ramkę odczytu |
Sekwencja 3' |
JEŻELI WYCHODZĄ NAM RÓŻNE TEMPERATURY TOPNIENIA DLA DWOCH STARTEROW TO BIERZEMY NIZSZA!
Enzym BamHi ma sekwencje: 5'GGATCC3'
3'CCTAGG5'
Wpisujemy to tak:
5'(6x ogon G/C) GGATCCNNstarter 3'
3'(6x ogon G/C)GGATCCNstarter5'
N- dowolony nukleotyd, wstawiamy aby uzyskac ramkę odczytu
6x ogon G/C- stwarza nam wieksza specyfike produktu
Ligacja- proces łączenia dwóch fragmentów lub końców nici DNA za pomocą ligazy. Proces ligacji wymaga energii w postaci ATP (eukariota) lub NAD (prokariota). Do ligacji potrzebujemy około 10-100ng wektora. Musi być zdefosforylowany- aby zapobiegać jego samoligacji. Temperatura optymalna dla ligazy- 37C. Temperatura ligacji nie może być zbyt wysoka bo DNA dąży ku rozdzieleniu nici, występuje przewaga pojedynczych i mogą się do siebie nie zbliżyć.
Wektor pGEX-2T = 4948 pz
Amp- gen kodujący oporność na ampicylinę
LacI- sterowanie ekspresją białka
Polilinker- miejsca cięć przez różne enzymy
Ptac- promotor
GST- metka, glutationo S-transferaza- pomaga oczyścić białko
PCR kolonijny- analiza bakterii lub drożdży po transformacji DNA. Szybko obrazuje plazmidy zawierające pożądany insert. Występuje początkowe podgrzanie w celu lizy komórek, metoda ta eliminuje konieczność robienia mikroanalizy przed jej zastosowaniem.
Zastosowanie:
- Określa obecność insertu w plazmidzie
- Dostarcza informacji na temat zarówno specyficzności oraz rozmiaru insertu, do określenia położenia insertu
Zalety:- łatwość wykonania oraz efektywność
Wady: - możliwość wystąpienia fałszywych pozytywnych wyników- resztki z hodowli lub niezligowane inserty.
Wyszukiwarka
Podobne podstrony:
II ekolo (1), Biotech PWr I stopien, Chemia fiz ćwiczenia
pytanianaegzamin, Biotech PWr I stopien, chemia fiz wykład Komorowski
IG notatki LAB, inżynieria genetyczna, laboratorium
pytania na inż. genetyczna, Biotechnologia PWR, Semestr 7, Inżynieria Genetyczna - Wykład, Inżynieri
genetyka notatki, Biotechnologia PWR, Semestr 2, Genetyka
Calka oznaczoxna, Biotechnologia PWR, Semestr 2, Analiza Matematyczna 2, Notatki
zadanie z pcr, Biotechnologia PWR, Semestr 7, Inżynieria Genetyczna - Laboratorium, Notatki
opracowane pytania MSI (1), Studia Zarządzanie PWR, Zarządzanie PWR I Stopień, V Semestr, Modelowani
Filtracja - sprawozdanie 1, Biotechnologia PWR, Semestr 7, Separacje i oczyszczanie bioproduktów - L
BIOLOGIA MOLEKULARNA Lista 3, Biotechnologia PWR, Semestr 5, Biologia Molekularna - Seminarium, List
Ćwiczenie 1 - oznaczanie stalej i stopnia dysocjacji, Biotechnologia PWR, Semestr 3, Chemia fizyczna
Biofizyka pytania z kola, Biotechnologia PWR, Semestr 5, Biofizyka - Wykład, Biofizyka - materiały
BIOFIZYKA- rozwiązania, Biotechnologia PWR, Semestr 5, Biofizyka - Wykład, Biofizyka - materiały
Chemia Ogólna - PROGRAM WPC1002w (Walkowiak), Biotechnologia PWR, Semestr 1, Chemia ogólna, Chemia o
TECHNIKA CYFROWA - sprawko lab 1, Studia, PWR, 4 semestr, Podstawy techniki mikroprocesorowej, labor
więcej podobnych podstron