Materiały do ćwiczeń laboratoryjnych z chemii - 2
Opracowali:
WODA W BUDOWNICTWIE
1. Wstęp
Woda stosowana w budownictwie do wytwarzania zaczynów, zapraw, betonów oraz mas ceramicznych jest nazywana wodą zarobową. Jako element środowiska naturalnego, woda oddziałuje na budowle, a tym samym ma wpływ na ich trwałość.
Pełni ona zasadniczą rolę w kształtowaniu właściwości technologicznych mieszanek, a także w procesach wiązania i twardnienia spoiw budowlanych.
Występujące w wodzie zarobowej związki mineralne i substancje organiczne wpływają na procesy wiązania. W przypadku hydratacji cementu substancje te mogą powodować obniżenie wytrzymałości betonu, występowanie plam na powierzchni betonu, a także doprowadzić do korozji zbrojenia w żelbecie.
Cząsteczka wody ma budowę polarną (jest dipolem), co jest powodem jej anomalnych właściwości.
Ulega dysocjacji elektrolitycznej, czyli rozpadowi na jony, według reakcji:
H2O = H+ + OH-
która jest pewnym uproszczeniem — w rzeczywistości woda rozpada się na jony OH- i H3O+ (jon oksoniowy).
Stała dysocjacji wody ma postać
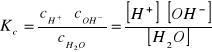
(1)
gdzie:
Kc — stała dysocjacji,
cH. — stężenie molowe jonów wodorowych,
cOH - — stężenie molowe jonów wodorotlenowych,
cH2O — stężenie molowe niezdysocjowanych cząsteczek wody.
W temperaturze 295 K stała dysocjacji wody Kc = 1,8-10-16 mol/dm3.
Oznacza to, że woda dysocjuje jedynie w minimalnym stopniu, a zatem można przyjąć, że stężenie cząsteczek niezdysocjowanych w przybliżeniu odpowiada całkowitemu stężeniu wody, czyli 55,56 mol/dm3.
Wartość ta wynika z prostego obliczenia:
jeden dm3 wody zawiera 1000 g wody,
jeden mol H2O = 18 g, zatem w jednym dm3 jest 1000g/18g/mol = 55,56 moli H2O.
Jak z tego wynika, iloczyn wartości stałej dysocjacji i mianownika (cH2O = 55,56 mol/dm3) we wzorze (1), jest też wielkością o stałej wartości, którą określa się jako iloczyn jonowy wody.
![]()
Ponieważ Kc = 1,8 • 10-16 i cH2O = 55,56 mol/dm3,
to iloczyn jonowy wody Kw równa się:
Kw = ch+ . coh- = 1,8∙10-16 . 55,56 = 10-14 (mol/dm3)2
Wartość tego iloczynu w roztworach wodnych (w określonej temperaturze) jest stała.
W praktyce dla określenia stężenia jonów wodorowych używa się pojęcia pH
pH = - logcH+ lub pOH = - logcOH-
gdzie cH+ — stężenie molowe jonów wodorowych w roztworze,
cOH - — stężenie molowe jonów wodorotlenowych.
Korzystając z tego wzoru oraz wartości iloczynu jonowego wody, można zawsze obliczyć pH roztworu oraz stężenie jednego rodzaju jonów znając stężenie drugiego, lub odwrotnie — znając pH obliczyć stężenia obu rodzajów jonów.
W zależności od stężenia jonów wodorowych odczyn środowiska można określić jako obojętny, zasadowy lub kwaśny. Odczyn obojętny występuje wtedy, gdy
cH+ = cOH-
Biorąc pod uwagę iloczyn jonowy wody łatwo obliczyć, że w takich warunkach stężenia obu rodzajów jonów wynoszą:
Ch+ = Coh- = (10-14)1/2 = 10-7 mol/dm3.
Jeśli występuje przewaga jonów OH-:
cOH- > 10-7 mol/dm3, tzn. pH > 7 i pOH < 7 i odczyn roztworu jest zasadowy,
jeżeli zaś w przewadze są jony H+:
cH+ > 10-7 mol/dm3, tzn. pOH > 7 i pH < 7 to odczyn środowiska jest kwaśny.
Wodę przed użyciem do zarabiania mieszanek należy poddać analizie chemicznej.
Wyjątek stanowi woda ze źródła objętego siecią wodociągową.
Składnikami, które w sposób zasadniczy wpływają na jakość wody zarobowej są:
jony siarczanowe (SO42-),
jony siarczkowe (S2-),
kwasy (jony wodorowe H+),
cukier,
substancje humusowe,
sole wapnia i magnezu, tzn. powodujące twardość wody.
SIARCZANY
jony siarczanowe reagują ze składnikami zaczynu cementowego, tworząc związki znacznie zwiększające swoją objętość i przez to mogą być przyczyną rozsadzania betonu.
Jeżeli agresja siarczanowa jest słaba, to tworzący się w reakcji z wodorotlenkiem wapnia gips (CaSO4.2H2O) — do pewnego stopnia — uszczelnia beton.
W przypadku dużego stężenia jonów siarczanowych w wodzie, gips reagując dalej, tworzy związki krystaliczne, z przyłączeniem dużej liczby cząsteczek wody, np. sól Candlota a w betonie nazywana etringitem: 3CaO.Al2O3.3CaSO4.32H2O, które powodują zniszczenie betonu.
SIARKOWODÓR I JEGO SOLE
Jony siarczkowe (S2-) ulegają stosunkowo łatwo reakcji utleniania i przechodzą w siarczynowe (SO32-), a następnie w siarczanowe (SO42-).
Szkodliwe działanie siarczanów zostało wyjaśnione powyżej.
siarkowodór może reagować również z wodorotlenkiem wapniowym tworząc Ca(HS)2 który jest łatwo rozpuszczalny w wodzie i nie wykazuje właściwości wiążących.
KWASY (JONY H+)
— są obecne w wodzie jako składniki naturalne, np.
kwas węglowy,
kwas huminowy,
produkty hydrolizy soli słabych kwasów lub słabych zasad.
— zanieczyszczona woda (ścieki) może zawierać również mocne kwasy mineralne,
jak solny
czy siarkowy.
Kwasy reagują ze składnikami cementu oraz z produktami jego uwodnienia tworząc łatwo rozpuszczalne związki.
Utrudnia to, a czasem uniemożliwia, wiązanie spoiwa.
CUKIER
jest związkiem organicznym zaliczanym do grupy węglowodanów. Głównym składnikiem cukru jest sacharoza (C12H22O11), która tworząc z wodorotlenkiem wapniowym cukrzany wapniowe utrudnia proces wiązania betonu i obniża jego wytrzymałość.
W niektórych przypadkach obecność cukru może całkowicie uniemożliwić wiązanie.
SUBSTANCJE HUMUSOWE
— występują głównie w gruntach, glebach bagiennych i torfowiskach. Powstają w wyniku rozkładu szczątków pochodzenia roślinnego i zwierzęcego.
Są to związki o charakterze kwasowym, np. kwas huminowy i charakteryzują się dużą masą cząsteczkową. Kwas ten reaguje z wodorotlenkiem wapniowym obecnym w zaczynie cementowym tworząc nierozpuszczalny huminian wapniowy.
Niebezpieczne działanie mogą wykazywać składniki towarzyszące kwasom humusowym, np. jony SO42- powstałe przez utlenianie pirytów.
Z tego względu stosowanie do betonu wody zawierającej zawiesiny jest niewskazane.
ZWIĄZKI POWODUJĄCE TWARDOŚĆ WODY
— określa zawartość w niej związków wapnia i magnezu w przeliczeniu na CaO ( stopnie twardości). Jednostką stosowaną do wyrażania twardości wody jest mmol/dm3.
Twardość 1 mmol/dm3 odpowiada obecności w wodzie 56 mg CaO lub równoważnej ilości 40 mg MgO.
Często stosowaną jednostką jest również stopień niemiecki (°n) i mval/dm3, przy czym 1 mmol/dm3 = 2 mval/dm3 = 5,6 °n.
Ze względu na rodzaj związków wapnia i magnezu występujących w wodzie rozróżnia się:
• twardość węglanową (przemijającą), wywołaną obecnością wodorowęglanów wapnia i magnezu. Twardość ta zanika w czasie ogrzewania wody, gdyż zawarte w niej wodorowęglany ulegają rozkładowi wg reakcji:
Ca(HCO3)2 = ↓CaCO3 + H2O + ↑CO2
Mg(HCO3)2 = ↓MgCO3+H2O+ ↑CO2
• twardość niewęglanową (trwałą), wywołaną obecnością innych soli wapniowych i magnezowych, np. chlorków, siarczanów, azotanów itp.
Całkowita twardość wody (twardość ogólna) jest sumą twardości węglanowej i niewęglanowej i określa całkowitą zawartość związków wapnia i magnezu w wodzie.
- Woda o zbyt dużej twardości utrudnia proces wiązania zaczynu cementowego, ponieważ zmniejsza rozpuszczalność składników cementu w wodzie, utrudnia proces przechodzenia do roztworu jonów wapniowych.
- Woda miękka natomiast jest przyczyną korozji ługującej betonu.
Wymagania techniczne dotyczące wody zarobowej
są określone w normie PN-88/B-32250 „Woda do betonów i zapraw".
W zależności od zastosowania rozróżnia się dwie odmiany:
I — woda do betonów, zapraw i zaczynów, w których jest spoiwo cementowe lub cementowo-wapienne,
II — woda do betonów i zapraw wykonywanych z innych niż w odmianie I spoiw mineralnych.
Tablica 1. Wymagania ogólne dotyczące wody zarobowej
wg PN-88/B-32250 ( norma nowa PN-EN 1008/2004)
Wymagania |
|
Barwa |
powinna odpowiadać barwie wody wodociągowej |
Zapach |
woda nie powinna wydzielać zapachu gnilnego |
Zawiesina |
woda nie powinna zawierać zawiesiny, np. grudek, kłaczków |
pH |
nie mniej niż 4 |
Tablica 2. Wymagania szczegółowe dotyczące wody zarobowej
wg PN-88/B-32250 ( norma zamiast PN-EN 206-1).
Zanieczyszczenia |
|
Odmiana wody |
|
|
I |
II |
|
Siarkowodór, mg/dm3, nie więcej niż |
20 |
+ |
- |
Siarczany, mg/dm3, nie więcej niż |
600 |
+ |
- |
Cukry, mg/dm3, nie więcej niż |
500 |
+ |
- |
Chlorki, mg/dm3, nie więcej niż |
400 |
+ |
- |
Twardość ogólna, mval/dm3, nie więcej niż |
10 |
+ |
+ |
Sucha pozostałość, mg/dm3, nie więcej niż |
1500 |
+ |
+ |
Obniżenie wytrzymałości zaprawy na zginanie lub ściskanie w stosunku do próbek zarobionych wodą destylowaną, % |
10 |
+ |
- |
Znaki + lub - oznaczają odpowiednio, że dana cecha dla danej odmiany wody jest lub nic jest wymagana |
|||
Rodzaj i stopień agresywności wody w stosunku do betonu w zależności od jej składu jest określony w normie PN-80/B-01800 „Klasyfikacja i określenie środowisk".
2. DYSOCJACJA ELEKTROLITYCZNA
Dysocjacja elektrolityczna jest to rozpad substancji na jony pod wpływem rozpuszczalnika. W takim procesie może zachodzić albo uwalnianie jonów z sieci krystalicznej albo rozpad cząsteczek polarnych (posiadających znaczny, trwały moment dipolowy) na jony.
Wiele substancji w stanie stałym ma budowę jonową. Należą do nich sole i niektóre mocne zasady, takie jak NaOH, KOH czy Ca(OH)2. W takich substancjach rozpuszczanie - szczególnie w rozpuszczalnikach polarnych, o wysokiej stałej dielektrycznej - polega na przechodzeniu do roztworu oddzielnych jonów. W tym przypadku dysocjacja polega na uwalnianiu jonów z sieci krystalicznej.
W związkach chemicznych, posiadających silnie spolaryzowane wiązania, takich jak np. kwasy organiczne (dotyczy wiązania pomiędzy atomami wodoru i tlenu w grupie karboksylowej -COOH) lub w większości kwasów nieorganicznych, pod wpływem polarnego rozpuszczalnika następuje rozrywanie cząsteczek i tworzenie oddzielnych jonów.
W przypadku amoniaku i zasad organicznych w czasie rozpuszczania w wodzie następuje przyłączanie jonów wodorowych do cząsteczek zasady i uwalnianie jonów wodorotlenkowych (pochodzących z wody). Taki proces - ściśle rzecz biorąc - jest jonizacją a nie dysocjacją, gdyż cząsteczki rozpuszczonego związku nie rozpadają się.
Ilościowo proces dysocjacji elektrolitycznej określają dwie wielkości: stopień dysocjacji i stała dysocjacji.
Stopień dysocjacji, oznaczany literą α, jest to stosunek liczby cząsteczek, które rozpadły się na jony nr (nie liczby jonów!) do całkowitej liczby cząsteczek danego związku wprowadzonych do roztworu nc. Wielkość tę można również wyrazić jako iloraz odpowiednich stężeń molowych:
![]()
(1.1)
Wartość stopnia dysocjacji podaje się często w procentach. Stopień dysocjacji zależy od stężenia substancji rozpuszczonej oraz od wielu innych czynników, takich, jak rodzaj rozpuszczonej substancji i rodzaj rozpuszczalnika, temperatura, obecność i stężenia innych substancji znajdujących się w roztworze. Stopień dysocjacji można określić na podstawie przewodności elektrycznej roztworu, jeśli w roztworze znajduje się tylko jedna substancja dysocjująca, lub na podstawie stężenia jednego z rodzajów jonów (na które rozpada się badana substancja) .
Stała dysocjacji jest to stała równowagi procesu dysocjacji słabego elektrolitu, określana jako stosunek iloczynu stężeń produktów dysocjacji (jonów danej substancji) do stężenia cząsteczek niezdysocjowanych.
KtnAnm ↔ nKtm+ + mAnn-
np. CH3COOH ↔ CH3COO- + H+
NH4OH ↔ NH4+ + OH-
Wyraża się ogólnym wzorem:
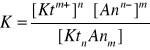
(1.2)
w którym Ktm+ i Ann- oznaczają odpowiednio kation i anion, KtnAnm - niezdysocjowaną cząsteczkę, a nawiasy kwadratowe - stężenia molowe substancji, których wzory znajdują się w nawiasach. Ze względu na bardzo szeroki zakres wartości stałej dysocjacji dla różnych substancji - często stosuje się funkcję K określoną jako pK i wyrażoną wzorem:
![]()
(1.3)
Dla elektrolitów jedno- wartościowych, to jest takich, których cząsteczka rozpada się na jeden kation jedno-dodatni i jeden anion jedno-ujemny istnieje prosta zależność pomiędzy stałą i stopniem dysocjacji:
NaCl = Na+ + Cl-
[Na+] = [Cl-] = c ∙α ; [ NaCl ] = c ∙ (1- α); ![]()
(1.4)
α - stopień dysocjacji, ilość cząstek zdysocjowanych do całkowitej ilości cząstek
zwana prawem rozcieńczeń Ostwalda.
Ponieważ K jest stałe, zatem α musi zmieniać się wraz ze stężeniem. Dokładne określenie tej zależności wymaga rozwiązania równania kwadratowego, jednakże w większości wypadków, gdy α « 1, można przyjąć, że 1 - α = 1, zatem ![]()
Na podstawie wartości stałej dysocjacji można następująco podzielić elektrolity:
Elektrolit |
K |
pK |
mocny |
>1 |
<0 |
średniej mocy |
1≥ K >10-3 |
0 ≤ pK < 3 |
słaby |
≤ 10-3 |
≥3 |
Dysocjują nie tylko substancje rozpuszczone, lecz także polarne rozpuszczalniki, np. woda.
Roztwory uważamy za rozcieńczone, jeżeli całkowite stężenie substancji rozpuszczonych nie przekracza l mol∙dm-3 ,
a dla substancji zdysocjowanych - l/z mol∙dm-3 (gdzie z - całkowity ładunek kationów powstających z jednej cząsteczki wyrażony w jednostkach elementarnych).
W czystej wodzie stężenia jonów wodorowych i wodorotlenkowych są jednakowe i wynoszą 10-7 mol∙dm-3 każde.
Zakresy wartości [H+], [OH-], pH i pOH dla roztworów kwaśnych, obojętnych i zasadowych zestawiono w tabeli 1.1
Tabela 1.1
Roztwór |
[H+] |
[OH-] |
pH |
pOH |
kwaśny |
l≥[H+]>10-7 |
10 7 > [OH-] ≥ 10-14 |
0 ≤ pH < 7 |
7 < pOH ≤ 14 |
obojętny |
[H +] = 10-7 |
[OH-] = 10-7 |
pH = 7 |
pOH = 7 |
zasadowy |
10 -7 > [H+] ≥ 10-14 |
1≥ [OH-]>10-7 |
7 < pH < 14 |
0 ≤ pOH < 7 |
Skala pH (podobnie jak pOH) obejmuje wartości od 0 do 14, przy czym
pH = - log [H+] (1.5)
pOH = - log [OH-] (1.6)
pH + pOH = 14 (1.7)
1.1. Obliczenia pH
W mocnym, całkowicie zdysocjowanym kwasie reakcja:
HzA → zH+ + Az- przebiega do końca. Stąd:
[H+] = cm ∙ z (1.1.1)
gdzie:
cm - stężenie molowe kwasu,
z - liczba jonów wodorowych oddysocjowanych z cząsteczki kwasu w procesie dysocjacji (zasadowość kwasu).
Analogicznie dla mocnych zasad:
Me(OH)z → Mez+ + z OH- (Me - metal)
[OH-] = cz ∙ z (1.1.2)
z tym, że tutaj z - liczba jonów wodorotlenkowych oddysocjowanych z cząsteczki zasady.
Słabe kwasy i zasady dysocjują tylko częściowo, dlatego do obliczania [H + ] i [OH-] należy stosować wzory na stałą dysocjacji. Dla słabego kwasu HA (gdzie A- - anion kwasu) stała dysocjacji Kk jest równa:
![]()
(1.1.3)
[HA] - stężenie niezdysocjowanego kwasu po ustaleniu się równowagi.
Jeżeli do roztworu oprócz słabego kwasu nie wprowadzono innych substancji dysocjujących na jony H+ lub A-, to
[H+] = [A-]
Stąd 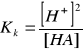
, zatem ![]()
W roztworach niezbyt rozcieńczonych dla większości słabych kwasów można przyjąć, że [HA] = cm (początkowe stężenie molowe kwasu).
Wówczas:
![]()
(1.1.4)
Analogicznie dla zasad
![]()
(1.1.5)
(indeks "k" oznacza kwas a "z" - zasadę).
Znając [H+] wartość pH dla kwasów oblicza się ze wzoru (1.5), natomiast
dla zasad znając [OH-] można najpierw obliczyć pOH ze wzoru (1.6), a następnie pH ze wzoru (1.7).
Przykład 1
Kwas azotowy(III) czyli azotawy dysocjuje zgodnie ze wzorem:
HNO2 ↔ H+ + NO2-
Stąd w ogólnym wzorze na stałą dysocjacji w miejsce [A-] podstawia się [NO2-], a zamiast [HA] podstawia się [HNO2] i otrzymuje:
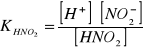
Znając wartość KHNO2 wynoszącą 4,5∙10-4 , oraz stężenie kwasu - np. 0,1 mol∙dm-3 można obliczyć ze wzoru (1.1.4) wartość [H+]:
![]()
Wartość pH wyniesie wówczas: — log 6,7∙10-3 =2,17.
2. PROCESY NA GRANICACH FAZ
2.1. ELEKTRYCZNA WARSTWA PODWÓJNA
Na granicy pomiędzy dwiema fazami ( np. metal zanurzony w roztworze wodnym) powstaje różnica potencjałów elektrycznych, wynikająca z nierównomiernego rozkładu ładunków elektrycznych. Takie rozmieszczenie ładunków nosi nazwę elektrycznej warstwy podwójnej. Występowanie różnic potencjałów na granicach faz jest w przyrodzie zjawiskiem powszechnym, a ich wielkość odgrywa istotną rolę w tak różnorodnych procesach jak np. tarcie, przenikanie substancji przez błony komórkowe w żywych organizmach, powstawanie napięcia w ogniwach galwanicznych.
Elektryczna warstwa podwójna utworzona jest z elektronów (od strony metalu) i równoważących ich ładunek jonów (od strony elektrolitu) oraz z obojętnych polarnych cząsteczek zaadsorbowanych na granicy faz.
W procesach elektrochemicznych nośniki ładunków elektrycznych (elektrony lub jony) muszą przechodzić z jednej fazy do drugiej przez podwójną warstwę elektryczną co związane jest z wykonaniem określonej pracy. Szybkość tych procesów jest więc uzależniona od budowy warstwy podwójnej i występującej w niej różnicy potencjałów.
2.2. ELEKTRODA I JEJ POTENCJAŁ
W elektrochemii rozpatruje się głównie wpływ różnorodnych czynników na napięcie na granicy faz układu przewodnik elektronowy/elektrolit. Układ taki nosi nazwę elektrody lub półogniwa, a wartość napięcia - potencjału elektrody. Określenie "elektroda" bywa również stosowane do samego przewodnika elektronowego, jednak dalej będzie stosowane jako synonim półogniwa.
Ogniwo galwaniczne jest to zatem połączenie dwóch elektrod przez jonowy kontakt ich elektrolitów.
Wiele metali po zanurzeniu do elektrolitu wykazuje tendencję do przechodzenia do roztworu w postaci jonów dodatnich pozostawiając elektrony w metalu, co można zapisać:
Me → Mez+ + z ∙ e-
natomiast jony tego metalu po zbliżeniu do granicy faz mają tendencję do redukcji przez pobranie elektronów z metalu:
Mez+ + z ∙ e- →Me
Szybkości tych dwóch procesów są początkowo różne. Jeśli przeważa wysyłanie jonów z metalu, to metal ładuje się ujemnie względem elektrolitu (metale nieszlachetne np. cynk).
Nadmiar ładunków ujemnych powoduje przyciąganie jonów dodatnich, co spowalnia ich wysyłanie przez metal, a przyspiesza przechodzenie z roztworu do sieci krystalicznej metalu. W przeciwnym wypadku, gdy jony szybciej przechodzą z elektrolitu do metalu (metale szlachetne np. miedź) - metal ładuje się dodatnio względem elektrolitu odpychając jony dodatnie i stopniowo zmniejszając szybkość tego procesu a przyspieszając proces przeciwny. Następuje zatem hamowanie szybszego procesu i przyspieszanie wolniejszego aż do wyrównania szybkości obydwu procesów czyli uzyskania stanu równowagowego, w którym występuje stała różnica potencjałów między metalem i elektrolitem zwana potencjałem równowagowym elektrody. Trzeba jednak podkreślić, że nie można zmierzyć bezwzględnej wartości potencjału elektrody, gdyż przy pomiarze należy jeden przewód woltomierza połączyć z metalem elektrody, a drugi zanurzyć do elektrolitu. Stanowiłby on łącznie z elektrolitem drugą elektrodę, a więc pomiar obejmowałby napięcia metal/elektrolit na dwóch elektrodach, a nie na jednej.
Z tego powodu wprowadzono elektrodę wzorcową, której potencjał w każdej temperaturze umownie przyjęto za równy zero. Jest to standardowa (inaczej - normalna) elektroda wodorowa.
STANDARDOWA ELEKTRODA WODOROWA zbudowana jest następująco: drut lub blacha platynowa, pokryta bardzo rozdrobnioną platyną (tak zwaną czernią platynową), zanurzona jest do roztworu kwasu siarkowego o stężeniu 1 mol∙dm-3 (aH+ = 1) i omywana strumieniem wodoru pod ciśnieniem jednej atmosfery (101325 Pa).
EoH ≡ 0
Potencjał elektrody (względny) definiuje się zatem jako napięcie równowagowe ogniwa zbudowanego z badanej elektrody i standardowej elektrody wodorowej. Wartość potencjału elektrody określa wzór Nernsta:
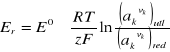
(2.2.1)
gdzie:
„+” - w przypadku stężeń jonów dodatnich (kationów)
„−” - w przypadku stężeń jonów ujemnych (anionów)
Er - równowagowy potencjał elektrody,
E° - standardowy potencjał elektrody mierzony w temperaturze 298 K, gdy aktywności
(patrz dalej) wszystkich reagujących substancji są równe jedności,
R - stała gazowa,
T - temperatura,
z - liczba elektronów biorących udział w elementarnej reakcji elektrodowej (np. liczba elektronów pobieranych przez jeden jon metalu przy jego redukcji do obojętnego atomu, równa ładunkowi jonu),
F - stała Faraday'a,
ak - aktywność składnika k, która dla czystych substancji stałych i ciekłych jest równa jedności;
w gazie - ciśnieniu parcjalnemu składnika k wyrażonemu w atmosferach;
a w roztworze - w przybliżeniu równa molowemu stężeniu tego składnika.
Dla rozcieńczonych roztworów wodnych aktywność wody przyjmuje się jako a H2O = 1,
vk - liczba stechiometryczna określająca liczbę jonów lub cząsteczek k-tej substancji, biorących udział w elementarnej reakcji elektrodowej;
indeksy "utl" i "red" oznaczają, że wyrażenie w nawiasie dotyczy wszystkich składników biorących udział w reakcji elektrodowej po tej stronie, gdzie występuje odpowiednio forma utleniona lub zredukowana składnika zmieniającego stopień utlenienia.
Wzór Nernsta przybiera znacznie prostszą postać, jeśli podstawić wartości liczbowe stałych oraz zależność między logarytmem naturalnym i dziesiętnym. Dla T = 298 K
![]()
(2.2.2)
Ponadto, w większości wypadków, zamiast ilorazów stężeń k składników występują stężenia pojedynczych substancji. Dla elektrody, w której metal wymienia jony z roztworem zgodnie z reakcją:
Me ↔ Mez+ + z ∙ e-
wzór ten można zapisać:
![]()
(2.2.3)
Podany w nawiasie zapis Mez+ / Me oznacza elektrodę zbudowaną z metalu Me i roztworu zawierającego jony Mez+ ( [Me] - stężenie formy zredukowanej =1)
2.3. PODZIAŁ ELEKTROD
Elektrody można podzielić na:
elektrody jonowymienne - gdy w procesie elektrodowym przez granicę faz przewodnik elektronowy /elektrolit przechodzą jony,
elektrody redoks - gdy przez tę granicę faz przechodzą elektrony. Trzeba podkreślić, że elektrony po przejściu do elektrolitu natychmiast łączą się z substancjami ulegającymi redukcji.
Elektrody jonowymienne dzielą się na:
elektrody pierwszego rodzaju, w których metal elektrody pozostaje w równowadze z jego jonami pochodzącymi ze zdysocjowanej soli tego metalu,
elektrody drugiego rodzaju, w których metal jest pokryty trudno rozpuszczalną solą tego metalu w roztworze zawierającym dowolną sól o anionie wspólnym z solą trudno rozpuszczalną.
Wśród elektrod redoks szczególny rodzaj stanowią elektrody gazowe, w których przewodnik elektronowy zanurzony jest w nasyconym gazem roztworze zawierającym jony powstałe w wyniku redukcji lub utleniania tego gazu.
2.4. WAŻNIEJSZE RODZAJE ELEKTROD I ICH ZASTOSOWANIE
Z uwagi na szybkość ustalania się równowagi reakcji elektrodowej tylko nieliczne elektrody pierwszego rodzaju, takie jak np. srebrowe, rtęciowe, miedziowe, kadmowe czy cynkowe mogą służyć do pomiarów stężeń kationów pozostających w równowadze z metalem elektrody jako tzw. elektrody wskaźnikowe.
Potencjały standardowe niektórych elektrod jonowymiennych pierwszego rodzaju zestawiono poniżej:
Elektroda |
Li+/Li |
Ca2+/Ca |
Na+/Na |
Mg2+/Mg |
A13+/A1 |
Zn2+/Zn |
E°,V |
-3,00 |
-2,84 |
-2,71 |
-2,38 |
-1,66 |
-0,76 |
Elektroda |
Cr3+/Cr |
Fe2+/Fe |
Cd2+/Cd |
Ni2+/Ni |
Sn2+/Sn |
Pb2+/Pb |
E°,V |
-0,71 |
-0,44 |
-0,40 |
-0,24 |
-0,14 |
-0,13 |
Elektroda |
H+/H |
Cu2+/Cu |
Hg22+/Hg |
Ag+/Ag |
Pt2+/Pt |
Au3+/Au |
E°,V |
0,00 |
+0,34 |
+0,79 |
+0,80 |
+0,2 |
+1,42 |
Szereg metali ustawionych w kolejności wzrastających wartości potencjałów standardowych zbudowanych z nich elektrod tworzy tzw. szereg napięciowy metali. Każdy z metali, jak również umieszczony w tym szeregu wodór, może wypierać z roztworu metale, którym odpowiadają elektrody o bardziej dodatnich (mniej ujemnych) potencjałach standardowych.
Elektrody drugiego rodzaju mogą służyć do pomiaru stężeń różnych anionów, najczęściej jednak - przy odpowiedniej konstrukcji - stosowane są jako elektrody porównawcze o stałym potencjale. Najważniejsze z nich to elektroda chlorosrebrowa i elektroda kalomelowa.
Elektrodę kalomelową najczęściej stosuje się jako elektrodę porównawczą, zawierającą nasycony roztwór KCl. Wówczas jej potencjał równowagowy w 298° K wynosi 0,2415V.
Elektrody redoks składają się zwykle z blaszki lub drutu platynowego i roztworu zawierającego jakiś pierwiastek na dwóch stopniach utlenienia, np. jony żelaza(II) i jony żelaza(III).
Potencjał elektrody redoks dla przykładowo podanej elektrody wyraża się wzorem:
![]()
Wśród elektrod gazowych największe znaczenie ma elektroda wodorowa.
Zbudowana jest z platyny omywanej strumieniem wodoru i roztworu wodnego, w którym zawsze znajdują się jony wodorowe. Na powierzchni platyny ustala się równowaga dla reakcji:
H+ + e- ↔ 1/2H2(g)
Potencjał tej elektrody wyraża wzór:
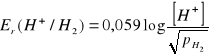
(2.4.3)
We wzorze na potencjał tej elektrody nie występuje potencjał standardowy, gdyż, jak podano uprzednio, jest on przyjęty za równy zeru. Jeśli ciśnienie wodoru omywający platynę jest równe ciśnieniu atmosferycznemu wówczas wzór przybiera jeszcze prostszą postać:
Er(H+/H2) = 0,059 log[H+] = -0,059pH (2.4.4)
Używana bywa do badań naukowych w wypadku niektórych pomiarów pH, szczególnie w roztworach silnie zasadowych, oraz do dokładnego cechowania innych elektrod.
Potencjały wielu rodzajów elektrod również zależą od stężenia jonów wodorowych. Powszechne zastosowanie do pomiarów pH jako elektroda wskaźnikowa znalazła elektroda szklana. Najważniejszą częścią elektrody szklanej jest bardzo cienka membrana, najczęściej w kształcie bańki, wykonana ze specjalnego gatunku szkła, przytopiona do grubszej rurki. W bańce znajduje się roztwór o stałym składzie i elektroda porównawcza - zwykle chlorosrebrowa. Na każdej stronie membrany szklanej powstaje potencjał liniowo zależny od pH roztworu kontaktującego z jej powierzchnią. Wewnętrzna część membrany oraz elektroda porównawcza mają stałe potencjały, natomiast zewnętrzna powierzchnia membrany uzyskuje potencjał zależny od pH badanego roztworu. Potencjał elektrody szklanej rozpatrywany jako różnica potencjałów pomiędzy metalem wewnętrznej elektrody porównawczej i roztworem na zewnątrz bańki szklanej wyraża się wzorem:
Eszkl. = E°szkl. - 0,059pH (2.4.5)
E°szkl. oznacza tu potencjał elektrody szklanej w roztworze o pH = 0 ( stężenie jonów wodorowych równa się 1).
2.5 PEHAMETRIA
Pehametria jest to zespół metod pomiaru wartości pH. W praktyce pH oznacza się albo metodami kolorymetrycznymi, albo potencjometrycznymi.
W metodach kolorymetrycznych stosuje się wskaźniki, które zmieniają barwę ze zmianą pH w zakresie około dwóch jednostek pH. Są to słabe, organiczne kwasy lub zasady, w których forma zdysocjowana ma inne zabarwienie od niezdysocjowanej. Zależnie od rodzaju wskaźnika zakres zmian barwy leży w różnych miejscach skali pH. Wskaźniki używa się albo w formie roztworu albo papierków wskaźnikowych, to jest nasyconych wskaźnikiem pasków bibuły, które zwilża się badanym roztworem i porównuje ze skalą barwną dołączaną do opakowania papierków. Niekiedy stosuje się mieszaniny wskaźników dla rozszerzenia zakresu pomiarowego. Dokładność takich pomiarów jest rzędu 0,2 jednostki pH.
W metodach potencjometrycznych wartość pH określa się najczęściej przy użyciu ogniwa składającego się z elektrod szklanej (wskaźnikowej) i kalomelowej (porównawczej). Pomiaru dokonuje się przy użyciu czułych miliwoltomierzy ze skalą w jednostkach pH, zwanych pehametrami. Przed pomiarem pehametr należy wycechować przy użyciu roztworu buforowego o dokładnie znanej wartości pH. Dokładność uzyskanych wyników może sięgać nawet 0,01 jednostki pH.
Pomiary pH służą do określania odczynu roztworu, do oznaczeń zawartości (lub stężenia) kwasów, zasad i niektórych soli w procesie miareczkowania pehametrycznego oraz do wyznaczania stałych dysocjacji słabych kwasów i zasad.
PRZEWODNOŚĆ WODY
Wskaźnikiem zawartości soli rozpuszczonych w wodzie jest jej przewodność. Przewodność właściwa (k) jest to odwrotność rezystancji słupa cieczy, zawartego pomiędzy elektrodami o powierzchni lcm3 w odległości l cm. W praktyce ze względu na trudności w wykonaniu naczyńka pomiarowego lub elektrody zanurzeniowej o dokładnie wymaganych wymiarach (S = lcm2, l = lcm) stosuje się układy pomiarowe o wymiarach zbliżonych;, które kalibruje się we wzorcowych roztworach (KCl). W wyniku kalibracji otrzymuje się współczynnik „k" zwany stałą naczyńka, który spełnia równanie:
K = k • Λ [μS∙ cm-1 ]
gdzie: Λ - przewodność badanej wody [μS] (mikrosimensy)
Pomiaru przewodności „dokonuje się przez przyłożenie do elektrod niewielkiego napięcia sinusoidalnego i wyznaczenie natężenia prądu płynącego przez roztwór. Natężenie to po wzmocnieniu, detekcji i kompensacji jest przekształcane w sygnał napięciowy proporcjonalny do mierzonej przewodności. Wielkość napięcia odczytuje się z miernika wyskalowanego w jednostkach przewodności. Woda destylowana używana w laboratorium powinna mieć przewodność w zakresie od kilku od kilkunastu mikrosimensów.
4. TWARDOŚĆ WODY
Właściwość wody zwana twardością wywołana jest obecnością rozpuszczonych w niej soli wapnia i magnezu oraz innych śladowych kationów wielowartościowych.
Właściwość tę można rozpatrywać na kilka sposobów.
Twardość całkowitą (ogólną) wody klasyfikuje się:
wg kationów: Tw.og = twardość wapniowa + twardość magnezowa.
lub wg anionów: Tw.og = twardość węglanowa + twardość niewęglanowa (stała)
Twardość wapniową powodują rozpuszczone w wodzie sole wapnia(II) i składa się ona z twardości węglanowej tj. Ca(HCO3)2 , CaCO3 , Ca(OH)2 i niewęglanowej, zwanej też twardością stałą np. CaCl2 , CaSO4 , CaSiO3
Twardość magnezową powodują analogicznie związki magnezu(II), które występują w wodzie w mniejszych ilościach niż wapnia.
Twardość węglanowa zwana jest niekiedy jako przemijająca, ze względu na rozkład wodorowęglanów podczas ogrzewania wody:
Ca(HCO3)2 −ogrzewanie → CaCO3↓ + H2O + CO2↑
Mg(HCO3)2 −ogrzewanie → MgCO3↓ + H2O + CO2↑
MgCO3 + H2O−ogrzewanie → Mg(OH)2↓ + CO2↑
Powstałe w wyniku tych reakcji osady są głównymi składnikami kamienia kotłowego.
Do ilościowego oznaczania twardości wody (tradycyjnie stosuje się tzw. milivale [mval∙dm-3]. Za jednostkę przyjęto twardość wody, jaką nadaje połowa milimola jonów wapnia lub magnezu w ldm3 wody.
W Polsce bywają jeszcze czasami używane tzw. stopnie niemieckie 1°niemiecki =10 mg CaO w l dm3 wody. (zawartość soli wapnia i magnezu przelicza się na tlenek wapnia).
Wykonanie ćwiczenia
Włączyć pH-metr . Do naczyńka pomiarowego z tworzywa wlać do 1/3 wysokości roztwór buforowy o pH = 4,0. Przycisnąć metalowy uchwyt i ostrożnie wyjąć elektrodę pomiarową z kolbki stożkowej. Opłukać ja wodą destylowaną z tryskawki podstawiając pod spód zlewkę. Następnie zanurzyć ją do naczyńka z roztworem buforowym i sprawdzić czy wyświetlacz wskazuje właściwe pH. Ewentualną różnicę skorygować potencjometrem „bufor” z dokładnością 0,05 jednostki. Po zakończeniu skalowania roztwór buforowy wlać z powrotem do butelki a elektrodę i naczyńko pomiarowe starannie przepłukać wodą z tryskawki.
Zmierzyć pH, przewodnictwo właściwe i twardość wody wodociągowej wlewając do naczyńka dla każdego pomiaru nową jej porcję. Pomiar zakończyć gdy wyniki trzech kolejnych próbek różnią się nie więcej niż 0,05 jednostki. Czas pomiaru każdej próbki ograniczyć do około 0.5 minuty. Twardość zmierzyć papierkami wskaźnikowymi.
Zmierzyć pH, przewodnictwo właściwe i twardość wody budowlanej postępując jak wyżej.
Z otrzymanych roztworów kwasu i soli sporządzić roztwory buforowe odmierzając starannie do małych kolbek stożkowych przy pomocy dwóch pipet o pojemności 5 cm3 (oddzielnej dla kwasu i soli) następujące objętości:
Kolbka Nr. |
Roztwór kwasu |
Roztwór soli |
I |
5 cm (jedna pipeta) |
20 cm3 (cztery pipety) |
II |
10 cm3 (dwie pipety) 10 cm (dwie pipety) |
10 cm3 (dwie pipety) |
III |
20 cm3 (cztery pipety) |
5 cm3 (jedna pipeta) |
Roztwory starannie wymieszać. Wlać niewielką porcję cieczy do naczyńka pomiarowego i przez zanurzenie przepłukać w niej elektrodę. Wylać zużyty roztwór, wlać do naczyńka jego nową porcję i zmierzyć pH.
Obliczyć stałą dysocjacji oddzielnie dla każdego roztworu posługując się wzorem:
![]()
gdzie: [![]()
] - stężenie soli, [HA] - stężenie kwasu.
Z otrzymanych wyników obliczyć średnia arytmetyczną i wpisać ją do tabelki sprawozdania.
Przykład obliczenia:
W kolbce I zmieszano jedna porcje kwasu i cztery porcje jego soli.
Stosunek stężenia soli do kwasu wynosi: ![]()
(zastanów się dlaczego?)
Z pomiaru pH tej mieszaniny otrzymano pH = 5,6
Z definicji pH = -log[H+]
Podstawia się w miejsce pH dane z pomiaru: -log[H+] = 5,6; log[H+] = - 5,6 czyli [H+] = 10-5,6
Obliczamy [H+] przy pomocy kalkulatora posiadającego funkcje 10x gdzie x = -5,6
Stężenie jonów wodorowych w kolbce I wynosi:
[H+] = 2,5∙10-6 stąd KI = 4 ∙ 2,5∙10-6
Podobnie oblicza się wartości KII i KIII dla roztworów z kolby II i III.
Średnią wartość oblicza się ze wzoru:
![]()
1
Wyszukiwarka
Podobne podstrony:
Przepisy porządkowe na ćw.lab, Ratownictwo Medyczne Studia, Giełda, 1. rok, Chemia ogólna, Biochemia
chemia-cwicz -lab -cz 3 1-cynkowanie, Chemia
MSIB Instrukcja do Cw Lab krystalizacja
Ćw lab nr 4 zagęszczalność gruntów
cw 3 lab, Imir imim, Semestr 3, Technologie wytwarzania
Materialy do cw lab biochemia
tematyka cw lab
cw lab 1
cw lab nr 5 schemat potencjalny sieci went k2
Cw lab pyt, 1. Stopień zawilżenia X powietrza kopalnianego wynosi 22 g/kg. Jaka będzie wilgotność wz
cw 2 lab, Imir imim, Semestr 3, Technologie wytwarzania
MiBM Reg. i wyk. ćw. Lab 2013 stacjonarne
HAR CW LAB EZ
Konspekt do cw. lab.-termowizja, Energetyka Politechnika Krakowska Wydział Mechaniczny I stopień, Mi
więcej podobnych podstron