43664 ksi ¬ki studia 9
pokolenie
pierwsze
pokolenie
drugie
pokolenie
trzecie
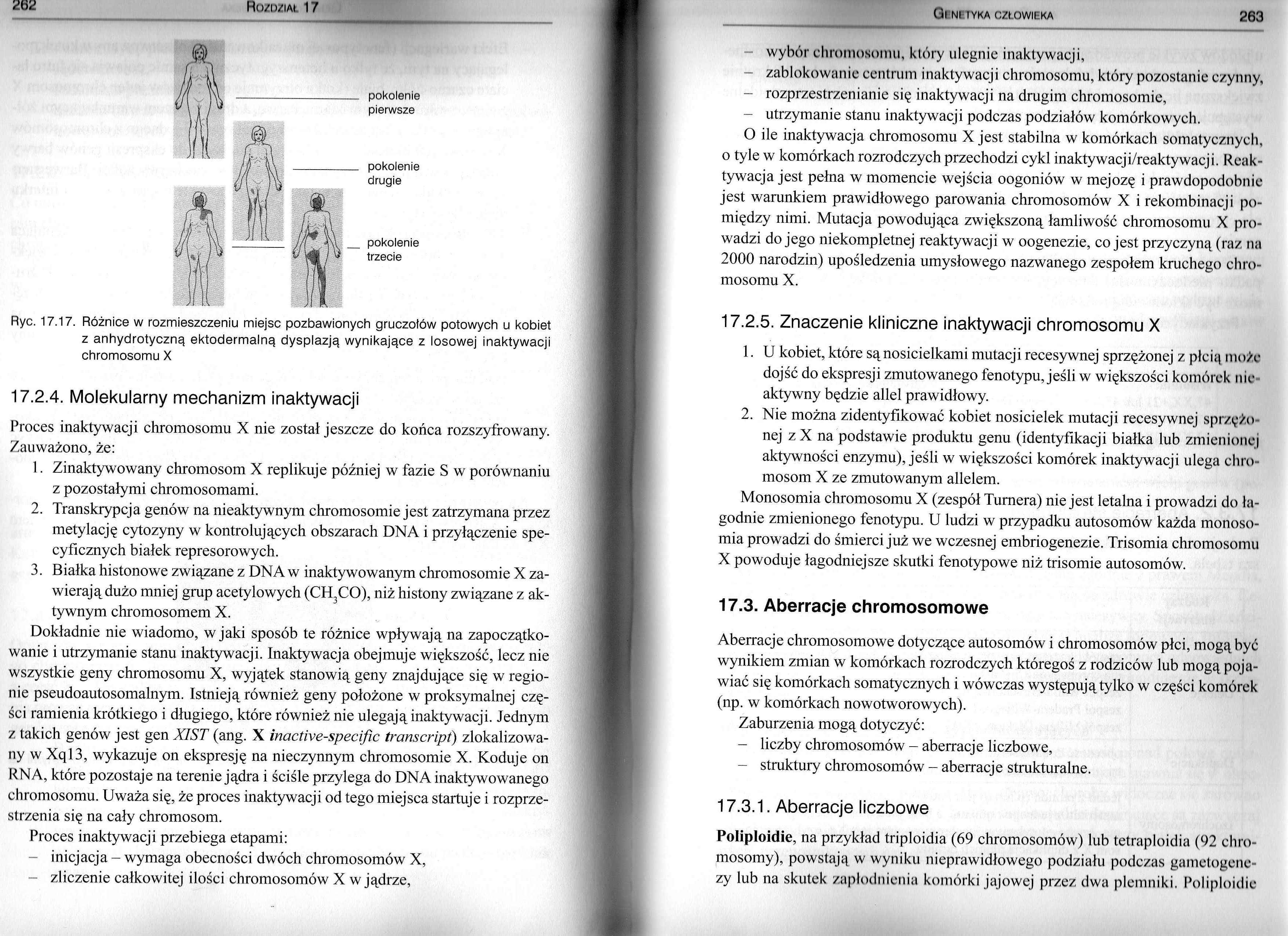
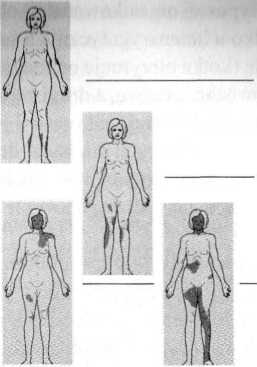
Ryc. 17.17. Różnice w rozmieszczeniu miejsc pozbawionych gruczołów potowych u kobiet z anhydrotyczną ektodermalną dysplazją wynikające z losowej inaktywacji chromosomu X
17.2.4. Molekularny mechanizm inaktywacji
Proces inaktywacji chromosomu X nie został jeszcze do końca rozszyfrowany. Zauważono, że:
1. Zinaktywowany chromosom X replikuje później w fazie S w porównaniu z pozostałymi chromosomami.
2. Transkrypcja genów na nieaktywnym chromosomie jest zatrzymana przez metylację cytozyny w kontrolujących obszarach DNA i przyłączenie specyficznych białek represorowych.
3. Białka histonowe związane z DNA w inaktywowanym chromosomie X zawierają dużo mniej grup acetylowych (CH3CO), niż histony związane z aktywnym chromosomem X.
Dokładnie nie wiadomo, w jaki sposób te różnice wpływają na zapoczątkowanie i utrzymanie stanu inaktywacji. Inaktywacja obejmuje większość, lecz nie wszystkie geny chromosomu X, wyjątek stanowią geny znajdujące się w regionie pseudoautosomalnym. Istnieją również geny położone w proksymalnej części ramienia krótkiego i długiego, które również nie ulegają inaktywacji. Jednym z takich genów jest gen XIST (ang. X inactive-specific transcript) zlokalizowany w Xql3, wykazuje on ekspresję na nieczynnym chromosomie X. Koduje on RN A, które pozostaje na terenie jądra i ściśle przylega do DNA inaktywowanego chromosomu. Uważa się, że proces inaktywacji od tego miejsca startuje i rozprzestrzenia się na cały chromosom.
Proces inaktywacji przebiega etapami:
- inicjacja - wymaga obecności dwóch chromosomów X,
- zliczenie całkowitej ilości chromosomów X w jądrze,
- wybór chromosomu, który ulegnie inaktywacji,
- zablokowanie centrum inaktywacji chromosomu, który pozostanie czynny,
- rozprzestrzenianie się inaktywacji na drugim chromosomie,
- utrzymanie stanu inaktywacji podczas podziałów komórkowych.
O ile inaktywacja chromosomu X jest stabilna w komórkach somatycznych, o tyle w komórkach rozrodczych przechodzi cykl inaktywacji/reaktywacji. Reaktywacja jest pełna w momencie wejścia oogoniów w mejozę i prawdopodobnie jest warunkiem prawidłowego parowania chromosomów X i rekombinacji pomiędzy nimi. Mutacja powodująca zwiększoną łamliwość chromosomu X prowadzi do jego niekompletnej reaktywacji w oogenezie, co jest przyczyną (raz na 2000 narodzin) upośledzenia umysłowego nazwanego zespołem kruchego chromosomu X.
17.2.5. Znaczenie kliniczne inaktywacji chromosomu X
1. U kobiet, które są nosicielkami mutacji recesywnej sprzężonej z płcu\ może dojść do ekspresji zmutowanego fenotypu, jeśli w większości komórek nie aktywny będzie allel prawidłowy.
2. Nie można zidentyfikować kobiet nosicielek mutacji recesywnej sprzężonej z X na podstawie produktu genu (identyfikacji białka lub zmienionej aktywności enzymu), jeśli w większości komórek inaktywacji ulega chromosom X ze zmutowanym allelem.
Monosomia chromosomu X (zespół Turnera) nie jest letalna i prowadzi do łagodnie zmienionego fenotypu. U ludzi w przypadku autosomów każda monosomia prowadzi do śmierci już we wczesnej embriogenezie. Trisomia chromosomu X powoduje łagodniejsze skutki fenotypowe niż trisomie autosomów.
17.3. Aberracje chromosomowe
Aberracje chromosomowe dotyczące autosomów i chromosomów płci, mogą być wynikiem zmian w komórkach rozrodczych któregoś z rodziców lub mogą pojawiać się komórkach somatycznych i wówczas występujątylko w części komórek (np. w komórkach nowotworowych).
Zaburzenia mogą dotyczyć:
- liczby chromosomów - aberracje liczbowe,
- struktury chromosomów - aberracje strukturalne.
17.3.1. Aberracje liczbowe
Poliploidie, na przykład triploidia (69 chromosomów) lub tetraploidia (92 chromosomy), powstają w wyniku nieprawidłowego podziału podczas gametogene-zy lub na skutek zapłodnienia komórki jajowej przez dwa plemniki. Poliploidie
Wyszukiwarka
Podobne podstrony:
ksi ¬ki studia 5 294 Rozdział 18 genów i genotypów nie zmieni się w następnych pokoleniach, o ile ni
ksi ¬ki studia&8 280 Rozdział 17 ! Najważniejsze wydarzenia podczas realizacji Projektu Badaniu
ksi ¬ki studia&9 282 Rozdział 17Wybrane informacje o zsekwencjonowanych chromosomach przedstawia pon
ksi ¬ki studia 2 288 Rozdział 18 Prawdopodobieństwo wystąpienia zjawisk wykluczających się jest sumą
więcej podobnych podstron