73794 str042 (4)

82 Ćwiczenie nr 10
W instalacjach destylacji ropy głównymi aparatami są piece rurowe i kolumny rektyfikacyjne zwane wieżami. Stąd używa się potocznego określenia: instalacja destylacji rurowo-wieżowej (DRW).
2.3. EKSTRAKCJA PERIODYCZNA CIECZY I CIAŁ STAŁYCH (ROZDZIELANIE SUBSTANCJI)
Ekstrakcja stanowi usuwanie jednego lub kilku składników z roztworu lub z ciała stałego za pomocą tzw. ekstrahenta tj. rozpuszczalnika selektywnego, w którym rozpusz-czają się w idealnym przypadku tylko składniki przeznaczone do usunięcia.
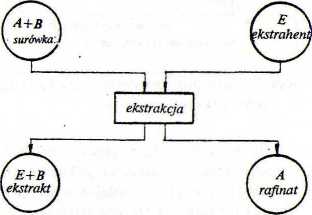
Zaleta ekstrakcji w porównaniu z innymi sposobami rozdzielania jest możliwość prowadzenia procesu w niskiej temperaturze. Aparatura jest stosunkowo prosta i daje się iatwo zautomatyzować. Operację ekstrakcji z udziałem trzech składników A, B, E ilustruje schemat na rys. 4.
Przyjęte oznaczenia: A - rozpuszczalnik pierwotny,
B - składnik, który należy usunąć z surowca,
E - rozpuszczalnik selektywny, będący ekstrahentem.
Rys. 4. Ideowy schemat ekstrakcji.
Przykłąd jest wyidealizowany gdyż zakłada wzajemna nierozpuszczalność rozpuszczalnika pierwotnego i ekstrahenta oraz całkowity rozdział rozpuszczalnika pierwotnego i składnika ekstrahowanego (dyfuzyjnie czynnego).
Rozdzielenie substancji między dwie fazy określa prawo podziału Nernsta. Według niego, jeśli do układu dwóch nie mieszających się lub słabo mieszających się cieczy tworzących dwie warstwy doda się pewną ilość substancji trzeciej rozpuszczalnej w obu cieczach to nastąpi podział wówczas podział tej substancji między dwie ciecze w taki sposób, że stosunek stężenia w jednym rozpuszczalniku do stężenia w drugim rozpuszczalniku jest wielkością stałą w stałej temperaturze co wyraża się zależnością:
= const = K
(współczynnik podziału Nernsta)
(2)
Tak więc współczynnik podziału Nernsta stanowi iloraz stężeń równowagowych (c) substancji rozdzielanej (B) w rafinacie (A) i ekstrakcie (E).
Ekstrakcja jest skuteczna wtedy gdy składnik B, który należy usunąć z surówki, rozpuszcza się dużo lepiej w rozpuszczalniku selektywnym (E) niż w rozpuszczalniku pierwotnym (A) tzn. współczynnik podziału K różni się od 1.
Ilość składnika B pozostałego w cieczy ekstrahowanej (rafinacie) wylicza się ze wzoru:
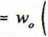
KV \n S + KV I
gdzie: w„ - ilość g substancji B po n ekstrakcjach w warstwie ekstrahowanej;
w„ - ilość g B w rozpuszczalniku pierwotnym A (surówce); K - współczynnik podziału Nernsta;
V - objętość warstwy ekstrahowanej w ml; n - ilość procesów ekstrakcji;
S - świeże porcje rozpuszczalnika selektywnego (w ml) użytego do ekstrakcji.
Sposób wykonania ekstrakcji (zadanie 4)
Ekstrakcję związków organicznych rozpuszczonych w wodzie lub innych ciężkich rozpuszczalnikach można prowadzić w sposób periodyczny (przerywany) lub ciągły. Ekstrakcję przerywana przeprowadza się w rozdzielaczu kulistym lub gruszkowym z krótką nóżką, którego pojemność winna być dwukrotnie większa niż objętość cieczy użytej do ekstrakcji. Korek i kran muszą być szczelne i nasmarowane smarem.
Roztwór lub zawiesinę wlewa się do rozdzielacza i dodaje rozpuszczalnika, którego ilość powinna stanowić 1/3 roztworu ekstrahowanego. Po zamknięciu rozdzielacza korkiem zawartość wstrząsa się ok. 2-3 min. wyrównując od czasu do czasu ciśnienie przez odwrócenie rozdzielacza do góry rurką spustowa i otwarcie na chwilę kranu. Następnie rozdzielacz umieszcza się w statywie w odpowiednim uchwycie i pozostawia do rozdzielenia dwóch faz. Po rozdzieleniu się
Soxhleta
warstw - warstwę dolną (wodną) przenosi się do drugiego rozdzielacza i ponownie ekstrahuje a warstwę rozpuszczalnika z wyekstrahowanym związkiem zlewa do odpowiedniego naczynia. Ekstrakcję powtarza trzykrotnie. Ekstrakty w rozpuszczalniku łączy się ze sobą, suszy, sączy przez sączek, oddestylowuje rozpuszczalnik i poddaje oczyszczeniu.
Ekstrakcję ciał stałych stosuje się aby uzyskać z nich zawarte substancje chemiczne.
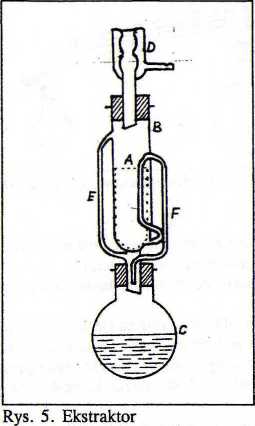
Ekstrakcję ciał stałych prowadzi się w aparacie Soxhleta pokazanym na rys. 5.
Substancję stalą wsypuje się do glizy A i umieszcza w rurze B aparatu Soxhleta. Aparat ten jest połączony z kolbą okrąglodenną C, w której znajduje się rozpuszczalnik podgrzewany do wrzenia. Pary rozpuszczalnika przechodzą rurką E do chłodnicy D zamontowanej na szczycie aparatu, skraplają się i spływają do gilzy, wypełniając rurę B aparatu. Po osiągnięciu górnego poziomu rurki bocznej F rozpuszczalnik z zawartym w nim związkiem przelewa się do kolby C. Po kilku godzinach ekstrakcji otrzymany ekstrakt suszy się i oddestylowuje z niego rozpuszczalnik. Znając masę próbki (gilzy) przed i po ekstrakcji można obliczyć ilość substancji wyekstrahowanej.
2.4. TEMPERATURA TOPNIENIA I KRZEPNIĘCIA
Temperatura topnienia (tj stałych związków organicznych to taka temperatura, w której pod ciśnieniem 1013 hPa związki te przechodzą ze stanu stałego w stan ciekły.
Dla czystych związków temperatura topnienia (ę) jest ostra, tzn przejście ze stanu stałego w stan ciekły zachodzi w zakresie 0,5-1 stopnia i praktycznie nie zależy od ciśnienia. Dla czystych związków również temperatura krzepnięcia ma tę samą wartość co temperatura topnienia. Temperaturę krzepnięcia określa się jako temperaturę, w której współistnieją obok siebie faza ciekła i stała pod sumarycznym ciśnieniem 1013 hPa.
Pomiar temperatury topnienia jest jednym z podstawowych parametrów w laboratorium służącym do identyfikacji związku chemicznego i oceny stopnia jego czystości. Obecność zanieczyszczeń na ogól powoduje obniżenie temperatury topnienia związku organicznego. Często w laboratorium "temperaturę topnienia mieszaniny” wykorzystuje się do określania czystości związku. Polega to na tym, że wzorcowy związek organiczny o określonej temperaturze topnienia zmieszany z tym samym związkiem otrzymanym w drodze syntezy w równych stosunkach wagowych, powinien wykazywać stalą temperaturę topnienia. Niższa temperatura świadczy o obecności zanieczyszczeń w produkcie.
Wyszukiwarka
Podobne podstrony:
str041 (4) 80 Ćwiczenie nr 10 mano przepływ wody. Destylacja ma zazwyczaj następujący przebieg: po p
img032 (44) AKADEMIA TECHNICZNO-HUMANISTYCZNA w Bielsku-Białej Ćwiczenie nr 10
str039 (4) 76 Ćwiczenie nr 9 76 Ćwiczenie nr 9(10) a - ilość 0,05 m roztworu wersenianu sodu zużyta
1a PJWSTKLaboratorium techniki cyfrowej Sprawozdanie z ćwiczeń nr: 10 Temat: System przerwań Imię
Układ Krążenia0010 Ćwiczenia nr 10 Vffacf drążenia i jego funkcjonowanie (cz.l). 1. &nbs
72143 str045 (4) 88 Ćwiczenie nr 10 Tabela 4. Temperatura topnienia i wrzenia wybranych substancji
Ćwiczenia nr 10: 1 godzina Doskonalenie poznanych umiejętności, tj. pływania stylem grzbietowym i st
(22) Ćwiczenie nr 10 Prosty sposób wyznaczania ciepła parowania ]. Wiadomości ogólne Ogrzanie względ
1a PJWSTKLaboratorium techniki cyfrowej Sprawozdanie z ćwiczeń nr: 10
75344 str040 (4) 78 Ćwiczenie nr 10 c) oznaczyć masę cząsteczkową związku, która określa rzeczywista
więcej podobnych podstron