
1
Prelekcja I:
Podstawowe określenia:
•
Genotyp – to materiał genetyczny danej osoby uporządkowany w
charakterystyczny układ alleli
•
Fenotyp – to końcowy efekt działania czynników genetycznych i
środowiskowych, ujawniający się w postaci określonego obrazu klinicznego
lub obserwowanej ekspresji genu. Badania korelacji genotypowo-
fenotypowej służą do oceny i powiązania obrazu klinicznego choroby ze
specyficzną mutacją genu.
•
Jeżeli allele w tym samym locus są identyczne, to osobę z takim układem
nazywa się homozygotą. Oba allele mogą być prawidłowe lub
nieprawidłowe.
•
Jeżeli allele w tym samym locus są różne to osobę z takim układem nazywa
się heterozygotą. Określenie to odnosi się zazwyczaj do sytuacji posiadania
jednego prawidłowego i jednego nieprawidłowego lub zmutowanego allelu.
Takie heterozygotyczne osoby nazywane są nosicielami.
•
Pojęcie „autosomalny” odnosi się do chromosomów (autosomów) nie
będących
chromosomami
płciowymi.
O
cechach
dziedziczonych
autosomalnie mówi się, gdy są one uwarunkowane genami zlokalizowanymi
w autosomach.
•
Sprzężenie z chromosomami X i Y odnosi się do genów mających loci na
chromosomie X lub Y. Z allelami sprzężonymi z chromosomami X związane
jest dziedziczenie dominujące lub recesywne. Określenie sprzężenie z płcią
jest synonimem używanym do przedstawienia dziedziczenia sprzężonego z
chromosomem X.
•
Cechy uwarunkowane dominująco występują zarówno u heterozygot,
jak i homozygot. Oznacza to, że obecność pojedynczego allelu danego genu
wystarcza do ujawnienia się cech.
•
Cechy uwarunkowane recesywnie występują tylko u homozygot, co
oznacza, że do ich wystąpienia konieczna jest obecność alleli recesywnych w
obu chromosomach. Pojęcia dominacji i recesywności dotyczą obrazu
klinicznego, a nie samych genów.
Dziedziczenia autosomalne dominujące:
•
Ponad połowa opisanych dotychczas cech jest dziedziczona dominująco:
około 1/3, a 1/10 jako cechy sprzężone z chromosomem X. Pojęcie choroby
dziedziczonej dominująco oznacza, że pojedynczy allel danej choroby ( jak u
heterozygoty) wystarcza do ujawnienia się jej objawów.

2
•
Kryteria dziedziczenia autosomalnego dominującego są następujące:
dana cecha przekazywana jest bez przeskoków z pokolenia na pokolenie.
przy skojarzeniu osoby chorej (heterozygoty) ze zdrową homozygotą
(najczęściej występująca sytuacja) każde dziecko ma 50% ryzyka
dziedziczenia zmutowanego allelu prawidłowego.
osoby obu płci chorują z jednakową częstością
Dziedziczenia autosomalne dominujące – cechy kliniczne:
•
Nowa mutacja:
Nowe mutacje są najczęściej przyczyną chorób o tak ciężkim przebiegu, że
zmniejszają
zdolność
do
reprodukcji.
Większość
przypadków
achondroplazji jest wynikiem nowej mutacji.
Późny wiek ojca może być związany ze zwiększeniem częstości nowych
mutacji w niektórych loci.
Częstość występowania nowych mutacji w niektórych chorobach
dziedziczonych dominująco jest znana i wykorzystywana w poradnictwie
genetycznym:
a)
zmniejszona penetracja – jeśli częstość ekspresji fenotypowej danego
genotypu jest mniejsza niż 100%, to mówi się o obniżonej penetracji.
O braku penetracji mówi się , gdy dany allel dziedziczony, lecz nie
wykazuje ekspresji fenotypowej.
b)
zmniejszona ekspresja – ekspresja jest zmniejszona w zależności od
tego, w jakiej postaci klinicznej lub w jakim stopniu poszczególny allel
ujawnia się w fenotypie danej osoby.
Nowa mutacja:
1.
Zmienność ekspresji jest powszechna w choroabach dziedziczonych
dominująco.
2.
Osoby chore, lecz zdolne do reprodukcji, wykazują zwykle mniejsze nasilenie
objawów choroby.
•
Przykłady kliniczne:
Choroba Huntingtona
Zespół Marfana
Dziedziczenie autosomalne recesywne:
•
W tym typie dziedziczenia charakterystyczny fenotyp obserwuje się tylko u
homozygot, a wtypowym rodowodzie występuje obok probanta chore
rodzeństwo obu płci oraz ich zdrowi rodzice.
•
Dziedziczenie recesywne podejrzewa się, jeśli rodzice są spokrewnieni, i

3
uważa się za pewne, jeśli stwierdza się niski poziom lub brak aktywności
określonego enzymu u każdego z obojga rodziców probanta.
•
Kryteria:
jeśli cecha jest rzadka, rodzice i krewni, w odróżnieniu od potomstwa są
zazwyczaj zdrowi.
jeśli recesywne geny są heterozygotami, to częstość segregacji w każdej
ciąży wynosi 25% zdrowych homozygot, 50% heterozygot i 25% chorych
homozygot.
choroba występuje z jednakową częstością u oby płci
jeśli cecha jest rzadka to istnieje duże prawdopodobieństwo, że rodzice są
spokrewnieni.
•
Cechy charakterystyczne:
częstość występowania heterozygot w populacji. Chore osoby są prawie
zawsze potomkami zdrowych heterozygot (nie homozygot).
nosicielstwo. Nosiciele to osoby zdrowe lecz u n iektórych z nich
występuje obniżenie o połowę w stosunku do normy aktywności
określonego enzymu.
•
Przykłady kliniczne:
Galaktozemia
Homocystynuria
Mukowiscydoza
Dziedziczenie sprzężone z chromosomem X:
•
Może być zarówno recesywne i dominujące.
•
Objawy choroby dziedziczonej recesywnie w sprzężeniu z chromosomem X
obserwuje się tylko u mężczyzn.
•
Dziedziczenie sprzężone chromosomem X podejrzewa się, jeśli dana cecha
występuje u kilku mężczyzn spokrewnionych poprzez kobiety. Ponieważ
mężczyźni mają tylko jeden chromosom X, są zatem homozygotami dla
genów sprzężonych z chromosomami X
•
Kryteria:
jeśli cecha jest rzadka, rodzice i krewni (wyłączając wujów ze strony matki
i innych męskich krewnych w linii kobiecej) są zazwyczaj zdrowi.
chorzy homozygotyczni mężczyźni nie mają ani chorych synów, ani
chorych córek.
heterozygotyczne kobiety nosicielki są klinicznie zdrowe, lecz przekazują
zmutowany gen 50% męskiego potomstwa. 50% córek to zdrowe
heterozygoty a 50% zdrowe homozygoty.
chore córki są najczęściej potomstwem heterozygotycznej kobiety i
chorego mężczyzny
każdy chory mężczyzna jest urodzony przez heterozygotyczną kobietę (z
wyjątkiem nowych mutacji)
jeśli cecha jest dominująca to wszystkie córki chorego mężczyzny są
chore.

4
•
Przykłady kliniczne
Dystrofia mięśniowa Duchenne'a (DMD)
Zespół leniwego chromosomu X [fra(X)]
Dziedziczenie mitochondrialne:
•
Ludzkie komórki mają setki mitochondriów rozpoznanych w cytoplazmie, z
których każde zawiera pewną liczbę kolistych cząstek DNA
•
Mutacje mitochondrialne DNA są odpowiedzialne za niewielką liczbę
schorzeń genetycznych.
•
Kryteria:
Każde mitochondrium zawiera pewną liczbę kopii kolistego genomu.
Niemal cały mitochondrialny DNA jest dziedziczony od matki
Określone tkanki mają różnż liczbę mitochondriów.
Duże rearanżacje genowe:
•
Delecje są łatwo wykrywane dzięki technologii rekombinacji DNA przez
stwierdzenie nieobecności lub zmianę wielkości fragmentu DNA. Z
wyjątkiem kilku zespołów są rzadkimi przyczynami mutacji w ludzkim
genomie.
•
Duplikacje sekwencji DNA są powszechne w procesie ewolucji i mogą być
spowodowane nieprawidłowym procesem koniugacji homologicznych
sekwencji DNA położonych blisko siebie, z duplikacją materiału
genetycznego, który jest zawarty wewnątrz genu. Mechanizm duplikacji jest
podobny do obserwowanego w balasemii. Duplikacje mogą zmienić ramkę
odczytu. Rodzinna hipercholesterolemia i DMD są przykładami chorób
genetycznych, które mogą być spowodowane duplikacjami.
•
Insercje są rzadkimi przyczynami mutacji w ludzkim genomie.
Transpozycja DNA jest jednak zjawiskiem powszechnym w ludzkim
genomie, lecz zwykle nie obejmuje sekwencji kodujących. Transpozycja może
niekiedy przerwać ciągłość genomu i być przyczyną nieprawidłowej ekspresji
uszkodzonego genu.
•
Mutacje
punktowe.
Zastąpienie
pojedynczego
nukleotydu,
podobnie jak obserwowane u bakterii i wirusów, jest najczęstszą przyczyną
mutacji w ludzkim genomie. Jeśli w kodonie dojdzie do zmiany
pojedynczego nukleotydu, to powstałe punktowe mutacje nazywa się
mutacjami zmiany sensu. Zaburzenia, które są skutkiem różnych mutacji
punktowych, to np. beta-talasemia, mukowiscydoza, fenyloketonuria i zespół
Taya-Sachsa.

5
Mutacje transkrypcyjne:
•
Mogą wystąpić w obszarze od 5’ końca początku kodonu inicjującego w
sekwencji DNA.
•
Miejsce to jest krytyczne dla regulacji transkrypcji.
•
Przykładem mogą być różne mutacje znalezione w sekwencji TATA, regionie
zlokalizowanym około 30 nukleotydów w kierunku końca 5’ od kodonu
inicjującego.
•
Ważną rolę w regulacji transkrypcji odgrywają także reszty na 3’ końcu genu
beta-globiny. Mutacje w tych dystalnych elementach promotora mogą być
przyczyną obniżenia aktywności transkrypcyjnej i spadkiem produkcji białka beta-
globiny.
Mutacje translacyjne:
•
Mutacje uszkadzające translację obserwowane są czasem w talasemii.
Opisane zostały także w innych chorobach takich jak fenyloketonuria.
•
Mutacje nonsensowne
•
Mutacje zmiany ramki odczytu.
Mutacje RNA:
•
Mutacje procesu cięcia RNA i jego stabilności mogą spowodować
nieodpowiednie cięcie wytwarzanego RNA w sekwencjach w kierunku do końca 3’.
Wynikiem tego jest powstanie RNA, który jest nienormalnie duży, niestabilny i
szybko ulega degradacji.
•
Mutacje procesu składania RNA.
Mutacje dynamiczne:
•
W ostatnich latach opisano nowy mechanizm powstawania mutacji u ludzi,
spowodowany wzrostem liczby trójnukleotydowych sekwencji powtarzalnych w
genie. Trój nukleotydy te mogą znajdować się w nie ulegającym translacji regionie
5’ genu, w regionie kodującym genu lub w nie ulegającym translacji regionie 3’
genu.
Negatywne mutacje dominujące:
•
Mutacja jednego z alleli w danym locus może dać efekt dominujący, jeśli
obecność tylko jednego funkcjonalnie prawidłowego allelu jest niewystarczająca do
wytworzenia odpowiedniej ilości produktu tego genu. Hipercholesterolemia
rodzinna jest chorobą będącą przykładem takiej mutacji.

6
Polimorfizm genetyczny
Polimorfizm i markery genetyczne
Definicje
•
Polimorfizm genetyczny – oznacza występowanie w populacji dwóch lub
więcej alleli w danym locus z częstością większą niż wynikająca z ogólnej częstości
mutacji. Polimorfizm to zrównoważenie genetyczne warunkujące zmienność
wewnątrz gatunku.
•
Polimorfizm. Wszystkie odmiany polimorfizmu są efektem zmian
zachodzących w sekwencji DNA, które mogą być wykryte metodami biologii
molekularnej. Polimorfizm można również wykazać w badaniach białek o
zmienionej funkcji, enzymów lub antygenów oraz nieprawidłowych cech
fizycznych organizmu. Polimorfizm może być zatem klasyfikowany zależnie od
metody jego wykrycia.
•
Polimorfizm DNA – jest identyfikowany poprzez bezpośrednie wykrycie
zmienionych sekwencji DNA.
•
Polimorfizm długości fragmentów restrykcyjnych (RFLP) –
polimorfizm krótkich tandemowych powtórzeń (STRP)
Znaczenie kliniczne polimorfizmu:
•
Większość rodzajów polimorfizmu nie wpływa na cechy kliniczne fenotypu.
•
Bez względu na ich efekt fenotypowy są one użytecznymi markerami
genetycznymi.
Markery genetyczne:
•
Markery
genetyczne
mogą
być
wykorzystane
do
określenia
prawdopodobieństwa asocjacji genów choroby w rodzinie lub u pojedynczych
chorych, a także do ustalenia pokrewieństwa oraz genetycznej zgodności krwi,
nasienia czy tkanki.
•
Bliźnięta. Z genetycznego punktu widzenia istnieją dwa rodzaje bliźniąt:
Jednojajowe (MZ) lub identyczne bliźnięta mają wszystkie geny takie same.
Pochodzą z jednej zygoty, która dzieli się na dwa embriony.
Dwujajowe (DZ) lub braterskie bliźnięta mają około połowy genów takich
samych. Rozwijają się z dwóch oddzielnych zygot i są tak samo genetycznie
spokrewnione, jak z pozostałym rodzeństwem.

7
Markery genetyczne:
•
Ustalenie ojcostwa:
Ustalenie ojcostwa wymaga pobrania próbek od dziecka, matki i
domniemanego ojca.
Badania z zastosowaniem markerów genetycznych mogą wykluczyć, ale
nie udowodnić ojcostwo. Są dwie możliwości wykluczenia ojcostwa:
a)
ojcostwo jest wykluczone jeśli żaden z dwóch alleli danego locus u
domniemanego ojca nie występuje u dziecka
b)
ojcostwo jest wykluczenie, jeśli u domniemanego ojca brakuje allelu,
który jest obecny u dziecka, a nie m ago u matki.
Im więcej markerów zostanie przebadanych i mniej wspólnych alleli
ustalonych, tym bardziej prawdopodobne, że ojcostwo danego mężczyzny
będzie wykluczone.
Prawdopodobieństwo ojcostwa może być obliczone na podstawie liczby
przebadanych markerów oraz częstości alleli, dla których dziecko jest
informacyjne.
•
Zastosowanie markerów genetycznych w sądownictwie obejmuje ponadto:
określenie pochodzenia krwi, nasienia lub próbek tkanek uzyskanych na
miejscu przestępstwa
małe ilości DNA mogą być użyte do określenia markerów genetycznych
charakterystycznych dla ofiary lub podejrzanego
identyfikacja zaginionych osób, w celu ustalenia rzeczywistego
pokrewieństwa biologicznego markery genetyczne osoby zaginionej
można pobrać z markeru jej rodziców i innych członków rodziny
Sprzężenie i mapowanie:
•
Sprzężenie to występowanie dwóch lub więcej loci genowych w tak bliskiej
odległości fizycznej w chromosomie, że bardziej prawdopodobne jest
przekazanie ich razem niż oddzielnie podczas mejozy.
•
Sprzężenie loci występuje, gdy położone są one w niewielkiej odległości od
siebie i crossing over zachodzi między nimi rzadko.
Mapowanie genów:
•
Jest przyporządkowaniem genów od określonego miejsca w chromosomie.
•
Ponad 6000 loci genetycznych zostało zmapowanych w poszczególnych
chromosomach człowieka.

8
•
Większość z tych loci to polimorfizmy krótkich tandemowych powtórzeń,
które są idealnymi markerami ze względu na ich olbrzymi polimorfizm i
łatwość w badaniach za pomocą reakcji łańcuchowej polimerazy PCR .
•
Powyższe badanie obejmuje ponad 450 genów.
•
Przeciętna odległość miedzy mapowanymi markerami wynosi mniej niż 1cM.
•
Wiele kolejnych loci mapuje się na bieżąco w ramach Projektu Badania
Genomu Człowieka.
•
Znanych jest kilka metod mapowania genów:
badania rodzinne
•
Istnieje wiele rodzajów map genetycznych zawierających różne, ale
uzupełniające się informacje. Przybliżona ich kolejność w skali wzrastającej
dokładności jest następująca:
mapa cytogenetyczna
mapa sprzężeń
mapa fizyczna
sekwencja DNA
Korelacja genotyp-fenotyp:
•
Korelacja genotyp-fenotyp jest związkiem specyficznych zmian w genomie z
charakterystycznymi objawami klinicznymi choroby genetycznej.
•
Wiele chorób genetycznych wykazuje istotną różnorodność w ekspresji czy
niekompletnej penetracji.
•
Przyczyny takich różnic zależą od rodzaju choroby i są zróżnicowane
międzyosobniczo.
•
Prawdopodobne przyczyny tej klinicznej zmienności objawów obejmują:
heterogenność locus
heterogenność alleli
somatyczne zmiany zmutowanego allelu
efekty epistatyczne
efekty epigeniczne
czynniki pozagenetyczne.
Genetyka molekularna chorób serca i naczyń:
•
W ostatnich dziesięcioleciach dokonał się znaczny postęp w rozumieniu, jaką
rolę odgrywają czynniki dziedziczne w procesach patologii człowieka, w tym
również w chorobach układu sercowo-naczyniowego.
•
Choroby genetyczne mogą być wynikiem nieprawidłowości pojedynczego
genu – tu rozróżniamy geny dominujące (wystarczy jeden gen do ujawnienia
choroby) i geny recesywne ( potrzebny komplet dwóch wadliwych genów do
pełnego ujawnienia choroby).

9
Prelekcja II:
Genetyka molekularna chorób serca i naczyń:
•
Choroby układu krążenia rozwijają się na wskutek czynników wrodzonych
(choroba obecna przy urodzeniu) lub nabytych, zależnych głównie od
naszego trybu życia.
•
Choroby genetyczne mogą być wynikiem nieprawidłowości pojedynczego
genu – tu rozróżniamy geny dominujące (wystarczy jeden do ujawnienia
choroby) i geny recesywne (potrzeba komplet dwóch wadliwych genów do
pełnego ujawnienia się choroby).
•
Jednogenowe choroby genetyczne serca to m.in. kardiomiopatia
(Przerostowa, rozstrzeniowa), kanałopatie (zespół wydłużonego, skróconego
QT, zespół Brugadów), choroby spichrzeniowe ( choroba Pompego,
Fabry'ego) zespół Marfana, choroba węzła zatokowego, hipercholesterolemia
rodzinna i inne.
•
Choroby genetyczne serca mogą się ujawnić już przy urodzeniu, w czasie
dorastania lub dopiero w wieku dorosłym. W niektórych przypadkach nie
dochodzi do ujawnienia się choroby. Zależne jest to od typu defektu, ale
także od wpływu dodatkowych czynników genetycznych i środowiskowych.
•
Choroby genetyczne serca niosą ze sobą ryzyko nagłej śmierci sercowej w w
przebiegu groźniej arytmii lub pęknięcia aorty. Dlatego przy rozpoznaniu
choroby uwarunkowanej genetycznie bardzo ważna jest ocena ryzyka
wystąpienia takiego powikłania.
•
Chorym w dobrym stanie z wysokim ryzykiem można zaproponować
zabiegowe postępowanie zapobiegawcze – wszczepienie kardiowertera w
przypadku arytmii czy profilaktyczną operację aorty w zespole Marfana.
Jednogenowe schorzenia układu sercowo naczyniowego.
Kardiomiopatia przerostowa:
•
Kardiomiopatia przerostowa jest jedną z najczęstszych chorób genetycznych
w kardiologii i najczęstsza przyczyną nagłego zgonu wśród dzieci i młodych
dorosłych.
•
Częstość występowania tej patologii ocenia się na ok 1 zachorowanie na 500
osób
•
Kardiomiopatia przerostowa dziedziczy się najczęściej jako cecha
autosomalna dominująca, rzadziej jako cecha autosomalna recesywna
sprzężona z chromosomem X lub choroba wynikająca z zaburzeń DNA
mitochondrialnego.
•
Schorzenie to spowodowane jest przez mutacje genów związanych głównie z
białkami aparatu kurczliwego mięśnia sercowego
•
Genetycy odkryli liczne mutacje genów przynajmniej 10 różnych elementów
białek sarkomeru, takich jak łańcuch ciężki sercowej beta-miozyny, sercowe

10
białko wiążące miozynę, sercowa troponina T, sercowa troponina I, alfa-
tropomiozyna, sercowe białko C i regulatorowe łańcuchy lekkie oraz sercowa
aktyna.
•
Poza typowymi zmianami sercowymi obserwowano w tych rzadkich
przypadkach np. dodatkowe szlaki przewodzenia prowadzące do
współwystępowania zespołu Wolffa-Parkinsona-White'a, głuchotę czuciowo
- nerowową, neurologiczne napięcia mięśni tułowia i encefalopatię.
•
Na obraz patologiczny kardiomiopatii przerostowej składa się przede
wszystkim znaczny przerost lewej komory serca, pogrubienie przegrody
międzykomorowej, powiększenie lewego przedsionka i zwykle mała jama
komory lewej
•
Przerost i dezorganizacja w układzie miocytów oraz włóknienie
śródmiąższowe występuje w całym mięśniu sercowym
•
Badania polimorfizmów genów kodujących angiotensynę II, aldosteron i
endotelinę, które mogą modyfikować fenotyp kardiomiopatii przerostowej
nie przyniosły jednoznacznych wyników.
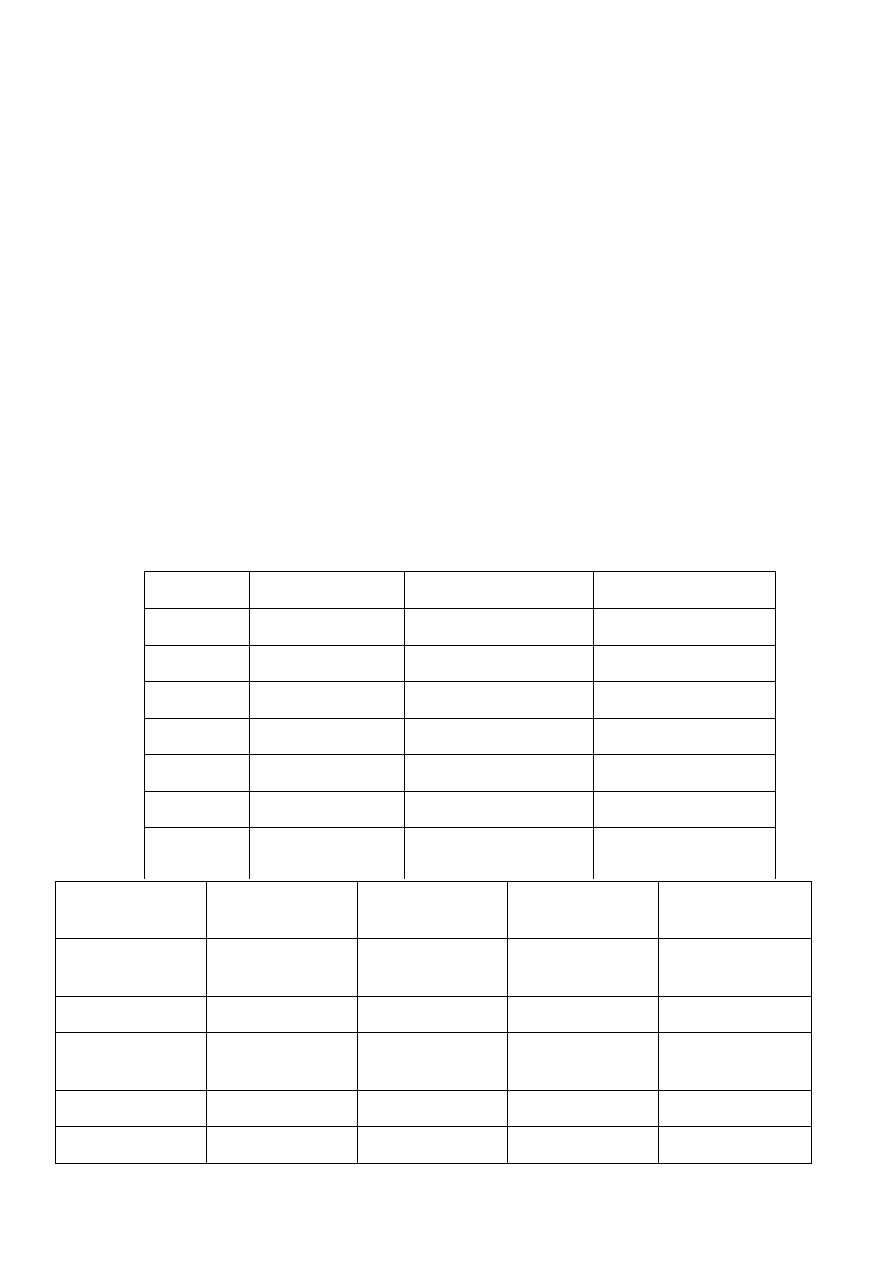
Geny, których mutacje najczyściej związane są z kardiomiopatią
przerostową:
Gen
Częstośc mutacji %
Obraz kliniczny i
rokowanie
MYH7 łańcuch ciężki
beta-miozyny
35-50
Mutacje R403Q ( często
łącznie z R719W i
R453C) – niekorzystne
rokowanie
Mutacje V1606M,
L908V, G256Q, P513C –
rokowanie dobre
MYBPC3
30-47
Późne objawy kliniczne,
duże ryzyko zgonu w
wyniku arytmii
TNNT2
6.05.2015
Łagodny przerost, duże
ryzyko nagłego zgonu w
wyniku arytmii
TMP1
3.05.2010
Pośredni i
niejednorodny poziom
przerostu
TMP1 alfa-tropomiozyny
Współwystępowanie
przerostu z zespołem
WPW, rzadko
występowanie rodzinne

11
Geny których mutacje najczęściej związane są z kardiomiopatią
rozstrzeniowią:
Do najczęstszych przyczyn rozstrzeni komór należą:
•
choroba wieńcowa,
•
przebyty zawał serca
•
wady zastawkowe
•
nadciśnienie tętnicze
•
zapalenie mięśnia sercowego i choroby układowe
Jednak w niektórych przypadkach identyfikacja czynnika sprawczego jest
niemożliwa. Choroba określana jest wtedy jako kardiomiopatia idiomatyczna.
Niekóre z tych przypadków mają podłoże genetyczne i występują w sposób
rodzinny lub sporadyczny. Kardiomiopatii rozstrzeniową dziedziczy się najczęściej
w sposób autosomalny dominujący, ale występują też przypadki dziedziczenia
autosomalnego recesywnego związanego z mitochondrialnym DNA i sprzężonego z
chromosomem X
Białka szkieletu komórkowego
DES (desmina)
Kardiomiopatii mogą towarzyszyć miopatia szkieletowa,
bloki przewodzenia w sercu, zaburzenia rytmu
SGCD, SGCB
Czysta postać kardiomiopatii kub w przypadku SGCB
dziedziczona autosomalnie recesywnie ciężka odmiana
dystrofii mięśniowej
DMD
Postać sprzężona z chromosomem X, małe stężenie
dystrofiny sercowej przy prawidłowych poziomach
dystrofiny mięśni szkieletowych
Białka Połączeń międzykomórkowych
VCL (winkulina),
DSP, CSRP3
Kardiomiopatia jest skutkiem zaburzenia interakcji
??????????( kontakt zasłania :/ )
Kardiomiopatia rozstrzeniowa typu left ventricular non-conpaction:
•
Wynika ona z zahamowania biogenezy w mięśniu sercowym i charakteryzuje
się przerostem lewej komory z głębokim beleczkowaniem i upośledzeniem
czynności skurczowej ze współistniejącą często rozstrzenia.
•
Podobne zmiany mogą dotyczyć również prawej komory. Kardiomiopatia
typu left ventricular non-conpation towarzyszą często różnego rodzaju wady

12
wrodzone wady serca np. ubytki w przegrodzie między przedsionkowej lub
międzykomorowej oraz zwężenie zastawki pnia płucnego.
•
Choroba może ujawnić się w niemowlęctwie lub w wieku późniejszym. Jej
przebieg jest zwykle niekorzystny z szybko postępującą dysfunkcją lewej
komory i jawną klinicznie niewydolnością serca
Zaburzenia rytmu serca:
•
Nagłe zgony związane z groźnymi zaburzeniami rytmu serca ze względu na
ich wielką liczbę stanowią bardzo istotny problem kliniczny w kardiologii.
•
Czynniki genetyczne mogą modyfikować ryzyko arytmii związane z typowym
podłożem patologicznym
•
Opisano geny sprzyjające zaburzeniom rytmu, co pozwoliło na pogłębienie
wiedzy na temat molekularnego podłoża różnych typów tych zaburzeń.
Zespół wydłużonego odstępu QT:
•
Zespół wydłużonego odstępu QT (zespół LQT) jest wrodzoną chorobą
arytmogenną
objawiającą
się
przede
wszystkim
nieprawidłowym
wydłużeniem okresu repolaryzacji, czyli odstępu QT, w powierzchniowym
elektrokardiogramie, nieprawidłowym kształtem załamka T oraz groźnymi
dla życia zaburzeniami rytmu serca.
•
Choroba występuje w strukturalnie prawidłowym sercu i jest związana z
dużym ryzykiem nagłego zgonu. Pierwsze objawy choroby, omdlenia lub
nagły zgon występują średnio w wieku 12 lat.
•
Opisano dwie główne formy fenotypowe choroby:
zespół Romano – Warda dziedziczony w sposób autosomalny dominujący
oraz rzadziej występujący;
zespół Jervella – Langego – Nielsena dziedziczony w sposób autosomalny
recesywny i współwystępujący z głuchotą czuciowo – nerwową;
Geny związane z występowaniem różnych postaci zespołu wydłużonego
odstępu QT:
Kan
ał
Gen (białko)
Postacie
choroby
Częstość
(%)
Obraz kliniczny
I
Ks
KNCQ1
(KvLQT1)
KCNE1 (MinK)
LQT1 lub JLN1
LQT5 lub
JLN2
50
2 – 3
LQT1 – zmniejszona
penetracja, łagodniejszy
przebieg, objawy
wywołane bezpośrednio
bodźcem adrenergicznym,
zwykle występują w czasie
wysiłku
I
Kr
KCNH2
LQT2
35 – 40
LQT2 – większa

13
(HERG)
KCNE2 (MiRP)
LQT6
<1
penetracja i bardziej ciężki
przebieg niż LQT1,
zwłaszcza u kobiet,
zaburzenia rytmu
wyzwalane bodźcem
dźwiękowym
LQT6 – niepełna
penetracja, łagodny
przebieg
I
Na
SCN5A
(Nav1.5)
LQT3
10 – 15
Gorsze rokowanie,
zwłaszcza u mężczyzn,
zaburzenia rytmu
występują w spoczynku
ANK2
(
ankrynina?
B)
LQT4
Towarzyszy bradykardia,
napadowe migotanie
przedsionków,
wielofazowe załamki T
I
Ca
CACNA1c
(Cav1.2)
LQT8
Zespół
Timothy-ego
Znaczne wydłużenie QT,
towarzyszy syndaktylia
skórna, blok AV 2:1,
wrodzone wady serca,
opóźnienie umysłowe,
autyzm, zaburzenia
metaboliczne, duża
śmiertelność
Zespół skróconego odstępu QT:
•
Charakteryzuje się on nieprawidłowo krótkim okresem repolaryzacji
objawiającym się w zapisie EKG odstępem QT < 300ms oraz wąskimi i
spiczastymi załamkami T.
•
Klinicznie zespół ten objawia się napadowym migotaniem przedsionków,
omdleniami i nagłym zgonem.
•
Pierwszym genem, którego mutacja została wskazana jako leżąca u podłoża
tego zespołu, był KCNH2 kodujący białko HERG.
•
Opisana mutacja powoduje wzrost aktywności ośrodkowego prądu
potasowego IKr.
•
Do tej pory opisano również mutacje w innych genach poza KCNH2
objawiające się fenotypem zespołu skróconego odstępu QT: KCNQ1, SCN5A,
KCNE2, KCNJ2.
14
Zespół Brugadów:
•
Jest to wrodzona choroba arytmogenna występująca w prawidłowym
strukturalnie sercu, dziedziczona w sposób autosomalny dominujący.
•
Wyraźnie częściej jest stwierdzana wśród mężczyzn niż u kobiet (8:1).
•
Zespół ten charakteryzuje się elektrokardiograficznie zupełnym lub
niezupełnym blokiem prawej odnogi pęczka przedsionkowo – komorowego
(Hisa) oraz uniesieniem odcinka ST w pierwszych trzech odprowadzeniach
przedsercowych (V1 –V3) z występowaniem groźnych dla życia tachyarytmii
komorowych.
•
Objawy kliniczne w postaci omdleń lub nagłego zatrzymania krążenia
ujawniają się zwykle w 3 – 4 dekadzie życia i występują zwykle w spoczynku
lub podczas snu.
•
W 20% przypadków podłożem molekularnym zespołu Brugadów jest
mutacja w genie SCN5A kodującego białko Nav1.5, składową kanału
sodowego.
•
W 80% przypadków udało się jedynie ustalić locus związane z zespołem
Brugadów zlokalizowane na ramieniu krótkim 3 chromosomu (3p22-25),
bez określenia genu odpowiedzialnego za chorobę.
•
Czynnościowo mutacje leżące u podłoża tego zespołu prowadzą do
upośledzenia prądu sodowego
•
Rozpoznanie stawia się na podstawie wystąpienia nagłego zatrzymania
krążenia (skutecznie reanimowanego), wywiadu rodzinnego (nagłe zgony w
młodym wieku) oraz badania EKG.
•
Leczenia zespołu polega na zabezpieczeniu chorego przed nagłym
zatrzymaniem krążenia. Wszczepia się kardiowerter-defibrylator.
Genetyka cukrzycy typu 1:
Skłonność do rozwoju cukrzycy typu 1 jest uwarunkowana
dziedziczeniem wielogenowym
•
Analiza sprzężeń 767 rodzin multipleksowych potwierdziła znaczenie 7
regionów w genomie mających związek z cukrzycą t.1:
chromosom 6 – IDM1 [LOD=65,8](3)
chromosom 6 – IDDM15 [LOD=2,36 – 4,8](4)
chromosom 11 – IDDM2 w miejscu genu insuliny [LOD=4,28](5)
chromosom 1q42 – [LO=2,2](6)
chromosom 2 – IDDM12 w regionie genu CTLA – 4 [LOD=2,62 –
3,22](7)
chromosom 10 – IDM10 [LOD=2,8](8)
chromosom 20 – w miejscu D16S3098 [LOD=4,13](6)
•
Wyodrębnia się monogenowe formy cukrzycy t.1.
zespół autoimmunologicznej poliendokrynopatii typu 1
15
zespół XPI (X-linked poliendocrinopathy, immune dysfunction, diarrhea)
związanych z chromosomem X
Mutacje dotyczące występowania tych zespołów dotyczą genów
kodujących czynniki transkrypcyjne: AIRE (autoimmune regulator) na
chromosomie 23 oraz FOX p3 na chromosomie X (33,34).
Genetyka cukrzycy typu 2:
•
Forma monogenowa:
konsekwencja rzadkich mutacji w pojedynczych genach; znane formy
monogenowe cukrzycy t.2 charakteryzują się istotnym defektem w
zakresie wydzielania insuliny bądź też głębokim upośledzeniem
wrażliwości na insulinę;
•
Forma poligenowa:
obraz kliniczny tych form choroby wynika z interakcji czynników
środowiskowych i genetycznych rozumianych jako wpływ wielu genów;
allele polimorficznych genów są obecne zarówno u zdrowych jak i u
chorych na cukrzycę, chociaż występują w tych grupach z różną
częstością;
Typ MODY
Wiek ujawnienia
cukrzycy
Zmiana pierwotna
Ciężkość cukrzycy
Powikłania
MODY1
młodzieniec, młody
dorosły
trzustka/inne
ciężka
często
MODY2
po urodzeniu
trzustka/inne
przebieg łagodny
rzadko
MODY3
młodzieniec, młody
dorosły
trzustka/inne
przebieg ciężki?
często
MODY4
młody dorosły
trzustka/inne
przebieg ciężki
nieznane
MODY5
?
trzustka/inne
przebieg ciężki
choroby nerek
Typ MODY
Lokalizacja zmiany
Nazwa genu
Częstość występowania
MODY1
20q
HNF – 4α (TCF4)
rzadko 1 – 2%
MODY2
7q
GCK
10 – 65%
MODY3
12q
HNF – 1α (TCF1)
20 – 65%
MODY4
13q
IPF – 1
rzadko
MODY5
17q
HNF – 1β (TCF2)
rzadko
MODY6
2q32
Neurod1
rzadko
MODY7
11q15.1
SUR1
E1506K
rzadko

16
Monogenowe formy cukrzycy typu 2:
•
mutacje
genu
insuliny
mogą
prowadzić
do
powstania
również
monogenowych form cukrzycy typu 2
•
w nielicznych przypadkach wykazano, że mutacje mogą dotyczyć części
kodującej genu
•
powodują one zmianę struktury insuliny i osłabienie lub zniesienie jej
czynności
•
u osób z mutacją genu insuliny pojawia się nietolerancja glukozy, której
towarzyszy
hiperinsulinemia,
prawidłowa
insulinowrażliwość,
bez
współwystępujących zaburzeń immunologicznych
•
w rzadkich przypadkach mutacje mogą dotyczyć punktów uchwytu
endopeptydaz przekształcających proinsulinę w insulinę
Monogenowe formy cukrzycy typu 2 z dominującą insulino
opornością:
•
Mutacje w genie receptora insuliny są bardzo rzadkie. Insulinooporność w
tym przypadku jest spowodowana mutacją w jednym lub dwóch allelach genu
receptora insuliny
Krasnoludkowatość (leprechaunizm) – charakteryzuje się nasiloną
insulinoopornością, cukrzycą, niższą masą urodzeniową, powolnym wzrostem.
Zespół ten źle rokuje.
Zespół Robsona – Mendeholla. Ujawnia się klinicznie w okresie dzieciństwa.
Charakteryzuje się szybkim wzrostem, hipertrofią paznokci i zębów,
przedwczesnym dojrzewaniem oraz wzrostem szyszynki.
Zespół insulinooporności typu A – dotyczy młodych kobiet. Charakteryzuje
się hiperandrogenizmem, zespołem policystycznych jajników oraz zmianami
skórnymi o typie acanthosis nigricans
MODY6
młody dorosły
trzustka
przebieg ciężki
nieznane
MODY7
dorośli
trzustka
przebieg łagodny
nieznane

17
Poligenowe formy cukrzycy typu 2:
a)
Kalpaina 10:
Zmniejsza uwalnianie insuliny pobudzane glukozą
Z drugiej strony inhibitory kalpainy hamują pobudzaną insuliną absorpcję w
adipocytach i miocytach
Kalpainy zmniejszają również pobudzaną insuliną syntezę glikogenu w
mięśniach. Niski poziom RNA dla kalpainy 10 w mięśniach prowadzi do
narastania insulino oporności
Gen dla kalpainy 10 jest zlokalizowany na chromosomie 2q. Ryzyko
zachorowania na cukrzycę typu 2 nie wiązało się z wariantem jednego
polimorfizmu genu kalpainy, ale wynika raczej z obecności kilku niekorzystnych
haplotypów tworzonych przez allele trzech SNP-ów o numerach: 19, 43 i 63.
Wszystkie te SNP-y zlokalizowane są w intronach, a więc nie wpłyną na strukturę
aminokwasową białka
Najprawdopodobniej mechanizmem patogenetycznym o podstawowym
znaczeniu jest jej wpływ na ekspresję genu kalpainy 10. Znaczenie kalpainy 10 w
patogenezie cukrzycy typu 2 jest różne w różnych populacjach
b)
Peroksysomalny aktywowany proliferacyjnie receptor γ (peroximose
proliferator activated receptor) PPAR – γ
Ostatnio opublikowane badania zarówno osób niespokrewnionych, jak i
rodzin bezspornie udowodniły znaczenie polimorfizm allelu proliny Pro12Ala w
patogenezie cukrzycy typu 2
Opublikowane dane pozwalają na powiązanie wariantu dziedziczenia Pro12
aminokwasu w PPARγ z patogenezą cukrzycy typu 2 (ryzyko rozwoju cukrzycy u
osób homozygot Pro12Pro PPARγ zwiększa się o 25%)
Ponieważ allel ten występuje z dużą częstością, około 85%, jego
umiarkowany efekt przekłada się na duże ryzyko odnoszące się do całej populacji i
może odpowiadać aż za 25% przypadków cukrzycy typu 2
•
W ostatnim okresie zidentyfikowano również u chorych na cukrzycę typu 2
na chromosomie 10q region o podwyższonym ryzyku rozwoju tej choroby
wykazano także linkage disequilibrium
•
W tym miejscu znajduje się gen czynnika transkrypcyjnego 7 jak 2
(transcription factor 7 like 2 – TCF7L2)
•
Jego obecność częściej spotyka się u chorych na cukrzycę typu 2
•
Znaczenie genu TCF7L2 w patogenezie cukrzycy typu 2 wiąże się z jego
wpływem na drogę przekazu sygnałów typu Wingless – WNT
•
Droga WNT odgrywa z kolei znaczną rolę w regulacji proliferacyjnego
różnicowania się komórek. WNT wiąże się ze swoim receptorem. W wyniku tego
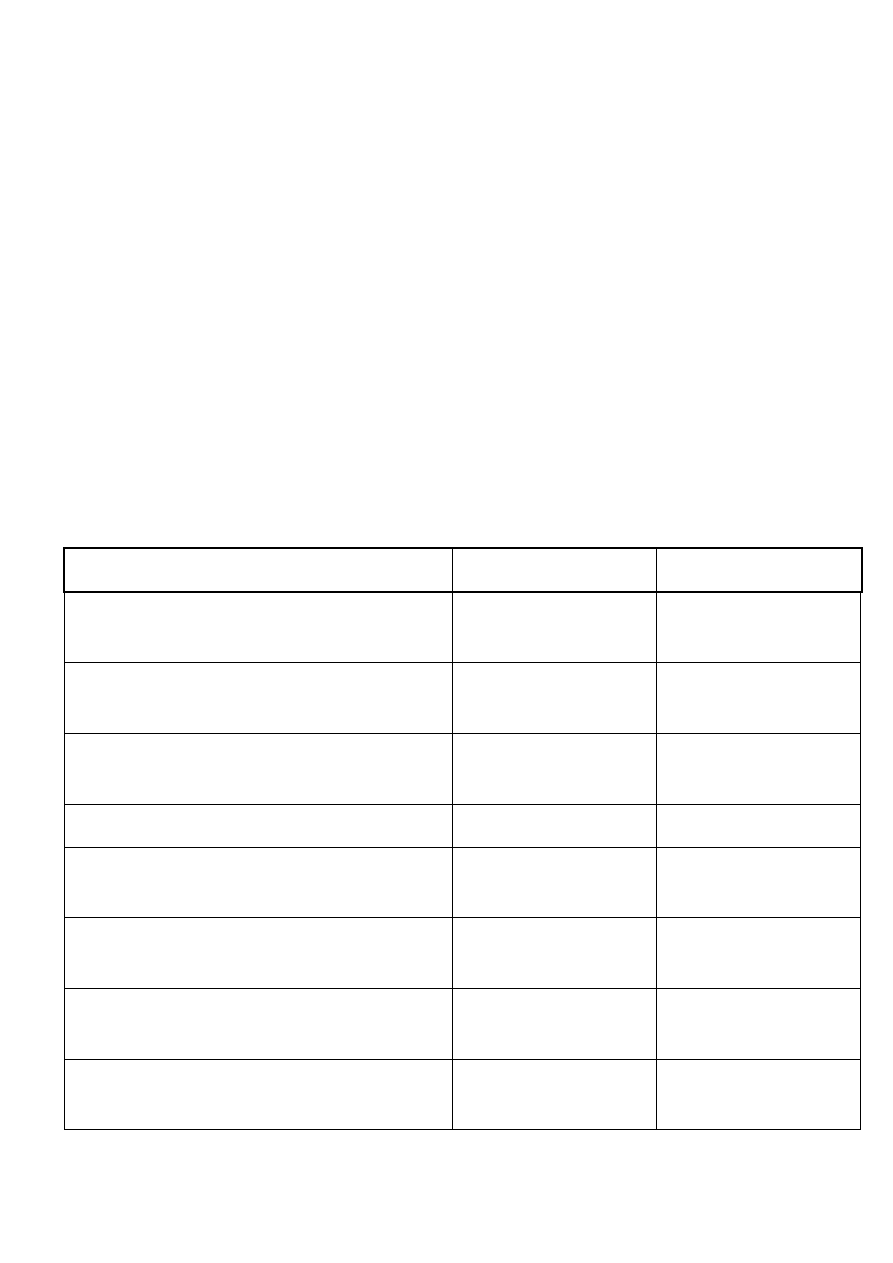
18
uwalniana jest β-katemina. β-katemina dostaje się do jądra, gdzie tworzy
heterodimer z TCF7L2
•
Gen TCF7L2 ma wielkość 215,9 kb. Jest zlokalizowany na chromosomie
10q25. Zidentyfikowano mikrosatelitarny marker DG10S478 zlokalizowany w 3
intronie tego genu, który ściśle wiąże się z ryzykiem rozwoju cukrzycy typu 2.
Autorzy zgenotypowali 5 polimorfizmów w obrębie tego genu. Wykazali ścisłe ich
powiązanie z markerem DG10S478 oraz ryzykiem rozwoju cukrzycy typu 2
•
Autorzy Zahng i wsp. potwierdzili ścisłą zależność pomiędzy pospolitym
wariantem TCF7L2 (RS 12255372 T/G), a wczesnym ryzykiem rozwoju cukrzycy
typu 2. Autorzy wykazali, że obecność allelu T wiąże się z 32% u kobiet wzrostem
ryzyka rozwoju cukrzycy typu 2, a u mężczyzn z 53% ryzykiem. U homozygot TT
ryzyko to było odpowiednio wyższe o 86% (? dziwne ale tak było na slajdzie) u
kobiet i o 11,5% u mężczyzn. W badaniu metaanalitycznym wykazali, że obecność
allelu T wiąże się z 48% większym ryzykiem rozwoju cukrzycy typu 2
Genetyka nadciśnienia tętniczego:
Choroba
Gen lub locus
Dziedziczenie
Niedobór 11-hydroksylazy steroidowej CPY111
Autosomalne
recesywne
Niedobór 17- hydroksylazy steroidowej CPY17
Autosomalne
recesywne
Zespól Liddle'a
SCNN18
Autosomalne
dominujące
SCNN16
Pozorny nadmiar mineralokortykoidów
AME
HSD11b2
Autosomalnie
recesywnie
Mutacja S810L genu receptora
mineralokortykosteroidów
NR3C2
Autosomalnie
dominująco
Rodzinny Hiperaldosteronizm Typ 1 (FH
I, GRA)
CYP11B1/B2
Autosomalnie
dominująco
Rodzinny Hiperaldosteronizm Typ 2 (FH
II)
7p22
Autosomalnie
dominująco
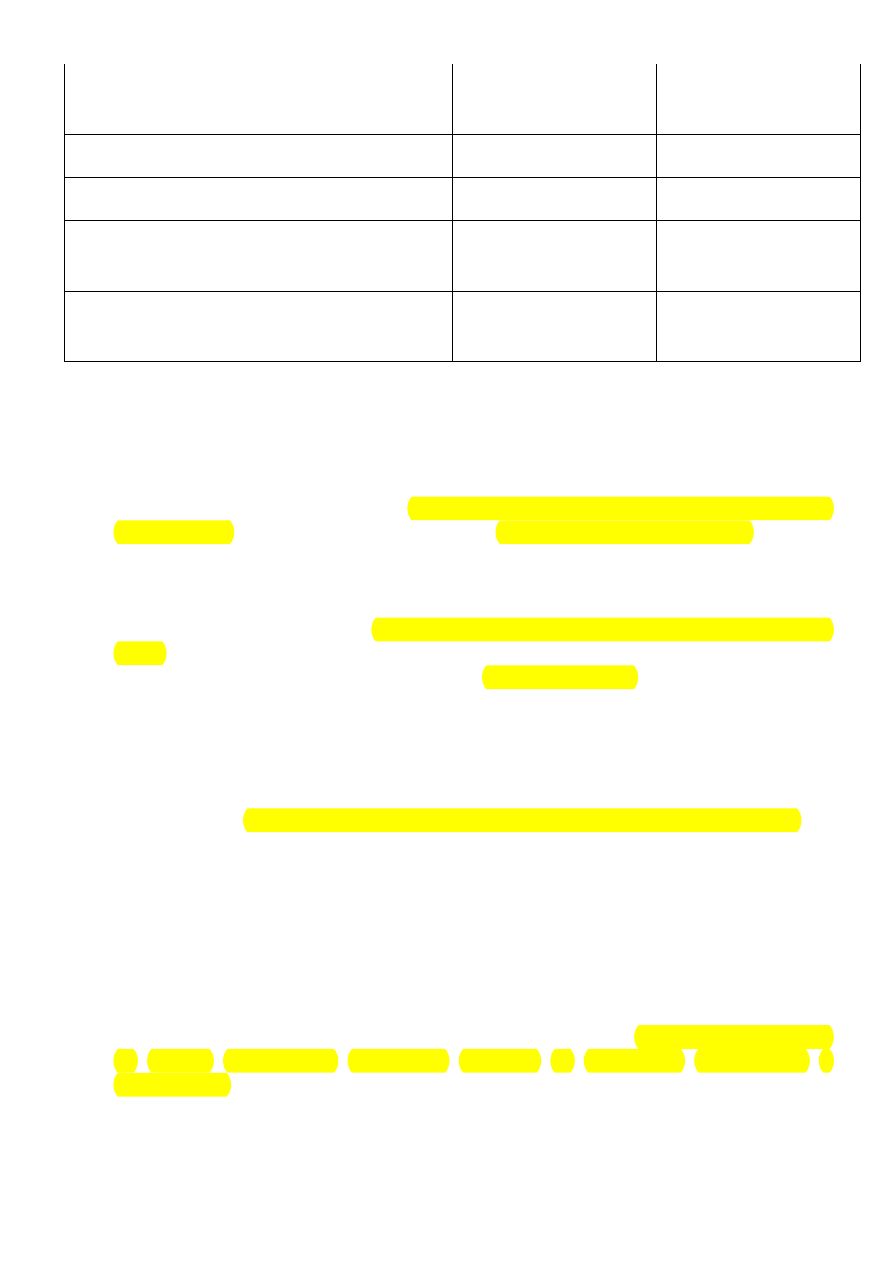
19
zespół Gordona (PAH II)
1q31-q42
Autosomalnie
dominująco (?)
PRKWNK1
PRKWNK4
Nadciśnienie z brachydaktylia
12p
Autosomalne
dominujące
Nadciśnienie z hipercholesterolemia i
hipomagnezemia
mtDNA
mitochondrialne
Choroby nerek wywołane mutacją pojedynczego genu:
Zespół Alporta:
•
Zespół Alporta jest wrodzona dziedziczną glomerulopatią spowodowaną
genetycznie uwarunkowanym zaburzeniem syntezy jednego z łańcuchów
kolagenu IV. Zespołowi temu towarzyszą często upośledzenia słuchu. Rzadko
wystepują zmiany w narządzie wzroku.
•
Przyczyną choroby jest zaburzenie syntezy błony podstwanej spowodowane
genetycznie uwarunkowanym brakiem łańcucha kolagenu IV. W 80-85%
przypadkach choroba jest dziedziczona w sposób dominujący związany z
płcią. Defekt dotyczy wówczas łańcucha 5 kolagenu IV, a zmutowany gen
jest położony na dystalnym ramieniu chromosomu X(COL4A5, Xq22). W
genie COL4A5 zidentyfikowano ponad 300 różnych mutacji. Są to mutacje
bezsensu, mutacje procesu składania RNA lub delecje poniżej 10 par zasad.
Mężczyźni chorzy na zepół Alporta (związany z chromosomem X) nie
przenoszą tej choroby na synów, lecz tylko na córki. Kobiety chore na zespół
Alporta (związany z chromosomem X) przenoszą chorobę w 1/3 do ½
przypadków. U kobiet zwykle jedynym objawem jest mikrohematuria. U
mężczyzn z delecjami w genie COL4A5 stwierdzono połączona z głuchotą
progresję do krańcowej niewydolności nerek w 2 lub 3 dekadzie życia.
•
Choroba przebiega łagodnie u kobiet, ale w sposób umiarkowany lub ciężki u
mężczyzn. W postaci dziedziczonej z sposób autosomalny recesywny defekt
dotyczy łańcuchów 3 i 4 kolagenu IV, a geny kodujące COL4A3 i
COL4A4 znajdują się na chromosomie 2 (2q36-37). Prawdopodobnie defekty
genetyczne mogą występować także na innych chromosomach (13?). Błona
podstawna z omówionych defektem na unikatowe właściwości antygenowe:
nie wiąże ona przeciwciał przeciwko błonie podstawnej. Wymienione zmiany
w błonie podstawnej występują również w naskórku, śliniankach i
soczewkach.
•
Rozpoznanie stawia się w oparciu o charakterystyczne objawy oraz wyniki
badań dodatkowych: rodzinne wystepowanie objawów klinicznych

20
wymienionych powyżej, wynik badania biopsyjnego nerki, a zwłaszcza
wygląd błony podstawnej w mikroskopie elektronowym, wykrycie mutacji
genowej po przeprowadzeniu badań genetycznych, i wzmożone wydalanie z
moczem produktów degradacji kolagenu.
•
W rozpoznaniu różnicowym należy wziąć pod uwagę stany chorobowe
przebiegające z krwiomoczem o charakterze rodzinnym.
•
Nawroty krwiomoczu mogą utrzymywać się wiele lat przed rozwojem
niewydolności nerek. Rozwój niewydolności nerek postępuje powoli. U
mężczyzn terminalna niewydolność nerek rozwija się w 4 i 5 dekadzie zycia.
U kobiet terminalna niewydolność nerek rozwija się rzadziej.
Choroby nerek wywołane mutacją pojedynczego genu
Zwyrodnienie torbielowate (Wielotorbielowatość) nerek:
•
Zwyrodnienie torbielowate jest częstą przyczyną krańcowej niewydolności
nerek. Aż >10% chorych leczonych nerkozastępczo to chorzy ze
zwyrodnieniem torbielowatym nerek. Wielotorbielowatość nerek to
schorzenie charakteryzujące się niezliczoną ilością różnej wielkości
torbieli, rozmieszczonych zarówno w korze jaki i w rdzeniu obu nerek.
Torbiele te powodują zniekształcenie i powiększenie narządu,
Molekularna i kliniczna charakterystyka ADPKD I ARPKD:
ADPKD
ARPKD
Charakterystyka
molekularna:
Autosomalnie dominująco
Autosomalnie
recesywnie
sposób dziedziczenia
PKD1(16p13.3)
PKHD1(16p21.1-12)
Gen
PKD2(4q21-22)
Fibrycystyna(polidacyna
) 4074 reszt
aminokwsowych
produkt genu-białko
Policystyna1(PC-1)- 4302
reszt aminokwasowych
Fibrycystyna(polidacyna
) zintegrowane białko
błonowe
struktura białka
Policystyna2(PC-2)- 968
reszt aminokwasowych
Fibrycystyna/polidacyn
a- nerki, wątroba,
trzustka
lokalizacja tkankowa
PC1L-integralne białko
błonowe
Fibrycystyna/polidacyn
a-białka
cytoplazmatyczne,
rzęski pierotne, ciałko
21
podstawne
lokalizacja komorkowa
PC-2 integralne białko
błonowe podobne do
kanału TRPC
Nieznana
Funkcja
PC-1, PC-2 heterogeniczne 1:6000--1:40000
Charakterystyka
kliniczna:
PC-1 błona
cytoplazmatyczna, rzeski
pierwotne
wczesne dzieciństwo
Czestość występowania
PC-2 reticulum
endoplazmatyczne, rzeski
pierwotne
kanaliki zbiorcze
Wiek wystąpienia EXRD
Pc-2 kanał jonowy
aktywowany przez Ca
Zaburzenia rozwoju
dróg żółciowych,
zwłóknienie wątroby,
nadciśnienie tętnicze,
nadciśnienie wtórne
Umiejscowienie torbieli
nerkowych
1:400-1:1000
objawy pozanerkowe
6 i 7 dekada zycia
wszystkie segmenty
nefronu
torbiele watrobowe,
trzustka, śledziona
tętniaki
wewnątrzczaszkowe,
nadciśnienie tętnicze

22
Prelekcja III:
Wybrane zagadnienia z genetyki chorób tkanki łącznej
Wprowadzenie:
•
Wrodzona podatność na chorobę tkanki łącznej ma znaczenie dla
wystąpienia, rozwoju i ciężkości jej przebiegu
•
Badania populacyjne, w tym badania nad rodzinnym występowaniem chorób
autoimmunologicznych, wykazały związek tych chorób z układem HLA
•
W przeciwieństwie do wielu chorób uwarunkowanych genetycznie przyczyną
chorób tkanki łącznej nie jest defekt 1 genu, lecz dysfunkcja wzajemnej relacji
wielu genów
Toczeń rumieniowaty układowy:
•
Toczeń rumieniowaty układowy (łac. lupus erythematosus systemicus, ang.
systemic lupus erythematosus, SLE ) – choroba autoimmunologiczna, rozwijająca
się na tle złożonych i niejasnych zaburzeń układu odpornościowego,
doprowadzający do procesu zapalnego wielu tkanek i narządów.
•
Zachorowalność szacuje się na 40-50 przypadków na 100 000, 10-krotnie
częściej chorują kobiety i szczyt zachorowań występuje w wieku 16-55 lat.
•
Choroba charakteryzuje się bardzo zróżnicowanym nasileniem i przebiegiem
naturalnym.
•
Przez dłuższy czas mogą dominować objawy z jednego narządu (co może być
niewystarczające do pewnego rozpoznania SLE), mogą pojawiać się częściowe
remisje i zaostrzenia. W miarę trwania choroby mogą pojawiać się objawy z
kolejnych narządów, jednak objawy uprzednio występujące nie ulegają wycofaniu.
•
Genetyczne badania epidemiologiczne dowiodły silnego związku TRU z
wrodzoną podatnością na chorobę
•
Stwierdza się 100 razy większe ryzyko występowania TRU u kolejnych
członków rodziny w porównaniu z ryzykiem stwierdzanym w populacji ogólnej
Toczeń rumieniowaty układowy – podgrupy:
•
Podostry toczeń skórny
•
Toczeń indukowany lekami
•
Toczeń noworodków
•
Toczeń z zespołem Sjögrena
•
Toczeń z zespołem antyfosfolipidowym
Kryteria Amerykańskiego Towarzystwa Reumatologicznego:
•
Zmiany skórne typu rumienia (często w kształcie motyla na twarzy), nigdy
nie przekraczają bruzd nosowo-wargowych.
•
Zmiany skórne rumieniowo-bliznowaciejące – rumień krążkowy (typu DLE).
•
Nadwrażliwość na światło słoneczne.
•
Nadżerki w jamie ustnej.

23
•
Zapalenie lub ból stawów – dotyczące co najmniej dwóch stawów, bez zmian
w obrazie RTG.
•
Zapalenie błon surowiczych – opłucnej (pleuritis) lub osierdzia
(pericarditis), stwierdzone w wywiadzie lub w chwili badania.
•
Zmiany w nerkach – utrzymująca się proteinuria (białkomocz lub obecność
wałeczków nerkowych w moczu.
•
Zaburzenia neuropsychiatryczne – szeroki wachlarz, najczęściej napady
drgawek lub psychoza.
•
Zaburzenia hematologiczne – niedokrwistość hemolityczna z retikulocytozą
lub limfopenia (poniżej 1500 w 1 mm
3
) lub leukopenia (poniżej 4000 w 1 mm
3
) lub
trombocytopenia (poniżej 100 000 w 1 mm
3
).
•
Zaburzenia immunologiczne – obecność komórek LE lub przeciwciał przeciw
dsDNA (natywne DNA), lub przeciwciał anty-Sm, lub fałszywie dodatnie
serologiczne odczyny kiłowe przy ujemnym teście na immobilizację krętków.
•
Przeciwciała przeciwjądrowe (ANA) – w mianie nie niższym niż 80 (zwykle
ANA>160), badane metodą immunofluorescencji lub inna odpowiednia jeśli nie
stosowano leków powodujących lekowe zespoły LE.
•
Za pewnym rozpoznaniem tocznia przemawia spełnienie co najmniej
czterech spośród 11 kryteriów, przy czym kryteria mogą być spełnione w chwili
badania lub w wywiadzie. Spełnienie dwóch lub trzech kryteriów pozwala na
rozpoznanie choroby toczniopodobnej.
•
U ok. 5% chorych nie wykrywa się obecności przeciwciał przeciwjądrowych.
Rozpoznaje się wtedy toczeń seronegatywny.
•
Nie można rozpoznać SLE, jeżeli chory nie spełnia żadnego z kryteriów
immunologicznych choroby, tzn. kryteriów 10 lub 11.
Toczeń trzewny układowy – leczenie:
•
Stosuje się leczenie objawowe, najczęściej farmakologiczne, oparte o leki
immunosupresyjne i przeciwmalaryczne.
•
Zwiększenie spożycia omega-3 wielonienasyconych kwasów tłuszczowych
może mieć korzystny wpływ na objawy choroby.
Twardzina:
•
Twardzina, sklerodermia (łac. Scleroderma) – rzadka, przewlekła choroba
charakteryzująca się stwardnieniem skóry i tkanek w wyniku nadmiernego
gromadzenia kolagenu. Choroba ta jest spowodowana występowaniem przeciwciał
przeciw topoizomerazie oraz centromerom (w CREST).
•
Częściej występuje w rodzinach, w których choroba już wcześniej się
pojawiała.
•
Występuje w dwóch postaciach:
Ograniczonej (skóra palców, przedramion i twarzy)
Układowej (zmiany w skórze, układzie naczyniowym, mięśniowym, kostnym
i narządach wewnętrznych).

24
•
Zmiany skórne charakteryzują się twardymi, wyraźnie odgraniczonymi
ogniskami barwy porcelanowej. Początkowo są one otoczone obwódką barwy
fioletowej, a następnie ulegają przebarwieniu i zanikowi.
•
Łagodną postacią twardziny jest zespół CREST (od Calcinosis -
ogniskowych wapnień, występowaniu objawu Raynauda, zaburzeń przełykowych –
Esophageal dysmotility, Sclerodactylia i Teleangiectasia).
•
Z czasem dochodzi do zwiększenia ilości i pogrubienia wiązek kolagenu,
zaniku odczynu zapalnego i zaniku przydatków skórnych „zatopionych” w
kolagenie.
Zapalenie skórno – mięśniowe:
Dermatomyositis (zapalenie skórno-mięśniowe, DM) – jest odmianą zapalenia
wielomięśniowego, w której zmiany dotyczą głównie mięśni obręczy barkowej i
biodrowej. Występuje dwa razy częściej u kobiet niż u mężczyzn. Czasami dochodzi
do wystąpienia zmian narządowych z zajęciem przełyku (trudności w połykaniu –
dysfagia) i pozostałych mięśni gładkich przewodu pokarmowego , mięśnia serca
(myocarditis), mięśni oddechowych. W przypadku zajęcia mięśni oddechowych,
może dochodzić do niewydolności oddechowej, która w ciężkich przypadkach
może doprowadzić do śmierci . U 20-40% dochodzi do śródmiąższowej choroby
płuc, która może doprowadzić do włóknienia płuc.
•
Objawy i przebieg:
Postępujące, symetryczne osłabienie siły mięśniowej, podwyższony poziom
enzymów mięśniowych (aldolazy i kinazy kreatynowej), zmiany w biopsji mięśnia
(zmiany zapalno-zwyrodnieniowe).
Towarzyszą temu zmiany skórne zlokalizowane najczęściej w obrębie dłoni
(rumienie, grudki nad drobnymi stawami- Objaw Gottrona), wybroczyny,
rumienie w obrębie wałów paznokciowych, hiperkeratoza na dłoniach („ręce
mechanika”) oraz zmiany rumieniowe w obrębie twarzy z nasilonym obrzękiem i
rumieniem w okolicy oczodołów („rzekome okulary”, „heliotrop”).
Przy długo trwającej chorobie, częściej u dzieci, pojawiają się złogi wapnia w
tkance podskórnej.
Dermatomyositis jest rewelatorem nowotworowym (w ok. 20-50%
towarzyszy nowotworom narządów wewnętrznych), zwykle u pacjentów, u których
choroba pojawiła się po 60. roku życia.
U dzieci DM nie jest związane z chorobą nowotworową.
Polimorfizmy genetyczne a predyspozycja do TRU, twardziny
układowej, zapalenia skórno-mięśniowego i wielomięśniowego:
•
Polimorfizm genów klasy III HLA (C2, C4A, C4B)
•
Nosicielstwo allela T1858 PTPN22 (protein tyrosine phosphatase)
•
Polimorfizm genu CTLA4 kodującego antygen związany z cytotoksycznym
limfocytem T

25
•
Polimorfizmy genów kodujących cytokiny: TNF-alfa, TNF-beta, IL-10, IL1A
•
Polimorfizm promotora genu MCP1 (monocyte chemotactic protein 1)
•
Polimorfizm genu fibrylaryny (FBN1) – związek tylko z TU
Polimorfizm genetyczny a autoprzeciwciała u chorych na TU,
twardzinę układową, zapalenie skórno-mięśniowe i wielomięśniowe:
•
Allele klasy II układu HLA są mocnej związane z autoprzeciwciałami niż
podatnością na chorobę lub jej obrazem klinicznym
•
Związek przeciwciał u chorych na TRU, TU, DM i PM z określonymi genami
wskazuje na znaczne zróżnicowanie w zależności od badanej rasy i kraju, w którym
badanie przeprowadzono
•
Opisano związek przeciwciał przeciw topoizomerazie I, ACA, PM-Scl,
przeciwciał przeciw fibrylarynie, U1-RNP z niektórymi genami
Polimorfizm genetyczny a zmiany narządowe u chorych na TU,
zapalenie wielomięśniowe i skórno-mięśniowe:
•
W niektórych badaniach wykazano związek między genami i zmianami
narządowymi u chorych na TU
•
Zwrócono także uwagę na powiązanie pewnych zmian narządowych w
zapaleniu mięśni z obecnością genów. Chorzy z przeciwciałami antysyntetazowi
mieli istotnie częściej zapalenie stawów, gorączkę, śródmiąższowe zapalenie płuc i
allel HLA-DRw52
•
Allel A (-308) TNFA związany jest z wapnieniem tkanek w młodzieńczym
DM i przewlekłym zapaleniu mięśni.
Genetyczne podstawy trombofilii i hemofilii:
Klinicznym obrazem niewydolności toru krzepnięcia jest hemofilia, a przejawem
nadmiernej aktywacji tego układu- trombofilia.
Hemofilia:
•
Hemofilie- grupa trzech uwarunkowanych genetycznie skaz krwotocznych,
których objawy wynikają z niedoborów czynników krzepnięcia:
VIII(hemofilia A),
IX (hemofilia B),
XI (hemofilia C)
•
Łączna częstość hemofilii A i B w populacji wynosi ok. 1 :12000. Hemofilia A
jest 4 do 8 razy częstsza niż hemofilia B.

26
Etiologia:
•
Hemofilia A i B są chorobami sprzężonymi z płcią. Geny, których mutacje
wywołują choroby, znajdują się na chromosomie X. Hemofilie dziedziczone są
wsposób recesywny, co oznacza, iż chorują jedynie osoby z pełną ekspresją
recesywnego genu;
•
Mężczyźni hemizygotyczni względem zmutowanego genu
•
Kobiety homozygotyczne względem zmutowanego genu.
•
Kobieta nosicielka zmutowanego allela genu posiada drugi chromosom X z
prawidłowym allelem i nie choruje.
•
Możliwe jest zachorowanie na hemofilie córki kobiety- nosicielki wadliwego
genu, w przypadku, kiedy ojciec takiej dziewczynki będzie chory na hemofilię:
wówczas obydwie kopie genu będą miały mutacje.
Schemat dziedziczenia hemofilii:
Typy hemofilii;
••••
Hemofilia A- spowodowana mutacja locus Xq28 powodującą niedobór
czynnika VIII krzepnięcia krwi (czynnika antyhemolitycznego):klasyczna
hemofilia
••••
Hemofilia B- mutacja w locus Xq27.1-q27.2, niedobór IX czynnika
krzepnięcia krwi (czynnika Christmasa)
••••
Hemofilia C- mutacja w locus 4q35, niedobór XI czynnika krzepnięcia krwi (
czynnik Rosenthala), najczęściej w populacji Żydów Aszkenazyjskich,
dziedziczenie autosomalne recesywne.
Postacie kliniczne:
•
Ciężka, stężenie czynnika VIII/IX w osoczu <2% normy
•
Umiarkowana, stężenie czynnika VIII/IX w osoczu 2-5% normy
•
łagodna, stężenie czynnika VIII/IX w osoczu 5-25% normy
•
utajona , stężenie czynnika VIII/IX w osoczu 25-50% normy

27
Objawy hemofilii:
•
krwawienia podskórne
•
wylewy do mięśni
•
nawracające wylewy do dużych stawów: obrzęk, ucieplenie, ograniczenie
ruchomości, bolesność, zniszczenie struktury stawu-artropatia hemofilowa
•
krwawienie
•
krwawienia z błony śluzowej jamy ustnej i nosa
•
krwioplucie okołoinfekcyjne
•
u niemowląt i małych dzieci wylewy na głowie
•
krwiomocz
•
krwawienie do OUNu (objawia się silnym bólem głowy)
•
krwawienie pozaotrzewnowe, do mięśnia biodrowo-lędźwiowego
Rozpoznanie:
•
wydłużony czas kaolinowo-kefalinowy (APTT)
•
obniżenie aktywności VIII lub IX
•
czas protrombinowy jest prawidłowy
•
czas krwawienia jest prawidłowy
•
ilość płytek krwi jest prawidłowa
•
prosty test róznicujący hemofilie A i B polega na dodaniu osocza pacjenta z
hemofilia B, jeśli badane osocze pochodzi od pacjenta z hemofilia A, dojdzie do
korekcji czasu kaolinowo-kefalinowego
Leczenie:
•
Leczenie polega na suplementacji preparatami czynników VIII lub IX.
Preparaty są rekombinowane (uzyskane metodami inżynierii genetycznej)
lub wysokooczyszczone, i na ogół są słabo immunogenne. Czynnik VIII ma
czas biologicznego półtrwania 10-15 godzin, czynnik IX około 24 godzin, stąd
w dawkowaniu czynnika IX podaje się go raz na dobę, a w celu utrzymania
prawidłowego stężenia czynnika VIII połowę dawki początkowej podaje się
co 12 godzin.
•
W zależności od postaci hemofilii leczenie może przebiegać profilaktycznie
bądź objawowo.

28
•
Leczenie profilaktyczne (zapobiegawcze) polega na okresowym,
powtarzanym uzupełnieniu brakującego czynnika krzepnięcia (dożylnie).
•
Leczenie objawowe polega na podawaniu dawek czynnika jedynie w
przypadku wystąpienia krwawienia i ma na celu jego zatrzymanie.
•
Leczenie profilaktyczne prowadzone jest dwa razy w tygodniu w przypadku
hemofilii B lub trzy razy w tygodniu w przypadku hemofilii A. Podawanie
leku chroni stawy dzieci przed uszkodzeniami i późniejszym inwalidztwem.
•
W łagodnych postaciach hemofilii A lekiem z wyboru jest pochodna
wazopresyny (DDAVP), uwalniająca czynnik VIII z rezerwy tkankowej jaką
są komórki śródbłonka. Po podaniu DDAVP stężenie czynnika VIII wzrasta
w osoczu 2-4 razy w ciągu 2-4 godzin. Po 3-4 dniach leczenia DDAVP należy
się spodziewać zjawiska tachyfilaksji.
Powikłania:
•
Najczęściej występującymi powikłaniami są postępujące zmiany
zwyrodniniowe (artropatia hemofilowa) na skutek długotrwałych,
nawracających wylewów dostawowych. Dzieje się tak gdy dzieci chore na
hemofilię nie są objęte leczeniem profilaktycznym, polegającym na
regularnym podawaniu czynnik krzepnięcia do 18. roku życia.
•
Powikłaniem leczenia koncentratem czynnika VIII (rzadziej IX) jest
wytworzenie przeciwciał skierowanych przeciwko temu białku. Dotyczy to
15-20% leczonych pacjentów.
Trombofilia:
Trombofilia – wrodzony lub nabyty zespół chorobowy charakteryzujący się
zwiększoną skłonnością do tworzenia zakrzepów żylnych lub rzadko tętniczych
spowodowaną zaburzeniami w ukł. krzepnięcia.
Przyczyny trombofilii wrodzonej:
a)
częste:
-
mutacja Leiden czynnika V (>20%)
-
niedobór białka C (6-9%)
b)
rzadkie:
-
niedobór antytrombiny III
-
niedobór białka S (kofaktora białka C)
Leiden V:
•
Mutacja pojedynczego nukleotydu w genie czynnika V (mutacja typu Leiden)
odpowiada za oporność na aktywowane białko C.

29
•
Czołowe miejsce pod względem częstości występowania na liście
genetycznych czynników ryzyka trombofilii, stwierdza się u 20-50%
pacjentów z zakrzepicą żylną.
•
Wśród kobiet stosujących doustną antykoncepcję hormonalną, u których
wystąpiła zakrzepica, 80% to homozygoty czynnika V Leiden.
Polimorfizmy innych białek a zwiększone ryzyko dziedzicznej
trombofilii:
•
Protrombina
•
Czynnik VII
•
Fibrynogen
•
Inhibitor aktywatora plazminogenu
•
Trombomodulina
•
Homocysteina
•
Plazminogen
•
Białko bogate w histydynę (HRPG)
•
Kofaktor II heparyny
Przyczyny trombofilii nabytej:
•
Zespół antyfosfolipidowy
•
Palenie tytoniu
Genetyka molekularna chorób układu endokrynnego:
MEN:
Mnoga
gruczolakowatość
wewnątrzwydzielnicza,
gruczolakowatość
wewnątrzwydzielnicza (ang. multiple endocrine neoplasia, MEN) może dotyczyć
różnych narządów, obejmuje trzy zespoły określane jako MEN I, MEN IIa, MEN
IIb, dziedziczone w sposób autosomalny dominujący. U krewnych chorych należy
przeprowadzić badania genetyczne, mające na celu wykrycie zmutowanych genów,
co pozwoli na wczesne wykrywanie guzów wchodzących w skład zespołów.

30
MEN I – zespół Wermera:
•
Przyczyna: defekt genu kodującego białko meninę zlokalizowanego na
długim ramieniu 11 chromosomu (11q13). W skład zespołu wchodzą:
Guz dominujący: nowotwór trzustki: insulinoma, gastrinoma, i inne
Pierwotna nadczynność przytarczycy
Guz przedniego płata przysadki
MEN II:
Mnoga gruczolakowatość wewnątrzwydzielnicza typu 2 (zespół gruczolakowatości
wewnątrzwydzielniczej typu 2, MEN II, ang. multiple endocrine neoplasia type II)
– rzadka choroba genetyczna objawiająca się predyspozycją do raka rdzeniastego
tarczycy, guza chromochłonnego nadnerczy i nadczynności przytarczyc, a także
innych rzadszych guzów i wad wrodzonych.
Etiologia MEN II:
MEN II wywołana jest przez mutacje w protoonkogenie RET. Dziedziczenie
mutacji jest autosomalne dominujące z wysoką penetracją.
Epidemiologia MEN II:
Na całym świecie rozpoznano około 500 – 1000 rodzin z MEN II. Szacuje się, że
częstość schorzenia wynosi 1 : 30 000. 80% przypadków stanowi MEN IIA, około
5% MEN IIB.
MEN II:
•
W zależności od obrazu klinicznego wyróżnia się dwa podtypy MEN II:
MEN IIA (zespół Sipple’a)
MEN IIB (zespół Williamsa – Pollocka, zespół Gorlina – Vickersa, zespół
Wagenmanna – Froboese’a)
MEN IIA - zespół Sipple’a:
•
Rak rdzeniasty tarczycy
•
Jedno lub obustronny guz chromochłonny nadnerczy (ponad 50%)
•
Nadczynność przytarczyc spowodowana gruczolakiem lub hiperplazją
komórek gruczołu (15 – 30%)
•
Liszajowate (ang. lichenoid) zmiany skórne, zlokalizowane najczęściej w
górnej części pleców
•
Choroba Hirschsprunga (rzadko)

31
MEN IIB – zespół Gorlina:
•
Szczególnie agresywny rak rdzeniasty tarczycy
•
Nerwiaki podśluzówkowe (np. warg, języka)
•
Nerwiakowłókniaki
•
Nerwiakowłókniakowatość przewodu pokarmowego (ganglioneuromatosis)
mogąca skutkować objawami niedrożnościowymi, okrężnicą olbrzymią
(megacolon), zaparciami lub biegunkami (około 40%)
•
Cechy marfanoidalne (wiotkość stawów, deformacje kostne, kifoskolioza lub
lordoza)
•
Widoczne włókna nerwowe rogówki
•
Guz chromochłonny (40 – 50%)
Rodzinny rak rdzeniasty tarczycy - (FMTC)-non-MEN:
W FMTC rdzeniasty rak tarczycy jest jedyną cechą zespołu
Nerwiakowłókniakowatość:
•
Nerwiakowłókniakowatości (neurofibromatozy, łac. neurofibromatoses) są
grupą chorób genetycznych dziedziczonych autosomalnie dominująco cechujących
się obecnością licznych, niezłośliwych nowotworów pochodzenia nerwowego
•
Znane są przynajmniej dwa typy choroby różniące się objawami i defektem
genetycznym
Nerwiakowłókniakowatość Typu 1:
•
Nerwiakowłókniakowatość typu 1 (neurofibromatoza typu I, choroba von
Recklinghausena, NF 1)- choroba genetyczna o dziedziczeniu autosomalnym
dominującym, ?należąca do grupy fakomatoz. W obrazie klinicznym występują
zmiany skórne, oczne, guzy wewnątrzczaszkowe i inne nowotwory o lokalizacji
pozaczaszkowej, a także zmiany kostne
•
Nerwiakowłókniakowatość typu 1 spowodowana jest mutacją w genie NF1
kodującym neurofibrominę 1.
•
Choroba ta jest nieuleczalna
Etiologia:
•
Choroba spowodowana jest odziedziczoną mutacją genu supresorowego NF1
w locus 17q11.2 kodującego białko neurofibrominę
•
Wskutek spontanicznej mutacji drugiego allela genu NF1 i tzw utraty
heterozygotyczności rozwijają się charakterystyczne dla choroby guzy
nowotworowe i hamartomatyczne

32
Epidemiologia :
NF1 jest stosunkowo częstą chorobą genetyczną i najczęstsza z fakomatoz
Częstość około 1:2500 żywych urodzeń i przynajmniej 1:4000- 1:5000 w populacji
Objawy duże:
•
plamy cafe au lait (>99%) na całym ciele, niekiedy już od urodzenia,
zazwyczaj pojawiające się w okresie niemowlęcym
•
piegowate nakrapiania średnicy 2-3 mm i przebarwienia skórne (70%) w
okolicach pachowych i pachwinowych (objaw Crowe'a), pojawiają się najczęściej w
okresie dojrzewania
•
guzki podskórne będące histologicznie nerwiakowłókniakami (>99%)
•
guzki Lischa (90-95%), ciemnożółte lub brązowe hamartomatyczne guzki
tęczówki, dobrze widoczne w lampie szczelinowej
Objawy wtórne i powikłania:
••••
Nerwiakowłókniaki splotowate (35%), o różnych lokalizacjach - tkanka
podskórna , narządy wewnętrzne
••••
upośledzenie umysłowe , deficyty psychospołeczne, nadpobudliwość, drobne
nieprawidłowości orientacji wzrokowo- przestrzennej(30%)
••••
padaczka (5%) zwykle pod postacią napadów częściowych złożonych lub
uogólnionych napadów toniczno -klonicznych
••••
guzy OUN
••••
glejak nerwu wzrokowego (1,5%)
••••
Nerwiakowłókniaki rdzenia kręgowego
••••
glejak ze stenozą wodociągu mózgu (1,5%)
••••
nowotwory złośliwe
••••
złośliwe guzy otoczki i nerwów obwodowych, MPNST (1,5 % ryzyko że u
pacjenta w ciągu całego życia rozwiana się te guzy wynosi 7-12%)
••••
mięsaki prążkowano komórkowe (1,5%)
••••
guz chromochłonny (0,7%)
••••
białaczki, zwłaszcza wczesnodziecięca białaczka nielimfocytowa (<1,0%)
••••
rakowiak dwunastnicy (1,5%)
••••
powikłania ortopedyczne: dysplazje i deformacje kostne, zwłaszcza skolioza
odcinka piersiowego kręgosłupa, dysplazja skrzydeł większych kości klinowej,
deformacje kości strzałkowej i piszczelowej, złamania patologiczne z tendecją do
tworzenia stawów rzekomych (25%)
••••
zwężenie naczyń nerkowych (1,5%), które może być spowodowane dysplazją
włóknisto - mięśniową i wywoływać nadciśnienie tętnicze nerkowopochodne
Problemy poznawcze
•
Najczęstszym problemem u pacjentów jest upośledzenie poznawcze oraz w
zdolności do uczenia się. Wykazano, że problemy poznawcze występują u około
80% dzieci z NF1 i mają znaczący wpływ na codzienne oraz szkolne życie.

33
•
ADHD występuje u około 38% dzieci z NF1.
•
Wykazano, że problemy poznawcze utrzymują się na stałym poziomie do
dorosłości i nie ulegają pogorszeniu, jak niektóre inne objawy fizykalne w NF-1.
Leczenie
•
NF1 jest chorobą nieuleczalną, możliwe jest jedynie leczenie objawowe.
•
Rokowanie zależy od stopnia nasilenia zmian narządowych i właściwego
prowadzenia pacjenta.
•
Główną przyczyną przedwczesnej śmierci chorych z NF1 są choroby układu
krążenia.
Nerwiakowłókniakowatość Typu 2:
•
Choroba genetyczna o dziedziczeniu autosomalnym dominującym,
przypominająca obrazem klinicznym nerwiakowłókniakowatość typu 1.
•
NF2 spowodowana jest mutacjami w genie NF2 w locus 22q12.2 kodującym
neurofibrominę-2, zwaną także merliną.
Epidemiologia:
NF2 jest rzadką chorobą; ocenia się,że w populacji europejskiej rozpoznawana jest
z częstością 1:210 000, a częstość żywych urodzeń wynosi 1:30 000 - 1:40 000.
•
W obrazie klinicznym NF2 można wyróżnić trzy grupy objawów, obejmujące:
zmiany nowotworowe (charakterystyczne są obustronne schwannoma nerwu
przedsionkowego - 68%)
zmiany skórne (plamy typu cafe au lait - 40%)
zmiany oczne (zaćma - 81%)
Rak jest chorobą genetyczną - nowotwór złośliwy jest w ostatecznym
rachunku chorobą związaną z wadami DNA.
Musi być zburzonych pięć czy sześć systemów regulacyjnych, by
normalna komórka zmieniła się w rakową. - R. Weinberg:
a.
Wzrost przy braku sygnałów inicjujących rozmnażanie.
Większość normalnych komórek czeka z podziałem na odpowiednią informację
z zewnątrz. Komórki rakowe często wysyłają swoje własne sygnały.
b.
Wzrost mimo zakazu.
Kiedy rozrastający się guz uciska sąsiadujące tkanki, wysyłają one chemiczne
sygnały, które mają powstrzymać komórki od dalszych podziałów. Nowotwór
ignoruje takie polecenia.

34
c.
Wyłączenie mechanizmów samozniszczenia.
W zdrowych komórkach uszkodzenia genetyczne przekraczające pewien poziom
krytyczny aktywują zaprogramowane samobójstwo. Komórki rakowe oszukują ten
mechanizm, choć niektóre elementy układu odpornościowego mogą je zmusić do
samozniszczenia.
d.
Umiejętność symulowania rozwoju naczyń krwionośnych
Aby się rozwijać, nowotwór potrzebuje tlenu i składników odżywczych. Uzyskuje
je, zmuszając przebiegające w pobliżu naczynia krwionośne do wytwarzania
nowych odgałęzień przenikających rosnący guz.
e.
Nieśmiertelność
Zdrowe komórki mogą się podzielić najwyżej 70 razy. Komórki nowotworowe
potrzebują większej liczby podziałów, by wytworzyć guz. Obchodzą więc
zabezpieczenia, takie jak telomery znajdujące się na końcach chromosomów. To
przełamuje barierę ograniczającą zdolność do rozmnażania.
f.
Zdolność tworzenia przerzutów i atakowania innych tkanek.
Rak staje się zwykle groźny dla życia, gdy zablokuje mechanizmy ograniczające
jego obecność do organu, w którym powstał. Pojawiają się przerzuty, które w
pewnym momencie zakłócają prawidłowe funkcjonowanie całego organizmu
Niektóre cechy komórki nowotworowej:
•
zmiany ładunku elektrycznego
•
utrata antygenów, nowe antygeny
•
maskowanie antygenów
•
Zaburzenia cytoplazmatyczne, zaburzona budowa i funkcja organelli
komórkowych
•
zmiany w składzie glikoprotein, glikosfingolpidów, utrata lub zmiana
struktury glikolipidów
•
zmieniona fagocytoza i endocytoza
•
obniżona adhezja i zniesienie hamowania kontaktowego wzrostu komórek
nowotworowych
•
zaburzenia transportu przez błonę komórkową, zmiany przepuszczalności
•
wzrost aglutynacji pod wpływem lektyn, zwiększenie ruchliwości cząsteczek
w błonie
•
zmiana aktywności enzymów głównie proteaz i glikozydaz
Klasyczne etapy nowotworzenia:
•
inicjacja
•
promocja
•
progresja
35
Teoria standardowa:
a.
Kancerogeny
Np. promieniowanie UV czy dym tytoniowy bezpośrednio wpływają na zmianę
sekwencji DNA w genach związanych z rakiem
b.
W wyniku mutacji genów supresorowych w komórce nie ma prawidłowo
działających białek które powstrzymają jej wzrost. Komórka dzieli się nadal choć
nie powinna.
c.
Równocześnie mutacje onkogenów powodują zwiększoną aktywność
onkoprotein. Stymulują one dodatkowe rozmnażanie komórek.
d.
Nadmiar onkoprotein i brak białek supresorowych nowotworów powoduje
szybki wzrost liczby zmutowanych komórek
e.
Po wielu cyklach mutacji i ekspansji jedna ze zmodyfikowanych komórek
pokonuje wszelkie ograniczenia utrudniające jej rozwój. Rak atakuję tkankę
przyległą do zajętego organu.
f.
W najbardziej zaawansowanym stadium komórki rakowe przedostają się do
krwi. W odległych miejscach organizmu tworzą wtórne ogniska choroby, czyli
przerzuty. W pewnym momencie zakłócają jego podstawowe funkcje życiowe
Onkogeny:
mechanizm aktywacji protoonkogenu w onkogen:
amplikacja
(zwielokrotnienie genu, mutacje punktowe, insercja promotora, translokacja)
protoonkogen onkogen
mRNA
mRNA
Białko białko zmienione pod względem struktury i funkcji
(onkoproteina)
funkcja fizjologiczna transformacja nowotworowa
Przykłady niektórych onkogenów i ich rola w procesie
nowotworowym:
Onkogen
Miejsce na
chromosomie
Przypuszczalna
funkcja
Typ nowotworu
Czynnik wzrostu
INT?
11q13
Czynnik wzrostu
fibroblastów
Rak żołądka
SIS
22q12
Podjednostka B
czynnika
wzrostu
pochodzącego z
Glioza
(nowotwór
mózgu)
36
płytek
Receptory
czynników
wzrostu
?
10q
Receptor kinazy
tyrozynowej
Liczne
nowotwory
układu
gruczołów
dokrewnych
?
Receptor
czynnika
wzrostu
naskórka
Glejak, rak piersi
?
17q11
Receptor
hormonu
tarczycy
Białaczka
promielocytowa
?
Receptor kinazy
białkowej
Nerwiak
niedojrzały
Białka
transdukcji
sygnału
?
11p15
GTPaza
Rak okrężnicy,
płuc, grasicy
?
12p12
GTPaza
Czerniak, rak
tarczycy, AML*
?
9q34
Kinaza białkowa Przewlekła
białaczka
szpikowa, ostra
białaczka
limfocytowa
Czynniki
transkrypcji
?
2p24
Białko wiążące
DNA
Nerwiak
niedojrzały, rak
płuc
?
6q22
Białko wiążące
DNA
Czerniak
złośliwy,
Chłonia,
białaczka
?
14q24
Wchodzi w
interakcje z
onkogenami jun
dla regulowania
transkrypcij
kostniakomięsak

37
GENY SUPRESOROWE- białko p53:
Zespół Li Fraumeni – brak supresorowego działania białka p53:
Mózg 12%, mięsak tkanek miękkich 12%, rak sutka 25%, nadnercza 1%, kości 6%
(mięsak kości), szpik kostny6% (białaczka)
Inne możliwe nowotwory: płuc, prostaty, trzustki, jelita grubego, Chłoniak,
czerniak
Retinoblastoma (białko Rb) – objawy:
1.
Biały refleks źreniczny
2.
Guz siatkówki
3.
Duży guz gałki ocznej
APC- gen polipowatości rodzinnej, jego rola w powstawaniu nowotworów jelita
grubego
38
Niektóre przykłady genów supresorowych i ich rola w powstawaniu
nowotworu:
Gen (
geny
pokrewne
w
nawiasach
)
Miejsce na
chromoso
mie
Funkcja produktu
genowego
Choroba wywołana
przez mutacje w liniach
komórek rozrodczych
RB1(p107,
p130)
13q14
Hamuje cykl komórkowy,
wiąże się z E2F
Retinoblastoma,
osteosarkoma
APC
5q21
Interakcje z β-kateniną na
szlaku sygnalizacyjnym Wnt
Rodzinna polipowatość
gruczolakowata
NF1
17q11
Zmniejszenie aktywności
białka ras
Nerwiakowłóknikowatoś
ć typu 1
NF2
22q12
Prawdopodobnie stanowi
połączenie pomiędzy
białkami błony komórkowej
i strukturalnymi
podporowymi komórkami
Nerwiakowłóknikowatoś
ć typu 2
P53
(p63,p73)
17p13
Czynnik transkrypcyjny;
indukuje zatrzymanie cyklu
komórkowego lub apoptozę
Zespół Li-Fraumeni
VHL
3p25
Reguluje elongację procesu
transkrypcji
Choroba von Hippla-
Lindaua (rak nerki)
WT1
11p13
Czynnik transkrypcyjny
typu palca cynkowego
Guz Wilmsa
P16 (p15)
9p21
Inhibitor CDK
Czerniak rodzinny
P16(p15)
BRCA1
17q21
Wchodzi w interakcję z
białkiem naprawczym DNA
RADS1
Rodzinny rak
piersi/jajnika
BRCA2
13q12
Wchodzi w interakcję z
białkiem naprawczym DNA
RADS1
Rodzinny rak piersi
PTEN
10q23
Fosfataza
Choroba Cowdena (rak
piersi i tarczycy)
AT
11q22
Regulator cyklu
komórkowego; reaguje na
uszkodzenie DNA;
interakcja z BRCA1
Ataksja teleangiektazja
CHC2?
22q12
Fosforylacje p53 i BRCA1
Zespół Li-Fraumeni
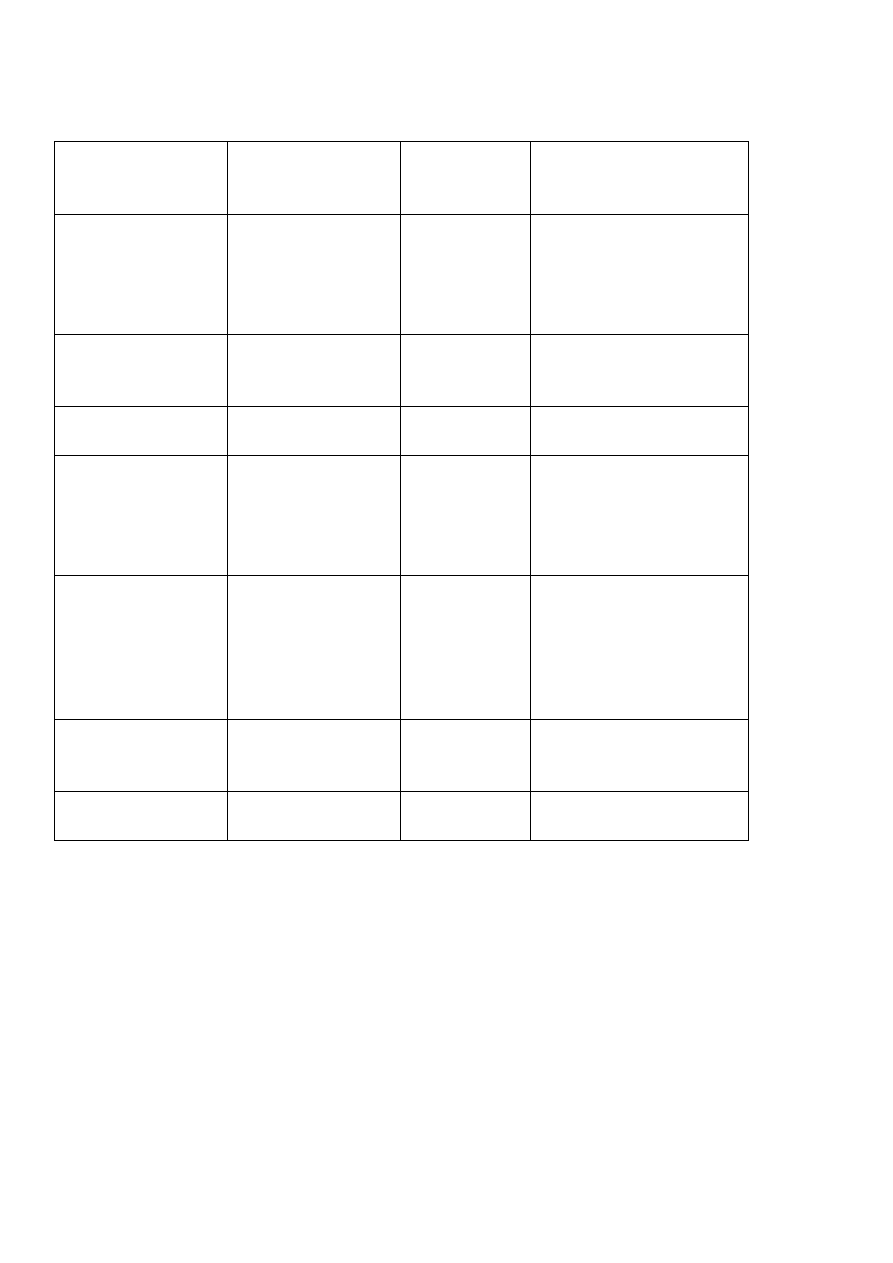
39
Geny, których zmutowane formy odgrywają rolę w nowotworzeniu:
Rodzaj genów
Funkcje w
komórkach
prawidłowych
Rodzaj
mutacji
Udział w
nowotworzeniu
Onkogeny
Proliferacja
komórkowa,
programowana
samo destrukcja
(apoptosis)
Dominujący Nie kontrolowana
proliferacja,
zahamowanie
różnicowania
Supresory
transformacji
nowotworowej
Proliferacja
Recesywna
Stała proliferacja
Geny związane z
angiogenezą
Unaczynienie
Recesywna
Wzrost unaczynienia
Geny związane z
inwazyjnością
Adhezja
komórek,
komunikacja
międzykomórko
wa
Dominujący Inwazyjność,
ruchliwość komórek
Geny związane z
przerzutowanie
m
Komunikacja
miedzykomórko
wa
Recesywna
Zmniejszanie
reaktywności na
sygnały
międzykomórkowe,
autonomizacja
komórek
Geny MHC
Kontrola
immunologiczna
Recesywna
Zmniejszenie
kontroli
immunologicznej
Antygeny
powierzchniowe
?
Dominujący
??
Geny fuzyjne jako przyczyna nowotworów- przewlekła białaczka
szpikowa:
•
Przewlekła białaczka szpikowa (CML):
podwyższona liczba mielocytów we krwi obwodowej
przebieg przewlekły
chorują dorośli
pochodzenie z pojedynczej komórki szpiku
u około 90% chromosom Filadelfia (Ph ¹)
(translokacja 22q na 9q)
gorsze rokowanie przy braku chromosomu
Filadelfia (Ph ¹)

40
•
Inne ostre białaczki z obecnością chromosomu Filadelfia Ph ¹ (+):
Podwyższona liczba limfocytów lub mielocytów
Przebieg ostry
20% dorosłych z ALL Ph ¹(+)
2% dorosłych z AML Ph ¹ (+)
5% dzieci z ALL Ph ¹(+)
Translokacja Filadelfia taka jak w CML
Gorsze rokowanie przy obecności Ph ¹(+)
CML – fuzja genów BCR/ABL:
Translokacja Ph
1
powoduje fuzję dwóch genów
Rak – proces chaotyczny, łączący działanie praw Murphy’ego i
Darwina:
jeśli coś ma się zepsuć, to na pewno się zepsuje, a w środowisku, w którym toczy
się walka o przetrwanie przeżyją i rozmnożą się najbardziej przystosowani.

41
Czynniki wzrostu – rola w kontroli podziałów komórek:
Ważne czynniki wzrostu i antagoniści:
1.
G
0
G
1
Naskórkowy czynniki wzrostu (EGF)
Czynnik wzrostu nerwu (NGF)
Czynnik wzrostu fibroblastów (FGF)
Płytkowopochodny czynnik wzrostu (POGF)
2.
G
1
S
Insulinopodobny czynnik wzrostu (IGF-1)
3.
Antagoniści
Transformujący czynnik wzrostu β (TGF-β)
Czynnik martwicy nowotworu (TNF)
Protoonkogeny o funkcji receptorów, lub aktywności fosfokinazowej:
Receptory błonowe, cytoplazmatyczne, jądrowe

42
Wykrywanie mutacji:
(odsyłam do poprzednich notatek, szczególnie z zakresu biologii)
Metody przesiewowe PCR- SSCP:
•
Analiza konformacji jednoniciowych DNA.
•
Polega na porównaniu konformacji badanych fragmentów kwasów
nukleinowych.
•
Umożliwia wykrywanie punktowych zmian w DNA, np. z. Marfana.
Metoda sekwencjonowania enzymatycznego Sangera:
Polega na enzymatycznej replikacji jednoniciowej matrycy DNA, rozpoczynającej
się od jednego startera, a kończącej wbudowaniem dideoksynukleotydu do nowo
powstałego łańcucha DNA.
Pirosekwencjonowanie – sekwencjonowanie w czasie rzeczywistym:
•
Wykorzystuje się pirofosfonian uwalniany podczas syntezy DNA.
•
W wyniku reakcji enzymatycznych dochodzi do emisji światła, jego
intensywność zależy od ilości uwalnianego pirofosfonianu, czyli od liczby
wbudowanych nukleotydów.
•
Stosowana do genotypowania poznanych wcześniej polimorfizmów.
Diagnostyka pośrednia:
•
Analiza asocjacji- porównuje rozkład alleli różnych polimorfizmów pomiędzy
grupą niespokrewnionych chorych a grupą kontrolną. Stosowana w chorobach
uwarunkowanych wielogenowo/ wieloczynnikowo.
•
Analiza sprzężeń- wykrywa sprzężenia pomiędzy markerami a
poszukiwanymi genami, wymaga badań rodzinnych. Stosowana w chorobach
jednogenowych.
Polimorfizm pojedynczego nukleotydu – SNP (single nucleotide
polymorphism):
Zjawisko zmienności sekwencji DNA, która polega na zmianie pojedynczego
nukleotydu (A, T, C, G) pomiędzy osobnikami danego gatunku lub drugim,
odpowiadającym chromosomem danego osobnika.
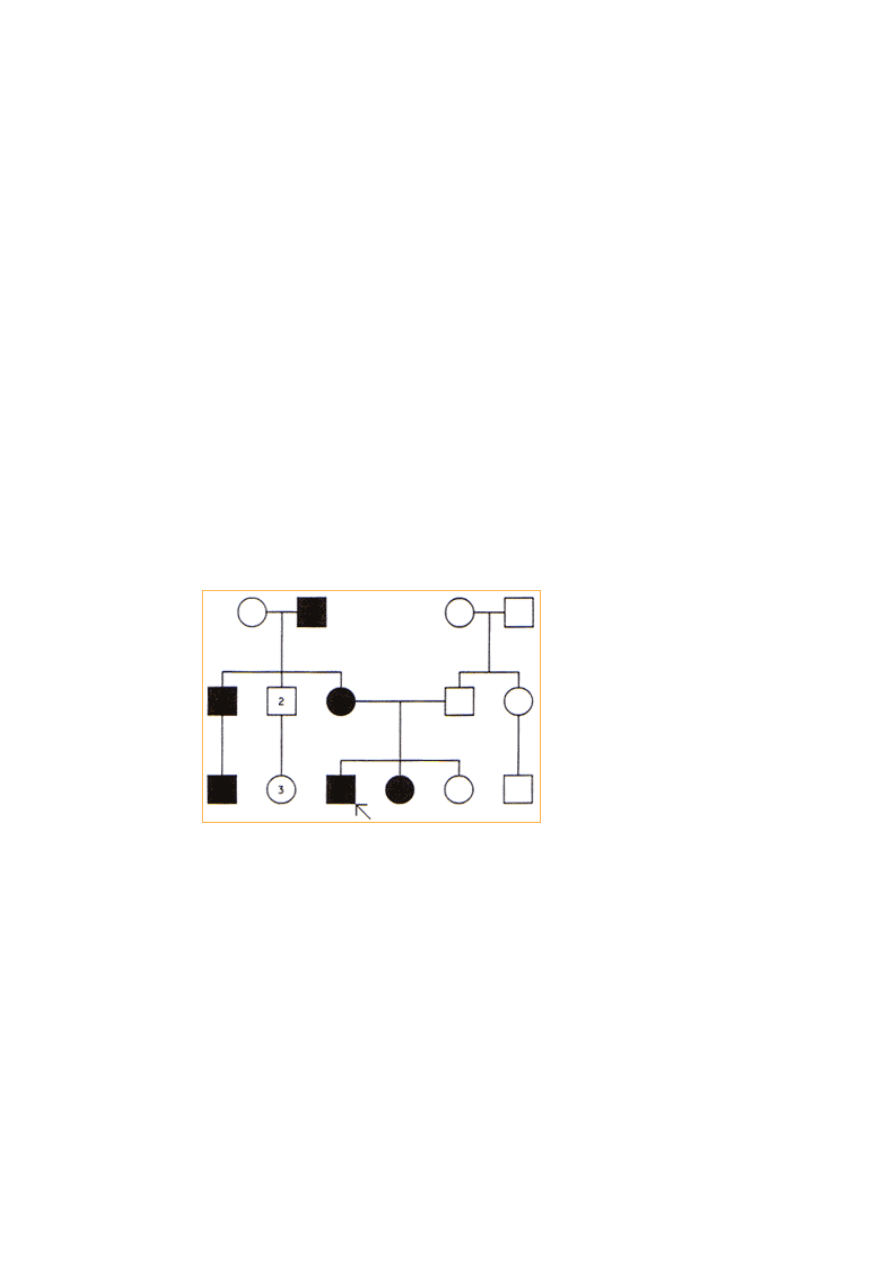
43
PrelekcjaIV:
Wskazania do skierowania pacjenta (rodziny) do Poradni Genetycznej:
•
każda choroba genetycznie uwarunkowana lub o podejrzanej etiologii
genetycznej
•
choroba o niewyjaśnionej etiologii powtarzająca się w rodzinie u dwóch lub
więcej osób
•
wrodzona wada rozwojowa lub zespół wad (także wówczas, gdy jest to
pierwszy przypadek wady rozwojowej w rodzinie)
•
upośledzenie umysłowe lub opóźnienie rozwoju psychomotorycznego (nawet
jeśli jest to pierwszy przypadek w rodzinie)
•
zaburzenia determinacji i różnicowanie płci oraz rozwoju płciowego
•
osoby w wieku rozrodczym, narażone na działanie szkodliwych czynników
mutagennych. Ciężarne eksponowane na czynniki teratogenne (np. infekcje
wirusowe, niektóre leki, alkohol i inne)
•
pary małżeńskie z niepowodzeniami rozrodu (dwa lub więcej poronienia
samoistne, martwe porody lub niepłodność małżeńska)
•
kobiety powyżej 35 roku życia, planujące potomstwo
Model dziedziczenia autosomalnego dominującego:
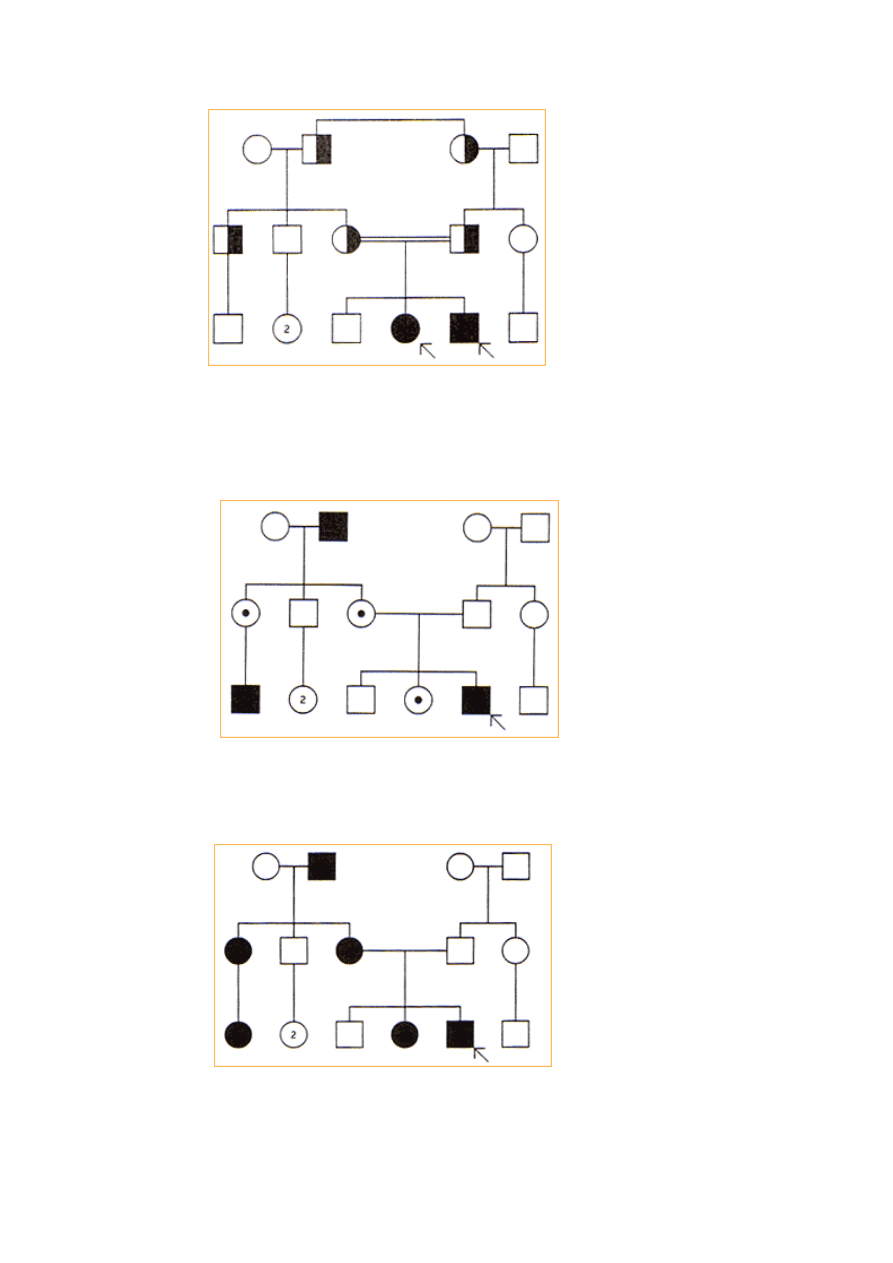
44
Model dziedziczenia autosomalnego recesywnego:
Model dziedziczenia sprzężonego z chromosomem X
(cechy sprzężone recesywnie):
Model dziedziczenia sprzężonego z chromosomem X
(cechy sprzężone dominująco):
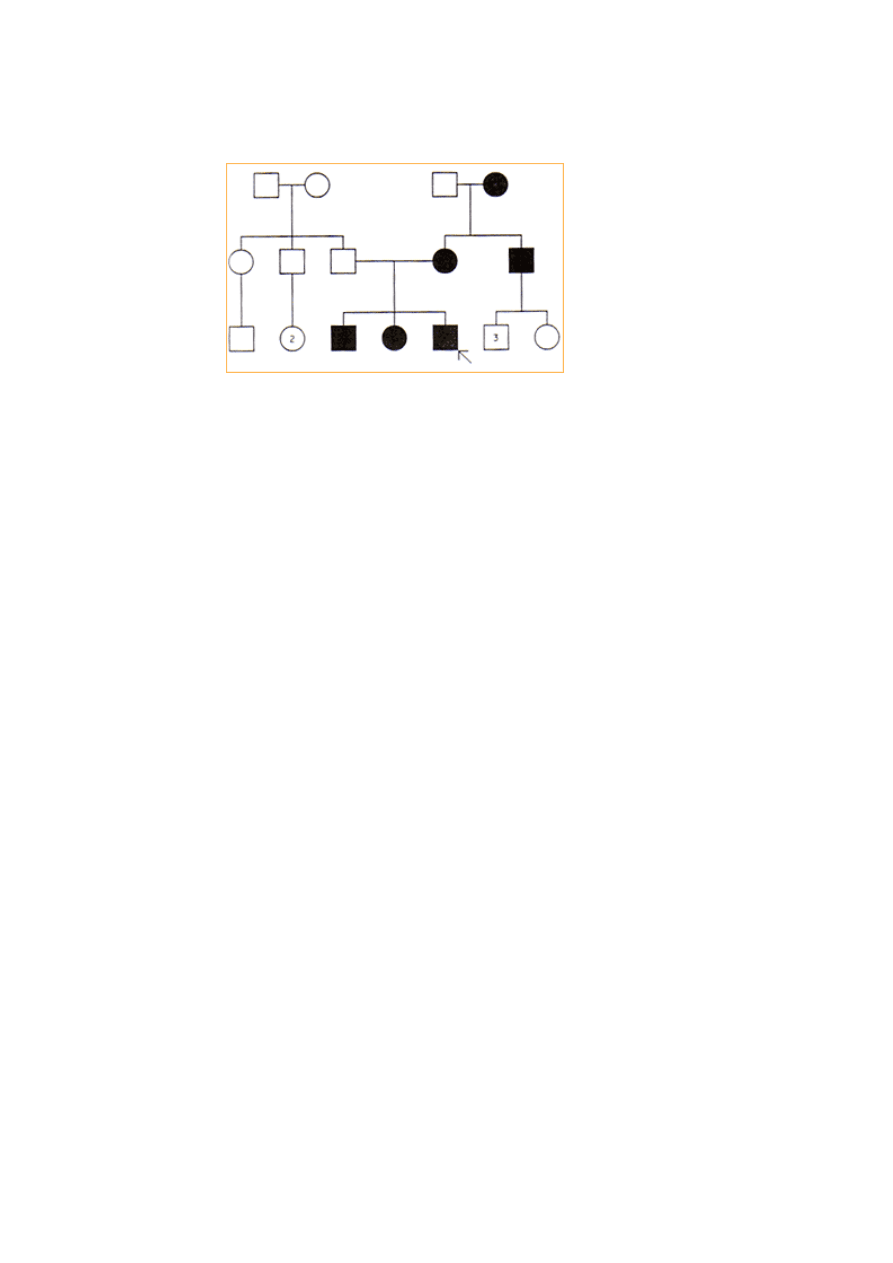
45
Model dziedziczenia mitochondrialnego:
Cele diagnostyki prenatalnej:
•
ocena stanu płodu
•
w ciążach podwyższonego ryzyka wykluczenie wady rozwojowej i / lub
choroby uwarunkowanej genetycznie
•
wykrycie wady rozwojowej i / lub choroby uwarunkowanej genetycznie, w
przypadku których interwencja lekarska w okresie życia wewnątrzmacicznego
stwarza szanse uratowania dziecka lub zmniejsza ryzyko powikłań okresu
okołoporodowego
•
wykrycie u płodu wad wrodzonych, w przypadku których istnieje szansa
uratowania dziecka pod warunkiem interwencji lekarskiej bezpośrednio po
urodzeniu
•
wykrycie wad letalnych
Metody nieinwazyjne:
Ultrasonografia:
Zaleca się przynajmniej 3-krotne wykonanie w czasie trwania ciąży:
•
11-14 tydzień
•
ok. 20 tygodnia
•
ok.30 tygodnia
Cele:
•
potwierdzenie wieku ciążowego
•
ocena żywotności płodu
•
ocena ilości płodów
•
diagnostyka wad płodu
•
ocena przezierności fałdu karkowego (11-14tydzień ciąży)

46
Przezierność karkowa (NT):
Rośnie wraz z wiekiem ciążowym, a tym samym długością ciemieniowo-
siedzeniowa
(CRL-crown-rump lenght)
Normy:
CRL= 45mm (11Hbd); mediana wynosi 1.2mm
CRL= 84mm (13+6Hbd); mediana wynosi 1.9mm
Ryzyko indywidualne obliczamy mnożąc wartość ryzyka wstępnego dla danej
pacjentki (wynikającego z jej wieku oraz wieku ciążowego) przez różnicę miedzy
wartością NT zmierzona a mediana dla danego CRL
Badanie NT:
pozwala zidentyfikować okolo 72% płodów z zespołem Downa
(odsetek wyników fałszywie dodatnich 5%):
NT= 3mm - ryzyko trisomii podwyższone ponad ryzyko wynikające z wieku matki
3 razy
NT= 4mm - ryzyko trisomii podwyższone ponad ryzyko wynikające z wieku matki
18 razy
NT= 5mm - ryzyko trisomii podwyższone ponad ryzyko wynikające z wieku matki
28 razy NT> 5mm - ryzyko trisomii podwyższone ponad ryzyko wynikające z
wieku matki 38 razy
Inne przyczyny zwiększenia grubości fałdu karkowego:
•
niewydolność płodowego układu krążenia związana z wada serca i/lub
dużych naczyń
•
zastój krwi żylnej spowodowany uciskiem
•
nieprawidłowy lub opóźniony rozwój ukladu limfatycznego
•
niedokrwistosc plodowa
•
hipoproteinemia
•
infekcje plodu powodujace niedokrwistosc lub niewydolnosc krążenia
Brak lub niedorozwój kości nosowej u płodu jako marker aberracji
chromosomowych:
Brak kości nosowej stwierdza się:
•
u 67% płodów z trisomią 21
•
u 55% płodów z trisomią 18
•
u 34% płodów z trosomią 13
•
u 11% płodów z monosomią X (zesp TURNERA)
•
u 7% płodów z triploidią

47
Wady wrodzone a aberracje chromosomowe:
Wskazania do wykonania inwazyjnej diagnostyki prenatalnej z ocena kariotypu
plodu:
•
zesp. Dandy’ego-Walkera (1/1000)- wystepuje w ok 50 zespolach
genetycznych (40% aberracje chomosomowe)
•
cystic hygroma (torbielowate struktury w okolicy potyliczno-szyjnej)- 75%
aberracje chromosomowe (gl. zespół Turnera)
•
małogłowie(1/1000)- 15% aberracje chromosomowe (trisomia 13, delecja 4p
i 5p)
•
brak ciała modzelowatego (1/1000) występuje w ok 100 zespołach
genetycznych w tym trisomii 13 i 18
•
przepuklina przeponowa (1/3000) 20% aberracja chromosomowe
•
przepuklina sznura pępowinowego (1/3000) 60% aberracje chromosomowe
•
wady serca (5-10/1000)- 5% aberracje chromosomowe
•
zarośniecie przełyku (1/3000)- 4% aberracje chromosomowe
•
hipotrofia płodu - 1% aberracje chromosomowe (trisomia 21, triploidia)
•
wodonercze - 3% aberracje chromosomowe
Ryzyko aberracji chromosomowych wzrasta wraz z ilością wad wrodzonych
Wyszukiwarka
Podobne podstrony:
Chemia kliniczna kontrola id 11 Nieznany
chemia kliniczna cw 1 2011 id Nieznany
Notatki na egzamin genetyka id Nieznany
arkusz Centrum handlowe 2007 id Nieznany (2)
farma kliniczna wyklad I id 168 Nieznany
GENETYKA WYKLADY PROPS id 18759 Nieznany
Parma centrum id 349488 Nieznany
Medicus Genetyka pomocnicze id Nieznany
IL BiolMed Genetyka2013 2014 id Nieznany
genetyka do nauki id 187516 Nieznany
Chemia kliniczna kontrola id 11 Nieznany
chemia kliniczna cw 1 2011 id Nieznany
Notatki na egzamin genetyka id Nieznany
cw 16 odpowiedzi do pytan id 1 Nieznany
Opracowanie FINAL miniaturka id Nieznany
How to read the equine ECG id 2 Nieznany
więcej podobnych podstron