
Skok do
strony tytułowej
serwisu
Z serwisu korzystało już
82201
osób
do strony tytułowej
Ogólne wprowadzenie
Chemia dla Ciebie
download
linki
|
mikro i makro
|
wyobraźnia
|
nowe spojrzenie
|
termodynamika
|
nie wszystko na raz
|
co los nam zdarzy
|
podsumowanie
|
Na czym polega trudność w studiowaniu chemii fizycznej?
Moim zdaniem, nie na trudności w zrozumieniu fizykochemicznego opisu zjawisk, lecz na niewłaściwym sposobie podania tej
wiedzy. Zrozumienie zjawisk fizykochemicznych, dość często złożonych i wymagających zazwyczaj znajomości matematyki,
przy klarownym wywodzie i logicznym powiązaniu przyczyn i skutków, co najwyżej może wymagać większej ilości czasu
i zwiększonego wysiłku intelektualnego. Problem polega głównie na tym, że ucząc chemii w XXI wieku, wiedząc, że
prawdziwych przyczyn makroskopowych skutków zjawisk szukać trzeba na poziomie cząsteczkowym (świat mikro), ciągle nie
potrafimy zerwać z podziałami i podejściem do zjawisk rodem z XIX wieku (głównie opis świata w skali makro). Ponadto
wykładowcy, autorzy podręczników i twórcy programów studiów (tzw. treści programowych) nie mogą się zdecydować, jak
traktować obiekt swoich działań, czyli studenta pierwszych lat studiów. Z jednej strony zakładają (niestety dość słusznie)
poziom wiedzy, każący tłumaczyć budowę materii na poziomie wywodów Daltona, ale za chwilę przechodzą do omawiania
równania Schrödingera w sposób, jakby to było prawo Pitagorasa czy Talesa. Chcąc wyłożyć chemię fizyczną przy założeniu
rzeczywistego poziomu wiedzy studiujących, trzeba by poświecić na to trzy-czterokrotnie więcej czasu niż dotychczas (co jest
mało realne). Sensownym zabiegiem byłoby włączenie fizykochemicznego opisu zjawisk do nauki różnych dziedzin chemii
(ogólnej, organicznej, nieorganicznej itp.) kosztem zredukowania objętości ich części opisowej. Wówczas przedmiot "chemia
fizyczna" należałoby potraktować jako uporządkowanie i pogłębienie wiedzy zdobytej wcześniej w trakcie nauki tych "innych
chemii". Także powiązanie konsekwentnego tłumaczenia wszelkich zjawisk zasadami termodynamiki z opisem prostych
modeli "makroskopowych" danych zjawisk, byłoby dla studiujących dużym ułatwieniem w percepcji wykładanego materiału.
Z całym szacunkiem dla przeszłości trzeba stworzyć nowy model wykładu chemii fizycznej, pozwalający uczynić z przedmiotu
rzeczywisty fundament wszelkich dziedzin chemii. A ponieważ prawdopodobieństwo wpływu mojego zdania na decyzje tych,
którzy rzeczywiście kształtują proces dydaktyczny, jest mniejsze od szansy trafienia szóstki w totka, niniejsza witrynka ma być
"protezą" ułatwiającą (przynajmniej niektórym) zrozumienie chemii fizycznej, w pewnym stopniu bazującą na powyższych
założeniach. Będzie to swoisty przewodnik po meandrach myślowych autorów podręczników do chemii fizycznej.
A oto dla przykładu krótki cytat z podręcznika akademickiego do chemii fizycznej (jednego z lepszych, moim zdaniem):
... operujemy często pojęciem orbitalu (rozumiejąc je ściśle, jako część kątową funkcji falowej
elektronu).
Orbital traktujemy często jako stan określony trzema liczbami kwantowymi n, l, m, który pomieścić może
maksymalnie dwa elektrony z przeciwnymi spinami
Zapamiętałem więc, że orbital to funkcja (tu już mam wątpliwości, czy chodzi o funkcję w pojęciu ogólnym, czy
o funkcję matematyczną), stan, który może pomieścić (niezbyt to szczęśliwe sformułowanie) elektrony. A już
kawałeczek dalej czytam:
Wszystkie powłoki elektronowe, złożone z poszczególnych orbitali stanowią w atomie tzw. osłonę
elektronową.
(...) Rozpatrzmy teraz ważny przypadek, gdy następuje przeskok elektronów z wyższych powłok
elektronowych na (...) orbital wewnętrznej powłoki ...
Przeskok elektronu (...) prowadzi do emisji dużego kwantu energii
Teraz zaczynam wątpić, czy to, co przed chwila zapamiętałem nie kłóci się z następnymi zdaniami, z których wynika,
że orbitale to fizyczne miejsca, przestrzeń, pomiędzy którymi mogą przemieszczać się elektrony i które stanowią
składniki większej struktury - powłoki. Ponadto mowa o wyższych powłokach, bez wyjaśnienia co to znaczy (wyższe od
czego?). W następnym miejscu czytamy:
Zapełnianie orbitali następuje kolejno według wzrastającej energii.
i kawałek dalej:
Elektrony mając do dyspozycji równocenne energetycznie orbitale ...
Czy to orbitale mają energię, czy chodzi o energię elektronów znajdujących się na tych orbitalach?
Tak oto dobry podręcznik "wyjaśnił" mi pojęcie orbitali i ich energii. Jak mam później zrozumieć nakładanie się orbitali,
tworzenie orbitali cząsteczkowych, orbitale wiążące i antywiążące, hybrydyzację itp. A wystarczyłoby parę zdań więcej
by sprawę uczynić łatwiejszą do zrozumienia.
Dla kogoś, kto ma już jakąś wiedzę na temat elektronów, orbitali, kwantowania energii itp. tego typu skróty myślowe
i brak precyzji sformułowań nie ma większego znaczenia. Taki styl wywodu jest nawet pożądany, bowiem pozwala
uprościć argumentację, dzięki czemu cały wywód staje się bardziej klarowny. Jeśli jednak czyta to ktoś, kto styka się
z tą problematyką po raz pierwszy - szybko nabierze przekonania, że chemia fizyczna to wiedza tak hermetyczna
i zagmatwana, że "uciec" od niej trzeba jak najszybciej.
A ponieważ nie da się "uciec" od chemii fizycznej i chemii w ogóle, bo to przecież opis tego, co dzieje się w nas i wokół
nas - postaram się w następnych podrozdziałach dotyczących energii i materii wyjaśnić, lub może raczej przybliżyć
problem i ułatwić zrozumienie tych pozornych niespójności w podręcznikach. Zapewne i mnie nie do końca uda się
uciec od niekontrolowanych skrótów myślowych i uproszczeń - przepraszam. Obiecuję jednak, że będę się starał, by
było ich jak najmniej.
Aby dobrze zrozumieć chemię (a chemię fizyczną przede wszystkim) trzeba pamiętać o kilku zasadniczych sprawach,
które teraz tylko krótko zasygnalizuję, a w kolejnych podrozdziałach postaram się rozwinąć myśli zawarte w poniższych
Zjawiska obserwowane i omawiane w skali makro (czyli w ilościach dostępnych naszym zmysłom: zlewka z roztworem,
kryształek soli widzialny gołym okiem itp.) są średnią statystyczną, wypadkową, zjawisk mikro (dziejących się na
poziomie cząsteczek i atomów). [
rozwinięcie tematu
]
Wielu pojęć dotyczących mikroświata nie da się zrozumieć, wyobrazić, bowiem nie mają one odpowiedników w znanym
nam makroświecie, a rzeczy, których nie poznaliśmy zmysłami nie potrafimy sobie wyobrazić. Stosujemy wówczas dość
ułomne porównania (np. elektron jako kuleczka) i branie tych porównań za "dobrą monetę" może prowadzić do wielu
nieporozumień. [
rozwinięcie tematu
]
Starajmy się patrzeć na świat, na zjawiska fizykochemiczne świeżym okiem. Wiele pojęć i określeń przyjęliśmy w szkole
"na wiarę" i z czasem podświadomie uznaliśmy je za oczywiste i więcej się nad nimi nie zastanawiamy - a szkoda.
Przemyślmy jeszcze raz te "oczywistości" (np. materia i energia czy ładunek elektryczny). [
więcej na ten temat
]
Rozpatrując różne zjawiska zachodzące w przyrodzie miejmy ciągle na uwadze, że w czasie ich trwania ścierają się
zazwyczaj dwie tendencje - spadek energii wewnętrznej, któremu przeciwstawia się spadek wartości entropii
("uporządkowanie" układu) lub w innym przypadku wzrost entropii (układ dąży do rozproszenia), któremu towarzyszy
zazwyczaj będący w opozycji wzrost energii wewnętrznej [
rozwinięcie tematu
]
Panta rei (wszystko płynie) - tę prawdę znali już starożytni. Nie zapominajmy, że materia nigdy nie trwa w idealnym
bezruchu. Oznacza to między innymi, że w czasie, gdy rozpatrujemy zjawisko czy proces
A
dzieją się jednocześnie
i wpływają wzajemnie na swój przebieg także zjawiska
B
,
C
,
D
... itd. Ta równoległość wzajemnie zależnych zjawisk
powoduje, że ich pełny opis byłby tak skomplikowany (jeśli w ogóle możliwy), że niczego by nie wyjaśniał. Ograniczamy
się więc zazwyczaj do opisu interesującego nas zjawiska, abstrahując od innych, dziejących się równolegle zjawisk,
chyba, że wpływają one na zjawisko obserwowane w stopniu na tyle znacznym, że pominąć ich się nie da. Ponieważ
jednak w różnych warunkach to samo zjawisko współdziała w sposób istotny z różnymi zjawiskami równoległymi, to co
jest prawidłowym opisem w jednej sytuacji, traci na aktualności w innej (np. dysocjacja substancji w roztworze o bardzo
małym stężeniu i w sytuacji wysokiego stężenia) [
rozwinięcie tematu
]
To samo zjawisko opisywane w skali makro i w skali mikro (atom - cząsteczka - kilka cząsteczek) będzie wyglądać
inaczej, co może sprawiać wrażenie, że są to różne rzeczywistości. Nigdy nie zapominajmy, że rzeczywistość jest jedna,
jest jaka jest (w naszym makroskopowym ujęciu świata), a tylko nasze punkty widzenia i związane z tym opisy się
różnią. [
więcej na ten temat
]
podpunktach.
Powyższa wyliczanka ani nie wyczerpuje tematu, ani kolejność tematów nie ma nic wspólnego z ich ciężarem gatunkowym. O istnieniu tych problemów (i innych też) pamiętać
trzeba zawsze, co nie znaczy, że zawsze wszystkie trzeba brać pod uwagę w rozpatrywaniu konkretnego zjawiska. Szczególnie ważne, by przypominać sobie o tych sprawach
wtedy, gdy "coś nam nie gra" w opisie zjawiska. Myślowe rozszerzenie zakresu interpretacji powinno pomóc w zrozumieniu odstępstw od reguły czy spodziewanego zachowania.
Innymi słowy, krótko można to ująć, że w przyrodzie
nie ma wyjątków
, prawa są niezmienne, tylko skutki mogą być różne w różnych sytuacjach (wpływ innych, równoległych
zjawisk).
Mikro i makro
Pierwotnym poznaniem i opisem zjawiska było poznanie i opis układu makroskopowego, dostępnego bezpośrednio naszym
zmysłom. Właściwości poszczególnych stanów skupienia, przejścia między nimi (topnienie, rozpuszczanie, parowanie itp.)
i reakcje chemiczne obserwowaliśmy na poziomie makro i z tych obserwacji powstawały pierwsze sformułowania reguł i praw
rządzących przyrodą. Dopiero później nauka odkryła atomową budowę materii i te same zjawiska zaczęły być rozpatrywane
na poziomie mikrozjawisk, zachodzących w skali mikro - między pojedynczymi cząsteczkami czy atomami. Ten opis zjawiska
na poziomie mikro pokazywał, jak złożonym był proces, który w skali makro wydawał się prostym (np. parowanie czy
rozpuszczanie). Pokazywał jednocześnie, że proces w skali makro nie jest sumą identycznych składowych mikro, lecz średnią
statystyczną, często dość różnych zjawisk elementarnych. Można powiedzieć, że ludzkość odkryła nie tylko fakt, że z pozoru
ciągły kawałek materii zbudowany jest z atomów, ale i że z pozoru proste zjawisko fizykochemiczne "zbudowane" jest z
różniących się między sobą zjawisk elementarnych. Tak więc chcąc zrozumieć np. dyfuzję, musimy sobie uświadomić
zjawisko bezładnych ruchów termicznych cząsteczek, zjawisko przypadkowości zderzeń tych cząsteczek, rozkładu ich energii
translacji (szybkości poruszania się) oraz rolę przypadku, rządzącego w tym świecie mikrozjawisk. Ze "statystycznego"
zsumowania tych wszystkich elementów powstaje zjawisko w skali makro, które często daje się opisać dość prostą zależnością
(cząsteczki z obszaru o większym stężeniu samoistnie przesuwają się do obszaru o stężeniu mniejszym aż do wyrównania
stężenia w całej objętości).
Ponieważ zjawisko makro jest "statystyczną sumą" wielkiej ilości dość różnorodnych zjawisk w skali mikro, jego przebieg
będzie zależał od ilości i rodzaju zjawisk elementarnych, a te z kolei będą zależeć od różnych parametrów zjawiska
(np. temperatury, stężenia, ciśnienia itp.). Przypomina to np. zjawisko mieszania barw. Poniżej zmieszano ze sobą kolor żółty z
czerwonym, uzyskując różne efekty, zależne od ilości jednego i drugiego składnika w mieszaninie. Pola skrajne różnią się
zasadniczo, środkowe są bardziej do siebie zbliżone, ale mało podobne do skrajnych.
Podobnie rzecz wygląda z procesem fizykochemicznym. Jeżeli składają się na niego procesy "czerwony" i "żółty", to
w warunkach, w których decydującą rolę gra mikroproces "czerwony", proces makro będzie przebiegał zdecydowanie inaczej,
niż gdy przewagę, ze względu na zmianę parametrów, uzyska proces elementarny "żółty". W przypadku prowadzenia procesu
w warunkach, gdy udział "żółtego" i "czerwonego" będą podobne, uzyskamy jeszcze inny przebieg procesu.
Jeżeli będziemy uczyć się o tym procesie "na pamięć", jak to ma miejsce w zdecydowanej większości przypadków, trudno
nam będzie zapamiętać, jak taka czy inna zmiana któregoś z parametrów procesu wpływa na jego przebieg. Szczególne,
że parametrów istotnych dla tego przebiegu jest zazwyczaj kilka. Jeżeli zrozumiemy i zapamiętamy podstawowy mechanizm
procesu - wpływ poszczególnych parametrów będzie logiczny i konsekwencje ich zmian oczywiste.
do strony
tytułowej
do góry
strony
Rozpuszczalność a temperatura
Badając zależność rozpuszczalności od temperatury, znajdujemy, że ciała stałe na ogół lepiej się rozpuszczają w wyższej
temperaturze (ale są wyjątki!), gazy w wyższej temperaturze rozpuszczają się gorzej, za to wpływ na ich rozpuszczalność ma
ciśnienie (ze wzrostem ciśnienia rozpuszczalność rośnie), zaś rozpuszczalność cieczy w innej cieczy czasem rośnie
z temperaturą, czasem maleje, a czasem najpierw rośnie a potem maleje. Niektóre ciecze mieszają się ze sobą w sposób
niczym nie ograniczony. I jak to wszystko spamiętać, nie pomylić się na egzaminie, skąd wiedzieć czy dana substancja to nie
"wyjątek" - głowa boli, choć problem (w skali całej chemii fizycznej) stosunkowo niewielki.
zagęszczenia cząsteczek gazu nad cieczą (ciśnienia gazu) - im większe, tym więcej cząsteczek będzie wnikać do wnętrza
roztworu;
powinowactwa gazu do cieczy (siły solwatacji) - im silniejsze, tym więcej cząsteczek gazu będzie tworzyć asocjaty
z cząsteczkami cieczy, co utrudnia opuszczenie roztworu;
ruchliwości cząsteczek gazu (temperatura) - im wyższa temperatura, tym większa ruchliwość cząsteczek gazu (co sprzyja
ruchowi w obu kierunkach), słabsze wiązanie cząsteczek gazu w cieczy (słabsza solwatacja, ułatwia opuszczanie cieczy
przez gaz) i większa ruchliwość cząsteczek cieczy, co też sprzyja "wyrzucaniu" gazu z roztworu. Ponieważ zmiana
temperatury wpływa jednocześnie na trzy procesy, efekt wypadkowy nie zawsze da się przewidzieć. Ponieważ jednak
siły solwatacji są zazwyczaj niewielkie a "zysk energetyczny" cząsteczek cieczy jest zazwyczaj większy niż cząsteczek
gazu, efekt końcowy to zazwyczaj zmniejszanie rozpuszczalności w miarę podwyższania temperatury.
Zastanówmy się zatem, co to znaczy "rozpuszczać się" i jaki jest mechanizm cząsteczkowy takiego procesu. Przejdźmy od
procesu w skali makro do składowych mikroprocesów. Rozpatrzmy na początek sytuacje wyjściowe głównych elementów
naszego procesu, czyli cieczy, gazu i ciała stałego w danej temperaturze.
Cząsteczki cieczy (rozpuszczalnika) poruszają się bezładnymi ruchami z różną prędkością i w różnych kierunkach,
przekazując sobie nawzajem energię kinetyczną (translacji) w procesach zderzeń. Ponadto na cząsteczki te działa siła
grawitacji (ziemskie pole grawitacyjne) i siły oddziaływań innych cząsteczek, głównie siły elektrostatyczne. Te ostatnie siły
mają bardzo duże znaczenie w cieczach polarnych (woda, alkohole) a minimalne w niepolarnych np. węglowodorach.
O sile grawitacji bardzo często zapominamy przy rozpatrywaniu zjawisk zachodzących z udziałem cieczy, a zawdzięczamy jej
istnienie ciśnienia hydrostatycznego.
Gaz - tu rodzaje oddziaływań są analogiczne, z tym, że grawitacja nie odgrywa tu większej roli (stosunkowo małe masy
cząsteczek gazu), a także oddziaływania międzycząsteczkowe, ze względu na duże odległości między cząsteczkami, są nikłe
i można nie zwracać na nie uwagi. Cząsteczki gazu zderzając się ze ścianami naczynia, w którym są zamknięte powodują
powstanie zjawiska ciśnienia, jednakowego we wszystkich kierunkach.
Ciało stałe, to cząsteczki o silnych oddziaływaniach międzycząsteczkowych, drgające w węzłach sieci krystalicznej
(energia kinetyczna) o rozkładzie energii drgań podobnym do rozkładu szybkości poruszania się cząsteczek cieczy.
Jak przebiega proces przechodzenia cząsteczek ciała stałego do roztworu
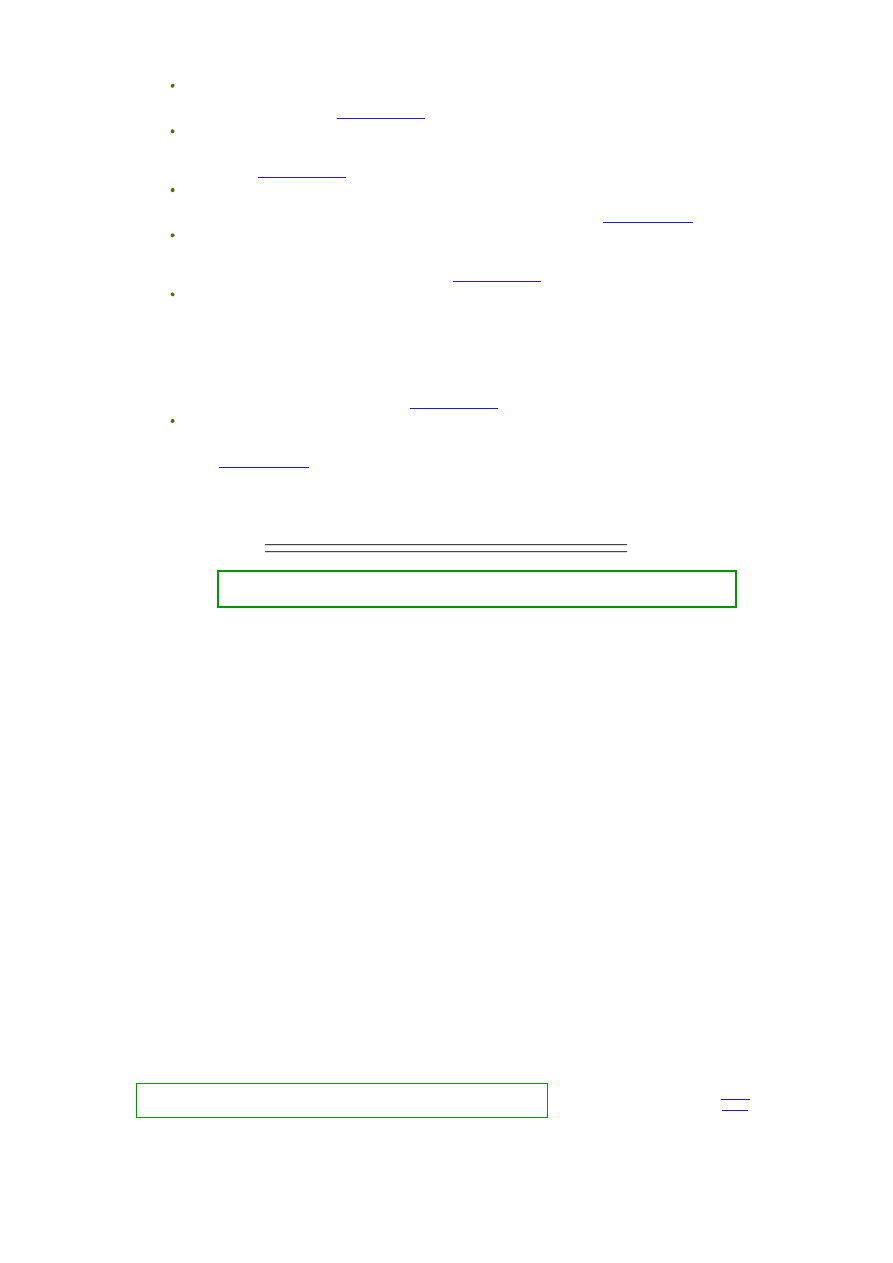
(to oczywiście bardzo skrócony i uproszczony opis).
Krystaliczne ciało stałe symbolizują niebieskie kółka powiązane siłami sieci
krystalicznej (przerywane niebieskie linie). Cząsteczki cieczy symbolizują
czerwone punkty, powiązane siłami oddziaływań międzycząsteczkowych
(przerywane czerwone linie).
Przypadkowe, co do kierunku i siły, drgania cząsteczek w sieci krystalicznej
(niebieskie strzałki) spowodowały "wypchnięcie" cząsteczki ciała stałego z sieci
krystalicznej. Żeby wniknąć pomiędzy cząsteczki cieczy cząsteczka ta musi mieć
energię wystarczającą nie tylko do pokonania sił spójności sieci krystalicznej ale
także do pokonania sił spójności cieczy, a ponadto, po wniknięciu do roztworu
musi się w nim utrzymać. To ostatnie będzie możliwe, jeśli siły oddziaływań między cząsteczkami cieczy, siły oddziaływań
cząsteczek cieczy z cząsteczkami substancji rozpuszczonej oraz siły przyciągania i wiązania cząsteczek w sieć krystaliczną
będą względem siebie odpowiednio równoważone.
Jeżeli teraz zastanawiamy się, jak zależeć będzie od temperatury proces rozpuszczania w skali makro, to rozważyć musimy, jak
zmienią się te trzy siły i jak zmienią się relacje ilościowe między nimi, bo to od nich zależy makroskopowy efekt
rozpuszczania. Najczęściej bywa tak, że podwyższenie temperatury zwiększa energie drgań cząsteczek w sieci (łatwiej się od
niej odrywają), zmniejsza siły oddziaływań międzycząsteczkowych cieczy (łatwiej wniknąć pomiędzy nie) ale zmniejsza
również solwatacje. W końcowym efekcie, tam gdzie proces solwatacji nie jest bardzo istotny, wzrost temperatury zwiększa
rozpuszczalność. Tam jednak gdzie solwatacja odgrywa zasadniczą lub nawet główną rolę - wzrost temperatury będzie
rozpuszczalność osłabiał.
Na podstawie procesu rozpuszczania ciała stałego w cieczy, dość prostego z pozoru i przedstawionego w uproszczony sposób,
wykazaliśmy, jak trudno przewidzieć przebieg procesu makroskopowego, jeśli nie zna się jego mechanizmu na poziomie
mikroświata. A trzeba pamiętać, że zazwyczaj dołączają tu zjawiska dysocjacji, asocjacji i sprawa komplikuje się jeszcze
bardziej. Uczenie się wszystkich przypadków na pamięć jest pozbawione sensu i w praktyce niewykonalne. Zapamiętanie
podstawowych mechanizmów procesu w powiązaniu z innymi wiadomościami (solwatacja, oddziaływania elektrostatyczne
miedzy jonami czy dipolami cząsteczkowymi, energia sieci krystalicznej itp.) pozwala nam dość swobodnie przewidywać
przebieg zjawiska w różnych układach, jeśli tylko znamy konieczne do tego parametry układu w którym proces przebiega.
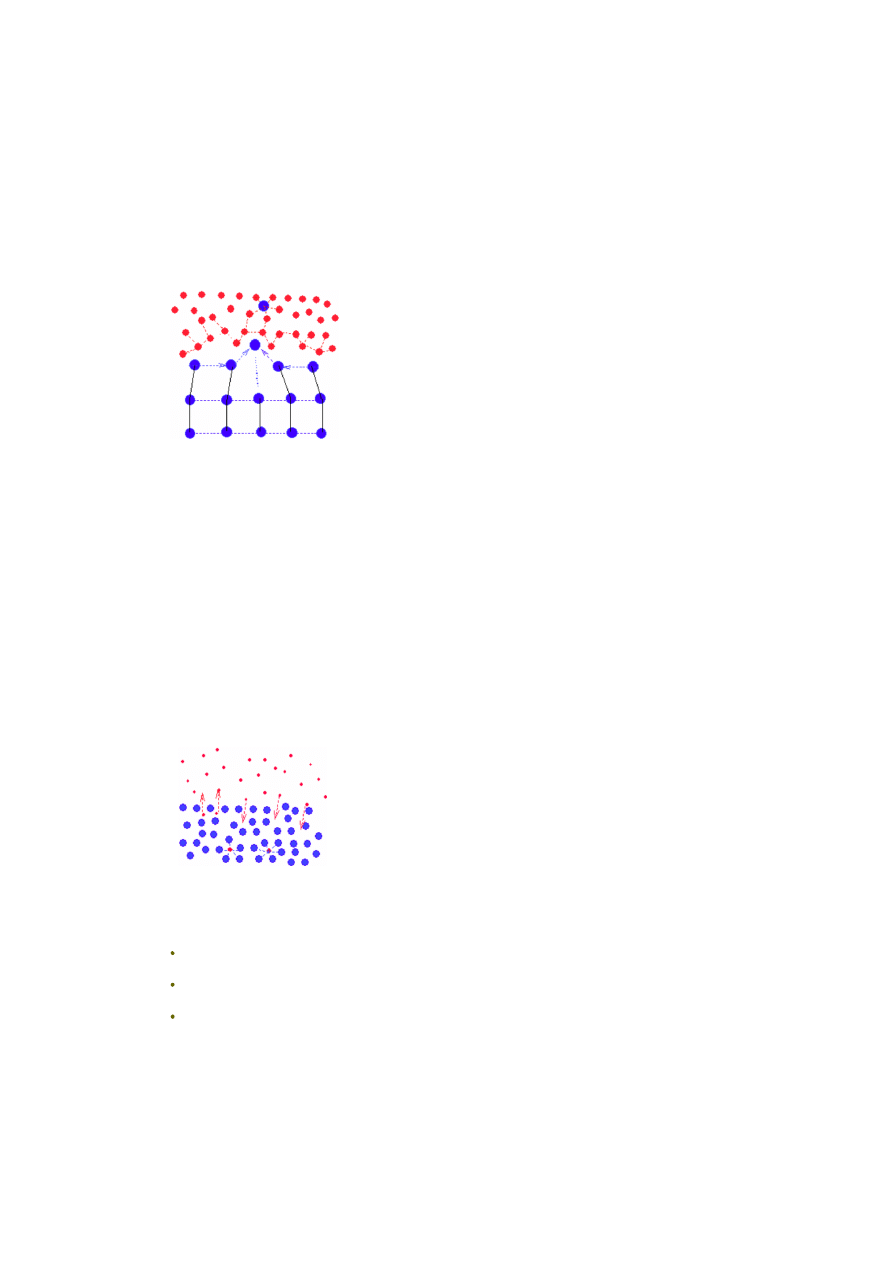
Prześledźmy teraz w podobny sposób zjawisko rozpuszczania się gazu w cieczy.
Gaz styka się z cieczą na granicy faz. Cząsteczki gazu (czerwone kropki) będące
w pobliżu tej granicy faz i zdążające w jej kierunku, mogą (jeśli posiadają
odpowiednią energię kinetyczną) pokonać siły napięcia powierzchniowego
(oddziaływań wiążących warstwę powierzchniową cząsteczek cieczy) i wniknąć
do wnętrza cieczy (niebieskie kropki). Tam, albo zostaną związane siłami
solwatacji przez cząsteczki cieczy, albo poprzez zderzenia z cząsteczkami ponownie
"wyrzucone" nad powierzchnię. Im więcej cząsteczek gazu w cieczy, tym więcej
z nich będzie z niej usuwanych (większe prawdopodobieństwo zderzeń). Po
pewnym czasie wytworzy się równowaga - tyle samo cząsteczek będzie się
rozpuszczać (wnikać do wnętrza cieczy) co opuszczać roztwór. Procesowi opuszczania cieczy przez cząsteczki gazu sprzyja
też ich stosunkowo mała masa, co w połączeniu z ruchliwością powoduje, że siła grawitacji ziemskiej nie ma tu praktycznie
znaczenia. Parametry tego stanu równowagi będą zależały od trzech czynników:
Tak więc najczęściej obserwowany spadek rozpuszczalności gazu w cieczy pod wpływem zwiększania temperatury nie jest
żadnym wyjątkiem od reguły, lecz wynikiem zmian w obrębie składowych procesów zjawiska. Także nikła zazwyczaj
rozpuszczalność gazów w cieczach wynika nie z jakichś ich specjalnych właściwości, a głównie z ich małej masy. Sprzyja ona
uzyskiwaniu dużych prędkości przez cząsteczki gazu po zderzeniu z "masywną" cząsteczką cieczy i ułatwiania w ten sposób
opuszczanie roztworu. Także niewielka zazwyczaj polarność cząsteczek gazu nie sprzyja solwatacji, która jest ważnym
czynnikiem utrzymującym gaz w cieczy. Gaz o dużej polarności (np. HCl) rozpuszcza się w polarnych cieczach bardzo dobrze
(HCl
aq
około 35%
w wodzie). Warto też zaznaczyć, że niska polarność gazów, to nie specyficzna ich właściwość, a raczej

odwrotnie - stan gazowy jest skutkiem niskiej polarności, bowiem cząsteczki nie wiążąc się ze sobą w asocjaty nie tworzą
elementów o dużej masie. Gdyby cząsteczki wody nie były polarne, woda w warunkach standardowych byłaby gazem o
właściwościach fizycznych zbliżonych do amoniaku bądź metanu.
Zajmijmy się teraz rozpuszczaniem cieczy, a dokładniej mieszaniem się cieczy, bowiem wskazać tu, co jest rozpuszczalnikiem
a co substancją rozpuszczoną, jest dość trudno. Na tym przykładzie można przy okazji zauważyć, jak zwodnicze i mało
precyzyjne są niektóre używane przez nas pojęcia (rozpuszczalnik, rozpuszczanie A w B itp.).
rys. 1
rys. 2
rys. 3
Rysunek po lewej symbolizuje dwie ciecze (cząsteczki "zielone" i
"czerwone"). Mieszanie będzie polegało na wnikaniu cząsteczek
zielonych między czerwone i czerwonych między zielone, aż do uzyskania
jednorodnej mieszaniny (ostatni rysunek).
Jeżeli jedna ciecz jest polarna, z silnymi oddziaływaniami
międzycząsteczkowymi, a druga ciecz jest niepolarna, to mieszanie takich
cieczy jest niemożliwe lub bardzo utrudnione. Mieszaniu się cząsteczek
mogą przeszkadzać siły międzycząsteczkowe, uniemożliwiające
wniknięcie "obcych" cząsteczek pomiędzy silnie przyciągające się
cząsteczki cieczy polarnej (rys. 2).
Jeżeli siły spójności cieczy polarnej pozwolą nawet na wprowadzenie
cieczy niepolarnej (np. przez silne wstrząsanie czy mieszanie), to siły
międzycząsteczkowe polarnych cząsteczek będą tworzyć powiązane
struktury i "wypychać" z nich cząsteczki niepolarne. Polarne cząsteczki,
które wnikną pomiędzy niepolarne, będą na tych samych zasadach
odtwarzać powiązane struktury łącząc się w większe agregaty i po chwili
obie ciecze będą znów rozdzielone.
Przy niezbyt silnych oddziaływaniach międzycząsteczkowych niewielka
część cieczy polarnej rozpuści się w niepolarnej i niewielka część cieczy
niepolarnej rozpuści się w cieczy polarnej. Otrzymamy dwie fazy ciekłe -
nasycony roztwór cieczy polarnej w niepolarnej i nasycony roztwór
cieczy niepolarnej w polarnej.
Jeżeli polarne będą obie ciecze, to mieszanie nastąpi dość łatwo, bowiem
oddziaływanie cząsteczek czerwona-zielona, czerwona-czerwona i
zielona-zielona będą podobne i zastąpienie w strukturach polarnych
cieczy jednych cząsteczek drugimi nie będzie stanowiło problemu
energetycznego.
Pewną rolę w mieszaniu się cieczy może odgrywać także wielkość
cząsteczek mieszanych substancji (na podobnej zasadzie, jak to
opisywaliśmy przy rozpuszczaniu gazów w cieczach).
Przy mieszaniu cieczy niepolarnych nie występują silne siły oddziaływań
międzycząsteczkowych i wymieszanie się cząsteczek następuje bez
większych problemów. Generalnie więc, zgodnie z zasadą "podobne
rozpuszcza się w podobnym", kłopoty z rozpuszczalnością pojawiają się
w miarę zwiększania się różnic między polarnościami mieszanych cieczy.
Wzrost temperatury, osłabiając oddziaływania międzycząsteczkowe,
zazwyczaj zwiększa rozpuszczalność (mieszalność cieczy), o ile nie
wpływa jednocześnie na inne zjawiska zachodzące wraz z rozpuszczaniem
(np. hydrolizę, dysocjację, solwatację itp.).
Tak więc zamiast próbować zapamiętać reguły dotyczące zjawiska rozpuszczania w skali makro, które są różne dla różnych
układów i różnie reagują na zmianę parametrów (np. temperatury), lepiej zastanowić się nad mechanizmem zjawiska (czy też,
jak już wiemy - zjawisk) w skali cząsteczkowej i na tej podstawie zbudować wyobrażenie o istocie procesu. Wówczas dla
każdej konkretnej sytuacji (a w najgorszym razie dla większości sytuacji) będziemy potrafili trafnie przewidzieć efekt makro
na podstawie analizy zjawisk na poziomie cząsteczkowym. Nawet korzystanie z popularnych reguł (np. owe przywołane już
wcześniej "podobne w podobnym") staje się bezpieczniejsze jeżeli znamy mechanizm (np. co rozumieć pod pojęciem
"podobne").
"Klasyczna" chemia fizyczna opisuje poruszany wyżej temat roztworów o wiele bardziej jednoznacznie i precyzyjnie
(termodynamiczne funkcje nadmiarowe, entalpie procesów np. solwatacji i rozpuszczania, entropia, roztwory idealne,
regularne, doskonałe, rzeczywiste itp.). Jeśli jednak zaczniemy studiować to "matematycznie doskonałe" ujęcie problematyki
roztworów po wcześniejszym przemyśleniu sprawy w taki uproszczony sposób (jak powyżej), skorzystamy z tego studiowania
o wiele więcej, niż gdy podejdziemy do tego bez "oswojenia" problemu i przygotowania.
do strony
tytułowej
do góry
strony
Skręcalność płaszczyzny polaryzacji światła spolaryzowanego
Izomeria optyczna sprawia zazwyczaj sporo kłopotów. Po pierwsze, dla swobodnego poruszania się wśród zagadnień w niej
omawianych potrzebna jest spora doza wyobraźni przestrzennej, a nie każdy ją posiada, zaś po drugie - nie bardzo wiadomo o
co tam chodzi. Co to za dziwna i tajemnicza cecha tych asymetrycznych cząsteczek, wywołująca skręcenie płaszczyzny drgań
fali światła. Omawianie zjawiska w skali makro sprowadza się do podziału substancji, na takie, które skręcają tę płaszczyznę,
bo mają asymetryczny atom węgla lub same są asymetryczne, oraz na takie, w których są atomy węgla asymetrycznego, ale
płaszczyzny polaryzacji nie skręcają (formy mezo), oraz na takie, które zawierają tyle samo cząsteczek skręcających w lewo
co i w prawo (racematy). Do tego wszystkiego dochodzą rozróżnienia diastereoizomerów i enancjomerów, konfiguracji L i D
oraz R i S, anomeryczne formy α i β ... i mętlik w głowie pełny i obezwładniający. Te ostatnie rozróżnienia, to sprawa
umowna, pewien fachowy język i nie ma wyboru - trzeba się go nauczyć. Jednak na samo zjawisko można spojrzeć nieco
inaczej niż to czyni większość podręczników - i może dla niektórych będzie to spojrzenie rozjaśniające sytuację.
Ostrzegam, że zamieszczone tu "wyjaśnienia" nie należą do oficjalnej nauki, są mocno popularno-naukowe (czasem bardziej
popularne niż naukowe) ale mam nadzieję, że ułatwią zrozumienie istoty zjawiska.
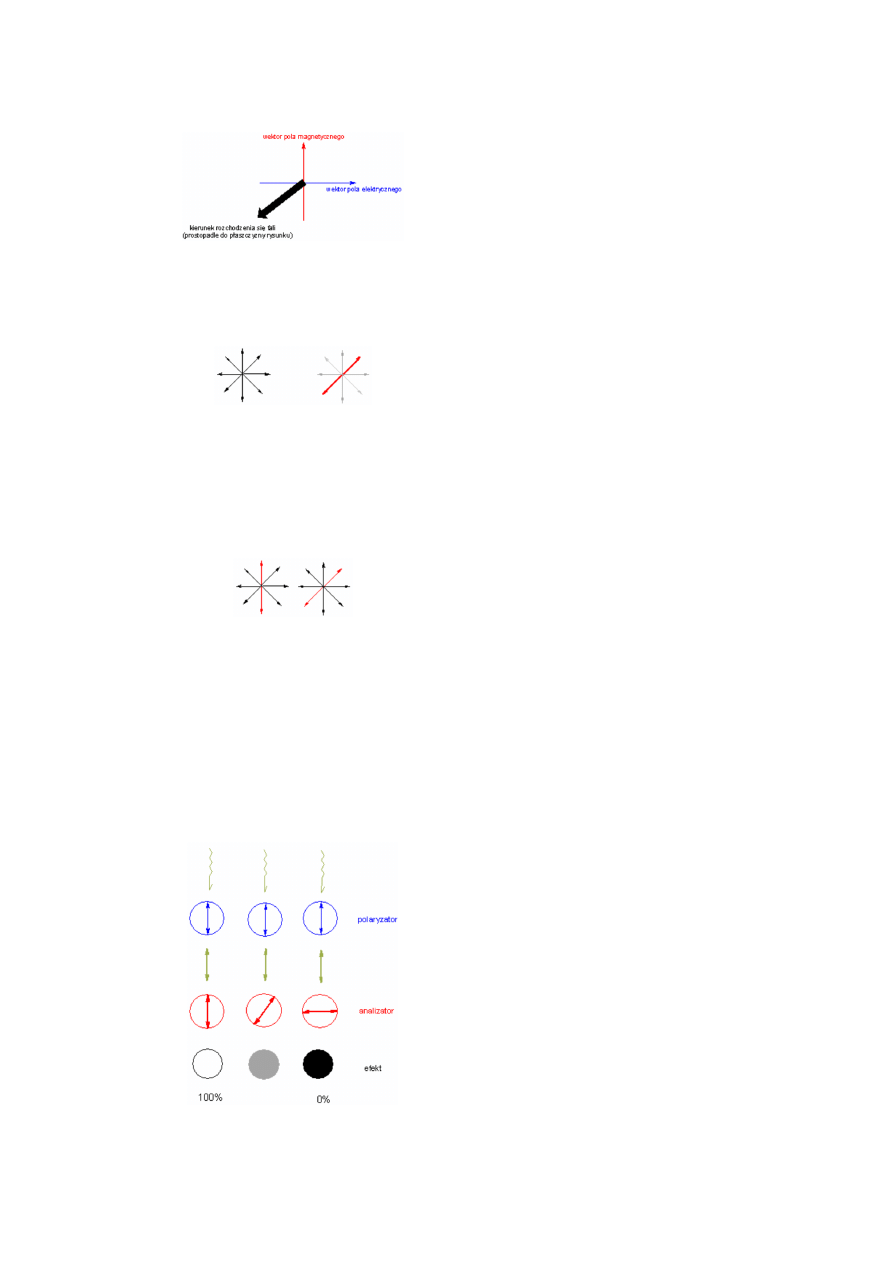
Zacząć trzeba od tego, że fala świetlna to kolejno po sobie
następujące zmiany natężenia pola elektrycznego i pola
magnetycznego (fala elektromagnetyczna). Wektory tych
pól (magnetycznego i elektrycznego) są do siebie
prostopadłe i jednocześnie prostopadłe do kierunku
rozchodzenia się fali. Tak więc patrząc na zbliżającą się do
nas falę elektromagnetyczną "widzimy" wektory tych pól
jako prostopadłe do siebie w płaszczyźnie naszego
widzenia. Promień świetlny wytworzony przez termiczne
źródło światła jest wiązką fal elektromagnetycznych o
bardzo różnych ułożeniach płaszczyzn wektorów
elektrycznych czy magnetycznych. W samej wiązce nie ma
wyróżnionego któregokolwiek kierunku drgań pola
(ułożenia wektora pola).
Jeżeli jednak taki "nieuporządkowany" promień (rysunek
po lewej) przepuścimy przez odpowiedni ośrodek, np.
dwójłomny kryształ szpatu islandzkiego (CaCO
3
), pod
odpowiednim kątem w stosunku do osi sieci krystalicznej,
to promień zostanie rozszczepiony na dwa promienie
o uporządkowanych kierunkach drgań. Każdy z tych
nowych promieni będzie składał się z wiązki fal drgających
w jednej płaszczyźnie (rysunek prawy, polaryzacja
w płaszczyźnie oznaczonej na czerwono, szare promienie
ulegają wygaszeniu). Wiązkę tak drgających fal świetlnych
nazywamy
światłem
spolaryzowanym.
Fala
elektromagnetyczna w wiązce spolaryzowanej nie różni się
niczym od fali w wiązce nieuporządkowanej, natomiast
światło spolaryzowane (wiązka fal o wyróżnionej
płaszczyźnie drgań) uzyskuje nowe właściwości,
wynikające z faktu uporządkowania. A właściwie nie tyle
uzyskuje nowe właściwości, co uzyskuje cechy
pozwalające zauważyć te właściwości. Jedną
z podstawowych "nowych" cech wiązki światła
spolaryzowanego jest wyróżniona płaszczyzna drgań.
Istnienie takiego wyróżnionego kierunku powoduje, że
wiązka przepuszczana przez ośrodki zawierające elementy
asymetryczności zmienia kierunek (płaszczyznę) drgań -
następuje skręcenie płaszczyzny polaryzacji promienia
spolaryzowanego. Takiemu samemu procesowi skręcenia
ulegają płaszczyzny drgań w wiązce niespolaryzowanej,
tylko wówczas nie ma sposobu by to zjawisko zauważyć.
Właściwości wiązki skręconej niczym nie różnią się od
właściwości tej wiązki przed skręceniem płaszczyzn drgań.
Gdyby na rysunku obok nie wyróżniono kolorem
czerwonym jednej fali, skręcenie całej wiązki o 45°
w prawo byłoby niezauważalne.
skręcenie płaszczyzn drgań wszystkich fal w promieniu w identyczny sposób
nie powoduje praktycznie żadnej zmiany w postrzeganiu tego promienia (jego
parametrów)
Jeżeli zbudujemy układ, w którym wiązka światła zostanie
najpierw spolaryzowana przez polaryzator (np. kryształ
szpatu islandzkiego) a potem przepuszczona przez
analizator (taki sam polaryzator jak pierwszy, tylko pełni w
tym układzie inną rolę) to ilość światła, która przejdzie
przez taki układ zależy od kąta między płaszczyznami
polaryzacji polaryzatora I i polaryzatora II (analizatora).
Jeśli kąt między płaszczyznami jest 0° - przechodzi całe
światło, jeżeli kąt ten wynosi 90° - wiązka zostaje
całkowicie wygaszona, w przypadkach pośrednich
ustawienia płaszczyzn efekt wygaszania jest częściowy.
Innymi słowy możemy powiedzieć, że jeśli spolaryzowana
wiązka światła pada na analizator w taki sposób, że jej
płaszczyzna polaryzacji jest zgodna z płaszczyzną
polaryzacji analizatora - przechodzi przez analizator
praktycznie bez strat. Jeśli wiązka pada tak na analizator,
że kąt między jej płaszczyzną polaryzacji a płaszczyzną
polaryzacji analizatora wynosi 90° - wiązka przez
analizator nie przechodzi (całkowite wygaszenie). Jest to
bardzo uproszczony schemat działania polarymetru, czyli
urządzenia pozwalającego określić wielkość kąta skręcenia
płaszczyzny polaryzacji światła spolaryzowanego.
Na rysunku poniżej zamieszczono schemat działania
polarymetru. Zasada działania tego urządzenia polega na
zniwelowaniu efektu osłabienia wiązki światła
spolaryzowanego, będącego konsekwencją skręcenia
płaszczyzny polaryzacji tej wiązki przez badany roztwór.
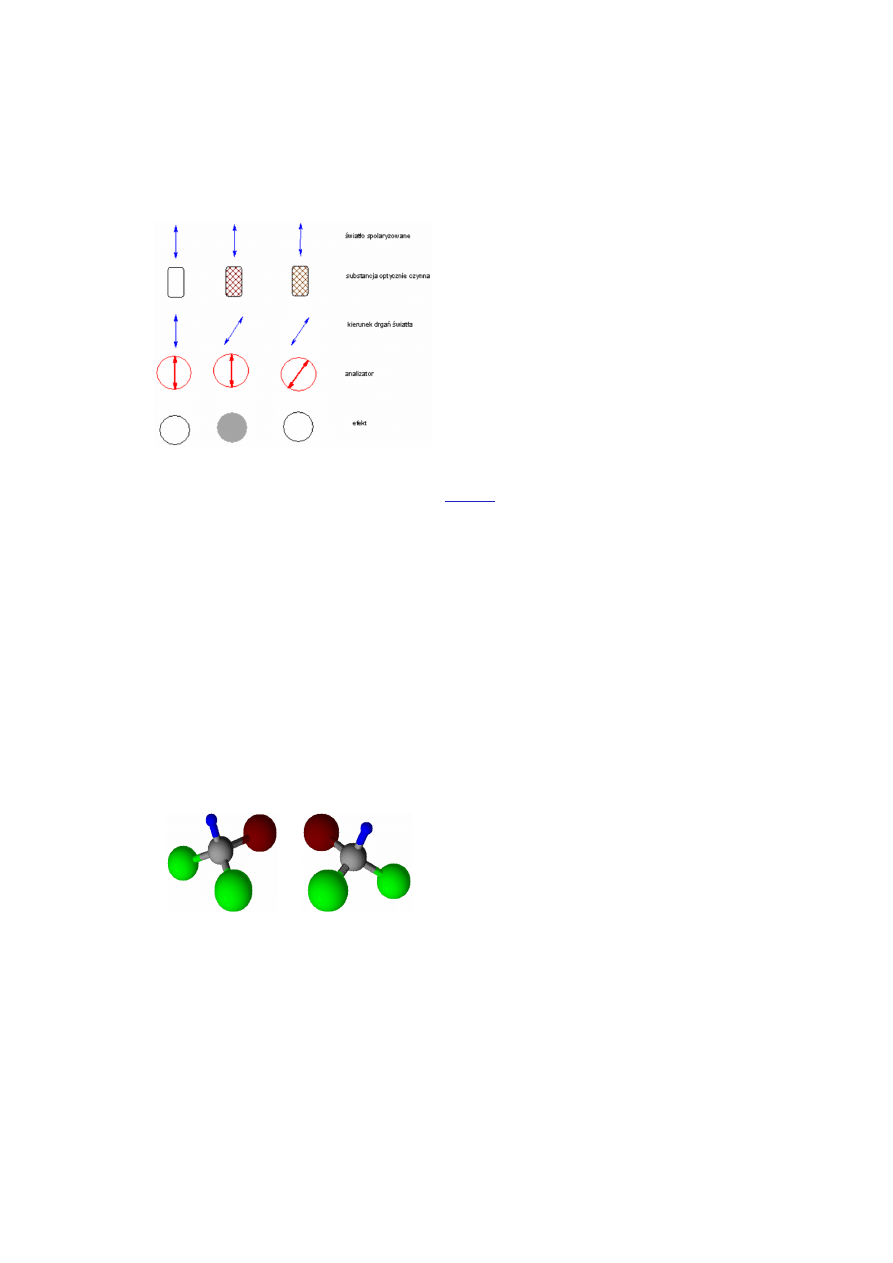
Praktyczne wykorzystanie polarymetru polega na tym, że
między polaryzator a analizator (ruchomy polaryzator)
wstawiamy naczynie z czystym rozpuszczalnikiem (woda,
ślepa próba) i ustawienie analizatora, przy którym nie
następuje osłabienie wiązki spolaryzowanej uznajemy za
zero. (Rysunek poniżej, pierwsza kolumna).
Następnie między polaryzator a analizator wstawiamy
naczynie z roztworem substancji optycznie aktywnej -
skręcającej
płaszczyznę
polaryzacji
światła
spolaryzowanego (kolumna druga rysunku obok). To
skręcenie powoduje, że światło padające na analizator drga
w płaszczyźnie tworzącej pewien kąt z płaszczyzną
polaryzacji (przepuszczania) analizatora. W konsekwencji
powoduje to osłabienie wiązki.
Jeżeli teraz przekręcimy analizator tak, żeby jego
płaszczyzna polaryzacji pokryła się z płaszczyzną
polaryzacji światła, które przeszło przez określoną warstwę
roztworu optycznie aktywnego, znów uzyskamy całkowitą
przepuszczalność. Na podstawie kąta, o jaki trzeba było
obrócić analizator dla uzyskania pełnej przepuszczalności
światła, wnioskujemy o kącie skręcenia płaszczyzny wiązki
spolaryzowanej przez badany roztwór.
Dla
światła
o określonej długości fali
(monochromatycznego) w danej temperaturze kąt
skręcenia płaszczyzny światła spolaryzowanego będzie
zależał od grubości warstwy roztworu i stężenia substancji
optycznie czynnej.
(patrz: też)
Tak mniej więcej wygląda sprawa z punktu widzenia makroskopowego opisu zjawiska. Nas jednak, jak zwykle, bardziej
interesuje molekularny aspekt sprawy - on bowiem pozwoli nam (tak przynajmniej przypuszczamy) na zrozumienie, co tak
naprawdę się dzieje i dlaczego tak się dzieje. Cząsteczki, z których zbudowana jest materia, składają się z atomów ułożonych
w przestrzeni w określonej dla danej cząsteczki konfiguracji przestrzennej. Atomy zawierają protony (ładunek dodatni) i
elektrony (ładunek ujemny), które wytwarzają wokół cząsteczki pole elektryczne, a będąc w ciągłym ruchu (spin, czyli
wirowanie, niewielkie przemieszczenia w obrębie wiązań - oscylacja, poruszanie się elektronów po orbitalach itp.) wytwarzają
zmienne pole elektryczne. To zmienne pole elektryczne jest przyczyną powstawania wokół cząsteczki zmiennego pola
magnetycznego. W sumie więc można powiedzieć, że cząsteczka generuje charakterystyczne dla siebie pole
elektromagnetyczne. Fala świetlna, będąc falą elektromagnetyczną, przebiegając obok cząsteczki sumuje się (wektorowo!)
z jej polem elektromagnetycznym, dając w efekcie nową jakość, nową falę, która od pierwotnej będzie różnić się (między
innymi) płaszczyzną drgań. Wielkość tego skręcenia płaszczyzny drgań zależeć będzie od składu i struktury przestrzennej
cząsteczki, od ilości cząsteczek, z którymi fala wejdzie w oddziaływania oraz od usytuowania cząsteczki w stosunku do
kierunku rozchodzenia się fali. Ponieważ w układach makroskopowych (a w praktyce te nas interesują) mamy ogromne ilości
cząsteczek współoddziałujących z falą świetlną, możemy uznać, że skręcalność takiego układu jest statystyczną średnią
wszystkich możliwych układów cząsteczki wobec promienia, a zatem jest charakterystyczna dla danego związku, jego
struktury. W układzie molekularnym skręcalność zależna byłaby nie tylko od budowy cząsteczki, ale także od ustawienia jej
w przestrzeni względem promienia. Jeżeli każda cząsteczka skręca płaszczyznę drgań fali świetlnej, to płaszczyzna drgań
światła spolaryzowanego powinna ulec skręceniu po przejściu przez każdy ośrodek przezroczysty, a w praktyce tak się nie
dzieje - skręcenie powodują tylko niektóre związki o szczególnej budowie cząsteczek.
Jeżeli cząsteczka o budowie i ustawieniu jak obok, po lewej,
skręca płaszczyznę np. o kąt +α , to cząsteczka po prawej
skręci płaszczyznę o kąt -α, bowiem jej struktura jest
identyczna, a ułożenie wobec promienia jest dokładnie
lustrzanym odbiciem ułożenia cząsteczki pierwszej.
Jednocześnie należy zauważyć, że są to identyczne
cząsteczki - wystarczy drugą obrócić w prawo o 120° wokół
osi pionowej aby otrzymać pierwszą. A ponieważ w układzie
makro występują praktycznie wszystkie możliwe ułożenia
cząsteczek, można uznać, że każdemu położeniu, które
skręca płaszczyznę polaryzacji o jakiś kąt w prawo
odpowiada ułożenie innej cząsteczki na drodze promienia,
które skręci o ten sam kąt płaszczyznę polaryzacji, ale
w lewo. Suma tych znoszących się na wzajem oddziaływań
spowoduje w końcowym efekcie brak zmiany położenia
płaszczyzny polaryzacji promienia wychodzącego w
stosunku do padającego. Substancje zawierające cząsteczki z
elementem symetrii nie powodują (w układach
makroskopowych) skręcenia płaszczyzny polaryzacji światła
przez nie przechodzącego.
Zestawienia cząsteczek o budowie asymetrycznej
Inaczej ma się sprawa w przypadku cząsteczek
asymetrycznych. Jeżeli w strukturze cząsteczki występuje
centrum asymetrii (najczęściej interesuje nas atom węgla
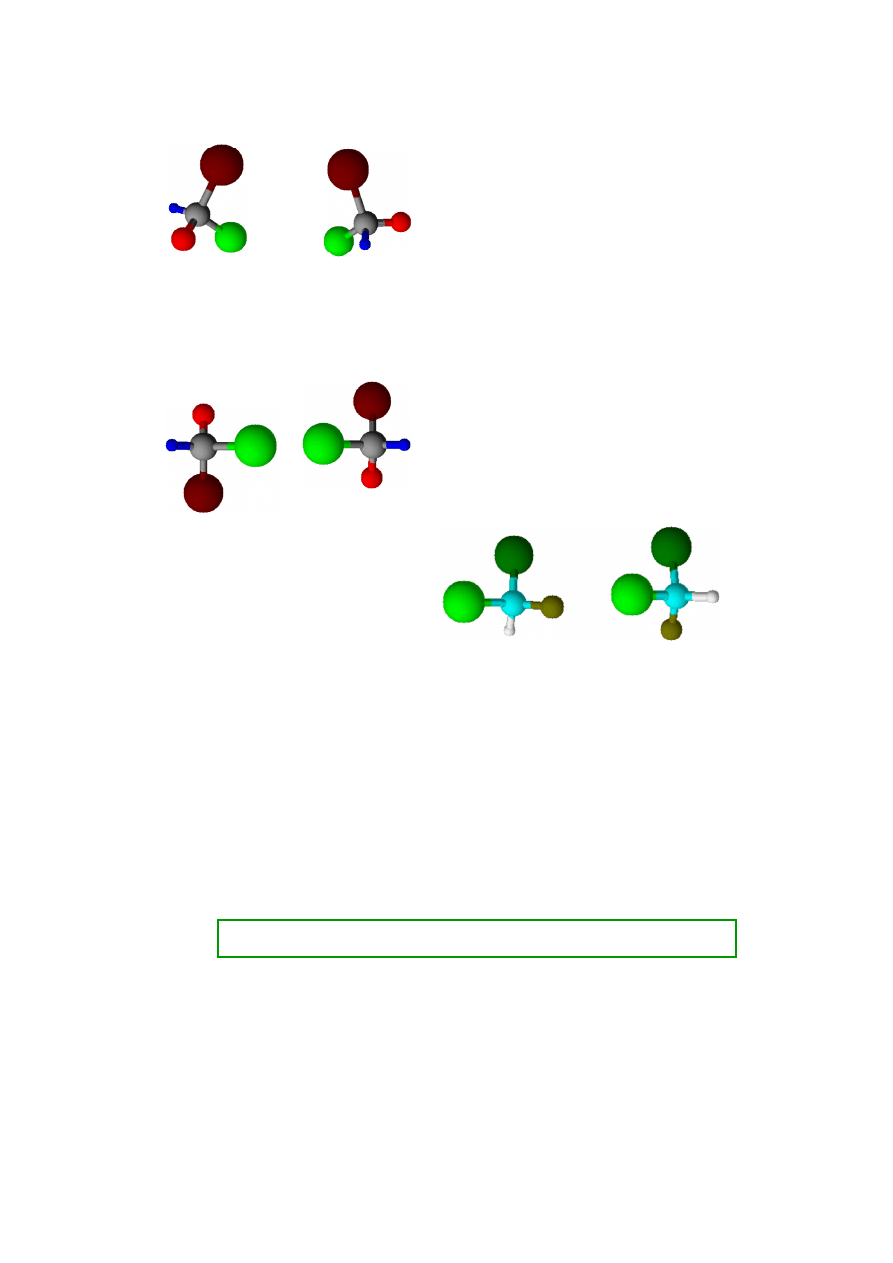
połączony z czterema
różnymi
podstawnikami) to żaden
obrót czy przesunięcie w przestrzeni nie da ułożenia, które
byłoby lustrzanym odbiciem tej cząsteczki (w przestrzeni
trójwymiarowej! Na rysunkach na płaszczyźnie dość
trudno to pokazać). Przykład (schemat) takiej struktury
obok. Każdy kolor czterech podstawników centralnego
atomu symbolizuje inny podstawnik. Jeżeli ustawimy
cząsteczki w stosunku do siebie tak, że dwa podstawniki
będą lustrzanymi odbiciami, to dwa pozostałe będą ułożone
różnie (górne i dolne zestawienie na rysunku obok). Znaczy
to, że nawet w nieskończenie dużym zbiorze cząsteczek
o asymetrycznej strukturze nigdy nie znajdzie się para
znosząca nawzajem swoje wpływy na promieniowanie
elektromagnetyczne (światło spolaryzowane). Zatem
promień spolaryzowany zawsze zostanie skręcony przez
takie cząsteczki o kąt charakterystyczny dla tej substancji.
Każdej strukturze cząsteczek danego związku o
asymetrycznej budowie odpowiada struktura będąca jej
lustrzanym odbiciem i skręcająca dokładnie tak samo
płaszczyznę polaryzacji światła, tyle tylko, że w przeciwnym
kierunku. Są to jednak inne cząsteczki, izomery, o tym
samym składzie atomowym ale
różnej strukturze
przestrzennej
.
Przez żadne przekształcenie w przestrzeni (bez rozrywania
wiązań) nie da się cząsteczki u dołu po lewej, przeprowadzić
w cząsteczkę po prawej. Takie dwie formy nazywamy
enencjomerami.
Enancjomery skręcają płaszczyznę polaryzacji światła
dokładnie tak samo, ale w przeciwnych kierunkach
(oznaczamy tę skręcalność jako + lub -).
Równomolowa mieszanina enancjomerów, zwana
mieszaniną racemiczną, jest optycznie nieczynna,
bowiem skręcalność w lewo zostaje zrównoważona
skręcalnością takiej samej ilości cząsteczek
w prawo. Wypadkowa skręcalność takiej
mieszaniny wynosi zero.
Jeżeli w cząsteczce związku jest więcej niż jedno centrum asymetrii (chiralności), to sprawy skręcalności nieco się komplikują.
Izomery optyczne, które nie są enancjomerami (lustrzanymi odbiciami trójwymiarowej konfiguracji) nazywamy
diasteroizomerami. W przypadku cząsteczek zawierających dwa identyczne co do składu centra asymetrii (patrz - kwas
winowy) może być układ (++); (--) lub (+-). Ta ostatnia forma jest optycznie nieczynna i nosi symbol mezo (np. kwas
mezowinowy). Brak skręcania płaszczyzny polaryzacji światła przez formy mezo wynika z tego, że skręcenie przez jedno
centrum asymetrii jest równoważone przez skręcenie w przeciwną stronę przez drugie centrum. Ale to są już sprawy izomerii,
chemii organicznej (szczególnie chemii cukrów) a nam chodziło tylko o mechanizm i zasadę zjawiska skręcalności.
Zatem w skali mikro każda cząsteczka skręca płaszczyznę polaryzacji światła, a wielkość i znak tego efektu zależy od
usytuowania cząsteczki względem kierunku rozchodzenia się promieniowania i jego płaszczyzny polaryzacji. Skręcalność jest
więc zjawiskiem powszechnym i niecharakterystycznym, jeśli chodzi o wielkość i kierunek zmian.
Natomiast w skali makro płaszczyznę polaryzacji skręcają tylko substancje posiadające centrum asymetrii, wielkość ta jest
dla danej substancji charakterystyczna, może stanowić nawet jej parametr identyfikacyjny, a także pozwala odróżnić różne
izomery tej samej substancji.
Zrobiło się trochę jaśniej? I o to chodziło. Teraz, mając ogólne wyobrażenie o zasadach zjawiska zabierz się do studiowania
pełnej, naukowej prawdy o polaryzacji światła i czynności optycznej związków. Zobaczysz o ile łatwiej Ci teraz będzie
zrozumieć wszystkie problemy związane z izomerią optyczną.
Czego wyobrazić się nie da
Im lepiej i głębiej poznajemy świat cząsteczek, atomów i cząstek elementarnych, tym bardziej staje się on dla nas ...
niezrozumiały. Rzecz w tym, że prawa i zasady obowiązujące w tym mikroświecie (mechanika kwantowa) nie przystają w
żaden sposób do naszego świata w wymiarze makro. Inaczej mówiąc, nie mamy doświadczeń życiowych, które pozwoliłyby
zjawiska mechaniki kwantowej opisać przykładami makroświata o mechanice newtonowskiej. A ponieważ elementów
mikroświata nie możemy obserwować bezpośrednio, za pomocą zmysłów, nie możemy również zrozumieć jak on działa. Dla
doraźnych celów dydaktycznych budujemy uproszczone modele w skali makro, które tłumacząc jedno - zaciemniają drugie.
Jeżeli w wyobraźni ucznia zaszczepimy pojęcie elektronu, jako maleńkiej kulki obdarzonej ładunkiem, trudno będzie mu
przyjąć później do wiadomości, że elektron jest falą i w dodatku może być w tym samym czasie w dwóch różnych miejscach.
Jeśli jednak nie podsuniemy mu żadnego rozwiązania, żadnego modelu z zakresu znanego mu świata makro, w ogóle nie
pojmie istoty elektronu i nie zrozumie najprostszych (jak na mikroświat) pojęć i zjawisk, jak choćby tworzenie wiązania
chemicznego. Dotarłszy jednak do pewnego poziomu wiedzy - np. kończąc naukę a zaczynając studiowanie - musimy
odważnie powiedzieć sobie DOŚĆ. Dość oszukiwania się, że elektron to kuleczka a wiązanie chemiczne to sprężynka, dość
oszukiwania siebie samego, że rozumiemy co to jest pole grawitacyjne czy elektryczne, że pojmujemy zjawisko zamiany
energii na masę i że w ogóle rozumiemy co to jest masa i energia. Przyjąwszy sokratesowskie "wiem, że nic nie wiem" (z
całym szacunkiem dla proporcji), zastanówmy się, czy przyznawszy się do niezrozumienia i niezdolności wyobrażenia sobie,
dość przecież podstawowych zjawisk i pojęć, musimy przyjąć niemożność poznania przez nas tego świata? Odwołajmy się tu

na moment do matematyki, która bazuje na aksjomatach (np. bezwymiarowość punktu czy jeden wymiar prostej, złożonej z
bezwymiarowych punktów - kto to ROZUMIE !?) i pojęciach też nie dających się wyjaśnić (nieskończoność, zero, wartość
nieskończenie mała ...) uchodzi (i słusznie) za królową nauk, i bez której dochodzenie do prawdy obiektywnej jest niemożliwe
w żadnej dziedzinie wiedzy, . Tak więc przyjmując coś "na wiarę" nie obniżamy rangi naukowego poznania, jeżeli tylko cała
konstrukcja zbudowana na tych aksjomatach jest spójna i skuteczna. Jeśli przyjmiemy "na wiarę" postulat fal materii de
Broglie'a, to dzięki temu będziemy mogli przewidzieć skutecznie przebieg wielu zjawisk, niewytłumaczalnych bez przyjęcia tej
teorii. Zauważmy, że bez względu na czyją korzyść rozstrzygnie się odwieczny spór materialistów z idealistami (czy pierwsza
była materia czy idea, lub trywializując: co było pierwsze - jajko czy kura) prawa fizyki nie zmienią się na jotę. Innymi słowy,
obojętnie czy przyjmiemy prymat jajka czy kury, opis jednego i drugiego pozostanie identyczny.
Te parę zdań powyżej to tylko wywołanie tematu, z którym każdy musi się sam uporać. Każdy sam musi przemyśleć sprawę
by zrozumieć głębie stwierdzenia "wiem, że nic nie wiem" i zrozumieć, że taka "niewiedza" to wiedza bardzo głęboka
i ułatwiająca pojmowanie nawet bardzo skomplikowanych i trudnych do przeniesienia w świat makro-modeli zjawisk
atomowo-cząsteczkowych.
Osobiście proponuję następujące podejście do sprawy:
Nie odrzucajmy porównań i przykładów spotykanych w podręcznikach i na wykładach, są to bowiem najczęściej dość dobrze
przemyślane i zazwyczaj "najlepsze z możliwych" makro-schematy mikro-zjawisk. Pamiętajmy jednak, że dotyczą one tylko
tego zjawiska (lub często nawet tylko jego elementu) i nie należy ich przenosić na inne zjawiska. Elektron jako naładowana
kulka może być dobrym przybliżeniem w tłumaczeniu podstaw istoty wiązania chemicznego czy dysocjacji, nie nada się
jednak zupełnie do wyjaśniania zjawiska rezonansu magnetycznego jąder (NMR). Tam bardziej przekonującym będzie
porównywanie elektronów do mgły czy chmury o różnej gęstości w różnych miejscach cząsteczki - taki model z kolei nie
bardzo się przyda przy objaśnianiu wiązań. Zatem nie - elektron jest kulką, czy - elektron jest chmurą, czy - elektron jest falą,
lecz - w zjawisku X możemy sobie przyjąć, że elektron jest kulką, ale w zjawisku Y musimy o tym "zapomnieć" i przyjąć,
że jest chmurą itp. Zaś czym jest elektron w rzeczywistości ? "Wiem, że nic nie wiem" - ale reakcje chemiczne potrafię
przeprowadzić zgodnie z założeniami i procesy fizykochemiczne wykorzystuję bez kłopotów w praktyce - a więc jednak coś
wiem?...
Spójrzmy na świat od nowa
Jak już pisałem na stronie tytułowej, kłopoty z chemią fizyczną biorą się między innymi stąd, że w szkole tego działu chemii
nie ma prawie wcale, a na studiach występuje on od razu w swojej "oficjalnej", precyzyjnej, "matematycznej" postaci. Czasu
na "zbędne" tłumaczenia podstawowych pojęć najczęściej nie ma i do wszystkiego trzeba dochodzić samemu. I tu czyha na
nas spore niebezpieczeństwo. Tak samo brzmiące słowa, pojęcia, co do znaczenia których w potocznym użyciu nie mamy
żadnych wątpliwości, w chemii fizycznej nabierają nowych, precyzyjniejszych znaczeń, których najczęściej nikt nam w porę
nie wyjaśnił (znów ten brak czasu). Tym sposobem, wykładowca używając terminu z zakresu chemii fizycznej ma co innego na
myśli niż słuchacz i problem z brakiem zrozumienia (lub gorzej - z niewłaściwym zrozumieniem) wykładanego tematu gotowy.
Dobrym przykładem takich niezgodności są stany skupienia. Od dziecka rozróżniamy ciała stałe, ciecze i gazy i taki podział
stanów materii utrwala nam później szkoła. W chemii fizycznej te trzy stany też występują, ale terminy te oznaczają teraz
trochę coś innego. Przez ciało stałe rozumiemy np. tylko stany krystaliczne, uporządkowane, zmieniające stan skupienia w
ściśle określonej temperaturze. Niektóre substancje, które uważamy w życiu codziennym za ciała stałe, wykazują bowiem
podstawowe cechy - stały kształt - dla fizykochemii są cieczami o bardzo dużej lepkości (szkło, parafina stała, wosk itp.).
W potocznym zrozumieniu ciała stałego też spotykaliśmy problemy z zakwalifikowaniem substancji do odpowiedniej grupy
(ciała maziste, gęste smary, plastelina), ale nikt nie robił z tego problemu. W chemii fizycznej, jak we wszystkich naukach
przyrodniczych, staramy się być bardzo precyzyjni i jednoznaczni, stąd o zakwalifikowaniu substancji do odpowiedniej grupy
nie decydują nasze subiektywne, zmysłowe odczucia a precyzyjna definicja (choć i tu pojawiają się czasem kłopoty), związana
z jakimś istotnym parametrem.
do strony
tytułowej
do góry
strony
Pięć stanów skupienia?
Przyzwyczailiśmy się do trzech stanów skupienia materii: ciało stałe, ciecz i gaz. W chemii fizycznej za ciało stałe uznajemy
ciała krystaliczne. Ciała stałe (w potocznym rozumieniu), które nie mają budowy regularnej, krystalicznej, zaliczamy do cieczy
o bardzo dużej lepkości. Podgrzewając ciała krystaliczne dochodzimy w pewnym momencie do określonej temperatury, w
której siły sieci krystalicznej nie są już w stanie utrzymać rozedrganych cząsteczek w ustalonym porządku i kryształ
przechodzi w ciecz (temperatura topnienia). Bezpostaciowe, amorficzne ciało stałe, które z punktu widzenia chemii fizycznej
jest cieczą o ogromnej lepkości, ogrzewane zmniejsza swoją lepkość, co oznacza, że energia kinetyczna poszczególnych
cząsteczek zaczyna być porównywalna z siłami oddziaływań międzycząsteczkowych, i ciało stałe poprzez stadium formy
mazistej przechodzi w "klasyczną" ciecz (o ile wcześniej nie ulegnie pirolizie - ale to dotyczy także krystalicznych ciał
stałych). Tak więc pojęcia - ciało stałe i ciecz, w chemii fizycznej znaczą co innego niż w języku potocznym, kryterium
podziału jest tu struktura cząsteczkowa a nie właściwości fizyczne.
Przyzwyczailiśmy się także traktować różne podziały i klasyfikacje zbyt formalistycznie, ostro rozgraniczając np. ciecze i gazy
czy ciała stałe i ciecze (w tym codziennym rozumieniu tych pojęć) - bowiem takie podejście "porządkuje" nam świat i daje
poczucie panowania nad mnogością informacji o nim. O tym, że istnieje niebezpieczeństwo "zgubienia" części świata przy
takim podejściu do problemu przypomina choćby wspomniany wyżej przypadek ciał mazistych, co do których nie mamy
pewności jak je zakwalifikować - do ciał stałych czy do cieczy. Także pozornie jednoznaczne i precyzyjne definicje potrafią
wprowadzić nas w błąd, jeśli niezbyt krytycznie podejdziemy do ich treści. Dla wygody i jasności sformułowań dość często
stosujemy różne skróty, nie dopowiadamy (np. opuszczamy informację o koniecznych warunkach) i jeśli ktoś potraktuje taką
"skróconą" definicję zbyt serio - kłopot gotowy. Na przykład dość często roztwór nasycony definiowany jest jako roztwór o
maksymalnym stężeniu - z lenistwa nie dodając, że w danej temperaturze. A potem nagle okazuje się, że są także roztwory
przesycone, tzn. takie, których stężenie jest większe niż nasyconych! Większe niż maksymalnie możliwe! I mętlik w głowie
pewny. Dla wyjaśnienia tej pozornej sprzeczności między definicjami szybko dodaje się, że roztwór nasycony to roztwór o

najwyższym możliwym stężeniu w danej temperaturze w stanie równowagi termodynamicznej, czy też termodynamicznie
trwały, czego większość studentów w początkowym okresie nauki i tak nie rozumie. I tak już przy dość prostych zjawiskach
można zacząć się gubić i przestać lubić chemię. Jak się przed tym uchronić? NIC NA PAMIĘĆ - WSZYSTKO NA ROZUM.
Trzy stany skupienia, do których już się przyzwyczailiśmy, a które w ujęciu fizykochemicznym dają ową piękną ostrość
podziału (znika nam problem ciał mazistych) nagle okazują się nie być jedynymi jakie zna przyroda. O istnieniu innych
najczęściej nie mieliśmy pojęcia, a trzeba w tym kontekście wspomnieć o stanie nadkrytycznym (fluid) i ciekłych kryształach.
Pierwszych w życiu codziennym nie spotykamy, bo występują w warunkach ciśnienia i temperatury na co dzień nie
spotykanych, te drugie zaś są stałym elementem naszej rzeczywistości, ale spotykamy się z nimi w postaci ekranów
i wyświetlaczy różnych urządzeń i nie mamy większego pojęcia o ich postaci w formie wolnej substancji. A jedne i drugie
łączą w sobie cechy znanych stanów skupienia. Ciekłe kryształy stanowią pomost między ciałami stałymi a cieczami, fluid jest
połączeniem cech gazu i cieczy. I znów, gdy pojawi się taka nowa jakość, wali się nam w gruzy miły porządek świata i ogarnia
nas frustracja. Żeby ustrzec się takich przykrych niespodzianek, pamiętajmy, że materia to cząsteczki zbudowane z atomów, a
te z kolei z cząstek elementarnych, i tylko to jest pewne, trwałe i stałe (choć i tu uczeni spierają się o detale). Wszystkie cechy
materii obserwowane w skali makro, to wypadkowe cech elementarnych składników materii i oddziaływań między nimi.
Trzeba sobie zatem odważnie powiedzieć - żadne charakterystyczne cechy materii w skali makro nie istnieją! To, co
opisujemy jako cechy materii (np. stany skupienia) to tylko przypadkowa wartość, będąca wypadkową cech składników w
danych warunkach. To, że mimo tego większość otaczającej nas materii potrafimy jakoś skwalifikować i zaszufladkować to i
tak cudowny przypadek. I nie jest to, wbrew pozorom, żadna rewolucyjna zmiana spojrzenia na świat - w wielu przypadkach
taki ogląd świata już od dawna akceptujemy, tylko nie zawsze zdajemy sobie z tego sprawę. Weźmy przykład wody.
Spotykamy ją w postaci oceanów, mórz, jezior, rzek, strumyków, deszczu i mgły, śniegu i lodu, lodowców i zamarzniętych
kałuż, kropli kapiących z kranu czy mokrej plamy na obrusie. I nikogo głowa nie boli o to, jak jednoznacznie odróżnić strumyk
od rzeczki i czym się różni kropla rosy od kropli deszczu. Wiemy, że od kropelki mgły aż po ocean to wszystko tylko zbiór
cząsteczek wody, a różne cechy różniące mgłę od oceanu, to nie jakieś specjalne, charakterystyczne cechy takiej postaci
wody lecz jedynie suma właściwości, dość ściśle zdefiniowanych, molekuł H
2
O. A że możemy zbiór cząsteczek wody
zwiększać cząsteczka po cząsteczce, to oczywiste, że nie istnieją żadne ścisłe rozgraniczenia między mgłą a oceanem, a
jedynie umowne podziały, zbudowane w oparciu o pewne cechy wynikające z wielkości zbioru cząsteczek wody. Skoro bez
większych zastrzeżeń akceptujemy takie podejście w stosunku do wody - to dlaczego mielibyśmy inaczej traktować różne
zbiory cząstek elementarnych i atomów? Choć z drugiej strony, dość trudno pozbyć się wręcz podświadomych skojarzeń, że
tlen to gaz, a woda to ciecz. Zatem, żadna postać materii nie jest cieczą, gazem czy ciałem stałym - każda substancja jest tylko
sobą. Może występować w postaci cieczy, ciała stałego bądź gazu, lub pod inną postacią. Musimy zatem myślowo oddzielić
substancję, cechy jej cząsteczek czy atomów, od cech fizycznych układów makroskopowych. Nie ma kropli wody na stole, nie
ma oceanu - są tylko cząsteczki wody tworzące bądź kroplę bądź ocean, albo kałużę, ale to zawsze tylko woda.
Prześledźmy teraz przemiany fazowe: ciało stałe - ciecz - gaz z punktu widzenia cząsteczkowego (oczywiście, jak zwykle
w dużym skrócie i uproszczeniu).
Cząsteczki dipolowe siłami elektrostatycznego oddziaływania ładunków ujemnych i dodatnich w cząsteczkach utrzymują
uporządkowaną strukturę sieci krystalicznej. Siły tego oddziaływania są większe niż energia kinetyczna drgań tych cząsteczek.
Wraz ze wzrostem temperatury rośnie energia pojedynczych cząsteczek, aż w pewnym momencie dochodzi do zrównania siły
wiązań cząsteczek w sieć z energią kinetyczną drgań i pękają wiązania sieci krystalicznej. Uporządkowana przestrzennie
struktura rozpada się - następuje topnienie substancji. Energia translacyjna cząsteczek w powstałej po stopieniu kryształu
cieczy jest większa niż siły oddziaływań międzycząsteczkowych, ale jeszcze siły międzycząsteczkowe są na tyle silne, że ciecz
stanowi układ dość spójny (lepkość, ciągłość strugi). Ponadto energia translacji (poruszania się cząsteczek) jest mniejsza od sił
grawitacji ziemskiej działającej na cząsteczki. Cząsteczki w cieczy poruszają się dość niezależnie (bezładnie), ale ciągle ich
zachowanie determinowane jest przez grawitacje i oddziaływania międzycząsteczkowe, którym próbuje przeciwstawić się
energia translacyjna cząsteczek. Wypadkowe tych trzech sił determinują główne cechy cieczy.
Podgrzewając układ dalej doprowadzamy do stanu, kiedy energia translacji przekroczy wartości potrzebne do zerwania
wszelkich oddziaływań międzycząsteczkowych oraz pokona siły grawitacji - wówczas ciecz przejdzie w gaz. W gazie siły
oddziaływań międzycząsteczkowych oraz siły grawitacji nie mają większego znaczenia i można na chwilę o ich istnieniu
zapomnieć. Cząsteczki gazu wypełniają więc równomiernie całą objętość naczynia w którym się znajdują i na wszystkie
ścianki wywierają takie samo ciśnienia (w cieczy istnieje w różnej odległości od powierzchni różne co do wielkości ciśnienie
hydrostatyczne).
Z tego co powiedziano powyżej wynika, że stan skupienia zależy głównie od wzajemnych relacji trzech czynników - energii
kinetycznej cząsteczek, wielkości siły grawitacji (co związane jest z masą cząsteczki) i wielkości sił oddziaływania
międzycząsteczkowego (co pośrednio związane jest ze składem i strukturą cząsteczki). Wyobraźmy sobie zatem substancję
w stanie krystalicznym, w którym wiązania sieci krystalicznej są tak silne, że cząsteczki nawet uzyskawszy energię
wystarczającą do pokonania sił grawitacji ciągle jeszcze nie mają wystarczającej energii potrzebnej do pokonania sił
utrzymujących je w sieci krystalicznej. W momencie osiągnięcia energii pozwalającej wyrwać się z układu kryształu,
cząsteczki te przechodzą bezpośrednio w stan gazowy, bowiem energię zdolną pokonać grawitację osiągnęły już dawno. Tak
duża energia translacji powoduje, że cząsteczki te poruszają się na tyle szybko, że oddziaływania międzycząsteczkowe tracą na
znaczeniu. Takie przejścia bezpośrednio z ciała stałego w gaz nazywamy sublimacją.
A teraz przyjrzyjmy się innemu zjawisku. Jeżeli mamy substancję w stanie gazowym, to chcąc przeprowadzić ją w stan ciekły
musimy obniżyć jej energię do stanu, w którym zaczną nabierać znaczenia siły grawitacji. To z kolei spowoduje zbliżenie się
cząsteczek do siebie na odległości, w których oddziaływania międzycząsteczkowe zaczną być znaczące. Jednym słowem, aby
skroplić gaz (doprowadzić do kondensacji par cieczy) trzeba zbliżyć cząsteczki gazu do siebie na odpowiednią odległość i
zadbać o odpowiednio niską energię translacji tych cząsteczek. Najłatwiej dokonać tego obniżając temperaturę (zmniejszenie
energii) i zwiększając ciśnienie (zbliżenie cząsteczek). A tak na marginesie - w "starym" spojrzeniu na świat nie zawsze
zauważamy tożsamość zjawiska kondensacji pary i skraplania gazu, często traktujemy je jako różne zjawiska, nie kojarząc, że
np. zwykła destylacja to także proces skraplania gazu!
A teraz wyobraźmy sobie sytuację, że zbliżenia cząsteczek gazu dokonujemy zwiększając ciśnienie i podgrzewając gaz,
zamiast go oziębiać. Możemy tym sposobem doprowadzić do sytuacji, w której cząsteczki substancji będą tak blisko siebie
(ciśnienie), że siły oddziaływań międzycząsteczkowych osiągną wartości charakterystyczne dla cieczy, a energia translacji
(wysoka temperatura), znacznie przewyższająca siły grawitacji i oddziaływań międzycząsteczkowych, spowoduje utrzymanie
podstawowych cech gazu. Taki stan będzie miał jednocześnie cechy gazu i cieczy i nazywany jest stanem nadkrytycznym
(fluidalny, płyn w stanie nadkrytycznym).
Ciekły kryształ - stan skupienia makroskopowo wyglądający jak ciecz, a strukturalnie jak ciało stałe, krystaliczne. Zachowuje
się w wielu zjawiskach fizycznych jak kryształ (wykazuje np. zjawisko anizotropii charakterystyczne dla układów
krystalicznych) ale wykazuje podstawowe cechy cieczy (np. przyjmuje kształt naczynia). Już sama nazwa wskazuje na
bezradność uczonych w zakwalifikowaniu go do którejś z "klasycznych" grup substancji. A sprawa jest stosunkowo prosta.

Niektóre substancje o specyficznej budowie cząsteczki i polarności, powodujących powstawanie dużych sił oddziaływań
międzycząsteczkowych, wykazują dość duży stopień uporządkowania molekuł. Jest on prawie tak regularny jak w kryształach,
skąd biorą się cechy krystaliczne takich substancji. Jednocześnie substancje te wykazują cechy zewnętrzne cieczy, bowiem
siły wiązań międzycząsteczkowych nie są aż tak silne, by unieruchomić wszystkie cząsteczki w sztywnej sieci. Jest to raczej
skupisko mikrokryształów, mogących przesuwać się względem siebie, ale także w dość uporządkowany sposób. Stąd płynięcie
takich substancji.
Czy w świetle powyższego warto upierać się przy trzech (czterech? pięciu?) stanach skupienia? Czy warto uznawać przejścia
fazowe gaz-ciecz-ciało stałe za "normalne", "naturalne" a sublimację za wyjątkowe zjawisko? Czy uczyć się na pamięć, że
gazu nie da się skroplić powyżej temperatury krytycznej, nie rozumiejąc jednocześnie dlaczego tak się dzieje? A może lepiej (i
mądrzej) pomyśleć, przemyśleć i zapamiętać, skąd takie czy inne właściwości się biorą?
do strony
tytułowej
do góry
strony
Forma substancji a reaktywność
Następne przyzwyczajenie, od którego dobrze jest się wyzwolić, to traktowanie symboli związków i samych związków jako
bytów tożsamych. Bardzo często zapominamy, że H
2
O to tylko symboliczny zapis cząsteczki wody a nie woda jako taka.
Informacje płynące z takiego zapisu nic nam nie mówią o rzeczywistości i na przykład niezrozumiałym jest, dlaczego
cząsteczki o masie molowej 18 tworzą ciecz a nie gaz, tak jak inne substancje o podobnej masie i strukturze - NH
3
; CO
2
, CH
4
czy H
2
S. Bardzo mało w czasie nauczania chemii mówi się o wpływie formy, postaci, w jakiej występują reagenty na
reaktywność substancji. Wyjątkiem, zazwyczaj jedynym, jest węgiel, na przykładzie którego nieco szerzej omawia się wpływ
struktury na reaktywność i właściwości chemiczne (grafit i diament). Omawia się jednak wówczas te różnice z punktu
widzenia alotropii pierwiastków i mało kto uogólnia i przenosi te wiadomości na inne pierwiastki i związki chemiczne. Nie
można, z praktycznego punktu widzenia, każdej reakcji opisywać pełną, wyczerpująca informacją, bo zagmatwalibyśmy tak jej
zapis, że stałby się całkowicie nieczytelny - ale trzeba ciągle powtarzać, że na przykład woda w reakcji, to nie H
2
O a skupiska
cząsteczek wody, powiązanych w przestrzenne formy wiązaniami wodorowymi ({H
2
O}
p
), że częściowo są to jony (minimalna
dysocjacja - ale jednak występuje) itp. itd.
Zapominając o złożoności realnej sytuacji reakcji nie zrozumiemy także, dlaczego raz ona zachodzi a innym razem nie, chociaż
w zapisie symbolicznym wygląda to tak samo. Na przykład niełatwo pojąć, dlaczego ketozy są reduktorami, choć ani grupa
alkoholowa ani ketonowa takich właściwości nie wykazuje i dlaczego glukoza jest reduktorem, skoro występując w postaci
cyklicznej też nie ma grupy aldehydowej. Wszelkie, nawet bardzo daleko idące uproszczenia w tłumaczeniu i zapisywaniu
chemii są konieczne i zazwyczaj przydatne - inaczej zapewne już na samym początku nauczania ucieklibyśmy z krzykiem
przerażenia - nie wolno jednak ani na chwilę zapominać, że są to uproszczenia, że rzeczywistość jest bardziej złożona. Na
nauczycielach wszelkich szczebli nauczania spoczywa obowiązek wyjaśniania, w odpowiednim momencie, co jest
uproszczeniem, o czym w danej chwili nie mówimy i co sprawia, że omawiany "wyjątek" jest wyjątkiem (czyli, jak już wiemy,
spełnieniem innej reguły, ważniejszej w danych warunkach). Zapisywanie glukozy we wszystkich omawianych reakcjach
wyłącznie wzorem C
6
H
12
O
6
trudno nazwać nauczaniem chemii - raczej jest to publiczna egzekucja tej pięknej nauki.
A dla uczących się uwaga - żeby zbyt szybko nie rozgrzeszali swojego intelektualnego lenistwa argumentem kiepskiego
nauczyciela - pamiętajcie, że liczy się tylko WASZA rzetelna wiedza na dany temat. Jeśli trafił wam się kiepski nauczyciel, to
wasz problem i kłopot, ale w niczym to was nie rozgrzesza. Dobry chemik zna dobrze chemię i w momencie kiedy musimy się
tą wiedzą wykazać, poziom naszych nauczycieli nie ma nic do rzeczy. Nauczyciel często nie wspomina o zawiłościach chemii,
bo go nikt o to nie pyta. Zanim wydamy opinię o naszym mistrzu, sprawdźmy najpierw jaki on rzeczywiście jest. Czyli
najpierw sami zacznijmy zastanawiać się nad problemami poruszanymi na wykładzie czy przeczytanymi w podręczniku, a
później wdajmy się w dyskusję z nauczycielem. Raz jeszcze powtórzmy sobie niepodważalną prawdę - chemii nie da się
nauczyć na pamięć, bez zrozumienia jej zasad, reguł i wzajemnych relacji między nimi. Dopiero widzenie zjawiska
w kontekście całej chemii i fizyki pozwala zrozumieć jego przebieg i jego istotę.
Podsumowując niniejszy podpunkt, pamiętajmy, że każda przemiana - tak reakcja chemiczna jak i zjawisko fizyczne
("fizykochemiczne") - odbywa się z pewnym efektem energetycznym. Jedne wiązania czy oddziaływania muszą zostać
pokonane, rozerwane (potrzeba energii) by mogły powstać nowe (odzysk energii). Zatem, czy i jak przebiegnie zjawisko
(reakcja), decydować będą etapy uwalniające lub pobierające energię. Żeby jakiś etap wymagający energii mógł zaistnieć,
układ musi mieć dostęp do "zapasu" energii. Ten zapas mógł być stworzony w poprzednim etapie, wyzwalającym energię, lub
takiego zapasu nie ma i proces się zatrzymuje. Tak więc o reakcji nie decyduje reguła, np. tlenki niemetali z wodą dają kwas,
lecz siła wiązań i oddziaływań. Dla CO
2
ta reguła jest prawdziwa, dla SiO
2
- już nie (siły wiązań tworzących krystaliczną
strukturę krzemionki - kwarcu na to nie pozwolą w "zwykłych" warunkach).
do strony
tytułowej
do góry
strony
Stany pośrednie i brzegowe
Wszyscy jesteśmy przyzwyczajeni, że roztwór opisujemy, charakteryzujemy, przede wszystkim jego stężeniem. Niekiedy
jednak zamiast stężenia pojawia się aktywność, czasem dla roztworów elektrolitów obliczamy siłę jonową roztworu. Wiemy,
że aktywność jest zawsze mniejsza od stężenia, zrównuje się z nim w przypadku roztworów o hipotetycznym stężeniu
"nieskończenie małym". Z kolei przy stężeniach dużych (co to konkretnie znaczy?) "obowiązkowo" posługujemy się
aktywnością zamiast stężenia, bo tu różnice między tymi wartościami są bardzo duże (czyli jakie?).
Obliczając pH roztworów silnego kwasu np. HCl
aq
, kwasu solnego, obliczamy po prostu logarytm dziesiętny ze stężenia HCl.
Jeśli jednak stężenie kwasu jest mniejsze niż powiedzmy 10
-5
mol/dm
3
, w obliczeniach musimy uwzględnić prócz stężenia
kwasu także inne czynniki (np. dysocjację wody), bo inaczej dla stężenia kwasu np. 10
-12
mol/dm
3
znajdziemy pH = 12 !!
I tak jest dla wielu zjawisk, procesów i reakcji. W pewnych zakresach parametrów zjawiska układ zachowuje się inaczej niż
w pozostałych. Najczęściej wyróżnić możemy praktycznie trzy takie zakresy - zakres pośrednich wartości, najszerszy (i o nim
głównie uczymy się w chemii, także fizycznej), oraz dwa zakresy brzegowe - parametrów bardzo dużych i bardzo małych.
W każdym z tych zakresów układ zachowuje się odmiennie, opisują go inne równania. Dzieje się tak z powodów, które już
były poruszane - złożoności każdego zjawiska. W każdym z tych trzech zakresów znaczenia nabierają inne elementy procesu.
Jak roztwór jest bardzo rozcieńczony, oddziaływania między jonami są na tyle słabe, że nie mają praktycznie żadnego wpływu
na zachowanie się roztworu i w praktyce spełniona jest równość c = a (aktywność jonów a można zastąpić stężeniem jonów
c). W roztworach stężonych, średnia odległość między jonami się zmniejsza, średnia siła oddziaływań jest na tyle duża, że o
zachowaniu roztworu decydować będzie aktywność ("użyteczne" stężenie a nie nominalne). Podobnie gazy rzeczywiste, przy
stosunkowo niskich ciśnieniach (kilku atmosfer) zachowywać się będą w praktyce jak gazy idealne (czyli takie hipotetyczne,

których cząsteczki nie mają objętości i nie oddziałują ze sobą), jednak przy ciśnieniach wysokich objętość ich cząsteczek jak i
siły oddziaływań międzymolekularnych będą musiały być wzięte pod uwagę dla dokładnego opisania zjawiska. Ponieważ
jednak dążymy zawsze do maksymalnej prostoty w opisie zjawisk (co jest wygodne ale nie zawsze bezpieczne) konstruujemy
wzory, które nie uwzględniają tych elementów procesu, które z PRAKTYCZNEGO punktu widzenia nie mają znaczenia.
Stąd dla różnych zakresów różne (pozornie) równania, które zazwyczaj dają się sprowadzić do jednego, opisującego pełny
zakres zjawiska, ale za cenę znacznego skomplikowania zapisu.
Konkluzja taka jak w pozostałych podpunktach - korzystajmy z uproszczeń, skupiajmy uwagę na tym, co w danych
warunkach najistotniejsze, ale ani na chwilę nie zapominajmy, że rzeczywistość jest o wiele bardziej złożona i uproszczeń nie
stosuje. To, że w tej chwili nie mówimy o procesie x, to nie znaczy, że go nie ma. Jest, i jak o nim zapomnimy, to on sam się w
odpowiednim momencie przypomni i może nas niemile zaskoczyć.
do strony
tytułowej
do góry
strony
Definicje, opisy, prawa, reguły ...
W świetle tego, o czym mówiliśmy wcześniej, dość oczywistym staje się, że w stosunku do sformułowań definicji, praw, reguł
itp. dotyczących opisu zjawisk i procesów obserwowanych w skali makro powinniśmy wykazywać "ograniczone zaufanie".
Jeżeli odnoszą się one do skali mikroświata - cząstek elementarnych, atomów, cząsteczek (choć tu już wzmożona ostrożność),
to owo zaufanie może być duże. Pamiętając o złożoności zjawisk obserwowanych w skali makro, oczywistym się staje, że
opisujące je reguły, definicje, ba, nawet prawa, powinny być obwarowane całym zestawem warunków (bo w różnych
warunkach suma zjawisk mikro jest różna). Jednak, gdybyśmy w sformułowaniu definicji czy reguły chcieli umieścić wszystkie
konieczne dla jej jednoznaczności warunki - stałaby się nieczytelna i nie do zapamiętania, czyli straciłaby sens. Pamiętajmy
więc, że wszystkie sformułowania dotyczące skali makro, nawet jeśli brzmią bardzo kategorycznie, najpewniej są tylko
przybliżeniami opisującymi zjawisko w warunkach najbardziej prawdopodobnych, najczęściej spotykanych. W warunkach
odbiegających od "normalności" to samo formalnie zjawisko przebiegać będzie zapewne nieco inaczej. Jednocześnie
przestrzegałbym przed dość popularnym podejściem do tego problemu, które zazwyczaj owocuje stwierdzeniem, że w danych
warunkach "substancja nie stosuje się do prawa ...". Takie sformułowania wyrabiają w nas podświadome przekonanie, że
prawa przyrody nie są absolutne i jedne substancje im podlegają a inne nie, a to nieprawda. Właśnie podleganie wszystkim
prawom przyrody powoduje, że substancja zachowuje się niezgodnie z naszym sformułowaniem prawa, ale nie niezgodnie z
prawem. Zjawisko przebiega zgodnie z prawem, a dokładniej zgodnie ze wszystkimi prawami, które w tej sytuacji mają
zastosowanie.
Dość częstym sposobem, stosowanym w podręcznikach, jest zawarcie zastrzeżeń dotyczących omawianego zjawiska we
wstępie do rozdziału. Często np. wstęp do rozdziału o gazach brzmi jakoś tak: Załóżmy, że mamy do czynienia z gazem
idealnym, tzn. takim, który ... i dalej biegnie długi rozdział o gazach. Już po paru stronach zapominamy, że opisy dotyczą
hipotetycznego gazu doskonałego i w pamięci zakodowują się nam informacje, jako dotyczące gazu jako takiego, czyli
rzeczywistego. A potem w opisie niektórych sytuacji dotyczących gazu rzeczywistego (np. skraplanie czy przejście w stan
nadkrytyczny) pojawiają się sformułowania sprzeczne z poprzednimi i problem gotowy. Tak więc bądźmy czujni i krytyczni
wobec wiedzy podręcznikowej - nie dlatego, że nie ufamy wiedzy jej autorów, ale dlatego, że ci autorzy, nie zawsze potrafią
przekazać swą wiedzę w sposób dla nas jasny i przejrzysty. Dlatego tak dobrym sposobem na rzetelną wiedzę jest
studiowanie
, czyli korzystanie z różnych źródeł, różnych autorów i samodzielne (a więc świadome) zestawianie tych
informacji w zasób wiedzy na dany temat.
Pamiętajmy też, że sporo różnych określeń, które w konkretnych przypadkach mają ściśle określone znaczenie, używanych
jest również w znaczeniu potocznym. W tym drugim przypadku ich znaczenie nie jest ściśle zdefiniowane i ma za zadanie
raczej wskazać na pewne elementy zjawiska niż precyzyjnie je określić, np. poziom energetyczny czy oddziaływanie
międzycząsteczkowe.
W opisie zjawisk chemicznych bardzo często korzystamy z określenia "wiązanie chemiczne". Mówimy o energii wiązania,
o jego długości, oscylacji wiązań, zerwaniu bądź utworzeniu wiązania. Takie częste korzystanie z tego pojęcia wyrabia w nas
podświadome odbieranie wiązania jako fizycznego elementu wiążącego dwa atomy, a pisanie wiązań we wzorach
strukturalnych jako kreski jeszcze bardziej to podświadome rozumienie utwierdza. Dla klarowności wywodu stosowanie
takiego pojęcia jest bardzo wygodne i nikogo nie namawiam by z niego zrezygnował - pamiętajmy jednak, że za tym pojęciem
nie kryje się żadna sprężynka czy patyczek, lecz jest to układ dwóch atomów i dwóch lub więcej elektronów, których
wzajemne oddziaływania elektrostatyczne i magnetyczne powodują, że tworzą one w pewnej konfiguracji układ dość trwały.
Pamiętajmy również, że jest to układ dynamiczny - położenie względem siebie poszczególnych elementów tego układu jest
zmienne, a jak "dołożymy" do tego nieokreśloność pojęcia elektron (kulka, chmura, fala ?) oraz nieprecyzyjność pojęcia
"orbital", to stanie się jasne, że pojęcie wiązania chemicznego w wielu przypadkach używane jest w tym "potocznym",
ogólnym znaczeniu.
Obłaskawiona termodynamika
Termodynamika - już sam dźwięk tego słowa budzi grozę i przerażenie wśród studentów młodszych lat. Co jest powodem, że
ten dział chemii fizycznej jest uważany za tak trudny?
Po pierwsze, dotyczy spraw dość trudnych do wyobrażenia, bo traktuje o przepływie energii pomiędzy różnymi formami
organizacji materii. Tak więc, bardziej intuicyjnie wyczuwana niż rozumiana energia, pojawia się tu w różnych postaciach -
energii kinetycznej, potencjalnej, chemicznej, wewnętrznej układu, zewnętrznej otoczenia, wewnętrznej cząsteczek,
przekazywana jest w sposób kwantowany lub ciągły, na sposób ciepła lub pracy itp. itd. Nie dość, że nie bardzo wiemy co to
jest ta energia, to jeszcze występuje w tak skomplikowanych kontekstach.
Po drugie, ponieważ opisuje sprawy trudne do wyobrażenia, aby być dokładną i precyzyjną w tych opisach musi posługiwać
się matematyką, czasem dość skomplikowaną - a tego wielu nie lubi. Często tylko poprzez przekształcenia matematycznych
opisów można wykazać i określić powiązania skutkowo-przyczynowe między zjawiskami mikroświata.
Po trzecie, pojawia się cała nowa nomenklatura, zestaw pojęć zazwyczaj precyzyjnie definiowanych, czyli konieczność
nauczenia się dość specyficznego i niełatwego języka. Często podobnie brzmiące terminy oznaczają dość różne pojęcia,
czasem odwrotnie - prawie identyczne pojęcia są opisywane zdecydowanie różnymi terminami.
Po czwarte (tu na szczęście bywają wyjątki - oby jak najliczniejsze) wykładowcy i autorzy podręczników ten dość trudny
materiał przekazują w literalnej zgodzie z rzeczywistością nie dbając specjalnie o komfort zrozumienia i percepcji słuchaczy i

{4Na + O
2
+ E
s
} +
(E
1
- E
2
)
= {2Na
2
O + E
p
}
czytelników. Czyli innymi słowy, mówią cała prawdę ale mało zrozumiale (tu, jak w całej chemii fizycznej - materiału do
wyjaśnienia dużo a czasu mało).
Poniżej postaram się w sposób bardzo uproszczony, nie wchodząc w szczegóły, przygotować grunt pod przyszłe zrozumienie
wszystkich zawiłości termodynamiki. Nauczyć się jednak niestety będziecie musieli sami.
do strony
tytułowej
do góry
strony
Energia wewnetrzna a entalpia
Cząsteczka to materia (atomy) i energia zawarta w wiązaniach chemicznych łączących te atomy oraz w oddziaływaniach
międzycząsteczkowych. Każda przemiana chemiczna (reakcja) oraz fizyczna ("fizykochemiczna") związana jest z przepływem
energii - energia bowiem nie powstaje i nie znika, zatem musi w odpowiedniej ilości być dostępna w układzie. Jeżeli kawałek
metalicznego sodu spalimy w tlenie, to dla każdego, początkującego nawet chemika będzie oczywiste, że musimy mieć co
najmniej cztery atomy sodu i cząsteczkę tlenu by przeprowadzić taką reakcję:
4Na + O
2
= 2Na
2
O
Włączmy do rozważań o przebiegu tej reakcji zmiany energetyczne. Rozbijmy myślowo ten proces na dwa etapy: rozerwania
wiązań w cząsteczce tlenu oraz rozerwania wiązań między atomami sodu w strukturze metalicznej (kryształ metaliczny), a
następnie połączenia atomów tlenu i sodu w dwie cząsteczki Na
2
O.
Tak więc oprócz materii (sód i tlen) i energii w niej zawartej (E
s
) potrzebna będzie dodatkowa energia E
1
(do
rozerwania
wiązań
substratów).
W analogiczny sposób możemy wyobrazić sobie przebieg reakcji odwrotnej, prowadzącej od tlenku sodu do pierwiastkowego
sodu i tlenu:
2Na
2
O = 4Na + O
2
Tu również najpierw musimy dostarczyć energii potrzebnej do rozerwania wiązań i przejścia w stan atomowy (E
2
). Energię
zawartą w cząsteczkach Na
2
O oznaczmy E
p
.
Obydwa te procesy prowadzą do identycznego stanu termodynamicznego, zawierającego 4 atomy Na, dwa atomy tlenu
i pewną ilość energii E:
4Na; O
2 {E
s
}
+ E
1
——>
Na, Na, Na, Na, O, O, {E}
<—— E
2
+
2Na
2
O
{E
p
}
Energia {E} układu "atomowego" będzie więc równa:
E = E
s
+ E
1
(E
s
- energia wewnętrzna substratów),
gdy reakcja biegnie od substratów 4Na i O
2
dotlenku, lub, rozumując analogicznie dla przejścia od tlenku sodu do "układu
atomowego":
E = E
p
+ E
2
(E
p
- energia wewnętrzna produktów).
Stąd równość: E
s
+ E
1
=
E
p
+ E
2
dająca po prostym przekształceniu: E
1
- E
2
= E
p
- E
s
Rozpatrując zatem reakcję, tak jak ją zapisaliśmy: 4Na + O
2
= 2Na
2
O , ale z uwzględnieniem również energii, możemy
napisać bilans:
{4Na + O
2
+ E
s
} +
E
1
= {2Na
2
O + E
p
} +
E
2
Z ilości energii E
1
pobranej w pierwszym etapie przez cząsteczki substratów została "zwrócona" ilość E
2
w czasie tworzenia
się cząsteczek produktów. W czasie reakcji została więc pobrana ilość energii ΔE =
E
1
- E
2
= E
p
- E
s
.
Możemy to wszystko ująć w następujący schemat:
4 Na; O
2
{E
s
}
+ E
1
——>
Na, Na, Na, Na, O, O, {E}
2Na
2
O{E
p
}
4 Na; O
2
{E
s
}
+ E
1
——>
Na, Na, Na, Na, O, O, {E}
Na, Na, Na, Na, O, O, {E}
- E
2
——>
2Na
2
O {E
p
}
4 Na; O
2
{E
s
}
+ E
1
——>
Na, Na, Na, Na, O, O, {E}
- E
2
——>
2Na
2
O {E
p
}
<——
E
2
+
który da się "rozłożyć" na dwa równania:
łącząc te dwa równania otrzymujemy:
Tak więc aby otrzymać z sodu i tlenu tlenek sodu musimy najpierw dostarczyć substratom, zawierającym energię E
s
,
dodatkową ilość energii E
1
, potrzebną do atomizacji substratów. Następnie część energii układu zostaje "zmagazynowana"
w postaci energii wewnętrznej produktu (E
p
) i pozostaje nadmiar energii (E
2
).
Całkowity wydatek energetyczny reakcji będzie zatem:
E
1
- energia włożona w rozbicie substratów na atomy, minus E
2
- energia zwrócona w czasie łączenia się atomów
w cząsteczki produktu. Tak więc wymieniona z otoczeniem w trakcie reakcji ilość energii wyniesie:
ΔE=
E
1
- E
2
= E
p
- E
s
.
Teraz zapis równania (pełniej odzwierciedlający rzeczywistość) będzie wyglądał następująco:

4Na + O
2
= 2Na
2
O + ΔE
Dlaczego wartość ΔE została zapisana z prawej strony równania? To następny "kłopot" - wiele symboli, punktów odniesienia
itp., które z logicznego punktu widzenia mogłyby być zapisywane różnie, w celu uniknięcia niejednoznaczności, zostało
przyjęte jako obowiązujące na zasadzie ogólnoświatowej umowy (podobnie jak np. układ SI). Na takiej właśnie zasadzie
wydatek energetyczny reakcji jest zapisywany po prawej stronie równania (jako "produkt", wynik reakcji). Oblicza się go
zatem jako E
p
- E
s
(energia wewnętrzna produktów - energia wewnętrzna substratów). Jeżeli znak wartości tej ΔE będzie
ujemny, tzn. ilość energii wewnętrznej zawarta w produktach E
p
jest mniejsza niż ilość energii wewnętrznej substratów E
s
,
reakcja jest egzoenergetyczna (egzotermiczna), oddaje energię do otoczenia, jeśli znak jest dodatni - reakcja jest
endoenergetyczna, pobiera energię z otoczenia.
Teraz już chyba pora na przybliżenie terminów - energia wewnętrzna i entalpia.
Jeżeli wyobrazimy sobie zamknięty, izolowany układ, który nie może wymienić z otoczeniem ani materii ani energii, to suma
wszystkich postaci energii zawarta w składnikach tego układu będzie jego energią wewnętrzną. W realnym świecie takim
praktycznym odpowiednikiem układu izolowanego może być bomba kalorymetryczna lub zwykły, szczelny termos. Trzeba
pamiętać, że pojęcie układ może oznaczać każdy element rzeczywistego świata, wyizolowany myślowo z reszty otoczenia.
Może to być zawartość naczynia reakcyjnego, ale może również być to pojedyncza cząsteczka w tym naczyniu. Granice
układu zakreślamy w zależności od potrzeb przeprowadzanych rozważań termodynamicznych czy obliczeń. Granicami układu
mogą być np. ścianki zamkniętego naczynia, ale też mogą to być granice całkowicie umowne, fizycznie nie istniejące. Tak
więc już samo pojęcie układu jest dość rozmyte. Układy dzielimy zazwyczaj na izolowane (jak wyżej), zamknięte, tzn. takie,
które nie mogą wymieniać z otoczeniem materii (zamknięte naczynie), lub najczęściej spotykane układy otwarte, w których
może ulegać wymianie zarówno materia jak i energia (np. zlewka z roztworem).
Każdy układ materialny "zawiera" w sobie parę "typów", rodzajów energii. Mamy tu zazwyczaj na myśli energię jądrową
(manifestuje swą obecność podczas przemian jądrowych, na co dzień o niej pamiętać nie musimy), energię elektronów, ze
szczególnym uwzględnieniem elektronów walencyjnych, wśród których rozróżniamy energię oscylacji (zmieniająca się w
czasie oscylacji wiązań) i energię elektronową (energię wiązań), energię rotacji cząsteczek i energię translacji (ruchu
postępowego cząsteczek). Do tego dochodzą energie oddziaływań międzycząsteczkowych - solwatacji, wiązań wodorowych,
oddziaływań międzyjonowych oraz innych oddziaływań typu dipol-dipol itp. W czasie różnego rodzaju przemian chemicznych
bądź fizycznych (przemiany fazowe) zmieniają się wartości poszczególnych typów energii, ale jeżeli układ jest izolowany
suma wszystkich energii pozostaje stała. Jeśli np. suma energii zmieniających się w czasie reakcji (np. energia wiązań,
solwatacji, wiązań wodorowych), po reakcji jest mniejsza niż przed reakcją, to "nadmiar" energii zamieni się na energię
translacji (wzrośnie temperatura układu).
Zmiany energii wewnętrznej, będące wynikiem procesu (reakcji) mogą zostać przekazane otoczeniu albo na sposób ciepła albo
na sposób pracy, albo na oba te sposoby jednocześnie. Najczęściej spotykanym sposobem prowadzenia reakcji chemicznych
i procesów fizykochemicznych jest proces pod stałym ciśnieniem (atmosferycznym). W takich procesach dochodzi najczęściej
do zmiany objętości układu (np. ze względu na zmianę temperatury), czyli zmiana energii wewnętrznej następująca w czasie
reakcji powoduje zmianę energii translacji (temperatura, przekazanie energii na sposób ciepła, energia pozostająca w układzie)
oraz równolegle zmianę energii na sposób pracy (p·ΔV - wykonanie pracy objętościowej, przekazanie energii do otoczenia na
sposób pracy). Funkcja termodynamiczna, która uwzględnia te dwa sposoby przekazania energii nazywa się entalpią. Czyli
innymi słowy entalpia (oznaczana jako H) ma taki sam sens jak energia wewnętrzna (oznaczana jako U); w przypadkach stałej
objętości układu (p·ΔV=0) jej zmiana jest równa zmianie energii wewnętrznej (ΔH = ΔU), zaś w warunkach izobarycznych
(p=const) ΔH = ΔU + p·ΔV co oznacza także, że ΔU = ΔH - p·ΔV. Zatem entalpia (różnica entalpii) oznacza całą energię
wymienioną w danym procesie, a energia wewnętrzna określa tylko energię, która pozostała w układzie.
I żeby się w tym wszystkim nie pogubić, pamiętajmy, że funkcje termodynamiczne pozwalają nam obliczać
różnice
między
dwoma stanami układu (np. przed i po reakcji) i mimo, że często mówimy np. entalpia procesu czy entalpia reakcji to na
myśli mamy różnice entalpii stanu początkowego i końcowego a nie bezwzględne wartości. Bezwzględne wartości energii
wewnętrznej czy entalpii nie są możliwe do obliczenia (podobnie jak potencjał czy wysokość mają sens praktyczny tylko w
przypadku określenia jakiegoś konkretnego punktu odniesienia). Tu warto przypomnieć, że umownie przyjmujemy, że entalpie
tworzenia pierwiastków w ich stanach stabilnych w warunkach standardowych (25°C, 1 atm) wynoszą zero (umowny punkt
odniesienia), co pozwala nam obliczać standardowe entalpie tworzenia związków (ponieważ wartość punktu odniesienia jest
umowna, także te wartości należy traktować jako umowne!). Więcej na ten temat w
rozdziale o budowie materii
.
do strony
tytułowej
do góry
strony
Entropia - chaos czy porządek
Drugie, równie ważne i równie trudne (albo i trudniejsze) do wyjaśnienia pojęcie to entropia. W najprostszym ujęciu entropia
jest funkcją opisującą stan uporządkowania materii. Tu trzeba zwrócić uwagę na dwa podejścia do słowa "uporządkowanie".
Jedni autorzy rozumieją słowo porządek, tak jak to ma miejsce w mowie potocznej (wszystkie elementy o podobnym
charakterze w jednym miejscu, blisko siebie, ułożone zgodnie z jakąś regułą), inni zaś podchodzą do tematu bardziej
filozoficznie, uważając za porządek stan, do którego dąży samoistnie natura, czyli w codziennym pojęciu "kosmiczny
bałagan", czyli wszystko wszędzie. My będziemy rozumieć porządek w sposób potoczny. Takie podejście powoduje mniejsze
zamieszanie pojęciowe w tym i tak niełatwym temacie.
Jeżeli doprowadzimy do układu energię, to wzrośnie energia wewnętrzna układu i przede wszystkim spowoduje to zwiększenie
ruchliwości cząsteczek (energii translacji). Taki wzrost ruchliwości to jednocześnie wzrost nieuporządkowania, czyli wzrost
entropii. Trzeba jednak zauważyć, że taki sam co do wielkości bezwzglednej wzrost nieuporządkowania (choć nie bardzo
wiadomo co to by miało znaczyć) jest względnie duży, jeśli dotyczy "idealnego" porządku, zaś może być uznany za niewielki,
jeśli dotyczy układu o bardzo niskim stopniu uporządkowania. Zamiana miejscami na półce dwóch książek, jeśli te książki są
tam ustawione od największej do najmniejszej, daje poczucie zabałaganienia i jest szybko zauważalna. Taka sama zmiana
wśród książek, które stoją bez planu, stanowi niewielki wzrost nieuporządkowania (a czasami może wręcz stanowić krok w
kierunku większego porządku).
Spójrzmy na diagram poniżej. Mamy tam pola białe i czerwone, w górnych wierszach "uporządkowane", w dolnych nastąpiła
zamiana elementu 3 z 24 i 6 z 25. Mamy wiec identyczny wkład energii potrzebnej do zamienienia pól miejscami i różny efekt
końcowy w sytuacjach A, B i C (różną zmianę stopnia uporządkowania, w ostatnim wiersz nie widać żadnej zmiany, choć
zamiana nastąpiła)
1
2
3
4
5
6
7
8
9
10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30
A

B
C
Ponieważ w przyrodzie stopień nieuporządkowania wiąże się zazwyczaj z wyższą temperaturą a porządek z niższą, entropią
nazywamy stosunek ilości przeniesionej energii na sposób ciepła ("bezwzględna wartość stopnia zabałaganienia") do
temperatury, w której to przeniesienie zostało dokonane ("standardowy poziom bałaganu"), czyli określamy względny wzrost
nieuporządkowania względem stanu "naturalnego", związanego z daną temperaturą.
Wzrost entropii, którą oznaczamy literą S wyznacza wzór ΔS = ΔQ/T
Jest to wzór, który definiuje entropię, jednak dotyczy stanów w przyrodzie niespotykanych, czyli
przemiany odwracalnej
,
zachodzącej w stałej temperaturze (a to oznacza, że układ i otoczenie jest w identycznej temperaturze i wówczas przepływa
energia, co jest sprzeczne z II zasadą termodynamiki). Jeżeli zastosujemy pojęcie entropii do stanów rzeczywistych, czyli
układu o temperaturze T
1
i otoczenia o temperaturze niższej T
2
, to przepływ energii na sposób ciepła między układem a
otoczeniem (od układu do otoczenia) o wielkości Q spowoduje zmianę zarówno entropii układu jak i otoczenia. Zmiana
entropii układu będzie równa ΔS
u
= -Q/T
1
, zaś zmiana entropii otoczenia będzie wynosić ΔS
o
= +Q/T
2
. Całkowita zmiana
entropii układu i otoczenia będzie sumą tych zmian i wyniesie:
Jak z powyższych wzorów wynika, spadek wartości entropii układu jest mniejszy niż wzrost entropii otoczenia. Zatem
całkowita zmiana entropii układu i otoczenia jest zawsze dodatnia. W momencie, gdy temperatura otoczenia i układu
zrównają się, następuje osiągnięcie stanu równowagi (niemożliwy jest przepływ energii) a ponieważ entropia sumaryczna
rosła, oznacza to, że w stanie równowagi osiągnęła maksimum.
Przepływ energii na sposób ciepła powoduje nie tylko prosty wzrost temperatury, ale przekłada się także na zrywanie
wszelkich wiązań, od solwatacji i wiązań wodorowych poczynając a na dysocjacji i rozrywaniu wiązań w reakcjach
chemicznych kończąc.
Konieczność rozpatrywania procesów z punktu widzenia skali makro i skali mikro każe nam również na pojęcie entropii
popatrzeć z tych dwóch punktów widzenia. Rozpatrując entropię na poziomie cząsteczkowym wiążemy wzrost entropii ze
wzrostem stanu nieuporządkowania, ale zastrzegamy, że to pojęcie ("nieuporządkowanie") niezdefiniowane. Aby być bliżej
stwierdzeń naukowych, ścisłych, powiążmy to intuicyjne pojęcie nieuporządkowania z matematycznym pojęciem
prawdopodobieństwa. Związek między wartością entropii S a tzw. prawdopodobieństwem termodynamicznym W podał
Boltzmann w postaci równania:
S = k· ln W
gdzie k - stała Boltzmanna.
Prawdopodobieństwo termodynamiczne jest proporcjonalne do prawdopodobieństwa matematycznego (W~P lub W=q·P) i tę
zależność wykorzystamy do wykazania powiązania między stanem nieuporządkowania a wartością entropii. Wyjaśnijmy
sprawę na modelu dyfuzji cząsteczek w roztworze (rysunek poniżej). Załóżmy, że stan początkowy tego układu (1), to 1 mol
cząsteczek znajdujących się w połowie A objętości tego roztworu. Stan końcowy (2) to równomierne rozproszenie wszystkich
cząsteczek w całej objętości roztworu. Ponieważ nie ma żadnych powodów, by dowolna cząsteczka znajdowała się akurat w
tej (A) a nie innej (B) części roztworu, prawdopodobieństwo "matematyczne" wystąpienia dowolnej cząsteczki w połowie A
wynosi 1/2, a jednego mola cząsteczek w jednej połowie roztworu związane jest z prawdopodobieństwem (1/2)
N
, gdzie N -
ilość cząsteczek w 1 molu. Przypominam, że prawdopodobieństwo wystąpienia n zdarzeń pozytywnych jednocześnie (każda z
cząsteczek zawartych w tym molu występuje w tej samej części roztworu) jest iloczynem prawdopodobieństw poszczególnych
zdarzeń, czyli w tym przypadku (1/2)
n
, gdzie n - ilość cząsteczek w molu, czyli N.
stan początkowy
stan końcowy
P = (1/2)
N
P = 1
N
Stan końcowy procesu dyfuzji to równomierne rozproszenie cząsteczek w całej objętości. Tu prawdopodobieństwo
wystąpienia dowolnej cząsteczki w objętości roztworu wynosi 1 (cząsteczka jest tu na pewno), czyli dla N cząsteczek jest to
1
N
= 1. Zatem stan końcowy jest stanem o wyższym prawdopodobieństwie niż stan wyjściowy. W trakcie spontanicznych
procesów układ dąży do stanu bardziej prawdopodobnego. Z wcześniej podanego równania i powyższych rozważań można
wyprowadzić wzór:
ΔS = k·ln q·P
2
- k·ln q·P
1
= k·ln (P
2
/P
1
) = k·ln (W
2
/W
1
)
A ponieważ
W
2
>W
1
to
k·ln (W
2
/W
1
)>0
czyli
ΔS>0
.
Uogólniając, prawdopodobieństwo stanu mniej uporządkowanego jest zawsze większe niż bardziej uporządkowanego.
W trakcie procesów spontanicznych układ dąży do stanu bardziej prawdopodobnego (mniej uporządkowanego), co wiąże się
ze wzrostem entropii.
Robiąc pewne uproszczenie można uznać, że przyroda dąży do rozproszenia zarówno energii jak i materii, co przekłada się na
wzrost entropii i spadek energii układu. Tak zachowują się wszystkie układy naturalne w procesach samorzutnych. Pytanie,
jakie pojawia się w tym kontekście: dlaczego istnieją trwałe struktury, skoro wszystko samoistnie dąży do rozproszenia?
Dzieje się tak dzięki temu, że zazwyczaj rozpraszaniu materii (wzrost entropii) towarzyszyć musi wkład energii, zaś obniżaniu
energii towarzyszy zazwyczaj większe uporządkowanie czyli spadek entropii. Zatem te dwa czynniki działają najczęściej
antagonistycznie, wyznaczając pewne zakresy i pułapy współdziałania, powodując może nieco chwiejną równowagę
w przyrodzie, ale jednak równowagę. Równowagę tę opisuje funkcja zwana entalpią swobodną.
do strony
tytułowej
do góry
strony
Entalpia swobodna a równowagi
Zmianę entalpii swobodnej w danym procesie (zwanej także potencjałem termodynamicznym) oznaczamy symbolem ΔG
i wyrażamy wzorem:
ΔG = ΔH - T·ΔS
Jak z powyższego widać, zmiana entalpii swobodnej bierze pod uwagę zarówno wymianę energii na sposób ciepła, wymianę
energii z otoczeniem na sposób pracy (te dwa elementy zawarte są w różnicy entalpii), jak również energię powodującą
rozproszenie materii (entropia).
Wyobraźmy sobie energetyczne konsekwencje pewnego procesu np. dysocjacji elektrolitycznej słabego elektrolitu (dajmy na
to kwasu octowego CH
3
COOH). Na początku procesu mamy w roztworze wodnym jedynie niezdysocjowane cząsteczki
CH
3
COOH. Po pewnym czasie, po kolejnych zderzeniach któraś z cząsteczek uzyskała energię pozwalającą na rozerwanie
najsłabszego wiązania, czyli dysocjację na CH
3
COO
-
i H
+
. Ten miniukład (dwa jony) ma energię wewnętrzną większą niż miał
przed dysocjacją, ale ma też większy stopień nieuporządkowania. Wzrosła zatem i entalpia (ΔH) i entropia (ΔS), zaś znak ΔG
będzie zależał od wzajemnych relacji wielkości zmiany entalpii, zmiany entropii i temperatury, w której toczy się proces. Już
wcześniej powiedzieliśmy, że dowolny proces będzie przebiegał tak długo, aż "zysk" z powodu zmiany wartości jednego
parametru (np. wzrostu entropii) zrównoważy "stratę" np. z powodu zmiany wartości drugiego parametru (np. wzrostu
energii). Tu zysk ze wzrostu entropii przeważa i cząsteczka pozostanie zdysocjowana. Takie minizjawisko powtórzy się dla
następnych cząsteczek, bowiem zawsze zysk ze wzrostu entropii przeważy stratę z powodu wzrostu energii. Jednak o ile
nakład energii na rozerwanie wiązania O-H jest w zasadzie zawsze taki sam ("straty" rosną liniowo, proporcjonalnie do ilości
zdysocjowanych cząsteczek w roztworze), to przyrost "zysku" spowodowanego wzrostem entropii jest z każdą zdysocjowaną
cząsteczką coraz mniejszy, bowiem taki sam efekt (rozpad cząsteczki na dwa jony) wprowadza względnie coraz mniejsze
zmiany w nieuporządkowanie roztworu. Przyrost ilości jonów o dwa, jeśli dotyczy sytuacji gdy ilość jonów rośnie od dwóch
do czterech jest przyrostem o 100%. Identyczny przyrost ilości jonów o dwa, jeśli dotyczy sytuacji wzrostu od 98 do 100
jonów, to przyrost tylko o około 2% zaś przyrost od 100 000 000 do 100 000 002 jonów jest już tylko "cieniem" tego
pierwszego. Tak więc "zyski" przyrastają coraz wolniej a "straty" rosną proporcjonalnie do ilości zdysocjowanych cząsteczek,
co powoduje, że w pewnym momencie, przy określonym stosunku ilości jonów do ilości cząsteczek niezdysocjowanych, te
dwie wartości dla układu makroskopowego (roztworu kwasu octowego o stężeniu c i temperaturze T) zrównają się. Dojdzie
do sytuacji, że ΔG = ΔH - T·ΔS = 0 i proces się zatrzyma. Uzyskamy stan równowagi termodynamicznej między jonami
CH
3
COO
-
i H
+
z jednej strony a cząsteczkami CH
3
COOH
z drugiej.
Zauważmy teraz, że podniesienie temperatury (ogrzanie roztworu) powoduje wzrost bezwzględnej wartości drugiego składnika
(T·ΔS), czyli ΔG<0 i proces rusza dalej w kierunku dysocjacji kolejnych cząsteczek, aż do uzyskania ponownie ΔG=0. O tym,
że stopień dysocjacji i stała dysocjacji rosną wraz z temperaturą wiedzieliśmy już wcześniej - tu jednak mamy wyjaśnienie w
oparciu o termodynamikę, czyli niejako wyjaśnienie rzeczywistego źródła takiego zachowania się słabych elektrolitów. Trzeba
też w tym miejscu przypomnieć, że uzyskana równowaga (makroskopowo opisywana stopniem i stałą dysocjacji) jest
równowagą dynamiczną, co oznacza, że na poziomie mikroukładów (pojedynczych cząsteczek) zachodzą procesy dysocjacji,
ale odbywają się one kosztem energetycznym równolegle przebiegających procesów łączenia się jonów w cząsteczki
niezdysocjowane. Statystycznie rzecz ujmując wydajność obu procesów (dysocjacji cząsteczek i asocjacji jonów) jest
identyczna i żadnych zmian w skali makro (w roztworze) nie obserwujemy.
Opisany powyżej proces dysocjacji słabego elektrolitu ilustruje schemat dolny-lewy. Przyrost wartości entropii układu jest
coraz mniejszy (coraz mniejsze są przyrosty procesów elementarnych, bo odbywają się "na tle" coraz większego
nieuporządkowania), zaś przyrost energii jest liniowy.
Schemat dolny po prawej ilustruje powiększony element lewego schematu, od punktu równowagi. Jasno z niego wynika, że
postęp w procesie makro (zwiększanie stopnia dysocjacji) jest niemożliwy, bowiem wówczas "koszt" (wzrost energii
wewnętrznej, opisany zmianą entalpii) jest zawsze większy od "zysku" (opisanego wzrostem entropii).
Jeżeli jednak stworzymy sytuację mieszczącą się w obrębie tego drugiego schematu, musi nastąpić natychmiastowa reakcja

układu. Dzieje się tak, kiedy do roztworu słabego kwasu w stanie równowagi (np. octowego, jak w tym przykładzie) dodamy
kwasu silnego (np. HCl). Kwas silny jest całkowicie zdysocjowany, tak więc w roztworze będziemy wówczas mieli o wiele
więcej jonów, niż wynika to z układu stanu równowagi kwasu octowego (matematycznie ze stałej równowagi dysocjacji tego
kwasu). Oznacza to z jednej strony wzrost entropii układu, ale z drugiej strony także wzrost energii wewnętrznej (entalpii). Jak
wynika z przebiegu wykresów tych dwu zmian, wzrost entropii będzie słabszy niż wzrost energii i układ dążąc do równowagi
spowoduje, że część anionów octanowych połączy się z protonami w cząsteczki niezdysocjowanego kwasu by obniżyć poziom
energii. Ponieważ tworzenie niezdysocjowanej cząsteczki słabego kwasu z jego anionu i protonu daje większy zysk
energetyczny (mocniej obniża energię) niż tworzenie niezdysocjowanej cząsteczki HCl, nastąpi cofnięcie dysocjacji kwasu
octowego (słabego) pod wpływem dodatku kwasu silnego. Proces już nam wcześniej znany, ale tu uzyskał logiczne
wyjaśnienie.
A jak jesteśmy przy równowagach, to tak "przy okazji" wspomnijmy i wyjaśnijmy sprawę procesów równowagowych,
odwracalnych w ujęciu termodynamicznym. Dowolny proces jest odwracalny, jeżeli ilość energii oddana do otoczenia podczas
przejścia układu od stanu A do stanu B jest identyczna z ilością energii potrzebną do przeprowadzenia układu ze stanu B
z powrotem w stan A. W rzeczywistości takie układy nie istnieją, są niemożliwe do zrealizowania. W praktyce np. dlatego,
że część energii wymieniana podczas zmiany stanu układu rozprasza się w otoczeniu. Najczęściej przywoływany proces
rozprężania i sprężania gazu, to nie tylko praca potrzebna do zmniejszenia objętości gazu, która później zostanie "oddana"
przez zwiększenie objętości gazu, ale też praca potrzebna na pokonanie siły tarcia tłoka, która ulega "rozproszeniu" w czasie
zarówno sprężania jak i rozprężania gazu. Innymi słowy można powiedzieć, że każdemu rozpatrywanemu procesowi
teoretycznie odwracalnemu towarzyszy inny (inne) proces, również powodujący wymianę energii i to w sposób
nieodwracalny. Wszystkie procesy samorzutne są procesami nieodwracalnymi, a odwracalność realnych procesów, o których
mówią podręczniki chemii fizycznej są stanami wyidealizowanymi, upraszczającymi rzeczywistość w celu jej łatwiejszego
zrozumienia (tak jak pojęcie gazu idealnego). Tak więc układy rzeczywiste mogą czasami praktycznie ulegać przemianom
odwracalnym (tak jak gazy rzeczywiste w pewnych stanach zachowują się praktycznie jak gazy idealne), jednak należy
pamiętać, że to tylko przybliżenie.
Za doskonałe przybliżenia takich odwracalnych procesów możemy uznawać procesy przebiegające na poziomie cząsteczek
i atomów.
Nie wszystko na raz
Jak już nie raz wspominaliśmy w tym opracowaniu, rozpatrując dane zjawisko nie wolno nam zapominać o toczących się
równolegle innych procesach, które mają wpływ na nasz rozpatrywany układ. Wpływ jest raz większy, raz mniejszy, w
zależności od rodzaju i wielkości oddziaływań między elementami jednego i drugiego układu. Z pewną przesadą można
powiedzieć, że w celu wyczerpującego opisu danego stanu czy procesu powinniśmy wziąć pod uwagę wpływ całego
wszechświata na nasz proces. Takie ortodoksyjne podejście do opisywania pełnej prawdy o układzie, z praktycznego punktu
widzenia nie jest ani możliwe, ani potrzebne, ani rozsądne. Opis byłby co prawda prawdziwy, ale tak zagmatwany, że
praktycznie nic by z niego nie wynikało. Z drugiej strony, zbytnio upraszczając sytuację możemy pozbawić się ważnych
informacji, których brak powoduje niezrozumienie zachowania się badanego układu. Jak więc znaleźć złoty środek? Tu
niestety gotowej recepty nie ma i być nie może. Umiejętności wyboru, co w danej sytuacji jest istotne, a co bez zbytniej straty
dla prawdziwego opisu zjawiska można pominąć, nabiera się z czasem, w trakcie zdobywania coraz głębszej wiedzy. Ktoś, kto
ma już sporą wiedzę o materii i prawach nią rządzących może dokonać szybkiego przeglądu i zadecydować, co w danym
układzie i w danej sytuacji jest najistotniejsze. Jednocześnie nie zgubi z pola widzenia tego, co mniej istotne a może mieć
znaczenie w szczególnych warunkach (warunki brzegowe). Będzie miał też na uwadze czynniki, które na obserwowany układ
wpływ mają tak mały, że w pierwszym przybliżeniu można je całkowicie pominąć, ale nie wolno zapomnieć, że istnieją i jakiś
jednak wpływ mają!
do strony
tytułowej
do góry
strony
Przykład pierwszy - dysocjacja
Rozpatrywaliśmy już przykład dysocjacji w przypadku słabego elektrolitu. Doszliśmy wówczas do wniosku, że nakład energii
na rozerwanie wiązań jonowych dość szybko równoważy zysk spowodowany wzrostem entropii, wynikłym z procesu
dysocjacji, co spowoduje zatrzymanie procesu w skali makro, czyli ustalenie się stanu dynamicznej równowagi. Dla
wyjaśnienia, czemu słabe elektrolity dysocjują tylko częściowo, taki uproszczony opis zjawiska zupełnie nam wystarczył.
Gdybyśmy chcieli jednak dokładniej wyjaśnić zjawiska związane ze słabymi elektrolitami - nie byłoby tak łatwo. Aby
zrozumieć te bardziej złożone problemy, np. istotę aktywności elektrolitów, zmiany stopnia dysocjacji w zależności od stężenia
roztworu i temperatury, niezależność stałej dysocjacji od stężenia, trzeba włączyć do opisu stanu układu (roztworu)
oddziaływania międzycząsteczkowe. Musimy wziąć pod uwagę energetyczne aspekty oddziaływań cząsteczek
rozpuszczalnika, cząsteczek związku rozpuszczonego, jonów powstałych z dysocjacji oraz wszystkich innych elementów
składających się na roztwór. Musimy te relacje między wszystkimi składnikami układu rozpatrzyć we wszystkich możliwych
konfiguracjach ("każdy z każdym") i odmianach (dipol-dipol, wiązania wodorowe, solwatacja, elektrostatyczne przyciąganie
i odpychanie itp.) z jednoczesnym uwzględnieniem zmian w wartościach dwóch najistotniejszych funkcji - entalpii i entropii.
Tak więc zbytnie uproszczenie opisu zjawiska powoduje niejasności (czemu stopień dysocjacji zależy od stężenia a stała
dysocjacji nie), zaś wzięcie pod uwagę wszystkich składowych układu i ich wzajemnych relacji tworzy trudną do całościowego
ogarnięcia sieć powiązań i wzajemnych wpływów. Żeby sobie umieć radzić z tego typu problemami trzeba ćwiczyć, uczyć się
(poznawać prawa i reguły) i znów ćwiczyć, ćwiczyć. A ćwiczyć radzę na początek na dość prostych przykładach: dokonać
najpierw uproszczonego opisu zjawiska, potem zastanowić się, jakie jeszcze inne czynniki teoretycznie mogłyby mieć wpływ
na obserwowane zjawisko i po kolei rozważać każde z nich w kontekście zjawiska głównego. Następnie wybrać te, co do
których nabraliśmy podejrzenia, że mogą mieć istotny wpływ na zjawisko (dwa, trzy najważniejsze) i jeszcze raz przemyśleć
całość procesu z uwzględnieniem tych wybranych wpływów. Dobrze jest też spróbować wyjaśnić zjawisko zarówno na gruncie
obserwacji makroskopowych (prawa, wzory, reguły) jak i na poziomie cząsteczkowym.
Można na przykład przeprowadzić takie rozważania:
Żeby konkretna cząsteczka kwasu octowego rozpadła się na jony, musi dojść do zdarzenia, które spowoduje uzyskanie przez
cząsteczkę takiej energii, że rozerwanie wiązania (pokonanie sił przyciągania elektrostatycznego jonów reszty kwasowej i
protonu) będzie możliwe. Można to sobie wyobrazić, jako np. zderzenie dipola cząsteczki wody o odpowiednio dużej energii
translacji z atomem wodoru ugrupowania COOH cząsteczki kwasu. W tym momencie trzeba sobie uświadomić, że różne
cząsteczki wody w tym roztworze mają różne energie translacji (temperatura roztworu określa średnią energię translacji

cząsteczek), tak więc nie każde zderzenie spowoduje oderwanie protonu. Warto też sobie przypomnieć, że wiązanie
chemiczne nie ma ustalonej długości, atomy związane wiązaniem oscylują (przybliżają się do siebie i oddalają) i stan, kiedy
wiązanie będzie wydłużone ułatwi oderwanie protonu. Od temperatury zależy i ilość cząsteczek wody o odpowiedniej energii
translacji, jak również temperatura ma wpływ na oscylacje wiązań. Stąd im wyższa temperatura, tym więcej cząsteczek wody
zdolnych dokonać dysocjacji kwasu octowego. Nie zapomnijmy też, że cząsteczki kwasu są otoczone cząsteczkami wody
(solwatacja, hydratacja), szczególnie w okolicy polarnego elementu cząsteczki kwasu czyli grupy -COOH, co stanowi swego
rodzaju "mur obronny" chroniący przed uderzeniami innych cząsteczek. Teraz już widzimy złożoność, prostego w pierwotnym
opisie, aktu dysocjacji.
Rozważmy drugi ważny element wpływający na dysocjację słabych elektrolitów - stężenie. Wiemy, że w danej temperaturze
stopień dysocjacji jest tym wyższy im stężenie elektrolitu niższe. Przypominając sobie, że stopień dysocjacji to stosunek ilości
cząsteczek, które uległy dysocjacji do ilości cząsteczek rozpuszczonych w roztworze, dochodzimy do konstatacji, że
dwukrotnie wyższy stopień dysocjacji może oznaczać taką samą ilość zdysocjowanych cząsteczek ale z dwukrotnie mniejszej
ilości cząsteczek rozpuszczonych (mniejsze stężenie). Możemy zatem zbudować sobie myślowy model, w którym bezwzględna
ilość cząsteczek zdysocjowanych zależy od rozpuszczalnika (jego "wysokoenergetycznej" części cząsteczek, zdolnych
sprowokować dysocjacje), stężenie substancji rozpuszczonej nie ma tu większego znaczenia, a wzrost stopnia dysocjacji to
wynik definicji tego parametru a nie zmiany właściwości substancji rozpuszczonej. Co na ten temat mówi doświadczenie?
Otóż zmieniając dziesięciokrotnie stężenie kwasu od c = 0,05 mol/dm
3
do c = 0,005 mol/dm
3
stopień dysocjacji rośnie nam od
2% do 6% (trzykrotnie) a ilość zdysocjowanych cząsteczek ... maleje ponad trzykrotnie. Tak więc widać, że nasz model
zbytnio nie odbiega od rzeczywistości.
Zastanówmy się teraz nad sytuacja "odwrotną". W roztworze są dodatnie protony i ujemne aniony reszty octanowej. Te dwa
odwrotnie naładowane elementy powinny łączyć się ze sobą, tym łatwiej i szybciej, że ich pola elektryczne będą "aktywnie
wyszukiwać" i przyciągać jony przeciwnego znaku. Spotkanie dwóch przeciwnie naładowanych jonów będzie tym łatwiejsze
im więcej ich będzie w roztworze (duże stężenie) i im wyższa będzie temperatura roztworu (szybko poruszające się jony
częściej będą się ze sobą spotykały). Z drugiej jednak strony, można sobie wyobrazić, że zbyt szybko poruszające się jony będą
zbyt krótko przebywać w polu wzajemnych oddziaływań i mimo spotkania do odtworzenia cząsteczki nie dojdzie. Tu widać, że
stężenie dość jednoznacznie wpływa na rozpatrywany proces łączenia się jonów: im większe - tym łatwiej się spotykają jony,
tym są bliżej siebie i tym silniej działają, ważne w tym przypadku, siły elektrostatyczne. Natomiast wypadkowy efekt wpływu
temperatury nie do końca jest jasny - zwiększona energia translacji powoduje częstsze spotkania jonów, ale też zwiększa
względna ilość spotkań nieaktywnych.
Ponieważ w stanie równowagi w układzie makroskopowym biegnie zjawisko dysocjacji w jedną stronę a proces łączenia się
jonów w drugą, wydajność tych dwóch procesów musi być równa. Zmiana parametru układu wpływa oczywiście na obydwa
procesy, na ich wydajności i w konsekwencji prowadzi do obserwowanych zmian. Temperatura zmienia i stopień i wartość
stałej dysocjacji, zmiana stężenia zmienia tylko stopień dysocjacji. To wiedzieliśmy już wcześniej, ale teraz wiemy - i to jest w
nauce najważniejsze - DLACZEGO tak się dzieje.
Który model zatem jest lepszy, prawdziwszy - ten wcześniejszy, termodynamiczny czy ten wykoncypowany przez nas przed
chwilą? W tym momencie nie ma to najmniejszego znaczenia. Jeśli do kogoś przemawia matematyka, rozumie ten język i
swobodnie się nim posługuje w rozumowaniu, model termodynamiczny będzie lepszy, prawdziwszy, bardziej naukowy. Jeśli z
matematycznych rozważań niewiele rozumiem, nie czuję pojęcia prawdopodobieństwo i nie rozumiem dlaczego wartość
entropii rośnie najpierw szybko a potem wolniej, zaś przemawia do mnie obrazowe zderzanie się cząsteczek, model drugi
będzie lepszy. Ważne, byśmy zapamiętali o co w tym zjawisku chodzi, dlaczego takie czy inne czynniki mają wpływ i jaki,
byśmy umieli powiązać przyczyny i skutki i na podstawie tej wiedzy szybko mogli ustalić wpływ jednych czynników na
drugie. Jak osiągniemy taki stan wiedzy na dany temat, to studiowanie i rozumienie tej ścisłej, "oficjalnej" wiedzy będzie o
niebo łatwiejsze i skuteczniejsze. Tak więc wychodząc od wyobrażeń nawet nie całkiem prawdziwych, szybko i skutecznie
będziemy mogli posiąść wiedzę ścisłą i w 100% prawdziwą (jeśli taka gdziekolwiek istnieje, ale to już zadanie dla filozofów).
Będziemy mieli i piątkę w indeksie i, co ważniejsze, rzetelną wiedzę, którą będziemy umieli zastosować w praktyce.
do strony
tytułowej
do góry
strony
Przykład drugi - rozpuszczanie
Rozpatrując sprawy związane z rozpuszczaniem substancji czy mieszaniem się cieczy - czyli z pozoru ze zjawiskami dość
prostymi - co i raz spotykamy "dziwne" zachowania, wyjątki od reguł. Szybko zaczynamy wątpić w jakiś porządek i istnienie
reguł rządzących tym procesem. Jedne substancje rozpuszczając się powodują ogrzewanie się roztworu a inne ochłodzenie,
a u większości nie zauważamy żadnego efektu cieplnego. Pewne substancje mieszają się ze sobą bez ograniczeń, większość
jednak ma określoną granicę rozpuszczalności tworząc roztwory nasycone. Okazuje się jednak, że można przejść poza zakres
rozpuszczalności, istnieją bowiem roztwory przesycone. Substancje najczęściej lepiej rozpuszczają się w wyższych
temperaturach ("wyjątek" gazy) ale np. Ca(OH)
2
rozpuszcza się w 60°C prawie o 30% gorzej niż w 20°C, a rozpuszczalność
NaCl praktycznie nie zależy od temperatury. I jak się w tym wszystkim połapać.
Taka "niekonsekwencja" w regułach związanych z procesem rozpuszczania sugeruje istnienie co najmniej paru równoległych
zjawisk, składających się na proces rozpuszczania (czy szerzej - tworzenia roztworu). Zastanówmy się, jakie procesy mogą
brać udział w tym zjawisku. Po pierwsze, energia wiązań kryształu czy silnych oddziaływań międzycząsteczkowych cieczy,
która musi zostać pokonana, żeby pojedyncze cząsteczki mogły wniknąć między cząsteczki rozpuszczalnika. Po drugie, musi
dojść do rozluźnienia oddziaływań międzycząsteczkowych rozpuszczalnika i następnie solwatacji cząsteczek substancji
rozpuszczonej. Jeżeli substancja jest elektrolitem, szczególnie jeśli jest silnym elektrolitem, nastąpi zjawisko dysocjacji.
Wszystkie te zjawiska powiązane są ze zmianą energii - rozluźnienie sił międzycząsteczkowych z pobraniem energii (rozpad
kryształu, dysocjacja), solwatacja z oddaniem energii. I tu już ktoś, kto ma choćby odrobinę doświadczenia w "myśleniu
fizykochemicznym" zauważa, że efekt końcowy (grzanie czy ochładzanie się roztworu), jak i wpływ temperatury na
zachowanie substancji, zależy od relacji ilościowych efektów energetycznych pobierania i oddawania energii na sposób ciepła.
Jeśli przeważą (co do wielkości wymienianej energii) procesy egzotermiczne (solwatacja) nad endotermicznymi
(rozpuszczanie się kryształu) cały proces będzie egzotermiczny. Jeśli te dwa typy procesów się bilansują, efekt termiczny
zjawiska tworzenia roztworu będzie bliski zera i efektu zmiany temperatury roztworu nie zauważymy.
Jeżeli rozpuszczamy w wodzie tiosiarczan sodu bezwodny (Na
2
S
2
O
3
), to roztwór bardzo silnie się rozgrzewa. W tym
przypadku silna solwatacja powoduje wydzielenie takiej ilości ciepła (energii na sposób ciepła), że znacznie przewyższa ona
ilość energii pochłoniętej przez sieć krystaliczną w celu uwolnienia cząsteczek i przez zjawisko dysocjacji, a właściwie
hydrolizy tiosiarczanu. Jeśli jednak rozpuszczać będziemy tiosiarczan pięciowodny (Na
2
S
2
O
3
*5H
2
O) to roztwór się bardzo
silnie ochłodzi. W tym przypadku proces hydratacji nie wystąpi, bowiem cząsteczki tiosiarczanu już są uwodnione. O
wielkości efektu zadecyduje proces zrywania wiązań sieci krystalicznej i wiązań jonowych, proces będzie endotermiczny.

prawdopodobieństwo znalezienia się konkretnej cząsteczki w części objętości A roztworu wynosi 1/4. Dla N cząsteczek
w roztworze, prawdopodobieństwo, że znajdą się wszystkie w tej części roztworu wyniesie (1/4)
N
, a więc będzie bardzo
małe. Stan, w którym cząsteczki skupione są w jednym miejscu jest bardzo mało prawdopodobny. Prawdopodobieństwo,
że konkretna cząsteczka znajdzie się w dowolnym miejscu roztworu wynosi 1, zatem prawdopodobieństwo
równomiernego rozłożenia N cząsteczek w roztworze wynosi 1
N
= 1. Tak więc układ samoistnie będzie dążył do stanu
bardziej prawdopodobnego. (Podobnie tłumaczyć możemy zjawisko wzrostem entropii);
inne tłumaczenie, też związane z prawdopodobieństwem zdarzeń losowych, ale bardziej działające na wyobraźnię, to
rozpatrywanie stanu roztworu o gradiencie stężenia (
c
1
>c
2
) jako zbioru cząsteczek rozpuszczalnika i substancji
rozpuszczonej, zderzających się przypadkowo ze sobą. Dla cząsteczek substancji rozpuszczonej usytuowanych na
granicy stężenia wysokiego i niskiego ilość zderzeń "od dołu do góry" będzie większa niż odwrotnie i więcej cząsteczek
będzie poruszać się w stronę mniejszego stężenia. Dla pełniejszego opisu takiej sytuacji trzeba by jeszcze rozważyć rolę
sił oddziaływania cząsteczek substancja-substancja oraz substancja-rozpuszczalnik.
W stanie równowagi wydajność procesu przemieszczania się cząsteczek między dwoma dowolnymi obszarami roztworu
jest identyczna, zatem roztwór makroskopowy wykazuje równowagę, jednakowe stężenie w każdym elemencie
objętości.
można również zaproponować podejście do tematu takie, jak w punkcie powyżej, ale wziąć pod uwagę nie samodzielnie
poruszające się cząsteczki substancji rozpuszczonej a asocjaty, elementy powstałe w zjawisku solwatacji (aktywny udział
cząsteczek rozpuszczalnika w tym procesie ).
Wydawałoby się, że już zapanowaliśmy na procesem rozpuszczania, a tu pojawia się nowy "kłopot". Jeśli proces rozpuszczania
jest egzoenergetyczny to wzrost temperatury powinien hamować i cofać ten proces. Wielu z nas tak by skojarzyło wcześniej
przyswojoną regułę dotycząca np. reakcji endo- i egzotermicznych. W tym przypadku to skojarzenie i zastosowanie takiego
schematu myślenia jest błędem. W przypadku reakcji chemicznych, temperatura decyduje o tym co jest bardziej
prawdopodobne - zamiana kompleksu aktywnego w produkt czy powrót ze stanu kompleksu aktywnego do substratów.
W przypadku procesu rozpuszczania temperatura ma bezpośredni dodatni wpływ na główny etap procesu - odrywanie
cząsteczek od sieci krystalicznej i ewentualną jonizację (dysocjację), natomiast wpływ ujemny może mieć na następujący
później proces solwatacji. Tak więc mimo, że proces całościowo może być egzoenergetyczny, to wzrost temperatury nie
będzie go osłabiał. Widać tu, jak bardzo zawodne może być korzystanie z ogólnych reguł, bez głębokiej znajomości zasad,
z których dana reguła została wywiedziona i granic, w których można ją stosować.
Jeżeli stan nasycenia potraktujemy jako układ znajdujący się w warunkach, w których szybkość (wydajność) procesu
przechodzenia cząsteczek z ciała stałego do roztworu zrównuje się z wydajnością procesu krystalizacji (przechodzenia
cząsteczek z roztworu do kryształu ciała stałego), to dla zrozumienia całości sprawy musimy przeanalizować, jak poszczególne
parametry tego stanu (głównie temperatura i związana z nią lepkość oraz translacja cząsteczek, tak rozpuszczalnika jak i ciała
rozpuszczonego) wpływają na wydajność jednego i drugiego procesu. Tylko wówczas będziemy w stanie ogarnąć całość
problemu i zrozumieć różne "anomalie" występujące w tych procesach. Ponadto musimy być świadomi, że inaczej parametry
stanu roztworu mogą wpływać na sam proces (szybkość rozpuszczania) a inaczej na moment osiągnięcia stanu równowagi,
czyli stężenie nasycenia (rozpuszczalność). W ujęciu termodynamiki statystycznej o stanie równowagi (nasyceniu) zdecyduje
zrównanie wzrostu energii (cząsteczki rozpuszczone, a szczególnie jony powstałe z dysocjacji będzie charakteryzować wyższa
energia niż elementy kryształu) ze wzrostem entropii. Zaś wielkość zmian energetycznych i zmian uporządkowania układu
(entropii) zależeć będzie od paru czynników (ciepło rozpuszczania i solwatacji, ilość cząsteczek i jonów w stanie nasycenia,
siła wiązań jonowych itp.) co w sumie daje trudny do przewidzenia a priori końcowy efekt makroskopowy.
Tak więc rozpuszczalność, stężenie stanu nasycenia, nie jest parametrem substancji rozpuszczanej lecz zależy zarówno od
cech substancji rozpuszczanej jak i rozpuszczalnika, jest wypadkową wzajemnych relacji między tymi cechami i związanymi
z tym zjawiskami. To też przyczynek do wcześniej postulowanego "nowego spojrzenia na świat". To, że cechy roztworu
zależeć muszą i od substancji rozpuszczonej i od rozpuszczalnika jest dla nas dość oczywiste, ale ponieważ tablice
fizykochemiczne podają wartości rozpuszczalności substancji, podświadomie często gotowi jesteśmy traktować
rozpuszczalność jako parametr charakterystyczny dla tej substancji, a nie całego roztworu.
do strony
tytułowej
do góry
strony
Przykład trzeci - dyfuzja
Dyfuzja - zjawisko polegające na samoistnym, równomiernym rozprzestrzenianiu się cząsteczek substancji rozpuszczonej w
całej objętości roztworu. Zjawisko to, jak wiele innych, objaśnić można na kilka sposobów:
Ponieważ zjawisko dyfuzji polega na przemieszczaniu się cząsteczek substancji rozpuszczonej w ośrodku rozpuszczalnika
można przy opisie tego procesu wziąć pod uwagę (choć ostrożnie - nie wszystko na raz!) także lepkość rozpuszczalnika,
stosunek masy cząsteczki rozpuszczalnika i substancji rozpuszczonej, siły grawitacji, oddziaływania międzycząsteczkowe
cząsteczek rozpuszczalnika a także kształty i rozmiary cząsteczek. Wszystkie te czynniki w większy czy mniejszy sposób mogą
wpłynąć na szybkość procesu dyfuzji. Trzeba też uzmysłowić sobie, że pojęcie "szybkość dyfuzji" nie ma żadnego realnego
sensu, dopóki nie zdefiniujemy go w sposób jednoznaczny. Wpływ wszystkich tych elementów jest zawarty w I i II prawie
Ficka, które pozwalają określić (przewidzieć) stężenie w każdym punkcie roztworu i w każdej dowolnej chwili. Nauczenie się
praw Ficka i zapamiętanie postaci matematycznego zapisu niewiele jednak przyczyni się do zrozumienia istoty zjawiska.
Natomiast, jeśli ktoś sprawnie posługuje się aparatem matematycznym (szczególnie rachunkiem różniczkowym), to
zrozumienie wyprowadzenia tych wzorów może być dobrym początkiem dla głębokiego zrozumienia problemu.
Mówiąc o uzyskiwaniu przez układ najniższych energii i najwyższych wartości entropii powinniśmy dodawać (a przynajmniej
o tym pamiętać), że najniższych czy najwyższych z możliwych do osiągnięcia w danej sytuacji. Jeżeli przedzielimy dwa

roztwory o różnych stężeniach błoną półprzepuszczalną, to układ będzie się starał osiągnąć równe stężenia po obu stronach tej
błony, ale jeżeli anion substancji rozpuszczonej będzie miał rozmiary nie pozwalające przeniknąć przez błonę (np. cząsteczki
białka), to będzie mogło dojść co najwyżej do wyrównania stężeń kationów. Tu pamiętać należy, że roztwór i każda jego część
musi być elektrycznie obojętna, nie może się więc zdarzyć, że na przykład.połowa kationów przejdzie na drugą stronę błony,
pozostawiając aniony. Doprowadziło by to do powstania ogromnego potencjału na granicy faz (po dwóch stronach błony). Ten
potencjał powstając już znacznie wcześniej zahamuje ruch kationów. To fizyczne ograniczenie możliwości ruchu niektórych
elementów układu podczas procesu dyfuzji przez błony leży u podstaw zjawiska ciśnienia osmotycznego i tzw. równowag
Donnana.
Rola przypadku w przyrodzie
W potocznym rozumieniu pojęcie przypadek kojarzy nam się zazwyczaj nie tylko z czymś nieprzewidywalnym, ale dodatkowo
mało prawdopodobnym, niespodziewanym. W matematyczno-fizycznym ujęciu przez przypadek, zdarzenie losowe,
rozumiemy wszystko to, czego dokładnie nie da się przewidzieć a priori, przewidzieć co do wartości, parametrów, ale że to
coś nastąpi jesteśmy najczęściej pewni. Warto sobie przemyśleć i uświadomić takie rozumienie przypadku, bo inaczej pojawią
się trudności w zrozumieniu wielu spraw związanych z fizykochemią.
Jeśli chodzi o rolę przypadku w przyrodzie możemy powiedzieć, że występujący w skali mikro przypadek, nieprzewidywalne
zdarzenie, zamienia się w skali makro w ... pewność, ścisłe prawo. Wyobraźmy sobie dwa naczynia z taką samą ilością gazu
(ilością cząsteczek), przy czym objętość jednego jest dwukrotnie większa od drugiego. Bez trudu wykażemy, że w warunkach
tej samej temperatury, ciśnienie w naczyniu większym będzie dwa razy mniejsze niż w naczyniu mniejszym (równanie
Clapeyrona). Tu nie ma cienia wątpliwości i o żadnym przypadku, żadnej przypadkowej wartości ciśnienia nie ma mowy.
Wiemy również, że ciśnienie to jest identyczne w każdym miejscu ściany naczynia. Wiemy także, że ciśnienie to jest skutkiem
uderzeń o ściany naczynia poruszających się bezładnie cząsteczek gazu. Jeśli więc wyobrazimy sobie, że śledzimy jedną
cząsteczkę gazu przez jakiś czas, to nie jesteśmy w stanie w żadnym momencie przewidzieć ani kierunku ruchu, ani szybkości
tej cząsteczki, ani nawet czy w najbliższej przyszłości uderzy ona o ściankę naczynia czy nie. Tak więc zachowanie każdej
z cząsteczek tworzących gaz jest nieprzewidywalne (losowe, przypadkowe) natomiast zachowanie się zbioru cząsteczek,
rozpatrywanych jako całość ("gaz") jest przewidywalne i ściśle określone. Suma przypadków tworzy pewność! To sobie trzeba
dobrze przemyśleć.
Wiemy, że rozpad jąder atomowych naturalnych izotopów promieniotwórczych odbywa się z bardzo dokładnie określonym
czasem półtrwania (czasem rozpadu promieniotwórczego połowy cząsteczek danego izotopu). Natomiast nie jesteśmy w stanie
określić, który atom ulegnie rozpadowi jako następny, ani nawet które atomy będą należały do tej połowy, która rozpadnie się
w tym czasie półtrwania. Każdy rozpad jest czystym przypadkiem, ale bez względu na to, które atomy i ile zgromadzimy w
jednym miejscu, zawsze dokładnie połowa z nich rozpadnie się po tym samym czasie.
Szybkość konkretnej reakcji chemicznej w konkretnych warunkach jest bardzo ściśle określona przez stałą szybkości reakcji k.
Szybkość ta określa ile cząsteczek substratu zamieni się w produkt w określonym przedziale czasu. Pojedyncze cząsteczki
substratu reagując ze sobą tworzą tzw. kompleks aktywny, twór pośredni między substratem a produktem. Jest to twór o
bardzo wysokiej energii i po powstaniu bardzo szybko musi zamienić się w produkt, lub z powrotem wrócić do postaci
substratu. Jeśli większość kompleksów aktywnych zamienia się w produkt, reakcja biegnie szybko, jeśli większość wraca do
postaci substratu, reakcja biegnie wolno. Tak więc szybkość reakcji określa suma zdarzeń pozytywnych (kompleks aktywny
przechodzi w produkt), natomiast to, czy konkretny kompleks aktywny zamieni się w produkt czy też wróci do stanu substratu
jest zupełnym przypadkiem. Tak więc znów suma całkowicie przypadkowych zdarzeń zamienia się w precyzyjnie i bez
żadnego przypadku określoną pewność.
Wielkość wypadkowego efektu dużej ilości zdarzeń przypadkowych, losowych, daje się wyznaczyć precyzyjnie przy pomocy
rachunku prawdopodobieństwa i podstawowych zasad statystyki. Warto zatem przypomnieć sobie, lub przybliżyć podstawowe
zasady tych działów matematyki. Bez podstawowej choćby znajomości matematyki dość trudno poruszać się w gąszczu
dokładnych opisów skomplikowanych zjawisk należących do fizykochemii.
Podsumowanie
W tym miejscu można już zrobić małe uogólnienie podsumowujące, pamiętając, że jak wszystkie uogólnienia, ma ono na celu
wyłącznie wstępne uporządkowanie wcześniej omówionych spraw a nie ogłoszenie prawdy niepodważalnej, jedynej i
ostatecznej.
Każde zjawisko można rozpatrywać w skali makro, bezpośrednio dostępnej naszym zmysłom. Na podstawie obserwacji
i doświadczeń znajdujemy uogólnione zasady i prawa wiążące poszczególne elementy i parametry procesu. Takie ujęcie,
będące bardzo często podstawowym ujęciem w szybkim kursie chemii fizycznej, pozwala przewidywać skutki, nie daje
jednak możliwości poznania prawdziwej istoty zjawiska. Aby zrozumieć zjawisko i móc przewidywać zachowanie się procesu,
bez względu na zakres zmienności wartości parametrów, musimy rozpatrzyć jego przebieg w skali cząsteczek i atomów.
Pamiętając, że najczęściej na rozważane zjawisko składa się kilka równoległych i nawzajem powiązanych procesów, starajmy
się wyodrębnić te najistotniejsze i na nie zwrócić przede wszystkim uwagę (nie wszystko na raz). Procesy mniej istotne
włączmy do rozważań później, gdy zrozumiemy działanie głównych sił sprawczych rozpatrywanego zjawiska. Pamiętajmy
jednocześnie, by nie "zapomnieć" o mniej ważnych, ale mających wpływ na rozpatrywane zjawisko, procesach.
Dla zjawisk, w których bierze udział duża ilość cząsteczek czy atomów, mimo losowego, przypadkowego charakteru zdarzeń
elementarnych, rachunek prawdopodobieństwa pozwala precyzyjnie i dokładnie przewidywać efekt końcowy, będący
statystyczną średnią efektów zdarzeń elementarnych. Nie zapominajmy, że wiele pojęć typu: pole elektromagnetyczne,
energia, ładunek, wiązanie chemiczne, orbital itp. mimo, że posługujemy się nimi tak, jakby oznaczały konkretną,
zdefiniowaną wartość materialną (procent wiązania jonowego, odbicie światła, energia orbitala) są tylko pojęciami,
niemożliwymi do wyobrażenia i bardziej wyczuwanymi intuicyjnie niż zrozumiałymi. Z pokorą zatem przyjmijmy, że
"wyjaśnieniem" niektórych zdarzeń z zakresu mechaniki kwantowej (poziom mikro) może być wyłącznie opis matematyczny.
Jednocześnie pamiętajmy, że ten matematyczny opis zjawiska pozwala obliczyć pewne parametry, przewidzieć zachowanie,
najczęściej jednak nie pozwala zrozumieć dlaczego tak się dzieje. Mówi nam co ale nie mówi dlaczego.
Tak więc starajmy się o następującą sekwencję poznawczą: ogólny opis zjawiska i powiązanie parametrów, zrozumienie istoty

zjawiska na poziomie cząsteczkowym i dopiero wówczas wzory i matematyka, jako precyzyjny opis. Nauczmy się też czytać
wzory, rozumieć definicje i prawa a szczególnie zdobądźmy umiejętność czytania wykresów i diagramów.
A oprócz tego, a może przede wszystkim, pamiętajmy o podstawowej zasadzie rozpraszania energii i materii - niech entalpia
i entropia, a szczególnie entalpia swobodna ( potencjał termodynamiczny), będą dla nas pojęciami zrozumiałymi, oczywistymi
i włączanymi na początku każdych rozważań.
Raz jeszcze przypominam. Te rozważania to przykład, jak można się starać przybliżyć zrozumienie, czasem rzeczywiście
niełatwych wiadomości z chemii fizycznej. To opracowanie nie ma rangi podręcznika, interpretacje zjawisk tu podawane mogą
czasem nawet znacznie różnić się od "oficjalnego" opisu a nawet mogą niekiedy być nie całkiem prawdziwe.
Zadaniem tego
opracowania nie jest nauczenie kogokolwiek czegokolwiek, a jedynie pokazanie drogi do łatwiejszego, rozumnego i
skutecznego sposobu przyswojenia sobie wiedzy z podręczników i wykładów
. Jest to tylko uzupełnienie, wprowadzenie do
tematu, na które zazwyczaj wykładowcom brakuje czasu. Jest to materiał do przemyśleń a nie szkolna wiedza podana do
wierzenia. Im bardziej nie będziesz się ze mną zgadzć - tym lepiej (dla Ciebie). Postaraj się jednak wyraźnie wyartykułować, z
czym się nie zgadzasz i dlaczego uważasz moje sugestie za gorsze od Twoich przemyśleń. O to tu chodzi - zacznijmy
samodzielnie myśleć! To nie tylko, że nie boli ale daje czasem dużą przyjemność i satysfakcję. Warto spróbować.
Jeżeli Twój opis zjawiska jest skuteczny, tzn. pozwala logicznie kojarzyć przyczyny i skutki, pozwala skutecznie i prawidłowo
przewidywać konsekwencje zmian parametrów układu, jest na tyle przejrzysty, że łatwy do zapamiętania - to już bardzo
dobrze. Nawet jeśli nie we wszystkim jest zgodny z oficjalną wiedzą. Jeśli w niczym nie kłóci się ze znanymi prawami
przyrody, to już super. A jeśli dodatkowo jest zgodny z oficjalną wiedzą na dany temat - zostań wykładowcą. Studenci będą
chodzić na Twoje wykłady niczym na najlepsze filmy.
do strony tytułowej
do góry strony
Wyszukiwarka
Podobne podstrony:
Wstęp do geografii fizyczne1
fizyczna-zaliczenie, Wstep do geografii fizycznej,Kostrzewski, WNGiG POZNAN
Wstep do gografii fizycznej, Geografia, Geografia fizyczna
(1) wstep do chemii kosmetycznej 2011 12 skrót do wysyłki
Zagadnienia do egzaminu z chemii fizycznej 13 zima
WSTĘP DO MAKRO, ogólny, UE Katowice BOND Finanse i Rachunkowość, Rok 1, Semestr 2, Makroekonomia,
chemia fizyczna wykłady, sprawozdania, opracowane zagadnienia do egzaminu Zagadnienia do egzaminu
Wstęp do psychopatologii zaburzenia osobowosci materiały
Tajemnica ludzkiej psychiki wstep do psychologii
Wstęp do Kulturoznawstwa 6 7
Wstęp do informatyki z architekturą systemów kompuerowych, Wstęp
Wstęp do XHTML
MTR 2009 Wstep do mechatr cz 3 (2)
recenzja filmu, pedagogika, semestr I, wstęp do pedagogiki, inne
Wstęp do teorii tłumaczeń 31.05.2010, moczulski
więcej podobnych podstron