
grupa7.med@gmail.com | 2008 - wdanie II, poprawione
1
LIPIDY
2008
Wydanie II, uzupełnione i poprawione

grupa7.med@gmail.com | 2008 - wdanie II, poprawione
2
SPIS TREŚCI
RZYCZYNY WTÓRNEJ HIPERLIPIDEMI

grupa7.med@gmail.com | 2008 - wdanie II, poprawione
3

grupa7.med@gmail.com | 2008 - wdanie II, poprawione
4
grupa7.med@gmail.com | 2008 - wdanie II, poprawione
5
1. Budowa i podział tłuszczowców
Lipidy (tłuszczowce) są estrami glicerolu i wyższych kwasów tłuszczowych
LIPIDY
PROSTE
ZŁOŻONE
1. Fosfolipidy
a. glicerofosfolipidy :
fosfatydyloetanoloamina (kefalina)
fosfatydylocholina (lecytyna)
difosfatydyloglicerol (kardiolipina)
b. sfingolipidy – zawierające sfingozynę
sfingomieliny
ceramidy
, które dalej dzielą się na:
cerebrozydy
gangliozydy
sulfatydy
fosfosfingozydy
2. Glikolipidy
3. Inne lipidy złożone – sulfolipidy, aminolipidy,
lipoproteiny
Kwasy tłuszczowe dzielimy w zależności od
- długośd łaocucha
- stopieo nasycenia
- miejsce syntezy
- lokalizacja wiązania nasyconego
Kwasy tłuszczowe
Nasycone - SFA
Nienasycone
- palmitynowy (C16)
- stearynowy (C18)
Wielonienasycone - PUFA
jednonienasycone – MUFA
-oleinowy i elaidynowy (C18)
Omega 6
omega 3
(Rodzina kwasu linolowego)
(rodzina kwasu alfa-linolenowego)
- linolowy (18:2)
- -linolenowy (18:3)
- arachidonowy(20:4)
- eikozapentaenowy
(20:5)

grupa7.med@gmail.com | 2008 - wdanie II, poprawione
6
2-3. Lipazy wewnątrz i zewnątrzkomórkowe
Lipazy należą do hydrolaz, możemy je podzielid na
1. Wewnątrzkomórkowe
2. Pozakomórkowe
a) wewnątrznaczyniowe
b) pozanaczyniowe
- lipazy przewodu pokarmowego (ślinianki, żołądka i trzustki)
- lipazy mleka
Lipazy zewnątrzkomórkowe:
a)lipazy przewodu pokarmowego
-lipaza kwasostabilna(językowa)
- wydzielana przez gruczoły Ebnera
- optymalne pH: 2,2-6;
- wyższe powinowactwo do kwasów średniołaocuchowych niż długołaocuchowych
- lepiej hydrolizuje estry kwasów nienasyconych
- nie hyrdolizuje fosfolipidów ani estrów cholesterolu
- działa w jamie ustnej i w żołądku
-lipaza żołądkowa
- optymalne pH: 2,2 – 7,4
- wydzielana przez komórki błony śluzowej trzonu żołądka
- wydzielane pobudzane przez substancje zwiększające produkcję soku żołądkowego
- powinowactwo do średnio i krótkołaocuchowych kwasów tłuszczowych
- duża rola w okresie noworodkowym i niemowlęcym
- hydrolizuje głównie wiązanie sn-3 estru – powstają 1,2-diacyloglicerole
-lipaza trzustkowa
- optymalne pH: 7,4-8,5
- wydzielanie podstawowe i stymulowane przez cholecystokininę i sekretynę
- dwie formy molekularne: S – slow i F – fast
- forma F jest w rzeczywistości kompleksem formy S, kolipazy i fosfolipidów
- wykazuje powinowactwo do kwasów tłuszczowych w pozycji alfa
- aktywacja pod wpływem Ca i kwasów żółciowych
kolipaza:
- glikoproteina
- syntezowana w trzustce jako postad nieaktywna
- aktywacja pod wpływem trypsyny oraz kwasów żółciowych i Ca
- ułatwia kontakt lipazy trzustkowej z triglicerydami
- pomaga w zakotwiczeniu lipazy na granicy fazy wodnej i lipidowej
- optymalne pH: 6
- wchodzi w skład formy F lipazy trzustkowej
-lipaza aktywowana solami kw. żółciowych - aktywują ją sole kw.żółciowych,
rozkłada triacyloglicerole, estry cholesterolowe, witaminowe. Występuje tez w mleku.
Synteza w trzustce.
-fosfolipaza A2- wydzielana jako proenzym aktywowany przez trypsyne i Ca2+.
Rozklada fosfolipidy. Synteza w trzustce.
grupa7.med@gmail.com | 2008 - wdanie II, poprawione
7
b) wewnatrznaczyniowe:
-LPL - lipaza lipoproteinowa
- odpowiedzialna za hydrolizę triglicerydów zawartych w chylomikronach i
VLDL – przekształca je w odpowiednio: remnanty chylomikronów i IDL
- występuje w naczyniach krwionośnych tkanek pozawątrobowych:
- mięśni szkieletowych
- tkanki tłuszczowej
- mięśnia sercowego
- aktywatory:
- apoC-II
- heparyna
- dożylne podanie heparyny powoduje uwolnienie enzymu z jego połączenia z
siarczanem heparanu i nagły wzrost lipolitycznej aktywności w osoczu
- substancje obniżające aktywnośd:
- apoC-III
- siarczan protaminy
-HTGL – lipaza TG wątrobowa
- zlokalizowana na śródbłonku zatokowych naczyo wątroby
- odpowiedzialna za katabolizm 1/3 remnantów VLDL oraz przekształcenie HDL
2B
w HDL
3
- substraty: triglicerydy i fosfolipidy
- aktywowana przez:
- insulinę
- hormony tarczycy
- do pełnej aktywności wymagane wysokie stężenie NaCl
- aktywnośd wyższa u mężczyzn
Lipazy wewnątrzkomórkowe:
-lipaza hormonowrazilwa -HSL- znajduje sie w adipocytach i służy do mobilizacji
zmagazynowanych kw. tłuszczowych w postaci TG. Odłącza ona kw. tłuszczowe w
pozycji C1 i C3 TG, następnie działają lipazy specyficzne w stosunku do monogliceroli.
HSL jest aktywowana poprzez kinaze białkową zależną od cAMP na drodze fosforylacji. Czyli
działa np. adrenalina -wiąże sie z receptorem czego efektem jest aktywacja
cyklazy adenylanowej, która z kolei przeksztalca ATP w cAMP, który aktywuje kinaze białkową
zależną od cAMP, która fosforyluje HSL będący aktywny właśnie w postacji ufosforylowanej. Tak
wiec insulina powoduje spadek cAMP i defosforylacje HSL czyli zahamowanie lipolizy.
grupa7.med@gmail.com | 2008 - wdanie II, poprawione
8
4. Wchłanianie tłuszczów
codzienne spożycie: 60-150 g – 90% to TG, reszta to cholesterol, fosfolipidy
WCHŁANIANIE DO ENTEROCYTÓW
Kwas tłuszczowy
H
2
0
Lecytyna
lizofosfatydylocholina
Fosfolipaza A2
Kw. tłuszczowy
kw.tłuszczowy
kwas
tłuszczowy + FABP
H
2
0
H
2
0
TG
2-monoacyloglicerol
Lipaza trzustkowa (+ko lipaza)
Kwas tłuszczowy
H
2
0
Estry Cholesterolu
esteraza lipidowa
Cholesterol
RESYNTEZA W ENTEROCYCIE
kwasy tłuszczowe najpierw aktywowane przez syntetazę acylo-S-CoA, następnie przenoszone przez
acylotransferazy na 2-monoacyloglicerol: 2-monoacyloglicerol + 2acylo-S-CoA = triacyloglicerol + 2CoA-SH
Lizofosfatydylocholina + Acylo-CoA ------------------ > lecytyna + CoA
pakowane do chylomikronów
Monoacyloglicerole + Acylo-CoA + AcyloCoA ---------------------------- > TG
Cholesterol + Acylo-CoA --------------------------------- > estry cholesterolu
Kwasy tłuszczowe do 10C -------------------------------- > żyłą wrotną do wątroby
grupa7.med@gmail.com | 2008 - wdanie II, poprawione
9
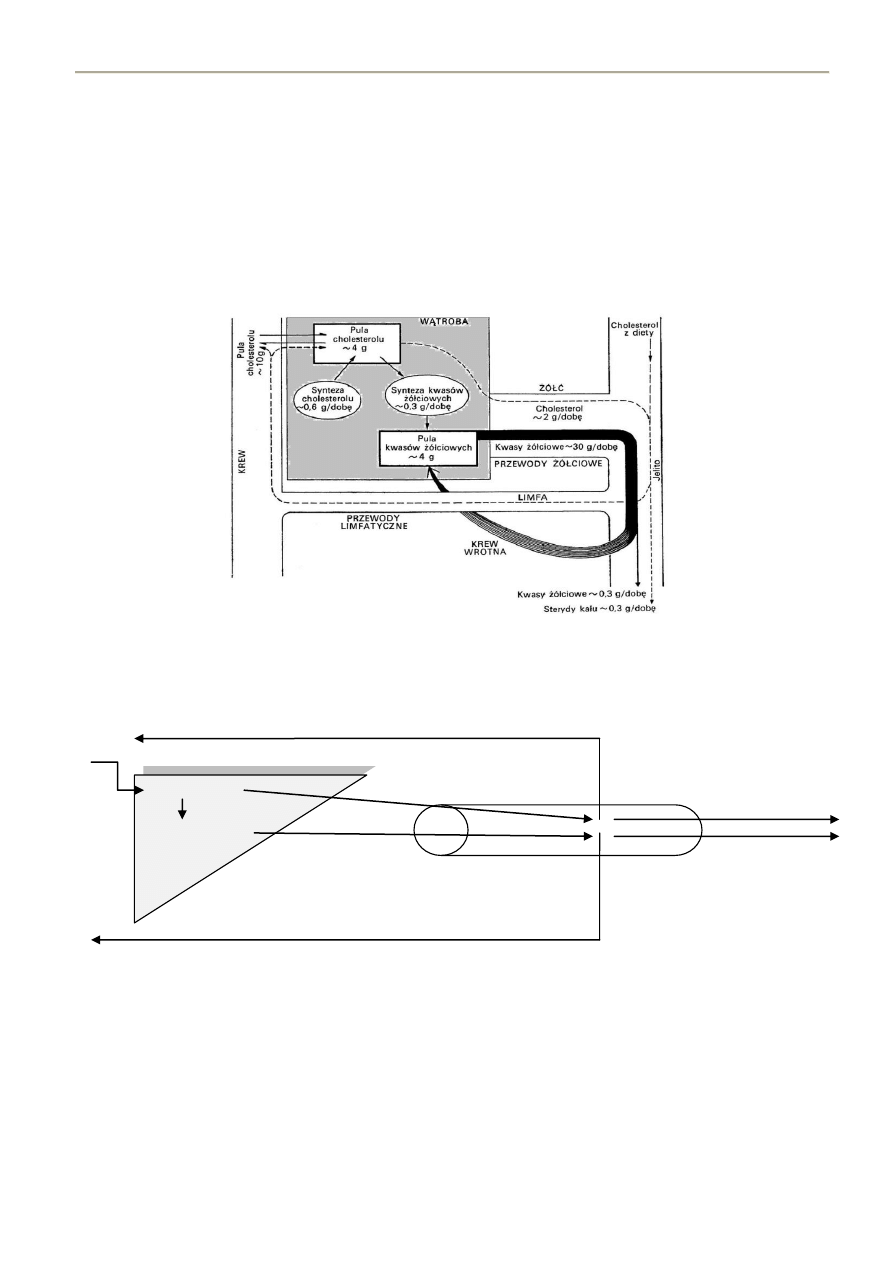
Krążenie wątrobowo-jelitowe cholesterolu
- całkowita pula kwasów żółciowych w organizmie: 3-5 g (średnio 4 gramy)
- ta pula przechodzi przez jelito 6-10 razy dziennie
- 97-99% kwasów żółciowych wydzielonych do żółci jest resorbowanych w jelicie krętym jako kwasy zarówno
pierwotne jak i wtórne – odbywa się to przy udziale błonowego białka IBAT (ileal bile acid transporter)
- kwas litocholowy jest resorbowany w mniejszych ilościach jako substancja trudniej rozpuszczalna
- wchłonięte kwasy powracają do wątroby z krwią żyły wrotnej, tam są ponownie wydzielane do żółci
- 1-2-3% kwasów żółciowych wydzielonych do jelita nie ulega wchłonięciu i jest wydalane z kałem jako sterole
kwaśne (0,3-0,4 g dziennie) – jest to główna droga eliminacji cholesterolu z organizmu
- z uwagi na tak niewielką dobową utratę kwasów żółciowych i synteza w wątrobie jest niewielka – sięga 0,3-0,4
g dziennie
- W przewodzie pokarmowym 2/3 to cholesterol endogenny (600-1000mg/dobę)
- 1/3 pochodzi z przyjmowanych pokarmów (200-500mg/dobę)
- Jedyną drogą wydalanie cholesterolu z organizmu jest żółd.
osocze
cholesterol
50%
ulega resorbcji
sterole obojętne
kwasy żółciowe
jelito
sterole kwaśne
99%
REGULACJA:
- dieta obfata w błonnik (jabłka, gruszki, otręby) hamuje wchłanianie cholesterolu w jelicie przez co zmusza
hepatocyty do pozyskiwania cholesterol (składnika żółci) z puli krążącej – obniża się tym samym TCh
- farmakologicznie – stosowanie żywic jonowymiennych (cholestyramina) – oddają one jon (np. chlorkowy w
przypadku cholestyraminy) i w to miejsce przyjmiją jon kwasu żółciowego uniemożliwiając jego wchłonięcie.
grupa7.med@gmail.com | 2008 - wdanie II, poprawione
10
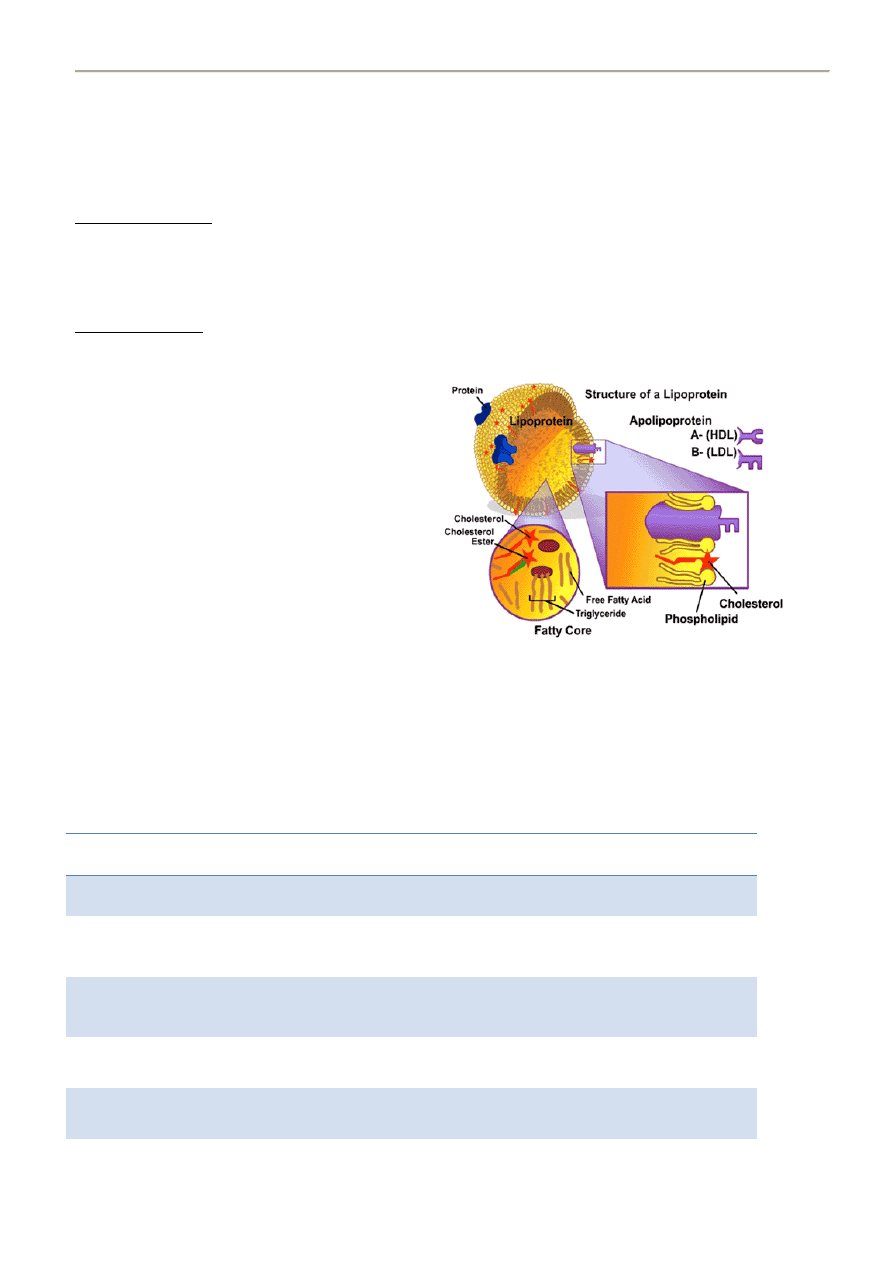
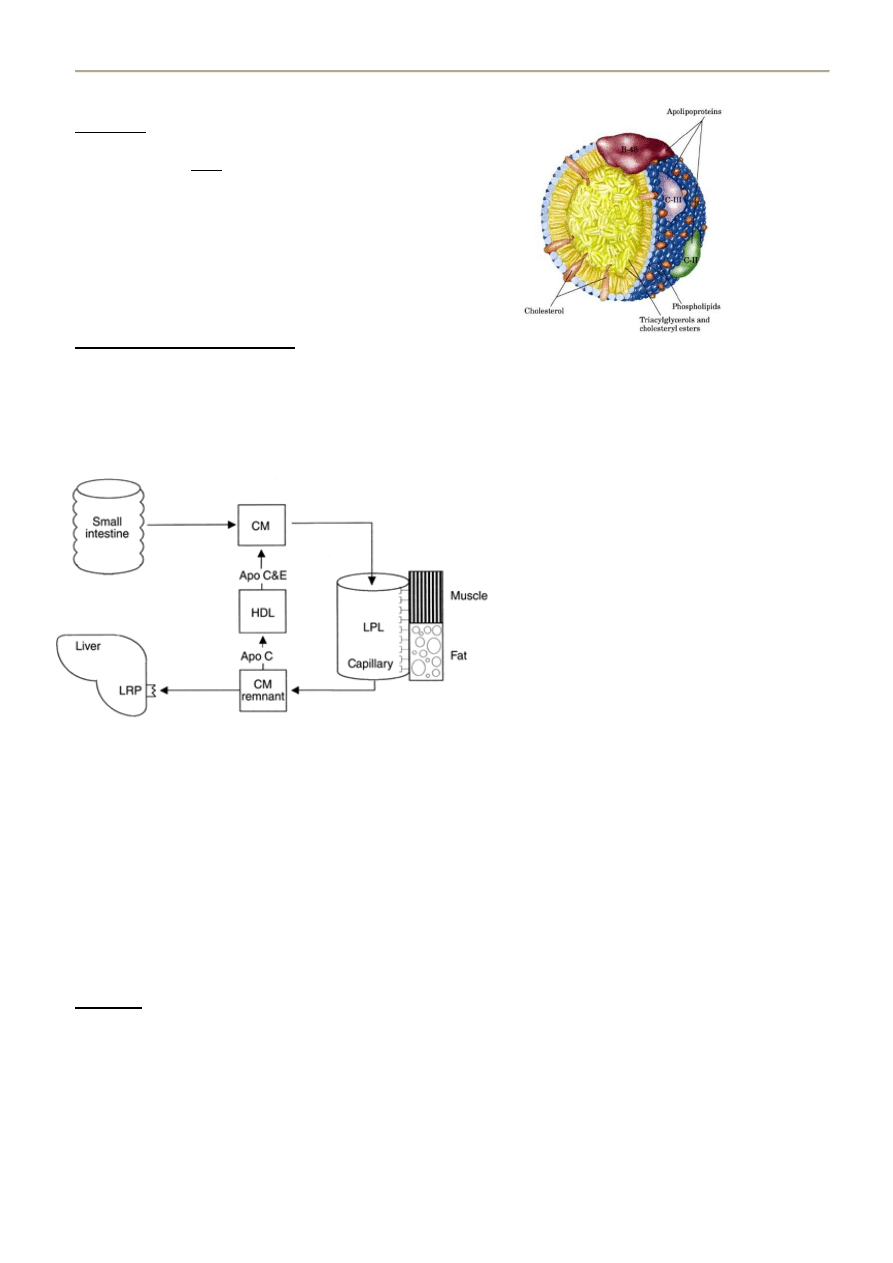
6. STRUKTURA LIPOPROTEIN
Związki tłuszczowe osocza, jako nierozpuszczalne w wodzie, transportowane są w kompleksie z białkami, tworząc
lipoproteiny. Wyjątkiem są wolne kwasy tłuszczowe, przenoszone w osoczu przez albuminy.
Lipoproteiny stanowią heterogenną grupę cząstek, różniącą się składem lipidowym, białkowym, miejscem syntezy i
metabolizmem. Podstawą klasyfikacji lipoprotein jest rozdział metoda ultrawirowania lub elektroforezy.
Lipoproteiny to cząstki – niestechiometryczne połączenia białkowo-lipidowe (ich masa i skład ulegają zmianie), w których
częśd białkowa jest połączona z częścią lipidową za pomocą:
- wiązao wodorowych
- sił Van der Waalsa
Budowa lipoprotein:
- cząstki sferyczne zbudowane z płaszcza i rdzenia
PŁASZCZ
- odpowiednie apoproteiny
- warstwa fosfolipidów (z hydrofilową główką na
zewnątrz)
- pomiędzy fosfolipidy wchodzi wolny cholesterol, który
może przechodzid do rdzenia jeśli zostanie
zestryfikowany przez LCAT - donorem reszty acylowej
jest zazwyczaj lecytyna (to sprawia że lipoproteina
zmienia swoją strukturę – as prof said: „Pantha rhei!”).
RDZEO – wysoce apolarny
- wewnątrz sferycznej cząstki znajdują się
triacyloglicerole (TG = glicerol + 3 reszty kw.
Tłuszczowych) oraz zestryfikowany cholesterol
LCAT
cholesterol + kw. Tłuszczowy -------------> ester cholesterolu (przechodzi z płaszcza do rdzenia)
Skład lipoprotein
CHYLOMIKRONY
VLDL
LDL
HDL
TG:
90-98%
50-65%
5-6%
7%
wolny
cholesterol:
1%
4-7%
6-15%
5%
estry
cholesterolu:
2-4%
8-14%
35-40%
10-20%
białka:
1-2%
7-10%
22-26%
45%
Fosfolipidy
(głównie lecytyna)
2-6%
12-16%
22-26%
25%

grupa7.med@gmail.com | 2008 - wdanie II, poprawione
11
LIPOPROTEINA a:
- budową przypomina LDL, wbudowane apo-B100
- dodatkowo posiada apo(a) – o strukturze przypominającej precle (tzw. Cringles)
- liczba domen przypominających precle uwarunkowana jest genetycznie
stąd w zależności od ilości białka stężenie w granicach 1 mg/dl do 100 mg/dl
(w 70% przypadków stężenie wynosi 20mg/dl)
- gęstośd w zakresie frakcji LDL < a > HDL
- apo(a) oddziaływuje z plazminogenem lub tPA
bo jest podobna do tPA i plazminogenu
- działanie - „a” jak aterogenne bo:
- trudno wiąże się z receptorem
- przez to ma wolny klirens, a jak jest jej dużo to łatwo ulega uszkodzeniu (oksydacji) i łatwo
gromadzi się w naczyniach
- przez podobieostwa do tPA/plazminogenu zaburza proces aktywacji układu fibrynolizy osocza.
Następuje przesunięcie na rzecz krzepnięcia
- stymuluje sekrecję PAI-1 (inhibitor aktywatora plazminogenu)
- zmniejsza się uwalnianie wolnej formy TGF-beta
(a TGF-beta ma działanie antyproliferacyjne na mm gładkie)
- podwyższenie LPa >30mg/dl - niezależny czynnik miażdżycy
- synteza: wątroba
- katabolizm: wątroba, jelito cienkie, śledziona
- u kobiet po 50r.ż następuje wzrost apo(a) – włączenie terapii estrogenowej obniża stężenie apo(a)
U kogo robid (drogie) badania na obecnośd apo(a)?
- osoby po zawale z prawidłowym lipidogramem
- osoby z b.wysokim LDL
- osoby z incydentami zakrzepowymi o niejasnej etiologii
LIPOPROTEINA X:
- patologiczna lipoproteina u pacjenta z cholestazą zewnątrz lub wewnątrz-wątrobową
- gęstośd jak LDL (czyli nie da się wykryd ultra wirowaniem, a jedynie w elektroforezie!)
- Rozróżnia się trzy rodzaje LpX (LpX1, LpX2, LpX3).
Ma ona kształt kulisty i jest dwuwarstwowym pęcherzykiem,którego ściana utworzona jest głównie z cholesterolu wolnego i
fosfolipidów w prawie równomolowych ilościach. Wnętrze pęcherzyka wypełnia faza wodna zawierająca białka osocza,
głównie albuminę:
- większośd to fosfolipidy i niezestryfikowany cholesterol
- białko, trójglicerydy i estry cholesterolu – łącznie mniej niż 12% lipoproteiny Lpx
- główny kwas żółciowy w Lpx – kwas litocholowy
- w polu elektrycznym przesuwa się w kierunku przeciwnym niż pozostałe frakcje
- LpX ma silne właściwości agregacyjne - wykazano, że enzymy zwykle związane z błoną komórkową łączą się także
z LpX (np. fosfataza zasadowa)
- obecna w surowicy niemowląt – cechy wrodzone genetycznie (niedobór enzymu LCAT)
- po likwidacji cholestazy poziom LPX bardzo szybko się stabilizuje
Powstawanie LpX - W powstawaniu LpX bierze udział flippaza (białko przenoszące lecytynę pomiędzy zewnętrzną i
wewnętrzną warstwą błony komórkowej; białko MDR2).
LpX w dużych ilościach występuje u pacjentów z pierwotną marskością żółciową wątroby. U pacjentów tych stwierdza się
wysokie stężenia cholesterolu, co jest właśnie wynikiem nagromadzenia się LpX. Mimo tego pacjenci ci cechują się niskim
ryzykiem rozwoju choroby niedokrwiennej serca. Wynika to najprawdopodobniej z antyoksydacyjnych właściwości LpX,
które nie tylko same nie ulegają oksydacji, ale także chronią przed oksydacją normalne cząstki LDL (dla przypomnienia- LpX
także jest frakcją LDL). Stężenie cholesterolu u pacjentów z pierwotną marskością żółciową wątroby może sięgad nawet
wartości 1400 mg% - 36mmol/l i nie powoduje to szybkiego rozwoju miażdżycy, lecz wiąże się z powikłaniem w postaci
zespołu nadlepkości i ewentualnych zmian niedokrwiennych.

grupa7.med@gmail.com | 2008 - wdanie II, poprawione
12
7. METODY ROZDZIAŁU LIPOPROTEIN
1) Elektroforeza
2) Ultrawirowanie
3) Metoda strąceniowa
Frakcje lipoprotein uzyskane metodą elektroforezy:
chylomikrony (nie wykazują ruchliwości elektroforetycznej – ze względu na małą zawartośd białka – 1-2%)
β-lipoproteiny (bo poruszają się w polu z prędkością B-globulin)
pre-β-lipoproteiny
α-lipoproteiny
Po przeprowadzeniu elektroforezy pasek wybarwia się na obecnośd lipidów za pomocą:
- sudanu 3
- czerwieni oleinowej
- czerwieni sudanowej
Ultrawirowanie jest jedną z najdokładniejszych metod – metoda referencyjna do pozostałych.
- trwa 24-48h
Frakcje uzyskane metodą ultrawirowania:
chylomikrony
VLDL
IDL (remnanty VLDL, nie powinno ich byd w prawidłowej surowicy)
LDL
HDL
VHDL
Metody strąceniowe – frakcje lipoprotein można wytrącid, stosując metody połączenia polianionów (heparyna, siarczan
dekstranu, dezoksycholan sodu) z kationem metalu (np. Mg
2+
, Mn
2+
)
Frakcje otrzymane:
- chylomikrony (białko : lipidy – niski stosunek stężenia polianionów)
- VLDL
- HDL (w wyższym stężeniu polianionów)
* Po szczegóły odsyłam do skryptu, nie chce mi się tego za bardzo przepisywad. Poza tym „to już powinni paostwo umied
bo to się w przedszkolu czy tam w szkole średniej robi” ;D

grupa7.med@gmail.com | 2008 - wdanie II, poprawione
13
8. STRUKTURA I FUNKCJA APOPROTEIN
Apoproteiny to białkowe składniki lipoprotein, które spajają składniki bardziej hydrofobowe i mniej hydrofobowe, przez co
umożliwiają kontakt lipoprotein z osoczem.
- funkcje:
- białko strukturalne lipoprotein
- udział w syntezie lipoprotein (np. apo-B48 bez której w jelicie nie może powstad prawidłowy
Chylomikron – powstaje szczątkowe ApoB48, tzw. trancated apoB48 - zaleganie TG w jelicie
- ligand receptora
- aktywują/hamują działanie enzymów
RODZINA APOLIPOPROTEIN A
- związana z metabolizmem HDL
- należą do niej apoA
I, II, IV
- syntezowane w wątrobie i jelicie
apoA-I
- stanowi około 70% białek HDL
- aktywuje białko ABC, które umożliwia przejście cholesterolu z komórki do HDL
- aktywuje LCAT
- acylotransferaza Lecytyna:Cholesterol
- umożliwia przejście cholesterolu z płaszcza lipoproteiny do rdzenia
- jest ligandem dla kubiliny
- wyraźny spadek stężenia w chorobie Tangierskiej (oprócz defektu ABCA1)
apoA-II
- stanowy około 20% białek HDL, chod występuje tylko w 2/3 cząsteczek HDL
- aktywuje HTGL ( lipaza wątrobowa triglicerydowa)
- oznaczanie stężenia w surowicy pozwala na ocenę zdolności do usuwania cholesterolu
apoA-IV
- razem z apoA-I aktywuje LCAT
RODZINA APOLIPOPROTEIN B
- rodzina związana z literą „L” („L” jak LDL, VLDL, Chylomikrony)
- wyróżniamy Apo-B100 i Apo-B48 które powstają z tego samego genu w wyniku modyfikacji - redagowania
- pełnią funkcję ligandu dla komórkowych receptorów apo B/E
- synteza w wątrobie (B100) lub enterocytach (B48)
apo B 100
- związana z całym złem tego świata: VLDL, LDL oraz Lp(a)
- ma miejsce wiążące heparynę
apo B
48 – związana z metabolizmem chylomikronów
RODZINA APOLIPOPROTEIN C
- związane z metabolizmem Lp bogatych w TG, chod wyjściowo związane z HDL (HDL oddaje
swoje apolipo-C innym Lp )
- syntezowane w wątrobie
- apo C I – aktywuje LCAT
- apo C II – aktywuje LPL (lipazę lipoproteinową)
- apo C III – hamuje LPL
RODZINA APOLIPOPROTEIN E
- synteza w wątrobie, neurogleju, śledzionie, nerkach i płucach
- wchodzi w skład chylomikronów, VLDL, IDL, i HDL, umożliwia katabolizm: chylomikronów ---> receptor LRP;
remnanty VLDL ---> receptor apoB/E
- występuje polimorfizm: genetyczny – chromosom 19 (patrz dalej) oraz
polimorfizm wynikający z posttranslacyjnej glikozylacji kwasem sjalowym

grupa7.med@gmail.com | 2008 - wdanie II, poprawione
14
9. GENETYCZNIE UWARUNKOWANA HETEROGENNOŚĆ APO-E
- Gen dla ApoE zlokalizowany jest na chromosomie 19, ma 3,7 kb długości i składa się z 4 egzonów.
- Występuje najczęściej w trzech allelach APOE2 APOE3 i APOE4.
U osób zdrowych ponad 10% między osobniczej zmienności w poziomie cholesterolu we krwi związane jest z posiadanym
allelem genu APOE. Wpływ genotypu APOE na poziom lipidów we krwi ma duże znaczenie dla ryzyka wystąpienia choroby
niedokrwiennej serca i miażdżycy tętnic szyjnych.
Obecnośd trzech alleli geno APOE warunkuje występowanie sześciu genotypów w populacji ludzkiej, w tym trzech
homozygotycznych (2/2, 3/3, 4/4) i trzech heterozygotycznych (2/3, 2/4, 3/4). Poszczególne izoformy apoE różnią się
powinowactwem do receptorów rodziny LDL(izoforma E2 charakteryzuje się znacznie mniejszym powinowactwem niż
izoforma E3, mniej jednoznaczne są dane dotyczące powinowactwa apoE4 chod mówi się, że ma największe powinowactwo
do receptora), ale także powinowactwem do lipidów - związane jest to przede wszystkimz obecnością lub brakiem arginy w
pozycji 112.
Genotyp APOE2/2 występuje z częstością (1:100)
- skorelowany jest z hiperlipoproteinemią typu III
APOE4 - jest głównym czynnikiem genetycznym związanym z występowaniem sporadycznej i rodzinnej formy
choroby Alzheimer'a . Istnieje dobrze udokumentowany związek późno ujawniającej się rodzinnej
formy AD z genotypem APOE4.
10. RECEPTORY DLA APOPROTEIN
ABC-A1:
a) ATP Binding Cassette Transporter A1, członek nadrodziny białek ABC
b) złożone z 2 podobnie zbudowanych kowalencyjnie ze sobą związanych połówek
c) każda połówka posiada:
- domenę wiążącą nukleotydy z dwoma konserwatywnymi motywami (Walker 1 i Walker 2)
- domenę transbłonową
- zewnątrzkomórkową (tworzącą glikozylowane pętle)
d) uczestniczy w transporcie lipidów z komórki na zewnątrz albo w mechanizmie bezpośrednim
tworząc kanał, albo pełniąc rolę regulatorową (dokładny mechanizm nieznany)
e) defekt genu ABCA1 jest przyczyną choroby tangierskiej
f) C-koniec (w cytozolu) oddziałuje z syntrofiną-β2 i utrofiną
g) ABCA1 odpowiada za transport:
- fosfolipidów (przede wszystkim); głównie fosfatydylocholiny
- cholesterolu
- innych substancji – α-tokoferolu, apoE, interferonu-1β
h) wzrost ekspresji genu następuje przy wysokich wewnątrzkomórkowych stężeniach cholesterolu
- powstające w komórce oksysterole tworzą kompleks z receptorami LXR i następnie aktywują
ekspresję genu ABCA1
Receptor LDL (LDLR):
a) receptor apoB/E zwany receptorem wysokiego powinowactwa
b) ligandy:
- apoB-100
- apoE
c) aktywnośd hamowana przez RAP – receptor associated lipoprotein
d) funkcje:
- tkanki pozawątrobowe: pobieranie (internalizacja) LDL
- wątroba: pobieranie różnych lipoprotein wyposażonych w apoB-100
e) obecny w zagłębieniach błonowych pokrytych klatryną
f) ekspresja genu receptora hamowana przez cholesterol

grupa7.med@gmail.com | 2008 - wdanie II, poprawione
15
g) struktura:
- domena wiążąca ligand (przy N-koocu) – liczne wiązania dwusiarczkowe
- domena zbliżona do Prekursora Czynnika Wzrostu EGF – EGFP
- domena bogato glikozylowana (bez znaczenia funkcjonalnego)
- domena transbłonowa
- domena cytozolowa przy C-koocu z sekwencją sygnałową
h) mutacje powodują hiperlipoproteinemię typu II
Scavenger:
Ligandy:
- lipoproteiny zmodyfikowane chemicznie
- polirybonukleotydy
- naturalne i zmodyfikowane polisacharydy
- fosfolipidy anionowe
- inne molekuły – azbest, siarczan poliwinylu, endotoksyny
Za jego pośrednictwem makrofagi „objadają” się zmodyfikowanymi lipoproteinami przekształcając się w foam
cells (kom. piankowate).
LRP – alfa2MR (receptor alfa-2-makroglobuliny):
- receptor remnantów chylomikronów i VLDL
- zlokalizowany na powierzchni hepatocytów i komórek kory nadnerczy
- lokalizacja w całej komórce: fibroblasty, makrofagi, SMC
Rola:
- bierze udział w metabolizmie remnantów chylomikronów i remnantów VLDL
- inną funkcją receptora jest regulacja aktywności proteinaz (np. serynowych)
- udział w katabolizmie białek macierzy
- regulacja fibrynolizy: wiązanie t-PA, u-PA, tPA-PAI1 oraz uPA-PAI1
- regulacja metabolizmu cytokin i hormonów - wiązanie kompleksów alfa-2-makroglobuliny z cytokinami
i hormonami
Ligandy LRP:
- apoE, remnanty lp zawierające apoE
- LPL
- kompleksy alfa-2-makroglobuliny-proteinazy, elastaza-alfa-1-inhiibtor proteaz (alfa-1-antytrypsyna)
- t-PA, u-PA, t-PA-PAI1, u-PA-PAI1

grupa7.med@gmail.com | 2008 - wdanie II, poprawione
16
11. RECEPTOR SRB-1 (scavenger receptor typ B1)
SR – scavenger receptor są grupą białek wiążących chemicznie lub oksydacyjnie zmodyfikowane lipoproteiny, polianiony i
komórki ulegające apoptozie. Istnieje co najmniej 6 klas SR (od A do F).
Receptory zmiatające odgrywają ważną rolę w wielu procesach fizjologicznych i patologicznych, w tym w
miażdżycy, adhezji komórkowej oraz wrodzonej nieswoistej reakcji odpornościowej skierowanej przeciw fragmentom błony
komórkowej mikroorganizmów.
Na podstawie dotychczasowych badao stwierdzono, że niektóre z receptorów SR w szczególny sposób są
zaangażowane w tworzenie komórek piankowatych. Należą do nich receptory: SR-A, CD36, CD68, LOX-1 (lectin – like ox-
LDL receptor), SREC (scavenger receptor expressed by endothelial cells), SR-PSOX (scavenger receptor for
phosphotidylserine and oxidized lipoprotein)
Scavenger receptor class A
SR-AI i SR-AII wiążą acetylowane i utlenione LDL, polianiony i martwe komórki.
Scavenger receptor class B
W skład receptorów klasy B wchodzą receptory CD36 i B1. CD36 jest receptorem fagocytującym i wiąże m.in. acetylowane i
utlenione LDL, fosfatydyloserynę i komórki apoptotyczne. SR-B1 jest receptorem lipoprotein o dużej gęstości HDL (high
density lipoprotein)
Struktura
Wyróżniamy częśd:
1) zewnątrzkomórkową w postaci pętli,
2) transbłonową, która kotwiczy białko do błony cytoplazmatycznej za pomocą kooca N- i C-
3) krótką cytoplazmatyczną, palmitylowaną/glikozylowaną przy C koocu
Dopiero niedawno odkryto 2 ważne białka, które pośredniczą w zwrotnym transporcie cholesterolu przez HDL. Jedno z nich
nazywa się transportem ABC-A1, drugie natomiast receptorem zmiatającym klasy B typ 1 (SR-B1).
Transport ABC-A1 występuje na komórkach i przenosi wolny cholesterol z wnętrza tych komórek na ich powierzchnię, z
której jest on pobierany przez bezcholesterolowe lub ubogie w cholesterol HDL.
SR-B1 jest obecny na hepatocytach i przejmuje cholesterol z HDL. Jest to tzw. bezpośredni zwrotny transport cholesterolu.
Do wnętrza hepatocytu zostaje wciągnięta cała cząsteczka HDL, następnie na cholesterol działa esteraza, a reszta cząsteczki
jest wydzielana poza komórkę (czyli nie tak jak w przypadku receptora wysokiego powinowactwa B/E – LDL weszło do
komórki, ApoB jest hydrolizowane do aminokwasów, cholesterol do wolnego cholesterolu. Tu zostają wyssane wyłącznie
estry cholesterolu)
Oprócz niego istnieje także pośredni transport zwrotny cholesterolu, w którym częśd cholesterolu z HDL (po estryfikacji) jest
przekazywana do lipoprotein IDL i LDL, w wymianie na trójglicerydy. Z kolei IDL i LDL przekazują cholesterol hepatocytom za
pośrednictwem receptora LDL.
Poza tym HDL po zadziałaniu na SRB1 stymuluje eNOS przez wzrost wewnątrzkomórkowego stężenia ceramidu:)
grupa7.med@gmail.com | 2008 - wdanie II, poprawione
17
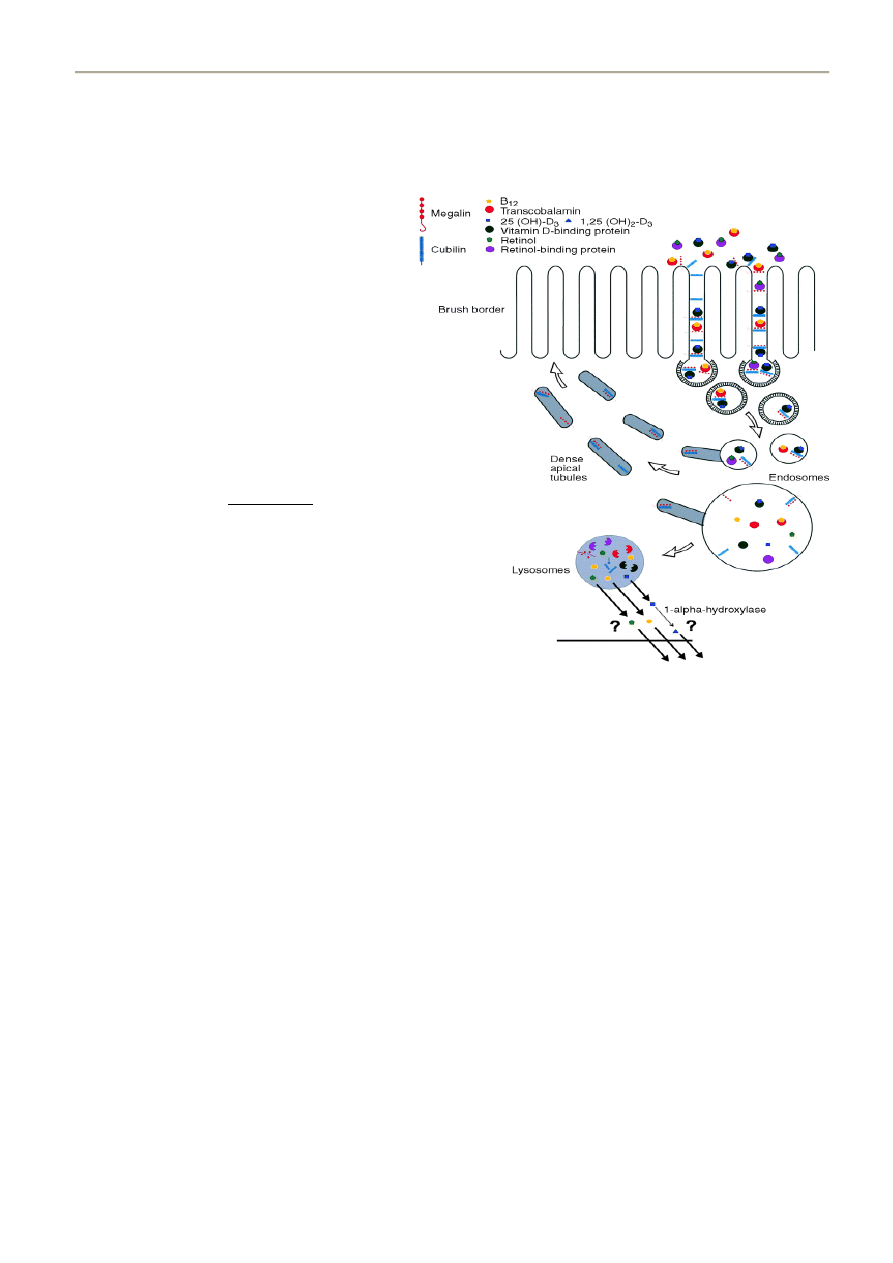
12. CUBILINA I MEGALINA – STRUKTURA I FUNKCJA
Słowem wstępu – megalina i kubilina są białkami odpowiedzialnymi za endocytozę w tarczycy, nerkach, jelicie itp.
Endocytoza rozpoczyna się od związania białkowego liganda z receptorem komórek nabłonka kanalikowego od strony
światła kanalika. Następnie receptory gromadzą się w opłaszczonych klatryną dołkach u podstawy mikrokosmków rąbka
szczoteczkowatego nabłonka kanalikowego. Uformowane dołki odrywają się w postaci pęcherzyków. W procesie tym bierze
udział klatryna, białka adaptorowe i inne białka wewnątrzkomórkowe, odpowiedzialne za powstawanie i dojrzewanie
endosomów. Dzięki działaniu błonowej pompy
protonowej, wnętrze pęcherzyków jest zakwaszane,
co powoduje oddysocjowanie liganda od receptora.
Wolne receptory powracają do błony komórkowej,
gdzie mogą wiązad kolejne cząsteczki. Wchłonięte
białka są w większości rozkładane przez enzymy
lizosomalne. Częśd z nich poprzez transcytozę może
byd uwalniana do krążenia po stronie podstawnej
komórek nabłonka. Dla wielu białek
niskocząsteczkowych zidentyfikowano dwa receptory
rąbka szczoteczkowatego nabłonka kanalikowego,
megalinę i kubilinę. Są to koreceptory, ulegające
ekspresji w bliskim sąsiedztwie i mające częśd
wspólnych ligandów. Te dwa współpracujące ze sobą
białka wykazują duże różnice strukturalne
Megalina
jest dużą transbłonową glikoproteiną
(zbliżoną budową do receptora LDL), zawierającą:
1) domeny podobne do EGF
2) domeny odpowiedzialne za dysocjację
liganda w zależności od pH
3) domeny kotwiczące
Kubilina
jest zewnątrzbłonową glikoproteiną o
masie cząsteczkowej około 460 kDa. W odróżnieniu
od megaliny nie ma domeny ransbłonowej..Zbudowana jest ze 110-aminokwasowego N-kooca, po którym następuje 8
fragmentów EGF-podobnych oraz 27 domen CUB (domeny CUB zawierają min.: fragmenty podobne do C1r i C1s układu
dopełniacza, fragmenty podobne do BMP1). Jedynie palmitylacja N-kooca umożliwia zakotwiczenie jej w błonie
Megalina, jako receptor transbłonowy, może działad samodzielnie, natomiast kubulina jedynie w połączeniu z megaliną.
Oba te receptory do swej pełnej aktywności wymagają jonów wapnia.
Kompleksy megalina-kubulina obecne są jedynie w:
- j. cienkim
- kanaliku proksymalnym nefronu
- łożysku (cytotrofoblast)
Sama megalina występuje w innych miejscach – płuca, tarczyca, etc
Megalina i kubilina są istotne w prawidłowej reabsorpcji białek w kanalikach proksymalnych nerek. W warunkach
fizjologicznych, dzięki aktywności tych receptorów, zostają odzyskane z moczu pierwotnego ważne biologicznie substancje,
m.in. aminokwasy, witaminy i minerały.
Dodatkowo przy pomocy megaliny/kubiliny następuje endocytoza 25(OH)D3 a następnie, wewnątrzkomórkowe
przekształcenie w 1,25(OH)D3 (przy udziale 1-hydroksylazy). Dodatkowo możliwe jest dzięki temu oddziaływanie witaminy
D w zakresie wchłaniania rozmaitych substancji
Ponadto przypuszcza się, że megalina która silnie wiąże Ca, może działad jak sensor w przytarczycach. Może również brad
udział w transporcie hormonów tarczycy. Cubilina i megalina wiążą lipoproteiny (odpowiednio HDL-kubilina i LDL-megalina),
ale ich rola w metabolizmie cholesterolu nie została do kooca ustalona, chod zwierzęta doświadczalne z
defektem/niedoborem kubiliny mają hipercholesterolemie.

grupa7.med@gmail.com | 2008 - wdanie II, poprawione
18
LIGANDY KUBILINY
- Apo A1 – połączenie HDL-apoA z kubilina (która jest dodatkowo połączona z megaliną)
uniemożliwia bezpowrotnego wydalenia ApoA-I (HDL)
- transferryna-Fe
- w jelicie cienkim – kompleks IF-B
12
LIGANDY WSPÓLNE dla kubiliny i megaliny
- DBP – Wit. D Winding protein
- łaocuchy lekkie immunoglobulin
- hemoglobina (udział w mechanizmie wydalania Hb)
- albumina
LIGANDY MEGALINY
- apoB100 (megalina w sensie swojej struktury zaliczana jest do receptorów LDL)
- apo H
- transkobalamina –witB12
- hormony peptydowe (np. PTH)
13.Receptor dla LDL (apoB/E)
- receptor wysokiego powinowactwa, występuje na wszystkich komórkach czerpiących cholesterol
- stała Michaelisa = 50mg/dl (mała)
BUDOWA
- 3 części: zewnątrzbłonowa, transbłonowa, cytozolowa
- 5 domen białkowych: wiążących ligand za pomocą apoB100 lub apoE , wykazującą homologię z EGF, cukrowej,
błonowej i cytoplazmatycznej.
SYNTEZA I DZIAŁANIE
Do prekursora białkowego receptora w AG dodawane są reszty cukrowe (adresowanie) a następnie receptor
wysyłany jest do zagłębieo cytoplazmatycznych. Po związaniu ligandu ulega internalizacji, fuzji z lizosomem,
którego wnętrze ulega zakwaszeniu. Po rozłożeniu LDL receptor wraca na powierzchnię.
WYSTĘPOWANIE WEWNĄTRZKOMÓRKOWE
-
związane jest z następstwami pobudzenia receptora, zmienia się w zależności od fazy endocytozy i
metabolizowania cholesterolu LDL, a także zapotrzebowania komórki na cholesterol
grupa7.med@gmail.com | 2008 - wdanie II, poprawione
19
NASTĘPSTWA POBUDZENIA:
1) receptory dla LDL znajdują się na powierzchni błony komórkowej we wgłobieniach („dołki opłaszczone”), które są
pokryte (opłaszczone) od strony cytoplazmatycznej białkiem klatryną,
2) po połączeniu receptora z cząstką LDL jest ona w całości endocytowana, a następnie rozkładana w lizosomach
(hydrolizie ulegają estry cholesterolu i apolipoproteiny kolejno do cholesterolu i aminokwasów), dzięki ATP-azie
błony pęcherzyka endocytarnego, która ma aktywnośd pompy protonowej, wytworzone w pęcherzyku kwaśne
środowisko sprzyja oddzieleniu się receptora LDL,
3) receptor LDL, który nie został zdegradowany wraca do błony komórkowej lub pozostaje we wnętrzu komórki w
zależności od zapotrzebowania komórki na cholesterol, zgodnie ze zjawiskiem „down regulation”,
4) uwolniony egzogenny cholesterol hamuje endogenna syntezę cholesterolu przez hamowanie reduktazy HMG-CoA,
hamuje syntezę receptora LDL na etapie genu i jego wydzielanie, aktywuje ACAT (acylotransferaza acylo-CoA :
cholesterol) by wolny cholesterol zestryfikowad i zdeponowad w postaci kropel cholesterolowych,
5) nadmiar wolnego cholesterolu jest utleniany do oksysteroli (hydroksy- i ketopochodne) – tzw. Aktywny
cholesterol, które hamują ekspresję genu receptora LDL łącząc się z nim w rejonie promotora,
KONSEKWENCJE NIEDOBORU LUB DYSFUNKCJI:
1. Hiperlipoproteinemia typu II (inaczej rodzinna hipercholesterolemia FCH)
Istnieją jej dwie postacie: receptoro-negatywna (brak receptora wysokiego powinowactwa ApoB/E) lub receptoro-defektywna (receptor
nie pracuje w 100% wydajnie). Drugi rodzaj podziału to podział ze względu na rodzaj dziedziczenia: homozygotyczna (oba geny są
nieprawidłowe, praktycznie zerowy wychwyt LDLi, występuje 1:1000000, śmiertelna, brak leczenia) lub heterozygotyczna (tylko jeden gen
nieprawidłowy, powolny wychwyt LDLi, występuje 1:500). Konsekwencją jest zaleganie remnantów VLDLi (mają taką samą siłę naciekania
ściany naczyo i indukcji miażdżycy jak LDLe) i LDLi prowadzące do choroby niedokrwiennej serca i miażdżycy (opis poniżej).
2. Hiperlipoproteinemia typu III (wtórna)
We wtórnej postaci, występującej najczęściej u kobiet w okresie menopauzy (50 lat +/- 3 m-ce) receptor wysokiego
powinowactwa ApoB/E (wątrobowy) jest czasowo „uśpiony” i można go z tego „snu” wybudzid. Konsekwencją tego jest
gromadzenie remnantów VLDLi oraz powstawanie miażdżycy naczyo wieocowych i obwodowych.
Zwracam uwagę na to, że w pierwotnej postaci nie dochodzi do defektu receptora ApoB/E, ale do genetycznego defektu
apolipoproteiny E znajdującej się na lipoproteinach! Patrz – Harper s. 367
3. Naciekanie ściany naczyo przez zmodyfikowane LDLe – powstawanie miażdżycy
Nie wyłapany przez wątrobę lub tkanki obwodowe LDL ulega np. oksydacji i powstaje oksydowany LDL, który wchodzi pod
śródbłonek i zostaje wyłapany przez makrofagi posiadające receptor niskiego powinowactwa – Scavenger Receptor (nie
podlega regulacji, zostaje przyjęta każda ilośd cholesterolu). Powstają w ten sposób komórki piankowate, które
przeładowane cholesterolem pękają i tworzą blaszkę miażdżycową, ponadto uwalniają PDGF, TNFά, FGF, TF,
metaloproteazy (MMP). Czynniki wzrostu powodują proliferację SMC itp. Metaloproteazy trawią czepiec włóknisty
powodując pęknięcie i nagłe wykrzepienie krwi. Proces ten lokalizuje się w naczyniach tętniczych mózgu, serca, kooczyn
dolnych.
4.bierze udział w internalizacji wirusa HCV w tkankach obwodowych
Wolny CHOLESTEROL w komórce
(oksysterole – keto i chydroksy pochodne)
ACAT
Blok reduktazy HMG-CoA
(magazynowanie estrów cholesterolu)
( tworzenia endogennego cholesterolu)
LDL-Receptor „down regulation”
-
gen dla receptora LDL (podobnie jak gen dla reduktazy HMG-CoA) jest regulowany przez białko SREBP,
które wiąże sterolowy element regulatorowy (SRE) i kontroluje tempo syntezy mRNA receptora LDL,
grupa7.med@gmail.com | 2008 - wdanie II, poprawione
20
14. Zależność między składem a gęstością cząsteczki
Lipidy mają niższą gęstośd i większą objętośd niż białko, a białko ma wyższą gęstośd i mniejszą objętośd (z uwagi
na uporządkowaną strukturę I, II, III rzędową). Dlatego te Lp które mają dużo białka (HDL) będą miały dużą
gęstośd i małą objętośd, a te, które białka praktycznie nie posiadają (np. chylomikrony) – będą największe, ale za
to będą miały najmniejszą gęstośd
średnica
(nm)
gęstośd
białko (%)
lipidy
całkowite
(%)
TG
cholesterol
całkowity
chylomikrony
remnanty
chylomikronów
VLDL
IDL
LDL
HDL
grupa7.med@gmail.com | 2008 - wdanie II, poprawione
21
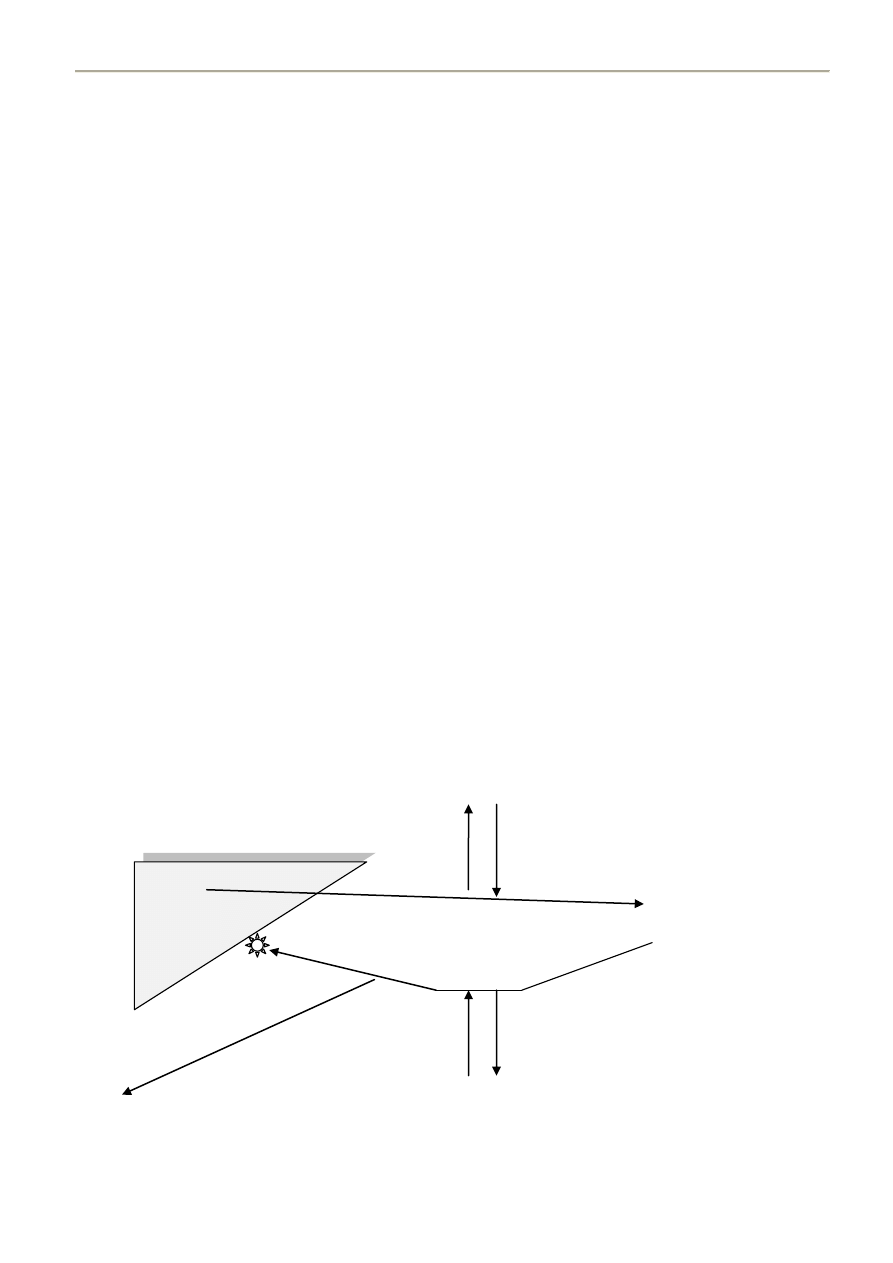
15. CHYLOMIKRONY – BUDOWA, BIOSYNTEZA, DEGRADACJA, FUNCJA
BUDOWA:
Apolipoproteiny: B-48
A-I,II
C-I,II,III
E
TG – 85-95%
Fosfolipidy – 4-6%
Cholesterol – 2-4%
BIOSYNTEZA I BIODEGRADACJA:
Powstają w ścianie jelita cienkiego po spożyciu pokarmu.
1) Glikozylacja apolipoprotein w RER, przemieszczenie ich do AG (apoB48 i apoA)
2) Synteza TG, fosfolipidów i cholesterolu w SER
Chylomikrony są wydzielane na drodze
egzocytozy.
Nie
wchodzą
do
krążenia
wrotnego, ale z chłonką trafiają do krążenia
ogólnego,
gdzie
poprzez
CETP
(białko
przenoszące estry cholesterolu) kontaktując się
z HDL – następuje przepływ apoC2, apoE i
estrów cholesterolu z HDL do chylomikronu a
TG w odwrotnym kierunku. Dopływają do
mięśni szkieletowych, serca, tk. tłuszczowej
gdzie wchodzą w interakcję z LPL (lipaza
lipoproteinowa, ektoenzym przytwierdzony do
ściany
naczyo
przy
pomocy
siarczanu
heparanu). Chylomikron za pomocą ApoC-II aktywuje LPL, która „otwiera” chylomikron i hydrolizuje TG.
Powstają WKT, które wiążą się z albuminą i trafiają do:
- mięśni – pozyskanie energii dzięki β-oksydacji,
- tk. tłuszczowej – resynteza TG,
- wątroby – resynteza TG, które trafią do VLDL albo stłuszczą wątrobę).
Następnie zubożały o TG chylomikron (remnant), oddaje HDL apoC-II i apoA a następnie zostaje
wyłapany przez wątrobę (receptor dla apoE - LRP).
FUNKCJA:
- nośnik lipidów pochodzenia pokarmowego,
- dostarczają substraty energetyczne tkankom,
- zawierają TG, które wątroba wbudowuje do VLDL,
* dodatkowo można powiedzied że chylomikron jest „transporterem” apoA (jest mu niepotrzebne, oddaje HDL) (?)
grupa7.med@gmail.com | 2008 - wdanie II, poprawione
22
16. VLDL- BUDOWA,BIOSYNTEZA, DEGRADACJA, FUNKCJA
Budowa:
VLDL - very low density lipoproteins - składa się z rdzenia lipidowego, zawierającego głównie niepolarne TG i
estry cholesterolu (ChE), otoczonego pojedynczą warstwą powierzchniową złożoną z amfipatycznych cząsteczek
fosfolipidy i cholesterolu.
Zawartośd apolipoprotein:
Apo B-100
Apo C-I,II,III
Apo E
Funkcja: Transportują triacyloglicerole zsyntetyzowane w wątrobie do tkanek pozawątrobowych.
Biosynteza i degradacja zachodzi w hepatocytach
1) Apo B-100 jest syntetyzowane przez rybosomy w ER, głównym stymulatorem apo B-100 jest insulina
2) W aparacie Golgiego następuje resynteza-asocjacja części lipidowej i białkowej.
3) Powstaje VLDL natywny, który trafia do układu krążenia na skutek fuzji pęcherzyka wydzielniczego z
błoną komórkową (odwrotna pinocytoza).
We krwi pobierają apo C-II, apo C-III, apo E oraz estry cholesterolu od HDL, oddając im TG (enzym: CETP).
Powstaje dojrzały VLDL, który podpływa do tkanek (tłuszczowej i mięśniowej), gdzie za pośrednictwem apo C-II
aktywuje LPL. Dochodzi do zmniejszenia ich średnicy i zwiększenia gęstości (hydroliza TG przez LPL).
Następnie zachodzi ponowna wymiana składników między zmniejszonymi VLDL a HDL: apo C-II, apo C-III, apo E
(częśd apoE) powracają do HDL, estry cholesterolu są przenoszone w kierunku odwrotnym od HDL do VLDL. W
wymianie tej również uczestniczy CETP. Dochodzi do względnego wzrostu cholesterolu w VLDL (względnego na
skutek przekazu TG i bezwzględnego poprzez zysk estrów cholesterolu).
Remnanty VLDL zawierają apo B-100 i apo E (bo częśd zostało) . W warunkach prawidłowych receptor apoB/E
preferencyjnie wyłapuje apo E. Remnant VLDL podpływa do zatoki wątrobowej, gdzie za pośrednictwem apo E:
2/3 remnantów VLDL zostaje wyłapana przez receptor apo B/E
1/3 nie wchodzi do komórki wątrobowej tylko jest hydrolizowana przez HTGL
Uwalnia się reszta kwasów tłuszczowych; wątroba włącza je do puli syntezy TG lub do spalania w procesie β-
oksydacji. Uwalniają się wszystkie białka poza apo B-100. W konsekwencji powstaje LDL (z 1/3 VLDL)
HDL
apoC2,3
TG
apoE
Estry Cholesterolu
VLDL
HYDROLIZA TG przez
LPL
Mięśnie, serce, tk.tłuszcz
rec. apoB/E
2/3
ChE
wraca apoC2,3,apoE
HTGL
1/3
HDL
LDL
(posiada jedynie B100)
grupa7.med@gmail.com | 2008 - wdanie II, poprawione
23
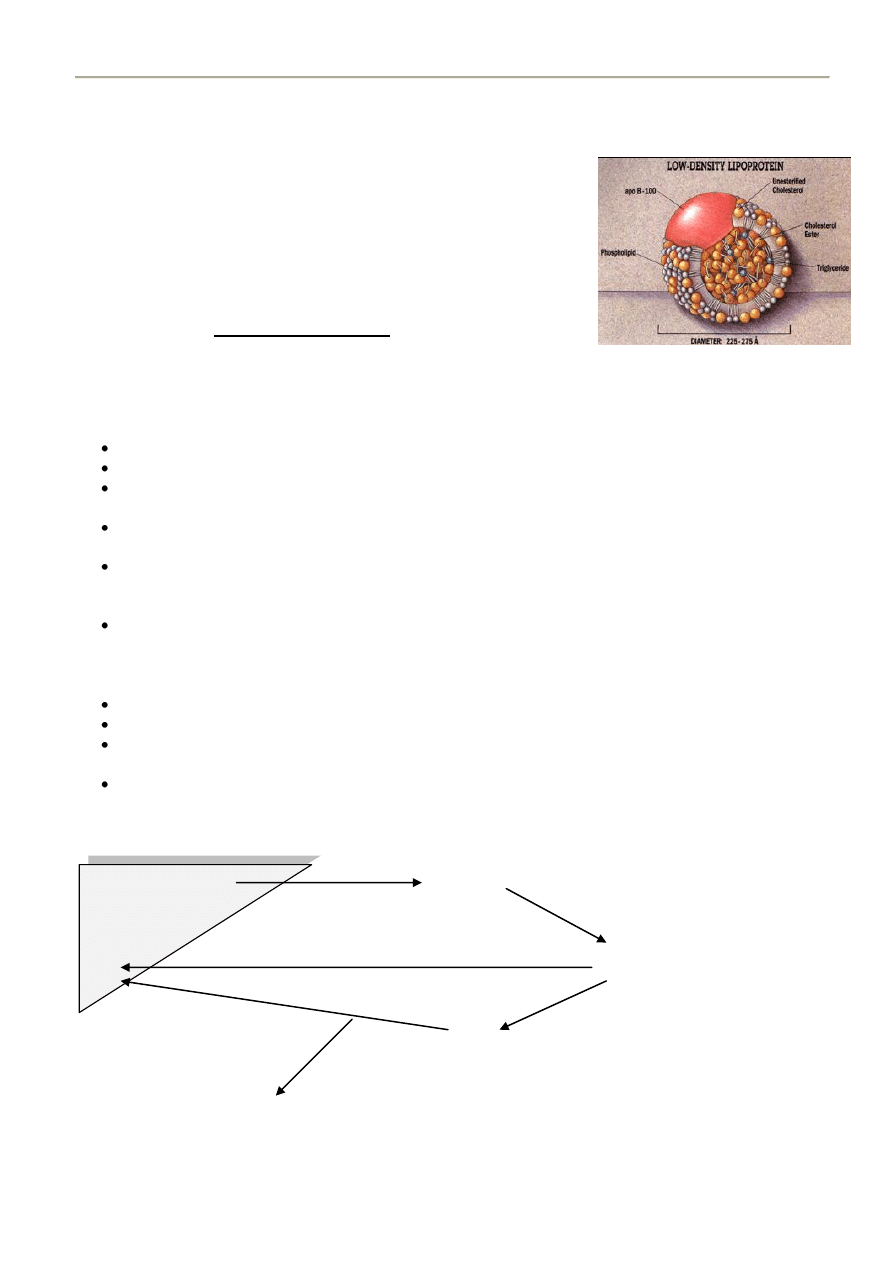
17. LDL- BUDOWA,BIOSYNTEZA, DEGRADACJA, FUNKCJA
Funkcja: transport cholesterolu z wątroby do innych narządów, głównie do nerek, mięsni, kory nadnerczy.
Budowa:
Zawierają znacznie mniej TG niż VLDL natomiast więcej cholesterolu i jego
estrów:
Triglicerydy 13%
Fosfolipidy 28%
Estry cholesterolu 48%
Cholesterol wolny 10 %
Białko 21% - Apo B-100 (jedyne białko)
Biosynteza i degradacja:
Większośd LDL powstaje z remnantów VLDL w zatokach wątroby
Cząstki LDL wytworzone z VLDL zachowują apo B-100, ale tracą inne apolipoproteiny na rzecz HDL
Dominującym wychwytem zarówno w wątrobie jak i tkankach obwodowych jest wychwyt przez receptor
apo B/E, który znajdujący się na powierzchni komórek docelowych.
LDL pobierane są na drodze endocytozy kierowanej receptorami. Po związaniu białka apo B-100 receptory
skupiają się w dołkach opłaszczonych klatryną i zostają wprowadzone do wnętrza komórek.
W lizosomach LDL ulegaja hydrolizie: estry cholesterolu są hydrolizowane do cholesterolu przez lipazę
lizosomalną, białka do odpowiednich aminokwasów). Cholesterol jest magazynowany w postaci estrów –
enzym estryfikujący to ACAT (acylotransferaza acyloCoA:cholesterolowa)
wysoki poziom cholesterolu w komórce:
Zmniejsza syntezę receptora LDL, co ogranicza pobieranie cholesterolu i hamuje syntezę cholesterolu w
komórce przez zahamowanie reduktazy HMG-CoA.
Ok. 3/4 LDL jest wychwytywane przez wątrobę
Ok. 1/4 LDL ulega tzw. Wychwytowi pozawątrobowemu-tkankowemu.
W receptor dla LDL wyposażone są wszystkie tkanki, które potrzebują cholesterolu do syntez chormonów
sterydowych – kora nadnerczy, jądro, jajnik, łożysko
Reszta cholesterolu wyłapywana jest przy pomocy receptora scavenger
VLDL
ApoB100
Wątroba produkuje
ApoE
ApoC
lipoliza (LPL)
Bezpośrednio
2/3
REMNANTY VLDL
3/4
lipoliza (HTGL) – 1/3 remnantów
LDL
1/4
LDL na obwód

grupa7.med@gmail.com | 2008 - wdanie II, poprawione
24
18. MAŁE, GĘSTE LDL
1. Wykrywanie małych gęstych LDL
elektroforeza w gradiencie pH
ultrawirowanie w gradiencie gęstości
NMR lipoproteid- pozwala oznaczyd stężenie samych cząstek LDL oraz ich wielkośd
Stężenie apo B-100
2. Małe, gęste LDL
Cholesterol LDL występuje w dwóch podfrakcjach:
A – duże lekkie
B – małe gęste
Wykazują bardziej aterogenny wpływ niż podfrakcja A
Ich obecnośd związana jest z insulinoopornością i nadprodukcją apo B-100, gdyż o
gęstości cząstek LDL decyduje właśnie apo B-100.
Czynnikiem indukującym wątrobę do produkcji apo B-100 jest insulina
Mechanizm zwiększenia cz. białkowej w zespole insulinooporności:
1) Insulina nie hamuje lipolizy w insulinoopornych adipocytach tk. tłuszczowej żółtej
2) zwiększoną zawartością FFA w osoczu i ich transportem do wątroby, w której obecne w dużej ilości FFA
stymulują syntezę VLDL hamując degradację apo B-100 (która uległaby degradacji przy niskim stężeniu
FFA)
3) insulina pobudza CETP
4) VLDL poprzez CETP wymienia z HDL triglicerydy oraz estry cholesterolu - powstały bogaty w TG HDL
podlega działaniu LPL/HTGL – powstaje mały gęsty HDL wydalany w nerkach (wychwyt przez cubilinę)
5) następuje patologiczna wymiana lipidów między VLDL a LDL (również poprzez CETP) – powstają bogate w
TG LDL, które podlegają działaniu LPL/HTGL –powstają małe gęste LDL
Aterogennośd polega na:
- małym powinowactwie do receptora LDL
- długim t
1
/
2
- Intensywnym naciekaniu ściany naczyniowej
- silnym wiązaniu z GAG/proteoglikanami
- przyspieszonej oksydacji
- intensywnym wychwycie przez makrofagi
19. HDL - BUDOWA, BIOSYNTEZA, DEGRADACJA, FUNKCJA
HDL2
białko 33%
TG 16%
fosfolipidy 43%
estry cholesterolu 31%
cholesterol wolny 10%
HDL3
białko 57%,
w części białkowej: apo A-I, apo E, wędrująca apo C-II
TG 13%,
fosfolipidy 46%,
estry cholesterolu 29%,
cholesterol wolny 6%
grupa7.med@gmail.com | 2008 - wdanie II, poprawione
25
BIOSYNTEZA
w hepatocytach, enterocytach i chłonce tkanek obwodowych powstaje HDL natywny zbudowany z dwóch
warstw fosfolipidow, pochodzących z rozpadu komórek oraz z apo A-I syntezowanego w wątrobie. Dzięki apo A-I
łączy sie z komórkami obwodowymi poprzez receptor ABCA1, który umożliwia przepływ lecytyny, tokoferoli i
cholesterolu z komórki do HDL. Cholesterol układa się w części płaszczowej, następnie enzym LCAT estryfikuje
cholesterol resztą kwasu tłuszczowego oderwanego od lecytyny. Estry Cholesterolu przechodzą do warstwy
rdzeniowej powodując większe upakowanie cząstki.
FUNKCJA
I działanie przeciwmiażdżycowe
1) usuwanie CH z tk. obwodowych i przekazywanie do wątroby
a)drogą bezpośrednia: przez rec. SR-B1 do hepatocytów wchodzi tylko CH
b)drogą pośrednią: przekazanie CH cząstkom VLDL i chylomikronów, które oddaję CH do
wątroby, w zamian za TG, dzięki białku przenoszącemu estry cholesterolu CETP
2) zmniejszenie ekspresji białek adhezyjnych
3) działanie antyoksydacyjne, dzięki wbudowanej paraoksonazie zmiatającej wolne rodniki
II magazyn apo C i apo E dla innych cząstek
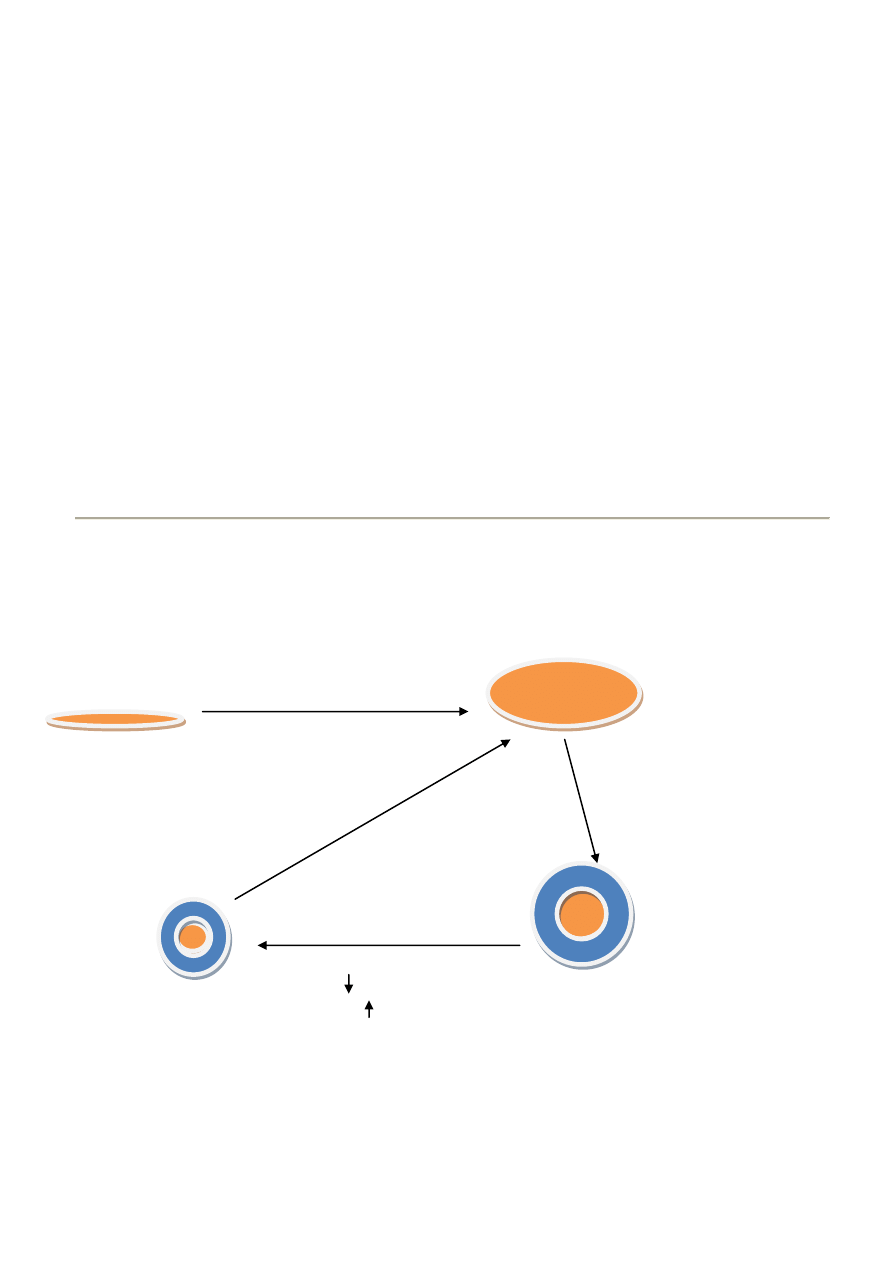
20. CYKL HDL, FRAKCJE HDL
Cykl HDLi polega na przemianach:
małe, gęste HDLe czyli HDL3 odbierają od tkanek Ch, LCAT estyfikuje wolny CH do CE, następuje upakowanie
cząstek, powstają HDL2a HDL2a pod wpływem działania CETP oddają estry cholesterolu oraz apoA-I cząstkom
VLDL i CHM przyjmują TG, powstają HDL2b HDL2b ulegają działanie HTGL, TG są hydrolizowane, powstają HDL3
natywny HDL odbiera cholesterol z tkanek (ABCA1) HDL3
HTGL
LCAT
HDL 2b
CETP
HDL 2a
cholesterolu
TG

grupa7.med@gmail.com | 2008 - wdanie II, poprawione
26
21. BEZPOŚREDNI I POŚREDNI TRANSPORT CHOLESTEROLU
- zachodzi dzięki cząstkom HDL odbierającym cholesterol z tkanek obwodowych; jest to możliwe dzięki białku
ABC-1 komórek obwodowych, aktywowanemu przez apo AI
a) droga bezpośrednia:
- zachodzi przy udziale receptora SR B1 – scavenger receptor typ B1
- całe cząstki HDL zostają wciągnięte do wnętrza do wnętrza hepatocytu
- estry cholesterolu są hydrolizowane przez esterazę
- HDL nie zostaje całkowicie strawiony - w „okrojone” formie (większą częśd stanowi apo AI)
zostaje wydzielony z powrotem z hepatocytu
b) droga pośrednia – jest najistotniejsza u człowieka:
- polega na wymianie lipidów między HDL a VLDL: HDL przyjmują triglicerydy przekazując częśd swojego
cholesterolu na VLDL
- VLDL jest następnie poddawany działaniu lipazy lipoproteinowej, w wyniku czego powstają IDL
- 2/3 remnantów VLDL internalizowane jest przez hepatocyty po związaniu przez receptor apoB/E
- 1/3 poddawana jest działaniu HTGL, przez co przekształca się w LDL, których 75% jest wyłapywane
przez wątrobę
cholesterol w wątrobie:
- obniża aktywnośd reduktazy β-hydroksy β-metylo glutarylo-S-CoA – hamuje syntezę
endogennego cholesterolu
- po przekształceniu w oksysterole hamuje ekspresję receptora LDL
- jest deponowany w hepatocycie dzięki ACAT
- służy do syntezy kwasów żółciowych
22. BIAŁKA ABC – FUNKCJA I STRUKTURA
Patrz „receptory” str. 14
Bialka ABC czyli białka posiadające kasetę wiążącą ATP np. białko SUR i białka MDR. Oddziaływują one z
apolipoproteiną apoA1 na HDL-ach. Białka te posiadają domenę wiążącą ATP i lokalizują się w błonie
cytoplazmatycznej komórek wątrobowych i obwodowych (pełnią funkcję wymiatacza cholesterolu z komórek i
przekazywania ich na HDL)
Defekt białka ABCa1 doprowadza do tzw. Choroby Tangierskiej, cechującej się goromadzeniem w komórkach
cholesterolu i niemożliwością przekazania go na obwód. Następuje zarazem silny hiperkatabolizm apoA1 wobec
czego spada jego stężenie i stężenie HDL
Ponadto ekspresja ABCa1 zachodzi w makrofagach dzięki czemu HDL może odebrad wolny cholesterol z
makrofagów i przez to zapobiega powstawaniu komórek piankowatych
grupa7.med@gmail.com | 2008 - wdanie II, poprawione
27
23. CHARAKTERYSTYKA ENZYMÓW METABOLIZUJĄCYCH LIPOPROTEINY
(LPL, HTGL, LAT, CEPT)
LPL – lipaza lipoproteinowa śródbłonka naczyo:
a) odpowiedzialna za hydrolizę triglicerydów zawartych w chylomikronach i VLDL – przekształca je w
odpowiednio: remnanty chylomikronów i IDL
b) występuje w naczyniach krwionośnych tkanek pozawątrobowych:
- mięśni szkieletowych
- tkanki tłuszczowej
- mięśnia sercowego
c) aktywatory:
- apoC-II
- heparyna (dożylne podanie heparyny powoduje uwolnienie enzymu i nagły wzrost aktywności)
- GAG wiążące eznym do ściany naczyniowej
e) substancje obniżające aktywnośd:
- apoC-III
- siarczan protaminy
HTGL – wątrobowa lipaza triglicerydowa:
a) zlokalizowana na śródbłonku zatokowych naczyo wątroby
b) odpowiedzialna za katabolizm 1/3 remnantów VLDL oraz przekształcenie HDL
2B
w HDL
3
c) substraty: triglicerydy i fosfolipidy
d) aktywowana przez:
- insulinę
- hormony tarczycy
LCAT - acylotransferaza lecytyna-cholesterol
a) syntezowana w wątrobie, uwalniana do krążenia
c) estryfikuje cholesterol w HDL biorąc udział w jego zwrotnym transporcie
d) dwa typy aktywności LCAT:
- alfa – estryfikacja cholesterolu HDL
- beta – estryfikacja cholesterolu w lipoproteinach zawierających apoB
e) jego aktywatory:
- apoA-I
- apoA-IV
- apoC-I
f) aktywnośd LCAT ostatecznie prowadzi do zwiększenia zawartości apoE i obniżenia zawartości apoC w VLDL
HSL – hormon-sensitive lipase:
a) hormonozależna wewnątrzkomórkowa lipaza adipocytów (HTL)
b) uwalnia wolne kwasy tłuszczowe z adipocytów w okresie międzyposiłkowym i przy zwiększonym
zapotrzebowaniu energetycznym
c) aktywnośd wzrasta wraz z wzrostem stężenia cAMP
d) aktywowana przez adrenalinę przez receptor beta-1-adrenergiczny
e) aktywnośd obniżana przez insulinę, która powoduje spadek poziomu cAMP

grupa7.med@gmail.com | 2008 - wdanie II, poprawione
28
CETP białko przenoszące estry cholesterolu
transportuje CE od HDL2a do VLDLi i CHM, w zamian przenoszone są TG, powstają HDL2b
1) INHIBITORY CETP:
- CETi-1 - szczepionka anty-CETP otrzymana poprzez połączenie epitopu przeciw CETP z limfocytów B z
epitopem przeciw toksynie tężca z limfocytów T
- Jtt-705 obie te cząsteczki inhibują CETP działając bezpośrednio przez wiązanie się z CETP co znosi
- torcetrapib aktywnośd przenoszącą estry cholesterolu
- alkohol etylowy - w umiarkowanych ilościach
Uzyskiwany efekt:
- spadek aktywności CETP
- spadek stezenia LDL
- wzrost stezenia HDL
2) Aktywatory CETP:
- apoE
- kwas 13-cis-retinowy
- kwas palmitynowy
- insulina
24. MODYFIKACJA CZĄSTKI LDL – MECHANIZMY, PRZYCZYNY, KONSEKWENCJE
zachodzą
a) w cz. cholesterolowej - powstają oksysterole, które blokują ekspresję rec. wysokiegopowinowactwa dla LDLi
oraz działają toksycznie na śródbłonek.
b) w cz. białkowej - apo B-100 ulega:
- oksydacji, powstałe nieprawidłowe białko, które ma postrzępioną strukturę i staje się obce antygenowo
- przyłączaniu glukozy w procesie glikacji – słabszy wychwyt przez receptory,
- tiolacji homocysteiną,
- angiotensyninowaniu – przyłączenie angiotensyny II
Konsekwencje: rozwój miażdżycy
LDL po oksydacji stają się ofiarą makrofagow, które wciągane są pod śródbłonek przez białko chemoatrakcyjne
MCP-1.W makrofagach znajduje się rec. niskiegopowinowactwa dla LDLi typu scavenger. Nie podlega on "down
regulation", więc cały dostępny CH jest przez nie wyłapywany i powstają kom. piankowate.
Wydzielają one PDGF, FGF, TNF alfa, a po pęknięciu metaloproteazy niszczące ścianę naczynia. Następuje
proliferacja SMC na zewnątrz naczynia, po uszkodzeniu ściany cholesterol z kom. piankowatych uwalniany jest
do światła naczynia, wydzielany jest także czynnik TF aktywujący proces krzepnięcia.

grupa7.med@gmail.com | 2008 - wdanie II, poprawione
29
25. OKSYSTEROLE – STRUKTURA, FUNKCJE
Są to utlenione pochodne CH, powstają gdy Ch jest za dużo
a) wewnątrznaczyniowo sa toksyczne dla śródbłonka
zwiększają ekspresję genów białek adhezyjnych
zwiększają produkcję TXA2 i endoteliny
zmniejszają produkcję NO i prostacykliny
b) wewnątrzkomórkowo - powstaje aktywny cholesterol, który na poziomie genu hamuje syntezę receptora dla
LDL. LDL nie wchodzą do komórek ich stężenie wzrasta w krwi.
Obie te drogi prowadzą do rozwoju miażdżycy, oksysterole są łatwiej wyłapywane przez makrofagi, poza tym
następuje wazokonstrykcja, metaloproteazy niszczą ścianę naczynia, uwolniony TF prowadzi do zwiększenia
krzepliwości krwi.
EFEKTY DZIAŁANIA OKSYSTEROLI
• cytotoksyczne - są inhibitorami wzrostu lub powodują śmierd komórek in vitro
• hamują biosyntezę DNA oraz biosyntezę cholesterolu
• są inhibitorami kalmoduliny
• wpływają na strukturę i funkcję błony komórkowej
• IMMUNOSUPRESJA
Hamowanie proliferacji i transformacji limfocytów
• DZIAŁANIE MUTAGENNE DLA MIKROORGANIZMÓW
ŻRÓDŁA:
1) dieta
2) oksydacja endogenna
•
autooksydacja
•
swoiste monooksygenazy (hydroksylacja 7 , 20 , 22 , 23 , 25, 26, 27), enzymatyczna lub nieenzymatyczna peroksydacja
lipidów
26. LP (a) – STRUKTURA, SYNTEZA, MECHANIZM DZIAŁANIA
27. LP (X) – STRUKTURA, SYNTEZA, MECHANIZM DZIAŁANIA
patrz str. 11
grupa7.med@gmail.com | 2008 - wdanie II, poprawione
30
28-29. Klasyfikacja hiperlipoproteinemii ze szczególnym uwzględnieniem
podziału Fredricksona.
Hiperlipoproteinemie- heterogenna grupa zaburzeo przemian lipidów lipidów, w których dochodzi do wzrostu
stężenia cholesterolu lub trójglicerydów w osoczu. Możemy wyróżnid HLp pierwotne i wtórne
Pierwotna – uwarunkowana genetycznie
Wtórna – zaburzenia lipidowe, będące powikłaniami schorzenia zasadniczego.
Podział wg EAS
Typ zaburzenia Stężenie cholesterolu całkowitego Stężenie triglicerydów
hipercholesterolemia 200 mg/dl
200 mg/dl
hipertriglicerydemia 200 mg/dl 200 mg/dl (>150 mg/dl)*
hiperiipidemia mieszana 200 mg/dl
200 mg/dl (> 150 mg/dl)*
*wgNCEPATPIII(2004)
Podział wg Fredicksona
Typ
Test zimnej flotacji
TCh
TG
Elektroforeza
aterogenność
I
Klarow na z kożuchem
norma
Chylomikrony
-
IIa
Klarowna
norma
LDL
+
IIb
mętna (opalizująca)
LDL i VLDL
+
III
Mętna z kożuszkiem
VLDL + IDL
+
IV
Mętna
norma
VLDL
- ?
V
Mleczna z kożuchem
VLDL + hylomikrony
-
Przyczyny wtórnej hiperlipidemi:
- cukrzyca
- choroby endokrynne (cukrzyca II,Cushing, akromegalia, niedoczynnośd tarczycy etc)
- choroby wątroby (marskośd, zapalenie, żółtaczka mechaniczna)
- choroby trzustki (przyczyna/skutek? ostre zapalenie trzustki wynika ze TG i samostrawienia)
- choroby nerek (zespół nerczycowy, przewlekła niewydolnośd nerek)
- choroby spichrzeniowe (von Gierke)
- ciąża ( naturalna hiperlipidemia)
- polekowa hiperlipidemia (wywołana m.in. glikokortkosterydami,
tiazydowymi lekami moczopędnymi, β-blokerami nieselektywnymi)
- inne (porfiria, toczeo układowy, jadłowstręt psychiczny, stres, otyłośd trzewna)
Diagnostyka Labolatoryjna Hiperlipoproteinemii
Badanie podstawowe – panel lipidowy (oznaczanie TCH, HDL, TG, wyliczenie ze wzoru friedewalda LDL)
Badania uzupełniające – Test zimnej flotacji, elektroforeza
Badania szczegółowe (oznaczanie Lp(a), ApoB, apoA1, fenotyp apoE, LDL, enzymy, genetyczne)
grupa7.med@gmail.com | 2008 - wdanie II, poprawione
31
Oznaczanie wtórnej hiperlipidemii
- wywiad
- badanie fizykalne
- oznaczenie TSH
- oznaczenie glikemii na czczo / OGTT
- enzymy wątrobowe (GGTP, FA, proteinogram, protrombina)
- oznaczenie kreatyniny
- badanie ogólne moczu
- oznaczenie aktywności amylazy w surowicy i w moczu
30. Hiperlipoproteinemia typu I
Hipertrójglicerydemia egzogenna, hyperhylomikronemia
chylomikrony
PRZYCZYNY (głównie genetyczne)
- defekt LPL (całkowity brak aktywności lub zmniejszona aktywnośd)
- defekt ApoC 2 (aktywator LPL)
- nadaktywnośd ApoC 3 (inhibitor LPL)
β (LDL)
- obecnośd przeciwciał przeciw heparynie (która jest aktywatorem LPL)
pre β (VLDL)
α(HDL)
OBRAZ:
- Częstośd występowania choroby – 0,3% populacji
- pacjenci nie leczeni umierają w dzieciostwie (samostrawienie trzustki)
- Dochodzi do gromadzenia chylomikronów
- Bardzo wysoka hipertrójglicerydemia (wartości rzędu 10 000 mg%)
- żółtaki - podskórne gromadzenie trójglicerydów – lokalizują się na wyprostowanych częściach
kooczyn ( łokcie, kolanach, w okolicach prostowników palców ), tworząc się zgrubienia w kolorze żółtym
- Lipaemia retinalis - gromadzenie złogów w siatkówce oka
- Odkładanie się chylomikronów w wątrobie, śledzionie, trzustce co prowadzi do
hepatosplenomegalii i uaktywnienia enzymów trzustkowych, samostrawienia trzustki i tym
samym do ostrego zapalenia trzustki (najczęstsza przyczyna zgonu w wieku dziecięcym).
DIAGNOSTYKA:
- symptomy: hepatosplenomegalia, żółtaki, lipemia retinalis
- badanie laboratoryjne (dieta „0”/16h) – ostry wzrost TG, normalny TCh
TCh/TG < 0,2 bardzo często <0,1
- w teście zimnej flotacji : surowica przejrzysta z kożuchem
- elektroforeza: pojawienie się paska chylomikronów
LECZENIE:
- DIETA – bez długołaocuchowych kw. Tłuszczowych (nie powstają chylomikrony)
- podawanie egzogennego apoC2
- okresowe transfuzje poprawiają stan chorego
- wektorowe wprowadzanie genów dla prawidłowej LPL (wektorem może byd wirus)

grupa7.med@gmail.com | 2008 - wdanie II, poprawione
32
31. Hiperlipoproteinemia typu II
Hipercholesterolemia rodzinna
PRZYCZYNY:
- forma recepto negatywna (brak receptora apoB/E)
- forma receptorodefektywna
β - LDL
TYPY: (ze względu na sposób dziedziczenia)
- homozygotyczna – raz na milion -człowiek umiera
- heterozygotyczna – raz na 500 - spowolniony wychwyt
pre β - VLDL
TYPY: (ze względu na wzrost frakcji)
- typ A - podwyższone stężenie TCh
- typ B - podwyższone stężenie TCh i TG
OBRAZ:
- remnanty VLDL i LDLi nie są wychwytywane (zwiększa się klirens LDL),
ulegają modyfikacjom, głownie oksydacji, co prowadzi do zmian miażdżycowych
- prowadzi to również do wzrostu cholesterolu wewnątrzkomórkowego - jeśli nie ma
wychwytu LDL przez komórki to nie jest hamowana reduktaza HMG CoA, co powoduje
zwiekszoną syntezę cholesterolu endogennego
- kępki żółte - złogi cholesterolu gromadzą się na powiekach (xanthelasma palpebrarum), w kącie nosa
- odkładanie cholesterolu w rogówce – rąbek starczy
- zółtaki w okolicach ścięgna Achillesa, prostowników, rzepki
DIAGNOSTYKA
- symptomy: żółtaki, kępki żółte, rąbek starczy, choroba wieocowa itd.
- badanie laboratoryjne (dieta „0”/16h) –TG normalny, wzrost TCh
Typ IIa TCh/TG > 1,5
Typ IIb TCh/TG = 1.0 – 1,5
- w teście zimnej flotacji :
IIa - surowica przejrzysta
IIb- surowica klarowana lekko opalizujaca
-elektroforeza:
typ IIa zwiększona ekstynkcja prążka β - lipoprotein
typ IIb zwiększona ekstynkcja prążka β- lipoprotein i preβ- lipoprotein
LECZENIE:
- typ IIa leczenia farmakologicznego nie ma.
przeszczep wątroby ( w Europie)
terapia genowa (w Ameryce)
pozaustrojowe oczyszczanie krwi z LDLi
- typ IIb leczenie farmakologiczne
Farmakologicznie podaje się statyny, które hamują reduktazę HMG-CoA – dochodzi do zablokowania szlaku
mewalonianowego, nie dochodzi do powstanie endogennego cholesterolu a tym samym następuje wzrost
ekspresji genu i synteza receptora LDL (wzrost syntezy nawet o 180%)
grupa7.med@gmail.com | 2008 - wdanie II, poprawione
33
32. Hiperlipoproteinemia typu III
Choroba szerokiego paska, choroba lipoprotein flotujących, hiperlipidemia mieszana
PRZYCZYNY:
- defekt genetyczny apolipoproteiny apoE (zamiast E3 występuje E2)
receptor apoB/E preferuje E3, więc jeśli nie ma E3 wyłapuje samo B
Stąd wychwyt VLDL(apoE) jest spowolniony a LDL zwiększony (apoB)
- okres menopauzy (wtedy jest to hiperlipoproteinemia wtórna)
OBRAZ:
- podwyższone stężenie trójglicerydów i cholesterolu
β - VLDL
- gromadznie remnantów VLDLi a stężenie LDL może byd obniżone
- remnanty VLDLi mogą tak jak LDLe naciekac sciany naczyniowe
i indukowad proces miażdżycowy
- powstaja na skórze zółtaki guzowate (inne niż w HLp I)
-występuje nietolerancja weglowodanowa, hiperurykemia
TYPY:
- pierwotna
-wtórna –najczęściej u kobiet podczas menopauzy
DIAGNOSTYKA
- symptomy: żółtaki guzowate, choroba wieocowa itd.
- badanie laboratoryjne wykazuje niewielki wzrost TCh (250-280mg%) i TG
- w teście zimnej flotacji : surowica mętna
- elektroforeza: szeroki pasek bo β lipoprotein i pre β lipoprotein się zlewają
(ponieważ remnanty VLDLi jako frakcja β -VLDL biegną miedzy β a pre β lipoproteinami)
33. Hiperlipoproteinemia typu IV
Trójglicerydemia endogenna – NADPRODUKCJA VLDL
TYPY IPRZYCZYNY:
β - LDL
- wtórnej- choroby wątroby i cukrzyca
- pierwotnej – brak informacji
pre
β - VLDL
DIAGNOSTYKA
- symptomy: , choroba wieocowa, powiększenie wątroby, śledziony itd.
- w teście zimnej flotacji : surowica mętna
-elektroforeza: zwiększona ekstynkcja prążka pre- β lipoprotein
OBRAZ:
- podwyższone stężenie trójglicerydów
- gromadznie VLDLi (ponieważ bardzo duża synteza a LPL ulega wysyceniu)
- VLDLe naciekają na wątrobę śledzionę trzustkę i na ścianę naczyniową – ryzyko miażdżycy zwiększone
grupa7.med@gmail.com | 2008 - wdanie II, poprawione
34
34. Hiperlipoproteinemia typu V
HLp 1 + 4 = 5
PRZYCZYNY:
- nadprodukcja VLDL
- podobnie jak w HLp I:
- defekt LPL
- defekt apoC II
- pojawienie się nieprawidłowej apoC II
- insulinoopornośd (insulina jako aktywator LPL).
Jeśli insulina działa, zazwyczaj mamy HLp typ IV, jeśli insulina przestaje działad, dochodzi nam dodatkowo
obniżenie aktywności LPL i typ IV przechodzi w typ V (i odwrotnie…)
OBRAZ:
- gromadzenie się VLDLi i chylomikronów (HLp 1+ 4)
- VLDLe naciekają na wątrobę śledzionę trzustkę – możliwe wystapienie ostrego zapalenia trzustki
DIAGNOSTYKA
- symptomy: powiększenie wątroby, śledziony itd.
- badanie laboratoryjne – wzrost TG i TCh
TCh/TG = 0,2-0,5
- w teście zimnej flotacji : surowica mleczna z kożuchem
-elektroforeza: zwiększona ekstynkcja prążka pre- β lipoprotein i
Pojawia się prążek chylomikronów
35-37 diagnostyka hiperlipoproteinemii, elektroforeza, test zimnej flotacji
Patrz skrypt

grupa7.med@gmail.com | 2008 - wdanie II, poprawione
35
38. ROLA ZABURZEŃ LIPIDOWYCH LIPIDOWYCH PATOGENEZIE MIAŻDŻYCY
TĘTNIC
Biorac pod uwagę rolę lipoprotein w procesie miażdżycowym można podzielid je na 3 grupy:
1.Lipoproteiny aterogenne
-LDL
-Lp(a)
-remnanty VLDL
-remnanty chylomikronów
2.Lipoproteiny nieaterogenne
-chylomikrony
3.Lipoproteiny antyaterogenne
-HDL-ale tylko gdy występuje w określonym stężeniu i gdy pełni swoją funkcję
(gdy jest funkcjonalnym HDL)
Modyfikacje LDL również odgrywają rolę w procesie miażdżycy ,zmodyfikowane LDL nie są
rozpoznawalne przez receptor wysokiego powinowactwa ,są wychwytywane przez makrofagi obdarzone
recptorem scavenger(receptor ten nie podlega regulacyjnemu hamowaniu zwrotnemu w miarę gromadzenia
cholesterolu w komórce).Po wchłonięciu LDL makrofagi uwalniają cytokiny m.in. czynniki wzrostowe
zapoczątkowywujace proliferację SMC,syntezę elementów łącznotkankowych oraz indukują ekspresję molekół
adhezyjnych w komórkach śródbłonka, które wiążą monocyty uniemożliwiając ich transfer do warstw
podśrodkowobłonkowych ,a wydzielone cyklokliny powodują ich przekrzształcnie do makrofagów. Dochodzi do
obiżenia syntezy prostacykliny , co pociąga za sobą zmniejszenie właściwości przeciwadhezyjnych komórek
sródbłonka.Zahamowaniu ulega również synteza EDRF.
grupa7.med@gmail.com | 2008 - wdanie II, poprawione
36
39. KRYTERIA PODZIAŁÓW KW. TŁUSZCZOWYCH
-długośd łaocucha
-stopieo nasycenia
-miejsce syntezy
-lokalizacja wiązania nienasyconego
KWASY TŁUSZCZOWE
NASYCONE (SFA) NIENASYCONE
(brak wiązao podwójnych)
( obecne wiązania podwójne w czasteczce)
*krótkołaocuchowe 2:0-6:0
*sredniołaocuchowe 8:0-10:0
*długołaocuchowe 12:0-16:0
*o bardzo długim łaocuchu 20:0-22:0
kaprylowy 8:0
kaprynowy 10:0
laurynowy 12:0
mirystynowy 14:0
palmitynowy 16:0
stearynowy 19:0
behemowy 22:0
lignocerynowy 24:0
(MUFA)
JEDNONIENASYCONE
(
PUFA)
(
jedno wiązanie podwójne)
WIELONIENASYCONE
np. oleinowy 18:1(cis)
(kilka wiązao podwójnych w cząstce)
elaidynowy 18:1 (trans)
Rodzina omega 6 (n-6) Rodzina omega 3(n-3)
(rodzina kwasu linolowego) (rodzina kwasu α-linolenowego)
linolowy 18:2 α-linolenowy 18:3
arachidonawy 20:4 eikozapentaenowy 20:5
dokozaheksaenowy 22:6
grupa7.med@gmail.com | 2008 - wdanie II, poprawione
37
40. KWASY TŁUSZCZOWE O RÓŻNYM STOPNIU NIENASYCENIA ŁAŃCUCHA –
WYSTĘPOWANIE ZNACZENIA W METABOLIZMIE, WPŁYW ICH WARTOŚCI W
DIECIE NA UKŁAD LIPIDOWY OSOCZA
- nienasycone kwasy tłuszczowe mają niższą temp. topnienia niż kwasy nasycone o tej samej długości łaocucha
-kwasy tłuszczowe i ich pochodne są tyle bardziej płynne im ich łaocuchy są krótsze i bardziej nienasycone
-zawierają 1-6 wiązao podwójnych, najczęściej o konfiguracji cis. Konfiguracja trans występuje rzadko
-obecnośd podwójnego wiązania typu cis sprawia ,że w miejscu tym następuje zagięcie długiej osi kwasu
tłuszczowego
-atomy wegla tworzące dwa lub więcej podwójnych wiązao są rozdzielone przez grupę *-CH2-]
a)Kwasy jednonienasycone MUFA(monoetenoidy, kwasy monoetenowe)
-zawierają jedno wiązanie podwójne
-oleinowy 18:1(cis)
występowanie: oleje:oliwkowy,rzepakowy,arachidonowy
-elaidynowy 18:1 (trans)
wystepowanie: margaryny twarde
-MUFA cis :↓LDL, bez wpływu na HDL
-MUFA trans:↑LDL,↓HDL
b)Kwasy wielonienasycone PUFA (kwasy polienowe)
-zawierają więcej wiązao podwójnych
1. omega-6 (n-6)
-linolowy 18:2
Występowanie: oleje: słonecznikowy,kukurydziany,krokoszowy
-arachidonowy 20:4
2. omega 3(n-3)
-α-linolenowy 18:3
oleje:lniany,sojowy,rzepakowy
-eikozapentaenowy 20:5
-dokozaheksaenowy 22:6
-PUFA ω-6 ↓LDL,↓TG,(↓ULDL)
-PUFA ω-3 ↓TG(↓ULDL),bez wpływu na LDL

grupa7.med@gmail.com | 2008 - wdanie II, poprawione
38
41. DZIAŁANIE METABOLICZNE PUFA – OMEGA -3
a)Należa tu wielonienasycone kwasy tłuszczowe szeregu omega-3
-kwas alfa-linolenowy
-kwas eikozapentaenowy-timinodonowy
-kwas dokozapentaenowy
-kwas dokozaheksaenowy-cerwonawy
-obecnie w dużych ilościach w olejach rybich
b)Działanie metaboliczne
1.Bezpośrednie działanie na ścianę naczyniową
-redukcja hiperplazji błony wewn. tętnicy przez hamowanie wydzielania PDGF
-hamowanie restenozy po angiplastyce
2.Wpływ na hemostazę i reologię(przepłw krwi)
-wydłużenie czasu krwawienia
-wzrost aktywności ŁPA
-wzrost aktywności antytrombiny III
-spadek stężenia fibrynogenu
-spadek poziomu PAI
-obniżenie lepkości krwi
3.Wpływ na lipidy krwi
-spadek stężenia TG
-spadek syntezy ULDL
-spadek syntezy kw. Tłuszczowych
-spadek syntezy apo B
-spadek LDL przy normalnym stężenia, przy podwyższanych stężeniach (hiperlipoproteinemia)
następuje wzrost frakcji HDL
-wzrost frakcji HDL
-nasilenie utlenienia kwasów tłuszczowych
4.↓RR,↑PGl
2
i PGl
3
-działanie antyarytmiczne i przeciwmiażdżycowe
grupa7.med@gmail.com | 2008 - wdanie II, poprawione
39
42. FABP – STRUKTURA, ROLA W WEWNĄTRZKOMÓRKOWYM METABOLIZMIE
KWASÓW TŁUSZCZOWYCH
Fatty acids binding proteins – białka wiążące kwasy tłuszczowe
Funkcje:
transport wewnątrzkomórkowy długołaocuchowych kwasów tłuszczowych
FABP preferują kwasy nienasycone (wyjątkiem jest FABP typu jelitowego)
powinowactwo zwiększa się wraz ze wzrostem hydrofobowego charakteru FA
decydują o prędkości pobierania FFA z osocza przez komórkę
transportują proliferaty peroksysomów z cytosolu do jądra, gdzie wchodzą w interakcję z PPAR
1 cząsteczka białka wiąże 2 cząsteczki kwasu
Typy FABP (co najmniej 8, różnią się powinowactwem do kwasów tłuszczowych i pełnioną funkcją):
1. wątrobowy
2. jelitowy
3. sercowy – doprowadza do wprowadzenia kwasu tłuszczowego w szlak
β-oksydacji i uzyskania energii
4. tkanki tłuszczowej – zajmuje się przenoszeniem kwasu tłuszczowego w kierunku
FATP (białko transportujące kwasy tłuszczowe) – kwasy tłuszczowe idą na eksport
5. mielinowy – zajmuje się głównie wprowadzaniem kwasu tłuszczowego do syntezy
ceramidu, a z niego przez obróbkę wytwarzana jest sfingomielina
i inne lipidy złożone.
6. Mózgowy
7. Adipocytarny
8. Naskórkowy
I-LBP – ileal lipid-binding protein – białko wiążące lipidy charakterystyczne dla jelita krętego
białko I-LBP wykazuje duże powinowactwo do skoniugowanych kwasów żółciowych (jelito kręte jest miejscem
resorpcji kwasów żółciowych)
43. POZAENERGETYCZNE DZIAŁANIE KWASÓW TŁUSZCZOWYCH
TŁUSZCZOWYCH KOMÓRCE
1. zaburzają strukturę i funkcję błon komórkowych jeśli chodzi o nadtlenki kwasów tłuszczowych
2. hamowanie aktywności pompy Na/K z konsekwencjami głównie dla komórek pobudliwych
3. hamowanie translokazy nukleotydów adeninowych
4. modulacja aktywności receptorów (T
3,
Ang II, glikokortykoidów, Epo)
5. regulacja funkcji kanałów K, Ca, Cl
6. modulacja ekspresji genów FABP, syntetazy acylo-CoA, desaturazy steroilo-CoA
grupa7.med@gmail.com | 2008 - wdanie II, poprawione
40
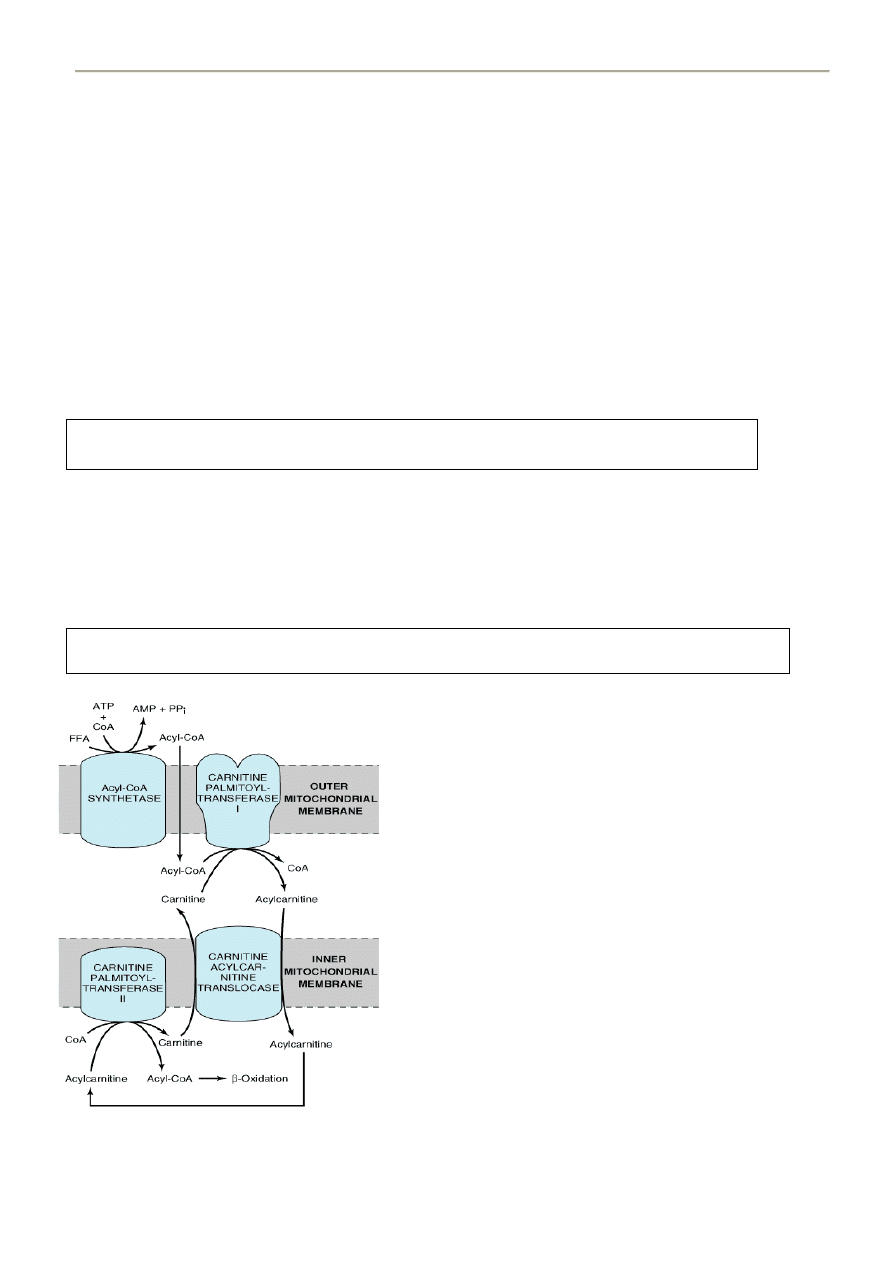
44. Beta oksydacja -
przebieg, energetyka, znaczenie w metabolizmie komórki
- utlenienie kwasów tłuszczowych zachodzi w mitochondriach. W każdym jego etapie uczestniczą
pochodne acylo-CoA i każdy etap jest katalizowany przez oddzielny enzym.
Całośd odbywa się kilku etapowo:
1) Aktywacja kwasu tłuszczowego w cytozolu
2) Przeniesienie go z cytozolu do mitochondriom
3) β oksydacja
AKTYWACJA
- podobnie jak w przemianie glukozy, kw. tłuszczowe muszą najpierw, w reakcji z ATP, zostad przekształcone w
aktywny metabolit żeby mogły reagowad z enzymami odpowiedzialnymi za ich dalszy metabolizm. Jest to
JEDYNY etap który wymaga nakładu ATP.
Miejsca – ER, peroksysomy, wewnątrz mitochondriów i na ich zew. błonie
Enzym – syntetaza Acylo-CoA swoista dla kwasu o określonej długości
Syntetaza ACYLO-CoA
Kwas tłuszczowy + ATP + CoA -------------------------------------- > Acylo-CoA + AMP + PPi
PRZENIKANIE WEWNĘTRZNEJ BLONY MITOCHONDRIALNEJ
- DŁUGIE kwasy tłuszczowe(np. FFA) przenikają jako pochodne karnityny (synteza z lizyny i metioniny w wątrobie
i nerkach, obficie występuje w mięśniach)
- Aktywacja niższych kwasów i ich utlenianie mogą zachodzid w mitochondriach niezależnie od karnityny
CPT I
Karnityna + AcyloCoA ------------------------------- > Acylokarnityna + CoA
CPT I – Palmitoilotransferaza Karnitynowa I
(ogólnie– acylotransferazy Karnitynowe I )
Translokaza Karnityna-AcyloKarnityna
Transport jednej acylokarnityny do wewnątrz i
jednej karnityny na zewnątrz
CPT II – Palmitoilotransferaza Karnitynowa II
CoA + Acylokarnityna -- > karnityna + AcyloCoA
(ogólnie– acylotransferazy Karnitynowe II )
Po przejściu CPT II powoduje odtworzenie Acylo-CoA w macierzy mitochondrialnej i uwalnia karnitynę.
grupa7.med@gmail.com | 2008 - wdanie II, poprawione
41
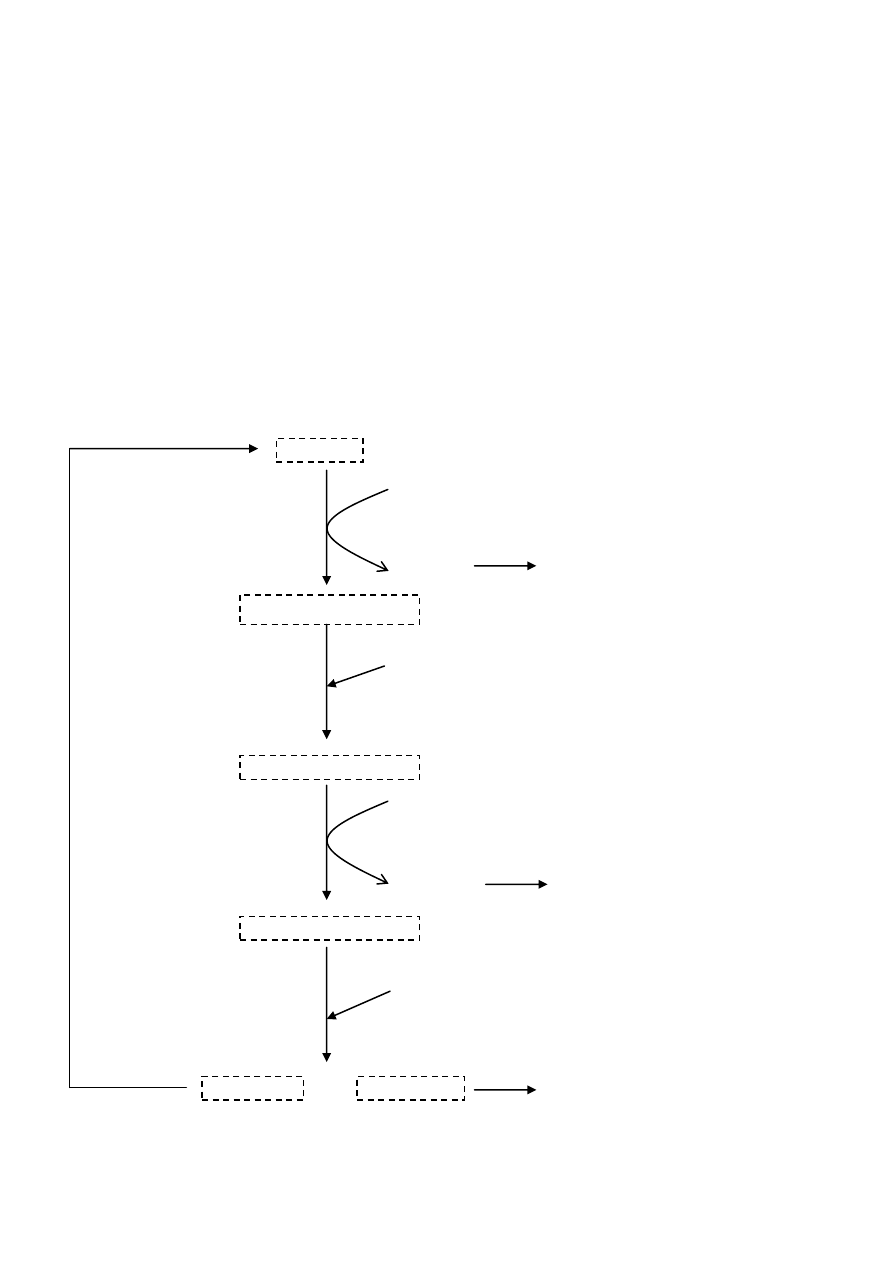
β-oksydacja
β-oksydacja to nic innego jak odczepianie, od kooca karbonylowego długiego kwasu tłuszczowego, kolejnych
acetylo-CoA. Łaocuch jest rozrywany między atomami węgla α i β – stąd nazwa.
Enzymy które biorą w tym udział noszą wspólnie nazwę „oksydaza kwasów tłuszczowych”
Przebieg:
1) Oderwanie dwóch atomów wodoru od C α i β katalizowane przez Dehydrogenazę Acylo-CoA
(koenzymem jest flawoproteina zawierająca jako grupę prostetyczną FAD)
W wyniku tej reakcji powstaje Δ
2
– trans – enoilo- CoA
2) Przyłączenie cząsteczki wody i wysycanie podwójnego wiązania - powstaje 3-Hydroksyacylo-CoA
3) Odwodorowanie na węglu 3 z utworzeniem 3-ketoacylo-CoA
4) Tiolaza rozrywa ketoacylo-CoA w pozycji 2,3 z udziałem acetylo-CoA
5) Produktem reakcji jest Acylo-CoA posiadający o 2C mniej niż wyjściowy związek, oraz cząsteczka acetylo
CoA. Powstały Acylo-CoA znowu ulega przekształceniom oksydacyjnym
Acylo-CoA
FAD
Dehydrogenaza
acyloCoA
FADH2
łaocuch oddechowy
Δ
2
– trans – enoilo- CoA
Hydrataza
H
2
O
Δ
2
enoiloCoA
3-Hydroksyacylo-CoA
NAD
+
Dehydrogenaza
3-Hydroksyacylo-CoA
NADH + H
łaocuch oddechowy
3-ketoacylo-CoA
Tiolaza
SH-CoA
(acetylotransferaza acetyloCoA)
Acylo-CoA + Acetylo-CoA
łaocuch oddechowy
grupa7.med@gmail.com | 2008 - wdanie II, poprawione
42
Parzystowęglowe kw. tłuszczowe dają w efekcie same Acetylo-CoA (2C)
Nieparzystowęglowe kw. Tłuszczowe dają w koocowej reakcji związek który ma 3 atomy węgla.
Powstają więc z niego cząsteczki acetylo-CoA i jedna cząsteczka propionylo-CoA (3C)
1) β –oksydacja z utworzeniem acetylo-CoA
2) Pozostał związek 3C przekształcany do propionylo-CoA
3) Propionylo-CoA jest następnie przekształcany do bursztynylo-CoA
Bilans utleniania palmitynianu (16 atomów węgla w łańcuchu)
Ilośd cykli beta oksydacji =
ilośd atomów
C
/
2
– 1
Ilośd ATP uzyskanej z NADH i FADH2 – z obu około 5 ATP (NADH = 3ATP, FADH2=2ATP)
Ilośd energii uzyskanej ze spalenia 1 cząstki acetylo-CoA – około 12 ATP
aktywacja palmitynianu do palmitoilo-CoA - 2 ATP
energia z NADH i FADH (7cykli ×5 ATP)
+ 35 ATP
spalenie acetylo-CoA (8 cząstek×12 ATP)
+ 96 ATP
Łącznie: + 129 ATP
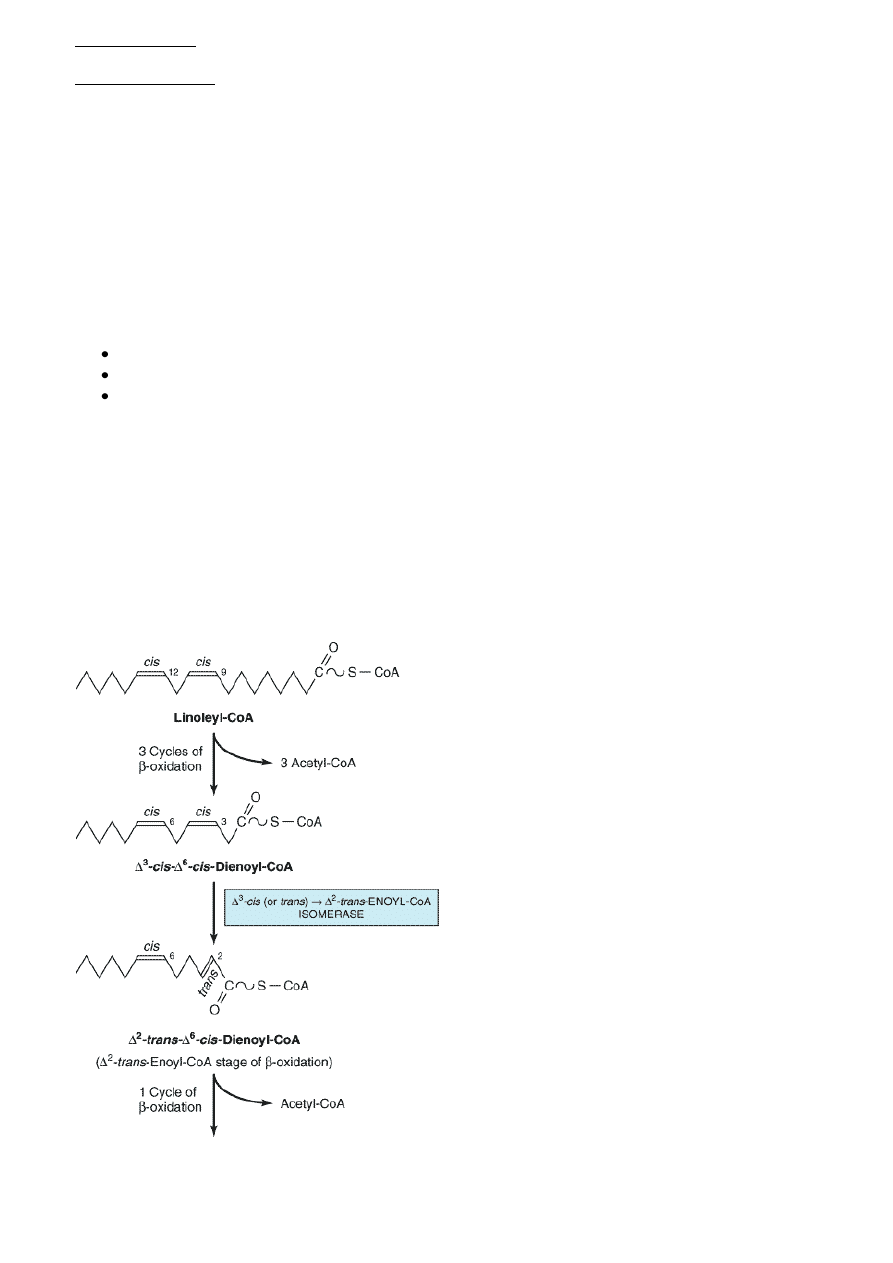
Utlenianie nienasyconych kwasów tłuszczowych
- odbywa się zmodyfikowanym szlakiem beta oksydacji
Mamy przykładowo linoleilo-CoA, ulega 3 cyklom beta
oksydacji, od kooca karbonylowego odrywany jest CoA i
w pewnym momencie natrafiamy na konformacje CIS.
Nasz Δ
3
-cis Δ
6
-cis Dienolio-CoA musi byd przekształcony
do czegoś, od czego można odłączyd Acetylo-CoA.
Konformacja Δ
3
cis bliżej kooca karbonylowego zostaje
przekształcona w Δ
2
trans, która bez przeszkód ulega
dalszej beta oksydacji, czyli hydratacji i dalszemu
utlenieniu (Δ
2
– trans – enoilo- CoA jest w zwykłej beta
oksydacji)
ENZYM – IZOMERAZA
Δ
3
cis (lub trans) -> Δ
2
trans- ENOILO-CoA
grupa7.med@gmail.com | 2008 - wdanie II, poprawione
43
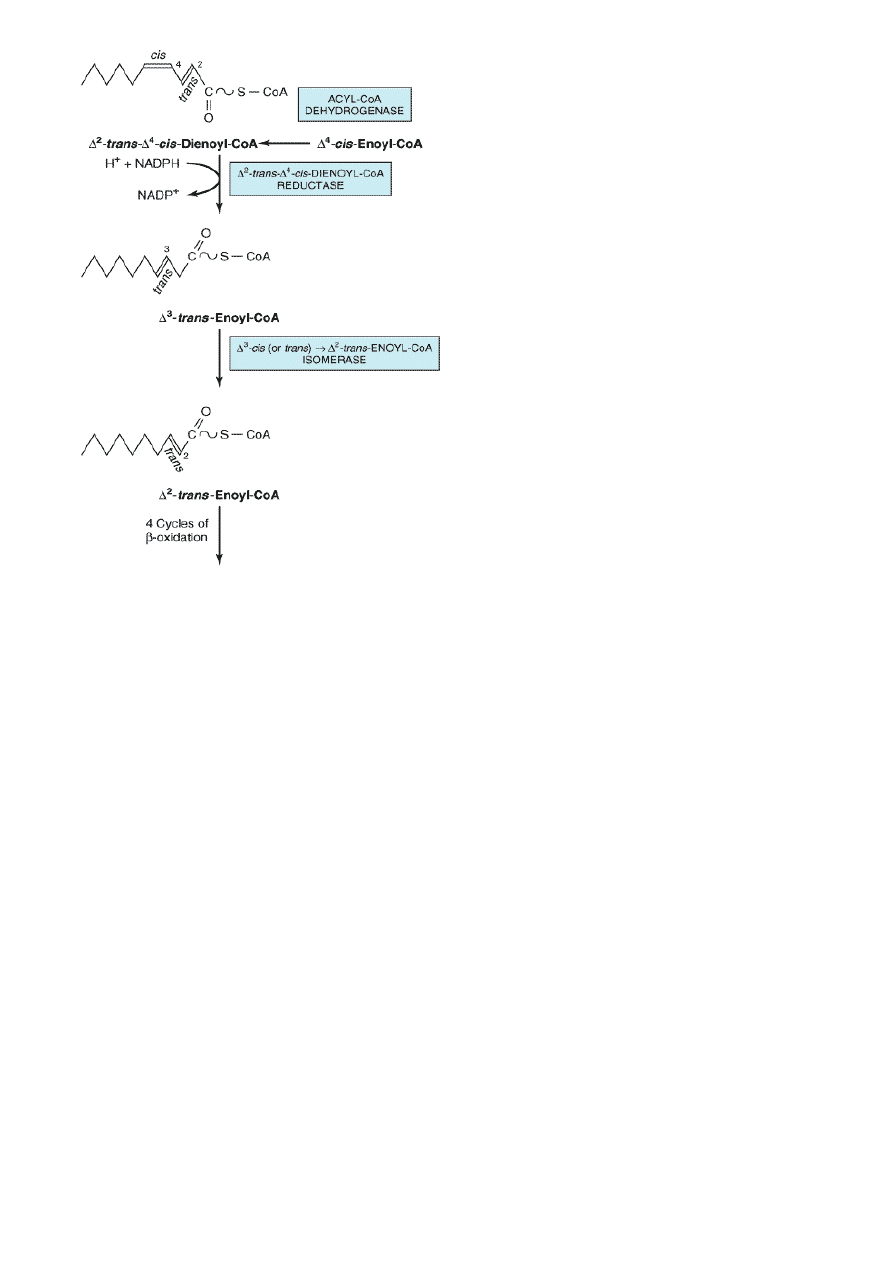
Każdy Δ
4
cis który został po wcześniejszych
przekształceniach (albo jakiś inny kwas który wszedł
do cylku i ma tylko wiązanie delta 4 cis) jest
przekształcany w wyniku działania dehydrogenazy
acylo-CoA do
Δ
2
trans Δ
4
cis dienoilo-CoA
Który w wyniku działania REDUKTAZY NADP-zależnej
jest przekształcony do
Δ
3
trans enoilo-CoA
a Δ
3
do Δ
2
w wyniku działania IZOMERAZY
ENZYMY:
1) Dehydrogenaza acyloCoA
2) Reduktaza Δ 2 trans Δ 4 cis dienoilo-CoA
3) izomeraza
Δ 3 cis (lub trans) -> Δ 2 trans- ENOILO-CoA
α
oksydacja
– w tkance mózgowej
– usuwanie po jednym atomie węgla począwszy od grupy karbonylowej
Polega na:
- hydroksylacji węgla C-alfa przy udziale hydroksylazy współdziałającej z:
1. cytochromem P-450
2. NADPH + H
+
3. Fe
2+
4. kwasem askorbinowym
- dekarboksylacji alfa-hydroksykwasu z równoczesnym utlenieniem
ω
oksydacja
- w siateczce śródplazmatycznej
- katalizowana przez enzymy układu hydroksylującego zawierającego cytochrom P-450
Grupa CH
3
jest przekształcana do CH
3
-OH i następnie utleniana do CH
3
-COOH przez co tworzy się kwas
dwukarboksylowy. Ten ulega beta oksydacji, zazwyczaj do kwasów adypinowego (C6) i korkowego (C8), które są
wydalane z moczem.
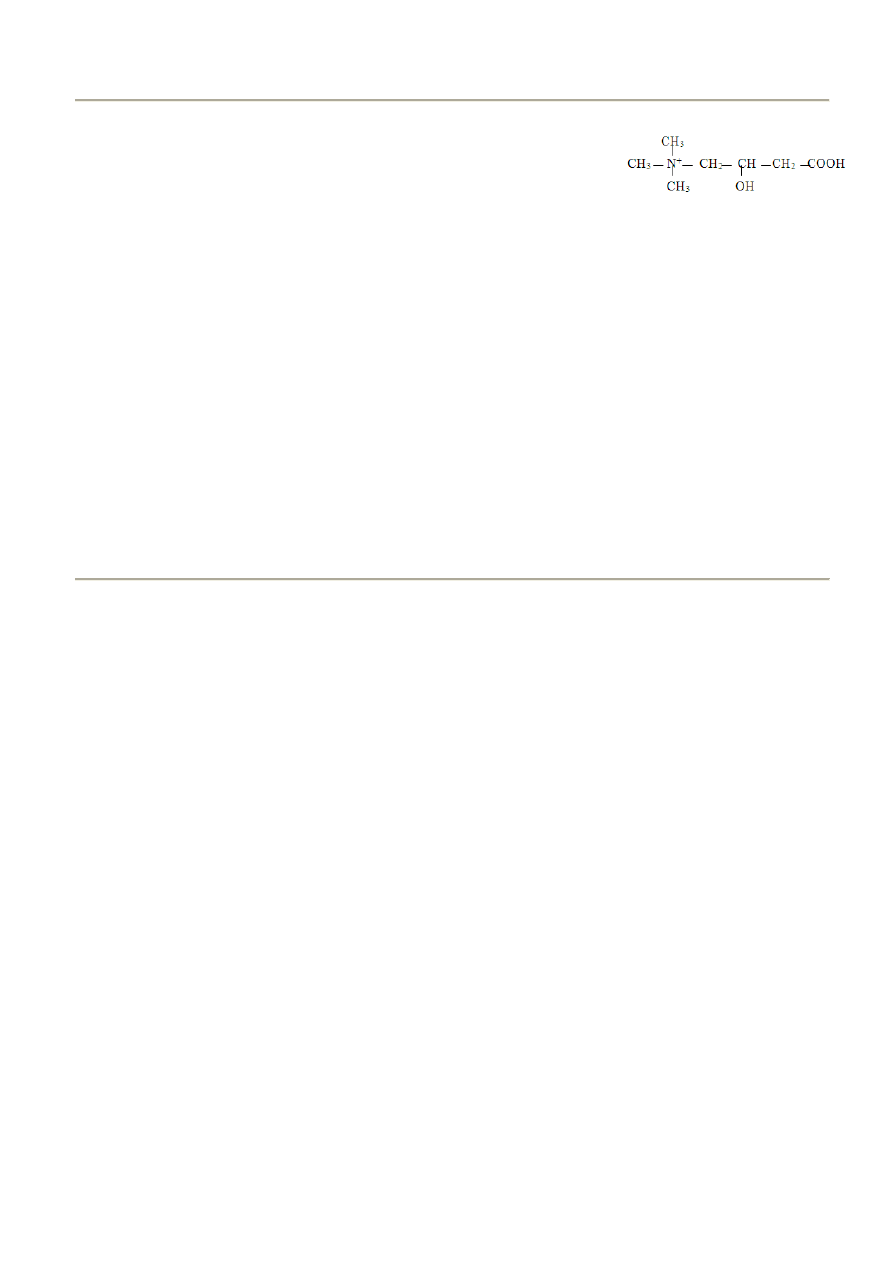
grupa7.med@gmail.com | 2008 - wdanie II, poprawione
44
45. KARNITYNA – STRUKTURA, WYSTĘPOWANIE ROLA W METABOLIZMIE
KWASÓW TŁUSZCZOWYCH
Struktura:
Karnityna to związek niskocząsteczkowy β-hydroksy-γ-trimetyloaminomaślan
Występowanie:
Jest szeroko rozpowszechniona, a szczególnie obficie występuje w mięśniach. Syntetyzowana
z lizyny i metioniny w wątrobie i nerkach.
Rola:
Umożliwia transport długołaocuchowych kwasów tłuszczowych przez wewnętrzną błonę mitochondrialną, dzięki
czemu mogą wejśd w proces β-oksydacji. Po przejściu długołaocuchowego kwasu tłuszczowego przez
zewnętrzną błonę mitochondrialną dochodzi do przeniesienia grupy acylowej na karnitynę z wytworzeniem
acylokarnityny. Reakcję tę katalizuje enzym palmitoilotransferaza karnitynowa I, obecny w zewnętrznej błonie
mitochondrium. Acylokarnityna przenika przez wewnętrzną błonę mitochondrialną przy udziale translokazy
karnitynoacylokarnitynowej. Wiąże się to z transportem jednej cząsteczki karnityny na zewnątrz. Acylokarnityna
reaguje z CoA, powstaje acylo-CoA i odtwarza się karnityna. Reakcja katalizowana jest przez
palmitoilotransferazę karnitynową II. Acylo-CoA wchodzi w proces β-oksydacji.
46. POWIĄZANIE PRZEMIANY TŁUSZCZOWEJ I CUKROWEJ. „TŁUSZCZE
SPALAJA SIĘ W OGNIU WĘGLOWODANÓW”
W wyniku β-oksydacji powstaje acetylo-CoA, który aby mógł byd dalej utylizowany w cyklu Krebsa potrzebuje
szczawiooctanu. Bardzo ważnym źródłem szczawiooctanu jest pirogronian.
Powstaje on w wyniku glikolizy, a następnie częściowo ulega dekarboksylacji oksydacyjnej do acetylo-CoA, a
częściowo ulega karboksylacji do szczawiooctanu. Reakcja karboksylacji pirogronian do szczawiooctanu odgrywa
tu kluczową rolę. Tłuszcze w ogniu węglowodanów nie spalają się wtedy, kiedy niczym nie zahamowane
następuje spalanie tłuszczów, przy niezrównoważonym spalaniu cukrów.
Dzieje się to:
o przy nie zrównoważonej diecie, która obfituje w tłuszcze, natomiast jest zubożona w cukry (tłuszcze się
spalają a cukry nie).
o głodówka – najpierw zużywane są cukry (to co się miało spalid to się spaliło), reszta glukozy
produkowana w okresie głodu nie jest przeznaczona na spalanie ale jest przesyłana do mózgu,
natomiast tłuszcze spalają się bez przeszkód.
o niewyrównana cukrzyca typu I – jeśli wprowadzi się insulinę egzogenną to strata ulega wyrównaniu.
o zatrucie ciążowe tzw. EPH – gestoza (E-edema-obrzęki; P-proteinuria-białkomocz;
H-hypertension-nadciśnienie) – w tych sytuacjach nie ma ognia węglowodanów, bo tu jest blok
metaboliczny – cukry się nie spalają
Przy braku ognia węglowodanów tj. przy braku szczawiooctanu tłuszcze się spalają, ale kooczą swój żywot na
acetylo-CoA. Nie ma kondensacji acetylo-CoA ze szczawiooctanem, czyli nie powstaje cytrynian. Cykl Krebsa
ustaje – nie ma energii i gromadzi się acetylo-CoA, co prowadzi do powstania ciał ketonowych.
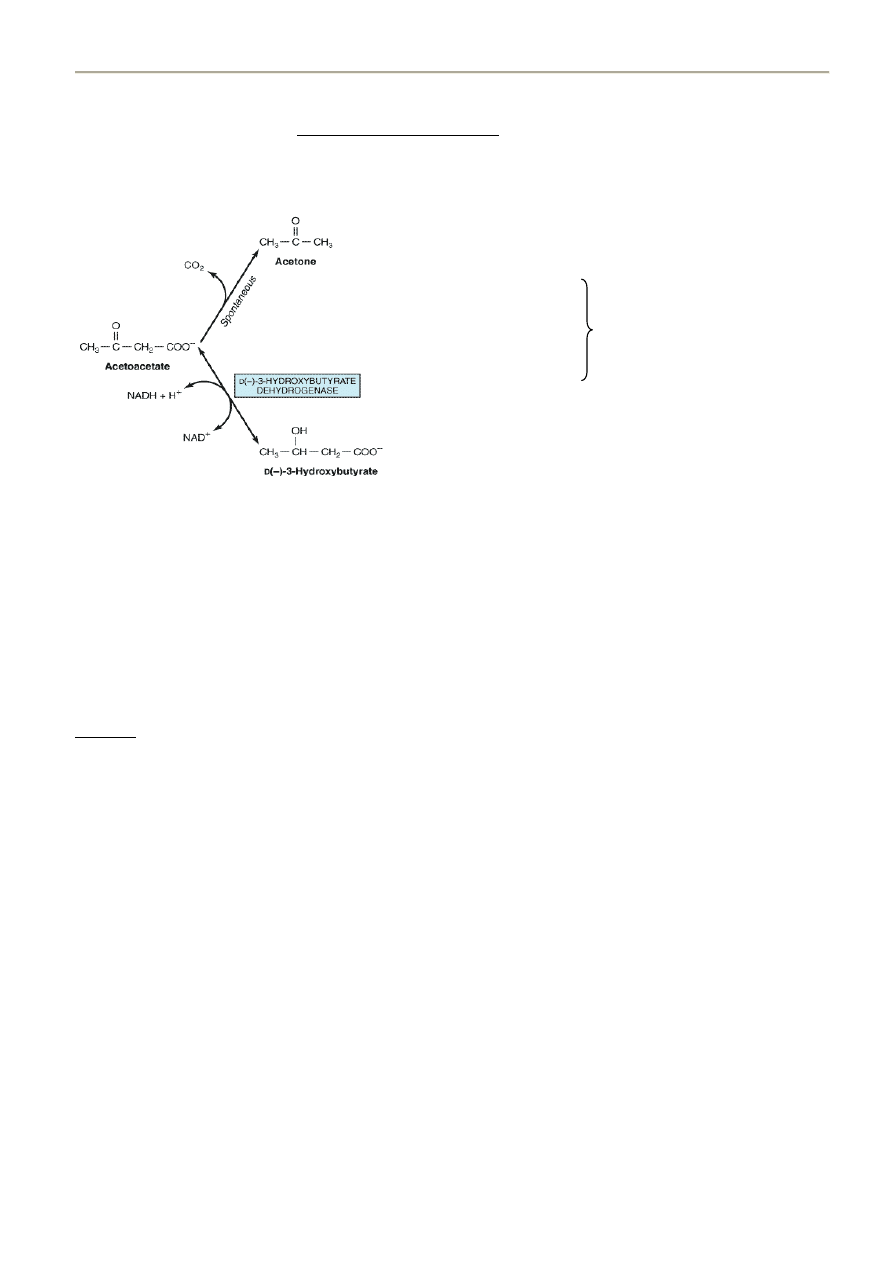
grupa7.med@gmail.com | 2008 - wdanie II, poprawione
45
47. Ciała Ketonowe – powstawanie, utylizacja, znaczenie patogenne
Wzmożone utlenianie kwasów tłuszczowych jest charakterystyczne dla stanu głodzenia i cukrzycy, co prowadzi
do powstania ciał ketonowych w mitochondriach wątrobowych (ketogeneza). Podczas głodzenia szczawiooctan
zamiast razem z acetylo-CoA wejśd do cyklu Krebsa, jest używany do glukoneogenezy. W tych warunkach z
acetylo-CoA powstają ciała ketonowe
Acetooctan
aceton
ciała ketonowe
β-hydroksymaślan
Acetooctan ulega ciągłej samoistnej dekarboksylacji do acetonu
β-hydroksymaślan i Acetooctam wzajemnie w siebie przechodzą
(w mitochondriach wielu tkanek, w tym w wątrobie)
Dehydrogenaza
3-hydroksymaślanowa (NAD)
β-hydroksymaślan < ----------------------------> acetooctan
Równowaga tej reakcji kontrolowana jest stosunkiem NADH do NAD (tak zwanym stanem redoksowym)
SYNTEZA
1) Dwa acetylo-CoA powstałe w beta oksydacji kondensują ze sobą, dając ACETOACETYLO-CoA
(taki proces jest odwróceniem reakcji katalizowanej przez tiolazę)
TIOLAZA
Acetylo-CoA + Acetylo-CoA <------------------- > AcetoAcetylo-CoA (acetooctan)
1) Kolejna cząsteczka Acetylo-CoA kondensuje z acetoacetylo-CoA dając HMG- CoA
Reakcja ta jest katalizowana przez mitochondrialną syntezę HMG-CoA.
Odszczepianie Acetylo-CoA od HMG-CoA jest z kolei możliwe dzięki działaniu mitochondrialnej liazy
HMG-CoA
HMG-CoA = 3 hydroksy 3 metylo glutarylo-CoA
SYNTAZA
AcetoAcetylo-CoA + Acetylo-CoA --------------------- > HMG-CoA
Acetooctan+acetylo-CoA
<----------------------
LIAZA
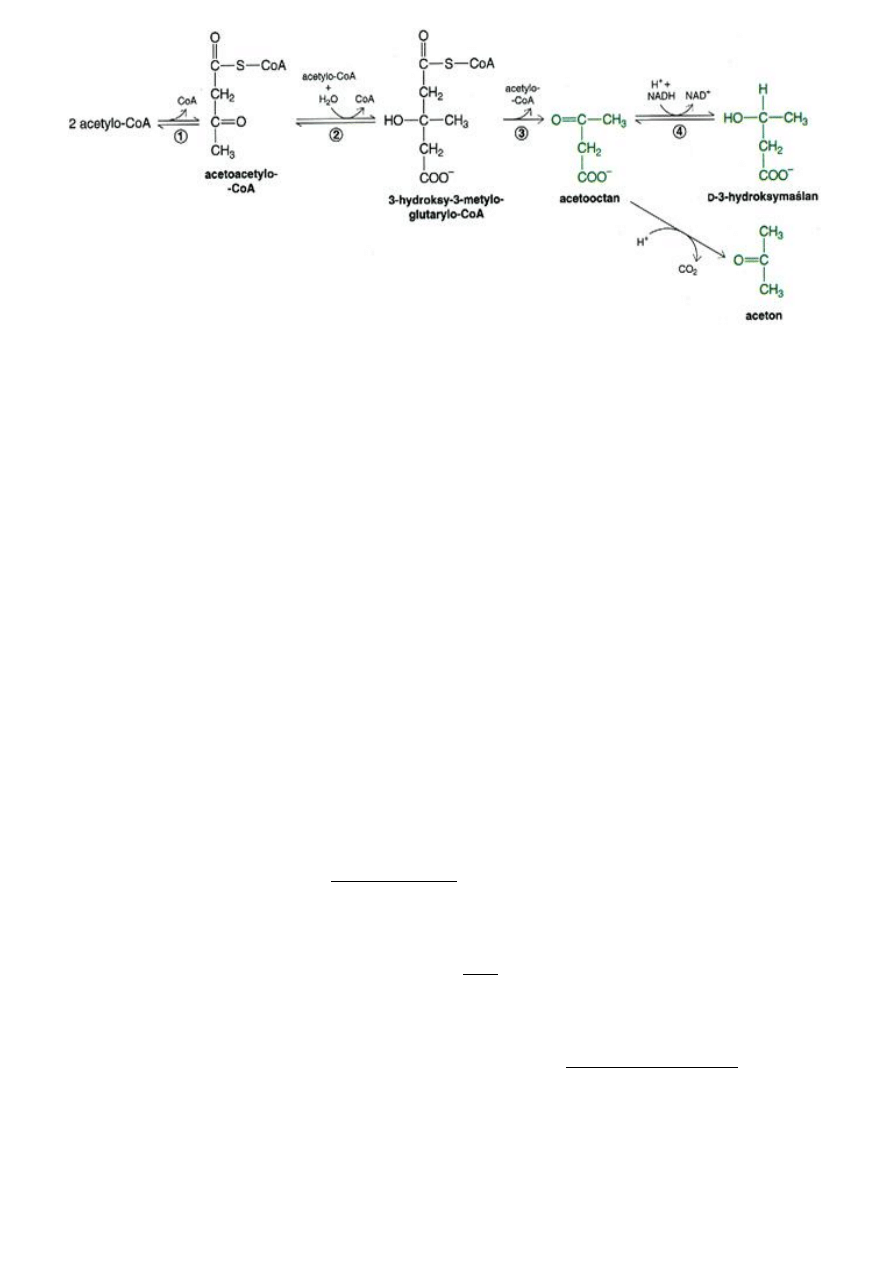
grupa7.med@gmail.com | 2008 - wdanie II, poprawione
46
1) Tiolaza
2) Syntaza HMG-CoA
3) Liaza HMG-CoA
4) Dehydrogenaza 3-hydroksymaślanowa
UTYLIZACJA ACETOOCTANU W TKANKACH POZAWĄTROBOWYCH
1) Acetooctan reaguje z skucynylo-CoA i CoA zostaje przeniesiony z wytworzeniem acetoacetylo-CoA i
pozostawieniem wolnego bursztynianu. Acetoacetylo-CoA wytworzony w tej reakcji jest rozkładan do
acetylo-CoA pod wpływem TIOLAZY i utleniony w cyklu Krebsa.
CoA-transferaza β ketokwasowa
Acetooctan + bursztynylo-CoA ---------------------------------------------- > AcetoAcetylo-CoA + bursztynian
2) β-hydroksymaślan jest utleniany przez dehydrogenazę β-hydroksymaślanową do acetooctanu; ten jest
rozkładany przez tiokinazę do 2 cząsteczek acetylo-S-CoA
Ketnemia jest spowodowana raczej zwiększonym wytwarzaniem ciał ketonowych w wątrobie a nie ich
zmniejszoną utylizacją na obwodzie.
a) MIĘSIEO SERCOWY I KORA NERKI – wykorzystują acetylooctan JAKO ŹRÓDŁO ENERGII
preferencyjnie nawet w stosunku do glukozy.
b) glukoza jest głównym związkiem odżywczym dla tkanki mózgowej i erytrocytów
REGULACJA KETOGENEZY
1) Pierwotna regulacja zależy od tkanki tłuszczowej i od tego ile FFA uwolnią do krwi z lipolizy TG
(FFA ---------> Acetylo-CoA -------- > ciała ketonowe)
Dlatego czynniki regulujące mobilizację FFA z tkanki tłuszczowej są ważne dla ketogenezy
2) Pierwsza przemiana FFA następuje pod wpływem CPT-I która reguluje wnikanie długołaocuchowych
kwasów.
- CPT I jest MAŁO AKTYWNA w stanie sytości (a aktywna w stanie głodu)
- silnym inhibitorem CPT I jest Malonylo-CoA (związek pośredni w biosyntezie
kw. Tłuszczowych, powstaje w wyniku działania karboksylazy Acetylo-CoA)
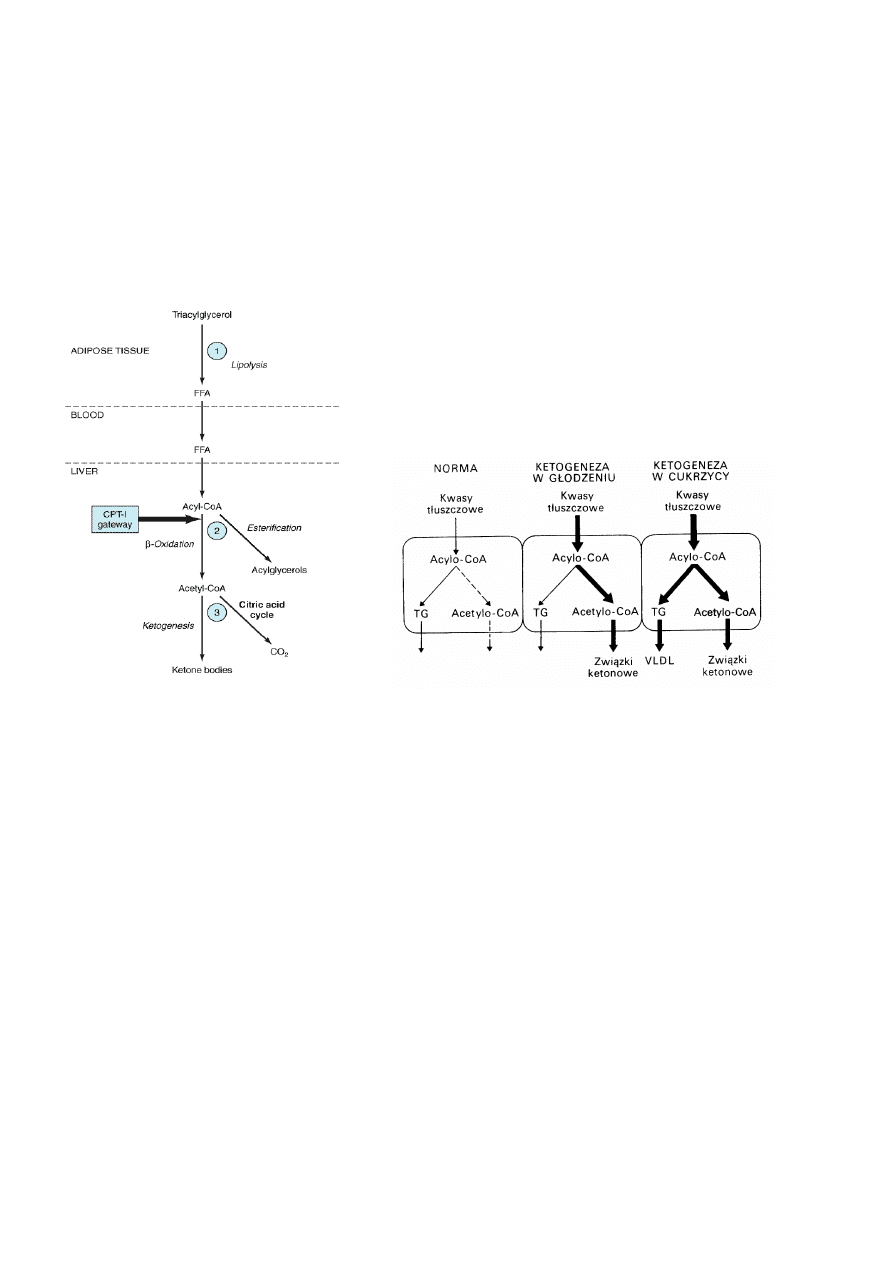
grupa7.med@gmail.com | 2008 - wdanie II, poprawione
47
w stanie sytości większośd FFA (uwolnionych w niewielkiej ilości przez tk. Tłuszczową) ulega przekształceniu do
acetylo-CoA i następnie estryfikacji do acylogliceroli i transportowane z wątroby w postaci VLDL
w stanie głodu tkanka tłuszczowa uwalnia dużo FFA, dużo acylo-CoA hamuje bezpośrednio karboksylaze
Acetylo-CoA i nie powstaje inhibitor CPT I ( malonylo-CoA ).
Dodatkowo w stanie głodu rośnie glukagon, który powoduje fosforylację karboksylazy acetylo-CoA a tym samym
ją hamuje.
3) acetylo-CoA powstały w beta oksydacji może wejśd do cyklu Krebsa lub w szlak ketogenezy
- regulacja ilością FFA (im więcej tym większa ketogeneza) i bilansem
energetycznym komórki. Ketogeneza jest takim mechanizmem, który
ozwala wątrobie utlenid zwiększające się ilości kwasów tłuszczowych
bez zwiększania całkowitego wydatku energetycznego
PATOLOGICZNE DZIAŁANIE CIAŁ KETONOWYCH
1) acetooctan jak i hydroksymaślan są dośd mocnymi kwasami; gromadząc się w surowicy wywołują
kwasicę metaboliczną
2) Wydalając się z moczem w postaci anionowej wiążą się z Na+ i K+ i tym samym zubożają organizm w sód
i potas.
3) Kwasica metaboliczna prowadzi do zaburzeo w OUN, czego efektem jest śpiączka cukrzycowa tzw.
śpiączka ketonowa
grupa7.med@gmail.com | 2008 - wdanie II, poprawione
48
48. CYKL RANDLE’A
Opisuje on powiązania wewnątrzkomórkowego metabolizmu kwasów tłuszczowych i glukozy w komórkach
mięśniowych. Często zachodzi przy cukrzycy.
Mechanizm działania:
Z powodu insulinooporności nie dochodzi do zahamowania działania enzymu HSL. Dochodzi do uwalniania dużej
ilości WKt, które płyną do mięśni. Tam dochodzi do β-oksydacji, w wyniku której powstaje wiele acetylo-CoA i
NADH.
Poza powyższymi działaniami kwasy tłuszczowe po wejściu do komórki jako acylo-CoA:
Acylo-CoA
Kinaza białkowa C
aktywacja
Białka IRS
fosforylacja
Kinaza serynowo-treoninowa
aktywacja
Zmniejszenie ekspresji GLUT4
Acetylo-CoA i NADH
Dehydrogenaza pirogronianowa
(odpowiedzialna za
dekarboksylację oksydacyjną, czyli
pirogronian→acetylo-CoA)
Wzrost st. produktów cyklu Krebsa-
szczególnie cytrynianu
-
fosfofruktokinaza
Gromadzenie się glukozo-6-fosforanu
-
heksokinaza
-
Spowolnienie glikolizy i zmniejszenie
zdolności komórki do gromadzenia
glukozy i gromadzenie się ciał
ketonowych
grupa7.med@gmail.com | 2008 - wdanie II, poprawione
49
49. LIPAZY WEWNĄTRZKOMÓRKOWE- PODZIAŁ, MECHANIZM DZIAŁANIA,
REGULACJA AKTYWNOŚCI
Podział:
1. HSL
2. lipazę diacylogicerolową
3. lipazę monoaacyloglicerolową
HSL- hormone sensitive lipase- lipaza hormonozależna
a. lokalizacja: tkanka tłuszczowa
b. działanie: rozkład TG na diacyloglicerol i kw tłuszczowy
o glicerol- nie może byd wykorzystywany w tkance tłuszczowej, więc przenika do krwi i Mozę byd
wychwytywany przez takie narządy jak wątroba i nerki
o WKT- w komórce przemieszczają się za pomocą FABP. Mogą byd ponownie przekształcane w
acylo-CoA i reestryfikowane z glicerolo-3-fosforanem, tworząc TG;
Czasem jednak szybkośd reestryfikacji nie jest dostatecznie szybka w stosunku do lipolizy.
Wówczas WKT uwalniane są do osocza, gdzie wiążą się z albuminą przyczyniają się do wzrostu
wolnych kwasów tłuszczowych w osoczu.
c. Regulacja aktywności za pomocą cAMP
o Aktywacja- wzrost cAMP co prowadzi do aktywacji kinazy białkowej A, która fosforyzuje HSL
Hormony: adrenalina, aminy katecholowe, glukoagon
o Obniżenie aktywności- obniżenie cAMP
Hormon: insulina
lipaza diglicerolowa
a. Mechanizm działania: z diacyloglicerolu uwalnia monoacyloglicerol i WKT
lipaza monoglicerolowa
b. Mechanizm działania: z monoacyloglicerolu uwalnia glicerol i WKT
Lipaza mono- i diacyloglicerolowa nie podlegają regulacji.
grupa7.med@gmail.com | 2008 - wdanie II, poprawione
50
50. WPŁYW HORMONÓW NA METABOLIZM LIPIDÓW
Lipolizę hamują:
Insulina:
- aktywacja fosfodiesterazy cAMP,
- hamowanie cyklazy adenylanowej,
- hamowanie HSL(przez ↓cAMP),
- pobudzanie fosfatazy HSL(inaktywacja),
- wzrost syntezy GLUT,
- aktywuje dehydrogenazę pirogronianową, karboksylazę acetylo-CoA,
reduktazę HMG-CoA, acylotransferazę glicerolo-3-fosforanową, LPL;
kwas nikotynowy, PGE
1
Lipolizę pobudzają (↑cAMP):
Glukagon:
- akt. cyklozę adenylanową→pobudza HSL
adrenalina i noradrenalina: (receptorβ1)
- utrudniają wiązanie insuliny z receptorem w błonie adipocytów;
ACTH:
- aktywacja cyklazy guanylanowej,
- stymuluje syntezę i transport receptora apoB/E ułatwiając pobieranie LDL z osocza przez korę
naderczy,
-aktywuje esterazę cholesterolową,
-pobudza syntezę kortyzolu;
MSH,
GH,
TSH,
Wazopresyna,
Hormony tarczycy,
b-LPH
Metyloksantyny
-pobudzają lipolizę przez zahamowanie fosfodiesterazy, co skutkuje wzrostem cAMP
Glikokortykoidy
-w obrębie twarzy, karku, górnej połowy ciała lipogenetyczne za pośrednictwem insuliny, gdzie
indziej katabolicznie(przez wzrost syntezy HSL- niezależnie od cAMP)
Mineralokortykoidy
- j. w. ,ale słabiej
Androgeny
- b. niekorzystny ↑LDL, ↓HDL
Estrogeny
-korzystny ↓LDL, ↑HDL
Gestageny
-niekorzystny-↑VLDL
CRH
-działanie podobne do kortyzonu
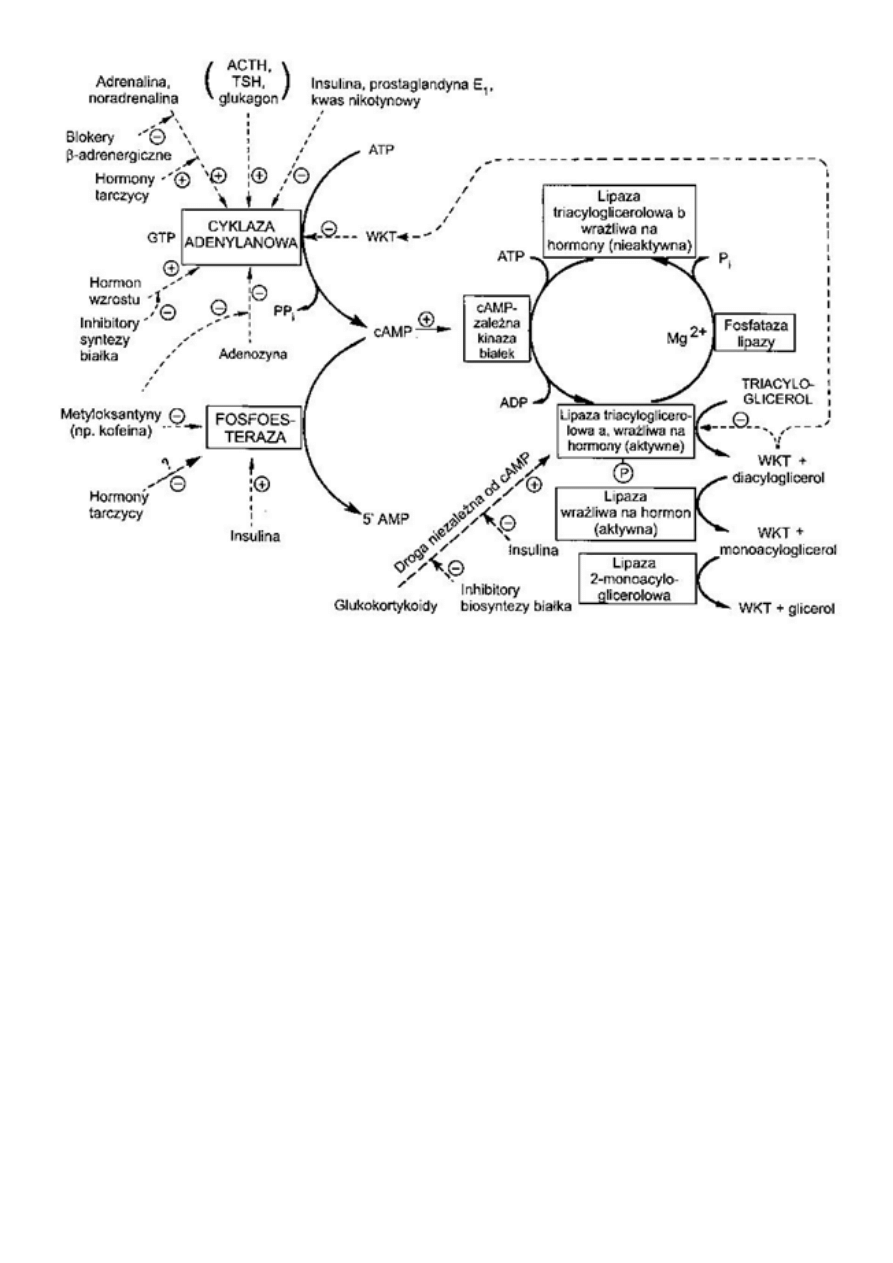
grupa7.med@gmail.com | 2008 - wdanie II, poprawione
51
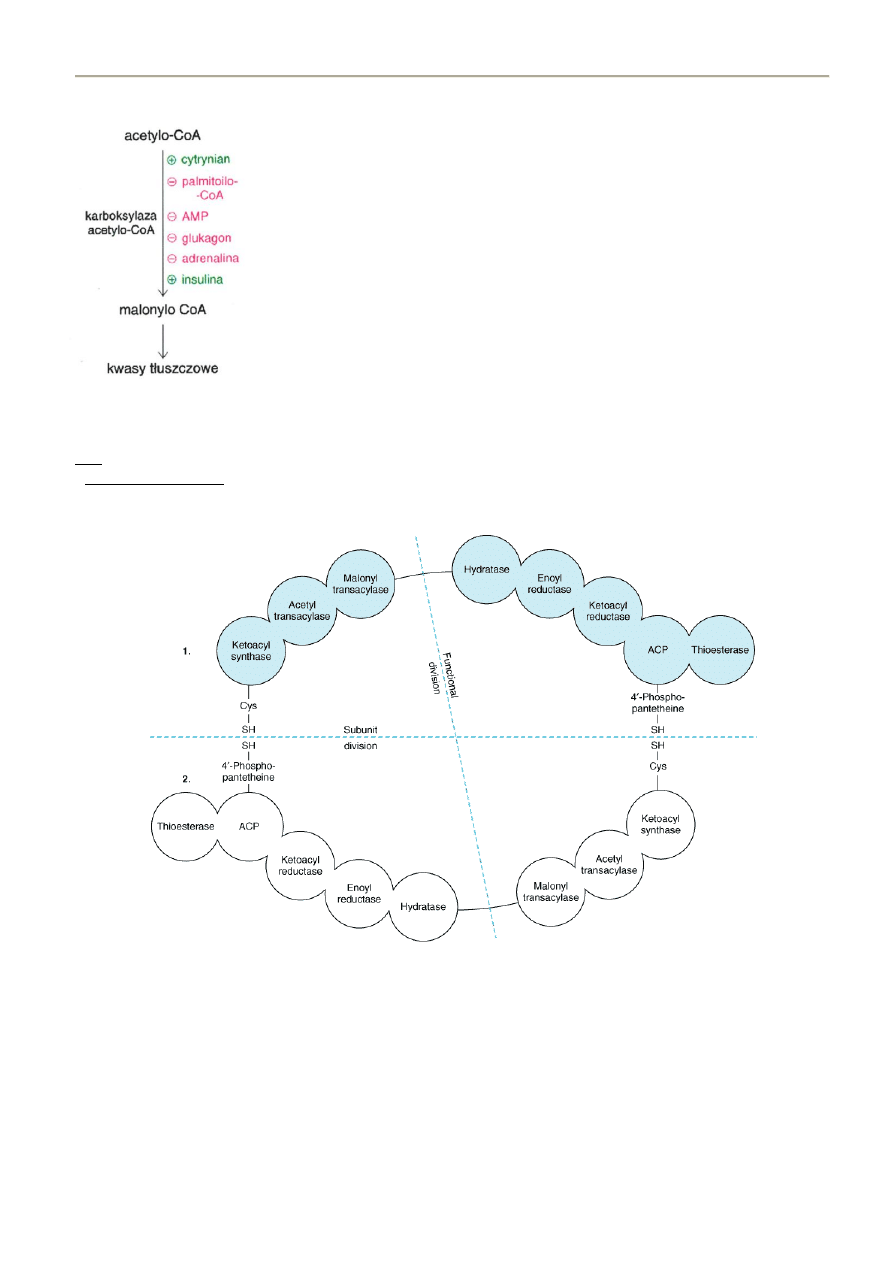
grupa7.med@gmail.com | 2008 - wdanie II, poprawione
52
51. Synteza i wydłużanie kwasów tłuszczowych
1) Początkowym etapem syntezy kwasu tłuszczowego jest wytworzenie
malonylo-CoA z acetylo-CoA przy udziale karboksylazy acetylo-CoA. Donorem CO
2
jest HCO
3
-
, dodatkowo do działania karboksylaza wymaga obecności biotyny (
wit. H)
2) Malonylo-CoA jest dalej przekształcany przez „Kompleks Syntazy Kwasu
Tłuszczowego”
Kompleks Syntazy jest dimerem, każda podjednostka posiada 7 aktywności
enzymatycznych w tym ACP. U niższych organizmów ACP = białko przenoszące acyl
pomiędzy podjednostkami całego kompleksu.
U człowieka stanowi „miejsce przyczepu” dla malonylo-CoA).
APC zawiera witaminę – kwas pantotenowy w formie fosfopanteteiny
APC jednej podjednostki łączy się przy pomocy wiązania FOSFOPANTETEINA-SH HS-cysteina
z syntazą ketoacylową drugiej podjednostki (na zasadzie „głowa do ogona”)
a) Na początku inicjująca cząsteczka acetylo-CoA łączy się z grupą SH-cysteiny co jest katalizowane
przez transacylazę acetylową
b) Malonylo-CoA łączy się z sąsiadującą grupą SH- fosfopanteteiny drugiego monomeru
Co jest katalizowane prez transacylazę malonylową i wytwarza się
ACETYLO(ACYLO)MALONYLOENZYM
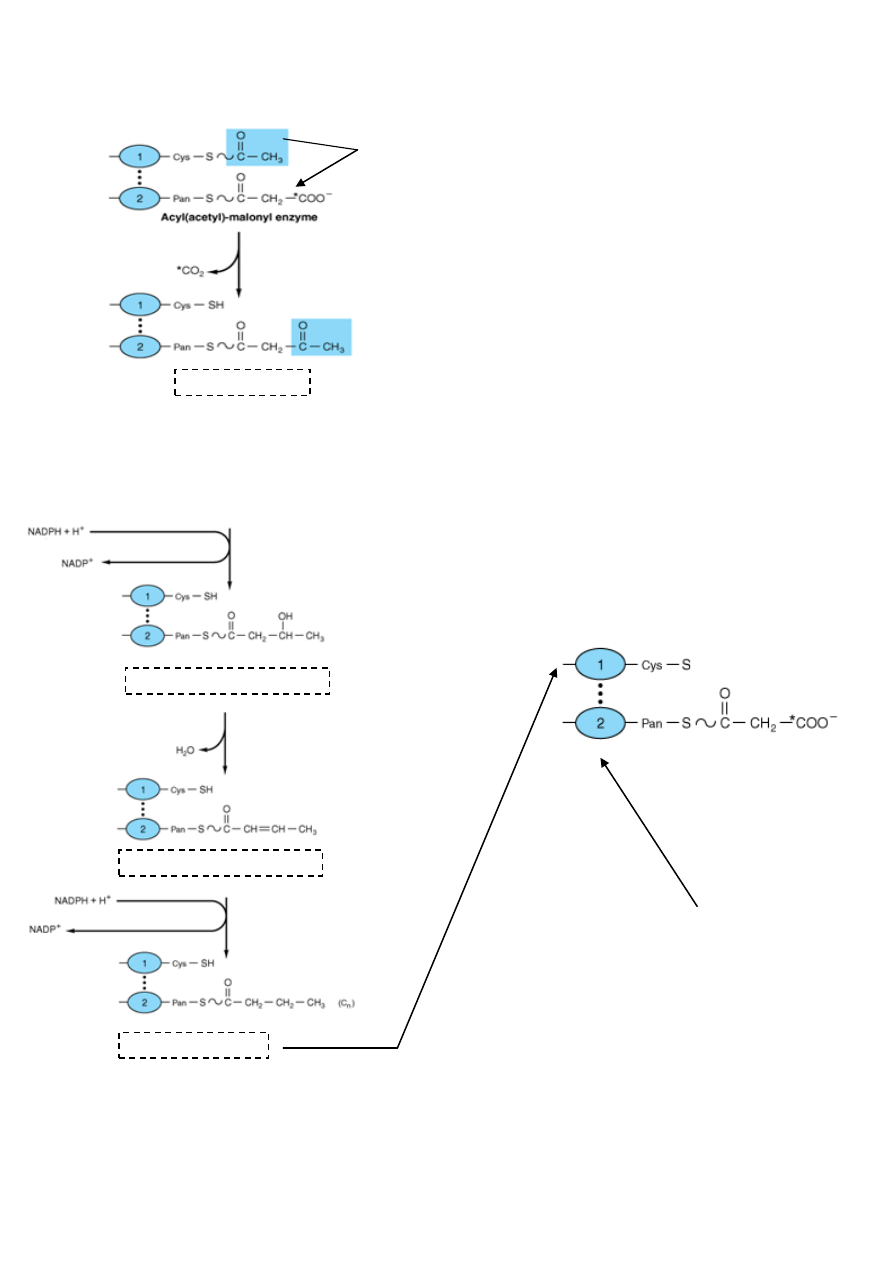
grupa7.med@gmail.com | 2008 - wdanie II, poprawione
53
c) Grupa acetylowa (zaznaczona w kwadraciku) atakuje grupę metylenową reszty malonylowej w
reakcji katalizowanej przez syntezę 3-ketoacylową z uwolnieniem CO
2.
Syntaza β-ketoacylowa
Acetoacetylo-ACP
d) To uwalnia grupę SH cysteiny. Powstała grupa Ketoacylowa zostaje następnie zredukowana,
odwodniona i ponownie zredukowana, a następnie przeniesiona na wolne miejsce, które
zostało po Acetylo-CoA (w miejsce nr 1)
Reduktaza 3-ketoacylowa
Hydroksy BUTYRYLO ACP
Dehydrataza
KROTONOILO ACP
Reduktaza enoilowa
Przyłączenie kolejnego malonylo-CoA
BUTYRYLO ACP
Przyłączenie nasyconego acylu zamiast Acetylo-CoA
e) Powyższa sekwencja powtarza się 6 razy, za każdym włącza się nowa cząsteczka malonylo-CoA
aż do powstania 16 węglowego rodnika (palmitynowego)
f) Powstały 16 węglowy rodnik jest uwalniany z udziałem tioesterazy (też jest w kompleksie)

grupa7.med@gmail.com | 2008 - wdanie II, poprawione
54
52. LIPIDY ZŁOŻONE – PODZIAŁ, STRUKTURA, METABOLIZM, FUNKCJA
1) Lipidy proste
2) Lipidy złożone – estry zawierające dodatkowe grupy funkcyjne
a. Fosfolipidy – zawierają resztę kwasu fosforowego i zasady azotowej lub aminoalkoholu.
1.glicerofosfolipidy – zawierające glicerol np.:
fosfatydyloetanoloamina (kefalina)
fosfatydylocholina (lecytyna)
difosfatydyloglicerol (kardiolipina)
2.sfingolipidy – zawierające sfingozynę
sfingomieliny
glikosfingolipdy, które dalej dzielą się na:
a.
cerebrozydy
b.
gangliozydy
c.
b. Glikolipidy
c. Inne lipidy złożone – sulfolipidy, aminolipidy, lipoproteiny
Struktura:
Estry kwasów tłuszczowych zawierające,oprócz alkoholu i kwasów tłuszczowych jeszcze grupy dodatkowe.
Funkcja:
o Fosfolipidy – składnik błon komórkowych i subkomórkowych
o Kwas fosfatydowy – główny związek pośredni w syntezie triacylogliceroli i fosfoglicerydów
o Lecytyna - rezerwuar choliny
o Dipalmitoilolecytyna – składnik surfaktantu
o Fosfatydyloinozytol - prekursor wtórnych przekaźników
o Glikolipidy - glikokaliks
o Gangliozydy - funkcja receptorowa
Metabolizm:
Glicerolofosfolipidy rozkładane przez fosfolipazy
sfingomielinaza
sfingomielina ------------------ >fosfatydylocholina+ceramid
ceramidaza
ceramid ----------------> sfingozyna+kw. Tłuszczowy
53. BIOSYNTEZA SFINGOZYNY

grupa7.med@gmail.com | 2008 - wdanie II, poprawione
55
54. CEREBROZYDY, SULFATYDY, GANGLIOZYDY – STRUKTURA,
KONSEKWENCJE ZABURZEŃ ICH DEGRADACJI
SFINGOLIPIDY z
budowane są z:
- sfingozyny – aminoalkoholu (nie zawierają glicerolu)
- długołaocuchowego kwasu tłuszczowego
ceramid
- ortofosforanu
- choliny
sfingozyna+kwas tłuszczowy = ceramid
GLIKOSFINGOLIPIDY
W grupie glikosfingolipidów występuje duża różnorodnośd (ponad 150 różnych typów),
z czego 1/3 zaliczana jest do typu ganglio. Najczęściej znajdują się w warstwach powierzchniowych
błon komórkowych i pełnią ważną rolę w interakcjach typu komórka-komórka.
Z tego powodu uważa się iż są ważnymi elementami takich procesów jak wzrost i rozwój komórek, w
tym i komórek nowotworowych oraz bakteryjnych podczas infekcji).
Glikosfingolipidy dzieli się na dwie grupy:
A. Obojętne glikosfingolipidy
Cerebrozydy = ceramid+cukier (glukoza/galaktoza)
Poszczególne cerebrozydy różnią się między sobą rodzajem kwasu tłuszczowego, który w nich występuje.
Powszechniewystępują przede wszystkim w tkance nerwowej mózgowia
Przykładowe cerebrozydy:
cerebron – obecny jest kwas cerebronowy;
nerwon – kwas nerwonowy;
kerazyn – kwas lignocerynowy
.
Najprostszymi cerebrozydami są:
1) glukozyloceramidy – glukozyloceramidy występują powszechnie w błonach tkanek pozanerwowych,
w małych ilościach w CNS
2) galaktozyloceramid – mogą przechodzid w sulfatydy (obecne w mielinie)
Galaktocerebrozyd -> R=H
Sulfatyd - > R=SO
4
grupa7.med@gmail.com | 2008 - wdanie II, poprawione
56
B. Kwaśne glikosfingolipidy
sulfatydy (jedna grupa siarczanowa na jednej reszcie cukrowej).
Gangliozydy, czyli sjalozylo-glikozylo-sfingolipidy, tym różnią
się od cerebrozydów, że zamiast pojedynczej reszty
monocukrowej, zwykle zawierają mniej lub bardziej
rozbudowany oligosacharyd, w którym zawsze występują
reszty kwasów sjalowych, tak jak np. w disjalogangliozydzie
przedstawionym poniżej. Najbardziej typowym kwasem
występującym w gangliozydach jest kwas stearynowy,
natomiast rzadziej występują kwasy nerwonowy i
lignocerynowy. Gangliozydy w znacznych ilościach są
obecne w substancji szarej mózgowia.
najprostszym gangliozydem występującym w tkankach jest G
M3
. Zawiera on ceramid, 1 glukozę, 1 galaktozę i
jedną cząsteczkę kwasu N-acetyloneuramidowego: G oznacza „ganglio” , M – monosialowy, cyfra oznacza
kolejnośd lokowania się na chromatograii
G
M1
– złożony gangliozyd, pochodna G
M3
– może służyd jako receptor dla toksyn cholery w jelicie cienkim:
Biosynteza cerebrozydów i sulfatydów:
UDP – Glukoza
Epimeraza UDP-Gal
UDP – galaktoza
UDP
PAPS (aktywny siarczan)
Ceramid
galaktozyloceramid
sulfatyd
(cerebrozyd)
Biosynteza gangliozydów
CMP-kwas N-acetyloneuraminowy
UDP – glukoza
UDP-galaktoza
( CMP-NeuAc )
UDP
UDP
CMP
Ceramid
glikozyloceramid
Cer – Glc – Gal
Cer-Glc-Gal
(Cer – Glc)
NeuAc
(G
M3
)
UDP-N-Acetylogalaktozamina
(UDP-GalNAc)
UDP
Cer-Glc-Gal-GalNAc-Gal
Cer-Glc-Gal-GalNAc
(G
M1
)
NeuAc
(G
M2
)
NeuAc
UDP-Gal
UDP
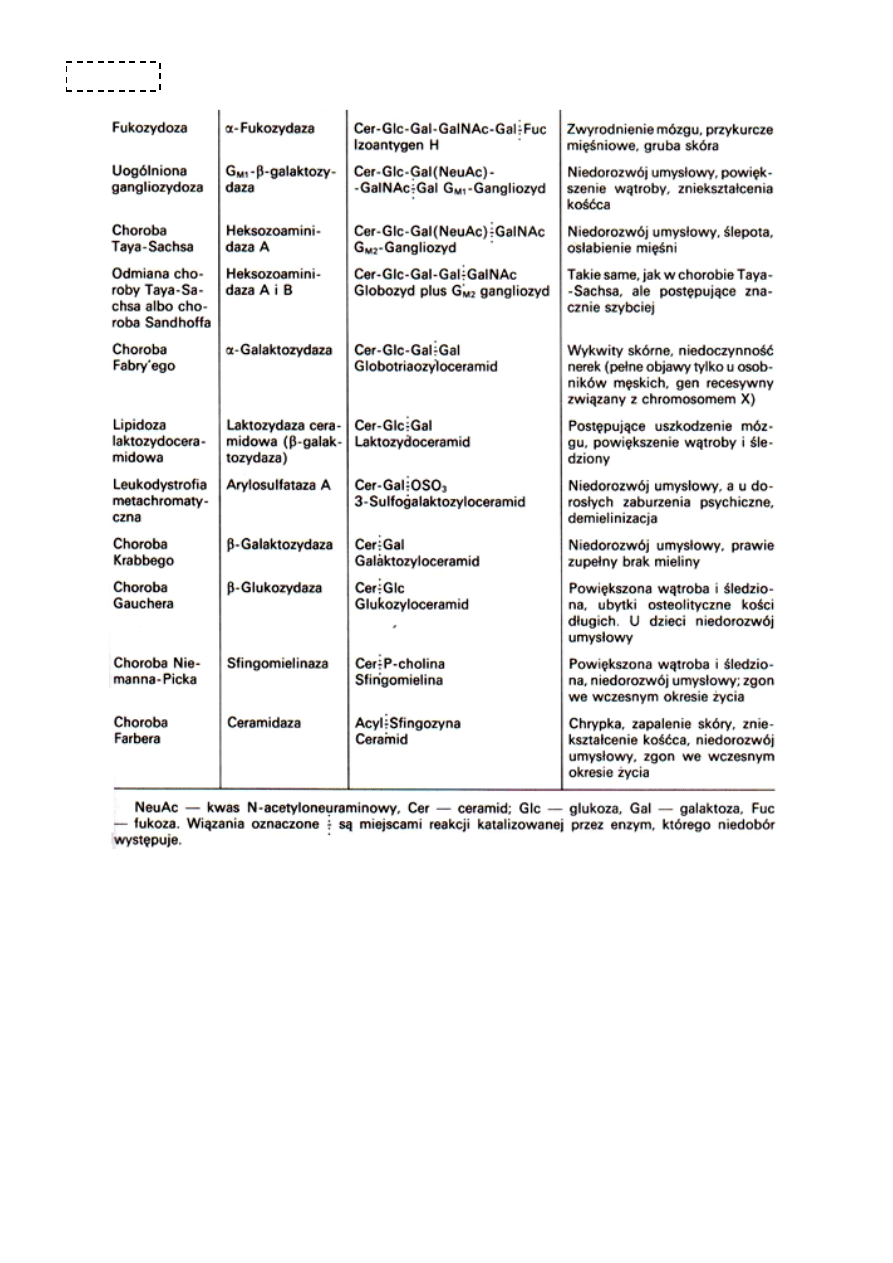
grupa7.med@gmail.com | 2008 - wdanie II, poprawione
57
PATOLOGIE

grupa7.med@gmail.com | 2008 - wdanie II, poprawione
58
55. FOSFOLIPIDY BŁONOWE JAKO DEPOT WTÓRNYCH PRZEKAŹNIKÓW
INFORMACJI
PLC
4,5-difosforan fosfatydyloinozytolu PIP
2
→diacyloglicerol(DAG)+1,4,5-trifosforanoinozytol(IP
3
)
PLD-substratem różne fosfolipidy błonowe-powstaje kwas fosfatydowy
Cykl lipidowy
CTP inozytol
Kinaza DAG ↓ ↓ fosforylacja
DAG→kwas fosfatydowy→CDP-DAG→fosfatydyloinozytol→PIP
2
↓ ↓
PP CMP
Cykl inozytolowy
IP
3
→IP
2
→IP→inozytol
IP
3
może ulec fosforylacji
IP
3
→IP
4
→IP
5
→IP
6
DAG-pozostaje w błonie, nierozpuszczalny w wodzie, aktywuje kinazę białkową C.
IP
3
-rozpuszczalny w wodzie, otwiera ROC
Kwas fosfatydowy, IP
4
-otwierają SMOC
IP
5,
IP
6
-prawdopodobnie funkcje regulatorowe w OUN
56. KWAS ARACHIDONOWY –
STRUKTURA, WYSTĘPOWANIE, ŹRÓDŁA, TORY METABOLICZNE,
INHIBITORY ENZYMÓW PRZEMIAN KWASU ARACHIDONOWEGO O ISTOTNYM ZNACZENIU KLINICZNYM
57. COX
3 izoenzymy:
- COX1
- COX2
- COX3
cyklooksygenaza – inaczej syntaza prostaglandynowa (PGHs):
- przekształca kwas arachidonowy w prostaglandynę PGH
2
(prekursor innych PG)
- aktywnośd:
- cyklooksygenazowa – przekształca kwas arachidonowy w PGG
2
- peroksydazowa – wykorzystuje hem jako grupę prostetyczną, redukuje PGG
2
do PGH
2
1) COX1:
- ekspresja konstytutywna
- aktywny przede wszystkim w układzie pokarmowym, nerkach, płytkach krwi, ścianie naczyniowej
- bierze udział w wytwarzaniu prostanoidów o znaczeniu ochronnym (regulacyjnym)
- ekspresja nie jest hamowana przez glikokortykoidy
2) COX2:
- ekspresja indukowalna w odpowiedzi na cytokiny prozapalne
- pełni istotną rolę w procesach zapalnych
- indukcja hamowana przez glikokortykoidy
- aktywnośd enzymu hamowana selektywnie przez rofekoksyb i celekoksyb
3) COX3:
- jego transkrypt powstaje w wyniku alternatywnego splicingu mRNA COX1
- obecna w mózgu – rola w percepcji bólu
- odwracalnie hamowana przez acetaminofen (paracetamol)
grupa7.med@gmail.com | 2008 - wdanie II, poprawione
59
58. PROSTAGLANDYNY, TROMBOKSANY, PROSTACYKLINA – STRUKTURA,
BIOSYNTEZA, EFEKTY DZIAŁANIA
Eikozanoidy
PODZIAŁ:
- prostaglandyny
- prostacykliny
prostanoidy
- tromboksany
- leukotrieny
Eikozanoidy powstają z kwasów C
20
ikozanowych pochodzących od kwasu linolowego, alfa-linolenowego lub
bezpośrednio od arachidonowego
1) kwas linolowy:
- traci dwa atomy wodoru przechodząc w gamma-linolenian
- zyskuje 2 atomy węgla przechodząc w 8,11,14-ikozatrienoan (dihomo-gamma-linolenian - DHLA) -
wątroba
- prostanoidy które powstają z 8,11,14-ikozatrienoanu (dihomo-gamma-linolenianu): PGE
1
, PGF
1
, TXA
1
- leukotrieny które powstją z 8,11,14-ikozatrienoanu: LTA
3
, LTC
3
, LTD
3
- ikozatrienoan może zostad odwodorowany przechodząc w 5,8,11,14-ikozatetraenoan (arachidonian):
przemiana DHLA w kwas arachidonowy
2) kwas alfa-linolenowy:
- odwodorowanie prowadzi do powstania oktadekatetraenoanu
- oktadekatetraenoan przyłącza 2C przechodząc w ikozatetraenoan, który tracą 2 atomy wodoru
przechodzi w:
- 5,8,11,14,17-ikozapentaenoan (który znajduje się też w pokarmie)
- 5,8,11,14,17-ikozapenatenoan jest surowcem do syntezy:
prostanoidów: PGD
3
, PGE
3
, PGF
3
, PGI
3
, TXA
3
leukotrienów: LTA
5
, LTB
5
, LTC
5
Wpływ na syntezę:
1) Wpływ na szlak wytwarzania aktywnych pochodnych kwasu AA mają:
a) glikokortykoidy – hamują fosfolipazę A
2
i indukcję COX2
b) NSAID – niesterydowe leki przeciwzapalne – ibuprofen, aspiryna – hamowanie COX1 i COX2
c) pochodne imidazolu – hamowanie syntazy tromboksanowej
d) kwas hydroperoksyeikozatetraenowy i inne nadtlenkowe pochodne kwasów tłuszczowych hamują
syntazę prostacyklinową
2) Fosfolipaza A
2
:
a) hamowana przez:
- glikokortykoidy – indukują lipokortynę, białkowy inhibitor
b) aktywnośd pobudzają:
- angiotensyna II
- bradykinina
- adrenalina
- trombina

grupa7.med@gmail.com | 2008 - wdanie II, poprawione
60
PROSTAGLANDYNY
1) struktura:
- kwasy tłuszczowe zbudowane z 20 atomów węgla
- zawierają w swej strukturze pierścieo cyklopentanowy
- pochodne kwasu prostanowego zawierającego cyklopentanowy pierścieo i dwa łaocuchy boczne: α i ω
- łaocuch α zakooczony grupą karboksylową od której rozpoczynamy liczenie atomów węgla
- łaocuch ω zakooczony grupą metylową
- wszystkie naturalnie występujące prostaglandyny mają w łaocuchu ω podwójne wiązanie między C13 i
C14 o konfiguracji trans i grupę hydroksylową w pozycji C15
różnice w budowie pierścienia cyklopentanowego między poszczególnymi klasami prostaglandyn:
- PGA – pierścieo zawiera jedno wiązanie podwójne (C10-C11), grupa ketonowa przy C9
- PGE – pierścieo nasycony, grupa ketonowa przy C9, hydroksylowa przy C11
- PGF – 2 grupy hydroksylowe: przy C9 i C11
2) SYNTEZA:
a) pierwszy etap polega na działaniu na kw arachidonowy 11-lipoksygenazy: powstaje 11-HPETE
(kwa 11-hydroperoksy eikozatetraenowy).
Kolejne metabolity powstają przy udziale syntazy prostaglandyny H (PGHs) –izoenzymy:
- PGHS-1 (inaczej COX1)
- PGHS-2 (inaczej COX2)
- oba izoenzymy PGHS mają aktywnośd cyklooksygenazy jak i peroksydazy
b) aktywnośd cyklooksygenazowa PGHS przekształca 11-HPETE w PGG
2
, które przez aktywnośd
peroksydazy przekształcany jest do prostaglandyny PGH
2
c) prostaglandyna PGH
2
(endoperoksyd) jest wyjściowym substratem do produkcji innych prostaglandyn oraz
tromboksanów
d) izomeryzacja PGH
2
prowadzi do powstania:
- PGD
2
- PGE
2
e) syntaza tromboksanowa (hamowana przez imidazol) przekształca PGH
2
w TXA
2
f) redukcja prostaglandyny PGH
2
oraz PGE
2
prowadzi do powstania:
- PGF
2alfa
g) syntaza prostacyklinowa przekształca PGH
2
w PGI
2
h) COX hamowana przez:
- aspirynę – w mechanizmie acetylacji enzymu
- inne leki NSAID: indometacyna, ibuprofen hamują PGHS przez kompetycję z arachidonianami
i) transkrypcja COX-2 ale nie COX-1 jest całkowicie inhibowana przeciwzapalnym działaniem kortykosterydów!!
synteza różnych klas prostaglandyn jest zróżnicowana narządowo:
- nerka i śledziona: PGE
2
i PGF
2α
- ściana naczyniowa: głównie PGI
2
- serce: jednakowe ilości PGE
2
, PGF
2α
i PGI
2
3) Mechanizm działania:
a) działają poprzez zwiększnie poziomu cAMP (zwykle), obniżają go w kanalikach nerkowych i tkance tłuszczowej
b) naturalne mediatory procesu zapalnego
- zaczerwienienie i wzrost temperatury:
- rozszerzenie małych naczyo krwionośnych
- wzrost przepuszczalności naczyo włosowatych
c) działanie kurczące na mięśnie gładkie macicy – wykorzystywane przy stymulacji akcji porodowej
d) hamowanie wydzielania soku żołądkowego – zmniejszenie produkcji HCl – przyspieszają gojenie się wrzodów
trawiennych
e) PGE
2
pobudza agregację płytek krwi
f) zatrzymanie Na
+
i wody
g) hamowanie lipolizy

grupa7.med@gmail.com | 2008 - wdanie II, poprawione
61
4) miejsca działania:
a) żołądek
- zwiększone wydzielanie śluzu (dlatego przy stosowaniu aspiryny
należy pamiętad że powoduje zmniejszenie wydzielania śluzu. Stąd wrzody)
- zwiększony przepływ krwi w błonie śluzowej
b) trzustka - wpływ na wydzielanie
c) wpływ na napięcie ścian naczyo
d) wpływ na płytki krwi
f) regulacja cyklu menstruacyjnego
g) neurotransmisja
5) Prostaglandyny powodują:
a) podwyższenie stężenia cAMP w:
- płytkach krwi
- tarczycy
- ciałku żółtym
- kościach płodu
- części gruczołowej przysadki
- płucach
b) obniżenie stężenia cAMP w:
- komórkach kanalików nerkowych
- tkance tłuszczowej
PROSTACYKLINA
= PGI
2
1) syntezowana w komórkach śródbłonka
2) działanie:
- wazorelaksacja
- antyadhezja
- antyagregacja
3) działa poprzez zwiększenie stężenia cAMP w komórce (mięśniowej), synergistycznie do NO
4) synteza katalizowana przez syntazę prostacyklinową:
- rozerwanie układu nadtlenkowego i utworzenie pierścienia furanowego
5) czas półtrwania – 3 minuty, potem przekształcana do 6-keto-PGF
1
, która jest niaktywna biologicznie
6) struktura:
- nasycony pierścieo cyklopentanowy
- dodatkowy pierścieo utworzony przez połączenie tlenu grupy karbonylowej przy C9 z węglem C6
- wiązanie podwójne C5-C6
7) działanie antagonistyczne do tromboksanu TXA
2
TROMBOKSAN
a) syntezowany z prostaglandyny PGH
2
w płytkach krwi - syntaza tromboksanowa
2PGH
2
= TXA
2
+ dialdehyd malonowy + związek siedemnastowęglowy
b) działanie przeciwstawne do prostacykliny i NO:
- wazokonstrykcja
- wzrost agregacji i adhezji płytek krwi
d) budowa nieprostaglandynowa (pierścieo oksonowy)
g) stosowanie aspiryny w niewielkich dawkach (40-70 mg/dobę) hamuje cyklooksygenazę płytkową zmniejszając
wytwarzanie tromboksanu, co wywiera pozytywny efekt na ścianę naczyniową i drożnośd naczyo
h) kwas 20:5, omega3 (ikozapentaenowy) jest wyjściowym związkiem do syntezy TX
3
i PG
3
, które hamują
uwalnianie kwasu arachidonowego i jego wykorzystanie do syntezy PG
2
oraz TX
2
; PGI
3
ma taką samą aktywnośd
jak PGI
2
, TX
3
jest natomiast słabszy pod względem działania od TX
2
; dlatego u osób spożywających duże ilości
olejów rybnych (Eskimosi) przeważa przeciwagregacyjne działanie prostanoidów
i) struktura: pierścieo sześcioczłonowy, w jego skład wchodzi atom tlenu; łaocuch omega ma podwójne wiązanie
C13-C14 i grupę OH w pozycji C15

grupa7.med@gmail.com | 2008 - wdanie II, poprawione
62
59. LEUKOTRIENY – STRUKTURA, BIOSYNTEZA, EFEKTY DZIAŁANIA
1) produkty przekształceo kwasu arachidonowego
2) synteza leukotrienów:
- powstają z kwasu arachidonowego pochodzącego z fosfolipidów błony okołojądrowej
- bodźce (np. alergenowe) powodują aktywację kinaz, która wyzwala kaskadę przemian katalizowaną
przez fosfolipazę Cγ: powstaje DAG i IP
3
;
- te wtórne przekaźniki prowadzą do aktywacji fosfolipazy A
2
, która przemieszcza się w kierunku otoczki
jądrowej, z której uwalnia kwas arachidonowy będący substratem dla 5-lipoksygenaz
- 5-lipoksygenaza katalizuje przemianę kwasu arachidonowego do kwasu5 hydroperoksyeikozatetraenowego
(5-HPETE), który przez peroksydazy lub samoistnie jest redukowany do 5-HETE ( kwasu 5 hydroksy
eikozatetraenowego
- 5-lipoksygenaza działa w kompleksie z białkiem FLAP (5-lipoxygenase activating protein) przy obecności jonów
Ca i ATP; flap jest integralnym białkiem otoczki jądrowej
- leukotrieny powstają z 5-HPETE w reakcji katalizowanej przez syntazę LTA4, która wprowadza wiązanie
epoksydowe do C5 i C6 - powstaje leukotrien LTA4
- LTC
4
powstaje w mastocytach oraz granulocytach zasadochłonnych i kwasochłonnych z LTA
4
przy udziale
transferazy glutationowej
- poza komórką, GGTP przekształca LTC
4
w LTD
4
- dipeptydazy cysteinyloglicynowe przekształcają LTD
4
w LTE
4
- w granulocytach obojętnochłonnych i makrofagach płucnych hydrolaza epoksydowa przekształca LTA
4
w LTB
4
4) leki przeciwleukotrienowe:
- zileuton, genleuton – przekształcanie HPETE w LTA
4
- cinalukast, montelukast, pranlukast, verlukast, zafirlukast – blokada receptorów leukotrienowych
Leukotrieny – funkcje:
a) skurcz mięśni gładkich oskrzeli – ich zwężenie:
- leukotrieny cysteinowe
b) skurcz naczyo krwionośnych
- LTD
4
, LTC
4
c) zwiększone wydzielanie śluzu:
- leukotrieny cysteinowe
d) chemotaksja – naciek zapalny:
- LB
4
, LTD
4
, LTE
4
e) wzrost przepuszczalności tkanek – obrzęk:
- leukotrieny cysteinowe
f) nadwrażliwośd oskrzeli:
- LTD
4
g) promowanie wzrostu komórek szpiku, fibroblastów, komórek nabłonka:
- LTC
4
, LTD
4
h) stmulacja limfocytów do wytwarzania IFN-γ:
- LTD
4

grupa7.med@gmail.com | 2008 - wdanie II, poprawione
63
60. LIPOKSYNY- STRUKTURA, BIOSYNTEZA, EFEKTY DZIAŁANIA
1) powstają przy udziale 12-lipoksygenazy i 15-lipoksygenazy
2) syntezowane przez:
- megakariocyty
- płytki krwi
- granulocyty – wzrost wytwarzania lipoksyn towarzyszy spadkowi syntezy LTB4
- makrofagi
- komórki szpiku kostnego
- komórki mezangium nerkowego
- nabłonek przewodów oddechowych
3) synteza przebiega 2 torami:
- substratem kwas arachidonowy: synteza prowadzi przez 15-HPETE lub 5-HPETE
(hydroperoksypochodne kwasu arachidonowego wytwarzane przez 15-LO i 5-LO), następnie 5,15-
diHPETE, wreszcie kwas 5(6)-epoksytetraenowy przekształcany w LXA4 lub LXB4
- substratem substancja z grupy leukotrienów: LTA4, przekształcany do 15-OH-LTA4 lub kwasu 5(6)-
epoksytetraenowego (np. płytki krwi syntezują LXA4 z LTA4)
4) metabolizm:
- LXA4 i LXB4 hydroksylowane przy udziale monooksygenazy zależnej od cytochromu P-450 i NADPH
5) mechanizm działania:
- związanie receptorem wpływa na metabolizm lipidów błonowych pobudzając bądź hamując uwalnianie
wtórnych przekaźników lipidowych
- powoduje to zmiany wewnątrzkomórkowego stężenia jonów Ca2+
6) działanie LXA4 i LX4:
- rola regulacyjna w procesie zapalnym
- hamowanie chemotaktycznej odpowiedzi neutrofili indukowanej przez LTB4
- immunosupresja – hamowanie uwalniania LTB4 z neutrofili
- szpik kostny – działanie synergistyczne z GM-CSF
- rozszerzanie naczyo krwionośnych
- wazodilatacja
- kurczące działanie na mięśniówkę przewodów oddechowych – bronchokonstrykcja
- 12-HPETE, 12-HETE oraz LXB4 hamują wydzielanie insuliny
7) rola lipoksyn w patologii:
- spadek syntezy u pacjentów z przewlekłą białaczką szpikową
- rola w patologii układu oddechowego (wpływ bronchokonstrykcyjny)
- wpływ na zjawiska immunologiczne związane z chorobami płuc

grupa7.med@gmail.com | 2008 - wdanie II, poprawione
64
61. LEKI OBNIŻAJĄCE
STĘŻENIE CHOLESTEROLU
1) INHIBITORY REDUKTAZY HMG- COA
Np. mewastatyna, lowastatyna
Zostaje zatrzymana synteza:
- ubichinonu
- cholesterolu
- dolichinonu
- geranylowanych i farnezylowanych białek
Inne funkcje:
- hamowanie proliferacji miocytów gładkich
- hamowanie nacieku makrofagów
- hamowanie ekspresji czynnika tkankowego
-hamowanie adhezji płytek
- hamowanie procesów wolnorodnikowych
- obniżenie stężenia Lp(a)
- modyfikacja reaktywności miocytów gładkich
- stabilizacja blaszki miażdżycowej.
2) ŻYWICE JONOWYMIENNE (Np. kolescypol, Cholestyramina)
Wiążą kwasy żółciowe, wiec nie mogą się wchłaniad. Komórki rozpoczynają syntezę endogenną
cholesterolu i następuje wyłapywanie cholesterolu z krwioobiegu (LDL i VLDL)
Zablokowanie reabsorbcji kwasów żółciowych wiec przemiana cholesterolu w kwasy żółciowe zostaje
znacznie zwiększona w wątrobie w dążeniu do utrzymania puli kwasów żółciowych na stałym poziomie.
Zachodzi zwiekszenie liczny receptorów LDL w wątrobie tym samym wzrost wyłapywanie LDL i w
konsekwencji obniżenie stężenia cholesterolu osocza.
3) FIBRATY – działają na PPAR
Funkcja:
- hamowanie syntezy VLDL
- obniżenie stężenia TG
- aktywacja LPL
- hamują CEPT- wzrost HDL ale jest to niekorzystne działanie fibratów ponieważ HDL
nie mogą na drodze pośredniej oddawad cholesterolu..
4)
KWAS NIKOTYNOWY
Działanie:
- hamowanie lipolizy w tkance tłuszczowej
- zmiejsza napływ WKT do wątroby
- hamowanie syntezy VLDL
- obniżenie stężenia TG
- hamują CEPT
5)
PROBUKOL
Wydaje się wzmagad katabolizm LDL drogą szlaków niezależnych od receptorów. Podobno ma
właściwości przeciwutleniacza co może byd ważne w patogenezie miażdżycy.
Pan profesor uważa, że to bzdura :)
6)
SITOSTEROL
-blokuje wchłanianie cholesterolu w przewodzie pokarmowym.
7)
KLOFIBRAT
i
GEMFIBROZIL
- zwrócenie drogi WKT napływających do wątroby ze szlaku estryfikacji na szlaki ich utlenienia
- zmiejszenie wydzielania przez wątrobe VLDL
- stymulują hydrolizę TGdów VLDLi pod wpływem LPL
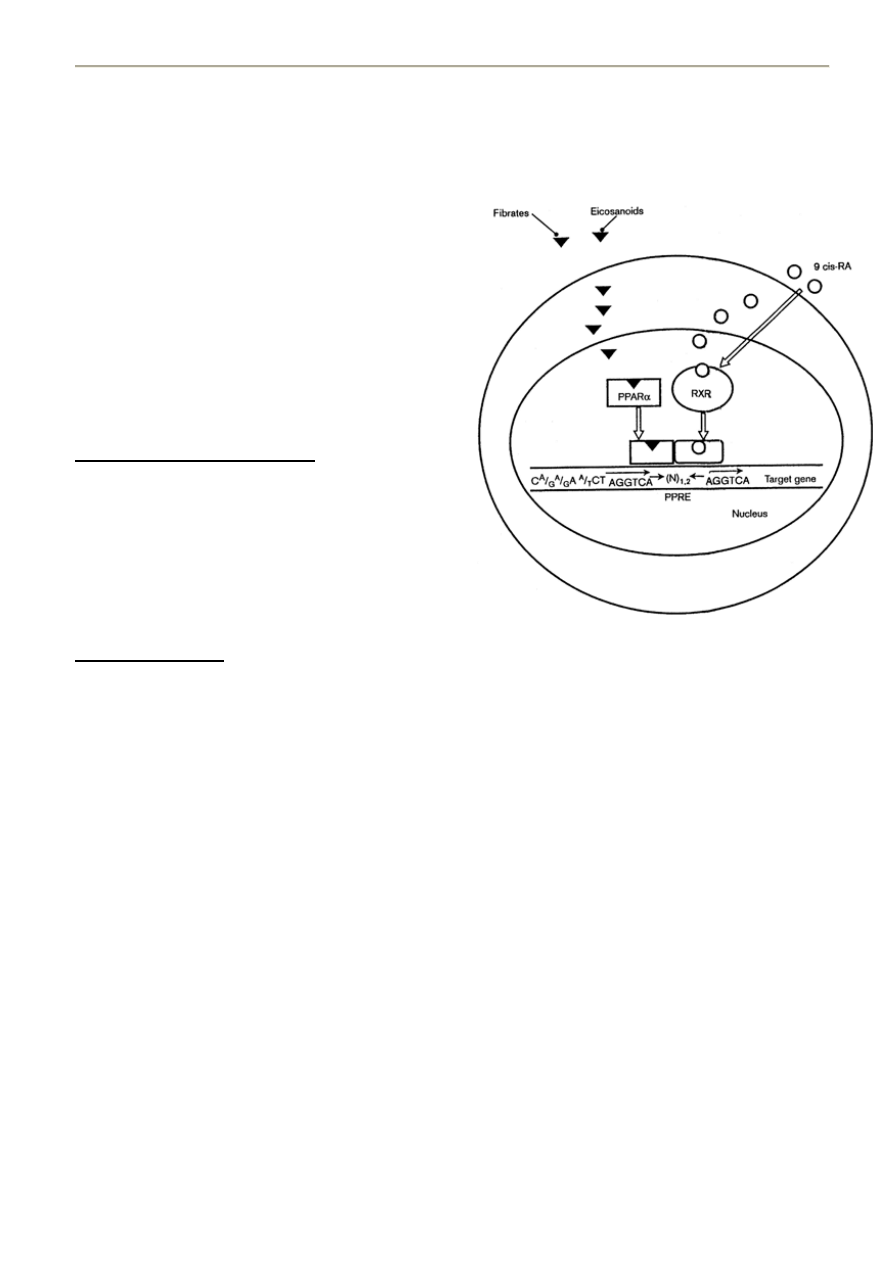
grupa7.med@gmail.com | 2008 - wdanie II, poprawione
65
62. PPAR – STRUKTURA, PODZIAŁ, FUNKCJE
PPAR- proxime proliferactor activated receptors- receptory aktywowane proliferantami peroksysomów
Należą one do rodziny receptorów jądrowych, do których należą także m.in.:
a. Receptor kwasu retinowego
b. Receptor hormonów tarczycy
c. Receptor witaminy D
d. Receptorprostanoidów
e. Receptor glikokortykoidów
f. Receptory sieroce
Zawieraja one charakterystyczne sekwencje AGAACA
i palce cynkowe
PPAR zbudowany jest z 6 modułów (A-F), które tworzą 4
domeny funkcjonalne:
o Domena A, B- aktywowana niezależnie od
ligandu
o Domena C- miejsce wiązania z DNA
o Domena D- bez funkcji
o Domena E, F- wiążąca się z ligandem
Mechanizm działania:
a) działanie z ligandem:
- działanie genowe (genomowe) - transaktywacja: PPAR związany z ligandem dimeryzuje w jądrze z RXR i
jako heterodimer łączy się z PPARre – PPAR response element w sekwencji DNA;
- działanie pozagenowe – transrepresja: PPAR połączony z ligandem hamuje inne czynniki transkrypcyjne
(Nf-κB, STAT) – działanie bez łączenia się PPAR z sekwencją DNA
b) działanie PPAR bez liganda:
- PPAR połączony z RXR (ale nie z ligandem) przyłącza się do PPARre w DNA;
- do tych dwóch czynników przyłącza się następnie kompleks represora wyposażony w enzym –
deacetylazę histonów; deacetylacja histonów prowadzi do przyjęcia przez DNA bardziej upakowanej
struktury i w efekcie hamuje ekspresję genów
- jeśli przyłączy się ligand, wtedy dołącza też kompleks aktywujący związany z acetylazą histonów, która
acetylując histony, stymuluje ekspresję genów

grupa7.med@gmail.com | 2008 - wdanie II, poprawione
66
Podział:
PPAR α (alfa):
a) występowanie:
- hepatocyty
- enterocyty
- kanaliki proksymalne nerki
b) PPAR α w połączeniu z ligandem powoduje:
- wzrost syntezy apoA-I, apoA-II – korzystnie na HDL
- wzrost beta-oksydacji w komórce wątroby
- przeciwzapalne hamowanie COX-2
- działanie pro- i przeciwapoptyczne w zależności od komórki
c) wpływ PPAR α na aterogenezę:
- spadek produkcji VLDL
- wzrost spalania FFA
- wzrost lipolizy
- zwiększenie ilości HDL
- wzrost transportu zwrotnego cholesterolu
- spadek stężenia małych gęstych LDL
- wpływ naczynioochronny – spadek ilości adhezyn śródbłonka
d) ligandami PPAR α są między innymi fibraty
PPAR δ lub β :
a) szeroko rozpowszechniony
b) działanie:
- indukcja syntezy HDL
- dojrzewanie oligodendrocytów
- tworzenie błon
- powstawanie raka jelita grubego
c) antagoniści:
- sulindak
d) agoniści:
- estry etylowe nasyconych i nienasyconych kwasów tłuszczowych
- fibraty (bezafibrat)
e) następstwa aktywacji PPAR δ:
- kardiomiocyt i mięśnie: wzrost transportu FFA oraz ich beta-oksydacji
- tkanka tłuszczowa: wzrost termogenezy
- odtłuszczenie naczyo: wzrost syntezy HDL
- wątroba: spadek glukoneogenezy, nasilenie cyklu pentozowego
- spadek odpowiedzi zapalnej komórek nacieku zapalnego
PPAR γ:
a) 3 podtypy:
1. – szeroko rozpowszechniony
2. – biała tkanka tłuszczowa
3. – makrofagi
b) defekty PPAR γ – jedna z przyczyn insulinooporności
WPŁYW PPAR γ NA TRANSDUKCJĘ SYGNAŁU PO POBUDZENIU RECEPTORA INSULINOWEGO:
a) działając przez szlak obejmujący czynnik MAPK, insulina powoduje:
- wzrost proliferacji miocytów gładkich
- wzrost syntezy PDGF
- wzrost syntezy angiotensyny II Ten szlak jest hamowany przez PPAR γ
- wzrost syntezy PAI-1
- wzrost syntezy endoteliny I
- wzrost ekspresji białek adhezyjnych
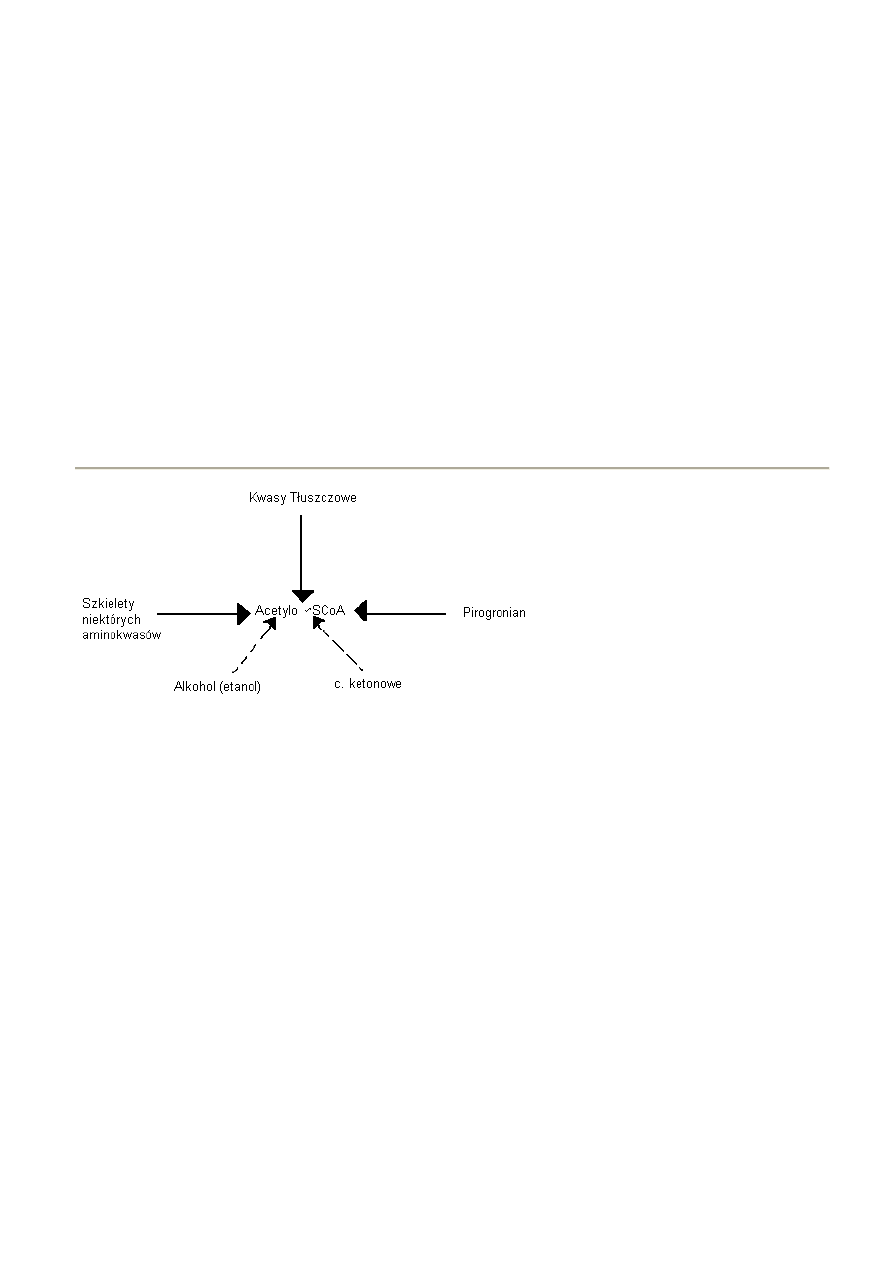
grupa7.med@gmail.com | 2008 - wdanie II, poprawione
67
b) PPAR γ aktywuje szlak, w którym uczestniczy 3-kinaza fosfatydyloinozytolu; jest to szlak korzystny, powoduje:
- wzrost ekspresji genu GLUT 4
- wzrost syntezy tlenku azotu
- spadek aktywności metaloproteaz (stabilizujący wpływ na płytkę miażdżycową)
- wzrost syntezy TIMP – tkankowych inhibitorów MMP
- hamowanie apoptozy np. endotelium
c) agoniści PPAR γ:
- prostaglandyny
- nienasycone kwasy tłuszczowe
- tiazolidenodiony: troglitazon, pioglitazon, parglitazon, ciglitazon, rosiglitazon – leki pozwalające na
przełamanie insulinooporności
63. OMÓWIĆ PODSTAWOWE PROCESY BIOCHEMICZNE, KTÓRYCH
PRODUKTEM JEST ACETYLO-COA JAKO PODSTAWOWY SUBSTRAT DO
TWORZENIA CHOLESTEROLU
Podstawowym zrodlem acetylo-S-CoA jest przemiana glukozy i kw tluszczowych.
Glukoza w procesie glikolizy zachodzacej w cytoplazmie przeksztalca sie do pirogronianu(w warunkach
tlenowych). Pirogronian ulega dekarboksylacji oksydacyjnej przy udziale kompleksu enzymatycznego zwanego
dehydrogenaza pirogronianowa w macierzy mitochondrialnej. Kompleks 3 enzymow wspoldzialajacych z 5
koenzymami prowadzi do wytrworzenia acetylo-S-CoA, ktory moze zostac wykorzystany m.in. do syntezy
cholesterolu.
Kolejnym szlakiem metabolicznym, w przebiegu ktorego powstaje acetylo-S-CoA jest proces B-oksydacji kw.
tluszczowych przebiegajacy w macuerzy mitochondrialnej(kwasy krotko i srednio o dlugolancuchowe) oraz w
peroksysomach(kwasy o bardzo dlugim lancuchu).
Metabolizm szkieletow weglowodorowych niektorych aminokwasow prowadzi do wytworzenia acety-S-CoA.
Rozpad cial ketonowych- acetoctanu i B-hydroksymaslanu w odpowiednich szlakach metabolicznych rowniez
prowadzi do wytworzenia tego zwiazku.
Meratabolizm alkoholu etylowego prowadzi rowniez do wytworzenia acetylo-S-CoA.
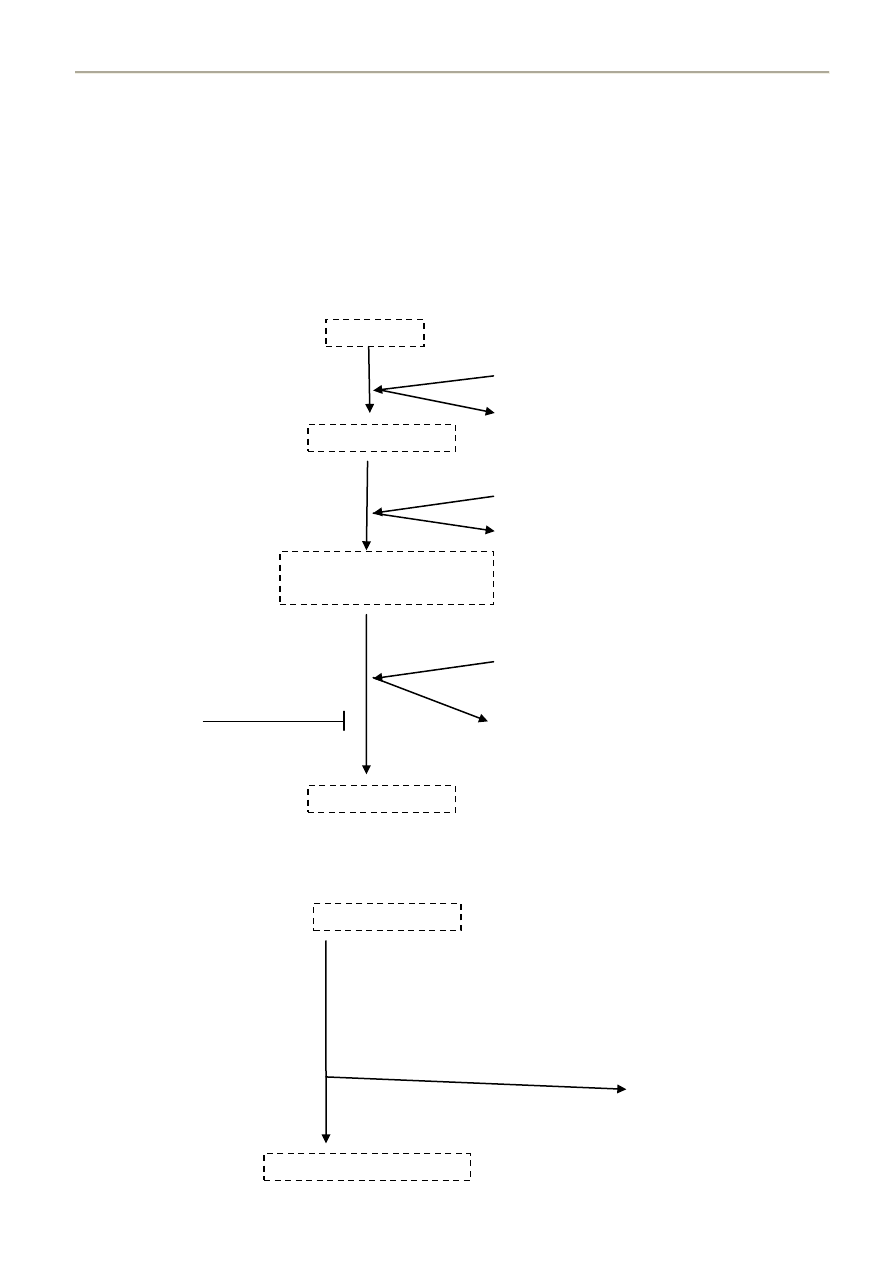
grupa7.med@gmail.com | 2008 - wdanie II, poprawione
68
64-67. Biosynteza cholesterolu, regulacja, gromadzenie w kom
Etapy:
1) Synteza mewalonianu z HMG-CoA (miejsce działania statyn)
2) Dekarboksylacja mewalonianu – powstają jednostki izoprenoidowe (difosforany izopentenylu)
3) Kondensacja 6 jednostek – powstaje skwalen
4) Cyklizacja skwalenu – powstaje macierzysty steroid (lano sterol)
5) Inne reakcje i utrata 3 grup metylowych – powstaje cholesterol
1) ketogeneza w mitochondriach
acetylo-CoA
tiolaza
acetylo-CoA
SH-CoA
acetoacetylo-CoA
syntaza HMG-CoA
acetylo-CoA
izoenzymy: cyto / mitochondrialny
SH-CoA
HMG-CoA
β(OH)- βmetylo glutarylo CoA
MIKROSOMALNA
reduktaza HMG-CoA
2 NADPH + 2H
STATYNY
2 NADP + SH-CoA
Mewalonian
2) powstawanie aktywnych jednostek izoprenoidowych przez fosforylację i dekarboksylację mewalonianu
Mewalonian
Fosfo mewalonian
Kinazy (+ATP )
- mewalonianowa
Difosfo mewalonian
- fosfomewalonianowa
- difosfomewalonianowa
Fosfo difosforan mewalonianu
Dekarboksylaza
Co2 + Pi
difosfomewalonianowa
pirofosforan izopentenylu
grupa7.med@gmail.com | 2008 - wdanie II, poprawione
69
3)
- kondensacja 2 cząstek jednostek izoprenoidowych z wytworzeniem difosforanu geranylu (10C)
- przyłączenie jeszcze 1 cząstki difosforanu izopentenylu - powstaje difosforan farnezylu (15C).
Dwie cząsteczki difosforanu farnezylu kondensują ze sobą dając najpierw difosforan preskwalenu (nie
tetrafosforan bo w międzyczasie odszczepia się jeden difosforan) a później następuje redukcja z udziałem
NADPH i oderwanie pozostałej reszty difosforanowej. Powstaje skwalen
pirofosforan izopentenylu (5C)
izomeraza
pirofosforan dimetyloallilu
Prenylotransferaza
PP
i
pirofosforan geranylu (10C)
pirofosforan izopentenylu
Prenylotransferaza
PP
i
pirofosforan farnezylu (15C)
DOLICHOL etc.
syntetaza skwalenu
NADPH + H
KONDENSACJA 2 CZĄSTECZEK
PIROFOSFORANU FARNEZYLU
NADP
2PP
i
SKWALEN (30C)
4) skwalen ma budowę przypominającą pierścieo stedydowy.
- Przed zamknięciem tego pierścienia skwalen przechodzi pod wpływem epoksydazy skwalenowej
(w siateczce cytoplazmatycznej) w formę 2,3-epoksy
- Cyklizacja zachodzi pod wpływem Lanosterolocyklazy oksydoskwalenowej i powstaje lanosterol
SKWALEN
Epoksydaza skwalenowa
NIEZBĘDNY TLEN MOLEKULARNY
Forma 2,3 epoksy
Lanosterolocyklaza
Oksydoskwalenowa
Lanosterol
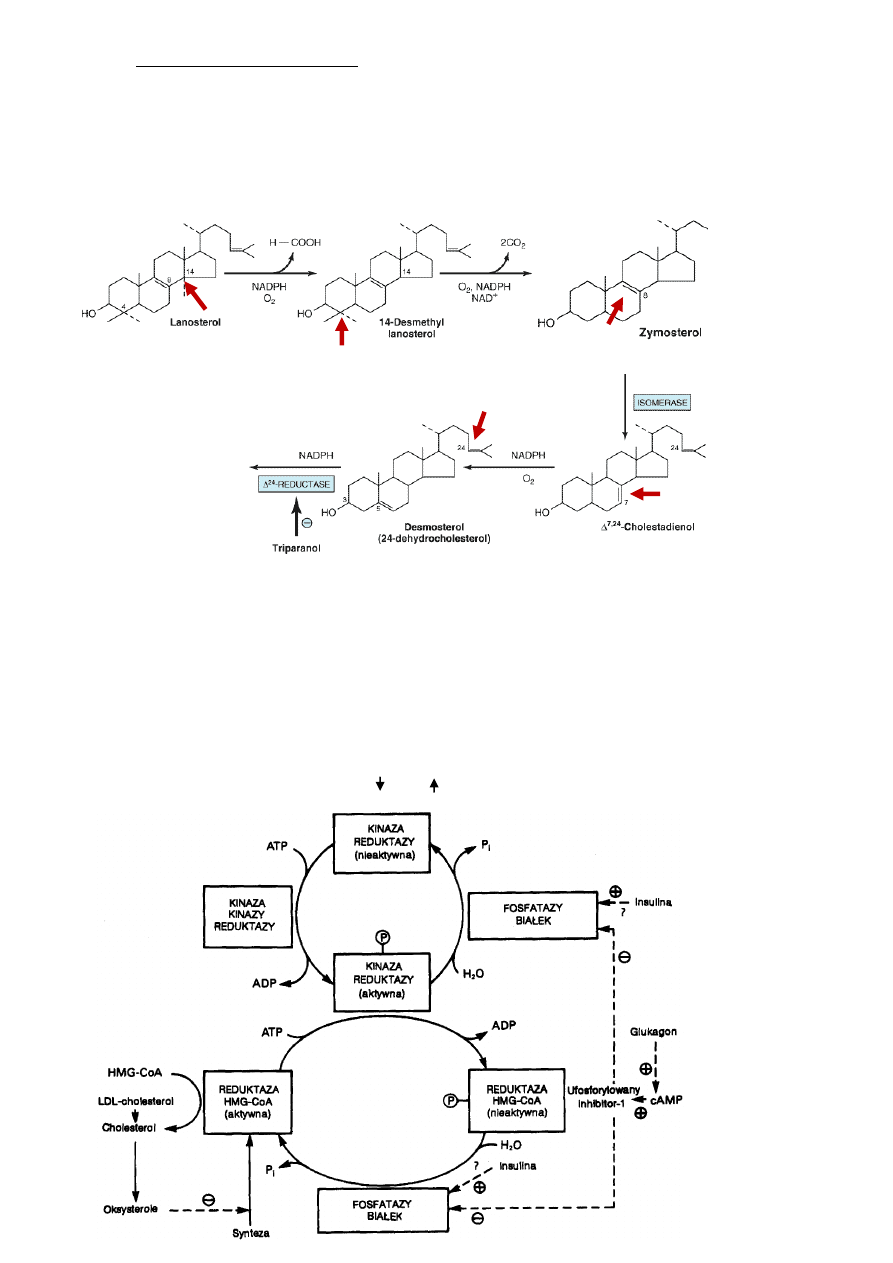
grupa7.med@gmail.com | 2008 - wdanie II, poprawione
70
5) przekształcenie Lanosterolu w cholesterol w błonach siateczki śródplazmatycznej.
- NIEZBĘDNY TLEN MOLEKULARNY (dlatego większośd prokariota nie ma cholesterolu)
- utlenienie grupy metylowej przy C14 – powstaje desmetylolanosterol
- usunięcie 2 grup metylowych przy C4 – powstaje zymosterol
- przesunięcie podwójnego wiązania w zymosterolu – powstaje Δ7,24 cholestedienol
- dalsze przesunięcia podwójnego wiązania w pozycję między C5 a C6 – tak jak w cholesterolu
- redukcja podwójnego wiązania w łaocuchu bocznym – powstaje cholesterol
Cholesterol
REGULACJA SYNTEZY CHOLESTEROLU
1) na etapie reduktazy HMG-CoA, która jest ATKYWNA w formie ZDEFOSFORYLOWANEJ
- oksysterole w komórce hamują ekspresję genu dla reduktazy HMG-CoA (SRE, SRE-BP)
- insulina i T3 aktywuje reduktazę HMG-CoA przez aktywację fosfatazy
- glukagon i glikokortykoidy hamują reduktazę
- przy niskim stężeniu ATP zanika synteza cholesterolu
- kinaza białkowa aktywowana AMP ( ATP = AMP – hamowanie reduktazy HMG-CoA)
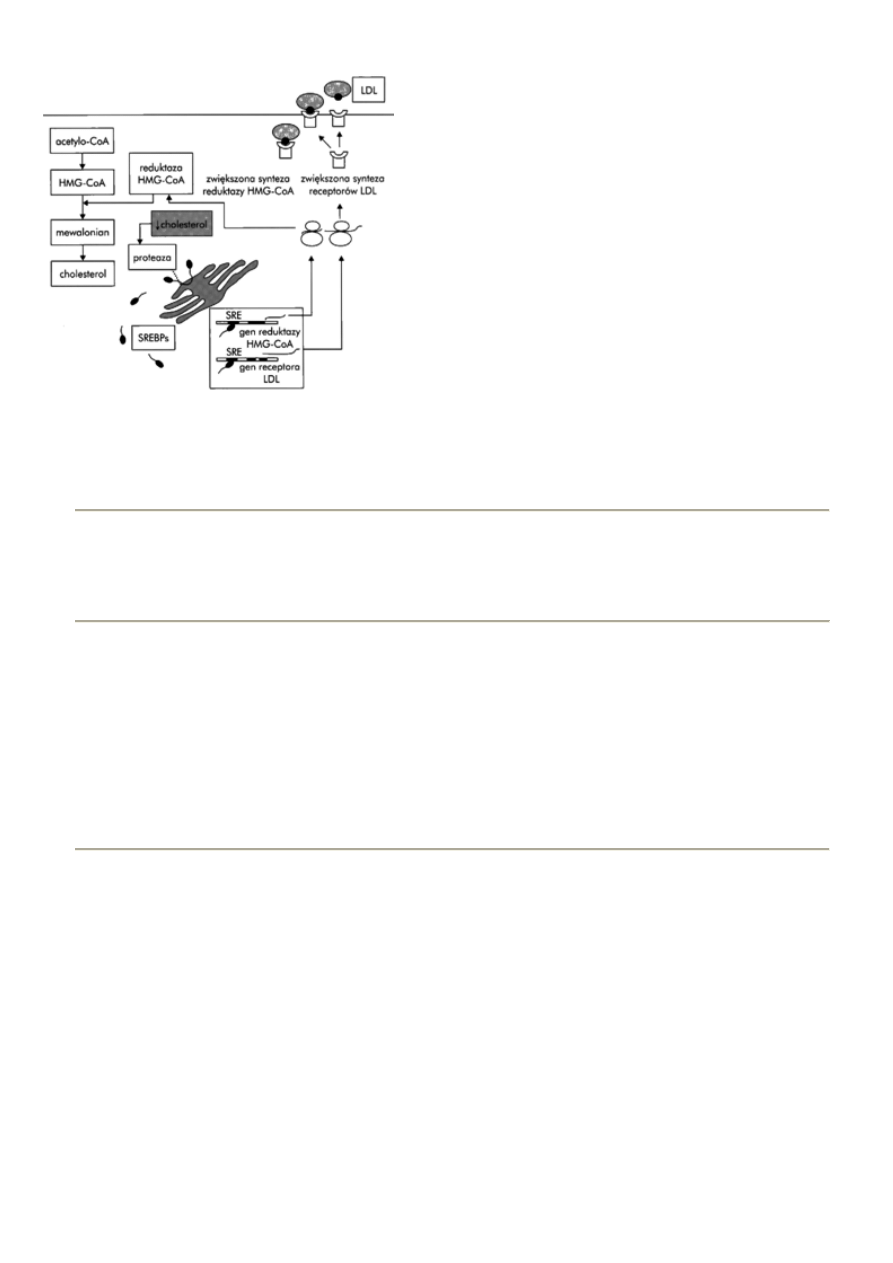
grupa7.med@gmail.com | 2008 - wdanie II, poprawione
71
2) przy wejściu LDL do komórki następuje wzrost stężenia
wewnątrzkomórkowego cholesterolu
- aktywacja ACAT (estryfikacja cholesterolu)
- hamowanie reduktazy HMG-CoA
- down regulation receptora LDL
SRE – steroid response element
66. METABOLIZM CHOLESTEROLU W POWIĄZANIU Z METABOLIZMEM
POSZCZEGÓLNYCH FRAKCJI LIPOPROTEIN
67. MECHANIZMY REGULUJĄCE GROMADZENIE CHOLESTEROLU W KOMÓRCE
Po przylaczeniu LDL z receptorem wysokiego powinowactwa nastepuje endocytoza kompleksu, ktory podlega
trawieniu lizosomalnemu. Uwolniony cholesterol wywiera dzialanie hamujace na reduktaze HMG-CoA (skoro
mamy cholesterol to po co go syntetyzowac i przesycac komorke). Powstaja oksysterole, ktore wywieraja
hamujacy wplyw na ekspresje receptora wysokiego powinowactwa ( jesli mamy wystarczajaca ilosc cholesterolu,
to po co go jeszcze imprtowac ). Cholesterol ulega zdeponowaniu w komorce w postaci estrow (enzym ACAT –
acylotransferaza acyloCoA:cholesterol - aktywnosc rosnie ze wzrostem stezenia cholesterolu)
68. FUNKCJA CHOLESTEROLU W ORGANIZMIE ORGANIZMIE JEGO WYDALANIE
Cholesterol pelni roznorakie funkcje w naszym oragnizmie:
- jest skladnikiem, elementem strukturalnym blon komorkowych
- bierze udzial w budowie plaszcza czastek lipoproteinowych
- jest substratem w syntezie witaminy D
- bierze udzial w syntezie hormonow steroidowych
- jest prekursorem kwaso zolciowych
- znaczenie patogenne w powstawaniu miazdzycy tetnic
- zaburzone proporcje w skladzie zolci miedzy solami kwasow zolciowych a cholesterolem skutkuja
wytracaniem sie kamieni zolciowych
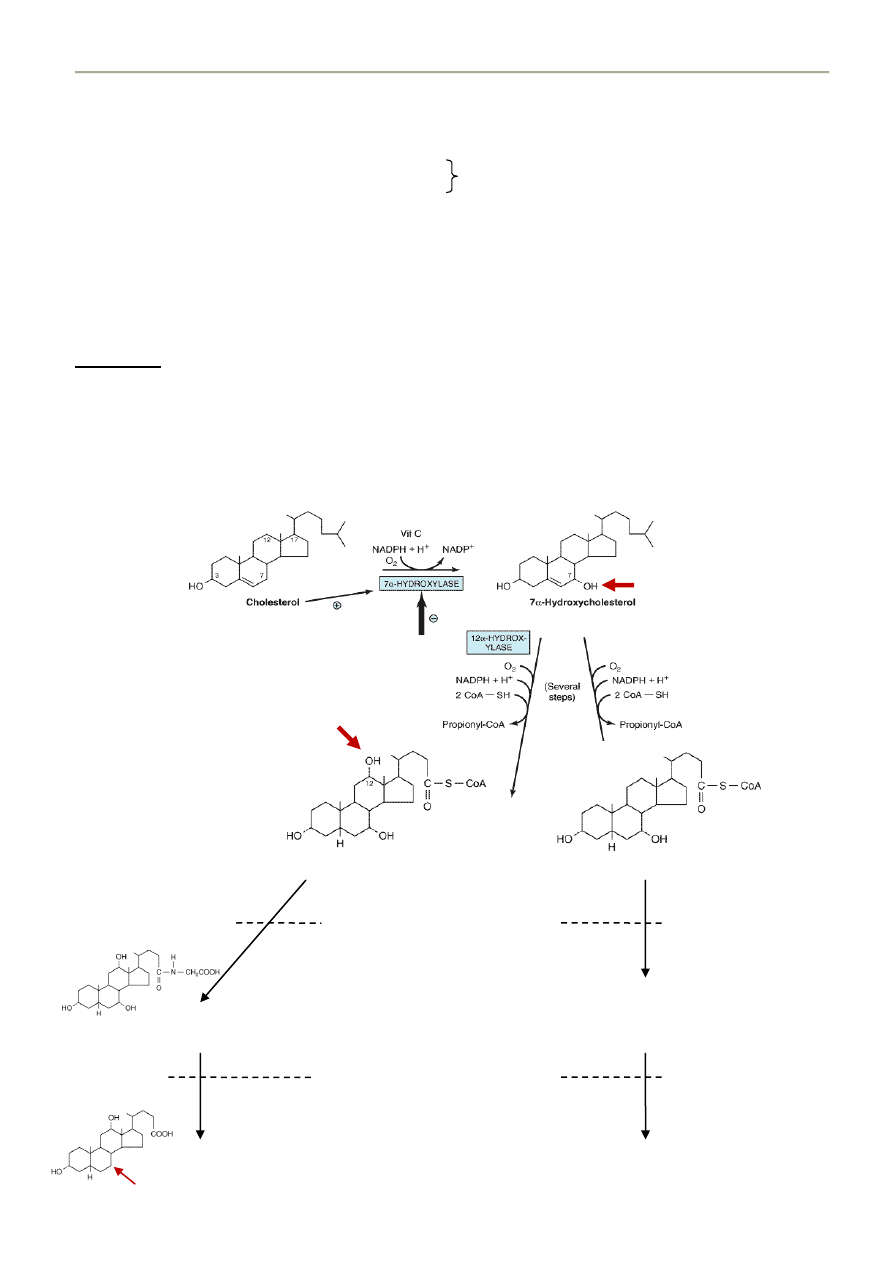
grupa7.med@gmail.com | 2008 - wdanie II, poprawione
72
69-71. Kwasy żółciowe – biosynteza, regulacja, połączenia w żółci, rola
Głównym składnikiem żółci jest lecytyna i sole kwasów żółciowych
Lecytyna – fosfatydylo cholina
Kwasy żółciowe - po 24C , bez wiązao podwójnych
- kwas cholowy
- kwas chenodeoksycholowy
PIERWOTNE
- kwas deoksycholowy
- kwas litocholowy
- wszystkie pierwotne kwasy żółciowe posiadają charakterystyczne cechy
- grupy OH przy C3 i C7 (dodatkowo mogą mied przy C12)
- całkowicie wysycony pierścieo steroidowy
- skrócony łaocuch boczny
BIOSYNTEZA
1) Hydroksylacja cholesterolu w pozycji 7 przez 7-hydroksylazę MIKROSOMALNĄ (wymaga tlenu, NADPH i
cytochromu P450)
2) Skracanie łaocucha bocznego i całkowite wysycenie wiązania podwójnego – jedyną różnicą między Choloilo-CoA a
Chenodeoksycholoilo-CoA to to, że u pierwszego dodatkowo jest hydroksylowana grupa C12
3) Kondensacja z tauryną lub glicyną z uwolnieniem SH-CoA
brak Wit C
kw. żółciowe
Pierwotne kwasy:
CHOLOILO-CoA
CHENODEOKSYCHOLOILO-CoA
Sprzęganie z glicyną lub tauryną
Uwalnianie SH-CoA
kwas glikocholowy lub taurocholowy
kwas tauro lub glikocholowy
(sól)
(sól)
dekoniugacja i 7dehydroksylacja
katalizowane przez bakterie w jelitach
Kwas deoksycholowy
kwas litocholowy
(wtórny kwas )
(wtórny kwas)

grupa7.med@gmail.com | 2008 - wdanie II, poprawione
73
Alfa 7 hydroksylaza:
-jest enzymem mikrosomalnym,
-enzym katalizujacy pierwszą reakcję przekształcenia cholesterolu w 7alfa-hydroksycholesterol,
-często jej aktywnośc jest regulowana razem z reduktazą HMG-CoA,
-ulega tak jak reduktaza HMG-CoA okołodobowym zmianom aktywności
Regulacja na poziomie 7 – hydroksylacji
- dostępnośd NADPH i witaminy C
- cholesterol pobudza, kwasy żółciowe hamują
Funkcje kwasów żółciowych
- rozpuszczalne w wodzie dzięki polarnym grupom karboksylowym i hydroksylowym,
przez co mogą działad na granicy 2 faz
- grupy metylowe są skierowane w jedną stronę płaszczyzny pierścienia, a grupy
hydroksylowe w drugą – dzięki temu cząsteczka kwasu żółciowego ma stronę
niepolarną skierowaną ku fazie tłuszczowej a polarną ku fazie wodnej
(właśnie dzięki tym właściwością kwasy żółciowe pełnią funkcję emulgatorów)
- wiążą się z cholesterolem przez co warunkują jego rozpuszczalnośd (nadmiar cholesterolu - >nie ma
wystarczającej ilości kwasów żółciowych i fosfolipidów go wiążących więc dochodzi do precypitacji w
drogach żółciowych i to może zapoczątkowac tworzenie kamieni żółciowych)
- biorą udział w transporcie tłuszczów w jelicie, wchłanianu witamin
- aktywują lipazy
70. POŁĄCZENIA KWASÓW ŻÓŁCIOWYCH W ŻÓŁCI
- kwasy żółciowe przechodzą do żółci w połączeniu z TAURYNĄ oraz GLICYNĄ
u ludzi stosunek tauryny do glicyny wynosi 1:3
- nowo wysyntetyzowane kwasy żółciowe w komórce wątrobowej występują jako tioestry-CoA tj:
* choloilo-CoA
* chenodeoksycholoilo-CoA
/te pochodne powstają przy udziale enzymu aktywującego/.
następnie działa enzym katalizujący połączenie pochodnych CoA z glicyną i tauryną, powstają:
* kwas glikocholowy
* kwas glikochenodeoksycholowy
* kwas taurocholowy
* kwas taurochenodeoskycholowy
Żółc jest mieszaniną związków organicznych i nieorganicznych. Jej głównymi składnikami są lecytyna i sole
kwasów żółciowych .

grupa7.med@gmail.com | 2008 - wdanie II, poprawione
74
71. PRZEMIANA PIERWOTNYCH KWASÓW ŻÓŁCIOWYCH WE WTÓRNE KWASY
ŻÓŁCIOWE
(→patrz synteza kwasów żółciowych schemat)
wtórne kwasy żółciowe powstają z pierwotnych pod wpływem aktywności bakterii jelitowych. Są to następujące
przemiany:
odszczepienie tauryny i glicyny
odszczepienie grupy 7alfa-OH
w efekcie powstają:
kwas deoksycholowy (z kwasu cholowego)
kwas litocholowy (z kwasu chenodeoksycholowego)
72. KRĄŻENIE WĄTROBOWO – JELITOWE KWASÓW ŻÓŁCIOWYCH, REGULACJA
WYDALANIA, SYNTEZY KWASÓW ŻÓŁCIOWYCH
Duża częśd wydalonych z żółcią kwasów żółciowych jest reabsorbowana do krążenia wrotnego, a następnie
wychwytywana przez wątrobę i ponownie wydalana z żółcią. Jest to na tyle wydajny proces ze codziennie
stosunkowo mała pula kwasów żółciowych może cyklicznie przejśc przez jelito 6-10 razy z niewielką utrata z
kałem (1-2%).Każdego dnia taka ilośc kwasów żółciowych jaka została wydalona z kałem jest syntetyzowana w
wątrobie. Z tego wynika, że ilośc kwasów żółciowych jest stała.
-wchłanianie kwasów żółciowych następuje w jelicie krętym (przez białko IBAT) a nie tak jak inne produkty
trawienia tłuszczów w jelicie czczym
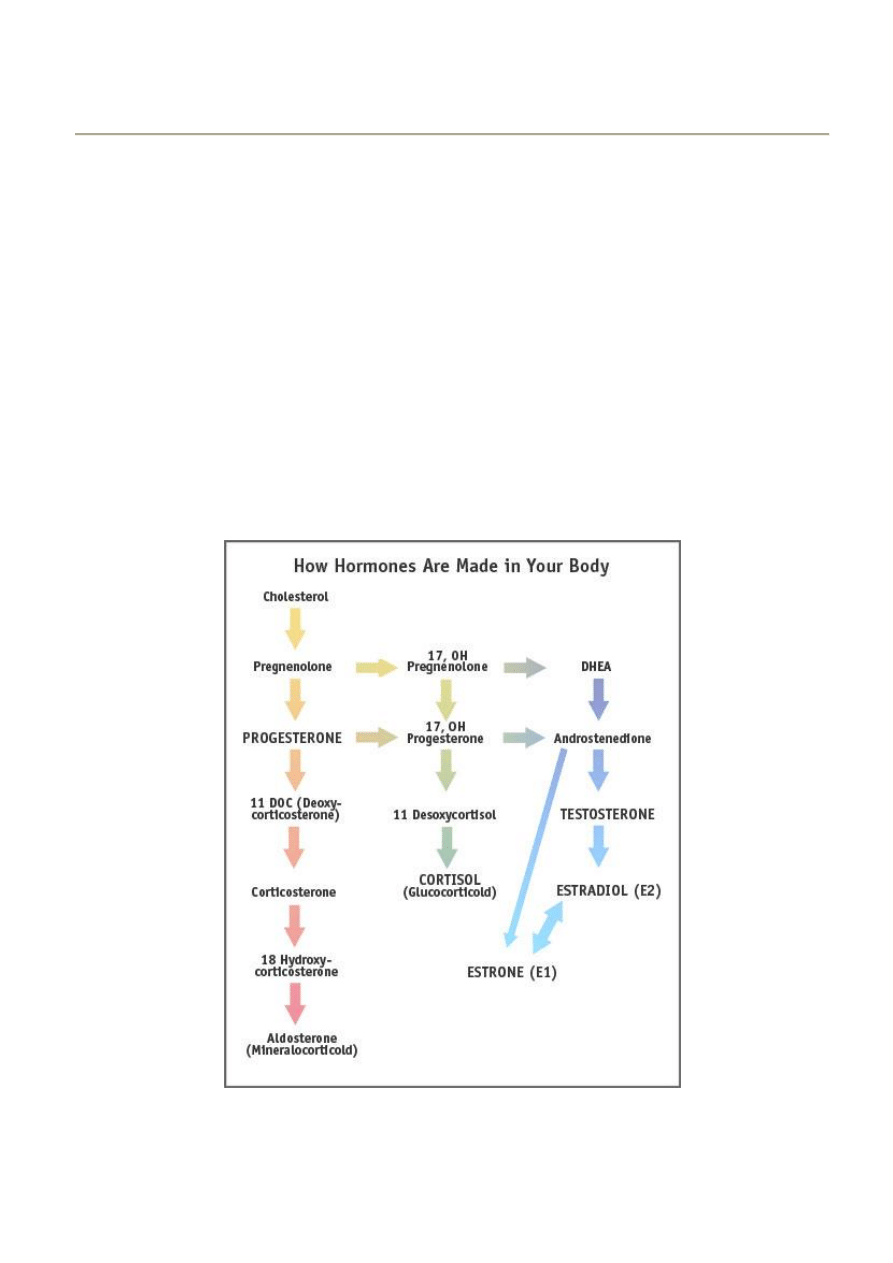
grupa7.med@gmail.com | 2008 - wdanie II, poprawione
75
73. HORMONY STERYDOWE. BIOSYNTEZA GAESTRAGENÓW,
GLIKOKORTYKOSTERYDÓW, MINERALOKORTYKOIDÓW, ANDROGENÓW,
ESTROGENÓW
Związkiem wyjściowym dla syntezy hormonów sterydowych jest cholesterol. W narządach, w których zachodzi
biosynteza hormonów sterydowych (są to narządy które mają rodowód mezodermalny), występuje układ
enzymatyczny, który zdolny jest ten cholesterol dalej przekształcad. Cholesterol jest albo przez te komórki
wyłapywany poprzez receptor wysokiego powinowactwa. W wyniku przekształcenia cholesterolu powstaje
układ cyklopentanoperhydrofenantrenowy (21-węglowy), czyli pregnan.
Tego typu układ potrafi syntetyzowad wyłącznie kora nadnerczy, w warstwie kłębkowatej i w warstwie
pasmowatej. Jeśli hormon sterydowy posiada układ pregnanu to mówi się, że jest to kortykosteroid właściwy.
Natomiast jeśli takiego układu nie ma należy do innych hormonów sterydowych, np. androgenów lub
estrogenów. Hormony sterydowe mogą się dzielid wg narządu, w którym są syntetyzowane, czyli hormony kory
nadnercza, jajnika, jądra, łożyska, ale znacznie lepszy jest podział, który mówi nam o funkcjach tych hormonów.
Wg tego podziału mamy kilka kategorii:
mineralokortykoidy w warstwie kłębkowatej kory nadnerczy
glikokortykoidy znalazły zastosowanie praktyczne jako leki
androgeny wytwarzane zarówno u mężczyzn i kobiet
gestageny
estrogeny
grupa7.med@gmail.com | 2008 - wdanie II, poprawione
76
Szlaki syntezy hormonów nadnerczy.
Glikokortykosterydy
Biosynteza zachodzi w warstwie pasmowatej kory nadnercza. Wyjściowym substratem dla glikokortykoidów jest
cholesterol, z niego powstaje pregnenolon, z niego – progesteron a następnie:
1) albo ulega utlenieniu w pozycji 21. – powstaje 11-dezoksykortykosteron
2) albo ulega hydroksylacji w pozycji 17. – powstaje 17-hydroksyprogesteron
11-dezoksykortykosteron ulega 11-hydroksylacji do kortykosteronu, a 17-hydroksyprogesteron ulega 21-
hydroksylacji do 11-deoksykortyzolu i 11-hydroksylacji do kortyzolu
Regulacja biosyntezy zależy od ACTH (adrenokortykotropina). Znajduje się on wpływem podzwgórzowego CRH.
Warstwa pasmowata znajduje się najsilniej pod wpływem ACTH, natomiast warstwa kłebuszkowata nie jest
wrażliwa na ACTH. Warstwa siatkowata tylko w wyjątkowych sytuacjach. Produktem kory nadnerczy w zakresie
glikortykoidów jest kortyzon który wywiera działanie hamujące na przysadkę mózgową.
Mineralokortykoidy
Ich synteza odbywa się w warstwie kłębuszkowatej kory nadnerczy w podobny sposób do poprzedniego, ale do
pewnego momentu.
21-hydroksylaza
11-hydroksylaza
18-hydroksylaza
progesteron 11-dezoksykortykosteron kortykosteron aldosteron
Głównym przedstawicielem mineralokortykoidów jest aldosteron oraz DOCA (dezoksykortykosteron).
Androgeny
Synteza androgenów odbywa się w szlaku bocznym po 17-hydroksylacji pregnenolonu. Po kolei powstają:
1) DHEA – dehydroepiandrosteron
2) Δ
4
(delta 4) – androstendion
3) testosteron
4) dihydrotestosteron (hydroepiandrosteron)
Pierwsze 3 są syntezowane w gruczołach dokrewnych, natomiast synteza dihydrotestosteronu
(najaktywniejszego) odbywa się w tkankach obwodowych, głównie w pęcherzykach nasiennych, gruczole
sterczowym oraz w skórze. Tylko w tych narządach obecny jest enzym 5-α-reduktaza, która przekształca
testosteron w di-hydrotestosteron.
Estrogeny
Powstają z męskich hormonów płciowych Kluczową reakcją dla powstawania estrogenów jest aromatyzacja
pierścienia A w układzie cyklopentanoperhydrofenantrenowym testosteronu w efekcie czego powstaje 17-β-
estradiol.

grupa7.med@gmail.com | 2008 - wdanie II, poprawione
77
Z androstendionu powstaje estron, ta frakcja jest szczególnie nasilona w tkankach obwodowych, głównie w
skórze, wątrobie i tkance tłuszczowej żółtej. Trzecią substancją z grupy estrogenów jest estriol, obecny u kobiety
ciężarnej - produkowany jest przez łożysko. W okresie ciąży wytwarzanie estriolu jest 1000 razy większe niż
poza jej okresem – dobry marker ciąży i rozwoju płodu.
estron (E
1
) – głównie pojawia się w menopauzie, powstaje również po owaroktomii
estradiol (E
2
) – główny estrogen jajnikowy
estriol (E
3
) – powstaje w okresie ciąży w łożysku, w wyniku konwersji androstendionu
Gestageny
Naturalnym gestagenem jest progesteron. Synteza zachodzi z cholesterolu poprzez pregnenolon.
Synteza:
jajnik
ciałko żółte – komórki luteinowe, produkują przez pewien okres ciąży progesteron, po czym od
pewnego momentu jego funkcja jest przejmowana przez
łożysko, które jest głównym narządem produkującym progesteron
kora nadnerczy – śladowe ilości, efekt uboczny syntezy mineralo- i glikokortykoidów, która biegnie przez
progesteron
75. MECHANIZM DZIAŁANIA HORMONÓW STERYDOWYCH
Z grubsza u wszystkich hormonów sterydowych jest taki sam. Przyjrzyjmy się mechanizmowi działania kortyzolu.
Hormony sterydowe jako substancje przenikające przez błonę cytoplazmatyczną, mają receptor nie w błonie ale
w cytoplazmie.
Receptor oznaczamy jako GR. W stanie natywnym jest on sparaliżowany, a jego „paralizatorem” jest grupa
białek HSP (białka szoku termicznego). Uniemożliwiają one połaczenie się receptora z DNA. Po połączeniu się
kortyzolu z receptorem, dochodzi w obrębie cytozolu do dysocjacji białek HSP i receptor z przyłączonym
hormonem jest w stanie aktywnym, ulega on dimeryzacji i taki dimer receptora dla kortyzolu przemieszcza się
do jądra komórkowego gdzie znajdują się elementy odpowiedzi dla niego. Jest to uniwersalny mechanizm
działania wszystkich hormonów sterydowych.
MOGĄ DZIAŁAD NA DRODZE TRANSAKTYWACJI LUB TRANSREPRESJI
TRANSAKTYWACJA – działanie genomowe – niekorzystne
GCS – glikokortykosteryd
GR – receptor glikokortkosterysowy, jest receptorem cytoplazmatycznym i w cytoplazmie
występuje w połączeniu z białkami szoku termicznego – HSP (Hot Shock Proteins)
Największe to HSP 90
Kiedy GCS połączy się z receptorem to HSP odłącza się, GR+GCS ulegają dimeryzacji i wędrują do jądra, gdzie
łączy się z GRE (Glikokortykoid Response Element) co wywołuje następujące skutki:
POZYTYWNE: dochodzi do wzrostu:
- IL-1
- LIPOKORTYNY-1, która hamuje fosfolipazę A2 (kw. arachidonowy się nie uwalnia - przeciwzapalne,
przeciwbólowe, przeciwalergiczne)
- inhibitora protezy leukocytarnej - hamowanie NFkB
- rybonukleaz – hamowanie GM-CSF i COX2
grupa7.med@gmail.com | 2008 - wdanie II, poprawione
78
NEGATYWNE: dochodzi do spadku:
- cytokin
- iNOS
- IL 1, 6, 11 , 13
- GM-CSF
- MMP-9 (metaloproteazy macierzowe)
TRANSAKTYWACJA
TRANSREPRESJA
TRANSREPRESJA – pozagenomowo – korzystne
GR+GCS nie ulegają dimeryzacji
Monomer GR+GCS szuka w cytozolu „ofiar”, z którymi może się połączyd i je dezaktywowad, są nimi:
- AP-1 (aktywator protein) który może sprzyjad apoptozie
- NFkB – zapalenie
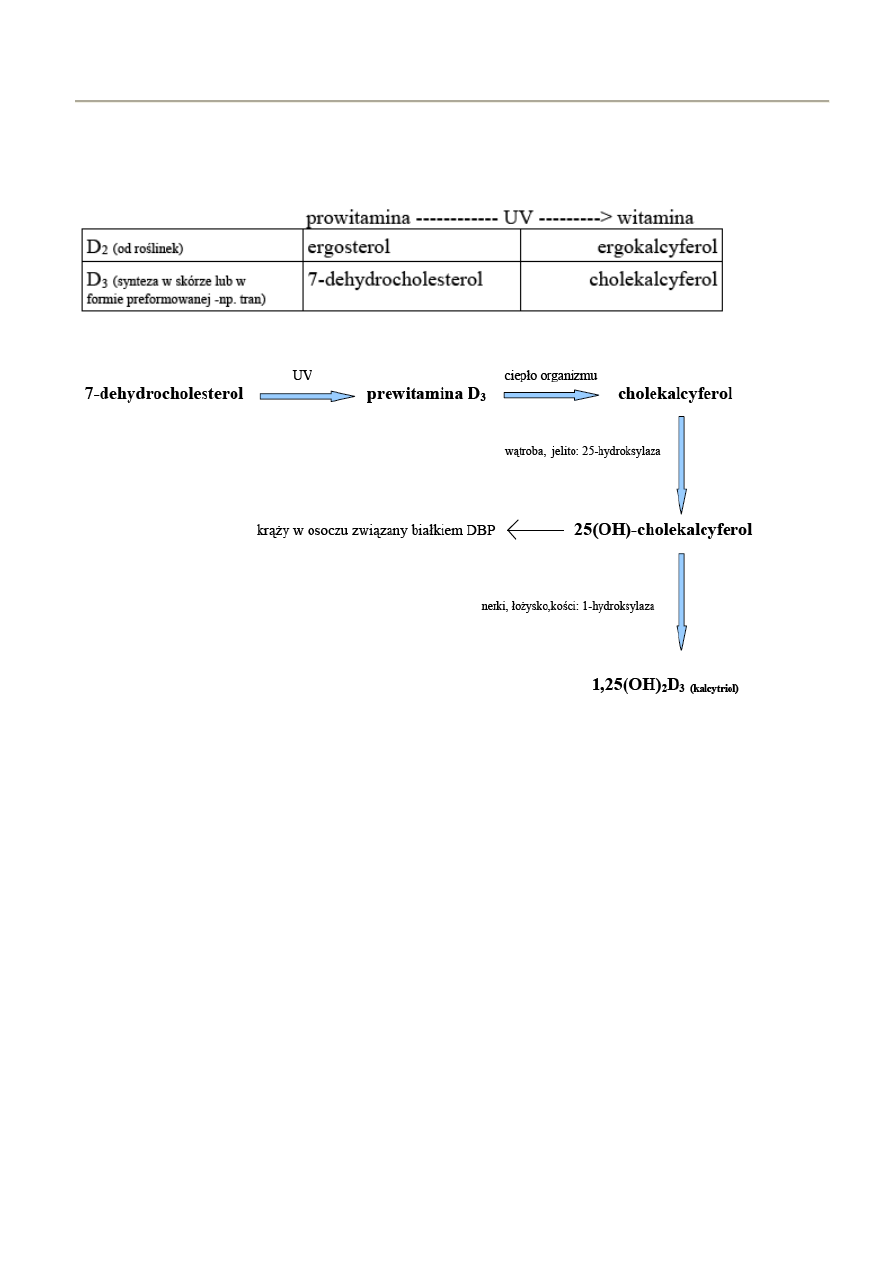
grupa7.med@gmail.com | 2008 - wdanie II, poprawione
79
76. WITAMINA D I JEJ HYDROKSYPOCHODNE. MECHANIZM DZIAŁANIA
WITAMINY D NA PRZEWÓD POKARMOWY, NERKI, UKŁAD KOSTNY.
Budowa:
kalcyferole to grupa związków chemicznych o charakterze steroidów (układ cyklo-pentano-perhydro-
F
enantrenowy
w wątrobie hydroksylacja przy 25C zachodzi zawsze, natomiast w nerkach
jest indukowana przez swoiste aktywatory (wymienione w kolejności „ważności działania”):
1. Hipofosfatemia (!)
2. hipokalcemia
3. PTH
4. spadek ilości 1,25(OH)2D3
5. estrogeny (estradiol)
6. androgeny
7. STH
8. T4
9. kortyzol
10. insulina
Inne hyrdoksylacje zachodzą przy różnych atomach węgla (24, 26, 27, 23) ale ich rola nie jest jeszcze poznana
(wiadomo tylko, że w jelicie i kościach powstaje 24,25(OH)2D3)
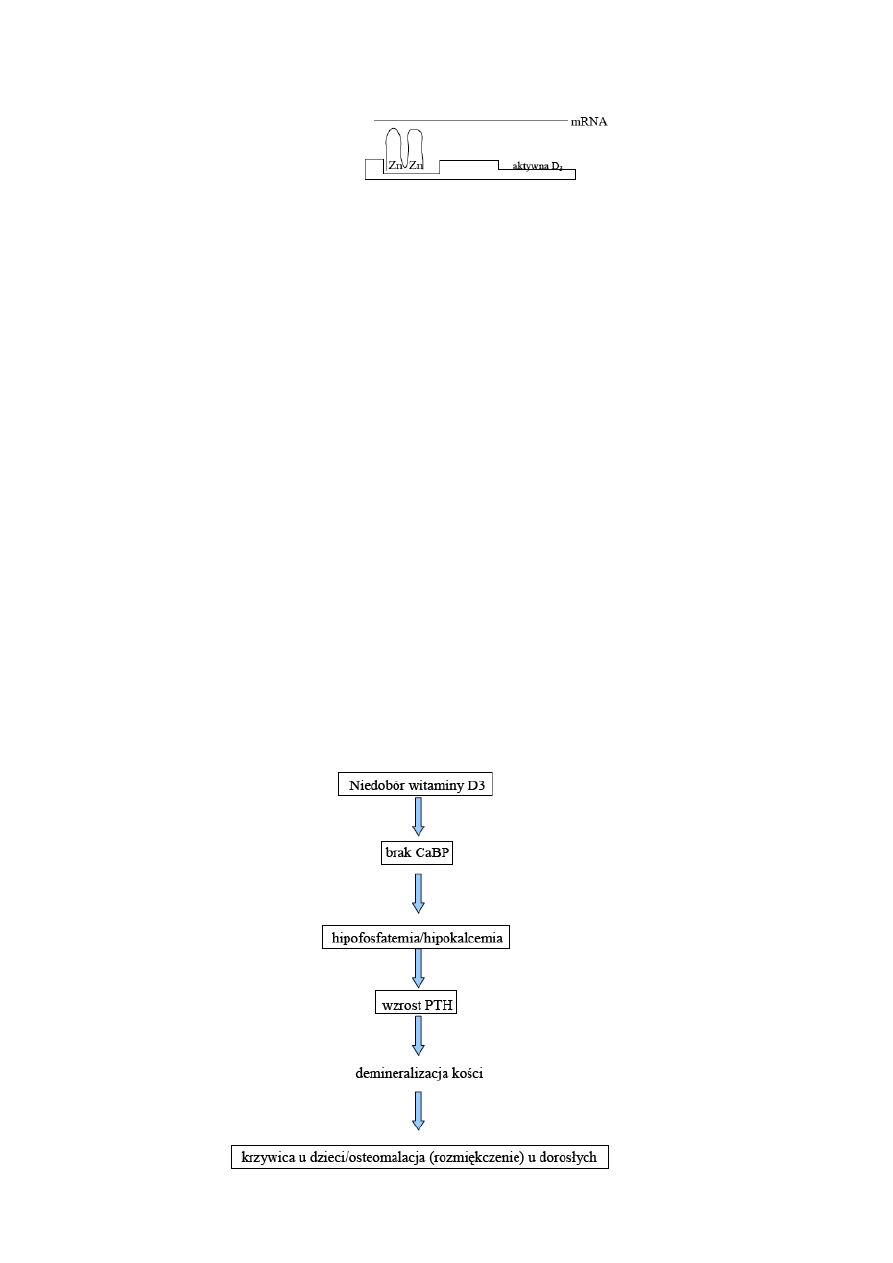
grupa7.med@gmail.com | 2008 - wdanie II, poprawione
80
Teraz krótko o aktywnej postaci witaminy D czyli 1,25(OH)2D3 .
Jest to cytokina działająca w głównej mierze na jelito cienkie, ale też i kości, nerki, przytarczyce i trzustkę.
Receptory dla witaminy D – VDR
Jelito – powoduje wytworzenie CaBP (Calcium binding protein) i jest to jedyny
hormon działający na jelito, który powoduje zwiększenie absorpcji Ca.
Pozostałe powodują jedynie zwiększoną osteolizę, albo wpływają na jelito za
pośdednictwem1,25(OH)2D3
Kości – indukuje syntezę osteokalcyny oraz powoduje wzrost osteolizy (remodeling)
Nerka – zatrzymuje wydalanie Ca
Przytarczyce – hamuje sekrecje PTH
Naczynia krwionośne - witamina D3 powoduje preferencyjne wiązanie wapna w
śródbłonku (przez wzrost ekspresji genów białek, które wiążą Ca w naczyniach
– na przykład osteokalcyna, osteopontyna i osteonektyna), przez co dochodzi
do zwapnienia. Ma więc działanie promiażdżycowe.
Niedobór:
– złe wchłanianie (np. cholestaza)
– choroby wątroby -> osteodystrofia wątrobowa
– choroby nerek -> osteodystrofia nerek
– brak receptora
– brak efektora
w tych ostatnich dwóch przypadkach pozostaje się jedynie modlid, gdyż nie znamy metod leczenia tych
niedoborów.
grupa7.med@gmail.com | 2008 - wdanie II, poprawione
81
inna
możliwość:
jeśli mamy pacjenta z uszkodzeniem nerek to nawet przy podaniu dużej ilości witaminy
D3 nie powstanie w jego organizmie forma aktywna
.
Bibliografia
Biochemia Harpera; wydanie 25, PZWL Warszawa 2006
E. Baokowski, Biochemia; Urban&Partner, Wrocław 2004
J.M Berg, J.L Tymoczko, L. Stryer, Biochemia; wydanie 5, PWN, Warszawa 2007
Wykłady prof. J. Gmioskiego - 2000, 2001, 2007, 2008
Rysunki i schematy: Harpers illustrated biochemistry - 27th edition
Wyszukiwarka
Podobne podstrony:
lipidy2 2
Lipidy do wywieszenia
lipidy skrot
Biochemia 4 Lipidy
Lipidy Teoria 14
LIPIDY I LIPOPROTEINY OSOCZA(1)
Lipidy w kosmetyc1
lipidy izoprenowe id 269491 Nieznany
ch zywnosci wyklad 4 lipidy low
Lipidy 6
2 Lipidy
lipidy - podział i metabolizm, Technologia żywności i żywienia człowieka, Biochemia
lipidy, Lekarski, FARMAKOLOGIA, 2. semestr, 2sem
biochemia lipidy
Lipidy 4
lipidy 2, Prywatne, Biochemia WYKŁADÓWKA I, Biochemia wykładówka 1, TESTY, testy
Lipidy
więcej podobnych podstron