
12
Prace IMŻ 3 (2010)
Michał KUBECKI
Instytut Metalurgii Żelaza
JAKOŚCIOWA I ILOŚCIOWA IDENTYFIKACJA
WIELOPIERŚCIENIOWYCH WĘGLOWODORÓW
AROMATYCZNYCH W GLEBACH POBRANYCH
Z TERENÓW POŁOŻONYCH W SĄSIEDZTWIE
HUTNICZYCH ZAKŁADÓW PRZEMYSŁOWYCH
Celem pracy było opracowanie i wdrożenie w Laboratorium Analiz Chemicznych Instytutu Metalurgii Żelaza metod
analitycznych identyfi kacji i oznaczania wielopierścieniowych węglowodorów aromatycznych (WWA) w glebach, za
pomocą chromatografu gazowego sprzężonego ze spektrometrem masowym (HRGC/HRMS). Przedmiotem badań były
wzorce z atestowaną zawartością wielopierścieniowych węglowodorów aromatycznych i gleby z terenów położonych w
sąsiedztwie hutniczych zakładów przemysłowych. Ustalono warunki pracy chromatografu gazowego i spektrometru
masowego, wdrożono metodę ekstrakcji WWA z próbek gleb z zastosowaniem techniki SPE oraz sporządzono odpo-
wiednie procedury analityczne.
Słowa kluczowe: związki organiczne, wielopierścieniowe węglowodory aromatyczne, gleba, metalurgia, przemysł
QUALITATIVE AND QUANTITATIVE ANALYSIS
OF POLYCYCLIC AROMATIC HYDROCARBONS IN SOILS
FROM THE NEIGHBOURHOOD OF METALLURGICAL
INDUSTRIAL PLANTS
The aim of the research was to develop and implement the new analytical methods for identifi cation and determi-
nation of polycyclic aromatic hydrocarbons (PAHs) in soil with the use of gas chromatography coupled with mass
spectrometry (HRGC/HRMS). Research were performed with the use of Certifi ed Reference Materials, with certifi ed
content of polycyclic aromatic hydrocarbons. Soil samples from areas situated in the neighbourhood of metallurgical
industrial plants were also used. HRGC/HRMS operating conditions were established, specifi c methods for prepara-
tion samples and analytical procedures were developed.
Key words: organic compounds, PAH, soil, metallurgy, industry
1. WSTĘP
Analiza zanieczyszczeń gleby i gruntów w otoczeniu
źródeł ich potencjalnego powstawania, umożliwia okre-
ślenie wpływu działalności przemysłowej człowieka
na środowisko naturalne, a także stanowi jeden z kie-
runków kontroli funkcjonowania zakładów przemysło-
wych.
Niejednokrotnie podstawowy zakres kontroli obej-
muje oznaczanie w glebach głównie metali ciężkich
i związków nieorganicznych, których najczęstszym
źródłem jest przemysł wydobywczy, odlewnictwo oraz
procesy hutnicze [1, 3, 5, 9]. Badania zawartości związ-
ków organicznych, z uwagi na brak sprecyzowanych
wymagań co do ich dopuszczalnych stężeń w glebach,
przez długi okres czasu realizowane były w znikomym
stopniu. Jednak coraz częstsze doniesienia na temat
szkodliwego wpływu na zdrowie człowieka szeregu
grup związków organicznych powstających w proce-
sach przemysłowych, oraz pojawianie się ich w śro-
dowisku naturalnym, przyczyniły się do poszerzenia
zakresu kontroli gleb o wybrane grupy związków or-
ganicznych.
Spośród wielu związków organicznych, do środowi-
ska naturalnego głównie w wyniku nieprawidłowego
funkcjonowania systemu oczyszczania gazów spali-
nowych czy wód poodpadowych, najczęściej trafi ają:
wielopierścieniowe węglowodory aromatyczne (WWA),
lotne związki organiczne (LZO) oraz węglowodory
chlorowane [2, 4, 6, 10]. Dotychczas nie prowadzono
stałego monitoringu tych substancji w próbkach gleb
z terenów położonych w sąsiedztwie hutniczych zakła-
dów przemysłowych. Dlatego też celem niniejszej pra-
cy było opracowanie metody identyfi kacji i ekstrakcji
wielopierścieniowych węglowodorów aromatycznych
z gleb, a następnie opracowanie metodyki ich ilościo-
wego oznaczania w tego typu próbkach.
Laboratorium Analiz Chemicznych Instytutu Meta-
lurgii Żelaza posiada wysokorozdzielczy chromatograf
gazowy (Trace GC Ultra) sprzężony z wysokorozdziel-
3-10.indb 12
21.09.2010 09:09:29
13
Prace IMŻ 3 (2010)
Jakościowa i ilościowa identyfi kacja wielopierścieniowych...
czym spektrometrem masowym (Finnigan MAT 95 XP)
umożliwiający analizę związków organicznych. W celu
przystosowania przyrządu do analizowania wielopier-
ścieniowych węglowodorów aromatycznych, konieczne
było przeprowadzenie odpowiednich prób eksperymen-
talnych, w toku których zostały opracowane nowe wa-
runki pracy urządzenia.
2. MATERIAŁ, ZAKRES I METODYKA
BADAŃ
Programy analityczne służące do oznaczania wielo-
pierścieniowych węglowodorów aromatycznych w gle-
bach, przygotowano bazując na pracach zrealizowa-
nych w Laboratorium Analiz Chemicznych w latach
2005–2008 [7, 8],
dostępnej literaturze [11], a także na
wskazówkach producentów kolumienek do ekstrakcji
do fazy stałej tzw. SPE (solid phase extraction). Opra-
cowano program do rozdziału WWA z zastosowaniem
kolumny chromatografi cznej Rtx-Dioxin (Restek) o dłu-
gości 60 m.
Badania prowadzono w oparciu o substancje wzor-
cowe z atestowaną zawartością WWA produkcji Accu-
Standards.
Materiał do badań stanowiły próbki gleb pobrane
przy pomocy laski glebowej do głębokości 0,2 metra,
z terenów położonych w odległości od 200 do 400 m od
granicy hutniczych zakładów przemysłowych.
2.1. ANALIZA JAKOŚCIOWA
WIELOPIERŚCIENIOWYCH WĘGLOWODORÓW
AROMATYCZNYCH
Istnieje kilka czynników ułatwiających identyfi kację
składników w badanej mieszaninie związków organicz-
nych. Najistotniejszym z nich jest czas retencji (RT –
retention time). Identyfi kacji dokonujemy poprzez po-
równanie czasu retencji badanego (szukanego) skład-
nika z czasem retencji wyznaczonym w identycznych
warunkach dla substancji wzorcowej.
W celu ustalenia parametrów pracy chromatografu
gazowego, przeprowadzono rozdział chromatografi czny
wzorca mieszaniny 17 wielopierścieniowych węglowo-
dorów aromatycznych. Do chromatografu wprowadza-
no 1
μl roztworu o stężeniu składników na poziomie
90
μg/ml. Przy kolejno zmienianych parametrach pra-
cy kolumny chromatografi cznej, ostatecznie uzyskano
program temperaturowy (T1), zamieszczony w tabli-
cy 1.
Tablica 1. Parametry pracy kolumny chromatografi cznej
w ramach programu temperaturowego T1
Table 1. Chromatographic column working parameters –
T1 temperature program
Przyrost temperatury,
o
C
Przedział czasowy, min
45 Æ 45
0–12
45 Æ 285
12–36
285 Æ 330
36–39
330 Æ 330
39–55
W trakcie analizy wzorcowej mieszaniny związ-
ków organicznych za pomocą systemu analitycznego
HRGC/HRMS (high resolution gas chromatography/
high resolution mass spektrometry) dla każdego z poja-
wiających się na chromatogramie pików, rejestrowano
pełne widmo masowe substancji opuszczającej kolum-
nę chromatografi czną. Po wyznaczeniu czasów retencji
i właściwym zinterpretowaniu widm masowych, przy-
pisano konkretnym pikom odpowiadające im związki
i wyznaczono dla nich najsilniejsze piki jonowe. Czasy
retencji oraz najsilniejsze piki jonowe dla WWA zesta-
wiono w tablicy 2.
Bazując na masach analizowanych jonów utworzo-
no program analityczny do oznaczeń WWA z pomocą
spektrometru pracującego w trybie wysokiej rozdziel-
czości (R = 10000). W trybie tym, w trakcie pomiaru
rejestrowane są pojedyncze jony.
Jako wzorzec masy jonów do bieżącej kontroli usta-
wień spektrometru masowego zastosowano fl uorowa-
ną naftę – PFK (perfl uorokerosene) producenta Alfa
Aesar.
Tablica 2. Czasy retencji i najsilniejsze piki jonowe WWA
Table 2. Retention times and the strongest ions peaks for
PAH
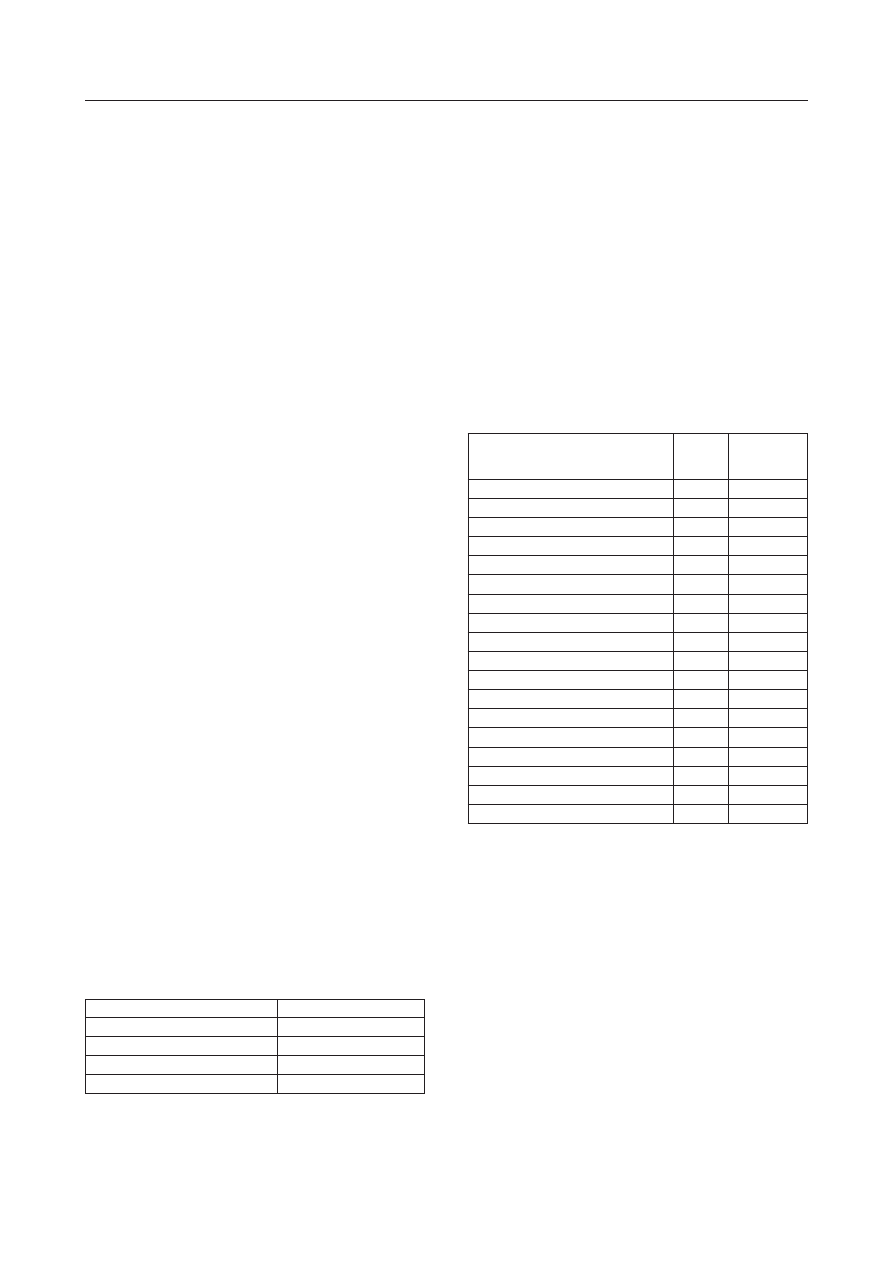
Analizowany związek
Czas
retencji
min
Najsilniejsze
piki jonowe
Naftalen
25,30
128,0548
Acenaftylen
29,65
152,0626
Acenaften
30,07
154,0783
Fluoren
31,40
166,0783
Fenantren
34,09
178,0783
Antracen
34,19
178,0783
Fluoranten
37,39
202,0783
Piren
37,96
202,0783
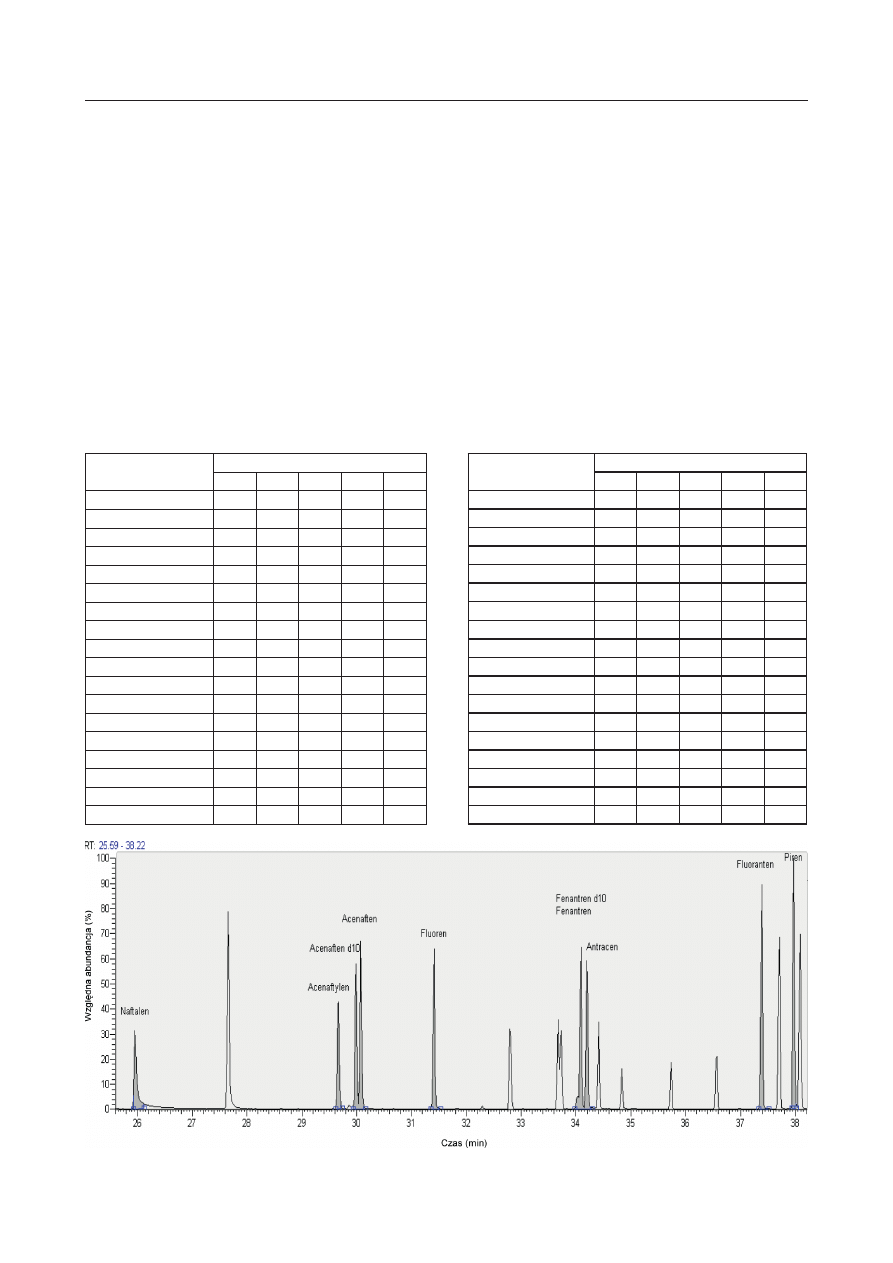
Benz[a]antracen
40,92
228,0936
Chryzen
41,02
228,0936
Benzo[b]fl uoranten
44,50
252,0939
Benzo[k]fl uoranten
44,50
252,0939
Bezno[a]piren
45,78
252,0939
Dibenzo[a,h]antracen
51,05
278,1096
Indeno[1,2,3]piren
51,61
276,1252
Benzo[g,h,i] perylen
53,47
276,0939
Wzorzec wewnętrzny – Acenaften d
10
30,01
164,1410
Wzorzec kontrolny – Fenantren d
10
34,03
188,1410
Tworząc program analityczny wprowadzono 11 okien
czasowych. W każdym z przedziałów czasowych wyko-
nywana jest sekwencja pomiarów polegająca na usta-
wieniu parametrów optyki jonowej (ustalenie warto-
ści pola magnetycznego) na podstawie odnalezionych
w widmie jonów PFK, a następnie poprzez zmiany pola
elektrycznego prowadzony jest pomiar intensywności
wybranych jonów analizowanego składnika.
2.2. ILOŚCIOWE OZNACZANIE
WIELOPIERŚCIENIOWYCH WĘGLOWODORÓW
AROMATYCZNYCH
Ilościowe oznaczenia wielopierścieniowych węglowo-
dorów aromatycznych w badanych próbkach zrealizo-
wano w oparciu o wzorcowe mieszaniny tych związków
w ampułkach zawierających około 1 ml roztworu, które
posłużyły do sporządzenia podstawowych roztworów
wzorcowych. Otrzymane na bazie dichlorometanu roz-
twory zawierały około 90 μg/ml każdego składnika.
Wykorzystując tak sporządzone roztwory przygotowa-
no serię 5 próbek kalibracyjnych. W tym celu do kolbek
3-10.indb Sek1:13
21.09.2010 09:09:29
14
Prace IMŻ 3 (2010)
Michał Kubecki
miarowych o pojemności 10 ml wprowadzono odpowied-
nio: 0, 100, 400, 600, 800 μl roztworu podstawowego.
Dla realizacji oznaczeń ilościowych do dichlorome-
tanowych roztworów wprowadzano stałą objętość (100
μl) roztworu wzorca wewnętrznego zawierającego ace-
naften d
10
oraz dodatek (100 μl) wzorca kontrolnego
(fenentren d
10
), pozwalający na kontrolę procesu eks-
trakcji i umożliwiający wyznaczanie odzysku analitu.
Stężenia WWA w poszczególnych roztworach wzorco-
wych zestawiono w tablicy 3.
Dla wymienionych roztworów wzorcowych zareje-
strowano chromatogramy z wykorzystaniem opisa-
nego wcześniej programu analitycznego. Na kolumnę
chromatografi czną wprowadzano 1 μl roztworu wzor-
cowego. Przykład zarejestrowanego chromatogramu
zamieszczono na rys. 1 i 2.
Zmierzono pola powierzchni pod pikami poszczegól-
nych związków z grupy WWA, a następnie wyznaczono
stosunki tychże pół do pola powierzchni wzorca kontro-
lnego dodanego w stałej ilości do roztworów kalibracyj-
nych (tablica 4).
Na podstawie obliczonego stosunku pola powierzchni
piku analitu do pola powierzchni wzorca kontrolnego
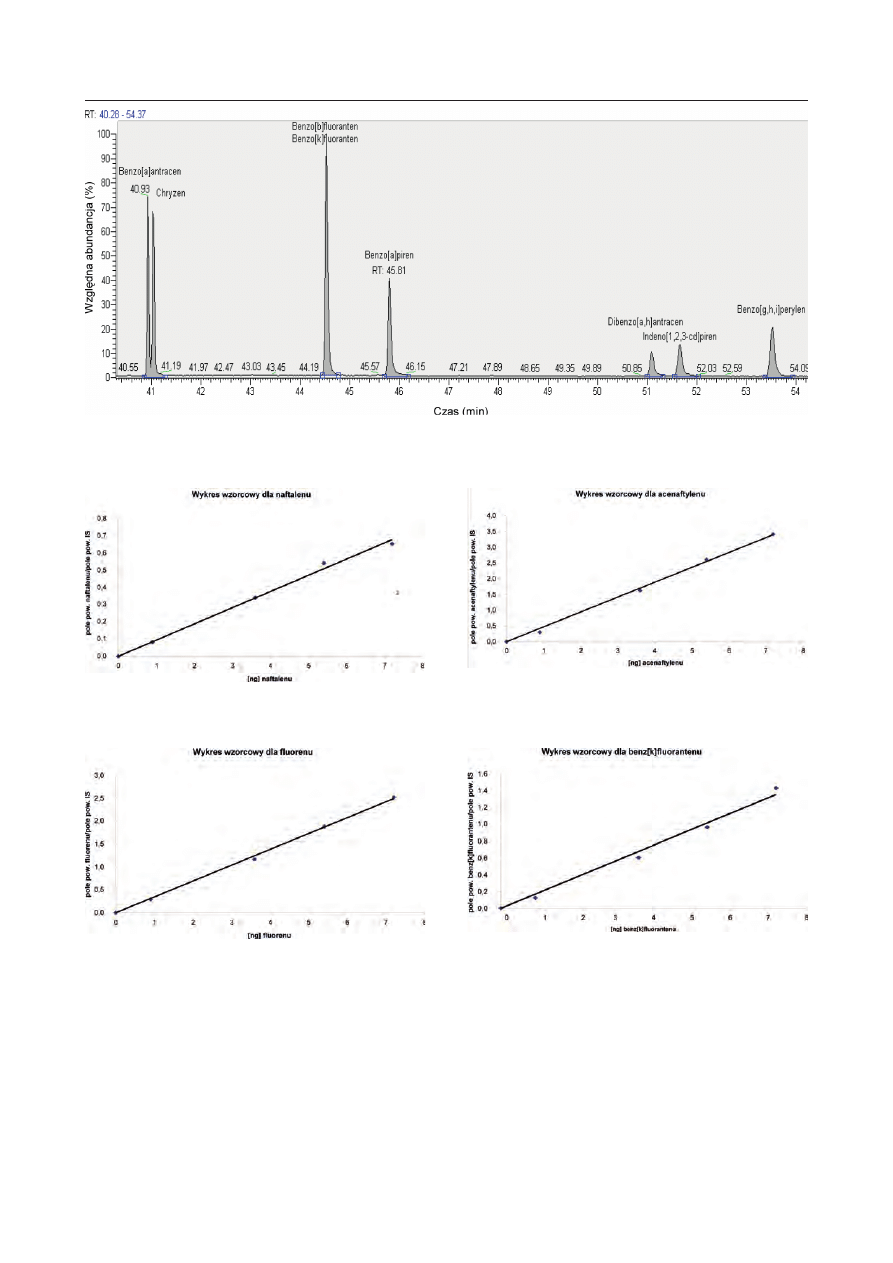
opracowano 16 wykresów wzorcowych. Przykładowe
wykresy sporządzone dla naftalenu, acenaftylenu,
fl uorenu i benzo[k]fl uorantenu w układzie współrzęd-
nych
oś X – ng analitu wstrzykniętego do chromatografu,
–
oś Y – stosunek pola powierzchni analitu do pola po-
–
wierzchni standardu wewnętrznego (Internal Stan-
dard),
zamieszczono na rys. 3–6.
Tablica 3. Stężenia WWA w roztworach kalibracyjnych
Table 3. PAH concentration in calibration solutions
Związek organiczny
Roztwór, μg/ml
a
b
c
d
e
Naftalen
0
0,9
3,6
5,4
7,2
Acenaftylen
0
0,9
3,6
5,4
7,2
Acenaften
0
0,9
3,6
5,4
7,2
Fluoren
0
0,9
3,6
5,4
7,2
Fenantren
0
0,9
3,6
5,4
7,2
Antracen
0
0,9
3,6
5,4
7,2
Fluoranten
0
0,9
3,6
5,4
7,2
Piren
0
0,9
3,6
5,4
7,2
Benzo[a]antracen
0
0,9
3,6
5,4
7,2
Chryzen
0
0,9
3,6
5,4
7,2
Benzo[b]fl uoranten
0
0,9
3,6
5,4
7,2
Benzo[k]fl uoranten
0
0,9
3,6
5,4
7,2
Benzo[a]piren
0
0,9
3,6
5,4
7,2
Indeno[1,2,3-cd]piren
0
0,9
3,6
5,4
7,2
Dibenzo[a,h]antracen
0
0,9
3,6
5,4
7,2
Benzo[g,h,i]perylen
0
0,9
3,6
5,4
7,2
Wzorzec wewnętrzny 1,8
1,8
1,8
1,8
1,8
Wzorzec kontrolny
1,8
1,8
1,8
1,8
1,8
Rys. 1. Przykładowy chromatogram uzyskany dla roztworu kalibracyjnego „d’’ i czasów retencji z zakresu od 26 do 39 min
Fig. 1. Exemplary chromatogram of the PAH calibration solution ,,d’’ for retention times in the range from 26 to 39 minute
Tablica 4. Stosunek pola powierzchni piku związku do
pola powierzchni piku wzorca kontrolnego
Table 4. Compound peak area to control standard peak
area ratio
Składnik
Roztwór
a
b
c
d
e
Naftalen 0
0,083
0,340
0,543
0,654
Acenaftylen
0
0,308
1,632
2,605
3,408
Acenaften
0
0,198
1,064
1,698
2,279
Fluoren
0
0,295
1,178
1,879
2,520
Fenantren
0
0,319
1,781
2,842
4,193
Antracen
0
0,223
1,387
2,213
3,449
Fluoranten
0
0,242
1,446
2,307
3,086
Piren
0
0,245
1,327
2,118
3,214
Benzo[a]antracen
0
0,086
0,432
0,690
1,092
Chryzen
0
0,145
0,785
1,252
1,898
Benzo[b]fl uoranten
0
0,127
0,592
0,945
1,332
Benzo[k]fl uoranten
0
0,126
0,592
0,965
1,426
Benzo[a]piren
0
0,092
0,422
0,673
0,729
Indeno[1,2,3-cd]piren
0
0,033
1,110
0,177
0,228
Dibenzo[a,h]antracen
0
0,047
0,160
0,256
0,414
Benzo[g,h,i]perylen
0
0,062
0,361
0,576
0,872
Wzorzec wewnętrzny
0,146
0,146
0,146
0,148
0,146
Wzorzec kontrolny
1,000
1,000
1,000
1,000
1,000
3-10.indb Sek1:14
21.09.2010 09:09:29
15
Prace IMŻ 3 (2010)
Jakościowa i ilościowa identyfi kacja wielopierścieniowych...
Rys. 2. Przykładowy chromatogram uzyskany dla roztworu kalibracyjnego „d’’ i czasów retencji z zakresu od 39 do 54 min
Fig. 2. Exemplary chromatogram of the PAH calibration solution ,,d’’ for retention times in the range from 39 to 54 minute
Rys. 3. Wykres wzorcowy dla naftalenu
Fig. 3. Standard diagram for naphthalene
Rys. 4. Wykres wzorcowy dla acenaftylenu
Fig. 4. Standard diagram for acenaphthylene
Rys. 5. Wykres wzorcowy dla fl uorenu
Fig. 5. Standard diagram for fl uorene
Rys. 6. Wykres wzorcowy dla benz[k]fl uorantenu
Fig. 6. Standard diagram for bezn[k]fl uoranthene
2.3. OPRACOWANIE METODY POBORU ORAZ
PRZYGOTOWANIA PRÓBKI ANALITYCZNEJ
W trakcie pobierania, jak i przechowywania próbek
do oznaczeń związków organicznych, głównym proble-
mem jest możliwość strat spowodowana ich parowa-
niem i migracją do materiału pojemnika. Dlatego też,
ważną rolę odgrywał zarówno odpowiedni dobór miej-
sca poboru próbek jak i wybór pojemników, w których
były one przechowywane.
Opracowując metodykę pobierania i przechowywania
próbek posłużono się m.in. normami PN-ISO 10381-1,
PN-ISO 10381-2 i PN-ISO 10381.
Przyjęto, że w celu zminimalizowania strat analitu,
próbki do badań należy pobierać do szczelnych pojemni-
ków. W przypadku większości prac zaleca się używanie
pojemników, które mogą pomieścić około 2 kg próbki.
Napełnianie i uszczelnianie pojemnika powinno odby-
wać się w taki sposób, aby pozostawić jak najmniejszą
3-10.indb Sek1:15
21.09.2010 09:09:30

16
Prace IMŻ 3 (2010)
Michał Kubecki
przestrzeń powietrzną nad próbką. Podstawową spra-
wą jest, aby pojemnik nie zanieczyszczał próbki i nie
adsorbował jej składników.
Dlatego w przypadku oznaczeń związków organicz-
nych konieczne jest użycie pojemnika z materiału
bardziej obojętnego niż tworzywa sztuczne. Do takich
celów używać można szerokoszyjnych pojemników
szklanych, pojemników aluminiowych zamykanych
nakrętkami, puszek z wtłaczanymi przykrywkami albo
pojemników z fl uorowanego polimeru np. PTFE.
Do strategii pobierania próbek wymagane jest roz-
ważenie, jaki stosować schemat, z jakiej głębokości po-
bierać próbki oraz jaka ma być ich wielkość.
Zaleca się, aby liczba miejsc pobierania próbek
w każdej potencjalnie zanieczyszczonej strefi e była pro-
porcjonalna do wielkości tej strefy, lecz zawsze z zacho-
waniem minimalnej liczby sześciu próbek. Szczegółowe
uwagi dotyczące schematów pobierania próbek podane
są w PN-ISO 10381-1.
W ramach prowadzonych badań zastosowano sche-
mat systematycznego pobierania próbek z siatką
o kształcie regularnym na obszarze wybranym losowo
z terenu położonego w określonej odległości od zakładu
hutniczego. Próbki pobierano przy pomocy laski glebo-
wej do głębokości 0,2 metra. Tak pobrane próbki na-
leżało w jak najkrótszym czasie przetransportować do
laboratorium, gdzie do momentu przygotowania próbki
analitycznej były przechowywane w lodówce w tempe-
raturze 4
o
C.
Dla realizacji badań pobrano 5 próbek gleb z terenów
położonych w odległości od 200 do 400 metrów od gra-
nicy hutniczych zakładów. Tereny te sklasyfi kowano
w odniesieniu do Rozporządzenia Ministra Środowiska
z dnia 9 września 2002 (poz. 1359), jako grunty zali-
czane do gruntów leśnych oraz zadrzewionych i zakrze-
wionych, nieużytków, a także gruntów zabudowanych
i zurbanizowanych (Grupa B). Tylko jedna z próbek
pochodziła z terenu zakładu przemysłowego, którego to
grunt sklasyfi kowano w grupie C.
2.4. BADANIA PROCESU EKSTRAKCJI WWA
Z GLEB
W celu wyizolowania wielopierścieniowych węglo-
wodorów aromatycznych z matrycy próbki, w Labo-
ratorium Analiz Chemicznych wykorzystano proces
ekstrakcji za pomocą rozpuszczalnika, wspomagany
ultradźwiękami. Proces ten polegał na poddawaniu 5
gramowej próbki gleby, umieszczonej w określonym
rozpuszczalniku organicznym, działaniu fal ultradź-
więkowych. Prowadzono go przez 30 minut, następnie
otrzymany ekstrakt oddzielano od próbki gleby przez
odwirowanie w wirówce laboratoryjnej przy 3000 obr/
min przez określony czas. Oddzielony ekstrakt oczysz-
czano na kolumienkach SPE w układzie próżniowym.
Związki zaadsorbowane na kolumience wymywano do
fi olki o objętości 2 ml odpowiednio dobranym rozpusz-
czalnikiem.
Ekstrakt zatężano w strumieniu azotu, a w ostat-
nim etapie dodawano roztworu wzorca wewnętrznego
(Syringe Standard) i kierowano do oznaczenia ilościo-
wego.
W pierwszym etapie, badania procesu ekstrakcji pro-
wadzono na próbkach syntetycznych – 5 g wyprażone-
go piasku z dodatkiem 400 μl wzorcowej mieszaniny
WWA i 200 μl wzorca wewnętrznego zawierającego
deuterowane związki acenaftenu, chryzenu, 1,4-dichlo-
robenzenu, naftalenu, perylenu i fenantrenu.
W wyniku przeprowadzonych eksperymentów, usta-
lono prowadzenie ekstrakcji w dwóch cyklach po 30
min, za każdym razem nową porcją rozpuszczalnika, za
który wybrano 2-propanol. Szybkość odwirowania po-
zostawiono bez zmian, natomiast czas ustalono na 10
minut. Do oczyszczania zastosowano kolumienkę SPE
z oktadecylową fazą stacjonarną, kondycjonowaną 3 ml
2-propanolu i 3 ml mieszaniny propanolu z wodą. Ozna-
czane związki eluowano z kolumny dwoma porcjami di-
chlorometanu po 500
μl każda. Ekstrakt zatężano do
objętości około 100
μl w strumieniu azotu. Dla kontroli
procesu ekstrakcji, do badanej próbki przed ekstrakcją
wprowadzano dodatek 200 μl wzorca kontrolnego.
3. WYNIKI I ICH DYSKUSJA
W oparciu o opracowaną metodykę (analizy i eks-
trakcji) przeprowadzono identyfi kację i oznaczenie za-
wartości wielopierścieniowych węglowodorów aroma-
tycznych w próbkach gleb pobranych z otoczenia hut-
niczych zakładów przemysłowych. Stwierdzono obec-
ność wymienionych substancji. Określono sumę stężeń
WWA – w 4 próbkach na poziomie dopuszczalnym, na-
tomiast dla gleby pochodzącej z terenu położonego przy
dawnej Hucie Bobrek (P1) stwierdzono czternastokrot-
ne przekroczenie dopuszczalnego stężenia (tablica 5).
Oszacowano, że granica wykrywalności wynosi około 7
do 80 fg substancji.
W tablicy 5 przyjęto następujące oznakowania:
P1 (Grupa B) – gleba z terenu położonego przy Hucie
Bobrek,
P2 (Grupa B) – gleba z terenu lasu położonego przy
Hucie Tlenku Cynku „Bolesław” w Bukownie,
P3 (Grupa C) – gleba pobrana z terenu położonego
w odległości 50 m od komina Huty Tlenku Cynku
„Bolesław” w Bukownie,
P4 (Grupa B) – gleba z terenu łąki położonej przy Hu-
cie Tlenku Cynku „Bolesław” w Bukownie,
P5 (Grupa B) – gleba pobrana z terenów położonych
w sąsiedztwie dawnej Huty Zabrze.
4. PODSUMOWANIE
W ramach pracy przeprowadzono cykl badań zawar-
tości wielopierścieniowych węglowodorów aromatycz-
nych w glebach. W ich wyniku wyznaczono za pomocą
pektrometru masowego widma masowe oraz dobrano
odpowiednie warunki rozdziału chromatografi cznego
badanych związków.
Na bazie roztworów wzorcowych wyznaczono wykre-
sy analityczne dla oznaczania grupy 16 wielopierście-
niowych węglowodorów aromatycznych w dichlorome-
tanowych ekstraktach z próbek gleb. Opracowano me-
todę ekstrakcji WWA z zastosowaniem ultradźwięków
oraz zaadaptowano, do oczyszczania uzyskanego eks-
traktu, technikę SPE (Solid Phase Extraction). Prze-
prowadzono próby z użyciem próbek syntetycznych.
Odzysk WWA wahał się w granicach 87–90%.
W oparciu o opracowaną metodykę (analizy i eks-
trakcji) przeprowadzono oznaczenia zawartości WWA
w próbkach gleby. Wszystkie przeanalizowane próbki
gleb wykazywały obecność 16 podstawowych związków
3-10.indb Sek1:16
21.09.2010 09:09:31
17
Prace IMŻ 3 (2010)
Jakościowa i ilościowa identyfi kacja wielopierścieniowych...
z grupy wielopierścieniowych węglowodorów aroma-
tycznych.
Największe ich stężenie stwierdzono w próbce P1
pochodzącej z terenu znajdującego się w bezpośrednim
sąsiedztwie niedziałającej już Huty Bobrek. Suma 10
WWA obecnych w tej próbce przekracza ponad czter-
nastokrotnie wartości dopuszczalne określone w rozpo-
rządzeniu Ministra Środowiska (tablica 5) dla gruntu
z grupy B. Sugerować to może konieczność ponowne-
go określenia stężenia WWA tym razem na większym
obszarze przylegającym do Huty Bobrek. Dodatkowo
w związku z losowym sposobem wyboru obszarów po-
boru próbek zastosowanym w przedstawionej pracy,
należy liczyć się koniecznością przeprowadzenia dal-
szych, bardziej precyzyjnych badań i opracowaniem
odpowiedniego systematycznego schematu pobierania
próbek. W konsekwencji tego powinna powstać szcze-
gółowa mapa zanieczyszczenia tego terenu związkami
organicznymi.
Tablica 5. Zawartość WWA w badanych próbkach gleb oraz ich wartości dopuszczalne stężeń
Table 5. PAH content in soil samples and their admissible concentration values
Składnik
Zawartość WWA, mg/kg
Wartości dopuszczalne
stężeń, mg/kg
P1
P2
P3
P4
P5
Grupa B
Grupa C
Naftalen
9·10
-5
0,002
0,003
0,001
0,001
0,1
40
Acenaftylen
0,09
2·10
-4
3·10
-4
5·10
-5
1·10
-4
-
-
Acenaften
0,11
3·10
-4
4·10
-4
9·10
-5
2·10
-4
-
-
Fluoren
1·10
-4
3·10
-4
4·10
-4
1·10
-4
3·10
-4
-
-
Fenantren
1,8
0,004
0,005
0,001
0,06
0,1
40
Antracen
0,4
0,001
7·10
-4
0,000
0,001
0,1
40
Fluoranten
5,9
0,01
0,007
0,002
0,02
0,1
40
Piren
4,7
0,005
0,006
0,001
0,01
-
-
Benzo[a]antracen
2,6
0,002
6·10
-4
0,0003
0,01
0,1
40
Chryzen
1,6
0,003
0,003
0,001
0,01
0,1
40
Benzo[b]fl uoranten
0,002
1·10
-5
8·10
-5
6·10
-5
0,01
0,1
40
Benzo[k]fl uoranten
0,002
1·10
-5
8·10
-5
6·10
-5
0,01
-
-
Benzo[a]piren
0,002
1·10
-5
5·10
-4
4·10
-5
0,01
0,02
40
Indeno[1,2,3-cd]piren
0,5
3·10
-4
1·10
-4
2·10
-4
3·10
-6
-
-
Dibenzo[a,h]antracen
4,2
0,002
2·10
-4
5·10
-4
8·10
-7
-
-
Benzo[g,h,i]perylen
2,1
0,001
2·10
-4
3·10
-4
2·10
-6
0,1
40
SUMA WWA
14,4
0,023
0,0018
0,0010
0,02
1
200
LITERATURA
Culbart E.B., Thorton I., Watt J., Wheatley, S., Moorcroft, S.
1.
and Thompson M.: Metal contamination in British Dusts and
Soils, J. Environ. Qual. (1988) 17, 226–234.
Dyke P.H., Foan C., Fiedler H.: PCB and PAH release from
2.
power stations and waste incineration processes in UK, Che-
mosphere (2003) 50, 469–480.
Guo-li L., Da-xue L., Quan-ming L.: Heavy metals contamina-
3.
tion characteristics in soil of different mining activity zones,
Trans. Nonferrous Met. Soc. China (2008) 18, 207-211.
Jiayin Dai, Muqi Xu, Jiping Chen, Xiangping Yang, Zhenshan
4.
Ke: PCDD/F, PAH and heavy metals in the sewage sludge
from six wastewater treatment plants in Beijing, China, Che-
mosphere (2007) 66, 353–361.
Krishna A.K., Govil P.K.: Soil contamination due to heavy me-
5.
tals from an industrial
area of Surat, Gujarat, Western India, Govil Environ Monit
6.
Assess (2007) 124, 263–275.
Smith J.N., Lee K., Gobeil C., Macdonald R.W.: Natural rates
7.
of sediment containment of PAH, PCB and metal inventories
in Sydney Harbour, Nova Scotia, Science of the Total Environ-
ment (2009) 407, 4858–4869.
Spiewok W.: Wdrożenie techniki GC/MS do praktyki Labora-
8.
torium Analiz Chemicznych, Sprawozdanie z pracy badawczej
Nr S0-0562, Gliwice 2005.
Spiewok W.: Identyfi kacja i oznaczanie związków organicz-
9.
nych WWA i PCB w pyłach hutniczych, Sprawozdanie z pracy
badawczej Nr S0-0564, Gliwice 2006.
Srinivasa Gowd S., Ramakrishna Reddy M., Govil P.K.: Asses-
10.
sment of heavy metal contamination in soils at Jajmau (Kan-
pur) and Unnao industrial areas of the Ganga Plain, Uttar
Pradesh, India, Journal of Hazardous Materials (2010) 174,
113-121.
White Paul A., Claxton Larry D.: Mutagens in contaminated
11.
soil: a review, Mutation Research (2004) 567, 227–345.
Wolska L., Gdaniec-Pietryka M., Konieczka P., Namieśnik J.:
12.
Problems of PAH quantifi cation by GC–MS method using iso-
tope-labelled standards, Talanta (2009) 78, 730-735.
Recenzent: prof. dr hab. Mariusz Holtzer
3-10.indb Sek1:17
21.09.2010 09:09:31
Wyszukiwarka
Podobne podstrony:
Poprawa wielkości liter za pomocą makra, excel
cw 4 Pomiary wielkości elektrycznych za pomocą oscyloskopu
Sprawozdania przerobione, POMIARY CZASU I CZĘSTOTLIWO¦CI, POMIARY WYBRANYCH WIELKOŚCI ELEKTRYCZNY
Pomiar wielkości fotometrycznych za pomocą walca fotometrycznego, wojtek studia, Automatyka, studia
cw 4, Pomiary wielkości elektrycznych za pomocą oscyloskopu
Sprawozdania przerobione, POMIARY FAZY i PAR. IMP, POMIARY WYBRANYCH WIELKOŚCI ELEKTRYCZNYCH ZA P
Pomiar wielkości komórek wątrobowca za pomocą mikroskopu
Sterowanie za pomocą wielkości nieelektrycznych
Czy rekrutacja pracowników za pomocą Internetu jest
Leczenie za pomocą MIBG
Instrukcja do ćw 06 Sterowanie pracą silnika indukcyjnego za pomocą falownika
Badanie za pomocą ankiety, Psychologia
Dziwny obiekt w okolicy Słońca uchwycony za pomocą koronagrafu SOHO, W ஜ DZIEJE ZIEMI I ŚWIATA, ●txt
Ćw 4; Wyznaczanie gęstości cieczy za pomocą wagi hydrostatycznej
Metoda projektowania układów regulacji za pomocą linii pierwiastkowych
Wyznaczanie przyspieszenie ziemskiego za pomocą wahadła matematycznego
4 Wyznaczanie gęstości cieczy za pomocą wagi hydrostatycznej
Identyfikacja roztworów związków nieorganicznych
więcej podobnych podstron