16.03.2009, Ćwiczenia nr 4., - Proteosomy, Peroksysomy, Lizosomy.
Proteosomy.
–
kompleks enzymów proteolitycznych umożliwiających pozalizosomową degradację białek
w cytozolu
–
struktury nieobłonione
–
biorą udział w destrukcji białek
–
złożone z 4 pierścieni, z których każdy zbudowany jest z 6 kompleksów białkowych
–
białka te tworzą formę kanału wzmocnionego od zewnątrz kwasami nukleinowymi ( w
postaci nieaktywnej 20 S) aby zaaktywować musza przyłączyć się czynniki CF1 i CF2 –
zwiększa się wtedy stała sedymentacji do 26 S
–
sygnałem do działania tych struktur są białka z ubikwityną(4,5):regulatorowe, uszkodzone
itp.
–
zachodzi tzw. ubikwitynizacja białek przeznaczonych do rozłożenia, jest w nich sygnał do
destrukcji – 10 aminokwasów, tam przyłącza się białko rozpoznające a ono powoduje
przyłączenie się ligazy ubikwitynowej i przyłącza się ubikwityna. Jedna jej jednostka
powoduje przyłączenie następnych do 4,5 i takie białko jest rozpoznawalne przez
proteasomy
Dwa rodzaje proteosomów.
–
20S – nieaktywna jednostka podstawowa
–
26S – aktywny kompleks enzymów degenerujących białka zkonigowanych z ubikwityną
Główne etapy proteolizy ubikwitynozależnej.
1) aktywacja ubikwityny z udziałem ATP
2) transestryfikacja – napiętnowanie ubikwityną białka przeznaczonego do degradacji
3) selekcja substratów białkowych skierowanych na drogę proteolizy
4) proteoliza zależna od ATP
5) uwolnienie ubikwityny
Lizosomy.
–
Końcowy szlak endocytarny stanowiący populację obłonionych pęcherzyków zawierające
kwaśne hydrolazy.
–
Pęcherzyki zawierające enzymy rozkładające białka, kwasy nukleinowe, węglowodany i
tłuszcze.
–
Łącznie w lizosomach jest obecnych ok. 40 różnych hydrolaz.
–
Obecność tych enzymów stwarza konieczność obłonienia lizosomów.
–
W lizosomie zachodzi nie tylko proces trawienia komórkowego wchłoniętych pokarmów,
ale także rozkład niepotrzebnych już cząsteczek.
–
W lizosomach zachodzą procesy degradacji między innymi białek glikolipidów i
sulfoglikolipidów oraz estrów wysokocząsteczkowych i estrów nieskocząsteczkowych.
–
Substraty mające ulec degradacji w lizosomach, trafiają tam głównie na drodze endocytozy
klatrynozależnej.
–
Zdecydowana większość enzymów hydrolitycznych stanowią białka rozpuszczalne obecne
w świetle lizosomów.
Białka ulegające proteolizie w lizosomach.
–
wymagają aktywacji i napietnowania ubikwityną
–
proces ich rozkładu zachodzi w wyspecjalizowanej, niebłonowej strukturze – proteosomie
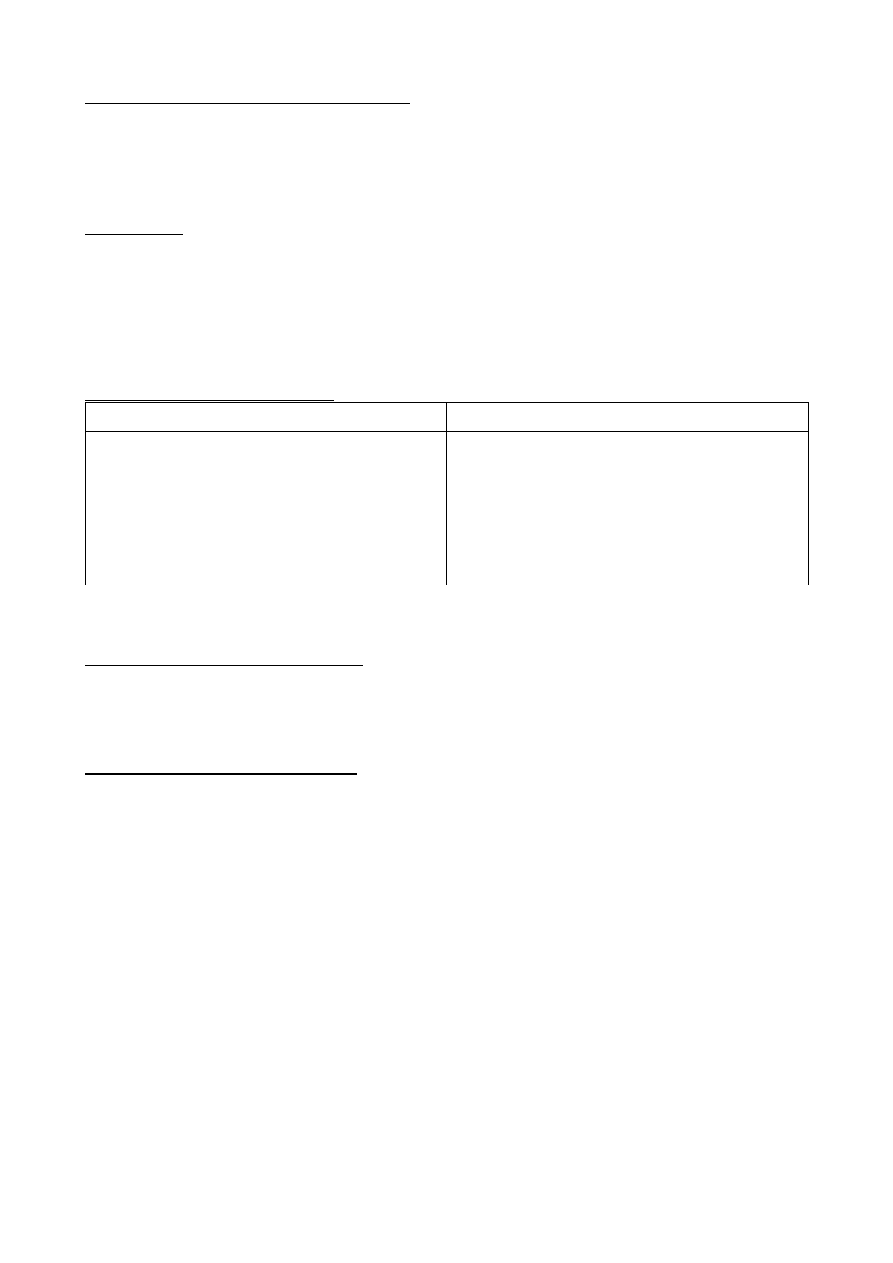
Białka ulegające degradacji w lizosomach.
–
nie wymagają naznaczenia ubikwityną
–
trafiają do obłonionego przedziału – lizosom, drogą endocytozy
–
są hydrolizowane z udziałem kwaśnych proteaz rozpuszczonych w macierzy lizosomów
–
w lizosomoach degradowane są też inne niż białka cząsteczki
Transcytoza.
Pęcherzyki przechodzą z jednej strony błony na drugą. Zjawisko transportowania danej substancji z
jednego bieguna komórki na drugi poprzez cytoplazmę. Z reguły substancja jest pobierana przy
udziale receptora na jednym biegunie i transportowana w pęcherzykach
wewnątrzcytoplazmatycznych. Po dotarciu pęcherzyka na drugi biegun komórki, błona pęcherzyka
zlewa się z błoną komórkową, a substancja uwalnia się do otoczenia. Przykładem może być
transport immunoglobulin A przez komórki nabłonkowe jelita.
Ważniejsze enzymy lizosomalne.
Nazwa enzymu
Substrat
Kwaśna fosfataza
Arylosulfataza
β
α
- Galaktozydyna
β
- Galaktozydyna
Katepsyna L
β
- Glukuronidaza
Heksaminidaza
Estry fosforanowe
Estry siarczanowe
Glikolipidy
Gangliozydy
Białka
Mukopolisacharydy
Glikozoaminoglikan
Proteoliza lizosomowa jest mniej selektywna niż cytozolowa.
Białka ulegają degradacji ponieważ.
–
jest to radzaj regulacji ich aktywności biologicznej
–
ulegają odnowie
–
są źródłem aminokwasów dla nowopowstających białek
Zaburzenia związane z lizosomami.
–
wynikające z braku enzymu (lizosomopatie)
–
mukolipidoza II; brak enzymu odpowiadającego za fosforylację mannozy
–
choroba Huntera (mukopolisacharydoza typu II (MPS II))
–
MPS I (zespół Hurler) - niedobór α-L-iduronizazy
–
MPS II (choroba Huntera) - niedobór sulfatazy iduronianu
–
MPS III (choroba Sanfilippo) - sulfataza-N-heparanu
–
spichrzenie – zaburzenie trawienia lizosomu
–
choroba Gauchera – AD; brak glukcerebrazy w komórkach żernych
–
kumulacja ceramidoglikozydów
–
choroba Niemanna – brak sfingomielidazy i gromadzenie sfingomieliny
–
choroba Taya i Sachsa – brak heksaaminidazy i gromadzenie gangliozyny GM2
–
choroba Fabry'ego – ciężko u mężczyzn; brak ceramido-3-heksodynazy
–
choroba Wolmana – brak kwaśnej lipazy i gromadzenie estrów glicerolu
–
choroba Sapchoffa i Jatzkewitza – tak jak u Taya i Sachsa, gangliozydaza GM1 →
niedorozwój umysłowy, zaburzenia kosci
–
lipogranolomatoza Farbera – gromadzenie cerkulazy
–
leukodystrofie metachromatyczna – metachromazja nerwów

–
glikogenoza
–
glikogenetyczne lizosomy – zmiany przy adaptacji komórek, jej ostre i przewlekłe
uszkodzenia
Gromadzenie wewnątrzkomórkowe prowadzi do przewlekłych uszkodzeń komórkowych.
–
astrocytoza – powstają ciałka resztkowe – w lizosomach zagęszczenie substancji
białkowych
–
autofagia – trawinie własnych struktur komórki; w warunkach fizjologicznych – zanik
gruczołu po laktacji
–
zanik pierwotny – brak energii w komórce – nie ma enzymów glikolitycznych
–
zanik brunatny – w lizosomach gromadzenie się lipidów, białek, fosfolipidów z rozkładu
cytoplazmy (przejaw wewnątrzkomórkowej martwicy)
Peroksysomy (mikrociałka).
–
organella komórkowa o średnicy 0,2 – 1 mikrometra
–
otoczone pojedynczą bloną białkowo-lipidową
–
wnętrze wypełnia jednorodna macierz
–
często zawierają inuluzje(?) krystalicznego białka
–
peroksysomy to głównie frakcja mikrociał, w której zachodzi rozkład H
2
O
2
; stąd pochodzi
nazwa tych organelli
–
zaangazowane w procesy utleniania, stąd ich główny system enzymów tworzą
ksydoreduktazy flawinowe i katalaza
–
produktem ubocznym aktywności peroksydazy jest toksyczny H
2
O
2
–
Jego rozkład (H
2
O
2
) zapewnia katalaza bądź peroksydaza
Rola peroksysomów.
–
detoksykacja komórek przez utlenianie metabolitów i ksenobiotyków
–
rozkład nadtlenku wodoru
–
oksydacja średniołańcuchowych i długołancuchowych kwsaów tłuszczowych do cząsteczek
8-węglowych (dalsze etapy zachodzą w mitochondriach)
–
biosynteza estrolipidów, chlesterolu
–
produkcja kwasów żólciowych
–
przemiana kwasu moczowego do alantoiny (oksydaza moczanowa)
–
metabolizm aminokwasów (alantoinian, gloksinian pierwszy)
Peroksysomy – pierwotne utleniacze:
–
pęcherzyki o średnicy 0,5-1,5 mikrometra otoczone pojedynczą błoną
–
występują w komórkach wątroby i nerek u ssaków, w fotosyntetyzujących komórkach
roślinnych; wystęopują nielicznie w większości komórek
Wnętrze peroksysomu wypełnia elektronowo-gęsta ziarnista macierz, której rdzeń zwany jest
nukleoidem. Strukturę tę stanowi krystaliczna postać oksydazy moczanowej (urykazy)
Organellum komórki eukariotycznej o średnicy 0,2-1,8 μm, otoczone jedną błoną, o kształcie
owalnym bądź sferycznym. W komórce roślinnej peroksysomy znajdują się w bezpośrednim
kontakcie z chloroplastami i mitochondriami i stykają się z powierzchniami ich błon.
U zwierząt występuje tylko jeden typ peroksysomu – zawierający enzym katalazę – uczestniczący
w procesie neutralizacji szkodliwego nadtlenku wodoru.

Metabolizm H
2
O.
RH
2
+ O
2
→
oksydacja, elektrony
R + H
2
O
2
Droga rozkład katalitycznego:
2H
2
O
2
→
katalaza
O
2
+ H
2
O
Droga rozkład peroksydacyjnego:
RH
2
+ H
2
O
2
→
elektrony
R + H
2
O
2
Powstający w wyniku rozkładu H
2
O
2
przez peroksydaza tlen zostaje wykorzystany do utlenienia np.
alkoholi, azotanów, a energia powstała ulega rozproszeniu w postaci ciepła powstaje ATP.
β
- oksydacja kwasów tłuszczowych
–
przebiega w komórkach roślin, grzybów i zwierząt
–
produktem jest acetylo – Co A
–
w komórkach zwierzęcych proces ten zachodzi też w mitochondriach
–
w komórkach zwierzęcych 25% do 50% rozkładu kwasów tłuszczowych zachodzi w
peroksysomach
–
dotyczy to kwasów tłuszczowych o długości łańcucha większej od 16 węgli
–
kwasy o dłuższych łańcuchach (16-20 czy 24-26 węgli) również są rozkładane w
peroksysomach
Metabolizm związków azotu.
Urikaza – oksydaza moczanowa przeprowadza w peroksysomach oksydację produktów przemiany
kwasów nukleinowych (puryny) i niektórych białek.
W metabolizmie związków azotowych są też inne enzymy peroksysomowe – aminotransferazy –
przenoszące grupy aminowe z aminokwasów na - ketokwasy. Peroksydazy biorą udział w
biosyntezie i degradacji aminokwasów.
W metabolizmie związków niezwykłych np. D-aminokwasów albo ksenobiotyków (alkany).
Oksydaza D-aminokwasowa jest być może dowodem, że peroksysomy są najstarszymi
endosymbiontami.
Procesy patologiczne związane z zaburzeniami funkcji peroksysomów (peroksysomopatie):
–
choroba Zellwegera (letalny zespół mózgowo-wątrobowo-nerkowy; zadka choroba
metaboliczna spowodowana zaburzeniem funkcji peroksysomów co powoduje gromadzenie
się w mózgu wielonienasyconych kwasów tłuszczowych o długości łańcucha C
26
-C
38
)
–
ciężka kamica nerkowa
–
adrenoleukodystrofia (choroba Siemerlinga-Creutzfeldta; Jest związana z zaburzoną
peroksysomalną beta-oksydacją kwasów tłuszczowych o bardzo długich łańcuchach co
prowadzi do nagromadzenia ich w różnych narządach)
Dysfunkcje dziedziczne = genetyczne:
–
zaburzenia związane z nieprawidłową biosyntezą peroksysomów, które nie powstają np.
kwasica hiperpi(?)tolowa
–
choroba Zellwegera
–
zaburzenia z dysfunkcją kilku enzymów – pozorny zespół Zellwegera, punktowa dysplazja
chrząstek, karłowatość lizomielicznna
–
defekt jednego enzymu – niedobór enzymu dwufunkcjonalnego, rzekomy zespól
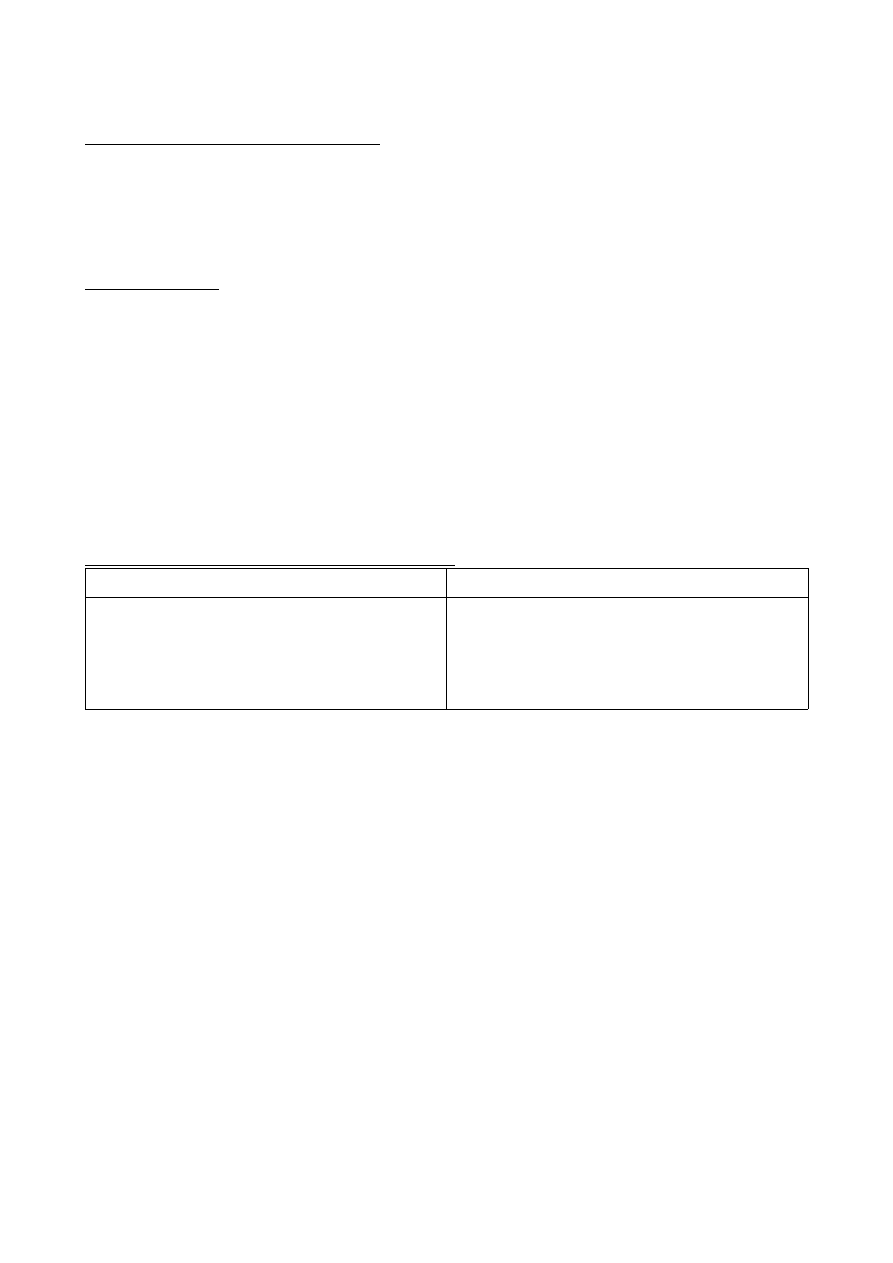
Zellwegera, katalaremia
Hipotezy powstawania peroksysomów.
1) „pączkowanie: błony gładkiej ER
2) podział już istniejących peroksysomów
Najbardziej prawdopodobna jest hipoteza druga, bo białka peroksysomów są syntetyzowane na
polisomach cytosolowych i importowane do już powstałych mikrociał.
Transport białek.
–
Białka są dostarczane potranslacyjnie.
–
Ich transport odbywa się z zużyciem ATP.
–
Muszą być wyposażone w trzyliterowy sygnał targetu - sekwencja SKL.
–
Jest ona zbudowana z 3 aminokwasów: small uncharged – seryna, prolina, alanina; kation
charged – lizyna
Pozbawienie sygnału SKL pozostawia białko perosysomowe w cytozolu. Dołączenie do obcego
bialka – wprowadza je do peroksysomu (uniwersalizm SKL). Uniwersalizm dotyczy białek
pochodzących z innych organizmów. Jako przykład może służyć zlokalizowana w peroksysomach
lucyferaza. Jest ona do nich transportowana w komórkach świetlików i transgenicznych roślin
(tytoń).
Ważniejsze enzymy peroksysomów zwierzęcych.
Enzym
Proces
Oksydaza moczanowa
Oksydaza D-aminokwasów
Acetylo CoA
Acetylotransferaza karnityny
Reduktaza 3-hydroksy-3-metyloglutaryulo-CoA
Katabolizm puryn
Utlenianie aminokwasów
Utlenianie kwasów tłuszczowych
Transport kwasów tłuszczowych
Synteza cholesterolu
________________________________________________________________________________
Odkrywcą peroksysomów jest Christian de Duve.
Peroksysomy różnią się od lizosomów wrażliwością na inny detergent - digitoninę – 10 razy więcej
digitoniny trzeba aby wyzwolić katalazę niż kwasnę fosfatazę. Gdyby były zlokalizowane w tych
samych pęcherzykach – wyzwalałoby je to samo stężenie digitoniny.
Gdy zawierają rdzeń krystaliczny – łatwo je odróżnić na zdjęciach TEM. Gdy go nie ma stosuje się
test DAB (reakcja z diaminobenzydyną).
W reakcji DAB po utlenieniu diaminobenzydyny przez katalazę powstaje polimer łączący się z
czterotlenkiem osmu.
Wyszukiwarka
Podobne podstrony:
0104 16 03 2009, cwiczenia nr 4 , Proteosomy, Lizosomy Paulina Szymczak
0104 16 03 2009, cwiczenia nr 4 , Proteosomy, Lizosomy Kamila Wawiernia(1)
0106 30 03 2009, cwiczenia nr 6 , Wrzeciono podziałowe Paul Esz(1)
0109 27 04 2009, cwiczenia nr 9 , Tkanka nabłonkowa Paul Esz(1)
0103 09 03 2009, cwiczenia nr 3 , Receptoryid 3139
0105 23 03 2009, cwiczenia nr 5 , Jąderko, budowa, funkcja, upakowanie DNA w chromosomy, metody bad
0209 29 04 2009, wykład nr 9 , Tkanka nabłonkowa Paul Esz(1)
0207 08 04 2009, wykład nr 7 , Cykl komórkowy Paul Esz(1)
16.03.2009 chirurgia, medycyna, giełdy, chirurgia, chirurgia
0205 25.03.2009, wykład nr 5.,, Jądro komórkowe
0203 11 03 2009, wykład nr 3 , Białka powierzchni komórkowej Cząsteczki adhezyjne
0202 04 03 2009, wykład nr 2 , Budowa i funkcje błony komórkowej oraz transport przez błony(1)
Historia PRL (5) 16-03-2009, Historia PRL Wykł
16-03-2009 med ret giełda, medycyna, giełdy, Medycyna ratunkowa
Termodynamika wyklad 4 [16 03 2009]
więcej podobnych podstron