METODY ANALIZY KWASÓW NUKLEINOWYCH
Jeżeli chcemy otrzymać DNA z jakiejkolwiek tkanki, to przed wyizolowaniem musimy tę tkankę najpierw:
zhomogenizować:
mechanicznie (homogenizator)
za pomocą ultradźwięków
LUB zniszczyć całą komórkę przy pomocy detergentów, np.:
SDS - sól sodowa siarczanu dodecylu - degraduje błony komórkowe, a dodatkowo denaturuje białka
Triton-X100 - tylko niszczy błony
oprócz detergentów konieczna jest jeszcze podwyższona temperatura i proteinaza K
jeżeli dodatkowo chcemy pozbyć się RNA z naszego preparatu to inkubujemy go w 37°C w obecności rybonukleazy A
Metody izolacji DNA:
ekstrakcja z użyciem mieszaniny fenolu i chloroformu:
kilkukrotne wytrząsanie lizatu z fenolem i chloroformem - usunięcie lipidów i białek (za denaturację białek odpowiedzialny jest fenol -zbierają się one na granicy faz)
wytrącenie DNA - konieczne są:
- izopropanol/etanol
- niska temperatura
- obecność jonów sodu
przemycie osadu etanolem (dalsze oczyszczenie)
rozpuszczenie osadu DNA w jałowej wodzie lub buforze TE o składzie:
- TRIS - zapewnia lekko zasadowe pH (bufor właściwy)
- EDTA - chelatuje jony, przez co DNA jest bardziej stabilny
adsorpcja na kolumnach wypełnionych krzemionką
podczas lizy w buforze z proteinazą muszą być jeszcze sole chaotropowe (KI, nadchloran sodu, guanidyny) - usuwają one wiązania wodorowe między DNA a wodą, dzięki czemu DNA może wiązać się z membraną krzemionkową
tiocyjanian guanidyny dodatkowo ułatwia lizę i inaktywuje nukleazy
przeniesienie lizatu na kolumnę z membraną krzemionkową
selektywna absorpcja DNA do membrany - czyli nic oprócz DNA się tam nie przyklei
odwirowanie kolumny i przemycie jej etanolem - wymycie soli i usunięcie ewentualnych zanieczyszczeń
DNA z kolumny wymywamy j.w. buforem TE lub jałową wodą
generalnie metoda jest prostsza, szybsza, bezpieczniejsza, ale mniej wydajna niż ekstrakcja
przy pomocy cząstek magnetycznych:
cząstki wykonane z tetratlenku żelaza (Fe3O4) o właściwościach magnetycznych miesza się z lizatem komórkowym - DNA specyficznie adsorbuje się na powierzchni cząstek
nie ma tak prosto, te cząstki nie są tylko żelazem; są pokryte:
- polistyrenem/krzemionką zwiększenie adsorpcji DNA
- LUB silanem (silanizacja)
przyłożenie pola magnetycznego w celu usunięcia lizatu
przemycie DNA buforem (tak, żeby go dodatkowo oczyścić) + przyłożenie pola, żeby ten bufor usunąć
elucja - wypłukanie DNA z cząsteczek za pomocą buforu wymywającego + ponowne przyłożenie pola magnetycznego, żeby „zabrać” roztwór DNA z cząsteczek
metoda jest prosta, szybka, wydajna, ale droga
Jak już wyizolujemy DNA, to sprawdzamy, czy nadaje się do dalszych analiz - określamy jego stężenie i ewentualne zanieczyszczenia (fenol, EDTA, heparyna). Korzystamy ze spektrofotometrycznej analizy widma w zakresie 230-280 nm
STĘŻENIE - pomiar absorbancji przy 260 nm (tam kwasy nukleinowe wykazują maksimum absorpcji) -
wartość ta jest wprost proporcjonalna do stężenia
jak przeliczyć absorbancję na stężenie: 1 OD (gęstość optyczna) = 50 μg/ml DNA
CZYSTOŚĆ - pomiar absorbancji przy: 280 nm (maksimum absorbancji białek) i 230 nm
stosunek A260/A280 - ocena stopnia zanieczyszczenia białkami; powinien się mieścić w granicach 1,8 - 2,0
stosunek A260/A230 - ocena stopnia zanieczyszczenia fenolami, EDTA, etanolem i polisacharydami - powinien wynosić ok. 1,8
PCR - Łańcuchowa reakcja polimerazy - służy do powielenia ściśle określonego, wybranego odcinka DNA
główne cechy:
specyficzność - powielamy tylko to co chcemy; można oczywiście powielić cały genom i nazywa się to wtedy WGA
przebiega poza komórką (in vitro)
konieczna obecność enzymów, ale innych niż w procesie replikacji in vivo
składniki reakcji PCR:
specyficzne startery
- jednoniciowe oligonukleotydy komplementarne do sekwencji, która sąsiaduje z tym fragmentem, który chcemy zamplifikować
- umożliwiają inicjację procesu - są „punktem przyczepu” dla polimerazy DNA
- muszą być dwa startery - po każdym dla jednej nici DNA - starter sensowny dla nici sensownej i starter antysensowny dla drugiej nici (i on jest jakby „na drugim końcu” powielanej sekwencji)
matryca - czyli nasz wyizolowany i sprawdzony DNA
rekombinowana polimeraza Taq - termostabilna polimeraza DNA, która katalizuje reakcję wydłużania starterów (czyli przyłącza po kolei kolejne „cegiełki”)
dNTP - trifosforany deoksyrybonukleotydów - substraty reakcji i źródło energii dla polimerazy
bufor - zapewnia odpowiednie pH dla aktywności polimerazy
Mg2+ - kofaktor reakcji polimeryzacji
(glicerol/DMSO) - dodawane w przypadku amplifikowana fragmentów bogatych w pary GC
przebieg procesu:
cały PCR składa się z 30-40 powtarzających się cykli:
TEMPERATURA |
CO SIĘ DZIEJE? |
95°C |
denaturacja DNA - rozplecenie helisy |
50-65°C (charakt. dla danej pary starterów) |
przyłączenie się starterów |
72°C |
wydłużanie starterów pod wpływem polimerazy |
próba kontrolna: mieszanina reakcyjna bez matrycy - sprawdzamy, czy do naszych buforów/starterów etc. nie dostał się oby materiał genetyczny
każdy cykl prowadzi do podwojenia ilości DNA
liczba końcowych kopii = 2x (x - liczba cykli)
UWAGA! Fragmenty o interesującej nas długości pojawiają się dopiero w drugim cyklu
Życie nie jest proste (a już zwłaszcza studenta… no chyba, że na UAMie), więc mamy jeszcze kilka wariacji na powyższy temat tzn. modyfikacji PCR
multipleks PCR - amplifikujemy na raz kilka różnych fragmentów
dodajemy 2-4 par starterów zamiast jednej
zamiast jednego produktu mamy ich kilka
jeżeli chcemy sobie te nasze fragmenty zidentyfikować za pomocą elektroforezy, to muszą one różnić się wielkością, bo inaczej wszystko się wymiesza i nie będzie wiadomo co jest co
Hot-start PCR - PCR z użyciem polimerazy typu Hot-start, która jest aktywowana dopiero w 95°C (czyli wtedy, kiedy denaturuje DNA)
nie powstają wtedy produkty niespecyficzne, ponieważ zaczynamy reakcję dopiero jak już wszystko jest dodane, jesteśmy gotowi, mamy dobry humor etc.
polimeraza jest zablokowana w niższych temperaturach, ponieważ jej centrum aktywne jest blokowane przez:
- przeciwciało
- LUB termolabilną grupę chemiczną, która odłącza się od polimerazy po ogrzaniu
metoda przydatna w amplifikacji malutkich fragmentów DNA
MSP PCR - metylospecyficzna reakcja PCR - umożliwia analizę profilu metylacji danego fragmentu (to jest to co robiliśmy na zajęciach i o niej powiem dokładniej niżej)
MSP PCR - cały myk polega na tym, że zanim zaczniemy powielać nasze DNA poddajemy je konwersji w obecności NaHSO3:
sekwencja zmetylowana nie ulegnie zmianie i nadal będzie zawierała cytozynę
sekwencja niezmetylowana ulegnie modyfikacji - niezmetylowana cytozyna zamieni się na uracyl
skoro mamy dwie różne „wersje” tego samego DNA, to trzeba użyć dwóch różnych par starterów
jeden dla sekwencji zmetylowanej w której są cytozyny i komplementarne do nich guaniny
drugi dla sekwencji niezmetylowanej, w której będzie uracyl i komplementarna do niego adenina
musimy jeszcze wrzucić coś w stylu „prób odniesienia”, żeby skontrolować, czy wyniki są wiarygodne (czy bufor był dobry, stratery nie za stare etc.)
kontrola pozytywna - DNA, który został CAŁKOWICIE zmetylowany dzięki bakteryjnej metylotransferazie
kontrola negatywna - DNA zupełnie niezmetylowany
i te kontrole powinny dać produkty z odpowiednimi dla siebie starterami - jeżeli nie, to mamy problem…
interpretacja wyników:
Starter dla zmetylowanego DNA |
Starter dla niezmetylowanego DNA |
Interpretacja |
Produkt |
Brak produktu |
Gen zmetylowany |
Brak produktu |
Produkt |
Gen niezmetylowany |
Produkt |
Produkt |
Tylko jeden z alleli genu jest zmetylowany |
zastosowanie:
farmakoepigenetyka, np. pacjent ze zmetylowanym genem MGMT nie będzie reagował na leki alkilujące i jego nowotwór będzie trzeba leczyć czymś innym
ELEKTROFOREZA DNA - metoda identyfikacji otrzymanych w PCR fragmentów DNA.
zasada metody:
DNA ma reszty fosforowe, czyli jest polianionem
w związku z tym, będzie go ciągnęło do dodatnio naładowanej elektrody, czyli ANODY (błagam, niech ktoś to przeczyta i skoryguje/ potwierdzi - zawsze mam z tym problem)
im mniejsza cząsteczka DNA, tym szybciej i dalej będzie migrowała w żelu po przyłożeniu napięcia - zależność odwrotnie proporcjonalna
nośnikami są:
żele agarozowe - rozdzielczość mniejsza, ale wystarczająca, zwiększa się wraz ze wzrostem stężenia agaru (które wynosi 0,7 - 3,0%)
żele poliakryloamidowe - rzadziej stosowane, ale mają wyższą rozdzielczość (tzn. fragmenty mogą się mniej od siebie różnić masą, żeby się ładnie rozdzieliły)
do przeprowadzenia elektroforezy konieczny jest też roztwór przewodzący prąd, na którym w sumie przygotowuje się żel:
bufor TBE - TRIS + kwas borny + EDTA
bufor TAE - TRIS + kwas octowy + EDTA
przebieg procesu:
rozpuszczamy agarozę w buforze z dodatkiem barwnika fluoryzującego (patrz niżej) na gorąco pilnując, żeby nam kuchenka mikrofalowa nie wybuchła xD
wylewamy żel do formy, żeby zastygł i wkładamy grzebienie, żeby zrobić w nim studzienki na DNA
taki gotowy żel wkładamy do aparatu do elektroforezy, w którym również jest już bufor
nanosimy próbki DNA, które:
- mają dodany barwnik, żeby było widać, kiedy i gdzie elektroforeza się skończyła
- zostały obciążone czymś zwiększającym lepkość (np. glicerolem), żeby przypadkiem nie wypłynęły ze studzienek zanim zaczniemy elektroforezę
wizualizacja dzięki barwnikom fluorescencyjnym (np. bromkowi etydyny), które są zdolne do interkalowania DNA, czyli związane z DNA wykazują jeszcze większą fluorescencję niż bez DNA - tam gdzie są paćki od fluorescencji, tam zatrzymał się DNA
identyfikacja fragmentów DNA z wykorzystaniem wzorca wielkości DNA - mieszanina fragmentów DNA o znanej wielkości - fragmenty o takiej samej wielkości będą dawały „paćkę” na elektroforegramie na tej samej wysokości
MIKROMACIERZE DNA - umożliwiają badanie wieeeeelu fragmentów DNA jednocześnie.
zastosowanie:
analiza poziomu ekspresji genów na poziomie RNA
ocena SMP - polimorfizmu pojedynczych nukleotydów (np. genów kodujących CYPy - test farmakogenetyczny AmpliChip)
określanie profilu metylacji DNA
zasada metody:
analogicznie do blottingu (pytanie, czy pamiętamy coś jeszcze z seminarek… xD), z tym, że tutaj używa się tysięcy sond zamiast kilku
poza tym barwnikami fluorescencyjnymi barwi się kwasy nukleinowe, a nie sondy
no i te sondy są przymocowane do stałego podłoża, na które nanosimy preparat
rodzaje mikromacierzy:
|
mikromacierze cDNA |
mikromacierze oligonukleotydowe |
sposób syntezy |
przy pomocy odwrotnej transkryptazy na matrycy mRNA |
chemiczny, np. fotolitografia |
znajomość sekwencji docelowej |
niekonieczna |
konieczna |
liczba sond na płytce |
kilkaset - kilkadziesiąt tysięcy |
kilkaset tysięcy |
co oceniamy |
wszystkie rodzaje RNA, które służyły do syntezy mikromacierzy |
? - jedna sonda jest zaprojektowana specyficznie do jednego transkryptu |
przygotowanie RNA |
1) próbkę też podajemy działaniu odwrotnej transkryptazy i znaczymy znacznikiem: czerwonym lub zielonym (osobny dla komórki zdrowej, osobny dla nowotworowej) 2) miesza się obie frakcje RNA („czerwoną” i „zieloną”) |
1) próbkę poddajemy działaniu odwrotnej transkryptazy 2) transkrypcja cDNA in vitro wyznakowane biotyną cRNA |
przebieg procesu |
|
Dwa eksperymenty - osobno dla próby badanej, osobno dla kontrolnej |
WYNIK |
WYNIK WZGLĘDNY porównuje się różnice w takich obrazach z komórki zdrowej i z komórki nowotworowej - im silniejsza fluorescencja tym silniejsza ekspresja |
WYNIK BEZWZGLĘDNY: Obraz miejsc i intensywność fluorescencji niesie informację o tym, które geny z jakim nasileniem uległy ekspresji (nie musimy odnosić się do próby kontrolnej, żeby stwierdzić, co się zmieniło) |
PCR W CZASIE RZECZYWISTYM
Ilościowe metody oznaczania RNA:
elektroforetyczne - porównanie rozdzielonego RNA ze znacznikiem masy
hybrydyzacyjne - Northern blotting
amplifikacyjne - PCR w czasie rzeczywistym (rt PCR)
PCR w czasie rzeczywistym - po każdym cyklu sprawdza się poziom fluorescencji, która jest wprost proporcjonalna do ilości skopiowanego w danym cyklu materiału genetycznego.
oznaczać w ten sposób można:
DNA
cDNA, które powstało na matrycy mRNA w procesie odwrotnej transkrypcji czyli de facto oznaczamy mRNA, co przydaje się do oceny ekspresji genów
etapy procesu:
izolacja RNA
reakcja odwrotnej transkrypcji (mRNA cDNA)
właściwy proces rt PCR - metoda jedno- lub dwuetapowa
interpretacja wyników:
ten wynik co go otrzymujemy jest liczbą cykli, które musiały zajść, żeby w sumie wyszła określona ilość materiału genetycznego
i mamy taką zależność:
- jest ekspresja dużo DNA mało koniecznych cykli MAŁY WYNIK
- nie ma ekspresji mało DNA trzeba dużo cykli, żeby go powielić DUŻY WYNIK
zastosowanie metody:
genotypowanie (np. określanie polimorfizmów poszczególnych genów)
oceny procentowej zawartości GMO w danym produkcie
określenie stopnia zainfekowania wirusem/bakterią
badanie oporności ustrojstw na leki
Izolacja RNA:
całe przedsięwzięcie jest problematyczne, bo RNA jest nietrwały:
podatne na hydrolizę wiązanie fosfodiestrowe w rybozie
łatwo rozkładane przez RNazy, które nie potrzebują do działania żadnych dodatkowych kofaktorów (w przeciwieństwie do DNaz)
dlatego konieczne są inhibitory RNaz, np. autoklawowana woda z DEPC
podobnie jak w przypadku DNA, izolacja RNA musi być poprzedzona homogenizacją:
rozdrobnienie w ciekłym azocie + homogenizacja (ręczna/mechaniczna)
niektóre tkanki wymagają nadtrawienia protezą K (np. mięśnie)
metoda Chomczyńskiego (AGPC) - umożliwienie izolacji RNA z wielu próbek jednocześnie
mieszanina chlorofmu i fenolu zawiera także sole chaotropowe (guanidyny)
ekstrakcja w mieszaninie chloroformu i etanolu rozdzielenie DNA (który idzie do organicznej fazy fenolowej) od RNA (z fazy wodnej) + denaturacja białek, które zostają na granicy faz
do fazy wodnej dodaje się jeszcze DNaz, bo zawsze trochę DNA zostaje
wytrącenie osadu RNA przemycie ponowne rozpuszczenie
metoda kolumienkowa
liza komórek solami chaotropowymi w temperaturze pokojowej
lizat nakłada się na kolumienkę
adsorpcja białek, resztek struktur komórkowych i innych syfów
w przesączu mamy RNA, DNA, resztki białek
przemywamy przesącz etanolem i wrzucamy na drugą kolumnę
tu adsorbuje się już RNA
wymywamy RNA z kolumny odpowiednim buforem
I znowu analogicznie do badań DNA - sprawdzamy, czy to nasze wyizolowane DNA w ogóle się do czegoś nadaje:
spektrofotometrycznie:
pomiar absorbancji przy 230, 280, 320 nm lub widmo w zakresie 200 - 350 nm
stosunek A260/A280 - stopień zanieczyszczenia preparatu, powinien mieścić się w granicach 1,8 - 2,1
- < 1,8 zanieczyszczenie białkami
- > 2,1 zanieczyszczenie rozpuszczalnikami organicznymi
elektroforeza:
na żelu agarozowym z czynnikiem denaturującym
RNA daje 2 wyraźne prążki od frakcji 18S rRNA i 28S rRNA
jak ich nie ma to znaczy, że DNA się zdegradowało i nie ma co ciągnąć dalej badania
przydaje się zwłaszcza w przypadku izolacji z tkanek roślinnych
To teraz dla odmiany coś, czego przy analizie DNA nie było - reakcja odwrotnej transkrypcji
ma na celu syntezę cDNA - jednoniciowej kopii RNA
do reakcji konieczne są:
odwrotna transkryptaza
startery - krótkie odcinki DNA
powstają hybrydy cDNA-RNA, które są substratami we właściwej reakcji rtPCR
rodzaje starterów:
specyficzne - synteza mRNA interesującego nas genu i niczego więcej
oligo(dT) - syntetyzują całe mRNA bez innych RNA, ponieważ mają zdolność do przyłączania się do ogonów poli-A na końcu 3' mRNA
losowe startery heksametryczne - synteza całego kodującego RNA (mRNA, tRNA, rRNA)
etap ten jest kluczowy dla całego procesu rtPCR
Właściwa reakcja PCR:
warunki procesu:
TEMPERATURA |
CZAS |
CO SIĘ DZIEJE? |
95°C |
jedna do kilkunastu minut |
rozplecenie helisy cDNA aktywacja polimerazy Taq |
50-60°C |
|
przyłączenie się starterów - tzw. hybrydyzacja odcinków starterowych |
72°C |
|
wydłużanie starterów pod wpływem polimerazy |
uwagi:
startery przyłączają się komplementarnie do specyficznej sekwencji mRNA, którą chcemy oznaczyć
ta sekwencja ZAWSZE będzie należała do rejonu intronowego DNA eliminacja powielenia DNA genomowego
etap dodatkowy, którego nie ma w klasycznym PCR - pomiar fluorescencji
względny wzrost fluorescencji jest wprost proporcjonalny do liczby kopii cDNA
poziom fluorescencji możemy oznaczyć metodą:
- nieswoistej detekcji
- swoistej detekcji
Metoda nieswoistej detekcji
używa się fluorochromów, które wykazują zdolność do interkalacji niespecyficznego dsDNA, np.:
bromek etydyny
SYBER Green 1:
- wzbudzenie przy 497 nm
- emisja światła o długości 520 nm
taki znacznik związany z cDNA, wykazuje większą fluorescencję niż bez cDNA
zalety: niski koszt
wady: może powodować błędne zawyżenie wyniku ekspresji, np. przez dimery starterów
żeby tego uniknąć wykonuje się analizę topnienia po zakończeniu reakcji amplifikacji:
- zwiększamy temperaturę mieszaniny tak o 0,5°C i mierzymy w tym czasie fluorescencję
- dsDNA będzie się „topił”, czyli rozplatał helisę, przez co znacznik będzie miał mniej cząsteczek, z którymi mógłby się związać i fluorescencja będzie spadać
- pojedynczy pik odpowiadający temperaturze topnienia świadczy o tym, że została powielona tylko jedna sekwencja i nie ma żadnych syfów (każde DNA ma swoją temperaturę topnienia i gdyby próba była czymś zanieczyszczona to wyszłoby więcej pików)
Metoda swoistej detekcji
zamiast znaczników fluorescencyjnych używa się:
sondy molekularnej
barwnika fluorescencyjnego przyłączonego do sondy
mechanizm działania sond hydrolizujących typu TaqMan
sonda składa się z barwnika oraz wygaszacza (cząsteczki wyciszającej fluorescencję)
po przyłączeniu do cDNA barwnik fluoryzuje, ale emitowane światło jest pochłaniane przez wygaszacz
w momencie, kiedy sekwencja się wydłuży i polimeraza „dojdzie” do sondy to ją hydrolizuje - barwnik się odłącza i już nic mu nie przeszkadza w emisji światełka wzrost fluorescencji
Porównanie obu metod:
Cecha |
Metoda niespecyficzna |
Metoda specyficzna |
koszt |
niższy |
wyższy |
specyficzność |
niska |
wysoka |
wygoda |
tak - uniwersalność dla każdej sekwencji |
nie - do każdej sekwencji trzeba projektować sondę |
wpływ mutacji w DNA na czułość |
nie |
tak |
wpływ niespecyficznych produktów na czułość |
zawyżenie wyników |
nie |
wpływ długości amplikonu na fluorescencję |
tak |
nie |
reakcja multiplet |
nie |
tak |
ocena specyficzności |
konieczna (elektroforeza lub krzywa topnienia) |
niekonieczna |
KRZYWA AMPLIFIKACJI REAKCJI rtPCR - wykres zależności intensywności sygnału fluorescencji od liczby cykli oznaczającej upływ czasu reakcji
na wykresie zaznaczamy względny wzrost fluorescencji po każdym cyklu (ΔRn) - różnice między względnym poziomem fluorescencji a poziomem tła
nachylenie tej krzywej jest zawsze takie samo i oznacza wydajność reakcji amplifikacji
wyznaczamy CT - liczbę kopii cDNA, występującą w momencie, gdy krzywa amplifikacji przecina linię progową wyznaczoną arbitralnie
im większa początkowo liczba kopii tym MNIEJSZA wartość CT, ponieważ potrzeba jest mniej cykli, żeby osiągnąć taką ilość kopii cDNA, która przekroczy linię progową
w krzywej amplifikacji wyróżniamy kilka faz:
pierwszą - „maskowaną” przez fluorescencję tła
drugą - amplifikacja powyżej tła (nieliniowa)
trzecią - liniowy wzrost fluorescencji
czwartą - fazę plateau - wysycenie i akumulacja produktu
Określenie wydajności reakcji rtPCR:
optymalna wydajność = 90-105%
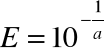
w celu jej określenia wykonujemy krzywą standardową - zależność CT od logarytmu dziesiętnego wyjściowego stężenia szeregu rozcieńczeń jeden próbki DNA;
wydajność zależy od:
oznaczanego genu
warunków reakcji
zastosowanego enzymu
metody detekcji
pochodzenia materiału do analizy
użytych starterów i termocyklera
Standardy kontroli - pozwalają na weryfikację przebiegu procesu:
WEWNĘTRZNE:
określenie poziomu przynajmniej jednego z genów homeostazy, np. gen β-aktyny
geny homeostazy w każdej komórce ulegają ekspresji na stałym poziomie, więc dlatego można je użyć jako geny referencyjne
jeżeli porównujemy dwa wyniki to nie porównujemy ich bezpośrednio, tylko najpierw dzielimy przez wynik dla genu homeostazy i porównujemy dopiero te ułamki
ZEWNĘTRZNE - dodanie znanej ilości cząsteczek DNA/RNA do każdej próby (w jednej probówce i dla jednej pary starterów)
Określanie ilości wyjściowego DNA - czyli to o co nam tak właściwie chodzi:
metoda BEZWZGLĘDNA:
nanosimy wartości CT na krzywą standardową
taką krzywą wyznacza się doświadczalnie na podstawie serii rozcieńczeń DNA o znanej liczbie kopii
metoda używana w przypadku określania:
- procentowej zawartości DMO w produkcie
- liczby ustrojstw w papu
- stopnia zainfekowania materiału biologicznego ustrojstwem
metoda WZGLĘDNA:
uzyskane wartości odnosimy do wartości CT dla kontroli
poziom ekspresji w kontroli uznajemy za 1 - i albo będzie większy albo mniejszy
pozwala nam to zaobserwować zmiany na poziomie ekspresji genu, ale nie wiemy ile tego materiału genetycznego było (w sumie to czasem nawet nie jest potrzebne).
ANALIZA BIAŁEK a'la BIOLOGIA MOLEKULARNA
PROTEOM - wszystkie białka ulegające ekspresji + informacje ich dotyczące
Techniki stosowane w analizie białek:
rozdzielenie białek na podstawie różnic w ich masach - filtracja żelowa
rozdzielenie białek z wykorzystaniem ładunku elektrycznego:
chromatografia jonowymienna
elektroforeza
ogniskowanie izoelektryczne
rozdzielenie białek z wykorzystaniem ich powinowactwa do określonych grup chemicznych - chromatografia powinowactwa
metody immunochemiczne (tym już badamy białka, które są oczyszczone powyższymi metodami:
ELISA
Western blot
spektroskopia mas - określenie tożsamości białka na podstawie jego masy, np. MALDI
Chromatografia powinowactwa:
białko specyficznie adsorbuje na ligandzie
ligand = substancja chemicznie związana z nieruchomym nośnikiem (i w ten sposób sama jest unieruchomiona)
ligandy wiążą określone białeczko spośród wielu wielu innych, przez co za jednym zamachem od razu je somie wydzielimy
rodzaje oddziaływań między białkiem a ligandem:
hormon-receptor
enzym-substrat/ enzym-inhibitor
przeciwciało-antygen/ przeciwciało-hapten
dopełniacz-przeciwciało IgG
odcinek kwasu nukleinowego - cukrowiec lub lektyna
nośniki - muszą być zaktywowane przed przeprowadzeniem procesu (np. przez reakcję z halogenocyjanami); są to pochodne:
dekstranowe
agarozowe
poliakrylamidowe
przebieg procesu
ligand (np. sefaroza z glutationem - substrat dla GST) wiąże się z nośnikiem i ulega unieruchomieniu
GST znajdujący się w roztworze badanym wiąże się ze swoim substratem (ligandem)
cała reszta, która się nie związała ulega wypłukaniu
elucja substratu (musimy go jakoś oddzielić od ligandu przecież) - czyli wypłukanie go z połączenia za pomocą dużego stężenia glutationu (enzym pójdzie tam, gdzie ma więcej substratu i zostawi nasz ligand)
taki eluat z naszymi izoformami GST będzie dalej badany bardziej specyficznymi metodami
ELEKTROFOREZA:
odpowiednie podłoża zapewniają połączenie dwóch technik:
sączenia molekularnego wykorzystującego różnice w masie poszczególnych białek
elektroforezy
rodzaje podłóż:
agarozowe - stosowane do sączenia większych cząsteczek - powyżej 200 kD (ma większe pory)
poliakrylamidowe - stosowane w technice PAGE (sączeniu mniejszych cząstek)
poliakrylamidowe + SDS (siarczan dodecylu) - stosowane w SDS-PAGE
SDS-PAGE:
SDS denaturuje białka i maskuje ich ładunek (sam jest silnie ujemny)
w tym momencie rozdział zachodzi tylko na zasadzie filtracji molekularnej, a o elektroforezie zapominamy
jeżeli użyliśmy markera masy możemy wyznaczyć masę białeczka
ograniczenie: analizujemy tak tylko białka proste lub pojedyncze podjednostki, ponieważ SDS niszczy wiązania pomiędzy podjednostkami, dzięki którym istnieją w ogóle białka złożone
chociaż to się czasem przydaje - można sobie sprawdzić z ilu podjednostek składa się białko
dodatek 2-merkaptoetanolu służy do sprawdzenia, czy poszczególne podjednostki są połączone wiązaniami disiarczkowymi
IEF - ogniskowanie izoelektryczne:
wykorzystanie zależności między ładunkiem cząsteczki białka a pH środowiska
przebieg procesu:
do nieruchomych nośników dodaje się amfolit uzyskanie gradientu pH
pH poniżej punktu izoelektrycznego dodatnio naładowane białko migracja w kierunku katody (-)
migruje, migruje, a pH wzrasta i wzrasta, aż w końcu osiągnie wartość pI (punktu izoelektrycznego) - białko nie ma ładunku i KONIEC migracji
Elektroforeza dwukierunkowa 2D (a niby czemu nie 3D, hm?) - połączenie IEF oraz SDS-PAGE:
w kierunku poziomym rozdziela się białka wg ich pI (metoda IEF)
tak rozdzielone białka rozdziela się w kierunku pionowym wg ich masy cząsteczkowej (metoda SDS-PAGE)
żeby było jeszcze ciekawiej można to połączyć z MS
Elektroforeza kapilarna - rozdział białek w kwarcowej/szklanej/plastikowej kapilarze wypełnionej buforem
metoda jest szybsza, łatwiejsza do interpretacji i jeszcze można ją zautomatyzować, żeby w ogóle było cudownie…
mała średnica = większe odprowadzenie ciepła możemy przyłożyć większe napięcia KRÓTSZY CZAS
w kapilarze może być:
bufor
żel SDS-PAGE
amfolit
jak zwykle wszystko ma swoją ciemną stronę (mocy…) i:
zaciera się ostrość rozdziału (no jak idzie za szybko to tak jest…)
można rak rozdzielać tylko małe próbki (większe się fizycznie do kapilarki nie wcisną ;P)
METODY IMMUNOCHEMICZNE - wykorzystują swoistą reakcję antygen-przeciwciało zachodzącą przez determinanty antygenowe (epitopy)
rodzaje przeciwciał:
poliklonalne - pochodzą od różnych populacji komórek i różnią się powinowactwem do antygenu, swoistością
monoklonalne - wytworzone przez pojedynczy klon komórek - identyczne powinowactwo u wszystkich
rodzaje znaczników używanych do identyfikacji kompleksu antygen-przeciwciało:
biochemiczne - biotyna i znaczniki enzymatyczne - enzymy, które katalizują reakcje, w których z bezbarwnego cosia powstaje barwny cosiek absorbujący światło, np. fosfataza alkaliczna, peroksydaza chrzanowa (o lol xD), β-galaktozydaza
radioaktywne - izotopy fosforu, siarki i jodu - identyfikacja metodą autoradiografii
fluorescencyjne, czyli fluorochromy, np. kumaryna, fluoresceina, rodamina
przykłady metod:
EIA - testy immunoenzymatyczne:
test immunoenzymatyczny ELISA
RIA - testy radioimmunologiczne - jeden ze składników reakcji (ten, który dodajemy, bo trudno oznakować coś co jest nieznane) jest znakowany izotopem (131I, 125I, 14C, 3H)
FIA - testy immunofluorescencyjne
immunoprecypitacja - określane białko wiąże się z przeciwciałem, a potem taki kompleks wytrąca się za pomocą np. sefarozy
Western blot
Test ELISA - służy do wykrywania i oceny ilościowej konkretnych antygenów lub przeciwciał
wykorzystuje się tu tworzenie kompleksu między antygenem, a jego przeciwciałem pierwszorzędowym (specyficznego dla danego antygenu)
możliwe jest również wykorzystanie tzw. przeciwciał drugorzędowych - wykrywa ono kompleks antygenu z przeciwciałem pierwszorzędowym, ale
w przeciwieństwie do wersji „tradycyjnej” to przeciwciało drugorzędowe jest znaczone i to na jego podstawie mamy wynik
czyli mówiąc prościej: za pomocą przeciwciała drugorzędowego możemy pośrednio oznaczyć przeciwciało pierwszorzędowe
test jednowarstwowy - przebieg:
w badanym materiale mamy antygen, który wiąże się z podłożem i czeka…
dodajemy przeciwciała swoiste do tego antygenu, który chcemy wykryć/oznaczyć - są one oznaczone konkretnym znacznikiem
powstają kompleksy antygen-przeciwciało
wypłukujemy te przeciwciała, które się nie przyłączyły lub zrobiły to niespecyficznie - jeżeli damy inny antygen, to skoro coś jest niespecyficzne to zostawi ten o który nam chodzi i ucieknie z tym dodanym
enzymy znacznikowe, które były połączone z przeciwciałami zaczynają produkować barwny produkt możemy oznaczać, bo mamy co zmierzyć (absorbancję)
test dwuwarstwowy:
wszystko jak wyżej - nasz antygen jest wykrywany przez przeciwciała i powstaje kompleks
tylko tutaj do antygenów pierwszorzędowych (zwykle monoklonalnych) przyczepiają się te drugorzędowe (zazwyczaj poliklonalne) i to właśnie one mają znacznik (enzym tworzący barwny produkt)
test pośredni („kanapkowy”) - unieruchomiony antygen reaguje z pierwszorzędowymi nieoznakowanymi przeciwciałami
lub odwrotnie - nieoznakowane przeciwciała wychwytują antygen z mieszaniny
a potem dodajemy oznakowane przeciwciała drugorzędowe
Test FIA
barwnik fluorescencyjny przyłączony jest kowalencyjnie do przeciwciała przez reszty lizyny
przykłady barwników:
TRITC i APC czerwone
FITC zielone
fikoerytryna (PE) pomarańczowe
rodzaje testów:
bezpośredni - wykrywamy obecność antygenu związanego ze znakowanym przeciwciałem pierwszorzędowym
pośredni - bardziej czuły; wykorzystujemy znakowane przeciwciała drugorzędowe (analogicznie jak wyżej)
Western blot
rozdzielone białka przenosimy na podłoże (błonę, nie żel):
białka po elektroforezie są na żelu
z tego żelu robimy „kanapkę”: żel-bibuła-błona-bibuła
„kanapkę” kładziemy między gąbki z buforem do transferu i przykładamy napięcie
ujemne białka migrują do anody i zatrzymują się na błonie
trzeba uważać, żeby nie transferować zbyt długo (białka przejdą poza błonę) ani za krótko (nie dojdą do błony)
rodzaje błon:
błona nitrocelulozowa - wiąże białka wiązaniami hydrofobowymi
błona nylonowa - wiąże białka wiązaniami hydrofobowymi i elektrostatycznymi
błona z PVDF (z poliwinylidenu) - do sekwencjonowania białek
dlaczego na błonę?
odporność mechaniczna
możliwość przeprowadzania reakcji barwnych
OK - jak już mamy białko na błonie musimy zablokować miejsca niespecyficznego wiązania (żeby nie przyłączyło nam się nic, tam gdzie nie chcemy)
roztwór mleka w proszku
surowica bydlęca
albumina + detergent
teraz inkubujemy naszą błonę razem z przeciwciałami pierwszorzędowymi, żeby dać im szansę znaleźć nasze białeczko i się z nim związać
jeżeli są one nieoznakowane (co zazwyczaj się dzieje), to będziemy musieli użyć jeszcze oznakowanych drugorzędowych
gdybyśmy oznakowali przeciwciało I-rzędowe to byłoby szybciej, ale drożej (a mówią, że czas to pieniądz…)
warunki: temperatura pokojowa, 1-12 h
przeciwciała I-rzędowe związane z NIP (nitrojodofenolem) ograniczenie niespecyficznych reakcji przeciwciał II-rzędowych
inkubacja z przeciwciałem drugorzędowym (o ile jest konieczność)
zawierają one enzymy katalizujące reakcje barwne
jeżeli oznakowano przeciwciała I-rzędowe biotyną, to do ich detekcji można użyć awidynę +odpowiedni enzym - większa czułość
odczyt wyniku:
klisza fotograficzna
urządzenie badające chemiluminescencję
MIKROMACIERZE BIAŁKOWE - badamy oddziaływania między białkami
mikromacierz - płytka na której są odpowiednio ułożone różne ligandy (przeciwciała, receptory, enzymy etc.) wyłapujące konkretne białka
wizualizacja - przeciwciała II-rzędowe znakowane:
fluorescencyjnie
chemiluminescencyjnie
metody kolorymetryczne oparte na wytrąceniu srebra
SPEKTROMETRIA MAS:
zasada metody:
badane białko przeprowadzamy w stan gazowy i jonizujemy laserem
mierzymy odchylenie jonów w polu elektromagnetycznym - zależy ono od masy cząsteczki
przebieg:
rozdział białek - elektroforeza dwukierunkowa
trawienie pojedynczego białka
odparowanie próbki razem z matrycą
taka płytka idzie do próżni, gdzie maltretuje się ją laserem jest gaz
faza gazowa idzie do spektrometru masowego, który wszystko mierzy (dokładniej - czas przelotu jonów)
wychodząc z maszyny cząsteczki rozdzielają się wg masy - mniejsze wychodzą szybciej
- antygen/przeciwciało sprzęga się z enzymem
- powstaje tyle barwnego produktu, ile powstało kompleksów antygen-enzym, a to już można oznaczyć spektrofotomerycznie
- najczęściej wykorzystuje się metodę kompetycyjną (wykorzystanie współzawodnictwa antygenu oznakonego z nieoznakowanym o wiązanie z przeciwciałami)
- oznacza się hormony, leki, witaminy czy antygeny nowotworowe
- po rozdziale mierzymy radioaktywność próbki
- potem się rozdziela i Western blottuje
- CHIP - izolacja białek histonowych zmodyfikowanych kowalencyjnie
![]()
Wyszukiwarka
Podobne podstrony:
Test z ćwiczeń 1-6, rok numer trzy, tpl, kolokwia
Test z ćwiczeń 7-10, rok numer trzy, tpl, kolokwia
Test z ćwiczeń 1-6 + odpowiedzi, rok numer trzy, tpl, kolokwia
Morfologia, rok numer trzy, farmakognozja, sprawdziany = kolokwia
spr 2(1), rok numer trzy, farmakognozja, sprawdziany = kolokwia
spr 1(1), rok numer trzy, farmakognozja, sprawdziany = kolokwia
klo farmakologia II(1), rok numer trzy, farmakognozja, sprawdziany = kolokwia
gnozja egzamin gr 2, rok numer trzy, farmakognozja, egzamin
Pytania - spr 3, rok numer trzy, farmakognozja, sprawdziany = kolokwia
pytania egzamin 2009, rok numer trzy, farmakognozja, egzamin
spr 4, rok numer trzy, farmakognozja, sprawdziany = kolokwia
Kolokwium 2 (sem) - pytania, rok numer trzy, chemia leków, seminaria
Gnozja - wykłady by me, rok numer trzy, farmakognozja, wykłady
TPL - wykłady z receptury jałowej, rok numer trzy, tpl, wykłady
Sprawdzian 1, rok numer trzy, farmakognozja, sprawdziany = kolokwia
spr 1, rok numer trzy, farmakognozja, sprawdziany = kolokwia
więcej podobnych podstron