
CHAPTER
9
Pediatric Brain
Tumors
Ann Marie Flannery
Farivar Yaghmai
The frequency, location, prognosis, and presenting signs and
symptoms of pediatric brain tumors are areas of frequent
misconception. Pediatric brain tumors are not rare; they
represent the most common solid tumor of childhood and the
second most common malignancy of childhood.
1
-
2
Three to
five new cases are seen per 100,000 children per year.
Pediatric tumors of the central nervous system (CNS) are
classically distributed with a posterior fossa preponderance.
However, in clinical practice, more than 50 percent of pedi-
atric tumors of the CNS are supratentorial.
3
'
4
This incidence
may reflect patterns of referral rather than change in inci-
dence.
The prognosis of pediatric brain tumors is better than is
generally assumed. The most common tumors can be treated
with surgery alone or with a combination of surgical and
adjuvant therapies.
CLINICAL PRESENTATION
Although numerous types of brain tumors occur during
infancy and childhood, the clinical presentations tend to be
rather nonspecific for a particular type of brain tumor. Signs
and symptoms of increased intracranial pressure predomi-
nate. In an infant with an open fontanel, the symptoms may
include irritability and failure to thrive. The signs include a
full fontanel and increasing head circumference.
3
The lesions causing this clinical presentation are di-
verse and include large supratentorial mass lesions or infra-
tentorial lesions that obstruct CSF pathways and cause resul-
tant hydrocephalus.
In older children, increased intracranial pressure is typi-
fied by headache, lethargy, and vomiting. The symptoms in
young children and infants are nonspecific and are fre-
quently mistaken for non-CNS problems, such as formula
intolerance, gastroenteritis, or school phobia. It is not rare
for a brain tumor to remain undiagnosed until the child
becomes extremely ill or focal localizing neurological signs
develop. This may lead to significant parental anger. Parents
should be reassured that the diagnosis of a pediatric brain
tumor can be extremely difficult to make without a very
high index of suspicion.
Other signs that may be seen with pediatric brain tumors
include papilledema, ataxia, and sixth cranial nerve palsy.
Signs such as hemiparesis or focal seizures may herald a
supratentorial brain tumor. Visual loss in the child and
inattention to visual stimuli suggest tumors associated with
the visual pathways, such as a craniopharyngioma, optic
nerve glioma, or hypothalamic glioma. Posterior fossa
tumors often cause ataxia and gait disturbances in addition
to signs of increased intracranial pressure. Brainstem glio-
mas have a characteristic presentation with cranial nerve
palsies, especially sixth and seventh, often associated with
ataxia. Unlike other pediatric brain tumors, signs and symp-
toms of increased intracranial pressure are not usually seen
until late in brainstem gliomas.
SURGERY
In most cases, the surgical approach to a pediatric brain
tumor is dictated by its location rather than the age of the
child. Most supratentorial brain tumors are treated as de-
scribed in the preceding chapter on adult brain tumors.
Craniopharyngiomas, tumors commonly seen in childhood,.,
are described in Chap. 12. •
Important principles in the surgical treatment of pediatric
brain tumors include attention to thermal regulation and
positioning. The pediatric circulating blood volume is ap-
proximately 75 cc per kilogram in children greater than 1
141
142
CHAPTER 9
year of age. Newborns have approximately 80 cc per kilo-
gram. Premature infants may have up to 105 cc per kilo-
gram. The circulating blood volume is relatively small;
therefore, meticulous hemostasis is an essential part of pedi-
atric neurosurgery.
5
Children have a larger surface-to-volume ratio than adults.
They have tendency to decrease their core temperature in a
cold operating room. Hypothermia can lead to complications
including cardiac arrhythmias and hypotension. As a result,
preservation of normal core temperature is very important
during the preoperative positioning, operation, and postoper-
ative periods.
Positioning of children for surgery is generally similar to
that used for adults. Surgical judgment is called for in
balancing the risks of pin fixation versus the benefits of
immobilization. Most pediatric neurosurgeons avoid the use
of the pin head holders in children less than 4 years of age
because of the relative fragility of the infant skull. Skull
fractures and epidural hematomas are known complications
of the use of the pin fixation in children. Often in a child, a
horseshoe head holder can be substituted.
A preponderance of posterior fossa tumors in the pediatric
age group dictates that posterior fossa craniotomy is among
the most common procedures performed for tumors in chil-
dren by pediatric neurosurgeons. Although the sitting posi-
tion has been employed for children, the risk of air embo-
lism and the effects of excessive loss of CSF have led to the
adoption of the prone position for most posterior fossa
operations in children.
The surgical approach—following prone positioning on
chest roles for most posterior fossa brain tumors in pediatric
patients, especially cerebellar astrocytomas, medulloblasto-
mas, and ependymomas as described below—includes a
midline incision from approximately the external occipital
protuberance (inion) to the upper or mfflcervical levels,
usually about C2-C3. Following retraction of skin and mus-
cles, the occipital bone is removed, by craniotomy, which
we prefer, or a craniectomy; the dura is visualized and
incised by a Y-shaped incision; and the cerebellum is ex-
posed. This approach allows adequate access to the cerebel-
lar midline, fourth ventricle, and hemispheres.
Approaches to the cerebellar-pontine angles are similar to
those described for adults. However, the surgical position is
likely to be the "park bench" or lateral decubitus position to
avoid the risks of the sitting position in this population.
CEREBELLAR ASTROCYTOMA
The most common pediatric brain tumor is among the be-
nign and treatable. The cerebellar astrocytoma may occur at
any age from infancy to adulthood; however, the classic
presentation is in the school-age child at about 5 to 10 years
of age. A slowly growing tumor, astrocytoma, frequently
presents as described in the clinical section, with signs and
symptoms of increased intracranial pressure including head-
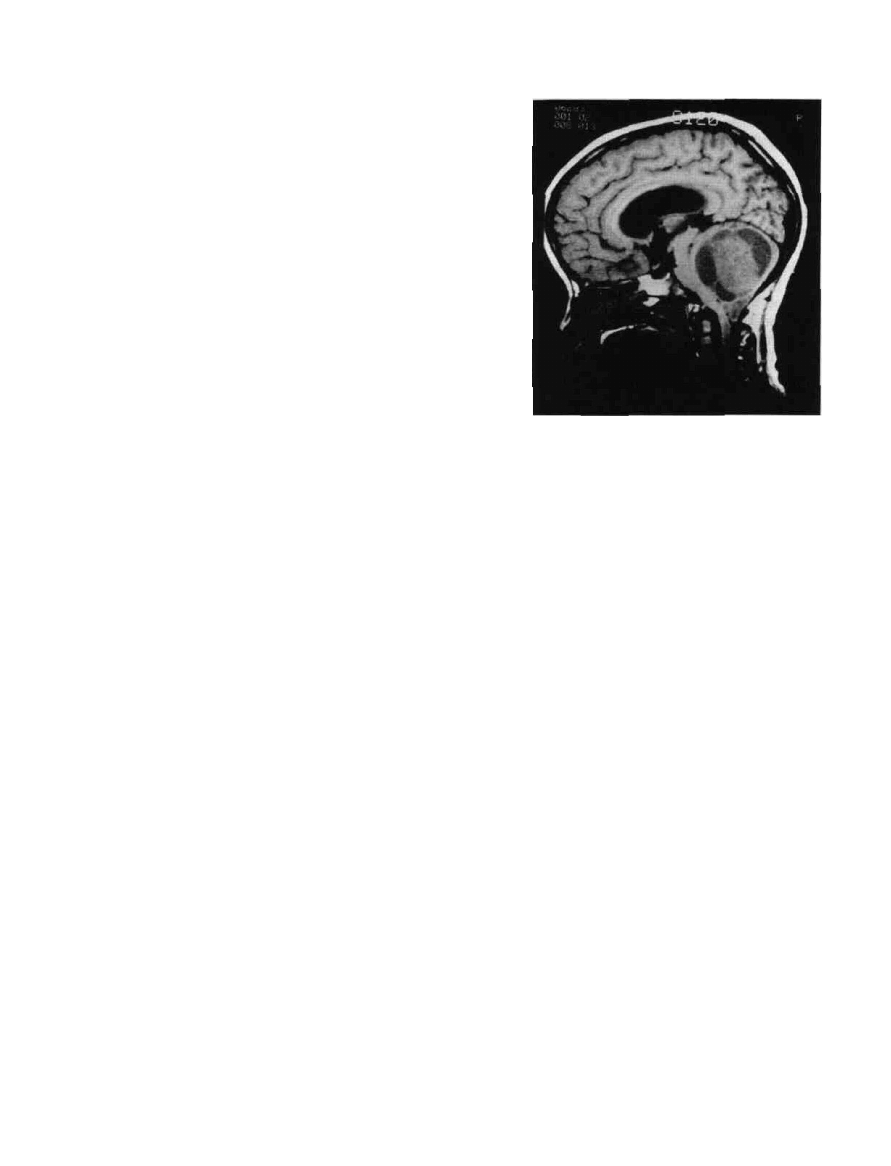
Figure 9-1 Cereblar astrocitoma,MRI.The low-signal cysts
outline the more dense tumor. The cerebellar tonsil has herniated
below the foramen magnum.
ache and vomiting, but the tumor is frequently not diagnosed
until ataxia and sixth nerve palsies herald the intracranial
location of the pathology.
Cerebellar astrocytomas are usually located in a hemi-
sphere, although they may be midline (Fig. 9-1). These
tumors may be either solid or cystic with an enhancing
nodule (Figs. 9-2 and 9-3). The most common histologic
pattern is pilocytic (Fig. 9-4).
Following the surgical approach through the midline, ef-
forts are made to resect the entire lesion. Complete resection
is usually possible and results in cures. Many patients with
cerebellar astrocytomas have been followed for periods of
over 25 years. Follow-up has shown that, although late
recurrences are possible, surgical cure may be expected.
7
Additional therapy is rarely indicated in the treatment of
cerebellar astrocytomas, although when tumors with ana-
plastic histologic features are found, adjuvant therapy is
indicated.
8
MEDULLOBLASTOMAS
The most common malignant brain tumor of childhood is the
medulloblastoma, sometimes referred to as the posterior fossa
primitive neuroectodermal tumor (PNET). As cerebellar astro-
cytomas, medulloblastomas may present at any age but are
commonly seen in children of preschool and early school
years. They present with clinical signs and symptoms, reflect-
ing the tendency of these midline posterior fossa tumors to
cause hydrocephalus, usually including headache, lethargy,
vomiting, papilledema, sixth nerve cranial palsies, and ataxia.
Medulloblastomas are highly cellular tumors composed of
relatively undifferentiated cells. Theoretically, this tumor is
PEDIATRIC BRAIN TUMORS
143
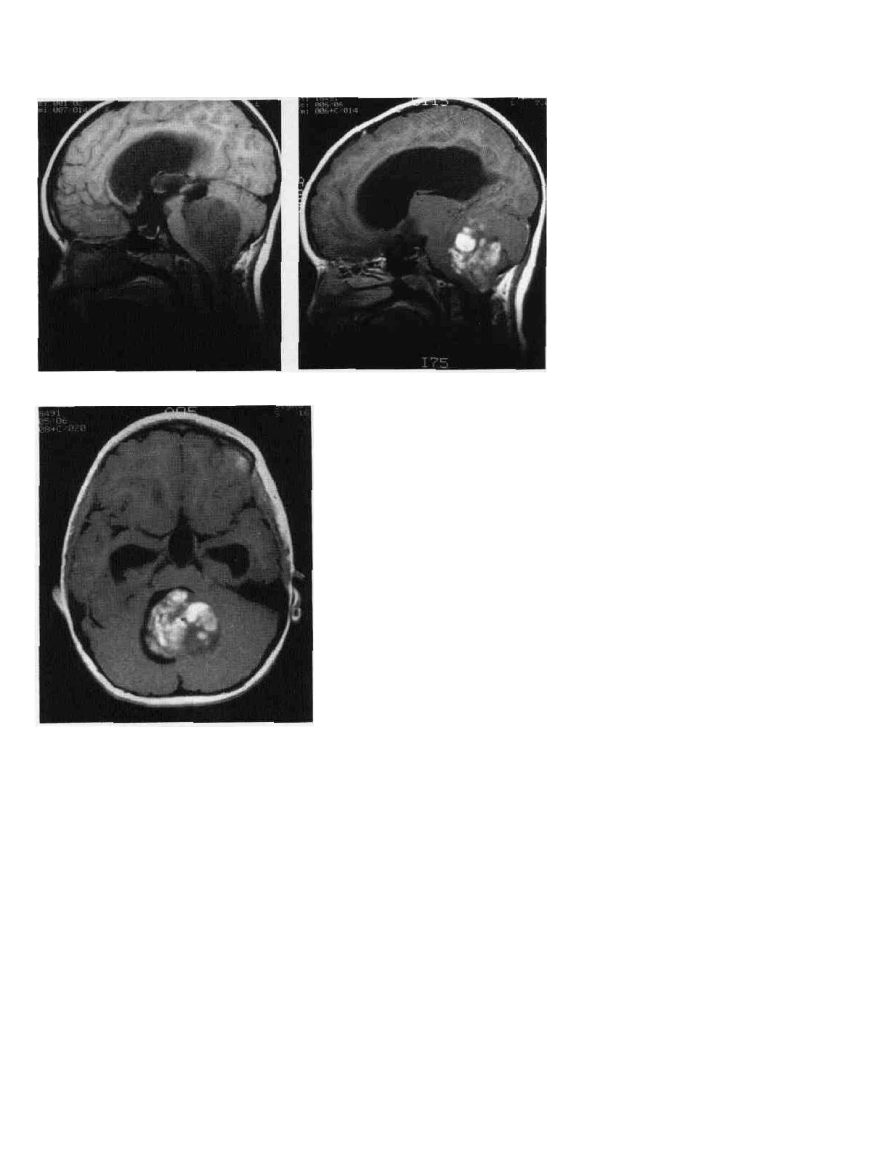
Figure 9-2 Cerebellar astrocytoma. A. MRI, sagittal section. This tumor is lower
signal than the brain. B. MRI, following gadolinium enhancement. The tumor enhances
mliomogeneously. C. MRI, same patient, cross section. The fourth ventricle is present
but displaced by the tumor, which arises from the cerebellum.
derived from the granular cell layer in the cerebellar vermis
(Fig. 9-5). These tumors tend to be midline (Figs. 9-6 and 9-7).
Patients who undergo total or near-total resection of the
tumor have an overall better outcome than similar patients
who receive biopsy only or very limited resection.
9
Medul-
loblastoma has a tendency to have spread by the time of
diagnosis. Cells are frequently transported along cerebro-
spinal fluid (CSF) pathways.
10
Following surgical resection, patients with medulloblasto-
mas are staged clinically. Staging depends on the size of the
primary tumor and the extent of its spread. Important factors
include the preoperative size of the tumor and whether or not
hydrocephalus is present, intraoperative findings including in-
volvement of the brainstem, and extent of resection (Fig. 9-8).
Postoperatively, CSF is examined for tumor cells. Evidence of
spread of disease is sought by examining bone marrow and by
looking for "drop" metastases along the spinal subarachnoid
space. Myelography has been used to detect metastatic disease;
however, in many medical centers, magnetic resonance imag-
ing (MRI) of the spine with gadolinium enhancement has been
found to be more sensitive and less invasive.
Since the period 1965-1970, survival with medulloblastoma
has improved significantly.
11
-'
3
Supplementation of surgery by
radiation therapy to the posterior fossa and craniospinal axis
provided the first improvement. The addition of adjuvant
chemotherapy has resulted in further prolongation of survival
without significant morbidity.
12
-
14
A variety of chemothera-
peutic regimens has shown some success, including: CCNU,
vincristine, and prednisone; CCNU, procarbazine, and predni-
sone (MOPP); and cisplatin and vincristine. 12,13,15,16
BRAINSTEM GLIOMAS
Brainstem gliomas frequently present in school-aged chil-
dren (6-12 years). Unlike many of the tumors discussed in
this chapter, brainstem gliomas do not usually present with
(A)
(B)
(C)

144
Figure 9-3 Cystic juvenile cerebellar astrocytoma. In this unfixed
surgical specimen, a large, cystic, thin-walled cavity (held at the
edges with surgical clamps) forms the main bulk of the tumor. The
cyst was filled with 15 ml of amber fluid that coagulated in a tube
at room temperature. The active parts of the tumor are two "mural
nodules" seen as the solid parts at the right (larger), and the left
(smaller) sides of the cystic cavity. The freestanding solid tumor
tissue is part of the larger mural nodule.
signs or symptoms of increased intracranial pressure. The
classic presentation includes cranial nerve palsies, especially
of the sixth and seventh nerves, often combined with signs
of cerebellar dysfunction such as ataxia and nystagmus. The
tumor usually infiltrates the pons; thus pontine cranial nerve
palsies are usually seen before signs of increased intracranial
pressure caused by obstruction of the fourth ventricle by the
expanding tumor mass (Fig. 9-9B).
On computerized tomography (CT), brainstem gliomas
are hypodense lesions usually in the region of the pons, and
enhancement by contrast media is variable. MRI demon-
strates the tumor clearly and is the preferred technique for
imaging. While the most common type of braiflstem glioma
Figure 9-5 Medullobiastoma. This is a transverse section of
cerebellum and brainstem at the midpons level. Notice the
infiltrating tumor that has greatly enlarged the surface area of
cerebellum. In the central parts of the cut surface of cerebellum,
bulk of pure tumor is seen, whereas in the periphery, infiltration
of cerebellar folia and the subarachnoid space is noticeable.
involves the pons, any part of the brainstem may be in-
volved. Others are radiographically classified exophytic, fo-
cally cystic, or at the cervicomedullary junction.
17
MRI
outlines these variations (Fig. 9-9A and B).
TREATMENT
Brainstem gliomas have a highly variable prognosis, de-
pending on location and tumor type. Improvements need to
Figure 9-4 Juvenile (pilocytic) astrocyloma of cerebellum. This
tumor is one of the most benign gliomas. Many examples have a
cystic component. In this photomicrograph, a microcystic area is
seen to one side. The surrounding astrocytes have round and ovoid
small nuclei. The other part of the tumor shows a denser
architecture with more prominent pilocytic elements. Minimal
surgical hemorrhage is noted. H&E xlOO.
Figure 9-0 MeuuiioDlastoma. in this example, the potential of
tumor for neuroblastic differentiation is noticeable. There is an
abundance of Homer-Wright rosettes. These are round and off-
round formations of tumor cells surrounding a fibrillar zone without
a lumen or vessel. H&E x200.
PEDIATRIC BRAIN TUMORS
145
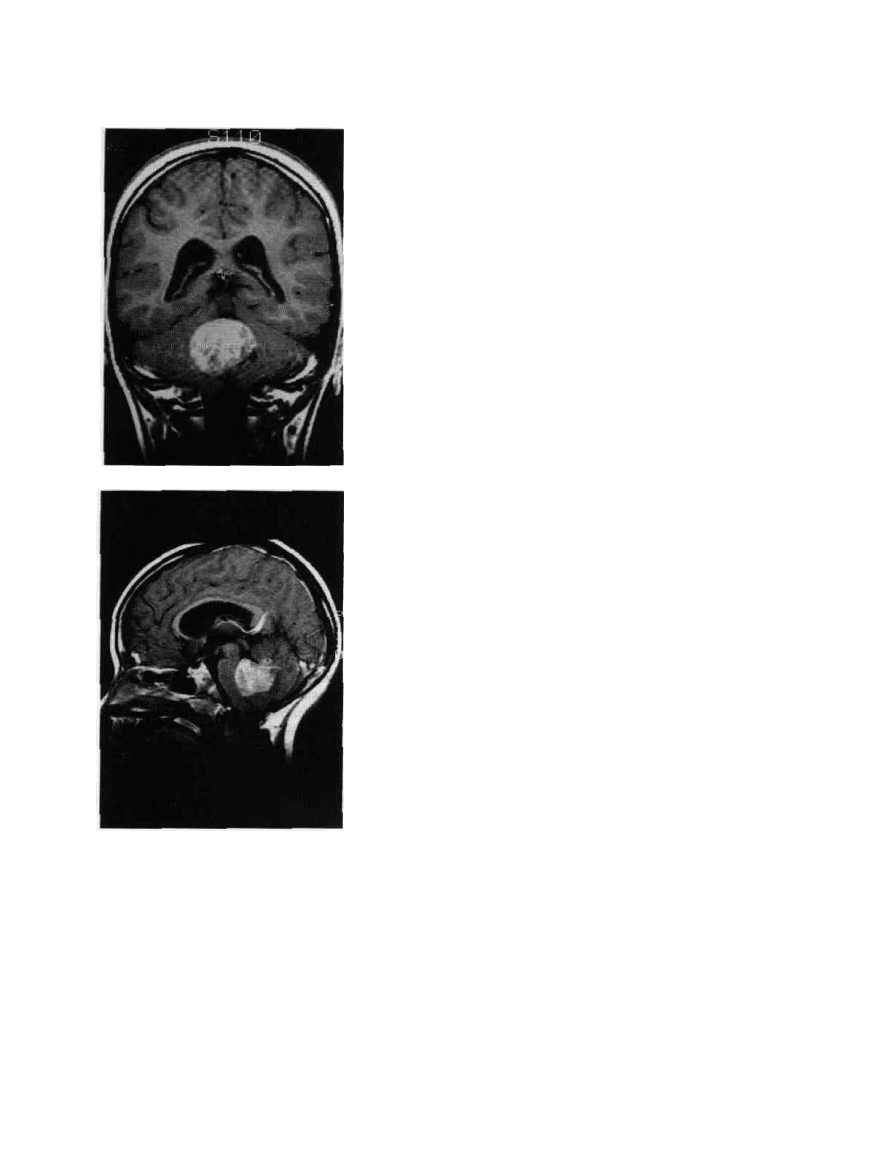
Figure 9-7 Medulloblastoma. A. MRI, coronal section. This
densely enhancing posterior fossa lesion is a medulloblastoma. The
midline position is a characteristic. B. MRI, sagittal section, same
patient. This tumor arises from the cerebellar vermis.
be sought in the measurement of the diffuse pontine and
malignant types.
Localization of brainstem gliomas and identification of
histology directs treatment plans and prognosis. Patients
with gliomas located outside the pons—including focal
cystic, exophytic, and cervicomedullary tumors—fre-
quently benefit from debulking. Prolonged survival is re-
ported.
18
~
20
Patients with diffuse gliomas which involve the pons and
other portions of the brainstem are not surgical candidates.
Stereotaxic biopsy may be indicated. Imaging by MRI, how-
ever, is usually diagnostic. Therapy usually includes conven-
tional radiation to the tumor. Experimental protocols include
twice-daily radiation therapy treatments to increase the toler-
ance without increasing toxicity. The outlook for diffuse
pontine gliomas, however, is poor, even with radiation ther-
apy. Only 30 percent of treated children survive for 1 year
and 5-year survival is less than 10 percent.
21
Chemotherapy
has improved survival in small trials.
22
If a brainstem glioma
has a malignant pathological picture on biopsy, survival will
be zero percent at 12 months, despite therapy.
PRIMITIVE NEUROECTODERMAL
TUMORS (PNET)
PNETs of children are supratentorial lesions which tend to
grow rapidly. Like other pediatric brain tumors, these lesions
often present with signs of increased intracranial pressure,
iincluding: increasing head circumference; emesis; lethargy;
Eand, most commonly in the older child, headache. Focal
neurological deficits and seizures may also occur.
The diagnosis is usually made by CT, MRI, or ultrasound
(Fig. 9-10). The hemispheral mass is often quite large by the
time of diagnosis. PNETs are usually enhanced with contrast
although the pattern of enhancement may not be homoge-
nous (Fig. 9-11).
23
PNETs are poorly differentiated tumors that occur in the
cerebral hemispheres but appear histologically similar to
medulloblastomas. Medulloblastomas are sometimes re-
ferred to as PNETs of the posterior fossa.
TREATMENT
When possible, a gross total resection is attempted in these
large cerebral lesions. Occasionally, involvement of deep
structures or functional areas precludes total excision. Post-
operatively, an evaluation similar to that in medulloblasto-
mas should search for metastatic lesions. Spread may be by
CSF pathways and evaluation includes a myelogram or MRI
of the spine with gadolinium. The CSF is sampled for tumor
cells postoperatively. The bone marrow is aspirated and
examined for tumor cells.
Following surgery and tumor staging, additional therapy
is probably beneficial. Radiation therapy has been useful in
the treatment of some PNET patients.
24
Radiation in young
children frequently causes developmental delay and is there-
fore avoided. Younger patients, especially those less than 3
years, may have prolonged survival when given chemother-
apy. Chemotherapy has also been used efficiently with radi-
ation therapy in children more than 36 months. The drugs
and dosages employed are similar to those utilized for me-
dulloblastomas. Data on the effectiveness of these ap-
proaches is currently under evaluation.
( A )
(B)
146
CHAPTER 9
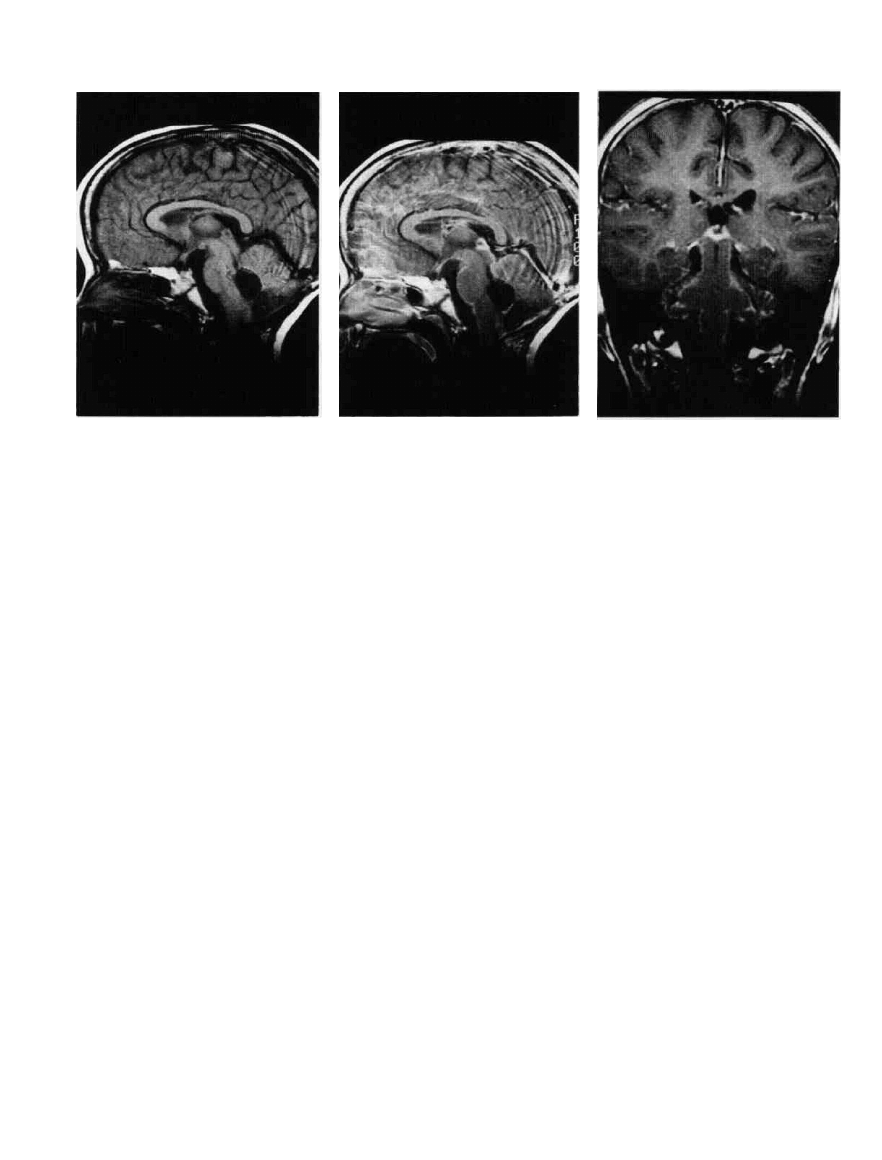
Figure 9-8 Postoperative medulloblastoma. A. MRI, sagittal
section. Removal leaves an enlarged fourth ventricle. B. MRI,
sagittal section following gadolinium. The enhancement in the
subarachnoid space outlines posterior fossae structures, especially
the pons. This enhancement is seen with metastatic spread ot
medulloblastoma. C. Coronal section, MRI, with gadolinium
enhancement. The subarachnoid spread of tumor outlines the
medulla, brachium pontins, cerebellar folia, and midbrain.
GLIOMAS OF THE OPTIC PATHWAYS TREATMENT
Gliomas of the optic pathways may involve optic nerves, the
optic chiasm, or the optic tracts. Often they extend onto the
hypothalamus. These anatomic involvements cause the clini-
cal presentation to include visual loss and endocrinological
dysfunction in addition to the classic findings of pediatric
brain tumors.
When such tumors occur in the first year of life, the
chiasm and structures posterior to it are often involved.
Affected infants have macrocephaly, irritability, and ocular
findings, including spasmus nutans. Older children may have
visual loss, and visual field defects can be documented.
Endocrinological dysfunction may include the diencephalic
syndrome and precocious puberty.
25
Tumors that are con-
fined to the orbit are often stable or very slowly growing
lesions which occasionally cause proptosis.
Both MRI and CT may show these tumors, but the resolu-
tion of involvement seen with an MRI scan is superior. The
MRI may show no contrast enhancement, variable enhance-
ment, or intense uniform enhancement. The contrast pattern
does not correlate with the pathological grade of the tumors
(Fig. 9-12).
The differential diagnosis of lesions for optic pathways
varies with the clinical situation. These tumors are fre-
quently associated with neurofibromatosis. In children with-
out neurofibromatosis (NF), the differential includes germin-
omas, craniopharyngiomas, and pituitary tumors. Children
with masses of the optic pathways, who do not have NF,
may need surgery for diagnostic purposes prior to starting
other therapy.
27
_____
Treatment options may include surgical debulking for large
lesions.
26
Radiation therapy has been useful in slowing
growth in tumors with proven histology that are very likely
to be gliomas such as those with progressive growth in
patients with NF. Chemotherapy has also shown promise in
small numbers of patients.
27
EPENDYMOMAS
Ependymomas are CNS tumors that may be found supraten-
torially and infratentorially. In children, the posterior fossa
location predominates. These tumors usually arise from the
fourth ventricle and spread directly by CSF metastasis
through the CSF pathways. The clinical presentation is very
similar to that of other midline posterior fossa tumors with
early nonspecific signs and symptoms, including headache,
irritability, emesis, and failure to thrive. Later developments
may include ataxia, papilledema, and cranial nerve palsies,
especially of the sixth cranial nerve (Fig. 9-13).
The age of onset of symptoms tends to be younger than
other posterior fossa tumors. The peak age of occurrence in
children is 1 year, the mean age at diagnosis is 5 years,
averaging just over 3 years.
26
Ependymomas also occur in
the adult population, where 23 years is the mean age of
presentation.
29
The overall mean age of presentation is about
16 years.
30
The relationship between histopathological appearance
(A)
(B)
(C)
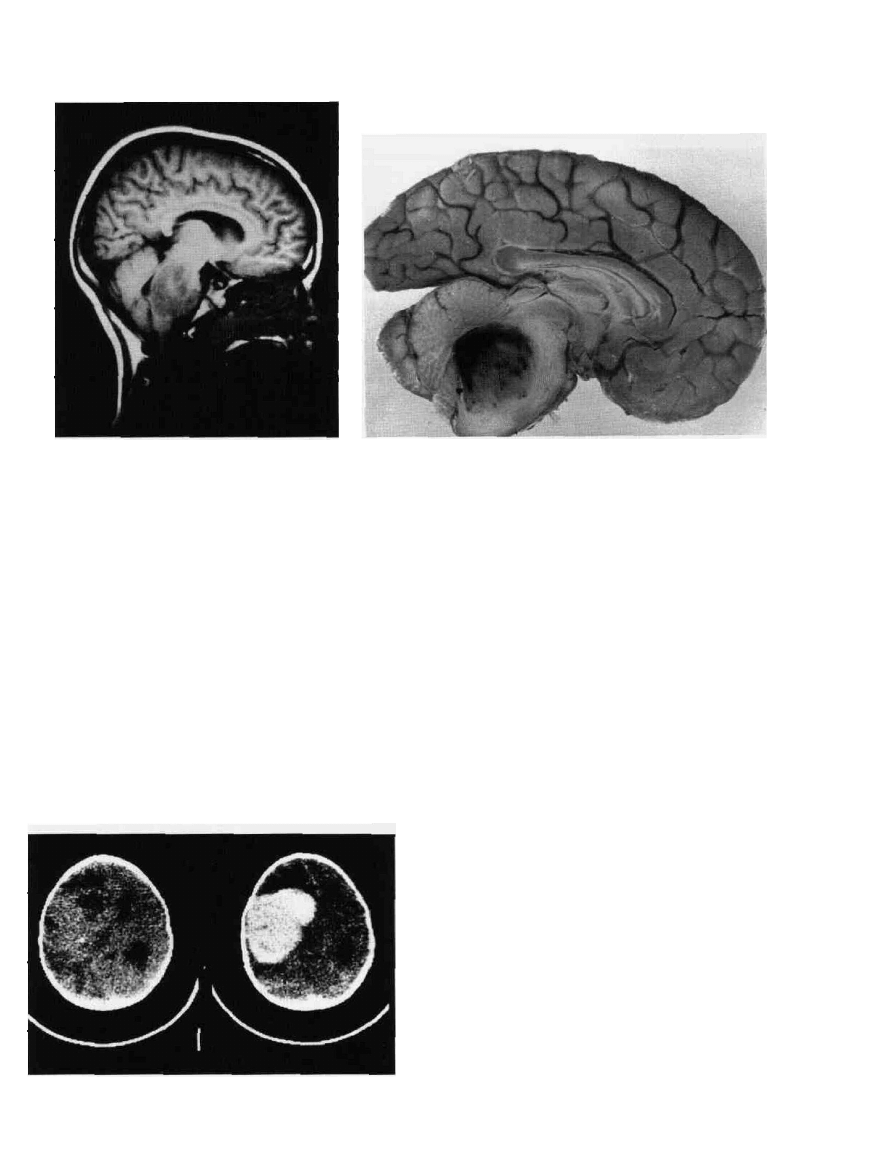
PEDIATRIC BRAIN TUMORS
147
Figure 9-9 Pontine glioma. A. Tl-weighted sagittal MRI. This
low-grade glioma has diffusely enlarged the brainstem from
diencephalon to lower pons. The medulla and cervical cord are
spared. Brainstem glioma. B. This is a midsagittal section of
brain at autopsy at approximately 8 months after the MRI in
Fig. 9A, showing the left cerebral hemisphere. Notice that the
infiltrating tumor has markedly enlarged the volume of pons and
(B)
medulla. The tumor in its anterior and inferior parts blends into
the structure of pons and medulla. In its central and posterior
parts, it has formed a rather pure tumor mass with variegated
texture (hemorrhage, necrosis) and has evolved into a markedly
anaplastic glioma (glioblastoma multiforme). Notice the slit-like
fourth ventricle.
and outcome has caused significant discussion. Evaluation
of the prognostic importance of histological features such as
anaplasia and the number of mitoses favors the use of the
latter rather than the former to predict survival.
31
Anaplastic ependymomas usually have a poor prognosis,
however. Even tumors with a more benign pathological
appearance may behave malignantly.
Figure 9-10 Right cerebral PNET. CT scan with and without
contrast. This tumor is isointense with brain before contrast but
enhances brilliantly and uniformly.
TREATMENT
The surgical approach to posterior fossa ependymomas is
similar to that for medulloblastomas and other midline poste-
rior fossa tumors. If possible, the tumor should be completely
removed. The outlook for this tumor is not as favorable as for
medulloblastoma, even with gross total resection.
32
Postoperatively, residual or metastatic tumor is sought by
checking CSF cytology, screening bone marrow aspirates,
and biopsy for tumor cells, as well as by use of MRI of the
spine with gadolinium or a myelogram to look for metastatic
deposits along the spinal subarachnoid space.
Treatment includes radiation therapy and may include
chemotherapy. A variety of radiation doses and protocols
has been used, as well as a number of chemotherapeutic
agents. To date, however, no therapy has been found to be
very effective. The 5-year survival rate is approximately 20
percent
35
TERATOMAS
Teratomas are seen most commonly in neonates and young
infants.
34
'
35
They are frequently found in the pineal region,
supratentorially, and in the sacrococcygeal region.
35
The
clinical signs and symptoms of the intracranial tumor reflect
(A)
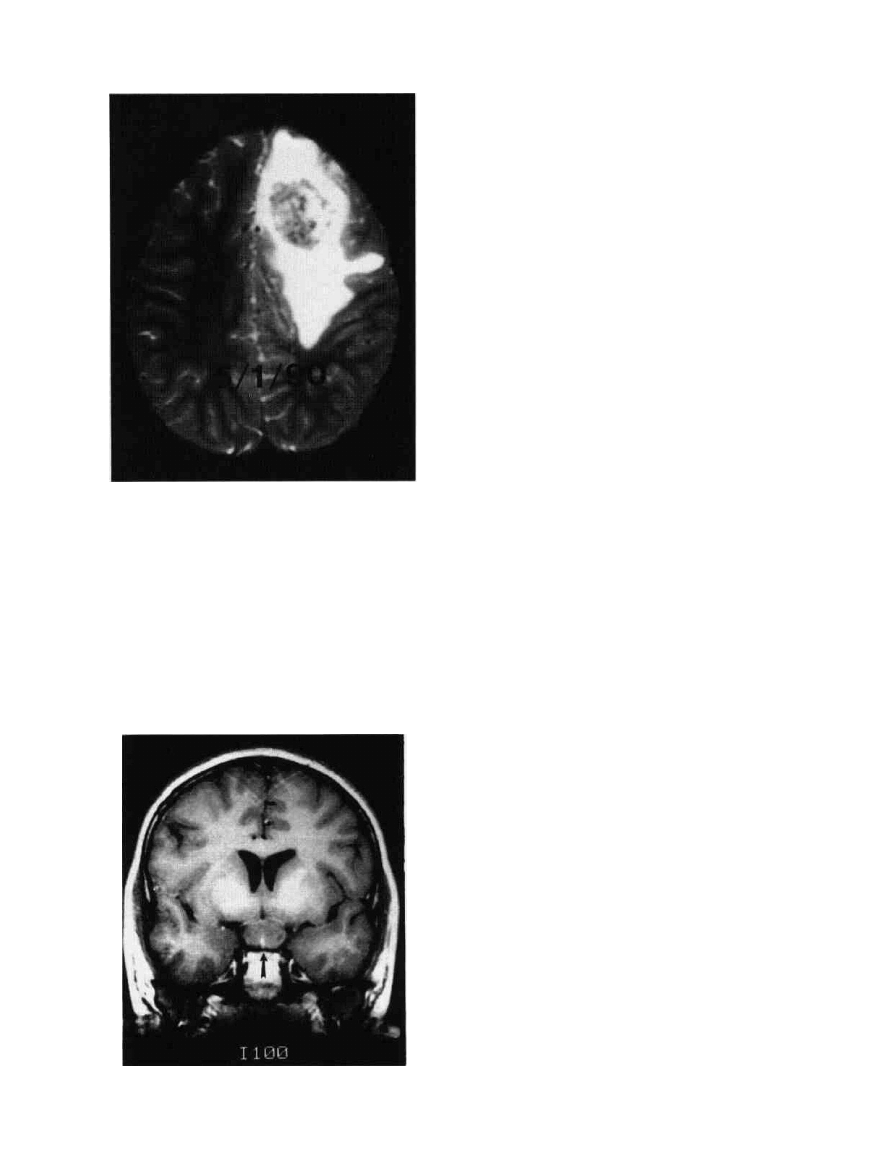
14ft
CHAPTER 9
Figure 9-1 PNET, MRI, transaxial, T-2 weighted. This left
frontopanetai P N E T surrounded by a large area 01 pentumoral
vasogenic edema.
the increased intracranial pressure and include macroce-
phaly, a full fontanel, irritability, and lethargy. Teratomas
include tissue from all three germ cell layers.
35
Teratomas are usually debulked and are completely re-
sected if possible. Benign teratomas have a favorable prog-
nosis after complete excision.
36
-
37
Malignant teratomas or
teratocarcinomas are less favorable, but, occasionally, in-
fants with malignant teratomas survive.
EPIDERMOID TUMORS
Epidermoids are frequently called "pearly tumors." The
glistening white appearance is due to the capsule of stratified
squamous epithelium. Derived from a single germ cell layer
of the developing embryo, epidermoid tumors grow slowly
and are located along the cisterns in the cerebellopontine
angle or in the parasellar area, but they may occur at other
locations including the fourth ventricle, lateral ventricles,
cerebrum, cerebellum, and brainstem (Fig. 9-14).
39
-
41
The CT appearance of epidermoids is that of a low-den-
sity lesion that does not enhance with contrast. The MRI
appearance is hypointense compared to brain in the Tl-
weighted image and hyperintense in the T2-weighted image
(Fig. 9-15).«
The clinical presentation reflects the slowly growing na-
ture of these tumors and their anatomical location. Symp-
toms are often gradual in onset and may include signs of
increased intracranial pressure, cranial nerve dysfunction,
endocrine dysfunction, aseptic meningitis, and seizures.
40
Treatment of epidermoid tumors is excision. The lesions
should be removed in toto whenever possible. The capsule
may be densely adherent to other structures, and viable
portions of capsule that are not removed reform tumors.
Residual epidermoid cells, however, grow slowly, and the
patient may remain asymptomatic for prolonged periods.
40
-
43
Figure 9-12 Optic chiasm glioma. T-l weighted, coronal
MRI. The optic chiasm (arrow) is diffusely enlarged by a
chiasmatic glioma. __
DERMOID TUMORS
Dermoid tumors are composed of epidermoid cells plus
dermal elements which may include hair. The tumors en-
large slightly more rapidly than epidermoid tumors and thus
commonly present in the first two decades of life. Dermoids
are characteristically midline tumors. They are also often
associated with sinus tracts that extend from the skin deep to
the tumor.
Symptoms of these tumors reflect their tendency to be
midline lesions and the resultant hydrocephalus. If a dermal
sinus is present, bouts of bacterial meningitis may occur.
Occasionally, the tumors cause chemical meningitis.
Imaging reflects the midline location and the high fat
content of these lesions. CT shows a hypodense lesion. The
signals on MRI reflect a higher fat content than that of the
brain.
Surgery for dermoids is similar to that for epidermoids.
Problems include the adherence of the capsule to intracranial
structures and the risk of spilling of the tumor contents.
Both inclusion tumors are benign, and the overall outlook
is generally good but dependent on the risks of, and outcome
from, the surgical resection.
39
-
43
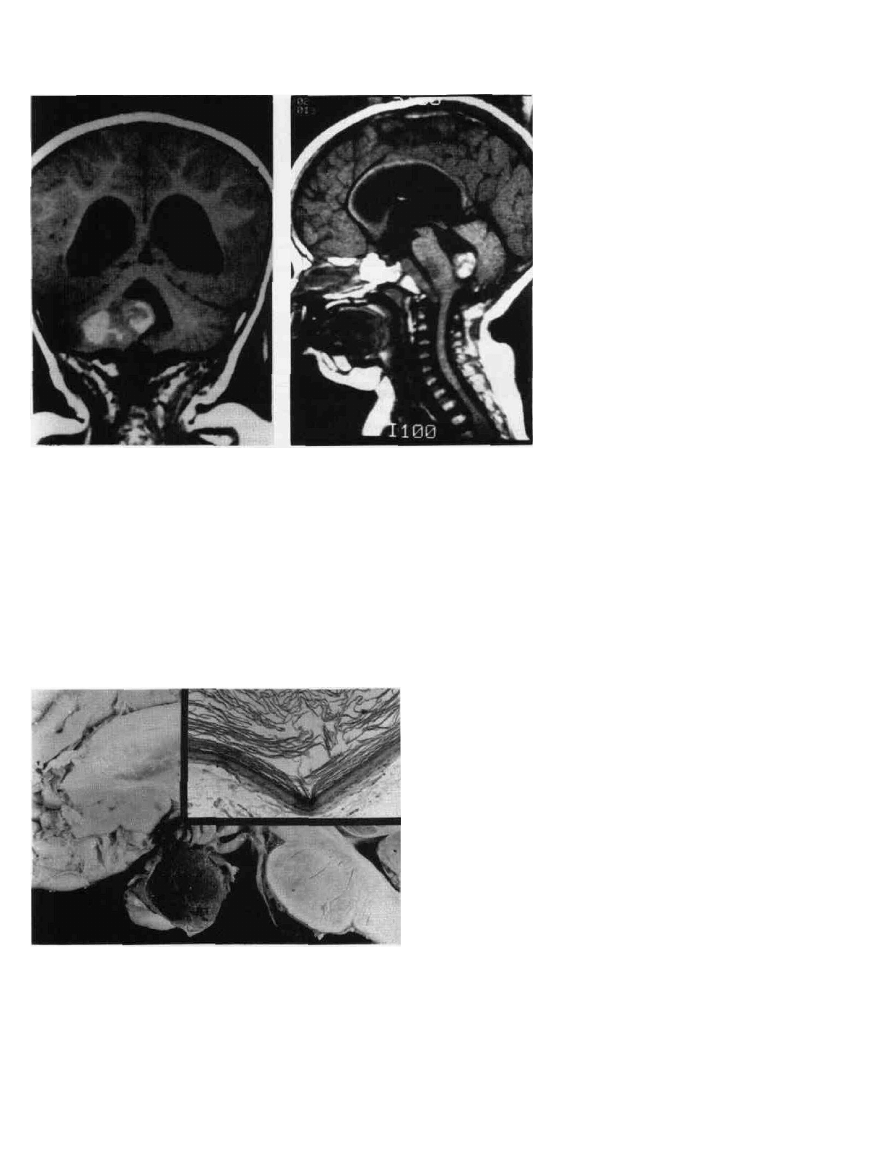
PEDIATRIC BRAIN TUMORS
149
Figure9-13 Ependymoma. T-l weighted
saginal (A) and coronal (B) MRI. The
tumor mass enhances inbomogeneously
with contrast and extends into the fourth
ventricle.
(15)
TUMORS OF THE PINEAL REGION
Tumors in the pineal region include tumors found in other
parts of the brain, such as gliomas, epidermoids, dermoids,
and meningiomas, all of which are described elsewhere.
Tumors that are unique to the pineal region are grouped into
two categories: (1) germ cell tumors, derived from nests of
primitive, totipotential germ cells that occur in midline
structures, and (2) pinealomas, including pineocytomas and
Figure 9-14 Epidcrmoid cyst of suprasellar region. In this sagittal
hemisection of brain, a large, round, thin-walled epidermoid cyst
is seen. The contents are friable keratinous material. This cyst was
an incidental autopsy finding in a middle-aged woman,
retrospectively with some hypothalamic-pituitary dysfunction. The
inset shows the typical wall structure of an epidermoid cyst. H&E
x63. There is an external fibrous matrix on which sits a thin,
compressed layer of squamous epithelium, slowly producing the
keratinous matter, filling the interior of the cyst. In contrast with
dermoid cysts, no skin appendages are found within the wall of the
cyst.
pinealoblastomas, which are believed to be derived from
parenchymal pineal cells.
44
Nonneoplastic pineal region cysts are common at au-
topsy.
45
MRI has also revealed that asymptomatic cysts
occur frequently (Fig. 9-16).
46
'
47
Occasionally a benign cyst,
because of size and location, can become large enough to
cause symptoms (Fig. 9-17). Symptoms will include head-
ache and gaze paresis, with or without hydrocephalus and
papilledema and pineal apoplexy with acute hydrocephalus.
Surgical excision of the cyst will usually result in improve-
ment.
4K
CLINICAL PRESENTATION
Obstruction to CSF flow results in hydrocephalus, making
the clinical presentation of tumors of the pineal region
similar to that of other tumors that present with increased
intracranial pressure. Pineal region tumors are associated
with Parinaud's syndrome, which includes "nystagmus re-
tractorius," paresis of upward gaze, inability to converge,
and midposition, unreactive pupils. Nystagmus retractorius
is the appearance of the globe being withdrawn into orbit
when an attempt is made to converge. These findings are
thought to be due to compression of the quadrigeminal
plate.
49
Pineal tumors may invade adjacent structures, including
the basal ganglia, hypothalamus, thalamus, internal capsule,
inferior collicular plate, and adjacent midbrain structures and
fornixes.
DIAGNOSIS
Computerized tomography and magnetic resonance imaging
show ventricular enlargement and the pineal region mass.
(A)
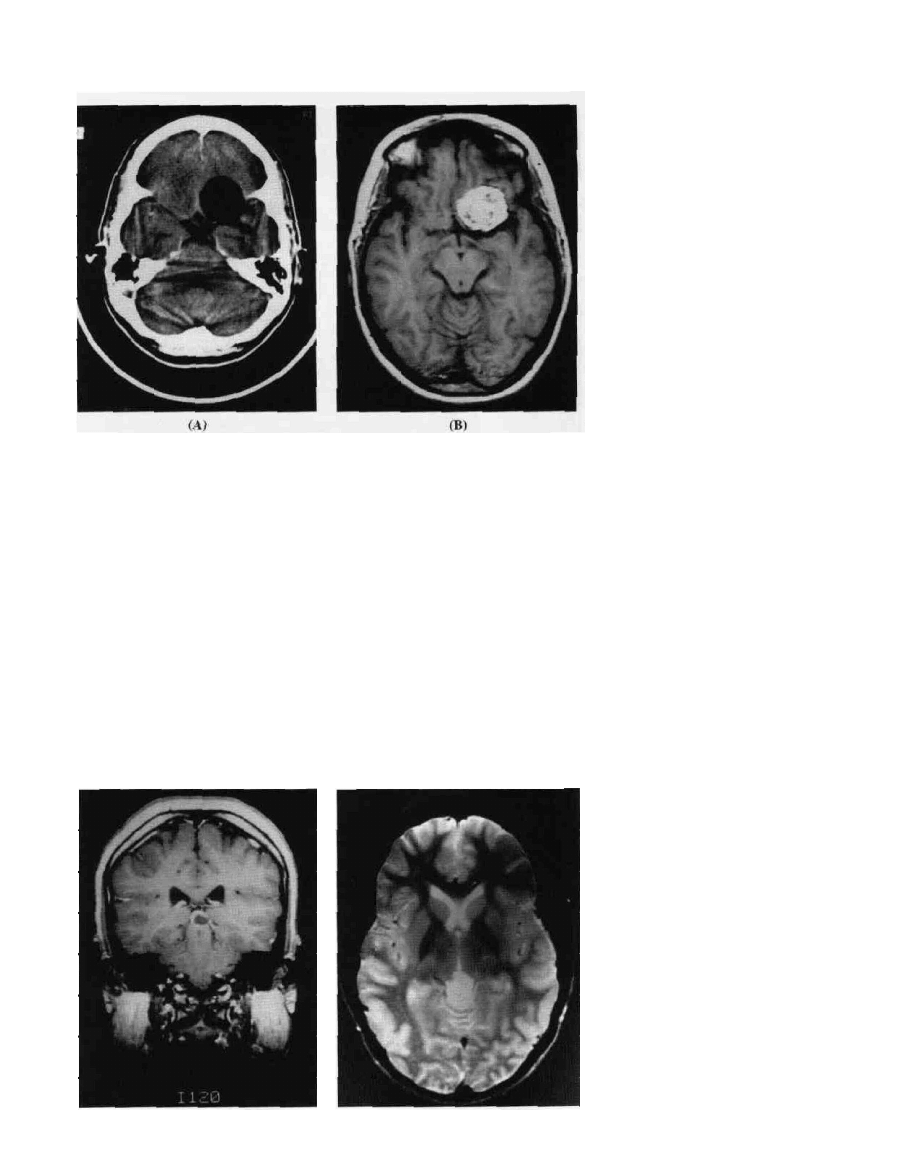
150
CHAPTER 9
Characteristic findings include a lesion that is hypointense or
isointense compared with brain prior to contrast. Contrast
enhancement usually shows a uniform uptake (Fig. 9-18).
THERAPY
In the past, the therapy of tumors of the pineal region was
limited because of the risks of surgery. Therapy advocated at
that time included only ventricular shunting and radiother-
apy.
35
Currently, recommended therapy includes resection
under direct vision. Resection is beneficial because there are
multiple types of tumors in this region, and improvements in
surgical technique, especially the use of the microscope, has
greatly enhanced the outcome. Tumors in the pineal region
may be totally removed.
The pineal region is approached by one of three ways.
The occipital transtentorial approach elevates the occipital
Figure 9-15 Epidermoid tumor. A. CT
scan. The low-density (black) mass on the
sphenoid wing is an epidermoid tumor. B.
MRI, same patient. The Tl-weighted MRI
reveals the fat density of the epidermoid
tumor and allows differentiation from a
CSF collection.
lobe and divides the tentorium to reach the pineal region.
50
The infratentorial, supracerebellar approach achieves access
by retracting the cerebellum down and working in the space
between the tentorium and the cerebellum.
51
Both of these
approaches were initially described with patients in the sit-
ting position. A modification of positioning, the "Concorde
position," may be more suitable for pediatric patients as they
can be operated on in a prone position, with a decreased
incidence of air embolism.
52
This modification is described
with the infratentorial approach.
A transcallosal approach has also been utilized in a pedi-
atric population with excellent results. The mortality, in the
period of CT and the operating microscope, is about 4.3
percent. This approach is similar to most transcallosal proce-
dures. The patient is positioned supine with the head ele-
vated and flexed.
53
The choice of approach to tumors of the pineal region
depends on the preference and the experience of the surgeon.
Figure 9-16 Pineal region cyst. Tl-
weighted coronal MRI. A. The thickened,
enhancing rim of this pineal region cyst
contrasts with its nonenhancing, low signal
center. The cyst has no mass effect or
edema. The T-2 weighted image is presented
inB.
(A)
(B)
PEDIATRIC BRAIN TUMORS
151
Figure 9-17 Cyst of pineal body. In this horizontal cut of the
region of midbrain, an incidental pineal body cyst is seen in the
lower midportion of this very low power pictomicrograph. A cyst
of this size is a common autopsy finding. Cysts of dimensions to
be demonstrable by imaging are seen in about 3-4 percent of
population; larger cysts should not be mistaken for genuine
neoplastic lesions.
Pineal tumors are very deep and surrounded by structures
that cannot be sacrificed or imperiled such as the vein of
Galen, the internal cerebral veins, and the basilar veins of
Rosenthal. The occipital transtentorial approach may give
better visualization of tumors straddling the tentorial notch
and extending above the tentorium. The infratentorial, supra-
cerebellar approach is probably best for tumors that extend
primarily below the tentorial notch,
54
avoiding the risk of
postoperative hemianopsia.
51
The transcallosal approach
allows excellent access to tumors that may have expanded
into the third ventricle.
Surgical therapy of pineal tumors improves the outcome
by decompressing large or malignant lesions, permitting
total excision of benign lesions, and establishing the patho-
logical identity of the tumor to allow for the safest, most-
effective postoperative therapy.
Additional therapy will be dictated by the pathological
identification of the tumor. Radiation therapy is particularly
important in the treatment of pineal germinomas, which are
very sensitive to relatively low doses of radiation therapy
(2000 cGy). Other tumors may be treatable by a combination
of radiation and chemotherapy.
CONCLUSION
The role of the neurosurgeon in the treatment of pediatric
brain tumors has become increasingly important and paral-
lels the improved outlook for children with brain tumors.
Although some tumors, such as intrinsic brainstem gliomas,
continue to have a poor prognosis, with others such as
cerebellar astrocytomas, medulloblastomas, and tumors of
the pineal region, surgery can be curative or provide the
basis for a multidisciplinary effort including radiation ther-
apy and chemotherapy, which is likely to lead to a cure or at
least long-term survival.
Figure 9-18 Pineal tumor. CT scan with
and without contrast. A. The pineal region
tumor is located posterior to the third
ventricle and is isointense with the brain. It
obstructs CSF flow from the third ventricle
and causes hydrocephalus, as demonstrated
by the enlarged ventricle. B. When iodinated
contrast is given, the tumor enhances and
becomes much more apparent.

1S2
CHAFFER r
REFERENCES
1. Freeman AI: Introduction. Cancer 56:1743-1744, 1985.
2. Duffner PK, Cohen ME, Freeman AI: Pediatric brain tumors:
An overview. CA 35:287-300, 1985.
3. DiRocco C, lanneli A, Caddia A: Intracranial tumors of the
first year of life. Childs Nerv Syst 7:150-153, 1991.
4. Rorke LB, Shut L: Introductory survey of pediatric brain
tumors, in McLaurin R et al (eds): Pediatric Neurosurgery.
Philadelphia, Saunders, 1989, chap 26, pp 335-337.
5. Iris-Hansen B: Body composition during growth. Pediatrics
47:264-274, 1971.
6. Schneider J Jr, Raffel C, McComb JG: Benign cerebellar
astrocytomas of childhood. Neurosurgery 30:58-63, 1992.
7. Garcia D, Latif H, et al: Astrocytomas of the cerebellum in
childhood. JNeurosurg 11:661-664, 1991.
8. Conway PD, Ochler HW, et al: Importance of histological
condition and treatment of pediatric cerebellar astrocytoma.
Cancer 67:277-285, 1991.
9. Albright AL, Wisoff J, et al: Current neurosurgical treatment
of medulloblastomas in children. Pediatr Neurosci 15:276-
282, 1989.
10. Flannery A, Tomita T, Radkowski M, McLone DG: Pediatric
medulloblastomas, post surgical evaluation with myelography
and cerebrospinal fluid cytology. J Neurooncol 8:149-151,
1990.
11. Belza M, Donaldson S, et al: Medulloblastoma: Freedom from
relapse longer than 8 years—a therapeutic cure? J Neurosurg
75:575-582, 1991.
12. Tarbell NJ, Loeffler JS, et al: The change in patterns of relapse
in medulloblastoma. Cancer 68:1600-1604, 1991.
13. Garton G, Schomberg P: Medulloblastoma-prognostic factors
and outcome of treatment. Mayo Clin Proc 65:1077-1086,
1990.
14. Krischer J, Ragab A: Nitrogen, mustard, vincristine, procarba-
zine, and prednisone as adjuvant chemotherapy in treatment of
medulloblastoma. J Neurosurg 74:905-909,J991.
15. Evans A, lenkin RD: The treatment of medulloblastoma. Re-
sults of a prospective randomized trial of radiation therapy
with and without CCNU, vincristine and prednisone. J Neuro-
surg 72:572-582, 1990.
16. Packer R, Sutton L, et al: Improved survival with the use of
adjuvant chemotherapy in the treatment of medulloblastoma. J
Neurosurg 74:433-440, 1991.
17. Epstein FJ, Wisoff J: Brain stem tumors in childhood: Surgi-
cal indications, in McLaurin R et al (eds): Pediatric Neurosur-
gery. Philadelphia, Saunders, 1989, chap 29, pp 357-365.
18. Epstein FJ, McCleary EL: Intrinsic brain stem tumors of child-
hood: Surgical indications. J Neurosurg 64:11-16, 1986.
19. Hoffman HJ, Becker L, Craven MA: A clinically and patho-
logically distinct group of benign brain stem gliomas. Neuro-
' surgery 7:243-248, 1980.
20. Stroink A, Hoffman H, et al: Transependymal benign dorsally
exophytic brain stem gliomas in childhood: Diagnosis and
treatment recommendations. Neurosurgery 20:439-444, 1987.
21. Langmuen et al: Management of pediatric pontine gliomas.
Childs Nerv Syst 7:13-15, 1991.
22. Pakish B, Urban C: Hyperfactionated radiotherapy and poly-
chemotherapy in brain stem tumors in children. Childs Nerv
Syst 8:215-218, 1992.
23. Figueroa R, El Gammal T, et al: MR findings of primitive
neuroectodermal tumors. J Comput Assist Tomogr 13:773-778,
1982.
24. Berger M, Edwards MSB, et al: Primary cerebral medullo-
blastoma: Long term follow up review and therapeutic guide
lines. J Neurosurg 59:418^23, 1983.
25. Menzes A, Bell W, Perret G: Hypothalamic tumors in childrer
Their diagnosis and management. Childs Brain 3:265-28-
1977.
26. Wisoff J, Abbott R, Epstein F: Surgical management of e.v>
phytic chiasmatic-hypothalamic tumors of childhood. / Neuro-
surg 73:661-667, 1990.
27. McCullough D, Johnson D: Optic nerve gliomas and other
tumors involving the optic nerve and cliiasm, in McLaurin et ai
(eds): Pediatric Neurosurgery. Philadelphia, Saunders, 1989,
chap 33, pp 391-398.
28. Dohrman G, Farwell J, Flannery J: Ependymomas and epend\-
moblastomas in children. J Neurosurg 45:273-283, 1976.
29. Maybon et al: Ependymomas. Mayo Clin Proc 24:65-71,
1949.
30. Kricheff I, Baker M, et al: Intracranial ependymomas: Factors
influencing prognosis. J Neurosurg 21:7-14, 1991.
31. Schiffer D, Chio A, et al: Histologic prognostic factors in
ependymoma. Childs Nerv Syst 7:177-182, 1991.
32. Kudo H, Oi S, et al: Ependymoma diagnosed in the first year
of life in Japan in collaboration with the International Socier.
for Pediatric Neurosurgery. Childs Nerv Syst 6:375-378, 199(
33. Goldwein JW, Corn B, et al: Is craniospinal irradiation re-
quired to cure children with malignant (anaplastic) intracranial
ependymomas? Cancer 67:2766-2771, 1991.
34. Albright L: Brain tumors in neonates, infants and toddlers
Contemp Neurosurg 7:1-10, 1985.
35. Ingraham F, Baily O: Cystic teratomas and teratoid tumors of
the central nervous system in infancy and childhood. J Neuro-
surg 3:511-532, 1946.
36. Ventureyra E, Herder S: Neonatal intracranial teratoma. /
Neurosurg 59:879-883, 1983.
37. Walker M, Pattisapu J, Fried A: Tumors of the cerebral hemi-
spheres in children, in McLaurin R et al (eds): Pediatric Neuro-
surgery. Philadelphia, Saunders, 1989, chap 31, pp 373-382.
38. Oi S, Tamaki N, et al: Massive congenital intracranial teratoma
diagnosed in utero. Childs Nerv Syst 6:459-461, 1990.
39. Andrews B, Halks-Miller M, et al: Neuroepithelial cysts of the
posterior fossa: Pathogenesis and report of two cases. Neuro-
surgery 15:91-95, 1984.
40. Guidetti V, Gagliardi FM: Epidermoid and dermoid cysts:
Clinical evaluation and late surgical results. J Neurosurg
15:91-95, 1984.
41. Ulrich J: Intracranial epidermoids: A study of their distribution
and spread. / Neurosurg 21:1051-1058, 1964.
42. Wagle W, Jaufmann B, Mincy E: Magnetic resonance imaging
of fourth ventricular epidermoid tumors. Arch Neural 48:438-
440, 1991.
43. Fornari M, Solero C, et al: Surgical treatment of intracranial
dermoid and epidermoid cysts in children. Childs Nerv Syst
6:66-70, 1990.
44. Burger PC, Scheithauer BW, Vogel FS: Surgical Pathology of
the Nervous System and Its Coverings, 3d ed. New York.
Churchill Livingstone, 1991, pp 386-398.
45. Hasegawa A, Ohtsubok Mori W: Pineal gland in old age:
Quantitative and qualitative morphological study of 168 human
autopsy cases. Brain Res 409:343-349, 1987.
46. Lum GB, William JP, Machen BC, et al: Benign cystic pineal
lesions by magnetic resonance imaging. J Comput Asst To-
mogr 11:223-235, 1987.

PEDIATRIC BRAIN TUMORS
153
47. Mamourian AC, Towfighi J: Pineal cysts: MR imaging. AJNR
7:1081-1086, 1986.
48. Wisoff J, Epstein F: Surgical management of symptomatic
pineal cysts. J Neurosurg 77:896-900, 1992.
49. Wolf JK: The Classical Brain Stem Syndromes. Springfield,
111, Charles C. Thomas, 1971, chap 5, pp 85-100.
50. Lazar M, Clark K: Direct surgical management of masses in
the region of the vein of Galen. Surgical Neurol 2:17-21,
1974.
51. Stein B: The infratentorial supracerebellar approach to pineal
lesions. J Neurosurg 35:197-202, 1971.
52. Kobayashi S, Sugitata K, et al: Infratentorial approach to the
pineal region in the prone position: Concorde position. / Neur-
osurg 58:141-143, 1983.
53. Hoffman HJ: Transcollosal approach to pineal tumors, in Ne-
walt E (ed): The Hospital for Sick Children Series on Pineal
Region Tumors: Diagnosis and Treatment of Pineal Region
Tumors. Baltimore, Williams & Wilkins, 1984, chap 11, pp
223-235.
54. Reid W, Clark WK: Comparison of the infratentorial and
transtentorial approaches to the pineal region. Neurosurgery
3:1-8, 1978.
STUDY QUESTIONS
I. A 3-month-old female infant is seen because of increasing
circumference of the head. She is alert, but the head circum-
ference measures 40 cm in greatest diameter and the infant
weighs 9 Ib. She had lost weight during the last 2 months. A
CT shows large lateral and third ventricles and a mass in the
fourth ventricle.
1. What is the differential diagnosis? 2. What type of
shunting procedure might be considered? 3. What complica-
tions might result from shunting? 4. How might definitive
(surgical) therapy be accomplished? 5. What long-term
prognosis might be appropriate with the various lesions
which might be encountered?
II. A 6-year-old male is admitted with a history of many
headaches and projectile vomiting of 2 months. He had
papilledema and "split" cranial sutures.
1. What imaging studies would be appropriate? 2. Assum-
ing a posterior fossa tumor, how should it be approached
surgically? 3. Assuming the lesion is medulloblastoma, how
aggressive should the surgery be? 4. What adjunctive ther-
apy(ies) might be administered? 5. What is the long-term
prognosis?
III. An 8-year-old boy is seen with right-sided loss of
hearing. There is a history of recurrent infections on the
right. A cystic mass with MR! evidence of cholesterol is
obtained.
1. What is the most likely diagnosis? 2. What might be
the relationship to the recurrent middle ear infection?
3. How should the lesion be treated? 4. What hazards of
resection should the surgeon anticipate? 5. What would be
the long-term prognosis?
IV. A 10-year-old male is seen because of failing vision and
proptosis on the right. MRI with gadolinium shows a mass
involving the chiasm and both optic nerves, larger on the
right. The mass is only mildly enhanced by gadolinium.
1. What diagnoses might be considered? 2. What surgical
therapy might be considered? 3. What are the chances of
hydrocephalus? 4. How could hydrocephalus occur? 5. What
are the possibilities of seizures?
V. A 2-year-old infant has hydrocephalus evidenced by a
large head. A CT with contrast shows an enhancing lesion in
the left lateral ventricle in the area of the trigone.
1. What is the most likely diagnosis? 2. How should the
patient be treated? 3. Should a shunting procedure be con-
sidered first? If so, why, or if not, why not? 4. What is the
ultimate prognosis? 5. Is radiation therapy an appropriate
consideration? Under what conditions?
Wyszukiwarka
Podobne podstrony:
brain tumor cap8
11 Brain Tumor
Fizjoterapia w pediatrii ortopedia
Prawne aspekty pracy pediatry
Propedeutyka Pediatrii wykłady dodatkowe
SEM01Wywiad-lekarski, studia, 5 rok, Pediatria (ex), 3 rok, blok
Urazy u dzieci, medyczne różne, pediatria
Przyczyny i rodzaje kaszlu, MEDYCYNA i RATOWNICTWO, Pediatria
egzamin z pediatrii 2009, Położnictwo, egzaminy
Genetyka V rok 2006resztapytan brak, MEDICINE cmuj, Pediatrics, egzamin
Splenomegalia, MEDYCYNA i RATOWNICTWO, Pediatria
Fizjoterapia oddechowa, pediatria
pediatria pytania koło, Pielęgniarstwo licencjat cm umk, II rok, Pediatria i pielęgniarstwo pediatry
więcej podobnych podstron