
Ćwiczenia laboratoryjne z chemii
organicznej
Praca zbiorowa, Wydział Chemii UJ
Strona tytułowa
Agnieszka Czarny, Bożena Kawałek, Anna Kolasa
Piotr Milart, Barbara Rys, Jarosław Wilamowski
Wydział Chemii Uniwersytetu Jagiellońskiego
Zakład Chemii Organicznej
ĆWICZENIA LABORATORYJNE
Z CHEMII ORGANICZNEJ
Zasady bezpieczeństwa,
aparatura i techniki laboratoryjne
Wydawnictwo Adamantan
Redakcja tekstu (bez instrukcji do ćwiczeń): Małgorzata Galus
Copyright © 2007 by Agnieszka Czarny, Bożena Kawałek, Anna Kolasa
Piotr Milart, Barbara Rys, Jarosław Wilamowski
Copyright © 2007-2008 by Grupa Wydawnicza Adamantan s.c. & skryptoszafa.pl
Żadna część tej publikacji nie może być kopiowana, powielana i reprodukowana
żadną metodą ani w żadnej formie, z wyjątkiem sytuacji określonych
w regulaminie serwisu internetowego SkryptoSzafa.pl.
Treść niniejszej publikacji podlega ochronie prawnej na podstawie
Ustawy o prawie autorskim i prawach pokrewnych z 4 lutego 1994 r.
Dz.U. 1994 nr 24 poz. 83.
ISBN: 978-83-7350-082-2 (printed)
ISBN: 978-83-7350-083-9 (e-book)
ISBN: 978-83-7350-084-6 (on-line)
Wydanie on-line
Warszawa 2007-2008
Wstęp
Skrypt, który oddajemy w ręce Czytelników, będący wynikiem wieloletnich doświadczeń
autorów gromadzonych w trakcie prowadzenia zajęć laboratoryjnych w Zakładzie Chemii
Organicznej Wydziału Chemii Uniwersytetu Jagiellońskiego, przeznaczony jest zarówno dla

studentów chemii, jak i studiujących na kierunkach, dla których chemia organiczna stanowi
jeden z przedmiotów podstawowych.
Powodzenie każdego eksperymentu jest uzależnione od umiejętności wykonywania
podstawowych czynności laboratoryjnych i operacji jednostkowych w sposób zgodny z
zasadami „dobrej praktyki laboratoryjnej”. Zasady te obejmują zarówno właściwy sposób
prowadzenia eksperymentu, jak i zagadnienia związane z bezpieczeństwem eksperymentatora
oraz uwzględniają fakt potencjalnego zagrożenia dla środowiska naturalnego. Wpojenie od
podstaw zasad „dobrej praktyki laboratoryjnej” jest jednym z podstawowych celów zajęć
odbywanych w laboratorium chemicznym i ten cel przyświecał nam przy opracowywaniu
skryptu.
W pierwszym rozdziale przedstawiamy zagadnienia związane z bezpieczeństwem pracy w
laboratorium, uwzględniając zagrożenia dla zdrowia eksperymentatora wynikające z kontaktu
z chemikaliami, sposoby postępowania w razie nieszczęśliwego wypadku oraz zasady
postępowania z odpadami. Kolejne dwa rozdziały poświęcone są prezentacji aparatury do
prowadzenia syntez organicznych, zasad właściwego jej doboru i montażu oraz omówieniu
podstawowych czynności laboratoryjnych. W rozdziale IV omówiono sposoby pomiaru
podstawowych parametrów fizycznych, a w następnym szczegółowo przedstawiono metody
izolowania i oczyszczania połączeń organicznych, a więc te operacje, które są najczęściej
stosowane w syntezie organicznej. Krótka informacja dotycząca ustalania składu
pierwiastkowego związków organicznych jest treścią rozdziału szóstego. Zasadniczą część
skryptu kończy krótki komentarz do ćwiczeń laboratoryjnych z chemii organicznej
przeprowadzanych w Zakładzie Chemii Organicznej Wydziału Chemii UJ, spis pozycji
literaturowych wykorzystywanych podczas tworzenia skryptu oraz wybrane informacje w
formie załączników, które są pomocne przy przygotowywaniu się do zajęć, wykonywaniu
eksperymentów i opracowywaniu ich wyników.
Integralną częścią skryptu jest interaktywna płyta CD lub materiały na stronie www, które
zawierają instrukcje do ćwiczeń wraz z charakterystykami wszystkich stosowanych związków
chemicznych oraz sposobami postępowania z odpadami powstającymi w trakcie
eksperymentu. Zamieszczone tam zdjęcia, przedstawiające szkło laboratoryjne i aparaturę do
prowadzenia reakcji oraz wykonywanie podstawowych operacji laboratoryjnych, pozwolą nie
tylko właściwie zmontować zestawy, ale będą również pomocne przy samodzielnym
planowaniu eksperymentu.
Intencją autorów było ograniczenie do niezbędnego minimum informacji dotyczących
podstaw fizykochemicznych poszczególnych operacji oraz mechanizmów reakcji
chemicznych i odesłanie Czytelników zainteresowanych tymi zagadnieniami do odpowiedniej
literatury.
Mamy nadzieję, że nasze opracowanie nie tylko pomoże opanować umiejętności prowadzenia
eksperymentu, ale sprawi również, że fascynujący świat związków chemicznych i ich
przemian będzie przyjazny i bezpieczny dla każdego studenta.
Autorzy
Kraków, lato 2007

I. Bezpieczeństwo pracy w laboratorium
chemicznym
I.1. Rozpoznawanie i unikanie zagrożeń
oraz zabezpieczanie się przed ich skutkami
Laboratorium, w którym prowadzone są eksperymenty z zakresu chemii organicznej, to
miejsce, w którym student zapoznaje się z zasadami prowadzenia eksperymentu chemicznego
oraz samodzielnie dokonuje syntez i analiz połączeń organicznych.
Praca w laboratorium chemicznym może jednak nieść ze sobą rozliczne niebezpieczeństwa.
Ich źródłem staje się kontakt ze stosowanymi chemikaliami, posługiwanie się wyposażeniem
technicznym i aparaturą znajdującą się w laboratorium. Prowadzona reakcja chemiczna, a
także jej produkty oraz powstające odpady mogą stanowić zagrożenie dla środowiska
naturalnego. Aby prowadzić doświadczenia w sposób bezpieczny dla eksperymentatora i
innych osób przebywających w laboratorium, przed rozpoczęciem pracy należy zapoznać się
z podstawowymi zasadami bezpiecznej pracy (podrozdz. I.4), szczegółowym regulaminem
wewnętrznym danej pracowni, kanonami wykonywania poszczególnych operacji (rozdz. III)
oraz sposobami postępowania w razie nieszczęśliwych wypadków (podrozdz. I.3). Powyższe
zasady muszą być ściśle przestrzegane przez każdą osobę przebywającą na terenie
laboratorium.
Przed przystąpieniem do wykonywania każdego doświadczenia konieczne jest
przeprowadzenie oceny ryzyka, które może nieść ze sobą dany eksperyment. Podstawowe
elementy tej oceny obejmują kolejno:
zapoznanie się z właściwościami związków chemicznych stosowanych w trakcie pracy i
powstających w wyniku prowadzonych reakcji;
analizę procedur stosowanych podczas wykonywania eksperymentu pod kątem
stwarzanych przez nie potencjalnych zagrożeń;
zaproponowanie sposobów zmniejszenia ryzyka (np. wybór odpowiedniego miejsca pracy,
zastosowanie właściwych środków ochrony osobistej itp.);
określenie sposobu postępowania z powstającymi odpadami (podrozdz. I.2).
Niebezpieczne substancje chemiczne
Pierwiastki, związki chemiczne i ich mieszaniny mogą być źródłem różnorodnych zagrożeń.
Zostały one podzielone na trzy grupy w zależności od charakteru powodowanych zagrożeń
(tabela 1). Do pierwszej należą połączenia, które ze względu na właściwości fizykochemiczne
mogą być przyczyną wybuchu lub pożaru, grupa druga to substancje o szkodliwym
oddziaływaniu na zdrowie człowieka, a ostatnia to połączenia niebezpieczne dla środowiska.
Każda substancja niebezpieczna jest scharakteryzowana przez podanie symbolu grupy
zagrożeń (E, O, F+, F, T+, T, C, Xn, Xi lub N) oraz odpowiadającego jej znaku graficznego

— piktogramu (rozdz. IX, Załącznik 1). Kolejny system to zbiór znormalizowanych zwrotów
R oraz zwrotów S (rozdz. IX, Załącznik 3). Zwroty R określają dokładniej ryzyko związane z
pracą z daną substancją. Na przykład, informują nie tylko o toksyczności związku, lecz
precyzują, jaką drogą może dojść do zatrucia organizmu (np. przez skórę, po połknięciu czy
przez drogi oddechowe). Zwroty S podają niezbędne środki bezpieczeństwa, jakie należy
zastosować podczas pracy, warunki przechowywania substancji itp.
Program zajęć laboratoryjnych z chemii organicznej został ułożony w taki sposób, aby
wyeliminować konieczność kontaktu z substancjami skrajnie niebezpiecznymi [np. bardzo
toksycznymi cyjankami, rakotwórczymi naftyloaminami, benzenem czy dodatkowo
stwarzającymi szczególne zagrożenie dla środowiska solami chromu(VI)]. Z drugiej jednak
strony, każdy absolwent studiów chemicznych powinien nie tylko umieć przewidywać
zagrożenia stwarzane przez poszczególne substancje, lecz również potrafić z nimi właściwie
postępować. Z tego względu w trakcie ćwiczeń studenci muszą pracować z różnego rodzaju
niebezpiecznymi związkami. Niezależnie od szczegółowego zapoznania się z właściwościami
każdej z substancji, używanych bądź otrzymywanych podczas poszczególnych
eksperymentów, należy znać szczególne zagrożenia stwarzane przez inne związki obecne w
pracowni chemii organicznej.
Popularnym, lecz skrajnie łatwo palnym rozpuszczalnikiem jest eter dietylowy
(temperatura wrzenia +34,6
o
C, temperatura zapłonu –40
o
C). Jego pary tworzą mieszaniny
wybuchowe z powietrzem, które mogą eksplodować nawet pod wpływem przypadkowych
wyładowań elektrostatycznych. Wszelkie operacje z eterem należy przeprowadzać z dala od
źródeł ognia, wyłącznie w wydzielonym pomieszczeniu do pracy z materiałami łatwo
palnymi. Niebezpieczeństwo pracy jest dodatkowo zwiększone ze względu na tendencję eteru
dietylowego (podobnie jak innych eterów — dioksanu, tetrahydrofuranu) do tworzenia
podczas przechowywania wybuchowych nadtlenków. Nieostrożne ogrzewanie,
odparowywanie lub destylacja rozpuszczalnika zawierającego nadtlenki może doprowadzić
do eksplozji. Większość rozpuszczalników niezawierających chlorowców (aceton, etery
naftowe, alkohole, octan etylu, toluen) i lotnych produktów reakcji jest wysoce łatwo palna,
a ich pary mogą tworzyć mieszaniny wybuchowe z powietrzem — wszystkie czynności
należy wykonywać z dala od źródeł otwartego ognia, w dobrze wentylowanym
pomieszczeniu. Wybuchowe związki polinitrowe, które mogą eksplodować w wyniku
ogrzania, uderzenia lub potarcia, nie są stosowane podczas ćwiczeń, poza niewielkimi
ilościami kwasu pikrynowego używanego do celów analitycznych, jednak na przykład
podczas nieostrożnego przeprowadzania reakcji nitrowania można przypadkowo otrzymać
takie połączenia jako produkty uboczne. Silne utleniacze [manganian(VII) potasu, dymiący
kwas azotowy(V)], mogą spowodować zapłon lub nawet eksplozję w reakcji z materiałami
podatnymi na utlenienie, nie wolno zatem dopuścić do ich przypadkowego kontaktu z
substancjami organicznymi.
Związki chemiczne mogą stwarzać zagrożenie dla zdrowia po wniknięciu do organizmu
drogą pokarmową (w wyniku połknięcia), oddechową (przez płuca) lub dermalną (przez
skórę). Negatywne skutki mogą mieć postać ostrego lub przewlekłego zatrucia, podrażnień,
poparzeń, reakcji alergicznej, a nawet nieodwracalnych zmian w organizmie (np. działania
rakotwórczego, mutagennego i innych). Wchłanianie substancji drogą oddechową jest
procesem szybkim i nie ogranicza się jedynie do gazów (np. tlenków azotu czy siarki) i par
lotnych substancji (np. rozpuszczalników organicznych), również aerozole lub pyły (np. pył
żelu krzemionkowego, włókna azbestu) mogą wnikać do organizmu tą drogą. Nie wolno
bagatelizować zagrożeń wynikających z kontaktu substancji niebezpiecznych ze skórą. Żrące

działanie mocnych zasad oraz kwasów nieorganicznych (w stanie czystym lub w postaci
stężonych roztworów), metali alkalicznych czy też chlorowców (np. bromu) jest sprawą
oczywistą. Jednakże należy pamiętać, że również m. in. kwasy organiczne (mrówkowy,
octowy), fenole i aminy alifatyczne są związkami żrącymi i niszczą wszystkie tkanki
organizmów żywych. Podobne działanie wykazują związki, które w wyniku hydrolizy
uwalniają żrące produkty (np. chlorki kwasowe, bezwodny chlorek glinu, chlorki fosforu
tworzą w reakcji z wodą chlorowodór). Skutkiem wniknięcia do organizmu substancji
toksycznej drogą dermalną jest również niebezpieczeństwo poważnych zatruć (nawet
śmiertelnych).
Spośród stosowanych podczas zajęć odczynników i otrzymywanych preparatów szczególną
ostrożność należy zachować przy pracy z nitrowymi i chlorowcowymi pochodnymi
węglowodorów, aminami aromatycznymi oraz związkami azowymi. Związki należące do
tych grup klasyfikowane są zazwyczaj jako szkodliwe lub toksyczne (np. mono- i
dinitrobenzeny, chloroform, chlorek metylenu, anilina, 1-fenyloazo-2-naftol), mogą wnikać
do organizmu przez skórę i drogi oddechowe, a w dużych dawkach przy długiej ekspozycji
mogą powodować zmiany nowotworowe. Pracując z tymi substancjami, należy bezwzględnie
stosować rękawice ochronne i wybierać dobrze wentylowane stanowisko. Podobnych
środków bezpieczeństwa wymagają wszelkie operacje z substancją, której charakterystyka
toksykologiczna nie jest podana w dostępnej literaturze.
Gdzie znaleźć bardziej szczegółowe informacje o zagrożeniach, które może powodować
substancja chemiczna? Na każdym opakowaniu zawierającym substancję chemiczną powinna
znajdować się etykieta zawierająca m. in. symbol kategorii zagrożeń, piktogram oraz
podstawowe zwroty R i S. Przed rozpoczęciem pracy z substancją chemiczną niezbędne jest
uważne zapoznanie się ze znaczeniem wszystkich symboli ostrzegawczych oraz zwrotów R i
S. Informacje takie są podawane również w instrukcji do każdego ćwiczenia.
Poszerzenie i uzupełnienie podstawowych informacji, które niosą oznakowania na
opakowaniach, znajdują się w Kartach Charakterystyki Substancji Niebezpiecznej. Każde
laboratorium posiada komplet tych kart dla wszystkich odczynników, które się w nim
znajdują. Karta charakterystyki w szesnastu punktach podaje szczegółowe informacje o
substancji chemicznej:
1. Identyfikacja substancji lub preparatu — nazwa połączenia chemicznego lub
preparatu handlowego zawierającego niebezpieczne składniki, jego przeznaczenie,
wytwórca i numer telefonu alarmowego.
2. Skład, informacja o składnikach — nazwy składników preparatu, synonimy nazw
składników, wzory chemiczne, numery CAS (ang. Chemical Abstracts Service
Registry Number) identyfikujące jednoznacznie każdą substancję chemiczną oraz inne
oznakowania numeryczne.
3. Identyfikacja zagrożeń — opis zagrożeń dla zdrowia i środowiska stwarzanych przez
substancję chemiczną podany za pomocą zwrotów R.
4. Pierwsza pomoc — opisuje środki i sposoby udzielania pierwszej pomocy.
5. Postępowanie w przypadku pożaru — opis dopuszczalnych i niedopuszczalnych
środków gaśniczych oraz sposobów gaszenia pożaru spowodowanego przez
substancję chemiczną.
6. Postępowanie w przypadku niezamierzonego uwolnienia do środowiska — opis
środków ostrożności oraz zasad postępowania po wprowadzeniu substancji
niebezpiecznej do środowiska (np. rozlania).

7. Postępowanie z substancją i jej magazynowanie — objaśnienie wymogów
dotyczących opakowania i przechowywania substancji.
8. Kontrola narażenia oraz środki ochrony indywidualnej — informacje o wymogach
stawianych stanowisku pracy z daną substancją oraz zalecanych środkach ochrony
osobistej.
9. Właściwości fizykochemiczne — dane o stanie skupienia, gęstości, barwie i zapachu,
temperaturach wrzenia, topnienia i zapłonu, właściwościach utleniająco-redukujących,
palności, właściwościach wybuchowych oraz prężności par.
10. Stabilność i reaktywność — opis warunków, których należy unikać oraz materiałów,
z którymi zetknięcie może powodować gwałtowną reakcję.
11. Informacje toksykologiczne — podane są objawy zatrucia oraz dawki je wywołujące
w zależności od drogi narażenia oraz informacje o skutkach narażenia na długotrwały
kontakt mogący wywierać wpływ na zdrowie, np. działanie rakotwórcze, mutagenne
czy też szkodliwie wpływające na zdolności rozrodcze organizmu.
12. Informacje ekologiczne — informacje o skutkach, które może wywierać na środowisko
produkt chemiczny, jeżeli znajdzie się w powietrzu, wodzie lub glebie.
13. Postępowanie z odpadami — objaśnienie sposobów neutralizacji i utylizacji odpadów
danej substancji i opakowania po niej.
14. Informacja o transporcie (nie dotyczy laboratorium).
15. Informacje dotyczące przepisów prawnych — o wymaganych prawnie oznaczeniach
każdego opakowania substancji (piktogramy, symbole zagrożeń, zwroty R i S).
16. Inne informacje — np. o źródłach informacji, z których korzystano przy tworzeniu
karty.
NOWY SYSTEM KLASYFIKACJI I OZNAKOWANIA SUBSTANCJI
CHEMICZNYCH
20 stycznia 2009 roku weszło w życie nowe rozporządzenie Parlamentu Europejskiego i Rady
WE w sprawie klasyfikacji, oznakowania i pakowania substancji i mieszanin nr 1272/2008 z
dnia 16 grudnia 2008 r.). Rozporządzenie CLP (Classification-Labelling-Packaging)
wprowadza w całej UE nowy system klasyfikacji i oznakowania chemikaliów oparty na
Globalnie Zharmonizowanym Systemie Klasyfikacji i Oznakowania Chemikaliów (tzw.
system GHS - Globally Harmonized System of Classification and Labeling of Chemicals),
uzgodnionym na forum ONZ w 2002 r., a wdrażanym obecnie przez blisko 70 krajów na
świecie. Wprowadzenie nowej legislacji ma na celu ogólnoświatowe ujednolicenie
standardów używanych w różnych krajach przy klasyfikacji i oznakowaniu substancji
chemicznych oraz umożliwienie szybkiej wymiany informacji o stwarzanych przez nie
zagrożeniach.
Rozporządzenie CLP zmienia sukcesywnie obowiązujący wcześniej system klasyfikacji i
oznakowywania substancji chemicznych i ich mieszanin, oparty o dyrektywy 67/548/EWG i
1999/45/WE, a na gruncie polskim – o ustawę o substancjach i preparatach chemicznych z
dnia 11.01.2001 r. z późniejszymi zmianami (tekst jednolity w Dz. U. Nr 152 poz. 1222. z
dnia 17.09.2009 r.) i opierającymi się na niej rozporządzeniami. Najważniejsze zmiany
wynikające z wdrożenia CLP dotyczące użytkowników końcowych produktów chemicznych
to:
- całkowita zmiana systemu obowiązujących piktogramów (załącznik 1B);
- zastąpienie tzw. zwrotów R - zwrotami H i EUH (rozszerzona lista zwrotów określających
rodzaj zagrożenia o zmienionym brzmieniu - załącznik 2B);
- zastąpienie tzw. zwrotów S - zwrotami P (rozszerzona lista zwrotów wskazujących na
niezbędne środki ostrożności o zmienionym brzmieniu - załącznik 2B);
- zmiana zawartości merytorycznej niektórych punktów kart charakterystyk substancji
chemicznych;
- zmiana i wprowadzenie nowych kategorii określających działanie toksyczne, żrące,
rakotwórcze, mutagenne itp.
Przykładowo: w miejsce dotychczasowych trzech klas rakotwórczości, mutagenności i
wpływu na rozrodczość (kategorii 1, 2 oraz 3 wprowadzono podział na dwie kategorie:
KATEGORIA 1 (odpowiednik wcześniejszych kategorii 1 i 2) - Istnieją dowody, że
substancja wykazuje działanie rakotwórcze/mutagenne/szkodliwe na rozrodczość.
Wyodrębniono tu dwie podkategorie: 1A – wykazano związek określonego wpływu w
oparciu o badania u ludzi oraz 1B – można przypuszczać, że istnieje określony wpływ
na zdrowie u ludzi w oparciu np. o badania na zwierzętach.
KATEGORIA 2 (odpowiednik wcześniejszej kategorii 3) - istnieją przesłanki, że
substancja może wykazywać działanie rakotwórcze/mutagenne/szkodliwe na
rozrodczość, brak jednak przekonujących dowodów na takie działanie u ludzi.
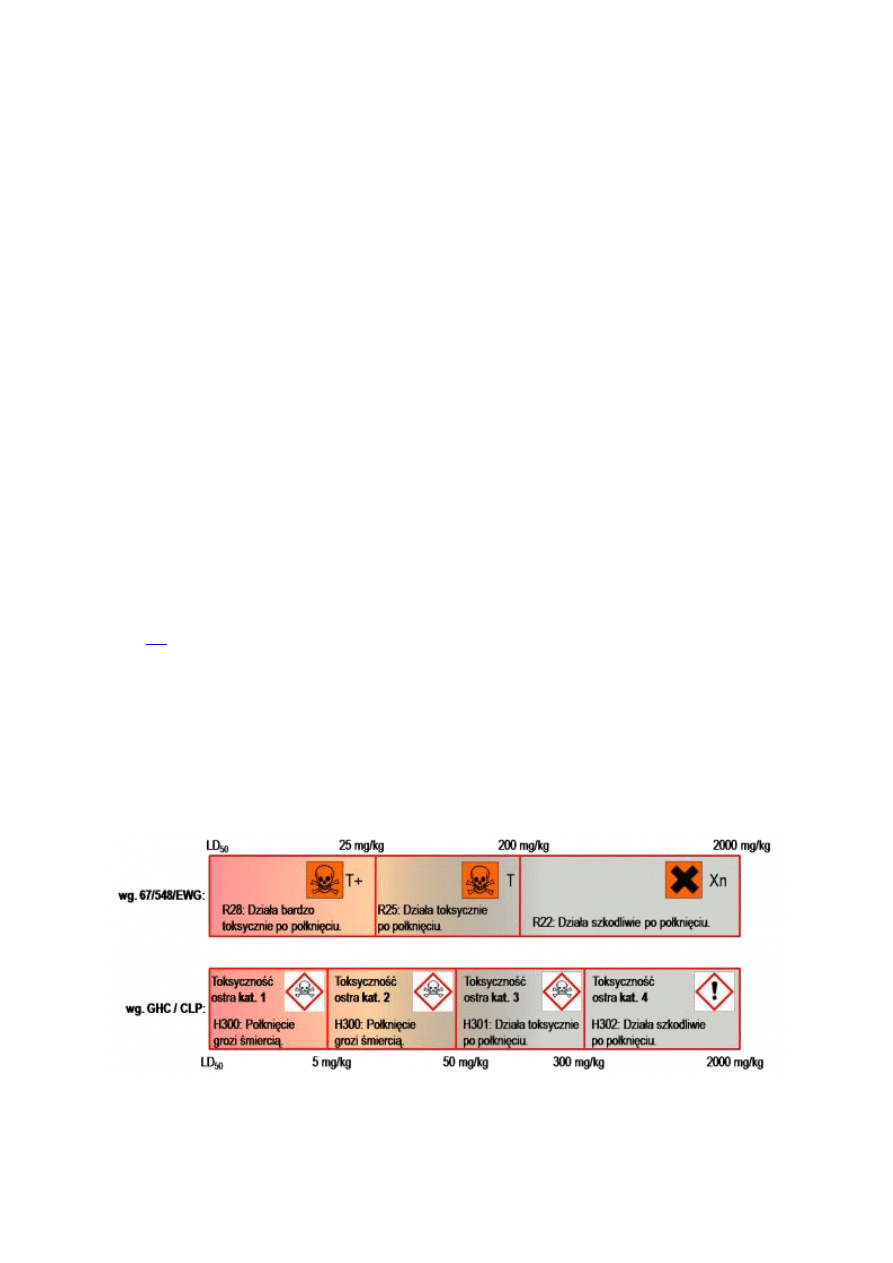
Wprowadzono też między innymi podział substancji na cztery kategorie toksyczności
ostrej
(w miejsce dotychczasowych trzech), podwyższając jednocześnie wartość LD
50
(ang.
Lethal dose, łac. dosis letalis – dawka substancji potrzebna do uśmiercenia 50% badanej
populacji osobników, wyrażana w miligramach na kilogram masy ciała). Przykładowo,
wcześniej granicą rozdzielającą substancje szkodliwe od toksycznych, w przypadku
toksyczności ostrej wywołanej spożyciem substancji jest LD
50
wynoszące 300 mg/kg, czyli
dla ok. 18 gramów dla człowieka o masie 60 kg. Zmiany w oznakowaniu substancji i ich
podział na poszczególne kategorii toksyczności ostrej przy zatruciu drogą pokarmową
przedstawiono na poniższym diagramie. Analogiczne zmiany zostały dokonane przy
klasyfikacji toksyczności substancji w wyniku narażenia drogą oddechową i dermalną.
Kategoryzacja substancji chemicznych ze względu na inne zagrożenia dla zdrowia (np.
działanie żrące, drażniące, alergizujące), zagrożenia fizykochemiczne (np. palność,
wybuchowość), zagrożenia dla środowiska itp. jest bardzo złożona (szczegóły zawiera
oryginalne rozporządzenie 1272/2008/WE:
lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2008:353:0001:1355:PL:PDF
Analizując nową klasyfikację substancji chemicznych, można wyciągnąć ogólny, racjonalny
wniosek: należy zachować szczególną ostrożność przy pracy (lub, jeśli to możliwe, unikać
kontaktu) ze związkami chemicznymi, które w którejkolwiek grupie zagrożeń zostały
zaklasyfikowane do kategorii 1, a w przypadku toksyczności ostrej – również kategorii
2.
Założony harmonogram wprowadzania przepisów CLP w ramach systemu GHS zakładał w
uproszczeniu, że:
- do 1 grudnia 2010 r. substancje (czyli związki chemiczne) klasyfikowało się, oznakowywało
i pakowało zgodnie z wcześniej obowiązującymi przepisami (dyrektywa 67/548/EWG);
- od 1 grudnia 2010 r. do dnia 1 czerwca 2015 r. pakowanie i oznakowanie fabrycznie
nowych, czystych związków (bez mieszanin, czyli według wcześniej obowiązującej
terminologii – preparatów) powinno odbywać się wyłącznie według przepisów
rozporządzenia CLP; natomiast klasyfikacja opiera się o przepisy zarówno rozporządzenia
CLP, jak i wcześniej obowiązującej dyrektywy 67/548/EWG (w karcie charakterystyki
obowiązuje podwójna klasyfikacja substancji); w konsekwencji w okresie przejściowym w
laboratoriach można używać substancje oznakowywane zarówno według starego, jak i
nowego systemu;
- od 1 czerwca 2015 zarówno do substancji, jak i mieszanin stosuje się wyłącznie przepisy
CLP.
Toksyczność ostra - to niekorzystne skutki występujące po podaniu drogą pokarmową lub po naniesieniu na skórę jednej dawki substancji (lub
też kilku dawek w przeciągu 24 godzin), albo po narażeniu inhalacyjnym trwającym 4 godziny.
Tabela 1. Klasyfikacja zagrożeń
stwarzanych przez niebezpieczne substancje
chemiczne
Rodzaj zagrożenia
Kategoria
Symbol zagrożenia
Z uwagi na właściwości
fizykochemiczne
wybuchowy
E
utleniający
O
skrajnie łatwo palny
F+
wysoce łatwo palny
F
łatwo palny
zwrot R10
Z uwagi na zagrożenie dla
zdrowia
bardzo toksyczny
T+
toksyczny
T
szkodliwy
Xn
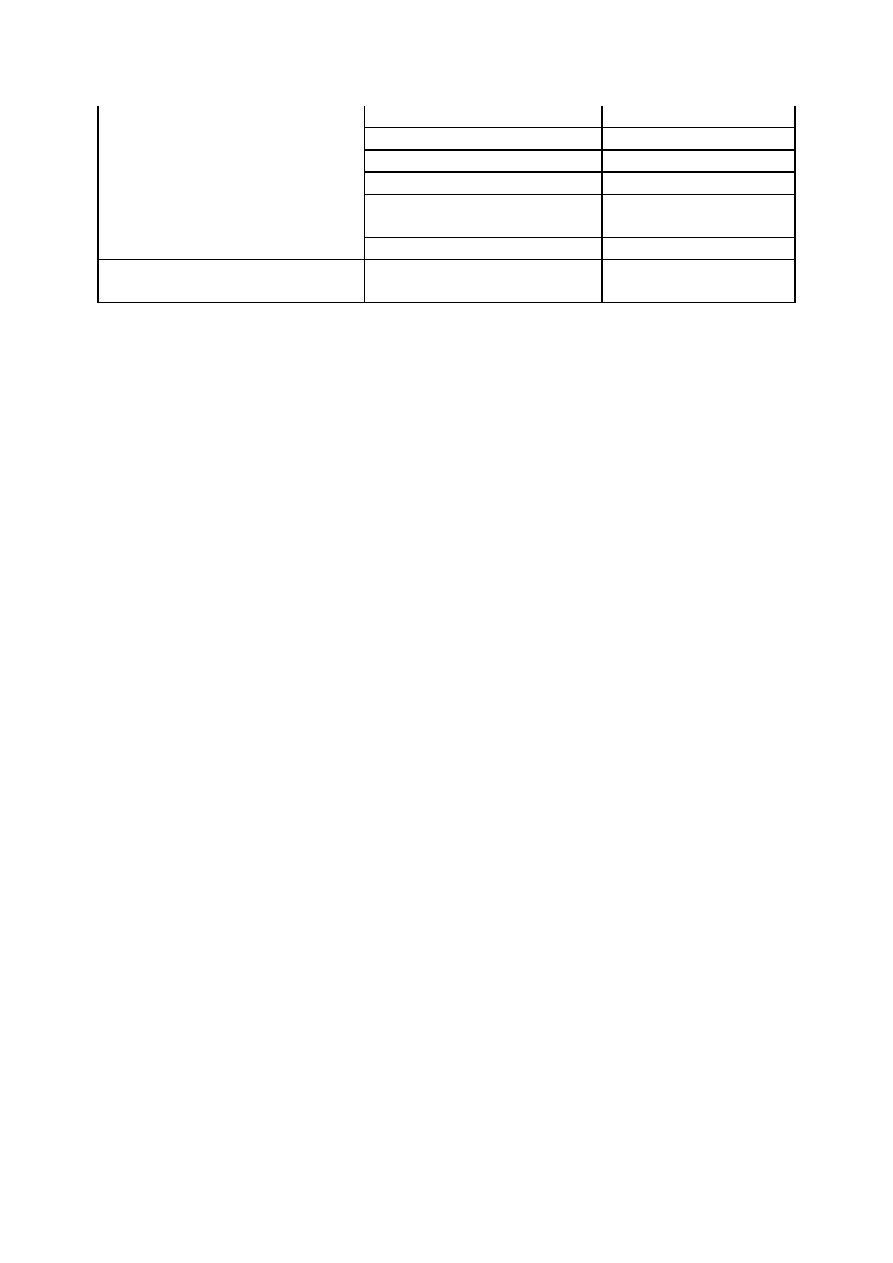
żrący
C
drażniący
Xi
uczulający
zwroty R42 i R43
rakotwórczy
zwroty R40, R45, R49
działający szkodliwie na
rozrodczość
zwroty R60, R61, R62,
R63
mutagenny
zwroty R46, R68
Z uwagi na zagrożenie dla
środowiska
niebezpieczny dla środowiska N oraz zwroty R52,
R53, R59
Bezpieczeństwo prowadzenia reakcji
chemicznej
Reakcje chemiczne mogą być źródłem różnorodnych zagrożeń. W trakcie reakcji mogą
wydzielać się toksyczne lub palne gazy, czy też powstawać niebezpieczne związki chemiczne.
W wyniku reakcji może wzrastać ciśnienie, stwarzając niebezpieczeństwo eksplozji, a
wydzielające się ciepło może prowadzić do gwałtownego wzrostu temperatury. Brak dozoru
nad wykonywaną reakcją doprowadza często do jej niekontrolowanego przebiegu, zagrażając
osobom przebywającym w pobliżu. Niewłaściwy montaż lub demontaż aparatury może
doprowadzić do jej zniszczenia, a w konsekwencji, na przykład, do rozlania zawartości
naczyń, zwarcia w instalacji elektrycznej, czy skaleczeń.
Aby uniknąć potencjalnych niebezpieczeństw, reakcje muszą być prowadzone zgodnie ze
standardami określonymi w procedurach opisujących sposób ich przeprowadzenia, w oparciu
o informacje zawarte w kartach charakterystyki substancji chemicznych i z uwzględnieniem
zasad poprawnej techniki laboratoryjnej. Pierwszym krokiem przed przystąpieniem do
przeprowadzenia reakcji chemicznej jest zaplanowanie eksperymentu, uwzględniające z
jednej strony typ reakcji i stosowane odczynniki, z drugiej zaś możliwe interakcje z
innymi wykonywanymi w pobliżu doświadczeniami.
Przy planowaniu eksperymentu należy dokładnie przestudiować opis i przeanalizować
momenty niebezpieczne, a następnie:
wybrać sprzęt odpowiedni do typu reakcji i jej skali — tzn. szkło laboratoryjne, źródło
ciepła, mieszadło, sprzęt monitorujący itp.,
wybrać odpowiednie stanowisko do przeprowadzenia reakcji blisko niezbędnych urządzeń
oraz w miejscu o właściwej wentylacji tak, aby nie zakłócać innych eksperymentów,
podjąć decyzję o zastosowaniu niezbędnych środków ochrony osobistej.
W przypadku wątpliwości co do sposobu prowadzenia reakcji należy konsultować się z osobą
prowadzącą ćwiczenia. Bezpośrednio przed rozpoczęciem syntezy należy sprawdzić
poprawność montażu aparatury (np. czy przewody doprowadzające wodę są właściwie
zamocowane, a przewody elektryczne zostały poprawnie podłączone). W trakcie
wykonywania syntezy należy stosować się ściśle do procedur dotyczących przeprowadzenia
reakcji i zwrócić specjalną uwagę na operacje, które mogą nieść ze sobą ryzyko. Naczynie, w
którym prowadzona jest reakcja chemiczna, musi być monitorowane bez przerwy — trzeba

pamiętać, że mogą wystąpić nieprzewidziane zdarzenia! Nigdy nie wolno pracować w
laboratorium, jeśli jest się jedyną osobą w nim przebywającą.
Po zakończeniu reakcji należy sprawdzić, czy wszystkie urządzenia są wyłączone, odczynniki
umieszczone we właściwych miejscach, zawory doprowadzające gaz i wodę są zamknięte,
aparatura szklana wymyta, a odpady chemiczne umieszczone we właściwym miejscu.
Minimalizowanie zagrożeń — środki
ochrony osobistej
Aby zapewnić możliwie wysoki poziom bezpieczeństwa i zredukować do minimum
konsekwencje ewentualnych wypadków, podczas pracy w laboratorium należy stosować
odpowiednie zabezpieczenia dotyczące zarówno stanowiska pracy, jak i osoby
eksperymentatora. Wskazane jest zapoznanie się ze wszystkimi znakami BHP umieszczonymi
w laboratorium i jego najbliższym sąsiedztwie (rozdz. IX, Załącznik 2).
Miejsce, w którym jest prowadzona reakcja, powinno mieć właściwą wentylację. Jedynie
proste operacje (odważanie, przesypywanie ciał stałych, sączenie) z użyciem nieszkodliwych
substancji lub roztworów wodnych o temperaturze pokojowej mogą być wykonywane na
stołach laboratoryjnych. Wszystkie pozostałe operacje muszą być wykonywane pod
sprawnie działającym wyciągiem. Wszelkie czynności ze skrajnie łatwo palnymi
rozpuszczalnikami (np. eterem dietylowym, eterem naftowym) lub z lotnymi i toksycznymi
substancjami (np. bromem) należy wykonywać jedynie w specjalnie dostosowanym
pomieszczeniu do pracy z materiałami łatwo palnymi, zwanym tradycyjnie pokojem
benzenowym lub eterowym. Pomieszczenie takie wyposażone jest w specjalną instalację
przeciwiskrową oraz wysokowydajne wyciągi.
Na stanowisku pracy może znajdować się jedynie sprzęt niezbędny do wykonywania reakcji
oraz odczynniki, w ilościach nie większych niż konieczne do przeprowadzenia syntezy.
W pobliżu powinien znajdować się sprzęt niezbędny do usuwania skutków ewentualnych
wypadków (np. sprzęt gaśniczy, środki adsorpcyjne i neutralizujące — zależnie od
wykonywanej reakcji — patrz podrozdz. I.2).
Pracę laboratoryjną należy wykonywać w skupieniu. Wymagana jest duża koncentracja na
przeprowadzanych czynnościach, gdyż błędy wynikające z roztargnienia mogą spowodować
poważne konsekwencje. Na przykład odkręcenie niewłaściwego zaworu gazowego przy
próbie zapalenia palnika kończy się zapłonem strumienia gazu u wylotu króćca ze ściany
dygestorium i poparzeniem eksperymentatora.
Każda osoba przez cały czas pobytu w laboratorium musi być ubrana w zapięty fartuch
ochronny i nosić okulary ochronne! Fartuch ochronny powinien być uszyty z włókien
naturalnych i zapinany na guziki. Nie wolno używać fartuchów wykonanych z łatwopalnych
zazwyczaj włókien syntetycznych. Okulary ochronne muszą mieć dużą powierzchnię szkieł
i osłaniać oko także z boku. Jeżeli ktoś korzysta z okularów optycznych, to musi nakładać na
nie okulary ochronne. Okulary optyczne nie chronią oczu w dostatecznym stopniu przed
działaniem substancji szkodliwych. Należy unikać stosowania szkieł kontaktowych — w
razie kontaktu chemikaliów z okiem utrudniają one szybkie i skuteczne przepłukanie gałki
ocznej i w konsekwencji zwiększają prawdopodobieństwo poważniejszych uszkodzeń oka.

Podczas szczególnie niebezpiecznych operacji (np. destylacji pod zmniejszonym ciśnieniem,
silnie egzotermicznych reakcji) zaleca się stosowanie specjalnych, szczelnie przylegających
gogli laboratoryjnych lub osłon chroniących całą twarz.
W trakcie pracy z odczynnikami żrącymi lub toksycznymi należy zabezpieczyć dłonie
rękawicami ochronnymi. Popularne, jednorazowe rękawice lateksowe zabezpieczają skórę
przed działaniem rozcieńczonych roztworów kwasów i zasad oraz niektórych związków
organicznych. Stężone kwasy, brom i inne agresywne chemikalia oraz rozpuszczalniki
organiczne mogą uszkodzić takie rękawice, stwarzając poważne zagrożenie dla skóry. Gdy
dojdzie do zanieczyszczenia wymienionymi czynnikami, rękawice należy jak najszybciej
zdjąć, a dłonie dokładnie umyć wodą z mydłem lub postępować zgodnie z opisem podanym w
następnym rozdziale. Lepszą ochronę dają rękawice wykonane z grubszych i trwalszych
materiałów (np. rękawice gospodarcze lub droższe rękawice laboratoryjne z tworzyw
vitonowych czy butylowych), które mogą służyć nawet jako rękawice wielokrotnego użytku.
Do wszelkich operacji z gorącymi naczyniami należy zakładać grube rękawice skórzane lub
odpowiednio impregnowane rękawice bawełniane.
Podczas przenoszenia (przesypywania, odważania) drażniących lub trujących substancji
stałych, wskazane jest stosowanie maseczek przeciwpyłowych. W trakcie ćwiczeń
studenckich nie ma konieczności używania bardziej złożonych środków ochrony osobistej
(np. fartuchów gumowych, masek ochronnych z filtrami przeciwchemicznymi).
I.2. Zasady postępowania z odpadami
chemicznymi
Nadrzędną zasadą obowiązującą przy postępowaniu z odpadami powstającymi w trakcie
pracy w laboratorium chemicznym jest zakaz wprowadzania substancji chemicznych do
odpadów komunalnych i ścieków. Zakaz ten, wynikający z ustawowego obowiązku ochrony
środowiska i aktów prawnych normujących postępowanie z odpadami, zapewnia jednocześnie
większe bezpieczeństwo pracy (ograniczenie emisji par, zmniejszenie prawdopodobieństwa
przypadkowych, niebezpiecznych reakcji w sieci kanalizacyjnej itp.) oraz umożliwia
regenerację niektórych rozpuszczalników.
Istnieje jednak niewielka liczba związków chemicznych, które w małych ilościach nie są
szkodliwe dla środowiska naturalnego i nie stwarzają innych zagrożeń. Odpady te mogą
zostać dołączone do odpadów komunalnych w postaci stałej lub wprowadzone do systemu
kanalizacyjnego w postaci rozcieńczonych roztworów wodnych, pod warunkiem, że
jednorazowy zrzut nie przekracza kilkudziesięciu gramów. Należy pamiętać jednak, że w
trakcie pracy w laboratorium rzadko ma się do czynienia z odpadem w postaci czystego
związku chemicznego, a obecność w mieszaninie związków nawet niewielkich ilości
substancji np. toksycznej, powoduje zakwalifikowanie całego odpadu jako odpadu
niebezpiecznego i uniemożliwia jego utylizację bezpośrednią. Związkami, które mogą
podlegać utylizacji bezpośredniej, są:
aminokwasy naturalne i ich proste sole oraz cukry,
bromki i jodki: sodu, potasu,
octany: amonu, sodu, potasu, wapnia,
chlorki, krzemiany i borany: sodu, potasu, magnezu, wapnia,
fosforany(V), siarczany(VI), węglany i wodorowęglany: amonu, sodu, potasu, magnezu,
wapnia.
Wszystkie pozostałe substancje odpadowe, w zależności od ich składu i postaci, zbierane są w
specjalnie w tym celu przygotowanych, opisanych pojemnikach, znajdujących się
w laboratorium (tabela 2). Sposób postępowania z każdym ubocznym produktem,
przesączem, odrzuconą frakcją itp. jest podany w instrukcji do każdego ćwiczenia.
Przed włączeniem substancji do zbiorczych pojemników z odpadami niezbędna jest jej
chemiczna dezaktywacja. Sposób dezaktywacji (np. redukcja resztek bromu bądź jodu,
rozcieńczenie stężonych kwasów, hydroliza nadmiaru chlorku benzoilu) jest podawany w
instrukcji do ćwiczenia. Kanistry z tworzyw sztucznych mogą być napełnione najwyżej do
4/5 objętości!
Do pojemników nie wolno wprowadzać:
trucizn i związków rakotwórczych klasy 1 (związków takich nie używa się podczas zajęć
ze studentami),
substancji stwarzających ryzyko przebiegu gwałtownych, egzotermicznych lub
wybuchowych reakcji zinnymi związkami [stężonych kwasów, manganianu(VII) potasu itp.],
substancji silnie drażniących i cuchnących (np. nierozłożonego chlorku benzoilu).
Wykonywane podczas zajęć laboratoryjnych reakcje mogą prowadzić do wytworzenia
szkodliwych substancji odpadowych w postaci par lub gazów (np. chlorowodoru, tlenku
azotu, siarkowodoru). Nie wolno dopuścić do wydzielania się takich substancji bezpośrednio
do atmosfery i należy je jak najszybciej dezaktywować chemicznie w odpowiednich
płuczkach lub skruberach wypełnionych cieczami absorpcyjnymi. Po zakończeniu reakcji
zawartość absorberów traktuje się tak jak odpowiednie odpady ciekłe.
Z wszelkimi wątpliwościami dotyczącymi postępowania z odpadami należy zwracać się do
prowadzącego zajęcia!
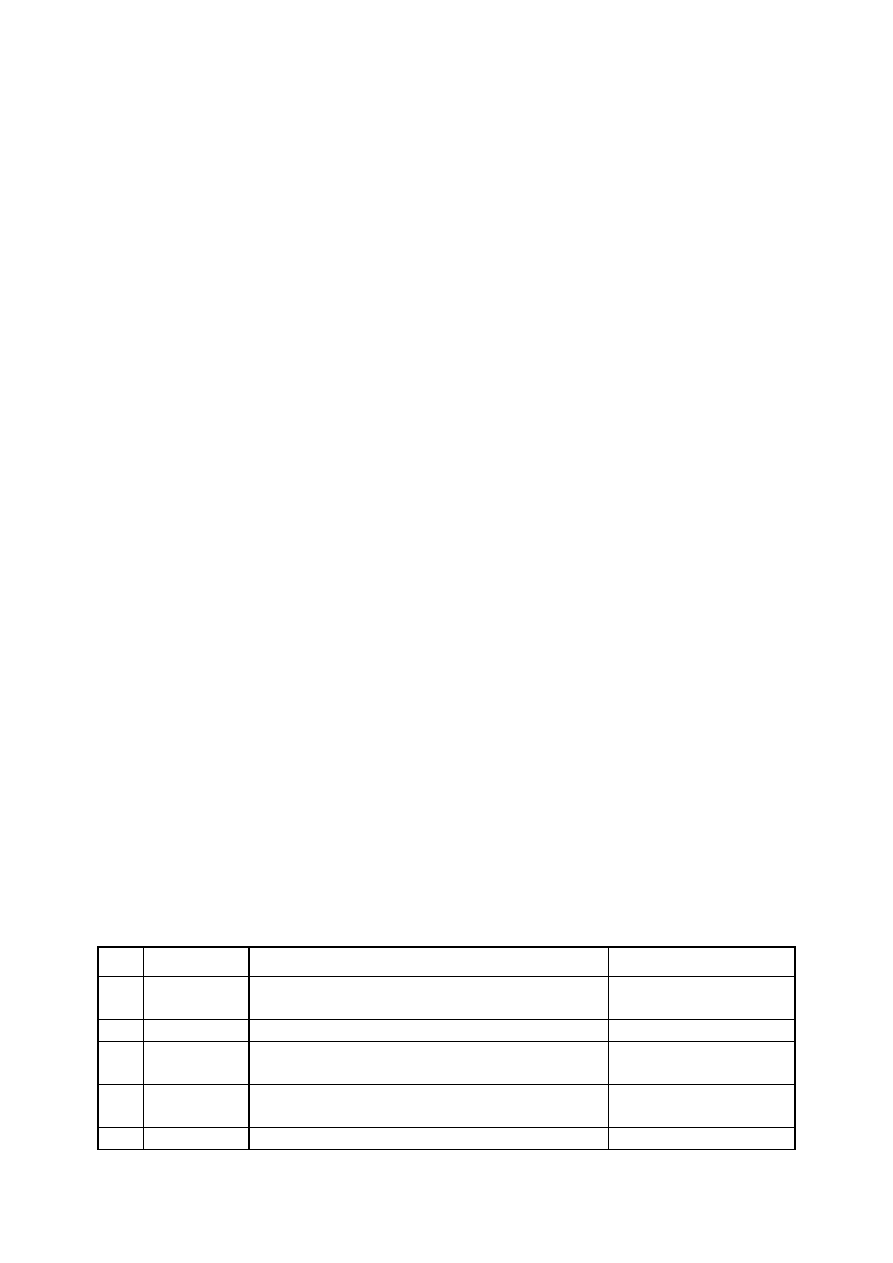
Tabela 2. Klasy odpadów zbierane w
pracowni studenckiej
L.p. Pojemnik
Zbierane odpady
Uwagi
1
A
zlewki acetonu po myciu szkła
(przeznaczone do regeneracji rozpuszczalnika)
nie wlewać odpadów
poreakcyjnych!
2
E
zlewki etanolowe i etanolowo-wodne
bez innych rozpuszcz.
3
O
ciekłe, palne (organiczne), bez fluorowców
o zaw. fluorowców <
3%
4
F
ciekłe, palne (organiczne), zawierające
fluorowce
o zaw. fluorowców >
3%
5
P
odpady stałe, palne (organiczne)
(też m. in. zużyte sączki po osadach
organicznych)
6
N
stałe, niepalne (w tym nośniki
chromatograficzne)
bez szkła
7
W-K
wodne roztwory rozcieńczonych kwasów
nieorganicznych
bez soli metali ciężkich
8
W-Z
wodne roztwory rozcieńczonych zasad
nieorganicznych
bez soli metali ciężkich
9
W-M
wodne roztwory soli Sn
2+
, Mn
2+
, Cu
2+
, Fe
2+
,
Fe
3+
10
W-S
roztwory zawierające jony siarczkowe
nie wolno zakwaszać
11
SÓD
odpady
ścinki sodu metalicznego
pojemnik z suchym
ksylenem
12
R
rtęć metaliczna i stłuczka szklana z rtęcią
(np. rozbite termometry)
13
SZKŁO
odpady
stłuczka szklana wolna od zanieczyszczeń
chemicznych (np. rtęci lub związków
chemicznych)
I.3. Wypadki w laboratorium chemicznym
Nawet w najlepiej wyposażonym i zorganizowanym laboratorium chemicznym zdarzają się
zupełnie nieoczekiwane wypadki przy wykonywaniu rozmaitych operacji. Aby ich
częstotliwość i skutki ograniczyć do minimum, należy ściśle przestrzegać przepisów BHP
obowiązujących na terenie laboratorium.
Każda osoba przebywająca w laboratorium musi znać lokalizację, zastosowanie i zasady
posługiwania się podstawowym wyposażeniem służącym do usuwania skutków wypadków, w
skład którego wchodzi:
sprzęt ochrony przeciwpożarowej (gaśnice śniegowa i proszkowa, koc gaśniczy, pojemnik
z suchym piaskiem),
zestaw adsorbentów i środków do neutralizacji rozlanych chemikaliów,
prysznic z bieżącą wodą do przemywania większych powierzchni ciała,
urządzenie do płukania oczu,
apteczka ze środkami pierwszej pomocy.
W przypadku ćwiczeń laboratoryjnych dla studentów osobą bezpośrednio odpowiedzialną za
bezpieczeństwo na sali ćwiczeń jest asystent prowadzący zajęcia. O każdym, choćby
najdrobniejszym, wypadku przy pracy musi być on natychmiast poinformowany przez
studentów i to on podejmuje decyzję o sposobie dalszego postępowania i, na przykład,
alarmowaniu odpowiednich służb ratowniczych. Gdy jednak rozmiar wypadku uniemożliwi
wezwanie pomocy przez osoby prowadzące zajęcia, należy natychmiast zawiadomić
odpowiednie służby ratownicze, korzystając z numerów telefonów eksponowanych w
widocznych miejscach laboratorium. Wzywając pomoc, trzeba poinformować szczegółowo,
jaki wypadek miał miejsce oraz dokładnie podać sposób dotarcia do miejsca wypadku od
głównego wejścia do budynku.

Należy zwrócić uwagę, że zdarzają się wypadki, podczas których każda chwila zwłoki
w podjęciu interwencji może rodzić poważne konsekwencje (np. zanieczyszczenie skóry bądź
oka żrącą substancją, zapłon rozpuszczalnika w zlewce). Aby zminimalizować skutki takiego
zdarzenia, należy umieć szybko podjąć właściwe działanie. Trzeba jednak pamiętać, że
nieumiejętna interwencja może spotęgować skutki wypadku!
Znajomość najczęstszych przyczyn wypadków, niebezpieczeństw towarzyszących
poszczególnym operacjom oraz sposobów reagowania w nagłych wypadkach należy zatem do
podstawowego kanonu wiedzy każdej osoby rozpoczynającej praktyczne działania
laboratoryjne.
Postępowanie w nagłych wypadkach
Rozlanie cieczy
Do rozlania cieczy może dojść w wyniku pęknięcia butelki lub innego naczynia
laboratoryjnego. Najczęściej jednak zdarza się to podczas nieostrożnego przelewania cieczy
lub wypuszczenia z ręki przenoszonego naczynia. Szczególną uwagę należy zwrócić na to, że
przy podnoszeniu cięższych butelek ręka (bądź rękawica gumowa) musi być sucha. W
przeciwnym razie butelka bardzo łatwo wyślizguje się z ręki. W przypadku przenoszenia
cięższych opakowań z odczynnikami na większą odległość należy używać do tego celu
plastikowego pojemnika z uchwytem (wiaderka). W przypadku rozlania cieczy lotnej
(acetonu, metanolu, chloroformu, a także roztworów wodnych amoniaku) standardową
procedurą jest ewakuacja ludzi z laboratorium, wyłączenie wszelkich źródeł ognia i
otworzenie okna. Ciecze trudno lotne wymagają mechanicznego usunięcia za pomocą
kawałków tkaniny lub ręcznika papierowego, a zużyte materiały należy umieścić w
pojemniku na odpady stałe palne (P). Szczególnym problemem jest usuwanie stężonych
kwasów. Należy je stopniowo bardzo ostrożnie rozcieńczać przez dodawanie wody i
jednocześnie zobojętniać stałym węglanem wapnia lub sodu. Po całkowitym zobojętnieniu
należy wytrzeć do sucha skażone miejsce, a zużyte materiały umieścić w pojemniku na
odpady stałe niepalne (N). Dogodnym sposobem usuwania rozlanych cieczy (także kwasów)
jest użycie substancji absorbujących (np. ziemi okrzemkowej). Także rozlana w laboratorium
woda wymaga szybkiego usunięcia, gdyż grozi to poślizgnięciem się i upadkiem.
Skaleczenie
Najczęstszą przyczyną skaleczeń w laboratorium chemicznym jest używanie uszkodzonej
(wyszczerbionej) aparatury szklanej oraz nieostrożne obchodzenie się ze szkłem
laboratoryjnym (np. pęknięcie lejka Büchnera wciskanego zbyt silnie do kolby ssawkowej,
nieumiejętne nakładanie węży na tubusy). Jeżeli zranienie jest niewielkie, to przemywa się je
wodą utlenioną (3-procentowym roztworem wodnym H
2
O
2
). Następnie należy obejrzeć ranę,
czy nie tkwią w niej odłamki szkła. Wolno usunąć za pomocą pęsety wyłącznie luźno leżące
szklane odłamki, nie wolno penetrować wnętrza rany. Zranienie zabezpiecza się jałowym
opatrunkiem. W przypadku ran głębokich może dojść do uszkodzenia większych naczyń
krwionośnych, nerwów i ścięgien. Oczywiście, najistotniejsze jest zahamowanie krwotoku,
jednak nie wolno stosować opasek uciskowych, bo może dojść do martwicy tkanek.
Zranioną rękę należy unieść do góry i tamować wypływ krwi w miejscu skaleczenia za
pomocą tamponów z waty owiniętej wyjałowioną gazą, przymocowanych lekko naciągniętym
bandażem. Należy bezwzględnie zwrócić się o pomoc do lekarza. Bardzo nieprzyjemne w

skutkach mogą być skaleczenia spowodowane wbiciem w dłoń szklanej kapilary. Na skórze
widoczny jest maleńki ślad (jak po wbiciu igły), ale wewnątrz rany mogą tkwić drobniutkie
odłamki szkła. Konieczna jest wówczas pomoc lekarska.
Oparzenie termiczne
Przyczyną oparzeń termicznych może być kontakt skóry z rozgrzanym sprzętem
laboratoryjnym, gorącą wodą, parą wodną, płomieniem palnika gazowego lub, w skrajnych
przypadkach, z ogniem podczas pożaru. Pierwsza pomoc w przypadku każdego oparzenia
polega na ochładzaniu uszkodzonego miejsca dużą ilością bieżącej zimnej wody, nawet przez
kilkanaście minut. Miejsca oparzonego nie wolno smarować maścią ani kremem, lecz
tylko zabezpieczyć jałowym opatrunkiem. Oparzenia otwartym ogniem są zazwyczaj
bardzo poważne (III lub IV stopnia) i wymagają natychmiastowej opieki lekarskiej Przy
oparzeniu II stopnia pojawiają się pęcherze wypełnione płynem, których nie wolno
samodzielnie przekłuwać. Jeżeli oparzona jest duża część ciała (np. cała dłoń), to należy
również zwrócić się o pomoc lekarską.
Oparzenie chemiczne
Oparzenia chemiczne mogą powstać w wyniku nieostrożnego obchodzenia się ze żrącymi
cieczami (patrz: rozlanie cieczy). Oparzenia bromem będą opisane w dalszej części tego
rozdziału. Większość oparzeń chemicznych jest wywołana stężonymi roztworami kwasów i
zasad. Pierwsza pomoc polega na natychmiastowym zmyciu żrącej substancji dużą ilością
bieżącej zimnej wody. Następnie, w celu zobojętnienia, należy przemyć oparzone miejsce 1-
procentowym roztworem kwasu octowego w przypadku oparzenia zasadą lub 1-procentowym
roztworem wodorowęglanu sodu przy oparzeniu kwasem. Nie należy stosować żadnych
maści, lecz zabezpieczyć oparzone miejsce jałowym opatrunkiem. Jeżeli oparzenie jest
głębokie lub rozległe, to trzeba skonsultować się z lekarzem, gdyż rany po oparzeniu
chemicznym goją się niekiedy bardzo trudno.
Jeżeli w trakcie wypadku doszło do oblania odzieży żrącą cieczą, to należy zdjąć wszystkie
zmoczone części garderoby. Nie wolno jednak odrywać ubrania przyklejonego do skóry (np.
rajstop), pozostawiając to fachowej opiece lekarskiej. Całą powierzchnię ciała, która mogła
mieć kontakt ze żrącą cieczą należy obficie spłukać wodą pod przeznaczonym do tych celów
prysznicem.
Ciało obce w oku
Jeżeli przestrzega się obowiązku przebywania w laboratorium w okularach ochronnych, to
prawdopodobieństwo dostania się do oka ciała obcego jest minimalne. W razie wypadku z
oczami zasada postępowania jest jedna: nie wolno samodzielnie podejmować żadnych
działań medycznych, lecz przemywać uszkodzone oko dużą ilością bieżącej wody przez
co najmniej 10 minut, przy szeroko rozwartej powiece, pamiętając jednak, aby strumień
wody nie był zbyt silny. Jeżeli poszkodowany nosi soczewki kontaktowe, to w trakcie
przemywania oka muszą zostać usunięte. Zdrowe oko powinno być w czasie przemywania
zamknięte (zasłonięte). Po przemyciu wodą należy skonsultować się z lekarzem okulistą,
który oceni skutki wypadku i zdecyduje o dalszym leczeniu. Do czasu konsultacji z lekarzem
należy osłonić uszkodzone oko opatrunkiem z jałowej gazy.
Zatrucie

Do zatrucia substancjami chemicznymi może dojść w wyniku wdychania ich par (zatrucie
przez drogi oddechowe) lub ich połknięcie (zatrucie przez przewód pokarmowy). Jeżeli
wykonuje się operacje laboratoryjne pod sprawnie działającym wyciągiem, to
prawdopodobieństwo zatrucia przez drogi oddechowe jest minimalne. Gdyby jednak doszło
do zatrucia parami substancji, należy wyprowadzić poszkodowanego z laboratorium i
ulokować go wygodnie w pobliżu otwartego okna. W cięższych przypadkach należy wezwać
pomoc lekarską. Przy rygorystycznym przestrzeganiu zakazu spożywania posiłków w
laboratorium, pipetowania ustami oraz próbowania substancji chemicznych możliwość
zatrucia przez przewód pokarmowy jest praktycznie wyeliminowana. Gdyby jednak
substancja dostała się do ust, to należy ją natychmiast wypluć i przepłukać usta wielokrotnie
wodą. Po połknięciu substancji toksycznej powszechnie zalecanym sposobem
postępowania jest podanie do wypicia czystej wody i wezwanie pomocy medycznej. Nie
wolno prowokować wymiotów po połknięciu kwasów, zasad lub większości
rozpuszczalników organicznych oraz gdy poszkodowany jest nieprzytomny. W każdym
przypadku należy postępować zgodnie z zaleceniami podanymi w karcie charakterystyki
substancji chemicznej. Lekarz powinien zostać poinformowany, jaką substancję połknął
poszkodowany.
Często lekceważona jest możliwość zatrucia przez wniknięcie substancji chemicznych
przez skórę. Zdarza się to na przykład podczas pracy bez rękawic ochronnych przy
przenoszeniu substancji, myciu szkła laboratoryjnego oraz wykonywaniu ekstrakcji
w nieszczelnym rozdzielaczu. Takie zatrucia są bardzo niebezpieczne, gdyż objawiają się
niekiedy po kilku godzinach. Po wystąpieniu pierwszych objawów trzeba skontaktować się z
lekarzem i podać, jakie substancje należy rozpatrywać jako ewentualne źródło zatrucia.
Niezmiernie istotne jest zatem staranne mycie rąk wodą z mydłem, jeśli tylko istnieje
podejrzenie, że skóra została zanieczyszczona. Pracując w rękawicach ochronnych, należy
okresowo kontrolować, czy nie doszło do przeniknięcia niebezpiecznych substancji do ich
wnętrza.
Niektóre osoby są szczególnie wrażliwe na kontakt z pewnymi substancjami chemicznymi.
Świadczy to o uczuleniu na te substancje. Objawami reakcji alergicznej może być
zaczerwienienie i swędzenie skóry, łzawienie, obrzęk błon śluzowych nosa i oczu, duszność
lub ból głowy. Jedyne środki, które można podać bez konsultacji z lekarzem, to preparaty
wapniowe (np. Calcium). W przyszłości należy unikać kontaktu z substancjami
wywołującymi wspomniane objawy.
Pożar
Potencjalną przyczyną pożaru w laboratorium chemicznym może być zapalenie się par
substancji lotnych w wyniku ich kontaktu ze źródłem ognia (płomieniem palnika, iskrą
elektryczną, wyładowaniem elektrostatycznym), ogrzanie par powyżej temperatury zapłonu
lub niekontrolowany przebieg reakcji chemicznej (np. sodu z wodą). W przypadku zaistnienia
pożaru ewakuuje się natychmiast ludzi z zagrożonego pomieszczenia, wyłącza źródła ognia
(instalację gazową i elektryczną), usuwa w miarę możliwości materiały łatwo palne oraz
zamyka okna i drzwi, aby ograniczyć dostęp tlenu.
Każde laboratorium wyposażone jest w różne środki gaśnicze. Są to gaśnice (na ogół
śniegowa — wypełniona sprężonym CO
2
, i proszkowa — wypełniona proszkiem
węglanowym lub fosforanowym), koce gaśnicze, piasek i oczywiście woda. Dobór

odpowiedniego sposobu gaszenia zależy od rodzaju płonącego materiału i wielkości pożaru.
Ze względu na rodzaj palącego się materiału pożary dzieli się na cztery podstawowe grupy:
pożar klasy A — płonące ciała stałe pochodzenia organicznego, podczas spalania których
występuje zjawisko żarzenia się (np. drewno, papier, węgiel);
pożar klasy B — płonące ciecze i topiące się podczas palenia ciała stałe (np.
rozpuszczalniki organiczne, wosk);
pożar klasy C — płonące gazy (np. gaz ziemny);
pożar klasy D — płonące metale (np. sód, magnez, glin).
Często spotykany symbol E oznacza pożar z grup A, B, C lub D obejmujący urządzenia
elektryczne pod napięciem.
Drobne pożary substancji w naczyniach (np. płonący w probówce lub w zlewce
rozpuszczalnik, względnie olej w łaźni olejowej) gasi się przez nakrycie siatką ceramiczną,
tkaniną szklaną, mokrą ścierką lub innym materiałem niepalnym. Palące się drewno, papier,
tekturę, tworzywa sztuczne, gumę itp. można ugasić wodą, pod warunkiem, że w pobliżu nie
znajdują się urządzenia elektryczne pod napięciem. Woda jest środkiem gaśniczym
stosowanym w przypadku pożarów typu A niezależnie od ich skali. Wodą nie wolno
natomiast gasić palących się cieczy! Wiele substancji ciekłych, które nie mieszają się z wodą,
wypływa na jej powierzchnię i tym samym pożar się rozszerza. Woda nie nadaje się także do
gaszenia urządzeń pod napięciem oraz reagujących z nią substancji (np. sodu, wodorków
metali). Niewielką ilość płonącego metalu można ugasić, zasypując go np. solą kuchenną.
W przypadku wystąpienia większych pożarów lub gdy obejmują one urządzenia elektryczne
pod napięciem, stosuje się odpowiednie gaśnice. Aby uruchomić gaśnicę, należy:
zerwać plombę i wyjąć zawleczkę zabezpieczającą,
skierować końcówkę węża (dyszę) w stronę źródła ognia,
nacisnąć dźwignię, cały czas trzymając gaśnicę pionowo.
Gaśnice śniegowe przeznaczone są wyłącznie do gaszenia pożarów typu B, C i E. Ciężki,
niepalny gaz skutecznie odcina dostęp tlenu do ognia, a ponadto silnie oziębiony w wyniku
rozprężania przy wylocie z gaśnicy chłodzi miejsce pożaru. W przypadku gaszenia
niewielkich pożarów nie ma konieczności zużycia całej ilości środka gaśniczego, a
uporządkowanie miejsca pożaru po ugaszeniu ognia nie sprawia problemów (inaczej niż w
przypadku użycia gaśnic proszkowych). Gaśnic śniegowych nie wolno stosować do pożarów
typu D, gdyż następowałaby egzotermiczna redukcja ditlenku węgla do trującego tlenku
węgla.
Większość gaśnic proszkowych nadaje się do gaszenia pożarów typów A, B, C i E, lecz są
one nieprzydatne do gaszenia palących się lub żarzących metali, bo środek gaszący może
wchodzić w reakcję z metalem.
Jeżeli w czasie pożaru zapali się ubranie na człowieku, to należy w miarę możliwości zrzucić
płonącą odzież. Nie wolno przy tym biegać, aby nie rozniecać dodatkowo ognia. Do gaszenia
można użyć koca gaśniczego (należy nim owinąć płonącego człowieka), ewentualnie wody z
prysznica. Nie wolno natomiast stosować gaśnicy śniegowej ani proszkowej!

Po ugaszeniu ognia trzeba bardzo starannie wywietrzyć pomieszczenie i usunąć pozostałości
po pożarze. Jeżeli środki gaśnicze w laboratorium są niewystarczające do ugaszenia pożaru,
to niezwłocznie wzywa się straż pożarną.
Porażenie prądem
Porażenie prądem zdarza się niezwykle rzadko w laboratoriach chemicznych. Gdyby jednak
do tego doszło, należy natychmiast wyłączyć instalację elektryczną przez wyłączenie
odpowiedniej grupy bezpieczników lub bezpiecznika głównego. Porażonego prądem należy
wygodnie ułożyć, a jeżeli nie oddycha, to należy wykonać sztuczne oddychanie (np. za
pomocą aparatu AMBU). Natychmiast należy wezwać pomoc lekarską.
Awaria sieci gazowej
W przypadku awarii sieci gazowej, np. gdy w pomieszczeniu wyczuwalny jest zapach gazu,
należy wyłączyć wszystkie źródła ognia, ewakuować ludzi z zagrożonego laboratorium
i otworzyć szeroko okna. W miarę możliwości należy zamknąć główny zawór gazowy.
Postępowanie w razie wypadku z wybranymi substancjami szczególnie niebezpiecznymi
Szczegółowe dane o zagrożeniach związanych z użyciem każdej substancji określają karty
charakterystyki substancji chemicznych. Pewne substancje używane podczas zajęć
laboratoryjnych są jednak tak niebezpieczne, że należy poświęcić im osobny komentarz.
Brom. Jest to brunatnoczerwona ciecz o wysokiej prężności pary. Powoduje zatrucia przez
drogi oddechowe oraz trudno gojące się oparzenia skóry. Praca z bromem musi być
wykonywana pod bardzo sprawnym wyciągiem, najlepiej w pomieszczeniu do prac z
materiałami łatwo palnymi. Student musi być ubrany w rękawice ochronne, a jego praca
powinna być nadzorowana przez asystenta od momentu pobrania bromu (w szczelnie
zamkniętym pojemniku) aż do umieszczenia go wewnątrz naczynia, w którym jest
prowadzona reakcja. Zestaw musi być uprzednio sprawdzony pod względem poprawności
montażu i szczelności (szczególnie kran wkraplacza). W bezpośredniej bliskości muszą
znajdować się pojemniki z 5-procentowymi roztworami disiarczanu(IV) sodu (do neutralizacji
przypadkowo rozlanego bromu) oraz wodorowęglanu sodu (do przemywania poparzonej
skóry). Puste naczynie po bromie należy przepłukać jednym z wymienionych roztworów lub,
gdy zawiera ono jedynie niewielką ilość par tego pierwiastka, położyć otwarte na boku
(dosłownie!) pod dygestorium, aby ciężkie pary bromu mogły wydostać się na zewnątrz do
instalacji wyciągowej. W razie oblania rękawicy gumowej bromem, należy ją niezwłocznie
zdjąć, gdyż brom uszkadza gumę i może przeniknąć do skóry. W przypadku oparzenia skóry
bromem trzeba natychmiast usunąć substancję za pomocą wody, etanolu lub benzyny (eteru
naftowego), a następnie przemywać oparzone miejsce 5-procentowym roztworem
wodorowęglanu sodu. Na oparzone miejsce nakłada się jałowy opatrunek. Jeżeli oparzenie
jest głębokie lub rozległe, to trzeba zwrócić się o pomoc do lekarza. W przypadku zatrucia
parami bromu należy przepłukać nos i gardło 0,5-procentowym roztworem tiosiarczanu sodu.
Poszkodowanemu można też podać do powąchania rozcieńczony roztwór amoniaku (bardzo
ostrożnie!), a do wypicia zimne mleko. Należy wyprowadzić go na świeże powietrze, a w
cięższych wypadkach wezwać pomoc lekarską.
Sód. Jest to szary miękki metal, reagujący gwałtownie z wodą (także z wilgocią z powietrza!)
oraz z wieloma innymi substancjami chemicznymi. Sód przechowywany jest pod warstwą

obojętnej cieczy (toluenu, ksylenu lub nafty). Kawałki sodu wydobywa się z naczynia za
pomocą pęsety i odcina potrzebną ilość, posługując się przy tym nożem i pęsetą. Nie zużyte
ścinki sodu muszą być ponownie umieszczone w naczyniu z cieczą obojętną. Nawet mały
kawałek sodu w zetknięciu z wodą lub inną substancją reaktywną może stać się przyczyną
pożaru. Skutecznym sposobem gaszenia płonącego sodu jest zasypanie go suchą solą
kuchenną lub suchym piaskiem. Jeżeli dojdzie do zetknięcia skóry ze stałym lub stopionym
sodem, to należy usunąć go za pomocą pęsety, a uszkodzone miejsce przemyć bieżącą zimną
wodą oraz 1-procentowym roztworem kwasu octowego lub 3-procentowym roztworem kwasu
borowego. Na miejsce oparzenia trzeba nałożyć jałowy opatrunek.
Rtęć. Jest to srebrzysty ciekły metal, którego pary mają stosunkowo wysoką prężność i są
nadzwyczaj toksyczne. Do skażenia laboratorium metaliczną rtęcią może dojść w wyniku
uszkodzenia termometru lub manometru rtęciowego. Rozlaną rtęć, najczęściej w formie
kulek, zbiera się za pomocą kartki papieru do szklanego naczynia, a w większych ilościach
przez wciąganie do kolbki ssawkowej za pomocą pompki wodnej. Absolutnie
niedopuszczalne jest zbieranie rtęci za pomocą odkurzacza, który rozbija ją na malutkie
cząstki i znacznie powiększa tym samym powierzchnię parowania i skażony obszar.
Pozostałości rtęci neutralizuje się przez posypanie sproszkowaną siarką (powstaje siarczek),
pyłem cynkowym (powstaje amalgamat cynku) lub jodowanym węglem drzewnym. Mało
znanym, a bardzo skutecznym sposobem jest kilkakrotne polanie skażonej powierzchni 10-
procentowym roztworem tiosiarczanu sodu. Analogicznie należy poddać dezaktywacji
aparaturę zanieczyszczoną metaliczną rtęcią. Odpady po dezaktywacji rtęci należy umieścić w
specjalnie oznaczonym pojemniku (R).
I.4. Podstawowe zasady bezpiecznej pracy
By praca lub tylko przebywanie w laboratorium chemicznym było bezpieczne, niezbędne jest
przestrzeganie podstawowych, uniwersalnych zasad bezpieczeństwa, które podane są poniżej
w formie przykazań laboratoryjnych:
Stosuj środki ochrony osobistej — noś zawsze fartuch i okulary ochronne, a w miarę
potrzeby również rękawice ochronne, dostosowane do potencjalnego niebezpieczeństwa.
Podczas pracy w laboratorium noś wygodne obuwie, najlepiej na podeszwie antypoślizgowej.
Zakazane jest używanie butów na wysokich obcasach. Osoby noszące długie włosy
powinny je krótko upiąć, gdyż istnieje ryzyko zapalenia włosów od palnika gazowego lub
zanieczyszczenia ich chemikaliami. Na czas pracy w laboratorium zaleca się zdjąć biżuterię
z palców. W razie oparzenia lub skaleczenia dłoni może ona utrudnić skuteczne udzielenie
pierwszej pomocy. Niewskazane jest też używanie tzw. tipsów.
Zapoznaj się także z lokalizacją w laboratorium sprzętu ratunkowego (sprzętu ochrony
przeciwpożarowej, zestawu adsorbentów, instalacji do płukania oczu i ciała, apteczki) oraz
dróg ewakuacji. Nie blokuj do nich dostępu!
Zgłoś prowadzącemu zajęcia swoje ewentualne problemy zdrowotne, które mogą mieć
znaczenie ze względu na spodziewany kontakt z substancjami niebezpiecznymi.
Nie jedz, nie pij, nie żuj gumy i nie pal w laboratorium. Nie stosuj kosmetyków.
Zanieczyszczenie chemikaliami jedzenia, napojów czy kosmetyków może być potencjalną
drogą wprowadzenia groźnych połączeń chemicznych do organizmu.
Nie pipetuj ustami. Do tego celu służą specjalne pompki.
Nie rozmawiaj głośno w czasie ćwiczeń. Rozprasza to uwagę i utrudnia pracę kolegom i
prowadzącym ćwiczenia. Jest to treść drugiego prawa Gumpersona, brzmiącego szczególnie

trafnie w języku angielskim: Maximize the labor and minimize the oratory in the laboratory.
Wyłącz telefon komórkowy na czas trwania ćwiczeń.
Przed przystąpieniem do pracy zapoznaj się z kartami charakterystyki substancji
niebezpiecznej, dotyczącymi danego ćwiczenia. Znajdziesz tam szczegółowe informacje na
temat postępowania z każdym z używanych odczynników.
Nie używaj szkła laboratoryjnego i sprzętu, które nie są w pełni sprawne. Przed
rozpoczęciem eksperymentu sprawdź każdy element aparatury, czy nie jest uszkodzony oraz
upewnij się, że instalacje wodna, gazowa i/lub elektryczna działają poprawnie.
Dobieraj odpowiednio miejsce pracy do wykonywanych czynności. Jeżeli korzystasz
zpalnika gazowego, to zorientuj się, co robi Twój sąsiad. Jeżeli pracuje on z substancją palną,
to musisz przenieść się w inne miejsce lub, jeżeli to niemożliwe, poczekać aż skończy pracę.
Nie rozpoczynaj pracy bez zezwolenia prowadzącego zajęcia. Nigdy nie pracuj
wlaboratorium, jeśli jesteś jedyną osobą w nim przebywającą.
W czasie wykonywania ćwiczenia ściśle przestrzegaj warunków opisanych wprzepisie.
Jeżeli uważasz, że coś można lub trzeba zmienić, skonsultuj to wcześniej zasystentem.
Utrzymuj na stanowisku porządek w trakcie pracy. Ogranicz ilość sprzętu
iodczynników do niezbędnego minimum. Natychmiast zbieraj i neutralizuj każdą rozlaną lub
rozsypaną substancję.
Odpady chemiczne składaj do właściwych pojemników.
Zamykaj dokładnie butelki i słoiki z odczynnikami. Zapobiegnie to parowaniu lotnych
rozpuszczalników, a także uchroni przed zniszczeniem odczynniki wrażliwe na działanie
wilgoci, na przykład środki suszące.
Odstawiaj wszystkie odczynniki tam, skąd je wziąłeś. Ułatwi to Twoim kolegom pracę
w laboratorium, a również Ty nie będziesz biegać po całej sali w poszukiwaniu każdego
odczynnika. To samo dotyczy sprzętu laboratoryjnego i innych materiałów do wspólnego
użytku.
Niczego nie wyrzucaj bez zastanowienia. Może to być „zapomniany&rdquot; preparat
Twój lub Twojego kolegi. Może to też być warstwa cieczy z rozdzielacza, którą trzeba dalej
przerabiać. A jeżeli jest to coś na pewno do wyrzucenia, to pomyśl, czy wylać to do zlewu
(zazwyczaj roztwory wodne), czy do odpowiednio oznaczonego pojemnika na odpadki ciekłe
lub stałe. W razie wątpliwości zapytaj asystenta. Jeżeli pozostawiasz w szafce laboratoryjnej
swój preparat, to opisz go czytelnie nazwiskiem i nazwą związku.
Nie wrzucaj do zlewu kamyczków wrzennych ani innych stałych substancji. Zatkany
zlew może być przyczyną zalania pomieszczeń na niższych piętrach.
Notuj na bieżąco wszystkie spostrzeżenia w trakcie wykonywania ćwiczeń. Notatki
będą niezbędne w czasie pisania sprawozdania.
Sprzątaj po sobie. Po zakończeniu ćwiczenia poświęć czas na wyczyszczenie stołu i
używanego przez Ciebie miejsca pod wyciągiem. Każdy z Twoich kolegów chce rozpoczynać
pracę w takich warunkach, w jakich rozpocząłeś ją Ty. Zwróć uwagę na to, aby używany
przez Ciebie sprzęt laboratoryjny (np. szkło, mieszadła, wyparka, zestaw do destylacji pod
zmniejszonym ciśnieniem) pozostał czysty po zakończeniu pracy.
Po zakończeniu ćwiczeń umyj ręce wodą z mydłem. Po co masz roznosić różne
substancje, które „przyczepiły się” w czasie eksperymentów, a co gorsze możesz je połknąć
wraz ze smacznym jabłuszkiem lub pączkiem.
Te zasady to jedynie elementarne reguły właściwego zachowania się w laboratorium, by
jednak w sposób bezpieczny dla eksperymentatora i innych osób pracujących w laboratorium
prowadzić doświadczenia, należy również przestrzegać właściwych technik pracy
laboratoryjnej, które zostaną omówione w kolejnych rozdziałach.

II. Podstawowe wyposażenie laboratorium
II.1. Przegląd szkła laboratoryjnego
Zdecydowaną większość operacji w pracowni chemii organicznej przeprowadza się przy
użyciu naczyń i innych części aparatury wykonanych z odpowiednich gatunków szkła
laboratoryjnego. Elementy aparatury, które są poddawane działaniu podwyższonej
temperatury lub żrących chemikaliów, powinny być wykonane ze szkła
borokrzemianowego, charakteryzującego się wysoką odpornością chemiczną i małą
rozszerzalnością cieplną. Wyposażenie pomocnicze, które nie będzie poddawane działaniu
wysokiej temperatury, ciśnienia ani agresywnych reagentów, może być wykonane z tańszego,
ale mniej trwałego szkła sodowego. Niektóre elementy wyposażenia, przeznaczone do prac w
temperaturach zbliżonych do pokojowej z użyciem roztworów wodnych i wybranych
rozpuszczalników organicznych (np. butelki, tryskawki, lejki), wykonuje się czasami z
tworzyw sztucznych, takich jak polietylen lub polipropylen. Drogi, ale niezwykle odporny
chemicznie i trwały do temperatury ok. 200
o
C teflon jest materiałem, z którego są
wytwarzane m. in. mieszalniki, korki i krany.
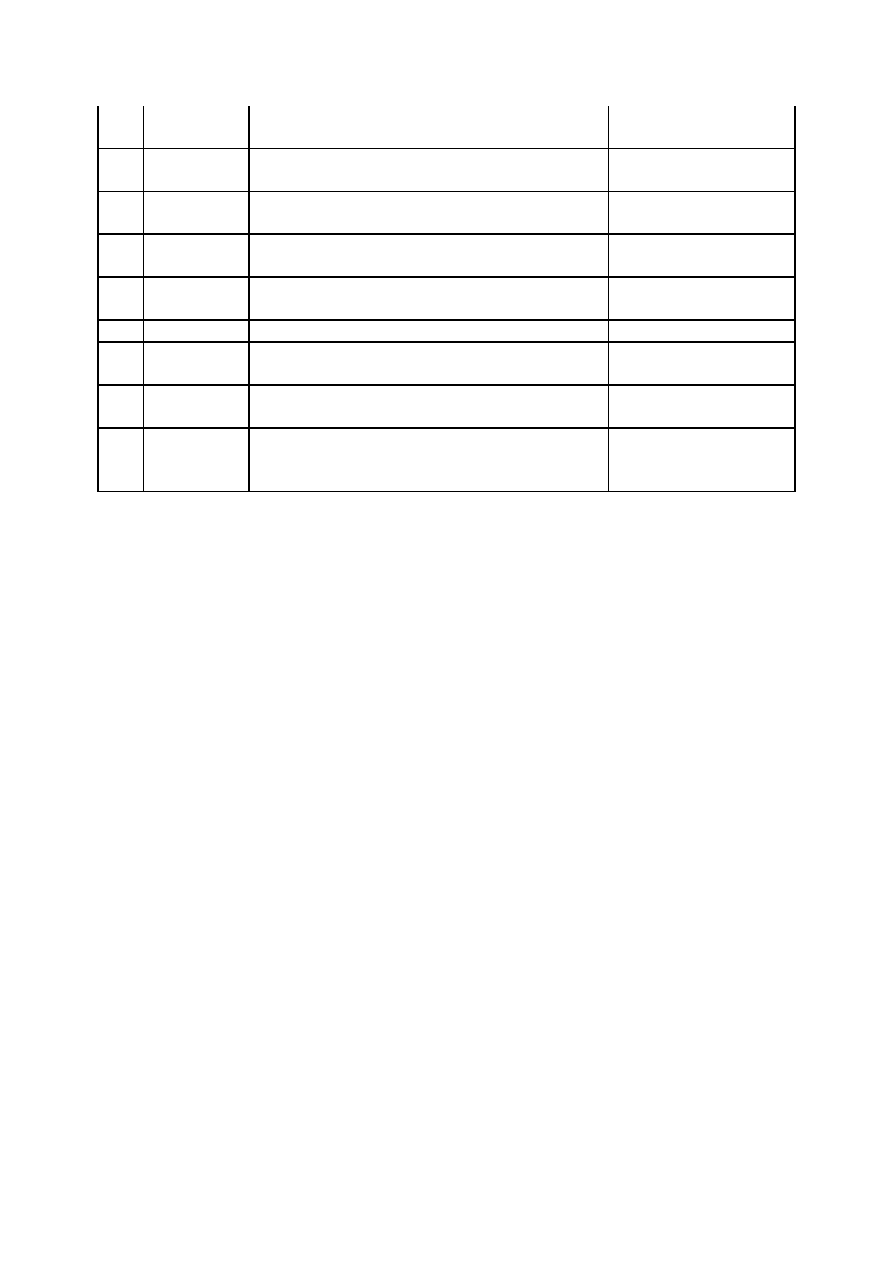
Łączenie części aparatury za pomocą korków z otworami (rys. 1a) jest obecnie stosowane
bardzo rzadko, np. do mocowania rurek czy niektórych typów lejków Büchnera. Większość
aparatury szklanej zaopatrzona jest w szlifowane i w konsekwencji zmatowione końcówki,
popularnie zwane szlifami. Jeden z łączonych elementów ma zmatowioną powierzchnię
zewnętrzną (szlif zewnętrzny), a drugi wewnętrzną (szlif wewnętrzny). Po wsunięciu
końcówki ze szlifem zewnętrznym w szlif wewnętrzny, o ściśle odpowiadającej geometrii,
otrzymuje się połączenie szczelne i bezpieczne (wolne od punktowych naprężeń). Większość
produkowanych szlifów ma ściśle znormalizowany kształt i wymiary. W praktyce
laboratoryjnej najczęściej spotyka się szlify stożkowe w trzech rozmiarach (rys. 1b–d): 29/32,
19/26 i 14/23, gdzie pierwsza liczba oznacza większą średnicę szlifu, a druga długość
oszlifowanej powierzchni w milimetrach. Jeżeli wymagana jest duża szczelność złącza,
wykorzystuje się szlify długie (np. 29/43), natomiast w przypadku, gdy połączenie nie może
być sztywne, używane są szlify kuliste (rys. 1e).
Jeżeli szlify poszczególnych części aparatury nie pasują do siebie, np. szlif wewnętrzny kolby
ma wymiar 29/32, a zewnętrzny chłodnicy — 19/26, można je połączyć za pomocą
odpowiednich złączek zwanych popularnie, choć nie całkiem poprawnie, reduktorami
szlifów. Są to krótkie szklane rurki, które na jednym końcu mają szlif zewnętrzny o innej
średnicy niż szlif wewnętrzny na drugim ich końcu. Jeżeli szlif zewnętrzny jest większy (rys.
1g), jest to złączka redukcyjna (właściwy reduktor, redukcja prosta), jeżeli natomiast
większy jest szlif wewnętrzny (rys. 1f), to jest to złączka ekspansywna (ekspander, redukcja
odwrotna). Często zdarza się, że zakończenie szlifowe aparatury powinno być zastąpione
gwintowym lub do otworu należy zamocować giętki wąż. Stosuje się wówczas odpowiednie
złączki (przejściówki) przedstawione na rys. 1h–j. W ostatnim przypadku drugi koniec złączki
powinien być zakończony „oliwką”, czyli pofałdowanym odcinkiem rurki, co utrudnia
zsuwanie się węży.
Rysunek 1. Elementy do łączenia części aparatury szklanej: a) połączenie korkowe: 1 —
korek z otworem; b), c), d) połączenie szlifowe stożkowe wraz z wymiarami i oznaczeniami:
2 — szlif wewnętrzny; 3 — szlif zewnętrzny; e) szlif kulisty; f), g) reduktory szlifów; h)
złączka szlif-gwint; i) złączka z jednym szlifem; j) złączka szlif-oliwka; k) kranik szklany
Szlify wykonuje się nie tylko na zakończeniach części aparatury szklanej, ale także
w szklanych kranikach (rys. 1k) czy korkach do butelek. Są to jednak przeważnie szlify
pasowane, czyli niewymienne, tzn. dwa wyszlifowane elementy aparatury tworzące daną parę
pasują tylko do siebie i nie można ich łączyć z innym elementami. Z tego względu
niedopuszczalna jest, na przykład, zamiana szlifowanych kraników szklanych z różnych
części aparatury. Poza wymienionymi spotyka się jeszcze szlify płaskie (w eksykatorach) i
cylindryczne (przy połączeniach dwóch części walcowych).
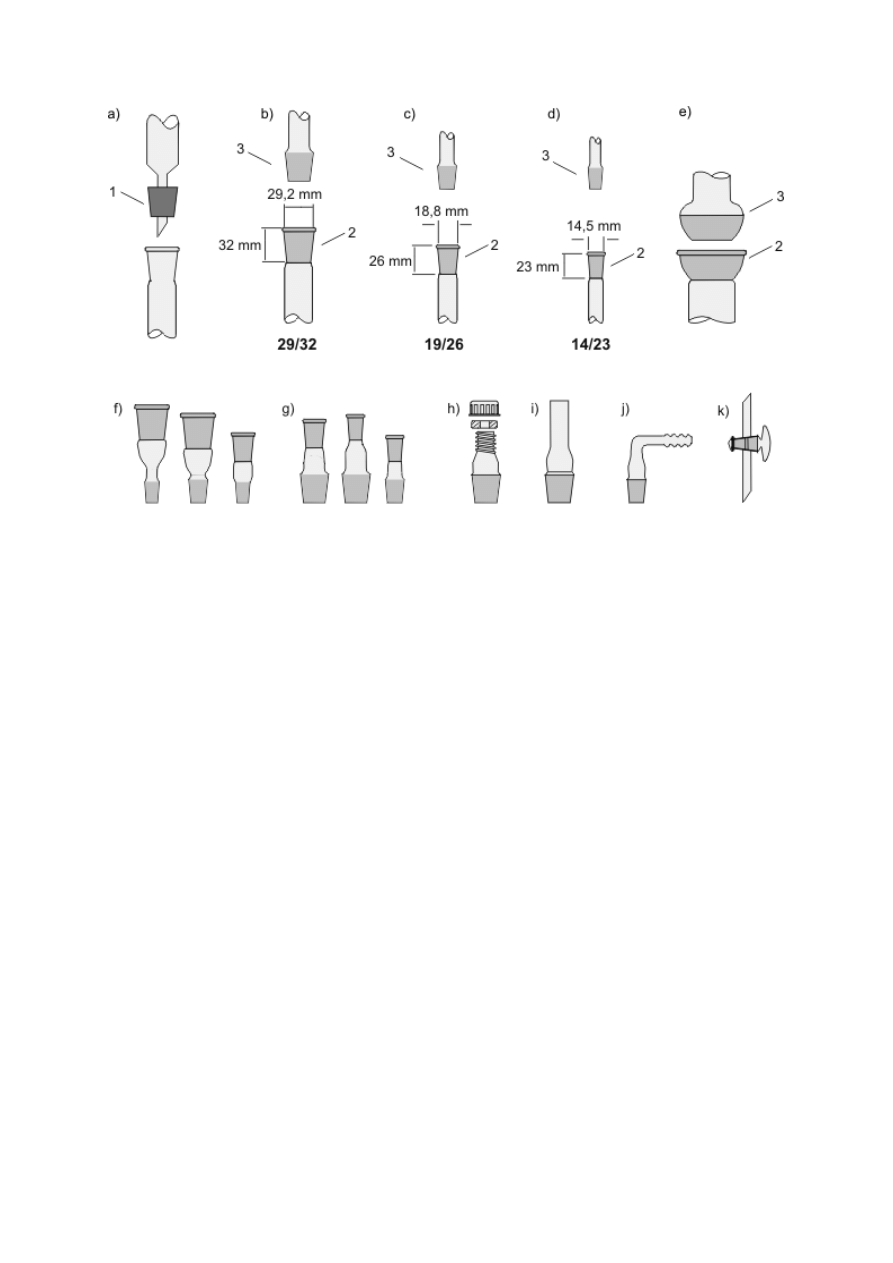
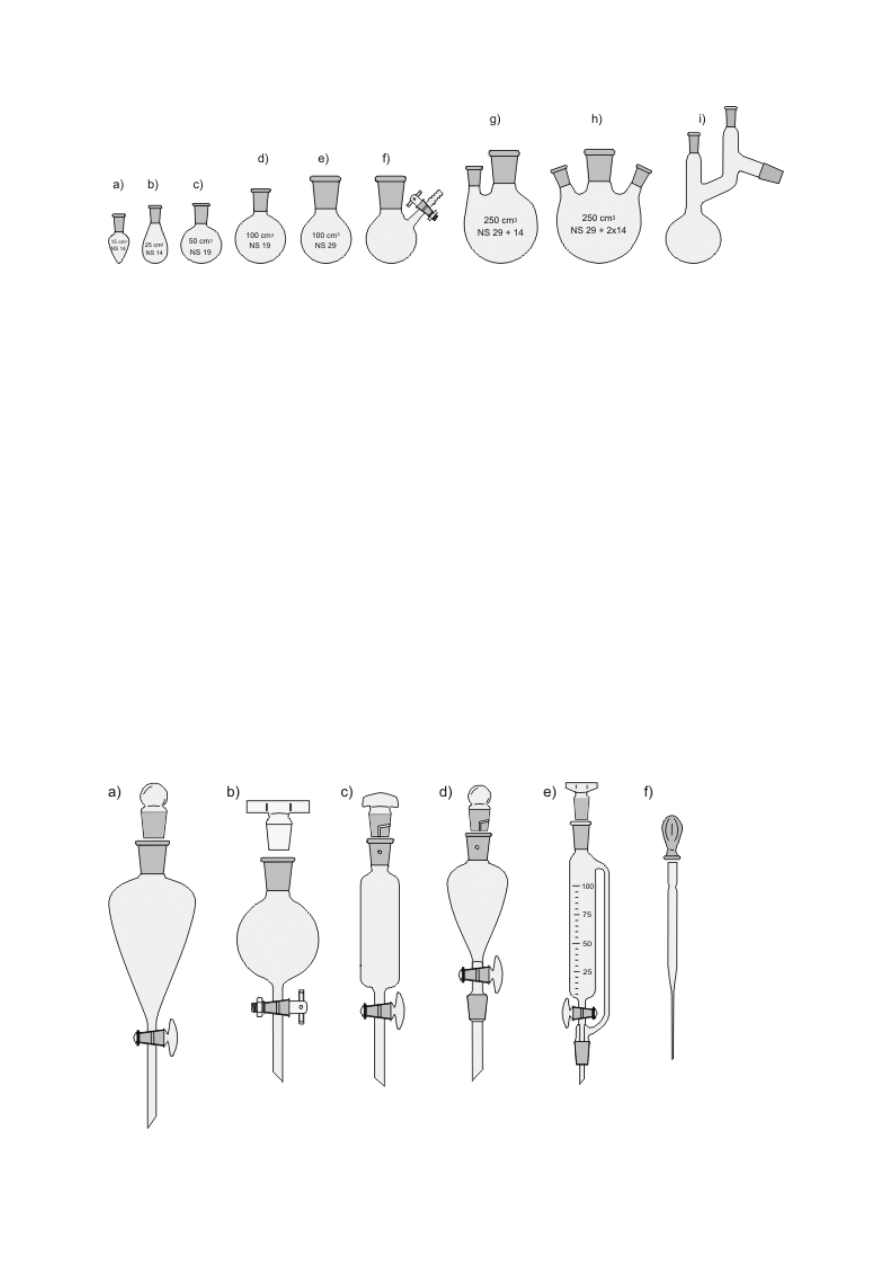
Standardowym wyposażeniem każdej pracowni jest zestaw zlewek różnego kształtu
i pojemności oraz kolb Erlenmeyera (kolb stożkowych), służących głównie do sporządzania
roztworów, rzadziej do przeprowadzania reakcji (rys. 2a–e). Należy jednak pamiętać, że
zlewki i kolby nie są naczyniami miarowymi i posługiwanie się podziałkami znajdującymi się
na ściankach może prowadzić do dużych błędów w szacowaniu objętości roztworu. Do
przenoszenia i przechowywania ciekłych substancji wykorzystuje się naczynia płaskodenne
zaopatrzone w szczelnie dopasowane korki na szlifie: kolby Erlenmeyera, butelki, kolby
płaskodenne (rys. 2f–h). Często są to naczynia wykonane ze szkła sodowego i nie nadają się
do ogrzewania!
Rysunek 2. a) Zlewka wysoka (wąska); b) zlewka niska; c) mała zlewka; d) kolba stożkowa
bez szlifu (kolba Erlenmeyera); e) kolba stożkowa o szerokiej szyi; f) kolba stożkowa ze
szlifem i korkiem; g) butelka z doszlifowanym korkiem; h) kolba płaskodenna ze szlifem i z
korkiem
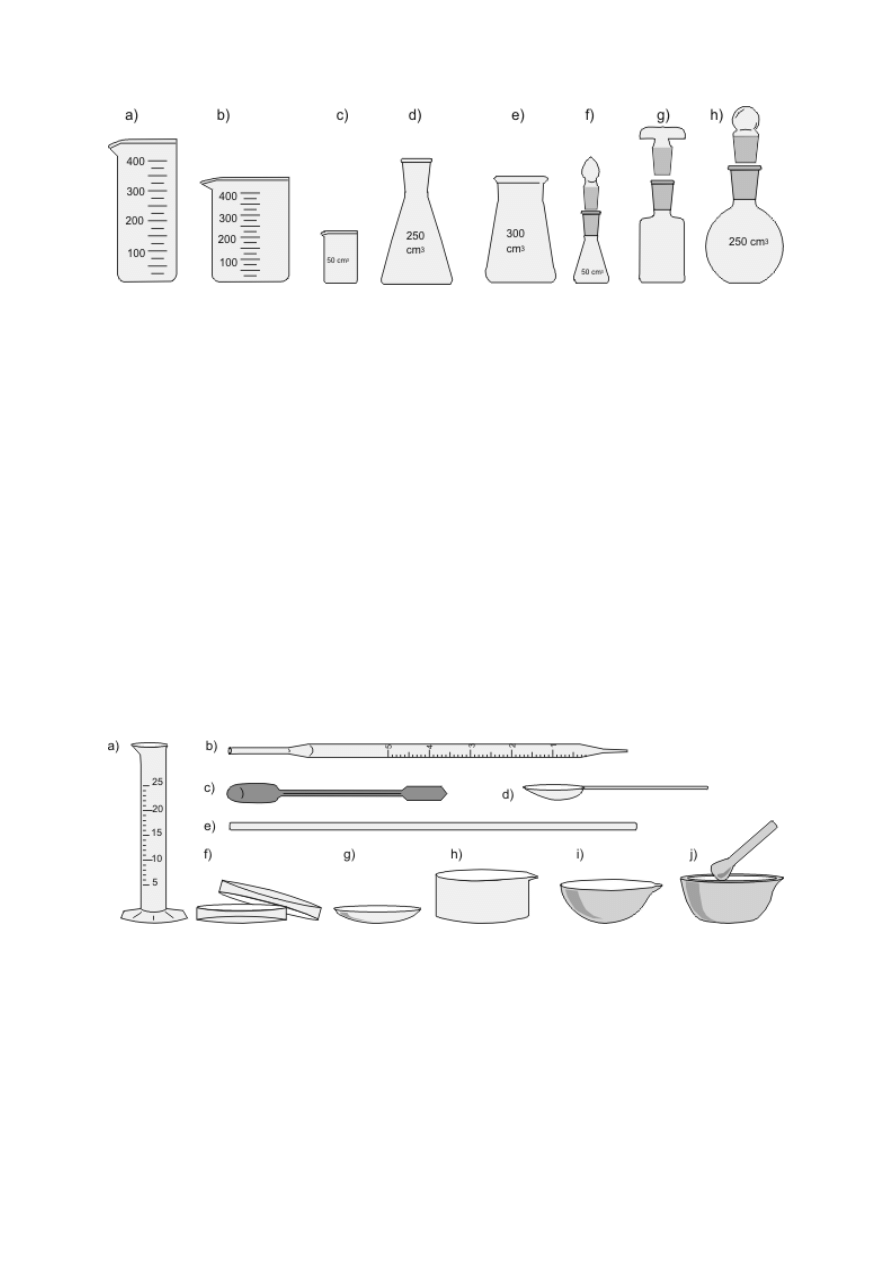
Do dokładniejszego odmierzania objętości służą cylindry miarowe (czyli menzurki,
o pojemności 10, 25, 50, 100 i więcej cm
3
) oraz pipety, najczęściej wyskalowane pipety
wielomiarowe, o pojemności 1, 2, 5, 10 lub 25 cm
3
(rys. 3a, b). Przy odważaniu
i przenoszeniu ciał stałych niezbędne są szpatułki metalowe o różnym kształcie, często
giętkie oraz łyżeczki metalowe lub polipropylenowe, przy przelewaniu i mieszaniu cieczy —
szklane bagietki (pręciki, rys. 3c–e).
Szalki Petriego i szkiełka zegarkowe (rys. 3f, g) służą przeważnie do suszenia
i przechowywania nieszkodliwych ciał stałych, czasami również do odparowywania
niewielkich objętości roztworów. Większe objętości roztworów wodnych nieszkodliwych
substancji można odparowywać w szklanych krystalizatorach i porcelanowych
parownicach (rys. 3h, i). Krystalizatory mogą również być wykorzystane do przygotowania
łaźni wodnej, lodowej lub olejowej. Rozdrabnianie i ucieranie ciał stałych przeprowadza się
w porcelanowych moździerzach, a niewielkich ilości cennych substancji — w moździerzach
agatowych (rys. 3j).
Rysunek 3. a) Cylinder miarowy (menzurka); b) pipeta wielomiarowa; c) łopatka metalowa
(szpatułka); d) łyżeczka metalowa, porcelanowa lub polipropylenowa; e) bagietka szklana
(pręcik); f) szalka Petriego; g) szkiełko zegarkowe; h) krystalizator; i) parownica; j)
moździerz z pistlem (tłuczkiem)
Zwykłe szklane lejki stożkowe służą głównie do przelewania lub, po założeniu sączka
z bibuły, do przesączania cieczy. Lejki polietylenowe z szerokimi krótkimi nóżkami ułatwiają
staranne przenoszenie ciał stałych (rys. 4a–c). We wszystkich lejkach przeznaczonych do
odsączania osadów pod zmniejszonym ciśnieniem na granicy korpusu i nóżki jest
zamocowana płytka z małymi otworami (lejek sitowy Büchnera) lub płytka ze spieku
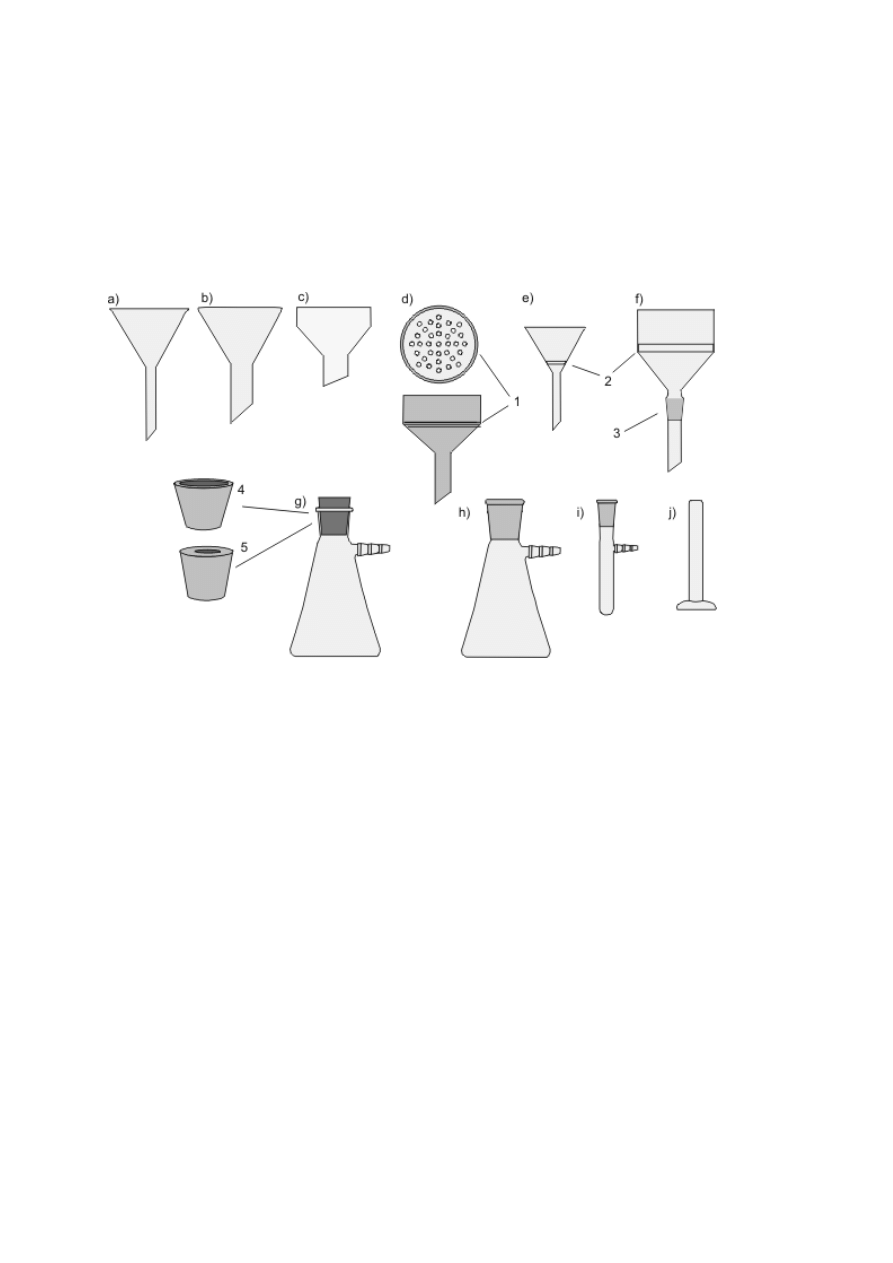
szklanego (rys. 4d–f). Lejki te mogą być rozmaitej wielkości i kształtu (walcowe bądź
stożkowe — Hirscha). Są wykonane z różnych materiałów (szkła, porcelany, polipropylenu),
a niektóre mają na nóżce zewnętrzny szlif. Omawiane lejki mocuje się w szyjce kolby
ssawkowej, czyli grubościennej stożkowej kolby z tubusem (rys. 4g, h). Jeżeli jeden z
elementów nie ma szlifu, nóżkę lejka należy osadzić w gumowym korku z otworem (5) lub
specjalnym gumowym kołnierzu (4). Zamiast kolby ssawkowej można też używać
grubościennej probówki z tubusem (rys. 4i). Odsączony osad odciska się za pomocą ubijaka
do osadów, który przypomina kształtem szklany gwóźdź z płaskim łbem (rys. 4j).
Rysunek 4. a) Lejek szklany zwykły; b) lejek z krótką, szeroką nóżką; c) polietylenowy lejek
do przesypywania osadów; d) porcelanowy lub polipropylenowy lejek Büchnera — widok z
góry i z boku, 1 — płytka z otworami w lejku sitowym; e) lejek Hirscha; f) szklany lejek typu
Büchnera ze szlifem, 2 — płytka ze spieku szklanego, 3 — szlif zewnętrzny na nóżce lejka;
g) kolba ssawkowa (stożkowa z tubusem) bez szlifu, 4 — kołnierz gumowy lub 5 — korek z
otworem; h) kolba ssawkowa ze szlifem; i) probówka ze szlifem i tubusem; j) ubijak do
osadów.
Większość operacji laboratoryjnych przeprowadza się w naczyniach o zaokrąglonym dnie,
zaopatrzonych w jeden lub więcej otworów zakończonych przeważnie szlifami
wewnętrznymi, czyli w kolbach gruszkowatych, sercowatych lub okrągłodennych (rys.
5a–e). Najczęściej w pracowni spotyka się kolby okrągłodenne o pojemności 50, 100, 250 i
500 cm
3
. Kolby wieloszyjne (rys. 5f–h), służące do prowadzenia reakcji wymagających
jednoczesnego podłączenia kilku elementów aparatury do jednego naczynia, różnią się liczbą
szyj (od dwóch do czterech), ich geometrią (szyje proste — gdy szyje są równoległe, lub
szyje skośne, gdy osie symetrii szyj przecinają się w punkcie leżącym we wnętrzu kolby)
oraz wielkością szlifów. W praktyce laboratoryjnej nie spotyka się kolb dwu- i trójszyjnych o
pojemności mniejszej niż 50 cm
3
. Kolba dwuszyjna, której jedna szyja zastąpiona jest rurką z
kranem do doprowadzania bądź odprowadzania gazów, jest nazywana kolbą Schlenka.
Zależnie od potrzeby kolby okrągłodenne mogą być scalone z różnego rodzaju nasadkami. Na
przykład do przeprowadzenia destylacji pod zmniejszonym ciśnieniem można zastosować
kolbę połączoną trwale z nasadką Claisena (rys. 5i).
Rysunek 5. a) Kolba sercowata; b) kolba gruszkowata; c), d), e) jednoszyjne kolby
okrągłodenne o różnej pojemności i rozmiarach szlifów; f) kolba Schlenka; g) dwuszyjna
kolba okrągłodenna o szyjach prostych; h) trójszyjna kolba okrągłodenna o szyjach skośnych;
i) kolba Claisena
Operacje rozdzielania dwóch niemieszających się ze sobą cieczy przeprowadza się przy
użyciu rozdzielaczy o różnej pojemności i kształcie: gruszkowatym, kulistym bądź
cylindrycznym (rys. 6a–c). Korki i krany mogą być wykonane z doszlifowanego szkła lub z
teflonu. To ostatnie rozwiązanie, mimo że droższe, zapewnia większą szczelność i eliminuje
konieczność smarowania tych elementów. Niektóre modele rozdzielaczy mają w górnym
szlifie naczynia mały otwór, a ich korki mają otwór lub nacięcie łączące ich dolną część z
boczną powierzchnią. Przy odpowiednim ustawieniu system taki pozwala wyrównać ciśnienie
wewnątrz naczynia z ciśnieniem atmosferycznym bez konieczności otwierania rozdzielacza.
Podobne naczynia służą też jako wkraplacze do dozowania ciekłych reagentów do kolb (rys.
6c–e). Często na ich nóżkach jest zamontowany szlif zewnętrzny, ułatwiający montaż, w
przeciwnym przypadku wkraplacze mogą być osadzane w szyjkach kolb na złączkach
przedstawionych na rysunku 1h lub, ostatecznie, na korkach z otworami. Jeżeli należy
wkroplić ciecz do zamkniętego naczynia, w którym panuje ciśnienie różniące się nieco od
atmosferycznego, należy użyć wkraplacza z wyrównywaczem ciśnienia, czyli rurką łączącą
jego nóżkę z górną częścią. Dodawanie/pobieranie niewielkich objętości cieczy do/z naczyń
otwartych można przeprowadzić, stosując pipetkę Pasteura, czyli jednostronnie wyciągniętą
w formie kapilary rurkę z gumowym kapturkiem (rys. 6f). W przypadku roztworów wodnych
można polecić pipetki Pasteura zintegrowane z naciągaczem, wykonane z polietylenu.
Rysunek 6. a) Rozdzielacz gruszkowaty ze szklanym korkiem i kranem; b) rozdzielacz
kulisty z teflonowym korkiem i kranem; c) rozdzielacz/wkraplacz cylindryczny ze szklanym
kranem i korkiem z otworem odpowietrzającym; d) wkraplacz gruszkowaty na szlifie z
korkiem z otworem odpowietrzającym; e) wkraplacz z wyrównywaniem ciśnienia; f) pipetka
Pasteura
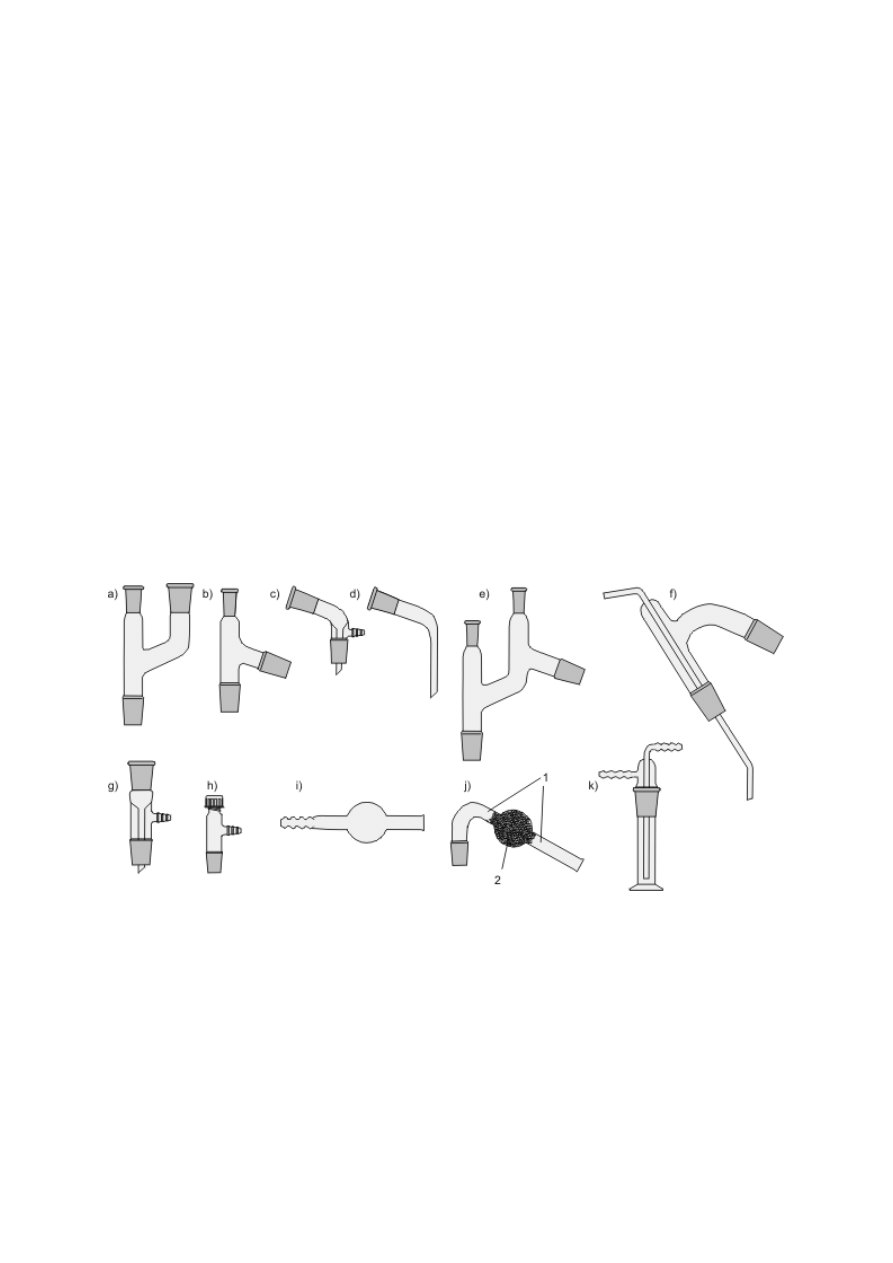
Konstruowanie złożonej aparatury ułatwiają różnego rodzaju nasadki, złącza i przedłużacze,
spośród których omówiono jedynie kilka najczęściej stosowanych (rys. 7). Nasadka „Y” (rys.
7a) pozwala na podłączenie dwóch elementów aparatury do kolby jednoszyjnej. Nasadki
destylacyjne (rys. 7b, e lub f) wraz z przedłużaczami (rys. 7c lub d) są stosowane do
konstruowania zestawów do destylacji prostej, pod zmniejszonym ciśnieniem, z parą wodną
itp. Złączki z bocznymi tubusami (np. 7g, h) wykorzystuje się w aparaturze pracującej pod
zmniejszonym ciśnieniem. Na przykład połączenie złączki (7h) z grubościenną kolbą
Erlenmeyera zastępuje kolbę ssawkową.
Nasadki o różnych kształtach (proste lub zgięte, zakończone szlifem lub oliwką), wypełnione
kolejno watą celulozową lub szklaną, higroskopijną substancją (często granulowanym
chlorkiem wapnia) i ponownie watą, służą do zabezpieczania aparatury przed dostępem
wilgoci z otoczenia (rys. 7i, j). Nazywa się je zwyczajowo „rurkami z chlorkiem wapnia”.
Pochłanianie niepożądanych składników z gazów można przeprowadzać też w płuczkach
(rys. 7k), wypełnionych odpowiednim adsorbentem.
Rysunek 7. a) Nasadka dwudrożna typu „Y”; b) nasadka destylacyjna; c) przedłużacz
destylacyjny z tubusem; d) przedłużacz destylacyjny typu fajka; e) nasadka Claisena do
destylacji pod zmniejszonym ciśnieniem; f) nasadka do destylacji z parą wodną; g) nasadka
prosta z tubusem (próżniowa); h) prosta nasadka gwintowana na szlifie z tubusem; i) rurka
zabezpieczająca przed dostępem wilgoci z połączeniem oliwkowym (pusta); j) rurka
zabezpieczająca przed dostępem wilgoci ze szlifem: 1 — wata, 2 — środek pochłaniający
wodę; k) płuczka
Ogrzewanie ciekłych substancji wymaga schładzania tworzących się par i zawracania ich do
naczynia bądź odprowadzania na zewnątrz. Pierwszemu celowi służą chłodnice zwrotne,
montowane w szyjach kolb za pomocą połączeń szlifowych (rys. 8a–e). Gorące pary, płynące

zazwyczaj wewnętrzną rurką chłodnicy, są oziębiane i skraplane przez chłodziwo poruszające
się w zewnętrznym płaszczu. W niektórych rozwiązaniach konstrukcyjnych stosuje się
również chłodzenie wewnętrzne (np. rys. 8d lub chłodnice w wyparkach). Najczęściej
stosowanym chłodziwem jest bieżąca woda (chłodnice wodne, rys. 8a–d), doprowadzana
dolnym, a odprowadzana górnym króćcem chłodnicy. Do schładzania par można wykorzystać
też powietrze (chłodnica powietrzna, rys. 8e) lub np. stały ditlenek węgla w chłodnicach
Dewara. Najprostszą chłodnicą wodną jest chłodnica Liebiga (rys. 8a) — o małej
efektywności schładzania par (odpowiednia do schładzania par cieczy o wysokich
temperaturach wrzenia), może być wykorzystana w konstrukcji np. zestawów do destylacji
(rys. 8f). Spośród innych chłodnic zwrotnych o zewnętrznym chłodzeniu wodnym
najpopularniejsze są chłodnice kulkowe (Allihna, rys. 8b, o umiarkowanej wydajności
chłodzenia) oraz chłodnice spiralne (Grahama, rys. 8c, mają najwyższą efektywność
chłodzenia par, lecz skropliny często zatykają spiralę; stosuje się je zatem głównie do
oziębiania par w zestawach destylacyjnych o chłodnicach pionowych). Górna część każdej
chłodnicy może być zakończona szlifem ułatwiającym montowanie innych elementów (np.
rurki z chlorkiem wapnia); jest to chłodnica z dwoma szlifami, w przeciwnym przypadku —
chłodnica z jednym szlifem (8b, e).
Schładzanie par, skraplanie ich i zbieranie poza naczyniem jest podstawą procesu destylacji
(patrz podrozdz. V.3), stąd urządzenia służące do chłodzenia przepływowego zwane są często
chłodnicami destylacyjnymi. Efekt ten można osiągnąć, konstruując zestaw z chłodnicy
Liebiga oraz odpowiednich nasadek i przedłużaczy (rys. 8f) bądź posługując się gotowymi,
zintegrowanymi urządzeniami (rys. 8g, h). Podobnie, jak w przypadku chłodnic zwrotnych,
chłodziwem może być woda (rys. 8g–i) lub powietrze (rys. 8j). W tym ostatnim przypadku,
element aparatury (j) nazywany jest powietrzną chłodnicą destylacyjną bądź, bardziej
formalnie, nasadką destylacyjną z boczną rurką bez szlifu. W małych zestawach do destylacji
lub sublimacji często używa się wodnych chłodnic palcowych, czyli tzw. palców
chłodzących, zwanych czasami „zimnymi palcami” (rys. 8i).
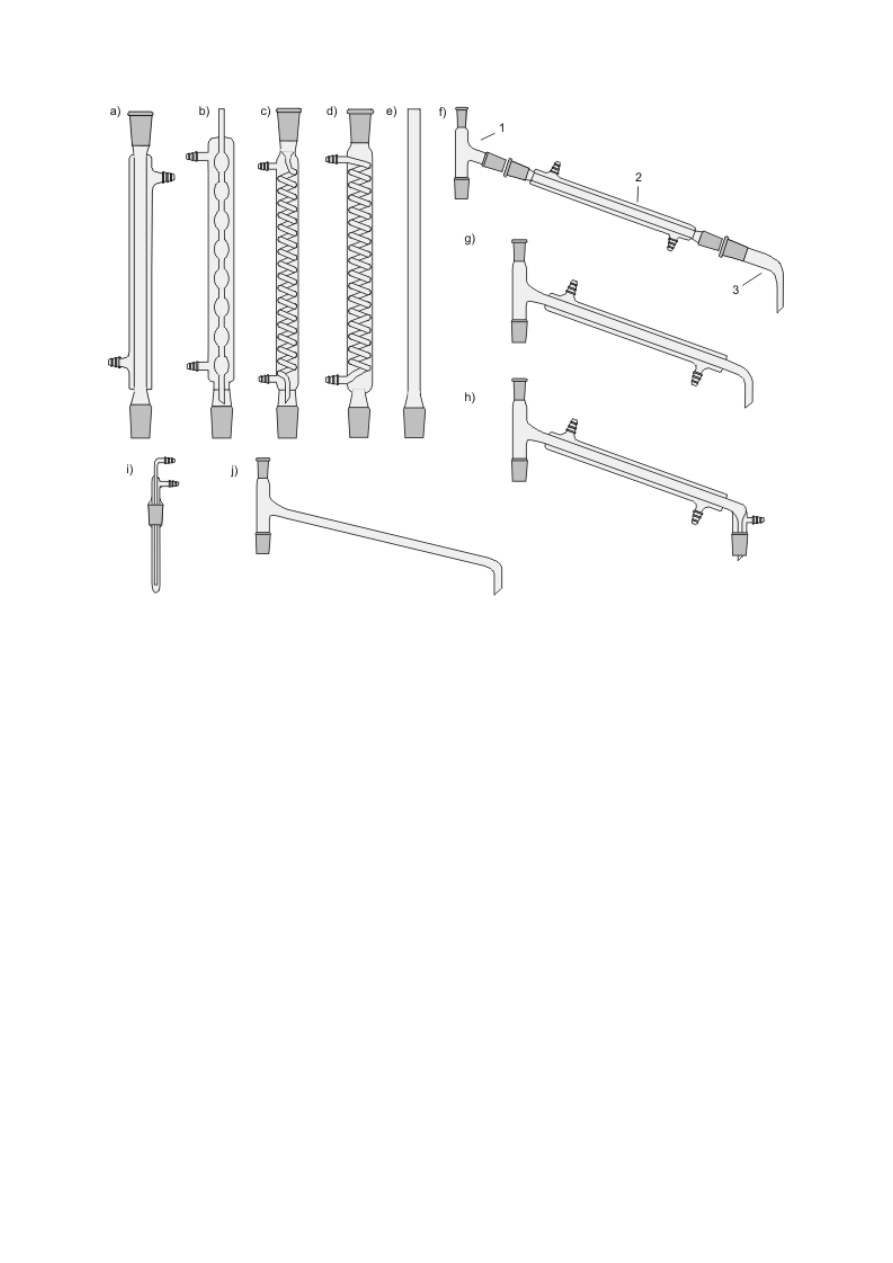
Rysunek 8. a) Chłodnica Liebiga z dwoma szlifami; b) chłodnica kulkowa Allihna z jednym
szlifem; c) chłodnica spiralna Grahama; d) chłodnica ze spiralnym chłodzeniem
wewnętrznym; e) chłodnica powietrzna z jednym szlifem; f) elementy tworzące zestaw do
destylacji: 1 — nasadka destylacyjna, 2 — chłodnica Liebiga, 3 — przedłużacz typu fajka; g)
chłodnica do destylacji w układzie otwartym (zintegrowany zestaw f); h) chłodnica do
destylacji zintegrowana z przedłużaczem ze szlifem i tubusem bocznym; i) chłodnica palcowa
(palec chłodzący, „zimny palec”); j) powietrzna chłodnica destylacyjna
W praktyce laboratoryjnej do pomiaru temperatury stosuje się dwa podstawowe rodzaje
termometrów: szklane (z wypełnieniem rtęciowym, alkoholowym lub toluenowym, rys. 9a–
d) oraz elektroniczne (cyfrowe) z czujnikiem termoparowym (rys. 9e). Termometry szklane
mogą być zaopatrzone w szlif stały (rys. 9b, c), szlif ruchomy (rys. 9d) lub nie posiadać szlifu
(rys. 9a). Część termometru od zbiorniczka do szlifu (bądź początku skali) jest zwana nogą
termometru. Termometry laboratoryjne są urządzeniami drogimi i podatnymi na
uszkodzenia, należy posługiwać się nimi z wyjątkową ostrożnością: nie wkładać do
rozgrzanych urządzeń, nie oziębiać gwałtownie, chronić przed mechanicznym uszkodzeniem.
Termometr zanurzony w otwartym naczyniu (np. w zlewce) musi być umocowany w małej
łapie. Zawsze należy dopilnować, aby termometr nie dotykał ścianek kolby ani nie zderzał się
np. z mieszalnikiem.
Stłuczenie termometru rtęciowego pociąga za sobą konieczność kłopotliwej neutralizacji
rozlanej rtęci (patrz podrozdz. I.3). Ze względów bezpieczeństwa należy starać się stosować
termometry wypełnione cieczami mniej toksycznymi niż rtęć lub różnego rodzaju termometry
elektroniczne. Termometry elektroniczne są bezpieczniejsze niż termometry szklane,
jednak należy pamiętać, że ich metalowych końcówek nie wolno zanurzać w żrących
roztworach (mocnych kwasach, stężonych zasadach, bromie), a głowica termometru nie
może być wystawiona na działanie wilgoci ani rozpuszczalników organicznych, gdyż może to
doprowadzić do zniszczenia części elektronicznych bądź obudowy!
UWAGA: Termometry elektroniczne sprzężone z mieszadłami magnetycznymi nie służą do
pomiaru temperatury w naczyniach do prowadzenia reakcji, lecz do kontroli temperatury w
łaźniach!
Rysunek 9. a) Szklany termometr bez szlifu; b) termometr szklany ze szlifem z krótką nogą,
1 — „noga” termometru; c) termometr ze szlifem z długą nogą; d) termometr z ruchomym
szlifem; e) termometr elektroniczny
II.2. Przygotowanie aparatury szklanej do
pracy
Aparatura szklana używana do eksperymentu musi być czysta, sucha i nieuszkodzona.
Niektóre zanieczyszczenia można usunąć przez staranne umycie szklanej części ciepłą wodą
z detergentem, za pomocą okrągłej szczotki. Bardzo wąskie rurki myje się specjalnymi
„czyścikami” (jak do czyszczenia fajek). W taki sposób nie da się jednak usunąć rozmaitych
rodzajów zanieczyszczeń organicznych. W takim przypadku naczynie spłukuje się
strumieniem acetonu z tryskawki nad pojemnikiem ze zlewkami acetonowymi, potem dopiero
w razie potrzeby myje wodą lub od razu pozostawia do wyschnięcia. Aceton jest względnie
tanim rozpuszczalnikiem, jednak droższym niż woda i toksycznym, należy więc używać go
oszczędnie. Ponadto zanieczyszczony aceton musi być poddany kosztownej utylizacji lub
regeneracji przed ponownym użyciem.
O ile reakcja lub krystalizacja nie jest prowadzona w wodzie, używana aparatura musi być
sucha. Wilgotne szkło można wysuszyć szybko, przemywając je niewielką ilością acetonu
i susząc w strumieniu ciepłego powietrza z ręcznej suszarki. Niektóre reakcje (np. ze
związkami Grignarda) wymagają bardzo starannego wysuszenia aparatury szklanej w
wysokiej temperaturze w suszarce elektrycznej lub w płomieniu palnika gazowego. Tak
wysuszone szkło nie powinno stygnąć na wolnym powietrzu, bo kondensująca się na nim
niewielka ilość pary wodnej może zakłócić przebieg eksperymentu. Powinno być suszone w
eksykatorze nad środkiem suszącym. Sprzęt szklany o większych wymiarach należy studzić
bezpośrednio w suszarce próżniowej po uprzednim wyłączeniu ogrzewania. Przed
rozpoczęciem pracy należy starannie obejrzeć szklane części czy nie są uszkodzone.
Wyszczerbione krawędzie szkła mogą spowodować groźne skaleczenie dłoni, a nawet drobne
pęknięcie może stać się przyczyną poważnego wypadku.
Aby zapewnić szczelność połączeń szlifowych i jednocześnie zabezpieczyć je przed
zacięciem (przypadkowym „sklejeniem się”, tzw. zapiekaniem), przed złączeniem
powierzchnie szlifu pokrywa się cienką warstwą odpowiedniego smaru silikonowego lub

mineralnego o dużej lepkości. Jest to niezbędne zwłaszcza podczas pracy pod zmniejszonym
ciśnieniem, przy ogrzewaniu, przy pracy z alkalicznymi roztworami, szczególnie jeśli złącza
szlifowe nie są wysokiej jakości. Smarowania wymagają również szlifowane krany, np. w
rozdzielaczach czy kolumnach chromatograficznych. Cienką warstwę smaru należy nałożyć
starannie, w kilku miejscach wzdłuż tulei na szlif zewnętrzny, złożyć go z wewnętrznym i
obracać, aż obie powierzchnie będą równomiernie pokryte smarem. Smar użyty w nadmiernej
ilości może w podwyższonej temperaturze spłynąć do mieszaniny reakcyjnej i zanieczyścić
ją. Szlifowane złącza uszczelnić można również za pomocą specjalnych teflonowych
kołnierzy lub teflonowej taśmy nawiniętej na szlif, używanie smaru jest wówczas zbędne. Nie
smaruje się również kranów i połączeń teflonowych.
Łączenie szklanych części w zestawy należy przeprowadzać z rozwagą, unikając naprężeń i w
efekcie uszkodzeń. Aparatura szklana jest mocowana do stabilnych statywów za pomocą łap
i odpowiednich złączek, czyli tak zwanych muf (rys. 10a). Poprawnie zamontowana na
statywie mufa powinna mieć pionowe wycięcie skierowane w stronę montującego, zaś
poziome umieszczone w górnej części mufy, tak aby montowana w nim następnie łapa
opierała się o mufę, a nie była „podwieszona” pod nią. Zależnie od rozmiaru i kształtu
aparatury do jej mocowania stosuje się łapy różnych wielkości (rys. 10b, c, d). Szkło
laboratoryjne o stożkowym kształcie (lejki, rozdzielacze gruszkowate) można zawieszać w
kółkach metalowych (rys. 10e).
Łapy przytrzymują szklane części aparatury, a ich liczba zależy od rodzaju zestawu (patrz np.
podrozdz. III.7). Na przykład pionowy zestaw do ogrzewania pod chłodnicą zwrotną wymaga
użycia dwóch łap, jedna obejmuje kolbę, druga chłodnicę. Nie wszystkie elementy pionowe
bardziej złożonych zestawów (np. nasadka destylacyjna) wymagają przymocowania łapami, o
ile przymocowany jest element leżący poniżej i powyżej. Konieczne jest przymocowanie
łapą chłodnicy destylacyjnej i innych cięższych części szklanych. Niektóre drobne elementy
zestawu mogą być przymocowane za pomocą plastikowych klamerek lub gumek
„recepturek”, jednak nie mogą znajdować się one zbyt blisko źródeł ciepła.
Łapę najbezpieczniej jest zaciskać na szklanej części jedną ręką, a drugą dokręcać śrubę, co
zmniejsza ryzyko zgniecenia szkła. Wszystkie śruby łap i muf najlepiej jest ostatecznie
dokręcić do lekkiego oporu po zmontowaniu całości zestawu.
Rysunek 10. a) Sposób mocowania mufy do statywu: 1 — miejsce do przykręcenia łapy; b)
łapa trójkątna z okładziną filcową — do małych części aparatury; c) łapa okrągła z okładziną
filcową — np. do szyjek kolb z szerokim szlifem; d) łapa czteropalczasta — do chłodnic; e)
kółko metalowe z punktowo nałożoną okładziną gumową do mocowania lejków, rozdzielaczy
gruszkowatych itp.

Po skończonej pracy szlifowane złącza należy szybko rozdzielić. Czynność ta ma zapobiec
tzw. „zapieczeniu się” szlifów. Otwieranie zapieczonych szlifów bywa niekiedy bardzo
kłopotliwe. Czasem wystarczy postukać w ich powierzchnię kawałkiem drewna (np.
trzonkiem jakiegoś narzędzia), jeżeli nie poskutkuje, delikatnie ogrzewa się je palnikiem
gazowym, ewentualnie zanurza w odpowiednich kąpielach. Przy otwieraniu zapieczonych
szlifów należy postępować bardzo ostrożnie, bo grozi to pęknięciem szkła i pokaleczeniem
eksperymentatora.
Jeżeli złącza szlifowe były pokryte smarem, należy dokładnie go usunąć za pomocą
miękkiego papieru jeszcze przed przelewaniem mieszaniny reakcyjnej, aby nie doprowadzić
do zanieczyszczenia produktu. Przed umyciem szklanych części po reakcji należy ponownie,
bardzo dokładnie oczyścić powierzchnię szlifów z resztek smaru. Aparatura szklana powinna
być umyta zaraz po zakończeniu eksperymentu, bo dużo trudniej jest usunąć stare, rozłożone i
spolimeryzowane resztki niż świeże zabrudzenia. Mycie osadów niewiadomego pochodzenia
należy przeprowadzać z wielką ostrożnością, bo na przykład w bezbarwnej, suchej masie
złożonej z wodorotlenku i węglanu sodu mogą kryć się resztki metalicznego sodu pozostałe
po osuszaniu jakiegoś rozpuszczalnika! Próba rozpuszczenia w wodzie takiej mieszaniny
doprowadzi do eksplozji.
Nieostrożne obchodzenie się z aparaturą szklaną może skończyć się pokaleczeniem.
Szczególną ostrożność zaleca się podczas łączenia lub demontażu części szklanych za
pomocą ruchów obrotowych oraz przepychania rurek czy termometrów przez gumowe korki z
dziurką. Dłonie należy wówczas zabezpieczyć grubą rękawicą skórzaną lub chwycić szkło
przez gruby materiał, zawsze możliwie jak najbliżej gumowego korka. Pęknięcie szkła
podczas takich czynności może spowodować bardzo niebezpieczne w skutkach skaleczenia,
połączone z uszkodzeniem ścięgien i naczyń krwionośnych dłoni.
Także drobny sprzęt metalowy znajdujący się w laboratorium wymaga sprawdzenia przed
przystąpieniem do montażu aparatury. Statywy metalowe powinny być stabilne. Śruby w
łapach i mufach powinny się lekko obracać, w przeciwnym razie należy je posmarować
smarem do metalu i „rozruszać” za pomocą kombinerek. To samo dotyczy podnośników
laboratoryjnych. Jeżeli miękkie okładki (korkowe, gumowe lub filcowe) w łapach uległy
zużyciu, można wsunąć między łapę a szklaną część kawałek tkaniny lub złożonego,
miękkiego papieru, aby nie dopuścić do bezpośredniego kontaktu metalu ze szkłem. Łapy
czteropalczaste powinny mieć końcówki zabezpieczone kawałkami węża gumowego,
podobnie jak kółka metalowe.
II.3. Użyteczne akcesoria laboratoryjne
W laboratorium znajduje się wiele innego sprzętu pomocniczego i materiałów, bez których
trudno wyobrazić sobie właściwe wykonanie eksperymentu. Sprzęt wielokrotnego użytku
należy utrzymywać w czystości i właściwym stanie technicznym, a materiały używać w
rozsądnych ilościach tylko do tego celu, do jakiego są przeznaczone. W szczególności:
Parafilm — rozciągliwa folia parafinowa służy do ochrony zawartości naczyń
laboratoryjnych przed wilgocią z powietrza. Kawałkami parafilmu owija się szczelnie korki i
zakrętki zamykające kolby i butelki.

Bibuła filtracyjna — specjalnie spreparowana bibuła (standardowo: średnia lub miękka)
służąca wyłącznie do sączenia, a nie do wycierania stołów laboratoryjnych i szkła,
odważania substancji czy też wykonywania notatek laboratoryjnych.
Papierki wskaźnikowe — najczęściej spotykane to papierki uniwersalne (do pomiaru pH,
skala zmiany barwy na opakowaniu), papierki Kongo (do pomiaru pH w roztworach silnie
kwasowych, przy pH < 3,0 barwa niebieska, pH > 5,2 barwa czerwona) oraz papierki
jodoskrobiowe nasycone jodkiem potasu i skrobią (w obecności utleniacza następuje
zabarwienie bezbarwnego papierka na kolor brunatnoniebieski). Przy korzystaniu z
papierków wskaźnikowych należy pamiętać, że do jednego oznaczenia wystarczy jego
pięciomilimetrowy odcinek, na który za pomocą bagietki nanosi się małą kroplę badanej
cieczy.
Węże z gumy naturalnej lub silikonowej i innych tworzyw sztucznych służą najczęściej
do doprowadzania wody chłodzącej do aparatury oraz doprowadzania i odprowadzania
gazów. Węże grubościenne są używane w zestawach pracujących pod zmniejszonym
ciśnieniem. Węże należy dobierać tak, aby dokładnie pasowały do tubusów aparatury
szklanej, końcówek kranów i innych urządzeń. Należy unikać łączenia ze sobą węży o różnej
średnicy — jeżeli jest to bezwzględnie konieczne, trzeba posługiwać się profesjonalnymi
łącznikami do węży. Jeżeli wąż jest zleżały i kruszy się, to nie nadaje się do użytku. Podczas
nakładania na króćce aparatury szklanej wewnętrzną część końcówki węża należy lekko
zwilżyć wodą lub gliceryną, a następnie jedną dłonią, drobnymi, ćwierćobrotowymi ruchami
nasunąć go na króciec. Druga dłoń powinna w tym czasie przytrzymywać część szklaną
możliwie blisko króćca. Jeżeli są problemy z nałożeniem węża, nie należy tego robić na siłę
(ryzyko rozbicia i poważnych skaleczeń), lecz wymienić wąż na inny, o większej średnicy lub
bardziej elastyczny. Zbyt luźno nasunięty wąż może zsunąć się po pewnym czasie, natomiast
zbyt naprężony — pęknąć.
Korki gumowe służą do zamykania kolb stożkowych i okrągłodennych. Nie powinno się
ich stosować do zamykania naczyń, w których znajdują się rozpuszczalniki i odczynniki
organiczne, powodujące rozkład gumy, pęcznienie korków i przyklejanie się ich do szkła.
Fiolki szklane z korkami polietylenowymi służą do przechowywania związków
organicznych. Można je wykorzystywać wielokrotnie, jednak liczne substancje reagują z
polietylenem, korki wówczas ciemnieją i powinny być wymienione na nowe.
Tkanina z włókna szklanego jest odporna na temperaturę, a jej kawałki stosuje się do
owijania szklanych fragmentów zestawów, na których następuje duża utrata ciepła. Zaleca się
stosowanie rękawiczek ochronnych — drobiny szkła wbijają się w naskórek i mogą go
podrażniać.
Klamry (spinki) do szlifów zapobiegają rozdzielaniu się części aparatury połączonych
złączem szlifowym. Klamry nakłada się, przytrzymując mocno jedną dłonią złączone szlify, a
drugą delikatnie wciska spinkę. Jeżeli złącze jest właściwie dopasowane, klamra uniemożliwi
jego przypadkowe otwarcie. Równie ostrożnie, przytrzymując jedną dłonią szklaną cześć
aparatury, po zakończonej reakcji zsuwa się spinkę. Klamry dostępne są w kilku rozmiarach,
odpowiadających standardowym wielkościom szlifów. Wykonywane są z kolorowego
plastiku lub metalu. Klamry plastikowe są odporne tylko do temperatury ok. 120
o
C,
stosowanie ich zatem w operacjach połączonych z ogrzewaniem wymaga dużej rozwagi.
Przeznaczenie i sposób użycia bardziej złożonego sprzętu laboratoryjnego (wag, płaszczy
grzejnych, mieszadeł, kriometrów, refraktometrów, wyparek, pomp itp.) zostanie omówione
w kolejnych dwóch rozdziałach przy omawianiu poszczególnych operacji laboratoryjnych.
III. Podstawowe czynności laboratoryjne

III.1. Przenoszenie i odmierzanie substancji
chemicznych
Jedną z najczęściej wykonywanych czynności w laboratorium chemii organicznej jest
odmierzanie ilości substancji. Należy pamiętać, aby podczas operowania każdym
pojemnikiem z odczynnikiem chemicznym mieć zawsze suche ręce (lub rękawice) i nie
dopuścić do wyślizgnięcia się pojemnika z dłoni. Większe butelki chwyta się zawsze dwiema
rękami — w okolicy szyjki i za dno. Niedopuszczalne jest trzymanie jedną dłonią butelki za
zakrętkę. Po otwarciu pojemnika i pobraniu substancji należy niezwłocznie zamknąć go, nie
dopuszczając do przypadkowej zamiany zakrętek lub korków! Przenoszenie butelek lub
słoików z chemikaliami na większą odległość (np. między pokojami) powinno odbywać się
po wstawieniu ich do odpowiedniego pojemnika z uchwytem (wiaderka).
W przypadku ciał stałych przeprowadza się ważenie substancji. Obecnie standardowym
wyposażeniem laboratorium są wagi elektroniczne, pozwalające na wyznaczenie masy
z dokładnością do 10 mg lub 1 mg, zależnie od modelu. Waga jest bardzo wrażliwym
urządzeniem, wymaga delikatnego postępowania, jest zatem ustawiona na stabilnym podłożu,
wypoziomowana i osłonięta szklaną klatką od gwałtownych ruchów powietrza. Nie wolno
stawiać na wadze przedmiotów o masie większej niż maksymalne dopuszczalne obciążenie
podane na obudowie wagi. Substancje stałe wolno ważyć w naczyniach szklanych (np. na
szkiełku zegarkowym, w szalce Petriego, zlewce itp.), w specjalnych pojemniczkach
wykonanych z tworzywa sztucznego lub w papierowych pudełeczkach wykonanych z
gładkiego papieru (rozdz. IX, Załącznik 6). Odczynnik pobiera się ze słoika za pomocą
metalowej szpatułki ewentualnie łyżeczki wykonanej z plastiku lub porcelany —
niedopuszczalne jest sypanie substancji wprost ze słoika! Po zakończeniu ważenia należy
szczelnie zakręcić słoik i odstawić go na właściwe miejsce. Jeżeli nieco substancji rozsypie
się poza naczynie, to należy wyłączyć wagę i usunąć zanieczyszczenia za pomocą papieru lub
pędzelka, a szalkę wagi przetrzeć ręcznikiem papierowym zwilżonym czystym acetonem lub
etanolem. Należy też zwrócić uwagę na porządek w otoczeniu wagi. Nie wolno odważać
substancji ciekłych. Jest to dopuszczalne jedynie wówczas, gdy ciecz jest zamknięta w
uprzednio starowanej fiolce szklanej lub zakorkowanej kolbce stożkowej.
Aby odmierzyć określoną ilość cieczy (podaną w jednostkach masy), należy przeliczyć jej
masę na objętość, korzystając z tablic fizykochemicznych zawierających dane dotyczące
gęstości cieczy. W większości przepisów potrzebne ilości reagentów ciekłych podane są w
jednostkach objętości, co znacznie ułatwia tok postępowania. Do pomiaru objętości cieczy
służą naczynia miarowe, a w szczególności cylindry miarowe (menzurki) oraz pipety o różnej
objętości. Podczas przelewania cieczy z butelki do innego naczynia (np. menzurki) należy tak
chwycić butelkę, aby etykieta skierowana była ku górze. Nie wolno dopuścić do spływania
kropli cieczy po zewnętrznej ściance butelki — można posłużyć się bagietką lub otrzeć
ostatnie krople o brzeg naczynia, do którego była przelewana ciecz.
Przy korzystaniu z pipet należy zwrócić uwagę na sposób pipetowania. Żadnych cieczy
w laboratorium chemicznym nie wolno pipetować ustami. Służą do tego celu specjalne
pompki z tłokiem, wykonane z plastiku, działające na zasadzie strzykawki lub gumowe
gruszki. Najwygodniejsze są gruszki gumowe wyposażone w trzy zawory. Do dolnego

wentyla przyłącza się pipetę. Jedną ręką naciska się górny zawór, a drugą gruszkę, aby
wycisnąć z niej powietrze. Następnie umieszcza się końcówkę pipety w odmierzanej cieczy i
manipulując zaworem dolnym i bocznym, nabiera się do pipety pożądaną ilość cieczy. Nie
wolno dopuścić, aby ciecz dostała się do pompki. Następnie umieszcza się pipetę nad
naczyniem i naciskając boczny zawór, spuszcza się z niej ciecz. Należy pamiętać o używaniu
tylko czystych i suchych pipet, aby nie zanieczyścić pobieranej cieczy. Po zakończeniu
odmierzania cieczy pojemnik niezwłocznie się zakręca a pipetę starannie myje (w zależności
od potrzeb acetonem lub wodą i acetonem), a następnie dokładnie suszy. Efektywny sposób
suszenia pipety polega na przyłączeniu jej do pompki wodnej i przepuszczaniu przez nią
powietrza w ciągu kilku minut. Do pobierania i dozowania substancji ciekłych można również
stosować strzykawki szklane lub plastikowe.
Niekiedy przepisy określają ilość cieczy w sposób przybliżony (np. „kilka kropli"). Wówczas
najkorzystniejsze jest użycie pipety Pasteura. Jest to szklana rurka bez podziałki, częściowo
wyciągnięta w formie kapilary. Na grubszy koniec nakłada się maleńką gumową pompkę
(kapturek, jak w zakraplaczu stosowanym przy lekarstwach) i naciąga się nieco cieczy do
pipetki. Następnie, naciskając delikatnie gumkę, bez trudu wkrapla się ciecz do naczynia.
III.2. Mieszanie
Mieszanie zapewnia efektywne stykanie się reagujących substancji i zabezpiecza przed
zjawiskiem przegrzania cieczy. W prostych przypadkach wystarczy wymieszać zawartość
zlewki szklanym pręcikiem, ale procesy długotrwałe, szczególnie połączone ze stopniowym
dodawaniem reagentów wymagają użycia mieszadeł. Najbardziej rozpowszechnione są
mieszadła magnetyczne. Napędzany elektrycznie, umieszczony pod ceramiczną lub
metalową płytką, wirujący magnes powoduje obroty niewielkiego, pokrytego teflonem
magnesika znajdującego się w mieszaninie reakcyjnej. Szybkość obrotów można precyzyjnie
regulować. Mniej efektywne są mieszadła, w których zamiast obracającego się magnesu
stałego, stosowany jest system indukujący rotujące pole magnetyczne.
Kształt mieszalnika musi być dostosowany do kształtu naczynia. Do kolb okrągłodennych
stosowane są mieszalniki elipsoidalne, do naczyń o płaskim dnie mieszalniki walcowate. W
obu przypadkach mieszalnik powinien być możliwie jak największy, aby wprawiać w ruch
obrotowy całą zawartość naczynia, jednak podczas obrotu nie może uderzać w inne części
aparatury (np. w termometr, jeśli ten jest zainstalowany w zestawie). Naczynie musi być
umieszczone koncentrycznie na płytce mieszadła, a ustawiona szybkość obrotów nie może
być zbyt duża, w przeciwnym razie mieszalnik nie będzie obracał się stabilnie, co może
doprowadzić do uszkodzenia zestawu. Z drugiej strony, zbyt mała szybkość obrotów nie
zapewni efektywnego mieszania.
Nie wolno wprowadzać mieszalnika do kolby znajdującej się nad mieszadłem magnetycznym
— grozi to rozbiciem naczynia. Najlepiej wprowadzać go do kolby lub zlewki przed
rozpoczęciem pracy, spuszczając po ściance pochylonego naczynia. Mieszalnik wyciąga się
z roztworu za pomocą teflonowego pręta z zatopionym w środku magnesem (popularnie
zwanym wędką magnetyczną), a wystudzoną płytkę mieszadła magnetycznego dokładnie
przeciera się ręcznikiem papierowym zwilżonym acetonem lub etanolem.
Mieszadła magnetyczne mogą współpracować z różnymi typami łaźni i płaszczy grzejnych
(bez metalowej obudowy), jeżeli tylko generują dostatecznie silne pole magnetyczne.
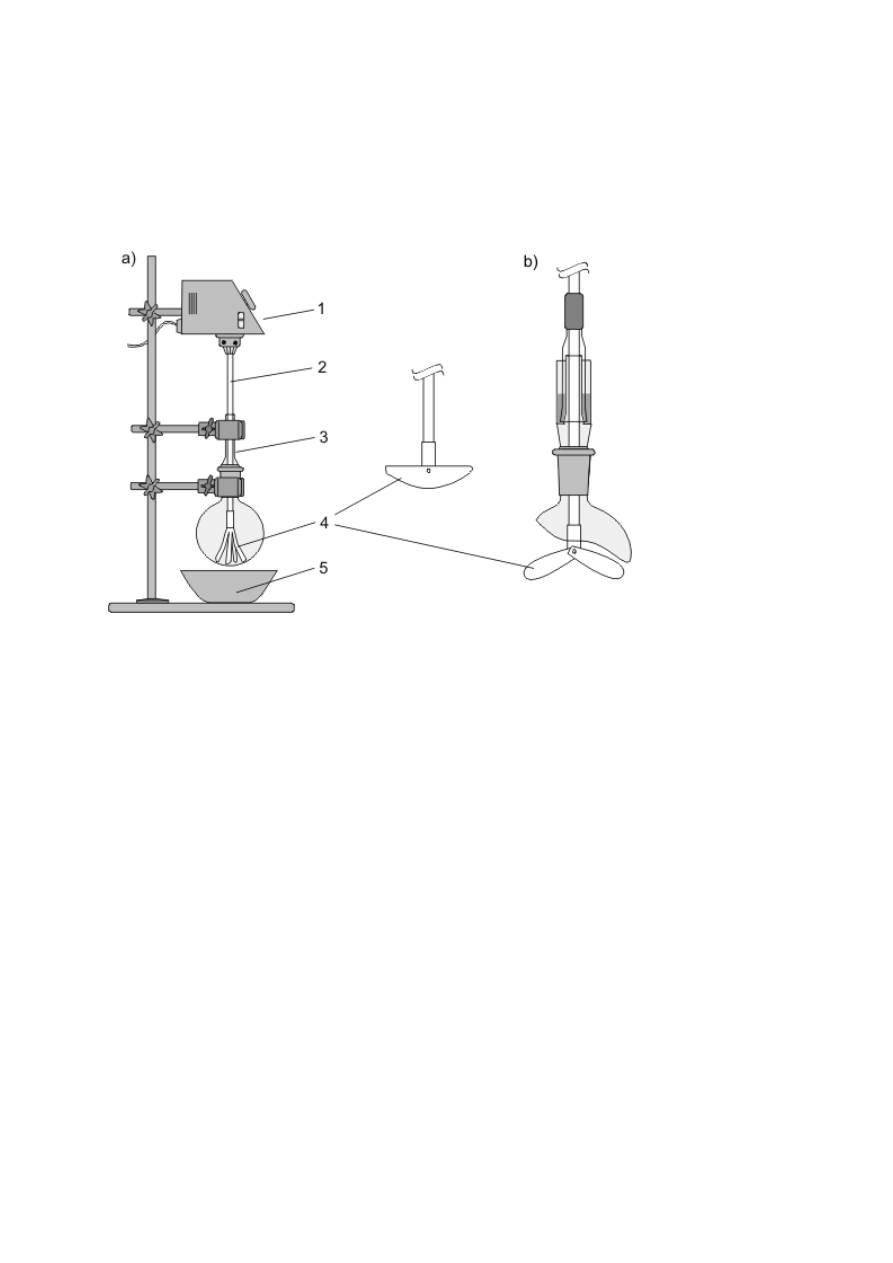
Powszechnie dostępne są mieszadła wyposażone fabrycznie w płytkę grzejną o regulowanej
temperaturze, często sprzężoną z termometrem elektronicznym (patrz podrozdz. III.2).
Używając mieszadła magnetycznego, należy zwrócić uwagę, aby przewody elektryczne nie
znajdowały się w pobliżu rozgrzanej płytki ani podczas pracy mieszadła, ani po jej
zakończeniu! Może to grozić przepaleniem izolacji, zwarciem w instalacji, zniszczeniem
sprzętu i porażeniem eksperymentatora.
Rysunek 11. a) Zestaw z mieszadłem mechanicznym bez uszczelnienia: 1 — silnik
elektryczny z regulacją obrotów, 2 — pręt mieszadła, 3 — prowadnica, 4 — przykładowy
kształt końcówki mieszadła, 5 — miska zabezpieczająca; b) uszczelnienie cieczowe do
mieszadła mechanicznego
Mieszadła magnetyczne nie działają w mieszaninach lepkich, zawierających duże ilości
ciężkiego osadu lub substancje ferromagnetyczne (pył żelaza). W takim przypadku należy
zastosować sprawniejsze, choć dużo bardziej kłopotliwe w użyciu, mieszadło mechaniczne.
Urządzenie to, napędzane silnikiem elektrycznym, obraca sztywny pręt, zakończony
szklanymi lub teflonowymi łopatkami, które zostały uprzednio umieszczone w naczyniu do
prowadzenia reakcji (rys. 11a). Często stosowane są elastyczne, teflonowe „wąsy”,
dopasowujące się do kształtu naczynia z mieszaną cieczą. Pręt mieszadła może przechodzić
luźno przez prowadnicę (rys. 11a) lub, jeżeli mieszanina reakcyjna wymaga izolowania jej od
otoczenia, musi być przeprowadzony przez specjalną nasadkę uszczelniającą. Może być nią
na przykład, dopasowana do szklanego pręta mieszadła prowadnica, przy czym stykające się
powierzchnie muszą być oszlifowane i pokryte cienką warstwą smaru o małej lepkości.
Innym rozwiązaniem jest zastosowanie uszczelnienia cieczowego (rys. 11b). Jest to szklany
dzwon umocowany do pręta elastycznym wężem gumowym i zanurzony w szerszej rurce
szklanej z cieczą (zazwyczaj olejem), przez środek której przechodzi kolejna rurka o średnicy
mniejszej od średnicy dzwonu, lecz nieco większej od średnicy pręta. Należy pamiętać, aby
podczas stosowania nasadki uszczelniającej umożliwić wyrównywanie ciśnienia wewnątrz
kolby okrągłodennej. Najprostsze rozwiązanie polega na użyciu kolby dwuszyjnej z
umieszczoną w szyi bocznej rurką ze środkiem suszącym.

Wszystkie te elementy wymagają bardzo precyzyjnego dopasowania. Jakikolwiek bowiem
boczny ruch pręta mieszającego może uszkodzić cały zestaw. Pręt mieszający musi być
zamocowany koncentrycznie w środkowej szyi naczynia, a jego końcówka nie może ocierać
się ani o ścianki, ani o termometr.
III.3. Ogrzewanie
Wszechobecne przez dziesiątki lat w laboratoriach chemicznych palniki gazowe Bunsena
służą obecnie głównie jako źródło płomienia w pracach pomocniczych związanych z obróbką
szkła lub podczas stapiania próbek substancji z metalicznym sodem, a jedynie wyjątkowo do
ogrzewania niepalnych mieszanin. Zastąpiły je elektryczne urządzenia ogrzewające. Wybór
odpowiedniego typu aparatu zależy od temperatury jaką chcemy osiągnąć, lotności
używanych rozpuszczalników oraz ewentualnej konieczności równoczesnego, intensywnego
mieszania składników. Pracując z gorącą aparaturą, należy zabezpieczać dłonie odpowiednimi
rękawicami ochronnymi!
Uniwersalnymi urządzeniami ogrzewającymi są płaszcze grzejne przystosowane do kształtu
kolb okrągłodennych różnych rozmiarów. W zależności od konstrukcji (gdy nie mają
zewnętrznej, metalowej obudowy) mogą też pracować z mieszadłem magnetycznym. Do sieci
elektrycznej włączane są zawsze przez autotransformatory lub inne urządzenia regulujące
(niezbyt precyzyjnie) dostarczaną moc prądu elektrycznego i tym samym temperaturę
ogrzewania (rys. 12a). Płaszcz grzejny może być umieszczony na podnośniku o regulowanej
wysokości, aby w razie potrzeby można go było szybko obniżyć i ewentualnie usunąć, np.
gdy reakcja przebiega zbyt gwałtownie. Należy zwrócić też baczną uwagę, aby przewody
elektryczne nie dotykały rozgrzanych części płaszcza.
Dodawanie substancji, zwłaszcza ciekłych, do naczyń ogrzewanych w płaszczu grzejnym
należy wykonywać bardzo ostrożnie, aby nawet niewielka ilość nie spadła na rozgrzaną
obudowę płaszcza, ani tym bardziej do jego czaszy grzewczej. Zanieczyszczone płaszcze nie
nadają się do użytku! Płaszcz powinien być dopasowany rozmiarem do używanej kolby i do
znajdującej się w niej ilości roztworu: zbyt mały nie zapewni efektywnego ogrzewania całej
objętości kolby, a zbyt duży będzie powodował bezpośrednie ogrzewanie górnej części kolby,
prowadząc do zesmolenia („przypiekania”) zawartości kolby. Z podobnego względu nie
należy wstawiać do płaszczy kolb zawierających małe ilości roztworu. Niedopuszczalne jest
wkładanie pustych kolb do gorących płaszczy grzejnych, ani żadnych kolb o
zanieczyszczonej powierzchni zewnętrznej.
Łaźnie olejowe umożliwiają równomierne ogrzewanie i precyzyjną regulację wymaganej
temperatury. W praktyce laboratoryjnej funkcję łaźni spełniają szklane krystalizatory lub
metalowe miseczki wypełnione olejem, najczęściej parafinowym. Olej parafinowy jest
użyteczny do temperatury ok. 180–200 ºC, ale jest palny! Olej silikonowy można stosować
do temperatury ok. 300 ºC, ale nie wyższej, ponieważ produkty rozkładu są toksyczne.
Łaźnię olejową umieszcza się najczęściej na płytce grzejnej mieszadła magnetycznego (rys.
12c). Używane są też łaźnie z zanurzoną w oleju spiralą grzewczą. Temperaturę oleju
kontroluje się zanurzonym w niej termometrem szklanym lub elektronicznym.
Najwygodniejszym rozwiązaniem są nowoczesne mieszadła magnetyczne zaopatrzone w
płytkę grzejną i połączone jednocześnie z termometrem elektronicznym, na którym można
ustawiać temperaturę, w jakiej ma być utrzymywana łaźnia znajdująca się na płytce tego
mieszadła. Aby było możliwe ogrzanie łaźni do zadanej temperatury, należy pamiętać, że
orientacyjna temperatura powierzchni płytki, ustawiana niezależnym od termometru
regulatorem, musi być przynajmniej o 50–100
o
C wyższa niż oczekiwana temperatura we
wnętrzu łaźni. Termometry te nie służą natomiast do pomiaru temperatur mieszanin
reakcyjnych! W celu zapewnienia równomiernego ogrzewania całej objętości łaźni olejowej
jej zawartość powinna być ciągle mieszana. Jeżeli łaźnia jest ustawiona na płytce mieszadła,
wystarczy umieścić w niej cienki pręt z materiału ferromagetycznego (często wystarcza
zwykły spinacz biurowy).
Kolba musi być uchwycona łapą, a cały zestaw zmontowany w taki sposób, aby było możliwe
obniżenie temperatury w kolbie przez szybkie uniesienie jej ponad powierzchnię rozgrzanego
oleju. Przygotowując łaźnię olejową, należy pamiętać, że po włożeniu do niej naczynia, a
następnie po ogrzaniu, poziom oleju znacznie się podniesie — nie wolno dopuścić do jego
przelania się przez brzegi łaźni! Poziom oleju w łaźni powinien być porównywalny z
poziomem cieczy w kolbie.
Po wyjęciu naczynia z łaźni olejowej należy odczekać kilka minut, aby większość oleju
spłynęła do łaźni, a jego resztę usuwa się z powierzchni naczynia ręcznikiem papierowym.
Każdą kroplę rozlanego oleju należy zetrzeć jak najszybciej, szczególnie jeśli spadła ona na
rozgrzaną płytkę mieszadła.
Nie wolno używać łaźni olejowej, do której dostała się woda. Gorący olej może się w takiej
sytuacji rozprysnąć, powodując oparzenia i pożar.
Rysunek 12. a) Płaszcz grzejny z regulatorem mocy; b) łaźnia wodna; c) łaźnia olejowa na
mieszadle magnetycznym z płytką grzejną z regulacją temperatury przez termometr
elektroniczny
Stosunkowo bezpiecznymi źródłami ogrzewania do temperatury 100 ºC są łaźnie wodne,
szczególnie przydatne do ogrzewania mieszanin zawierających bardzo lotne, nisko wrzące
rozpuszczalniki (rys. 12b). Para wodna z wrzącej łaźni kondensuje się na zewnętrznych
ściankach kolby, należy więc możliwie starannie dopasować pierścienie ograniczające
przestrzeń między kolbą a łaźnią. Przed użyciem łaźni wodnej należy sprawdzić, czy jest w
niej wystarczająca ilość wody. Temperaturę łaźni wodnej pracującej z wyparką próżniową
należy dostosować do temperatury wrzenia oddestylowywanego rozpuszczalnika.
Do krótkotrwałego, łagodnego ogrzewania można też zastosować gorące powietrze
wytworzone przez ręczną suszarkę elektryczną lub promiennik podczerwieni.

Istotnym zagadnieniem jest dobór odpowiedniego naczynia, w którym prowadzi się
ogrzewanie substancji. Ze względu na ryzyko pęknięcia nie wolno ogrzewać
grubościennych naczyń wykonanych ze szkła o dużej rozszerzalności cieplnej (szkła
sodowego). Nawet kolby ze szkła borokrzemianowego mogą pęknąć po gwałtownym
ogrzaniu (lub ochłodzeniu). Nie wolno zatem wkładać części aparatury szklanej, na przykład,
do rozgrzanej łaźni olejowej. Gorące szkło laboratoryjne (zlewki, kolby) musi być odstawiane
na odpowiednie podkładki lub podstawki (drewniane, korkowe, z wełny mineralnej), nigdy
bezpośrednio na płytki ceramiczne lub szyby na stołach laboratoryjnych! Geometria naczynia
powinna być dostosowana do kształtu elementu grzewczego. Na przykład, nieefektywne jest
ogrzewanie kolb okrągłodennych na płaskich płytkach grzejnych, a bardzo ryzykowne —
ogrzewanie zlewek w płaszczach grzejnych.
Ogrzewanie roztworów do wrzenia
Jeżeli zachodzi potrzeba ogrzewania dowolnego roztworu związku organicznego do wrzenia,
to bez względu na stosowany rozpuszczalnik nie wolno prowadzić takiego procesu
w otwartym naczyniu, gdyż następowałoby wówczas wydzielanie się do atmosfery palnych
lub szkodliwych par. Zastosowanie chłodnicy zwrotnej pozwala ograniczyć to zjawisko.
UWAGA: W każdym przypadku gdy ogrzewa się roztwór do wrzenia, zawartość naczynia
musi być intensywnie mieszana lub do roztworu należy dodać 2–3 tzw. kamyczki wrzenne.
Zapobiega to niebezpiecznemu przegrzewaniu się cieczy powyżej temperatury wrzenia i w
konsekwencji gwałtownemu podrzucaniu zawartości kolby, gdy cała objętość cieczy
przegrzanej przechodzi nagle w stan pary. Kamyczki wrzenne są to kawałki niepowlekanej
porcelany lub innego porowatego materiału, które inicjują wrzenie m. in. dzięki
mikroskopijnym banieczkom powietrza wydobywającym się z ich porów podczas
ogrzewania. Kamyczków wrzennych nie wolno wrzucać do rozgrzanych roztworów — grozi
to wyrzuceniem cieczy z naczynia. Po jednokrotnym użyciu lub po przerwaniu wrzenia tracą
swoje właściwości. Jeżeli z jakiegoś powodu proces wrzenia zostanie przerwany, przed jego
wznowieniem należy wrzucić do kolby nowy kamyczek wrzenny.
Ogrzewanie pod chłodnicą zwrotną jest jedną z najczęściej stosowanych w syntezie
organicznej operacji jednostkowych, zapewniającą utrzymanie stałej temperatury procesu —
jest nią temperatura wrzenia cieczy lub temperatura termostatowanej łaźni. Pary
rozpuszczalnika lub ciekłego reagenta są skraplane i zawracane do kolby w chłodnicy
zwrotnej umocowanej w szyjce kolby.
Ciecze o temperaturze wrzenia poniżej 150
o
C skraplane są w chłodnicy zwrotnej z płaszczem
wodnym (rys. 13a), do skraplania cieczy wrzących w temp. ponad 200 ºC stosuje się
chłodnicę bez płaszcza wodnego, tzw. powietrzną (rys. 13b). Do ogrzewania cieczy
o pośrednich temperaturach wrzenia wykorzystuje się chłodnice wodne z zatrzymanym
obiegiem wody lub bardzo długie chłodnice powietrzne. W obu zestawach (rys. 13a, b), przed
rozpoczęciem ogrzewania należy podnieść znajdujące się na podnośnikach urządzenia
grzewcze (lub zsunąć niżej kolby wraz z chłodnicami), tak aby bezpośrednio ogrzewana była
cała objętość cieczy w kolbie.
Ogrzewanie do wrzenia pod chłodnicą zwrotną prowadzi się w kolbie okrągłodennej o takiej
pojemności, aby ciecz wypełniała ją mniej więcej w połowie, lecz nigdy więcej niż w 2/3
objętości. Szyjka kolby oraz chłodnica zwrotna (w połowie wysokości) powinny być
uchwycone łapami i umocowane do tego samego statywu. Nie należy zbyt mocno dokręcać
łapy na chłodnicy, zapobiegnie to zgnieceniu chłodnicy oraz zapewni możliwość delikatnego
poruszania całego zestawu w linii pionowej. Jeżeli ogrzewa się roztwór w małej kolbce (o
pojemności 100 cm
3
lub mniejszej), można ją przypiąć do chłodnicy za pomocą odpowiedniej
spinki do szlifów — nie ma wówczas potrzeby mocowania kolbki do statywu. Należy jednak
pamiętać, że spinki wykonane z plastiku ulegają zniszczeniu po ogrzaniu do temperatury
wyższej niż 120
o
C!
Rysunek 13. a) Zestaw do ogrzewania na płaszczu grzejnym pod chłodnicą zwrotną z
płaszczem wodnym: 1 — podnośnik, 2 — płaszcz grzejny, 3 — kamyczki wrzenne, 4 —
kolba okrągłodenna z ogrzewanym roztworem, 5 — łapy mocujące zestaw do statywu, 6 —
chłodnica zwrotna, 7 — doprowadzenie wody wężem z kranu, 8 — odprowadzenie wody
wężem do zlewu. b) Zestaw do ogrzewania w termostatowanej łaźni olejowej pod chłodnicą
powietrzną: 1 — mieszadło magnetyczne z płytką grzejną sprzężone z termometrem, 2 —
łaźnia olejowa, 3 — mieszalnik w łaźni, 4 — mieszalnik magnetyczny w kolbce, 5 —
termometr elektroniczny sprzężony z mieszadłem, 6 — chłodnica zwrotna (powietrzna)
Chłodnicę podłącza się do sieci wodnej za pomocą szczelnych, giętkich węży, zwracając
uwagę, czy są nałożone dostatecznie ciasno na wyloty chłodnicy i kranów. Woda z kranu
doprowadzana jest do chłodnicy zwrotnej zawsze dolnym przyłączem (króćcem,
tubusem z oliwkami), a odprowadzana — górnym. Powoduje to całkowite wypełnienie
płaszcza chłodzącego cieczą i zapewnia efektywne, przeciwprądowe wychładzanie
zawracanych do kolby skroplin. Podczas nakładania węży na króćce chłodnicy należy
postępować zgodnie z opisem podanym w podrozdz. II.3, pamiętając o właściwym doborze
średnicy węża, aby nie dopuścić do jego zsunięcia lub pęknięcia. W obu przypadkach może
dojść do zalania wodą laboratorium, uszkodzenia aparatury, zwarć w instalacji, porażenia
prądem itp.! Podobne efekty może mieć nagłe zerwanie węża gumowego z wylotu kranu pod

wpływem zbyt dużego ciśnienia wody, dlatego też przepływ wody powinien być niezbyt
gwałtowny, ale wystarczający do skraplania par wrzącej cieczy nie wyżej niż w połowie
wysokości chłodnicy. Nałożone na chłodnice węże najlepiej jest zabezpieczyć specjalnymi
metalowymi obejmami (ściągaczami). Wstępną kontrolę szczelności i ustawienie strumienia
wody należy wykonać jeszcze przed podstawieniem pod zestaw źródła ciepła, aby uniknąć
zalania wodą czy to mieszadła, czy łaźni olejowej lub płaszcza grzejnego.
Kończąc ogrzewanie, należy najpierw wyłączyć i usunąć źródło ciepła, a dopiero gdy
wrzenie ustanie, wyłączyć ewentualne mieszanie i zamknąć przepływ wody w chłodnicy.
Kolbę można zdemontować (rękawice ochronne!), jeśli nie obserwuje się już skraplania par
cieczy w chłodnicy. Nie wolno dopuścić do spływania skroplin z chłodnicy na pozostawiony
płaszcz grzejny lub na blat roboczy. Demontaż samej chłodnicy rozpoczyna się od zdjęcia z
kranu węża doprowadzającego wodę, a dopiero po opróżnieniu płaszcza wodnego, odłącza się
węże od chłodnicy. Zdejmując węże ze szklanych części aparatury, należy uchwycić dłonią
cały obwód węża znajdujący się na króćcu i delikatnymi ruchami zsunąć wąż, drugą ręką cały
czas trzymając pozostałą część aparatury jak najbliżej króćca. Jeżeli wąż nie chce się zsunąć,
najbezpieczniej jest odciąć go bezpośrednio przy wylocie chłodnicy lub kranu, a pozostałą
część naciąć wzdłuż i dopiero wówczas delikatnie zdjąć.
III.4. Chłodzenie
W przypadku wielu reakcji egzotermicznych wymagane jest intensywne chłodzenie naczynia,
w którym przeprowadzany jest taki proces. Naczynie zawierające mieszaninę reakcyjną
zanurza się wówczas w łaźni chłodzącej, tzn. krystalizatorze lub miseczce z tworzywa
sztucznego wypełnionej mieszaniną oziębiającą. Aby chłodzenie było jak najbardziej
efektywne, należy dopilnować, aby poziom cieczy w naczyniu znajdował się poniżej poziomu
mieszaniny oziębiającej. Często podczas ochładzania zawartość zlewki lub kolby powinna
być intensywnie mieszana. Wówczas łaźnię oziębiającą umieszcza się na (zimnej!) płytce
mieszadła magnetycznego.
Skład mieszaniny dobiera się w zależności od wymaganej dla danego procesu temperatury.
Starannie rozdrobniony i wymieszany z wodą lód służy do oziębiania do ok. 0 ºC. Lód
rozdrobniony i zmieszany z chlorkiem sodu w stosunku 3 : 1 obniży temperaturę do ok. –20
ºC, a monohydrat chlorku wapnia z lodem w stosunku 1,4 : 1 nawet do ok. –50 ºC.
Utrzymanie niskich temperatur przez dłuższy czas wymaga usuwania stopionego lodu i
dosypywania świeżego. Jeszcze niższą temperaturę osiągnąć można przez zmieszanie
rozdrobnionego stałego ditlenku węgla (tzw. „suchego lodu”) z acetonem (–78 ºC).
Przygotowanie takiej mieszaniny polega na powolnym dodawaniu acetonu do rozdrobnionego
„suchego lodu”; odwrotna kolejność nie jest wskazana ze względu na intensywne wydzielanie
gazowego ditlenku węgla. Temperatura wrzenia ciekłego azotu wynosi –196 ºC i jest on
rzadko stosowany do ochładzania mieszanin reakcyjnych, natomiast często używa się go do
wymrażania par np. podczas destylacji pod zmniejszonym ciśnieniem (podrozdz. V.3).
Mieszaniny o tak niskiej temperaturze muszą być umieszczane w naczyniach Dewara
(termosach).
Należy pamiętać, aby nigdy nie zanurzać naczynia z gorącym roztworem w mieszaninie
oziębiającej, gdyż może to doprowadzić do pęknięcia naczynia! Jeżeli zachodzi konieczność
pozostawienia roztworu w obniżonej temperaturze (w granicach od –15 do +5
o
C) na dłuższy
czas, należy przenieść go do szczelnie zamkniętego, stabilnego i podpisanego naczynia i
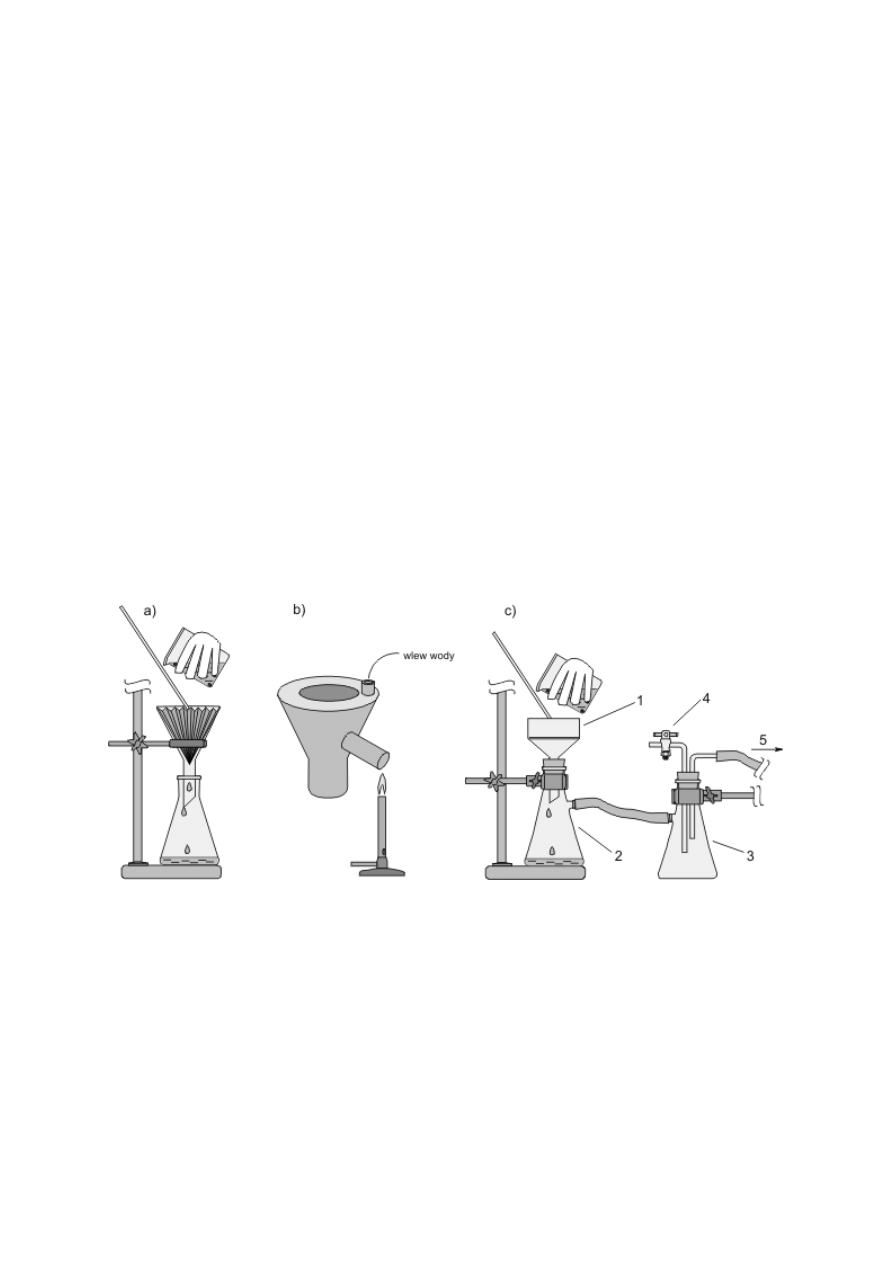
postawić w specjalnej lodówce laboratoryjnej. W żadnym przypadku nie wolno wstawiać do
lodówek mieszanin zawierających skrajnie łatwopalne rozpuszczalniki (etery)!
III.5. Sączenie
Jednym z podstawowych procesów w preparatyce chemicznej jest rozdzielanie mieszanin
ciało stałe-ciecz. Jeżeli osad ciała stałego jest ciężki i przylega do ścianek naczynia, można
ciecz znad niego ostrożnie zlać, czyli zdekantować.
Znacznie jednak częściej dokonuje się takiego rozdzielenia za pomocą sączenia. W procesie
tym albo oddziela się stałe zanieczyszczenia od cieczy, albo oddziela stały produkt od ciekłej
mieszaniny reakcyjnej lub ługu pokrystalizacyjnego. W pierwszym przypadku mieszaninę
przesącza się przez fałdowany sączek z bibuły umieszczony w szklanym lejku stożkowym
(rys. 14a). Zaletą tej metody jest szybkość sączenia, zwłaszcza jeśli zastosowana zostanie
bibuła miękka lub, co najwyżej, średnia (uniwersalna). Roztwór szybko przenika przez całą
powierzchnię bibuły, co ma kluczowe znaczenie, gdy trzeba szybko przesączyć roztwór
gorący lub sporządzony w lotnym rozpuszczalniku. Taka sytuacja ma miejsce, na przykład,
podczas krystalizacji (na sączku pozostają wówczas nierozpuszczalne w gorącym roztworze
zanieczyszczenia — podrozdz. V.1) oraz podczas procesu osuszania cieczy za pomocą stałego
środka suszącego (na sączku zostaje niepotrzebny już osad środka suszącego razem z
zaabsorbowaną wodą — podrozdz. III.6). Sączenie gorących roztworów jest ułatwione, jeżeli
lejek umieści się w specjalnym, uprzednio ogrzanym płaszczu do lejków (rys. 14b).
Rysunek 14. a) Zestaw do sączenia przez sączek fałdowany; b) płaszcz miedziany do
ogrzewania lejków; c) zestaw do odsączania osadów pod zmniejszonym ciśnieniem: 1 —
lejek Büchnera, 2 — kolbka ssawkowa, 3 — płuczka zabezpieczająca, 4 — zawór
odpowietrzający, 5 — wąż łączący zestaw z pompką wodną lub membranową
Istnieje kilka sposobów składania sączka fałdowanego (zwanego też sączkiem karbowanym).
Najprostsza w opisie, podana poniżej metoda zaczerpnięta została z Preparatyki organicznej
Vogla [11]. Krążek bibuły o promieniu porównywalnym z długością „boku” stożkowej części
lejka (czyli tworzącej stożka) składa się na pół, a otrzymane półkole ponownie na pół. Po
rozłożeniu otrzymuje się półkole z zaznaczoną zgięciem linią 2–4 (rys. 15a). Następnie składa
się krawędź 1–2 do linii 2–4 oraz krawędź 2–3 do linii 2–4. Po rozłożeniu powstają nowe
fałdy 2–5 i 2–6. Ponownie składa się w tym samym kierunku krawędź 1–2 do linii 2–5 oraz
2–6, otrzymując po rozłożeniu linie 2-7 i 2-8. Podobnie postępuje się z prawą krawędzią 2-3,
zaginając ją do linii 2–6 i 2–5, co doprowadza do powstania karbów 2–9 i 2–10. Otrzymuje
się wówczas półkole podzielone na osiem równych segmentów, zagiętych w tym samym
kierunku (rys. 15b). Ostatnią czynnością jest zrobienie fałdy w połowie każdego segmentu,
ale tworząc zagięcie w kierunku przeciwnym niż pierwsza seria fałd. Powinno to doprowadzić
do uzyskania harmonijki (wachlarza) podobnego do przedstawionego na rys. 15c. Rozłożenie
pofałdowanego tak półkola daje regularnie fałdowany sączek (rys. 15d). W podobny sposób
można wykonać sączek fałdowany z kwadratowego arkusza bibuły o boku dwukrotnie
dłuższym niż „bok” lejka. Przed rozłożeniem wachlarza wystarczy odciąć nożyczkami
wystające rogi bibuły. Fałd nie można dociskać zbyt mocno, szczególnie w pobliżu punktu 2,
gdyż grozi to przedziurawieniem sączka. Przy pewnej wprawie można spróbować składać
sączek w taki sposób, żeby fałdy nie zbiegały się dokładnie w jednym punkcie, lecz były
rozsunięte na odcinku ok. jednego centymetra. Tak przygotowany sączek fałdowany najlepiej
jest, przed umieszczeniem w lejku, odwrócić na drugą stronę („przenicować”). Zapobiega to
przedostawaniu się do przesączonego roztworu włókien bibuły oddzielonych od sączka
podczas jego karbowania. W przypadku przesączania niewielkich ilości cieczy (kilka cm
3
)
zamiast sączka fałdowanego można użyć luźno zwinięty kłębek waty wsunięty w nóżkę lejka.
Sposób ten nie nadaje się jednak do oddzielania bardzo drobnych zawiesin.
Rysunek 15. Kolejne etapy składania sączka fałdowanego
Lejek z sączkiem fałdowanym umieszcza się w kółku umocowanym do statywu, a pod nóżkę
lejka podstawia się odpowiedni odbieralnik (kolbkę Erlenmeyera lub zlewkę). Uproszczony
zestaw można skonstruować wstawiając lejek bezpośrednio w szyjkę kolby, jednak aby
w naczyniu, do którego przesączana jest ciecz, nie powstało zwalniające proces sączenia
nadciśnienie, należy pomiędzy lejkiem a ścianką naczynia pozostawić małą szczelinę.
Aby zminimalizować straty związane z wsiąkaniem roztworu w bibułę, przed sączeniem
bibułę sączka należy zwilżyć czystym rozpuszczalnikiem. Często spotykanym błędem jest
niewłaściwy sposób przelewania roztworów z kolby na sączek, który prowadzi do
zanieczyszczania zewnętrznych powierzchni kolb sączonym roztworem. Podczas przelewania
roztworu ostatnie jego krople należy zebrać bagietką, tak aby nie ściekały na zewnątrz
naczynia. Po zakończeniu sączenia nierozpuszczalną pozostałość wraz z sączkiem należy
przemyć dokładnie małymi porcjami czystego rozpuszczalnika.
Jeżeli produkt, który należy wyodrębnić jest ciałem stałym, wówczas odsącza się go pod
zmniejszonym ciśnieniem na lejku Büchnera (rys. 14c). Istnieje wówczas możliwość
dokładnego przemycia i odciśnięcia osadu oraz ogranicza się stopień zanieczyszczenia
produktu włóknami bibuły. Na płaskim dnie szklanego, porcelanowego lub plastikowego
lejka Büchnera umieszcza się krążek bibuły filtracyjnej. Krążek musi dokładnie przykryć
otworki w dnie lejka, ale nie może być od niego większy i zaginać się na ściankach.
Plastikowe (najczęściej polipropylenowe) lejki Büchnera nadają się doskonale do sączenia

roztworów wodnych i alkoholowych, są bezpieczne w pracy i łatwe do demontażu i umycia.
Nie są jednak zalecane przy pracy z innymi rozpuszczalnikami! Jeżeli zachodzi konieczność
odsączenia niewielkiej ilości osadu z dużej objętości roztworu, zamiast walcowatych lejków
Büchnera należy zastosować lejki stożkowe (tzw. lejki Hirscha). Aby wyeliminować
zanieczyszczenia osadu włóknami bibuły, zamiast klasycznego sitowego lejka Büchnera (z
płytką z otworami), stosuje się lejki z płytką ze szkła porowatego o małej średnicy porów, nie
wymagające stosowania bibuły.
Lejek Büchnera osadza się za pomocą gumowego kołnierza (uszczelki) lub gumowego korka
z otworem w wylocie grubościennej kolby ssawkowej. Rozmiar uszczelnienia musi być
dopasowany do szyi kolby i nóżki tak, aby lejek pod własnym ciężarem nie przechylał się ani
nie wypadał. Z drugiej strony kołnierz (korek) nie może wsuwać się całkowicie w szyjkę
kolby, gdyż po włączeniu pompki wodnej zostanie zassany gwałtownie do środka.
Alternatywą może być zastosowanie lejka Büchnera i kolby ssawkowej zaopatrzonej w szlify
tej samej wielkości. Kolbę należy przymocować do statywu za pomocą łapy i połączyć
krótkim kawałkiem grubościennego węża gumowego z pustą kolbą zabezpieczającą z
zaworkiem, a tę z kolei z pompką wodną (aspiratorem). Pompka wodna podłączona jest na
stałe do sieci wodociągowej, a zatem po odkręceniu silnego strumienia wody w układzie
zostaje obniżone ciśnienie. Nagłe zmiany szybkości przepływu wody mogą spowodować
błyskawiczne przerzucenie wody do zestawu sączącego. W takim przypadku woda zgromadzi
się w kolbie zabezpieczającej i nie przedostanie się do kolby ssawkowej.
Przy zastosowaniu pompki wodnej można obniżyć ciśnienie w zestawie nawet do ok. 10 hPa
(zależnie od konstrukcji aspiratora, szczelności układu, temperatury wody itp.). Jest ona
jednak urządzeniem mało wydajnym i dość kosztownym przy częstym użytkowaniu (zużycie
wody). Coraz częściej w celu obniżenia ciśnienia m. in. przy sączeniu na lejku Büchnera
stosuje się również inne typy pomp (np. pompy membranowe), które zapewniają podobne
obniżenie ciśnienia, jednak są bardziej wydajne i cechują się niższymi kosztami eksploatacji.
Przed rozpoczęciem sączenia należy zwilżyć bibułowy sączek kilkoma kroplami
rozpuszczalnika, a następnie odkręcić kran pompki wodnej. Sączoną mieszaninę wylewa się
porcjami na lejek Büchnera, przestrzegając zasady, aby bibuła przylegała ściśle do dna lejka i
była stale pokryta sączoną mieszaniną. Osad pozostały jeszcze w naczyniu przenosi się na
sączek, przerywając na chwilę sączenie i zawracając do niego nieco przesączu, zmywa się go
ze ścianek naczynia i sączy powtórnie. Czynność tę powtarza się tyle razy, aż cały
praktycznie osad znajdzie się na sączku (przynajmniej na tyle, na ile jest to możliwe). Przy
przerywaniu sączenia i wyłączaniu pompki wodnej należy odłączyć ją od kolby ssawkowej
przed zakręceniem kranu, w przeciwnym razie woda może dostać się do przesączu.
Działanie podciśnienia jest skuteczne tylko wówczas, gdy zawiesina, a następnie osad
pokrywa dokładnie całą powierzchnię dna lejka. Sączenie prowadzi się, dopóki przesącz
kapie do kolby ssawkowej. Proces ten, czasem długotrwały, można przyspieszyć,
przyciskając preparat na lejku Büchnera płaską powierzchnią uchwytu dużego szklanego
korka lub specjalnymi odciskaczami (szklane gwoździe z płaskimi łebkami, ubijaki do
osadów). Dokładne odciskanie osadu na lejku jest konieczne, jeśli produkt jest
bezpostaciowy lub drobnokrystaliczny i ma konsystencję gliny. Jeżeli zalecane jest przemycie
osadu niewielką porcją rozpuszczalnika, powinno się go wcześniej ochłodzić, aby
zminimalizować ewentualne rozpuszczenie się części osadu. Pozostawienie na lejku przez
kilka minut odmytego i dokładnie odciśniętego osadu przy włączonej pompce wodnej lub
membranowej znacznie przyspiesza wysuszenie osadu.

Osad zdejmuje się ostrożnie z sączka za pomocą szpatułki i w celu całkowitego wysuszenia
przenosi na podpisane szkiełko zegarkowe lub szalkę Petriego.
III.6. Suszenie
Konieczność osuszania substancji podyktowana jest faktem, że wiele substancji organicznych
to związki wrażliwe na wodę. Obecność wody przeszkadza też w prowadzeniu niektórych
reakcji organicznych, a że wilgoć jest wszechobecna, stosuje się rozliczne metody osuszania
substancji organicznych. Do usuwania wody używa się najczęściej substancji pochłaniających
wodę, takich jak:
bezwodne sole nieorganiczne, łatwo tworzące hydraty (Na
2
SO
4,
MgSO
4
, K
2
CO
3
, CaCl
2
),
niektóre tlenki metali (CaO, BaO) i niemetali (P
4
O
10
), które, wchodząc w reakcje z wodą,
dają odpowiednio wodorotlenki lub kwasy,
higroskopijne stężone kwasy nieorganiczne (H
2
SO
4
),
żel krzemionkowy,
sita molekularne (glinokrzemiany sodu, potasu i wapnia),
metaliczny sód (Na) i wodorki metali (CaH
2
lub NaH), które, reagując gwałtownie z wodą,
mogą być użyte do skutecznego usuwania nieznacznych ilości wody, np. z mało reaktywnych
rozpuszczalników organicznych (węglowodory, etery),
higroskopijne wodorotlenki metali (NaOH, KOH).
Czasami środki suszące są zaopatrzone w dodatek barwnego wskaźnika (np. soli kobaltu),
który zmienia swoją barwę w zależności od stopnia uwodnienia.
Dobór odpowiedniej metody zależy od stanu skupienia oraz właściwości chemicznych
suszonej substancji. Powinien on przede wszystkim wykluczać możliwość reagowania ze
sobą substancji suszonej i substancji suszącej. Dokładne dane dotyczące doboru środka
suszącego w zależności od klasy związku można znaleźć w podręcznikach preparatyki
organicznej i poradnikach fizykochemicznych.
Suszenie gazów
Suszenie gazów przeprowadza się, przepuszczając je przez kolumny szklane, wypełnione
stałym środkiem suszącym, często zmieszanym ze stałym wypełniaczem, co zapobiega
zbryleniu i zatykaniu się kolumny. Dobrze nadają się do tego zeolity, czyli sita molekularne.
Czasami stosuje się szeregowy układ kilku kolumn wypełnionych różnymi środkami
suszącymi, nierzadko z dodatkiem barwnego wskaźnika reagującego na zawartość wilgoci
(rys. 16).
Gazy obojętne lub kwaśne (np. HCl) można też osuszać, przepuszczając je przez płuczkę ze
stężonym kwasem siarkowym(VI), zabezpieczoną z obydwu stron ustawionymi szeregowo
pustymi płuczkami.
Wielokrotnie w trakcie suszenia substancji lub podczas pracy z substancjami, których
trwałość zagrożona jest w przypadku zetknięcia się z wilgocią z powietrza, istnieje
konieczność zabezpieczenia aparatury przed dopływem pary wodnej z zewnątrz. Jako
zamknięcie aparatury z zachowaniem możliwości wyrównywania ciśnienia stosuje się
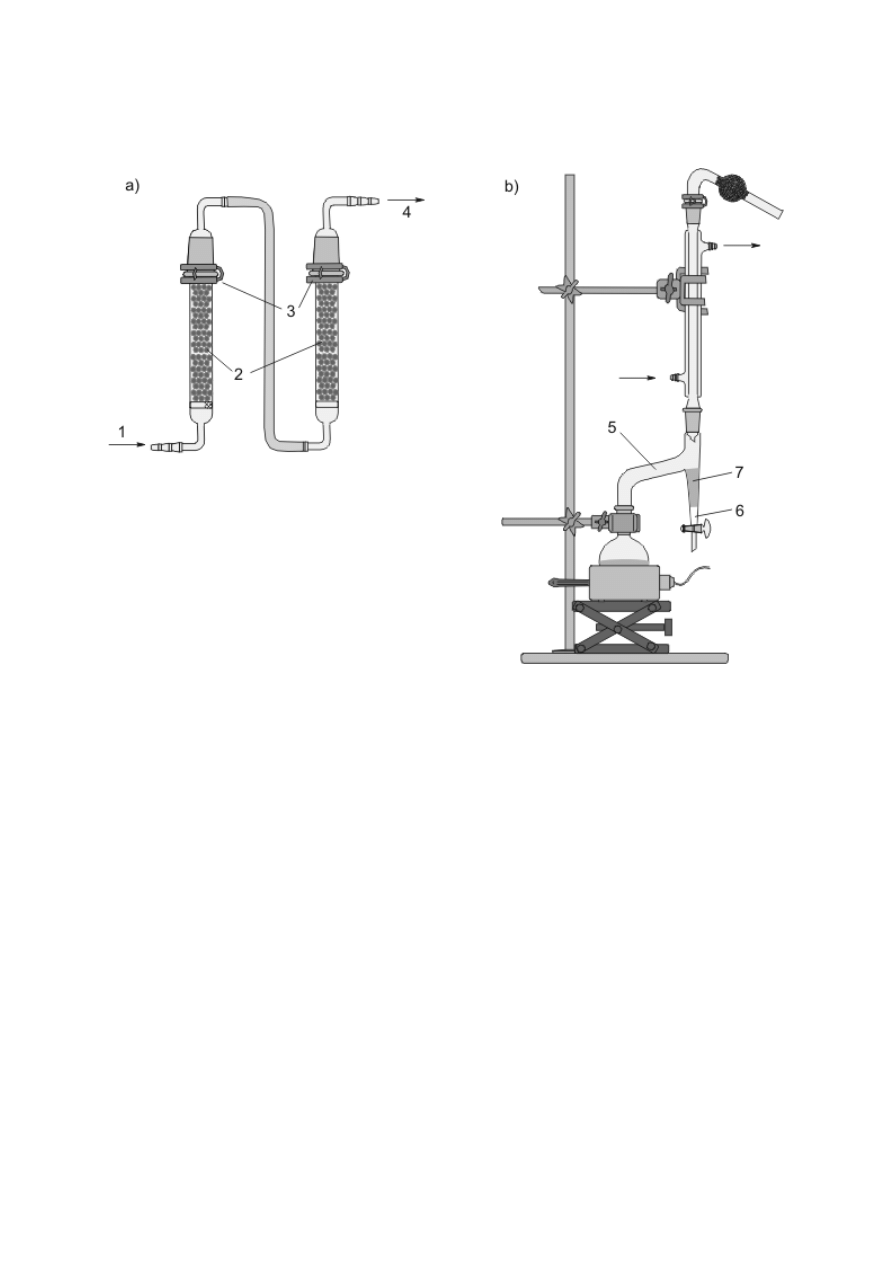
wówczas nasadki wypełnione środkiem higroskopijnym (tzw. „rurki z chlorkiem wapnia”,
podrozdz. II.1, rys. 7i, j).
Rysunek 16. a) Połączone szeregowo płuczki do suszenia gazów: 1 — wlot gazu (np. z butli),
2 — higroskopijny wypełniacz, 3 — połączenie szlifowe utrzymywane spinkami do szlifów,
4 — wylot gazu (np. do aparatury); b) zastosowanie nasadki azeotropowej: 5 — nasadka
Deana-Starka, 6 — wydzielająca się woda, 7 — rozpuszczalnik o gęstości mniejszej od
gęstości wody
Suszenie cieczy
Ciecze organiczne, a więc czyste rozpuszczalniki, odczynniki, produkty reakcji lub ich
roztwory (np. powstające w wyniku ekstrakcji roztworów wodnych rozpuszczalnikiem
organicznym, podrozdz. V.4), mogą zawierać rozpuszczoną w nich pewną ilość wody.
Usunięcie wody z cieczy organicznej, czyli suszenie cieczy, wykonuje się, mieszając suszoną
ciecz z odpowiednio dobranym stałym środkiem suszącym w szczelnie zamkniętym naczyniu
szklanym o możliwie dużej powierzchni dna. Nie każda higroskopijna substancja nadaje się
do osuszania roztworów — środek suszący musi spełniać kilka podstawowych warunków:
nie może wchodzić w reakcję z osuszaną substancją ani katalizować jej przemian,
powinien być praktycznie nierozpuszczalny w osuszanej cieczy,
proces przyłączania wody musi być względnie szybki i efektywny (zachodzić szybko
wcałej objętości środka osuszającego, w miarę możliwości w sposób nieodwracalny).

Warunki te spełniają niektóre higroskopijne, bezwodne, obojętne sole nieorganiczne, zdolne
do tworzenia hydratów, trwałych w temperaturze pokojowej, np. bezwodny siarczan(VI) sodu
(Na
2
SO
4
) czy też droższy, lecz szybciej działający i bardziej efektywny, siarczan(VI)
magnezu (MgSO
4
). Tani, o dużej zdolności pochłaniania wody, chlorek wapnia, ma zdolność
do wiązania się ze związkami organicznymi, które zawierają grupy hydroksylowe i
karbonylowe, można go zatem stosować tylko do osuszania węglowodorów, eterów i
niektórych ich pochodnych halogenowych. Do suszenia amin lub ich roztworów w
rozpuszczalnikach węglowodorowych lub eterowych można również stosować wodorotlenek
sodu lub potasu w postaci granulek lub pastylek względnie węglan potasu.
W celu usunięcia resztek wilgoci ciekłą substancję umieszcza się w suchej kolbie stożkowej
ze szlifem i dodaje do niej małymi porcjami środek suszący. Po każdym kolejnym dodaniu
środka suszącego kolbę zamyka się korkiem, wstrząsa energicznie zawiesinę i bacznie
obserwuje skutki tej operacji. Jeśli środek suszący tworzy z wodą zawartą w rozpuszczalniku
roztwór, należy go oddzielić (najlepiej w rozdzielaczu) i kontynuować osuszanie następną
porcją środka suszącego. Jeżeli środek suszący ulega zbryleniu, należy dodać nową jego
porcję i ponownie wytrząsnąć. Energiczne wstrząsanie jest szczególnie ważne w przypadku
rozpuszczalników o gęstości większej niż gęstość wody, gdyż bez niego środek suszący
pozostanie na dnie naczynia, a woda wypłynie na powierzchnię rozpuszczalnika. Dopiero jeśli
kolejna porcja dodawanego środka suszącego pozostaje sypka, należy uznać jego ilość za
wystarczającą. Nadmiar użytego środka suszącego nie jest wskazany ze względu na straty
substancji osuszanej, wynikające z adsorpcji substancji na powierzchni kryształów. Szybkość
pochłaniania wilgoci zależy od rodzaju substancji higroskopijnej. W przypadku użycia
siarczanu magnezu roztwór należy pozostawić w szczelnie zamkniętej kolbce na
przynajmniej 15–20 minut, okresowo wstrząsając jej zawartość.
Po zakończeniu suszenia ciecz należy oddzielić od środka suszącego. Można tu wykorzystać
dekantację lub lepiej sączenie przez sączek fałdowany umieszczony w lejku szklanym
(podrozdz. III.5). Środek suszący pozostały w kolbce i znajdujący się na sączku należy
przemyć dokładnie niewielką ilością czystego i najlepiej osuszonego rozpuszczalnika.
Często jako środki suszące do różnych rozpuszczalników (np. węglowodorów, eterów,
estrów, ketonów, DMSO i DMF) stosuje się sita molekularne, czyli zeolity typu 3A, 4A lub
5A (symbole te odpowiadają jednolitej średnicy porów w zeolicie wyrażonej w angstremach
[Å]), które mogą absorbować cząsteczki o różnych wielkościach. Mają one tę cenną
właściwość, że działają także w podwyższonych temperaturach oraz że można je
regenerować, czyli usunąć z nich pochłoniętą wodę w wyniku ogrzewania w suchej
atmosferze do temp. 150–300
o
C (zależnie od rodzaju sit).
Specjalnym przypadkiem suszenia cieczy jest suszenie metalicznym sodem lub wodorkami
wapnia lub sodu. Przy stosowaniu tych środków należy zachować szczególną ostrożność,
gdyż reagują one burzliwie nawet z niewielką ilością wody, a wodór wydzielający się w
trakcie tej egzotermicznej reakcji może ulec zapłonowi. Suszenie sodem wykonuje się,
stosując ten metal w postaci drutu otrzymanego w specjalnej prasie lub w postaci
rozdrobnionej. Można to uzyskać, stapiając sód pod warstwą suchego toluenu lub ksylenu,
doprowadzając te rozpuszczalniki do wrzenia pod chłodnicą zwrotną, zabezpieczoną przed
wilgocią. Po stopieniu sodu chłodnicę zastępuje się korkiem i tak zamkniętą kolbę mocno się
wstrząsa aż do ochłodzenia rozpuszczalnika i zastygnięcia sodu w postaci drobnych kuleczek.
Po zastygnięciu sodu kolbę należy otworzyć w celu wyrównania ciśnienia, a korek zastąpić
rurką z chlorkiem wapnia. Suszenie rozpuszczalników sodem lub wodorkami metali można

zrealizować na zimno, pozostawiając zawiesinę w kolbie kulistej lub kolbie stożkowej
zamkniętej rurką z chlorkiem wapnia, lub na gorąco, ogrzewając zawiesinę do wrzenia
rozpuszczalnika w kolbie okrągłodennej z chłodnicą zwrotną, na wylocie której umocowana
jest rurka z chlorkiem wapnia. Suchy rozpuszczalnik destyluje się następnie w zestawie z
chłodnicą również zabezpieczoną rurką z chlorkiem wapnia (podrozdz. V.3). Przy suszeniu
sodem lub wodorkami metali dobrze jest wstępnie osuszyć substancję, np. bezwodną solą
nieorganiczną. Pamiętać też trzeba, że sód i wodorki metali nadają się przede wszystkim do
suszenia węglowodorów i eterów, absolutnie nie można ich używać do suszenia kwasów,
alkoholi, związków karbonylowych i fluorowcowanych. Problem stanowią też odpady po
suszeniu, które mogą zawierać resztki właściwych substancji suszących, zwłaszcza sodu,
ukryte pod warstwą tlenków i wodorotlenków. Resztki sodu użytego do suszenia należy
zbierać w zamkniętym słoiku pod warstwą suchego rozpuszczalnika. Nigdy nie wyrzuca się
resztek sodu do pojemnika na odpady stałe, ani tym bardziej do zlewu. Grozi to
poważnym pożarem, a nawet eksplozją. Wodorki pozostałe po suszeniu, jak i niewielkie
ilości sodu, należy rozłożyć alkoholem (np. izopropylowym), przeprowadzając je w
bezpieczniejsze alkoholany.
Suszenie ciał stałych
Suszenie ciał stałych otrzymywanych na ogół w wyniku reakcji lub krystalizacji polega na
usunięciu rozpuszczalnika. Często, zwłaszcza w przypadku lotnych rozpuszczalników,
następuje to już w trakcie sączenia substancji przez lejek Büchnera. Proces suszenia można
zakończyć, umieszczając substancję znajdującą się na szalce Petriego lub szkiełku
zegarkowym pod promiennikiem podczerwieni lub też w ogrzewanej suszarce szafkowej. Jest
to znacznie bardziej skuteczne niż suszenie na powietrzu. Nie wolno suszyć związków w
papierowych pudełkach! W przypadku analitycznie czystych substancji lub gdy należy usunąć
trudno lotny rozpuszczalnik, a także w przypadku hydratów (tworzą je także związki
organiczne!), najlepiej jest posłużyć się ogrzewaną suszarką próżniową. Istnieje wówczas
możliwość precyzyjnego ustawienia zarówno temperatury, jak i ciśnienia oraz czasu suszenia.
We wszystkich procesach suszenia termicznego (także pod lampą) należy pamiętać
o skorelowaniu parametrów suszenia z temperaturą topnienia związku. Temperatura, w jakiej
suszymy związek, powinna być zawsze znacznie niższa od jego temperatury topnienia. Na
przykład związki o temperaturze topnienia ok. 100
o
C lub niższej nie powinny być
umieszczane pod promiennikiem cieplnym, ponieważ istnieje ryzyko stopienia się, a nawet
rozkładu substancji. Bardzo ostrożnie należy suszyć również związki wykazujące znaczną
prężność par w temperaturze pokojowej lub podwyższonej (np. lotne z parą wodną), gdyż
podczas łagodnego ogrzewania, a nawet w temperaturze pokojowej mogą sublimować.
Związek przeznaczony do suszenia w podwyższonej temperaturze nie może zawierać
znaczących ilości rozpuszczalnika, gdyż po ogrzaniu mógłby się po prostu zacząć w nim
rozpuszczać. Pod lampą nie można suszyć związków, które są wrażliwe na światło i w tych
warunkach ulegają reakcjom fotochemicznym, co objawia się przebarwieniem lub
ciemnieniem substancji.
Czasem, ze względu na brak odporności termicznej związku, wskazane jest suszenie
w temperaturze pokojowej. Można to zrealizować, susząc związek na powietrzu lub lepiej
w eksykatorze. Jest to grubościenne naczynie szklane z doszlifowaną pokrywą, w którym na
dnie znajduje się środek suszący (najczęściej chlorek wapnia, wodorotlenek potasu lub żel

krzemionkowy), a nad nim, na porcelanowej półeczce z otworami, umieszcza się suszoną
substancję na szkiełku zegarkowym lub szalce Petriego.
Eksykator można zastąpić szczelnie zamykanym słoikiem szklanym typu twist, na którego
dno wprowadzono środek suszący. Jeśli eksykator jest urządzeniem próżniowym, czyli ma
rurkę odprowadzającą powietrze i zamkniętą kranem, to po wypompowaniu powietrza z
eksykatora za pomocą pompy próżniowej można prowadzić proces suszenia pod
zmniejszonym ciśnieniem. Pamiętać należy, że ze względu na możliwość implozji
eksykator próżniowy powinien być w trakcie pracy zabezpieczony z zewnątrz tkaniną.
Istnieją także inne fizyczne metody osuszania substancji organicznych. Zaliczamy do nich
azeotropowe usuwanie wody oraz sublimację zamrożonej wody.
Woda tworzy z niektórymi rozpuszczalnikami (np. węglowodorami, tetrachlorometanem,
alkoholami) azeotropy, które wrą w postaci mieszaniny o stałym składzie procentowym
w temperaturze niższej niż czyste rozpuszczalniki (podrozdz. V.3). Oznacza to, że jeśli
rozpuszczalnik zawiera niewiele wody, to przy destylacji frakcję najniżej lotną stanowi
mieszanina azeotropowa tego rozpuszczalnika z wodą i frakcja ta destyluje jako pierwsza aż
do wyczerpania się wody. Niekiedy stosuje się dodatek trzeciego składnika, który wspomaga
usuwanie wody, gdyż azeotrop trójskładnikowy wrze w najniższej temperaturze. Taka metoda
z użyciem benzenu jako trzeciego składnika była stosowana do odwadniania etanolu. Kolejne
frakcje stanowiły: azeotrop trójskładnikowy, który wrzał aż do wyczerpania się wody,
następnie azeotrop benzen/etanol, który wrzał aż do wyczerpania się benzenu i wreszcie
bezwodny etanol. Benzen o potwierdzonym działaniu rakotwórczym został wycofany z
użycia, można go jednak zastąpić cykloheksanem o zbliżonej temperaturze wrzenia, który
także z wodą tworzy mieszaninę azeotropową.
Jeżeli rozpuszczalnik tworzy z wodą mieszaninę azeotropową, nie miesza się z nią i ma
gęstość mniejszą niż 1 g/cm
3
(np. toluen), można stosunkowo łatwo usunąć z niego resztki
wilgoci lub wykorzystać go do osuszania innych substancji, stosując nasadkę Deana-Starka
(tzw. nasadkę azeotropową). Również w przypadku gdy w trakcie przeprowadzanej syntezy
wydziela się woda, która zgodnie z regułą przekory powinna być usuwana z układu, stosuje
się zestaw przedstawiony na rys. 16b. W aparaturze o takiej konstrukcji w chłodnicy skrapla
się jako pierwsza frakcja najniżej wrzący azeotrop rozpuszczalnika organicznego i wody.
Skropliny złożone z dwóch faz: wody i rozpuszczalnika organicznego wypełniają nasadkę, w
której woda gromadzi się na dnie, a nadmiar rozpuszczalnika wraca do kolby. Po dłuższym
czasie większość wody zostaje usunięta z rozpuszczalnika lub roztworu znajdującego się w
kolbie. Pozwala to więc nie tylko w sposób ciągły odprowadzać wodę z mieszaniny, ale także
kontrolować jej wydzieloną objętość, co jest szczególnie istotne, na przykład, podczas
kontrolowania przebiegu reakcji.
Jedną z metod suszenia substancji bardzo wrażliwych na działanie podwyższonych
temperatur jest sublimacja wymrożonej wody czyli liofilizacja. Substancję osuszaną oziębia
się, stosując na przykład zestalony ditlenek węgla, a następnie, stosując technikę sublimacji
próżniowej (podrozdz. V.2), odparowuje się wytworzony w trakcie oziębiania lód.
III.7. Zasady projektowania i montażu
zestawów do prowadzenia reakcji

Większość przepisów, zamieszczanych w renomowanych preparatykach organicznych oraz
np. w instrukcjach do ćwiczeń studenckich, zawiera dość szczegółowy opis proponowanych
zestawów do prowadzenia reakcji chemicznych. Niestety, nie zawsze sugerowana tam
aparatura jest dostępna w laboratorium. Ponadto opisy syntez w innych źródłach, np. w
oryginalnych czasopismach chemicznych, ograniczają się do podania jedynie podstawowych
parametrów reakcji — ilości reagentów, czasu trwania reakcji, temperatury itp. Aby zatem
powtórzyć przedstawiony w nich proces, trzeba samodzielnie odtworzyć szczegóły techniczne
syntezy. Umiejętność projektowania prostych zestawów do prowadzenia syntez organicznych
należy więc do kanonu podstawowych umiejętności każdego chemika.
Na ostateczny kształt zestawu odpowiedniego do przeprowadzenia określonej syntezy
organicznej ma wpływ szereg czynników, które trzeba przeanalizować, udzielając odpowiedzi
na następujące pytania:
Czy obecność wilgoci bądź tlenu może spowodować rozkład substratów lub produktów
względnie w innych sposób zakłócić przebieg reakcji?
Czy stosowane reagenty, rozpuszczalniki i powstające produkty są lotne w warunkach
reakcji? Czy ich pary muszą być zawracane do naczynia, czy też odprowadzane na zewnątrz i
pochłaniane?
W jakiej temperaturze ma być prowadzona reakcja? Czy będzie wymagane ogrzewanie do
temperatury wrzenia rozpuszczalnika, czy też wskazane jest użycie łaźni grzejnych lub
chłodzących o określonej temperaturze?
Czy istnieje konieczność monitorowania procesu (pomiaru temperatury procesów
egzotermicznych, pobierania próbek do testów analitycznych itp.)?
Czy wszystkie odczynniki dodaje się na początku procesu, czy należy dozować (wkraplać,
dosypywać itp.) któryś z substratów w trakcie reakcji?
Czy konieczne jest mieszanie? Czy mieszadło magnetyczne będzie efektywne?
Jakie ilości reagentów będą użyte? Jaka musi być w konsekwencji pojemność kolb,
wkraplaczy, rozmiar mieszalników itp.?
Czy reagenty lub warunki procesu (temperatura) nie uszkodzą proponowanej aparatury (np.
metalowych końcówek termometrów, mieszalników w powłoce polietylenowej, spinek do
szlifów wykonanych z tworzyw sztucznych)?
Jak długo trwa reakcja? Czy można przerwać proces, a jeśli tak, to w którym momencie i
jak należy zabezpieczyć zestaw?
Należy pamiętać, że zestaw musi być dostosowany do przeprowadzenia w nim wszystkich
kolejnych etapów syntezy, do etapu rozkładu mieszaniny reakcyjnej włącznie! Odpowiedź na
pierwsze dwa z powyższych pytań pozwoli określić, czy reakcję można wykonywać w
reaktorach otwartych, czy trzeba użyć układu półotwartego czy też izolowanego od otoczenia.
Zestawy otwarte
Jeżeli syntezę prowadzi się w temperaturze zbliżonej do pokojowej lub niższej, w jej trakcie
nie stosuje się lotnych bądź wrażliwych na obecność tlenu lub wilgoci reagentów
i rozpuszczalników, a w reakcji nie wydzielają się szkodliwe gazy, które należy
neutralizować, to reakcję można prowadzić w otwartym naczyniu (zlewce, kolbie
Erlenmeyera lub kolbie okrągłodennej). Jest to wygodny, lecz niestety rzadko możliwy do
wdrożenia w praktyce, sposób wykonywania syntezy. Mieszanie, dozowanie reagentów,
monitorowanie reakcji nie wymaga montowania złożonych zestawów.

W praktyce zawartość każdego naczynia do prowadzenia reakcji powinna być mieszana, by
zapewnić jednakowe warunki w całej objętości. Pewnym odstępstwem od tej zasady mogą
być syntezy prowadzone przez długi czas w temperaturze pokojowej, w układach
jednofazowych i nie wymagające dozowania reagentów. Najwygodniejszym sposobem
mieszania jest umieszczenie naczynia w pojemniku (krystalizatorze, misce) stojącym na
płytce mieszadła magnetycznego, przy czym można używać mieszadeł bez płytki grzejnej,
jeśli reakcja nie wymaga ogrzewania. Pojemnik ten można wypełnić mieszaniną oziębiającą,
jeżeli reakcja ma być prowadzona w niskiej temperaturze, lub pozostawić pusty, jeśli reakcja
zachodzi w temperaturze pokojowej. Będzie on zabezpieczał mieszadło przed zalaniem w
razie przewrócenia bądź pęknięcia naczynia. Przed umieszczeniem na mieszadle w zlewce lub
kolbce Erlenmeyera umieszcza się cylindryczny element mieszający. Wielkość mieszalnika,
szybkość obrotów i położenie naczynia na płytce (możliwie na środku płytki) należy dobrać
tak, aby cała zawartość naczynia była wprawiana w jednostajny ruch. Jeżeli mieszalnik
zacznie „rzucać się" po naczyniu, wyłącza się mieszadło i ponownie włącza, ustawiając nieco
wolniejsze obroty i próbując znaleźć optymalne położenie naczynia na płytce.
Pewnym problemem, zwłaszcza w przypadku zlewek i kolbek o małej pojemności, jest
zapewnienie stabilności naczynia w łaźni wodnej lub lodowej. Jeżeli nie ma możliwości
uchwycenia, np. małej zlewki łapą, poleca się zastosować kolbkę Erlenmeyera i założyć na jej
szyję obciążający gumowy pierścień.
Jeżeli reakcja wymaga monitorowania temperatury, to pomiar przeprowadza się
termometrami szklanymi bez szlifu lub termometrami elektronicznymi. Te ostatnie są w tym
przypadku wygodniejsze i bezpieczniejsze w użyciu, jednak nie można ich używać, gdy
zachodzi ryzyko uszkodzenia metalowej końcówki (stężone kwasy, zasady, brom). Każdy
termometr musi być przymocowany łapą do statywu. Niedopuszczalne jest swobodne
wstawianie termometrów, zwłaszcza szklanych, do naczyń, w których prowadzona jest
reakcja chemiczna. Zestaw należy zmontować w taki sposób, aby mieszalnik magnetyczny
nie uderzał o szklaną końcówkę termometru! Zanim zamontuje się termometr, należy
wcześniej znaleźć optymalne ustawienia dla równomiernego mieszania.
Reagenty stałe można bez problemów dosypywać za pomocą szpatułki w trakcie całej
syntezy, pomagając sobie w przypadku kolbek Erlenmeyera plastikowym lejkiem do osadów.
Odczynniki ciekłe dodaje się bądź za pomocą prostych wkraplaczy bez szlifu, bądź pipetką
Pasteura. Ten drugi sposób jest polecany przy dozowaniu małych objętości nielotnych
odczynników. Przed dodaniem kolejnej porcji odczynnika należy się upewnić, że poprzednia
już przereagowała. W naczyniach otwartych nie prowadzi się syntez wymagających
wprowadzania reagentów gazowych.
Przykładowe zestawy, skonstruowane zgodnie z podanymi wcześniej wytycznymi,
przedstawiono na rys. 17a i b. Oba służą do przeprowadzania syntez w obniżonej,
kontrolowanej w ciągły sposób temperaturze, wymagających stopniowego dodawania
ciekłych reagentów i mieszania mieszadłem magnetycznym. W pierwszym wariancie (rys.
17a) reakcję wykonuje się w zlewce, dodając reagenty z wkraplacza i posługując się
termometrem elektronicznym — taki zestaw wykorzystuje się na przykład w niektórych
reakcjach diazowania. Zimny, rozcieńczony kwas chlorowodorowy stosowany w tej syntezie
nie powoduje uszkodzenia metalowej końcówki termometru. W drugim przypadku (rys. 17b)
synteza przeprowadzana jest w kolbie Erlenmeyera obciążonej gumowym pierścieniem,
zamiast wkraplacza użyto pipety Pasteura, a temperaturę mierzy się szklanym termometrem.
Wariant taki polecany jest m. in. w niektórych reakcjach nitrowania.
Rysunek 17. Przykładowe zestawy do prowadzenia syntez w naczyniach otwartych: a), b)
różne możliwości prowadzenia reakcji w obniżonej temperaturze, przy ciągłym mieszaniu,
stopniowym dodawaniu ciekłych reagentów i stałej kontroli temperatury; 1 — łaźnia lodowa;
c) zastosowanie mieszadła mechanicznego do reakcji prowadzonej w temperaturze
pokojowej, tempo wkraplania reagenta uzależnione jest od zmian temperatury, 2 —
krystalizator zabezpieczający
Gdy mieszanina reakcyjna ma dużą lepkość lub powstaje znaczna ilość osadu, względnie
stosuje się substancje ferromagnetyczne lub wymagane jest bardzo intensywne mieszanie,
należy zastosować mieszadło mechaniczne. Aby ograniczyć możliwość rozchlapywania
zawartości naczynia, zaleca się stosowanie kolb okrągłodennych, przy czym pręt mieszadła
umieszcza się zawsze w środkowej szyi. Jeśli reakcja może być prowadzona w naczyniu
otwartym, nie ma konieczności stosowania uszczelnień do mieszadła, a dodatkowe elementy
wprowadzane do kolby nie muszą być osadzone w szlifach. Przykład takiego zestawu
przedstawiono na rys. 17c. Umożliwia on przeprowadzenie reakcji w temperaturze pokojowej
(krystalizator stanowi jedynie zabezpieczenie na wypadek pęknięcia kolby), jednak przy
wewnętrznej kontroli temperatury, która może wzrastać przy dodawaniu reagenta.
W niektórych przypadkach optymalny kontakt reagentów można zapewnić przez intensywne
wstrząsanie mieszaniny substratów, np. w przypadku reakcji w układzie dwufazowym,
zwłaszcza gdy produkt wydziela się w postaci osadu. Wytrząsanie prowadzi się na
mechanicznych wytrząsarkach, w naczyniach zamkniętych, technika ta nie nadaje się zatem
do procesów egzotermicznych lub wymagających dozowania reagentów. Jeśli reakcja jest
szybka, a substraty względnie bezpieczne, można ostatecznie ich mieszaninę wytrząsać
ręcznie, np. w szczelnie zamkniętej korkiem kolbie lub butelce, jednak ze względów
bezpieczeństwa technika ta nie jest polecana.

Zestawy półotwarte z ograniczeniem emisji
W przypadku większości syntez wymagane jest częściowe odizolowanie mieszaniny
reakcyjnej od otoczenia. Jednym z głównych powodów jest konieczność ograniczenia emisji
szkodliwych substancji z naczynia, w którym przeprowadza się reakcję. Dzieje się tak, jeżeli
stosowany rozpuszczalnik lub przynajmniej jeden z reagentów jest w temperaturze
pokojowej substancją lotną i niebezpieczną; dotyczy to praktycznie wszystkich popularnych
rozpuszczalników organicznych;
reakcja przebiega w podwyższonej temperaturze, co prowadzi do powstawania par
rozpuszczalników bądź reagentów, które muszą być zawracane do naczynia; czasami
zachodzi też konieczność odprowadzania i skraplania par poza naczyniem (np.
oddestylowywanie produktu);
w reakcji powstają gazy, które muszą być pochłaniane (np. chlorowodór, tlenki azotu);
jednym z substratów jest gaz (jego nadmiar też musi być pochłaniany)
Oczywiście, nie wolno wykonywać tych reakcji w hermetycznie zamkniętych układach, a w
szczególności ich ogrzewać — wzrost ciśnienia wewnątrz układu może doprowadzić do
eksplozji! Zasada ta nie dotyczy profesjonalnych, wysokociśnieniowych autoklawów
reakcyjnych. Naczynie, w którym zachodzi jakikolwiek proces z udziałem lotnej cieczy,
zwłaszcza w podwyższonej temperaturze, musi być zaopatrzone w chłodnicę zwrotną. Jeżeli
w reakcji powstają lub są używane niebezpieczne gazy, układ musi być ponadto podłączony
do zestawu do ich pochłaniania. Wszystkie pozostałe otwory w naczyniu powinny być
zamknięte lub szczelnie połączone z innymi, potrzebnymi częściami aparatury (wkraplaczem,
termometrem itp.). Jako naczynia stosuje się zatem kolby okrągłodenne jedno-, dwu- lub
trójszyjne, a czasem nawet reaktory o czterech lub pięciu otworach.
Jeżeli cała synteza sprowadza się do zmieszania substratów w kolbie z odpowiednim
rozpuszczalnikiem, a następnie do ogrzewania mieszaniny w temperaturze wrzenia tego
rozpuszczalnika, można wykorzystać zestaw przedstawiony na rys. 13a. Dopóki nie wydziela
się duża ilość osadu bądź drugiej fazy ciekłej, nie ma konieczności dodatkowego mieszania.
Należy jednak pamiętać o dodaniu kamyczków wrzennych! Jeżeli reakcja wymaga
ogrzewania w temperaturze niższej niż temperatura wrzenia rozpuszczalnika, zaleca się użyć
termostatowanej łaźni olejowej z jednoczesnym mieszaniem zawartości kolby. Stosuje się
mieszalnik właściwy dla kolb okrągłodennych, czyli owalny („jajo”), a kamyczki wrzenne
stają się wówczas zbędne (rys. 13b). Jeżeli reakcja ma być prowadzona w temperaturze nieco
niższej od 100
o
C, to medium grzewczym może być wrząca łaźnia wodna. Ze względu na
brak możliwości mieszania jest to sposób efektywny tylko w przypadku jednorodnych
mieszanin reakcyjnych.
Jeżeli reakcja wymaga kontroli temperatury, stopniowego dozowania reagentów lub na
przykład pobierania próbek w trakcie jej trwania, konieczne jest użycie naczynia dwu- lub
trójszyjnego. Szklany termometr na szlifie mocuje się w bocznej szyi, tak aby jego
zbiorniczek był zanurzony w mieszaninie, lecz nie dotykał żadnych elementów aparatury,
można dodatkowo przypiąć go spinką do szlifów. Reagenty ciekłe dodaje się z wkraplacza ze
szlifem, umiejscowionego w następnej szyi. Małych wkraplaczy nie trzeba przytrzymywać
łapą, jeśli ich mocowanie jest zabezpieczone spinką. Alternatywnie, ciekłe reagenty można
dodawać z wkraplacza bez szlifu z długą nóżką, który umocowany jest w łapie u górnego
wylotu krótkiej chłodnicy zwrotnej. Jest to sposób dopuszczalny przy reakcji w małej skali,
gdy brak jest na przykład dwuszyjnych kolbek o pojemności 50 cm
3
. Odczynniki stałe
powinno się w miarę możliwości dodawać do kolby na samym początku reakcji lub też
rozpuszczać je w odpowiednim rozpuszczalniku i w takiej postaci wprowadzać stopniowo do
kolby. Gdy jest to niemożliwe, stały substrat można dodawać przez lejek z szeroką nóżką
umieszczony w bocznej szyjce kolby, a po dodaniu każdej porcji lejek wyjąć, a szyjkę
zamknąć dopasowanym korkiem. Można również połączyć naczynie zawierające stały
substrat (np. małą erlenmeyerkę lub probówkę) z boczną szyjką kolby za pomocą gładkiego
kołnierza gumowego i przechylając pojemnik wsypywać odczynnik we właściwym tempie.
Obie te techniki zawodzą jednak, jeżeli mieszanina reakcyjna musi być utrzymywana cały
czas w stanie wrzenia.
Niekiedy powstający w wyniku syntezy lotny produkt główny lub produkty uboczne muszą
zostać oddestylowane z naczynia, w którym prowadzona była reakcja. Wówczas zamiast
chłodnicy zwrotnej w centralnej szyjce kolby montuje się chłodnicę destylacyjną. Tego
rodzaju technikę stosuje się czasami, na przykład, w reakcjach estryfikacji. Rysunek 18
ilustruje omówione techniki przeprowadzania reakcji z ograniczeniem emisji par reagentów.
Rysunek 18. Przykładowe zestawy do prowadzenia syntez z ograniczeniem emisji par,
umożliwiające: a) mieszanie w stałej, podwyższonej temperaturze i jednoczesne
wprowadzanie ciekłego substratu; b) chłodzenie, okresowe wprowadzanie stałego substratu i
kontrolę temperatury; c) dodawanie ciekłego substratu z jednoczesnym oddestylowywaniem
produktu
Gdy w wyniku syntezy powstaje niebezpieczny gaz (chlorowodór, ditlenek siarki, tlenki azotu
itp.), należy odprowadzać go z aparatury do odpowiedniego absorbera, w którym następuje
jego chemiczna neutralizacja. W górnej części chłodnicy (lub w szyjce kolby, jeśli reakcja
zachodzi w niskiej temperaturze) mocuje się nasadkę zakończoną oliwką, na którą nakłada się
elastyczny wąż (rys. 19a). Drugi koniec węża podłącza się do absorbera zawierającego
roztwór neutralizujący &mdash w przypadku wymienionych gazów może to być roztwór
wodorotlenku sodu. Najprostszy pochłaniacz składa się z lejka stożkowego, umieszczonego
szerszym końcem tuż nad powierzchnią cieczy w zlewce. Lejek powinien być umocowany w
łapie i przechylony pod kątem kilku stopni, aby zmniejszyć prawdopodobieństwo zassania
roztworu ze zlewki do naczynia, w którym prowadzona jest reakcja.
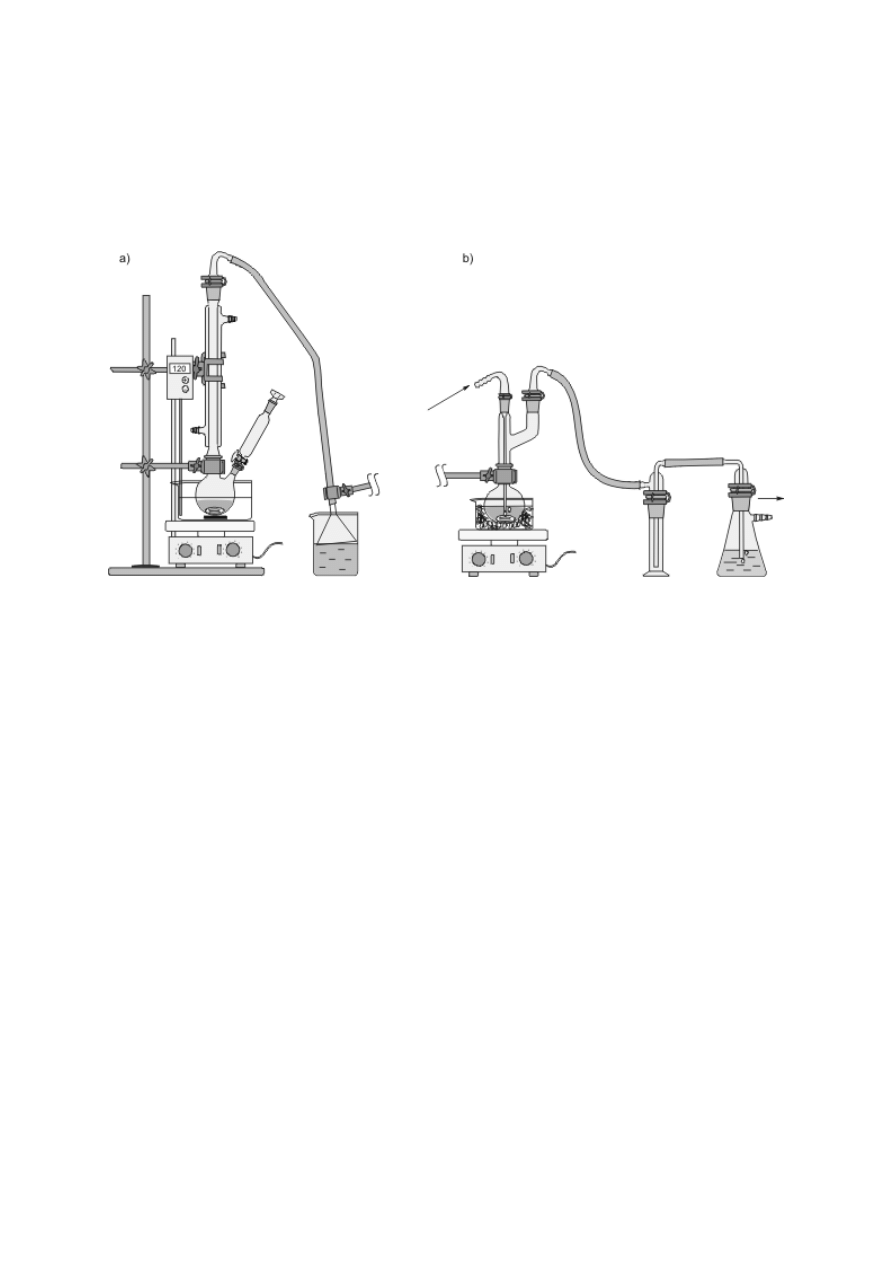
Układ do pochłaniania gazów musi być też zainstalowany, jeżeli przez mieszaninę reakcyjną
jest przepuszczany strumień agresywnego, gazowego reagenta. Gaz wprowadza się tuż pod
powierzchnię mieszaniny lub, jeżeli odczynnik jest słabiej absorbowany, nieco głębiej
i intensywnie miesza się roztwór (rys. 19b). Pochłanianie nadmiaru gazu wymaga użycia
bardziej rozbudowanego pochłaniacza. Gaz jest przepuszczany przez płuczkę zabezpieczającą
do naczynia zawierającego większą objętość roztworu neutralizującego.
Rysunek 19. Przykładowe zestawy do prowadzenia syntez z udziałem gazowych reagentów:
a) gaz wydzielający się w reakcji jest odprowadzany do uproszczonego pochłaniacza przez
górny wylot chłodnicy; b) gazowy substrat jest przepuszczany przez oziębioną mieszaninę
reakcyjną, a jego nadmiar odprowadzany przez płuczkę zabezpieczającą do pochłaniacza;
zastosowanie nasadki „Y„ pozwala przeprowadzić ten proces przy użyciu kolby jednoszyjnej
Zestawy półotwarte z ograniczeniem
dostępu wilgoci
Wiele odczynników stosowanych w syntezach organicznych ulega rozkładowi w kontakcie z
wodą (np. sód metaliczny, bezwodny chlorek glinu lub żelaza, bezwodnik octowy). Ponieważ
woda obecna jest w postaci pary w powietrzu oraz adsorbuje się na powierzchni szkła, reakcje
z udziałem odczynników wrażliwych na wilgoć wymagają starannego przygotowania suchej
aparatury (patrz podrozdz. II.2) i zabezpieczenia jej przed dostępem wilgoci z powietrza. Aby
zestaw nie był hermetycznie zamknięty, a jednocześnie do wnętrza aparatury nie
przedostawała się wilgoć z otoczenia, należy każdy otwór połączyć z nasadką zawierającą
substancję higroskopijną (rys. 7i, j). Nasadka zabezpieczająca może być mocowana
bezpośrednio w szyjce naczynia, jeśli reakcja przebiega w temperaturze pokojowej lub niższej
(rys. 20a), względnie w górnym otworze wodnej lub powietrznej chłodnicy zwrotnej, jeśli
mieszanina jest ogrzewana (rys. 20b). Gdy należy wkraplać odczynnik wrażliwy na wilgoć,
a nie są dostępne wkraplacze z wyrównywaniem ciśnienia, korek wkraplacza również
zastępuje się rurką z chlorkiem wapnia. Do podłączenia nasadek można wykorzystać łączniki
z bocznymi tubusami, a przez rurki można też odprowadzać powstające w reakcji gazy do
odpowiednich absorberów, tak jak przedstawiono to na rys. 20c. Metoda ta jest wskazana w
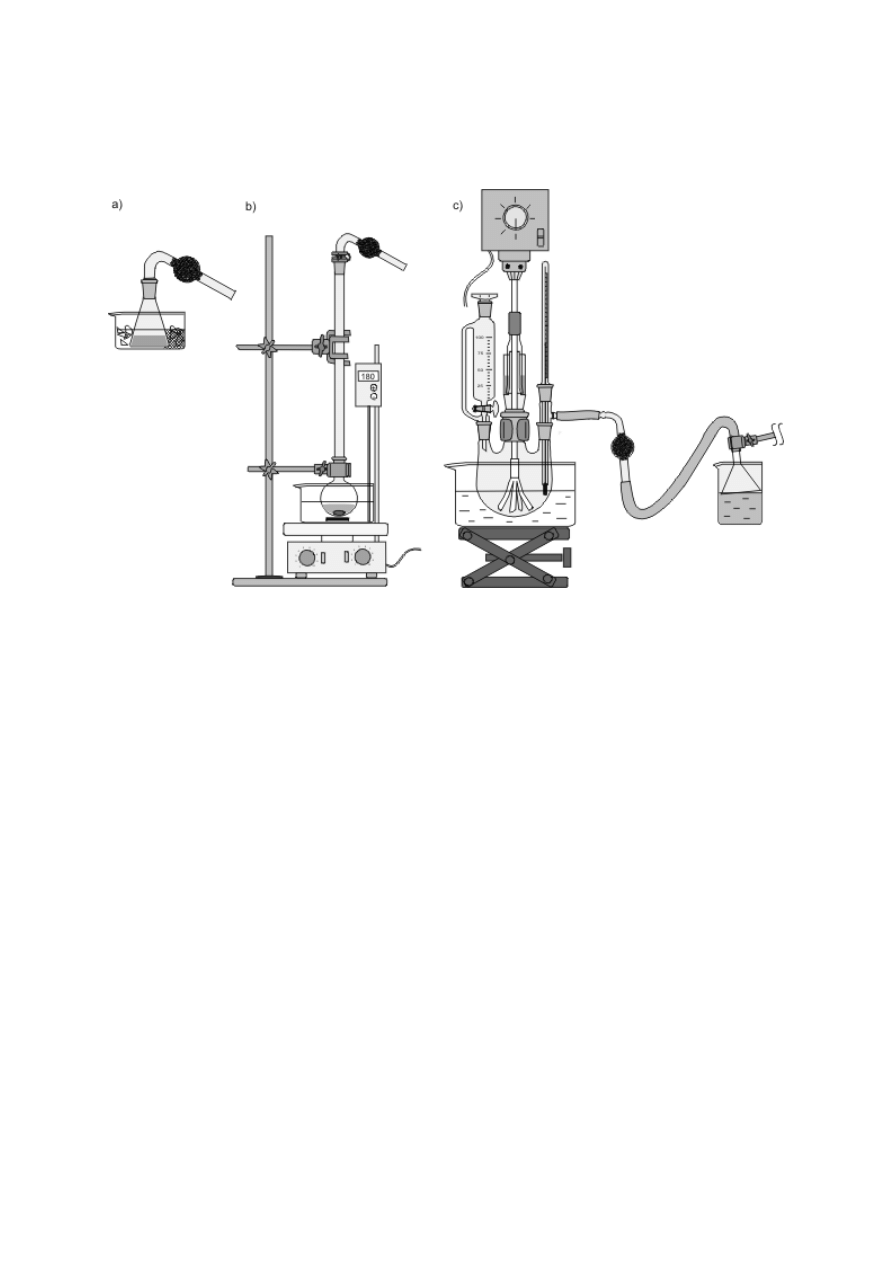
przypadkach konieczności montowania rozbudowanych zestawów, na przykład do procesów
acylowania Friedela-Craftsa, które ze względu na konsystencję mieszaniny reakcyjnej
wymagają użycia mieszadeł mechanicznych.
Rysunek 20. Sposoby zabezpieczania zestawów przed dostępem wilgoci za pomocą nasadek
ze środkiem suszącym: a) nasadka umocowana w szyi naczynia; b) nasadka w górnym szlifie
chłodnicy; c) odprowadzenie gazów do pochłaniacza przez rurkę ze środkiem suszącym
podłączoną do bocznego tubusa
Aby podjęte środki były efektywne, układ trzeba chronić przed dostępem wilgoci również
innymi sposobami — należy unikać ogrzewania zestawu na łaźni wodnej, wszelkie operacje
związane z przenoszeniem odczynników czy otwieraniem zestawu wykonywać możliwie
najszybciej. Odczynniki gwałtownie reagujące z wodą (np. tetrahydrydoglinian litu, wodorki i
amidki metali itp.) wolno otwierać, odważać i przenosić do innych pojemników tylko w
suchej, neutralnej atmosferze, najlepiej w specjalnej komorze wypełnionej gazem obojętnym
(„dry-box”).
Zestawy izolujące mieszaninę reakcyjną od
otoczenia
Niektóre reagenty bądź produkty pośrednie, a czasem nawet ostateczne produkty syntez, są
wrażliwe na działanie tlenu atmosferycznego. Wszelkie operacje z ich udziałem należy
przeprowadzać zatem w zestawach, w których powietrze zastąpiono gazem obojętnym —
azotem lub argonem. Przykładami takich przemian są reakcje, w których uczestniczą m.in.
niektóre związki metalo-, boro-, fosforoorganiczne, połączenia o charakterze wodorków
metali, złożone związki koordynacyjne itp. Reakcje takie nie wchodzą w zakres
podstawowego programu laboratorium chemii organicznej i nie będą w tym miejscu
szczegółowo dyskutowane. Warto zdawać sobie jednak sprawę, że nawet tak pozornie trudne
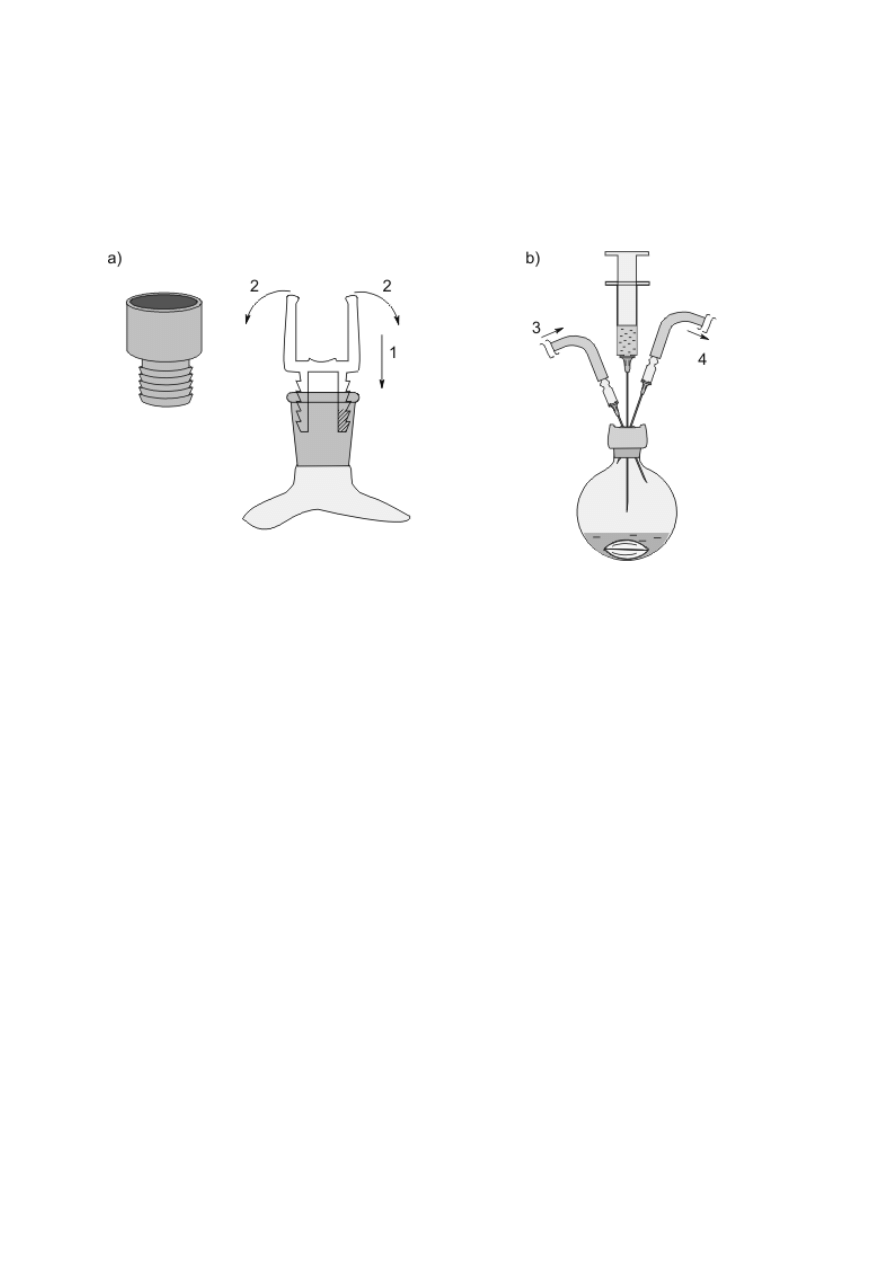
przemiany można przeprowadzić na małą skalę bez konstruowania złożonej aparatury.
Dużym ułatwieniem jest zastosowanie specjalnego kapsla, wykonanego z gumy naturalnej
bądź silikonowej, znanego powszechnie pod nazwą septum (rys. 21a). Korki takie
produkowane są w rozmiarach dopasowanych do wszystkich znormalizowanych wielkości
szlifów. Karbowaną część korka wsuwa się w szyjkę kolby, a następnie wywija się jego
kołnierz tak, aby ściśle przylegał do zewnętrznej ścianki szyjki.
Rysunek 21. a) Gumowy kapsel typu septum, widok z boku i sposób zakładania: 1 — wsunąć
do szyi, 2 — odwinąć kołnierz na zewnętrzną stronę szyi; b) zestaw z kapslem septum do
prowadzenia reakcji w atmosferze gazu obojętnego przy jednoczesnym dozowaniu ze
strzykawki ciekłego reagenta: 3 — wlot gazu, 4 — odprowadzenie gazu do płuczki
zabezpieczającej
W środkową część kapsla można łatwo wbić igłę od strzykawki. Układ jest tak szczelny, że
odsysając powietrze przez tę igłę, można obniżyć ciśnienie w kolbie do ok. 10 hPa.
Posługując się w podobny sposób drugą igłą, połączoną przez odpowiednie złączki ze
źródłem gazu obojętnego, do kolby wprowadza się, na przykład, odtleniony i osuszony argon.
Po kilkakrotnej wymianie gazów w kolbie można rozpocząć właściwą reakcję w obojętnej
atmosferze przy lekkim nadciśnieniu gazu obojętnego. Potrzebne odczynniki dodaje się przy
użyciu strzykawki przez kolejną igłę (rys. 21b). Wbijanie kilku igieł w jeden kapsel powoduje
znaczne zmniejszenie szczelności układu, jednak w większości przypadków nie wpływa to na
przebieg samej reakcji.
Zamknięcia typu septum można stosować też w opisanych wcześniej zestawach. Pewnym
ograniczeniem jest jednak cena tych kapsli, a z założenia powinno traktować się je jako
akcesoria jednorazowego użytku.
Zasady montażu i demontażu aparatury
Aby bezpiecznie i sprawnie przygotować zestaw, a następnie wykonać przy jego użyciu
syntezę, należy przestrzegać następujących wskazówek.

Przed przystąpieniem do montażu zestawu należy sprawdzić, czy w wybranym miejscu
laboratorium działa właściwie wentylacja oraz czy jest sprawna, zależnie od potrzeb,
instalacja wodna, elektryczna i/lub gazowa. Ponadto należy się upewnić, czy przeprowadzane
doświadczenie nie będzie kolidowało z eksperymentami sąsiadów (np. stosowanie palnika w
sąsiedztwie palnych rozpuszczalników lub ogrzewanie na łaźni wodnej w pobliżu zestawów
zabezpieczanych przed wilgocią).
Trzeba zbadać, czy sprzęt elektryczny nadaje się do użytku (np. czy kable nie mają
uszkodzonej izolacji, czy płaszcze grzejne nie są wilgotne). Wskazane jest, aby działanie
mieszadeł, łaźni wodnych, regulatorów mocy i płaszczy grzejnych przetestować jeszcze przed
zmontowaniem aparatury.
Wszystkie części aparatury szklanej trzeba dokładnie obejrzeć — jakiekolwiek pęknięcia
(rysy, gwiazdki) czy też uszczerbki dyskwalifikują uszkodzone elementy.
Należy sprawdzić, czy poszczególne elementy aparatury (połączenia szlifowe, korki, rurki)
pasują do siebie, dając szczelne łącza. W przeciwnym przypadku trzeba je wymienić lub
zastosować odpowiednie łączniki redukcyjne. Złącza szlifowe i krany należy nasmarować.
Węże łączące części aparatury lub np. chłodnicę z kranem muszą szczelnie przylegać do
króćców i oliwek, ale powinny dać się nakładać i zdejmować bez użycia nadmiernej siły
(podrozdz. II.3 oraz III.3).
Montaż aparatury należy przeprowadzać ściśle według wskazówek podanych w podrozdz.
II.2. Należy dopilnować, aby węże doprowadzające wodę nie krzyżowały się zprzewodami
elektrycznymi oraz aby połączenia te nie były napięte. Ani węże, ani przewody elektryczne
nie mogą znajdować się w bezpośredniej bliskości elementów grzejnych! Najdroższe
części aparatury (termometry) mocuje się w miarę możliwości na końcu. Nie wolno
wprowadzać mieszalników do naczynia znajdującego się nad mieszadłem
magnetycznym — grozi to rozbiciem dna!
Przed rozpoczęciem reakcji należy dokonać próbnego uruchomienia aparatury „na sucho„.
Ustawia się wówczas właściwy strumień wody w chłodnicy, obroty mieszadła, upewnia się,
czy mieszalnik nie uderza w termometr itp.
Po zakończeniu reakcji, o ile w instrukcji nie podano inaczej, należy doprowadzić
mieszaninę reakcyjną do temperatury pokojowej (usunąć płaszcz grzejny lub np. unieść kolbę
nad łaźnię olejową, nie przerywając mieszania). Dopiero po ostudzeniu zakręcić wodę w
chłodnicy i wyłączyć mieszanie.
Jeżeli reakcja była prowadzona w łaźni na mieszadle magnetycznym, kolbę należy oczyścić
z resztek oleju, a mieszadło odstawić w bezpieczne miejsce do ostudzenia. Zarówno
mieszadło jak i płaszcz przed schowaniem (zwrotem) muszą być wystudzone i oczyszczone!
Kolejnym etapem jest demontaż najcenniejszych części aparatury (termometrów,
wkraplaczy na szlifie), dokładne spuszczenie wody z chłodnicy przez zdjęcie węża z kranu, a
następnie odłączenie chłodnicy od kolby. Dopiero po usunięciu resztek smarów ze szlifów
można przystąpić do dalszej obróbki mieszaniny reakcyjnej.
IV. Oznaczanie stałych fizycznych
IV.1. Temperatura wrzenia
Najlepszą metodą oznaczenia temperatury wrzenia (tw.) jest metoda destylacyjna. Przy
zastosowaniu możliwie najmniejszego zestawu do destylacji wymaga ona jednak
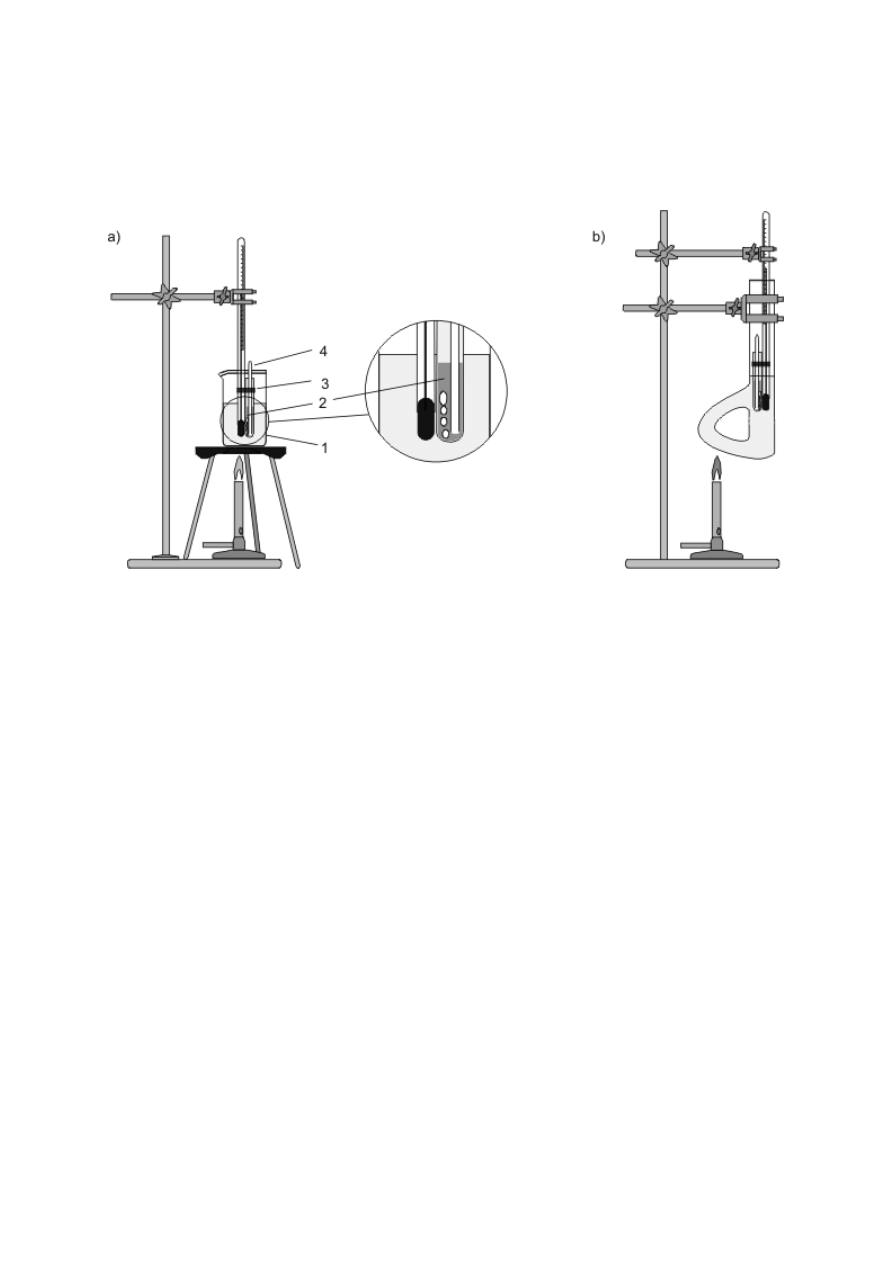
przedestylowania kilku mililitrów cieczy (2–3 cm
3
). Dopiero wówczas ustala się właściwa
temperatura wrzenia z błędem nie większym niż 1–2 °C. Jeżeli nie dysponuje się taką ilością
cieczy, to wykorzystuje się mikrometody, wymagające nie więcej niż 0,5 cm
3
substancji.
Najczęściej stosowana jest metoda Siwołobowa.
Rysunek 22. a) Zestaw do pomiaru temperatury wrzenia metodą Siwołobowa: 1 — zlewka z
olejem, 2 — mikroprobówka z próbką badanej cieczy, 3 — miejsce mocowania
mikroprobówki do nóżki termometru, 4 — jednostronnie zatopiona kapilara z otwartym
końcem skierowanym ku dołowi; b) zestaw z aparatem Thielego
W celu przeprowadzenia pomiaru mikroprobówkę o średnicy około 5 mm (identyczną ze
stosowanymi do stapiania substancji organicznych z sodem) napełnia się badaną cieczą do
wysokości około 1 cm (0,3–0,5 cm
3
cieczy) i przymocowuje do termometru (rys. 22a).
Wieloletnie doświadczenia wykazały, że do zamocowania fiolki najlepiej nadaje się mocna
nić bawełniana. Dno fiolki powinno znajdować się na poziomie końca nóżki termometru.
Termometr przymocowuje się do statywu za pomocą małej łapy. Nóżkę termometru wraz z
fiolką umieszcza się w małej zlewce wypełnionej olejem lub wodą, w zależności od
przewidywanej temperatury wrzenia. Zlewka ustawiona jest na płytce ceramicznej i
ogrzewana palnikiem gazowym. W fiolce umieszcza się odwróconą dnem do góry zatopioną
kapilarę (taką jak do pomiaru temperatury topnienia).
W trakcie ogrzewania obserwuje się powolne wydzielanie drobnych banieczek
rozprężającego się powietrza. Wskazane jest zamieszanie co pewien czas zawartości łaźni za
pomocą bagietki szklanej, aby zapewnić równomierne ogrzewanie termometru i próbki.
W momencie kiedy łaźnia osiągnie temperaturę wrzenia, bańki wydzielają się gwałtownie i
mają zwiększoną objętość z uwagi na przechodzenie cieczy w stan gazowy. Wtedy odstawia
się palnik, ale badana ciecz ulega przegrzaniu na skutek bezwładności cieplnej układu
ogrzewającego. Po krótkim czasie temperatura zaczyna opadać i w pewnym momencie ciecz
z fiolki zostaje wciągnięta do kapilary. Odczytuje się wówczas wstępną, orientacyjną
temperaturę wrzenia. Wciąganie cieczy jest spowodowane skropleniem się jej par po
obniżeniu temperatury i wytworzeniem się podciśnienia wewnątrz kapilary. Czeka się, aż
temperatura łaźni obniży się o 8–10 °C w stosunku do orientacyjnej temperatury wrzenia.

Następnie zaczyna się powoli ogrzewać łaźnię (ok. 1 °C na minutę) aż do ponownego
pojawienia się ciągłego strumienia pęcherzyków gazu. Wówczas notuje się temperaturę
(pierwszy odczyt) i przerywa ogrzewanie. Ostatni pęcherzyk „waha się” przez moment przy
wylocie kapilary, a po chwili następuje zasysanie cieczy do jej wnętrza. W tym momencie
odczytuje się ponownie temperaturę (drugi odczyt). Różnica między pierwszą a drugą
odczytaną wartością nie powinna być większa niż 1–2 °C. Jest to zakres zmierzonej
temperatury wrzenia cieczy. Pomiar należy powtórzyć dwukrotnie, a przy braku wprawy
nawet więcej razy. Jeżeli badana ciecz jest stosunkowo czysta, to wyniki nie powinny się
różnić o więcej niż 2–3 °C. Kolejne pomiary można wykonywać na tej samej próbce, chyba
że zaobserwuje się wyraźne ślady rozkładu substancji (ciemnienie, dymienie). Jeżeli w czasie
kolejnych prób objętość cieczy ulegnie znacznemu zmniejszeniu, to można ją uzupełnić za
pomocą pipety Pasteura bez konieczności demontażu zestawu.
Zamiast zlewki można użyć napełnionego olejem aparatu Thielego w kształcie probówki
z bocznym „uszkiem" ogrzewanym palnikiem (rys. 22b). Zastosowanie tego aparatu zapewnia
równomierne ogrzewanie oleju w pobliżu termometru i badanej próbki.
IV.2. Współczynnik załamania światła
Jednym z parametrów pozwalających ocenić czystość cieczy, w tym także ciekłego produktu
reakcji, jest współczynnik załamania światła. Jego pomiar opiera się na zjawisku fizycznym
załamania światła przy przejściu z jednego ośrodka do drugiego. W praktyce chodzi tu
o załamanie światła na granicy faz powietrze-analizowana ciecz. Do pomiaru współczynnika
załamania światła służy między innymi refraktometr Abbego. Jest to proste w obsłudze
urządzenie, które ma szereg zalet bezcennych w codziennej praktyce laboratoryjnej. Pozwala
na wyznaczenie współczynnika załamania światła z dokładnością ok. 0,0002 jednostki,
wymaga użycia zaledwie kilku kropli cieczy oraz, dzięki wyposażeniu w kompensator
Amiciego, umożliwia pomiar dla światła słonecznego (białego). Wartość współczynnika
załamania światła zależy od długości fali świetlnej i od temperatury. W tablicach
fizykochemicznych podawane są zazwyczaj wartości dla czystych cieczy wyznaczone w
temperaturze 20 °C dla światła sodowego (żółte światło, linia widmowa D, λ = 589,3 nm). Na
przykład dla wody taka wartość jest zapisana w następujący sposób: n
D
20
= 1,3330.
Kompensacja Amiciego pozwala na porównywanie wartości n
D
z wartościami wyznaczonymi
dla światła słonecznego. Przy bardzo precyzyjnych pomiarach refraktometr powinien być
podłączony do termostatu, utrzymującego stałą temperaturę części pomiarowej aparatu, ale
niestety jest to dość kłopotliwe rozwiązanie w laboratorium chemii organicznej. Zazwyczaj
temperatura w laboratorium waha się o kilka stopni w stosunku do „wzorcowej” 20 °C. Jak
wynika z pomiarów temperaturowych, wartości współczynnika załamania światła zmieniają
się w zależności od rodzaju cieczy o 0,0003–0,0006 wraz ze zmianą temperatury o 1 °C.
Można więc przyjąć, że różnica o 0,002–0,003 między wartością zmierzoną w tych
warunkach a wartością tablicową świadczy o dostatecznej czystości cieczy.
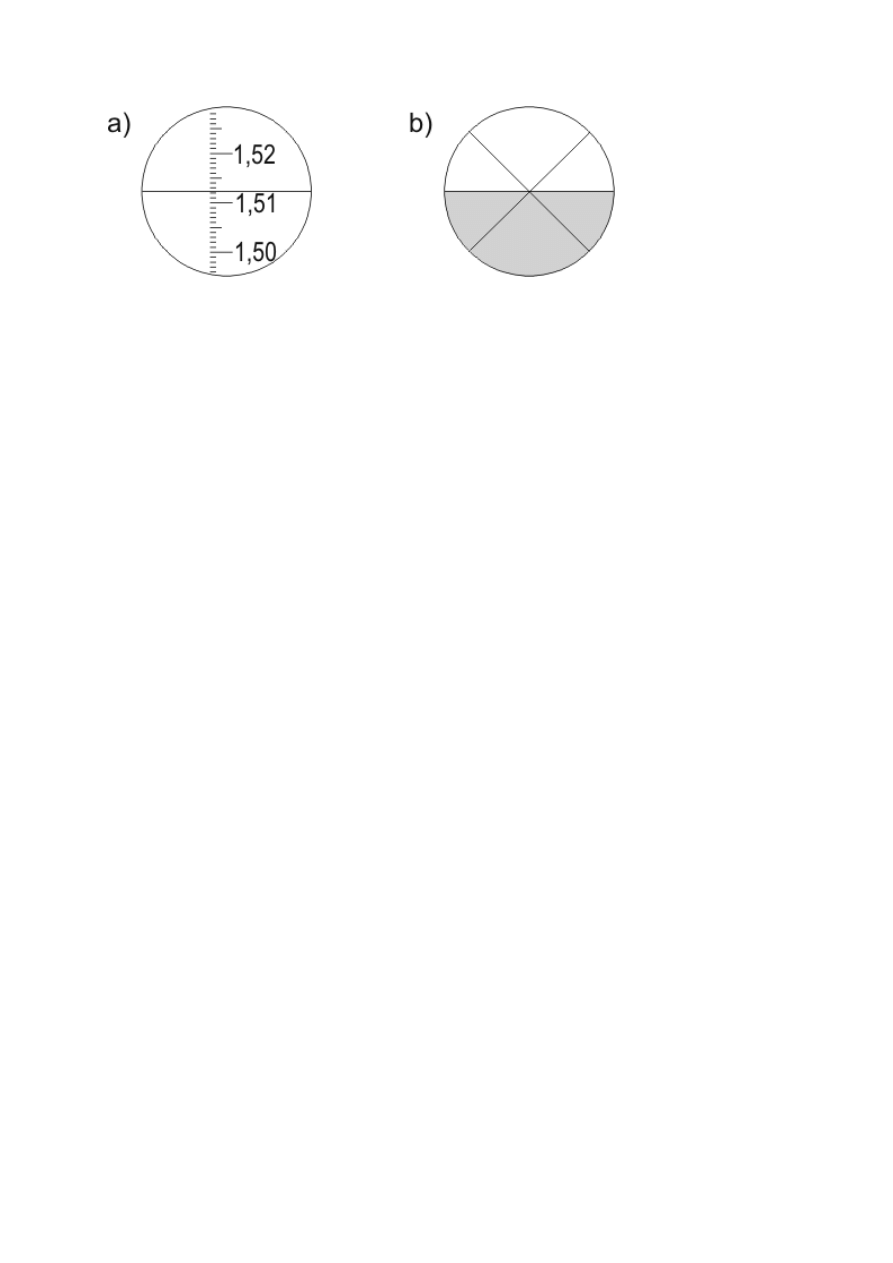
Rysunek 23. Widok w okularach refraktometru Abbego: a) lewy okular: skala do
odczytywania wartości współczynnika załamania światła; b) prawy okular: ostra granica
przechodząca przez skrzyżowanie nitek aparatu
Pomiar współczynnika załamania światła jest stosunkowo prosty. Na czyste i suche płytki
szklane refraktometru nanosi się kilka kropli badanej cieczy i płytki dociska się do siebie.
Pokrętłami aparatu ustawia się w polu widzenia okularu ostrą granicę między częścią jasną
i ciemną. Granica między częściami obrazu nie powinna być podbarwiona ani na czerwono,
ani na niebiesko i powinna przechodzić przez punkt przecięcia (skrzyżowanie) nitek okularu
(Rysunek 23). W drugim okularze odczytuje się wartość współczynnika załamania światła z
dokładnością do trzeciego miejsca, szacując wartość czwartego miejsca po przecinku — n
D
odczytane z rysunku 23 wynosi zatem 1,5122. Należy pamiętać, aby po zakończeniu pomiaru
oczyścić płytki szklane z resztek analizowanej cieczy i przemyć je watą nasączoną etanolem.
IV.3. Temperatura topnienia
Jednym z najprostszych sposobów charakteryzowania stałej substancji organicznej jest
pomiar jej temperatury topnienia (tt.). Uzyskane wyniki mogą być pomocne zarówno przy
identyfikacji związku, jak też przy określaniu jego czystości. Czysty, krystaliczny produkt ma
zazwyczaj stałą, ściśle określoną temperaturę przejścia z fazy stałej w fazę ciekłą. To
przejście fazowe następuje najczęściej w przedziale temperatur mniejszym niż 1
o
C. Jednak
obecność nawet nieznacznej ilości zanieczyszczeń w próbce powoduje zazwyczaj zauważalne
obniżenie jej temperatury topnienia w stosunku do czystej substancji — jest to tzw. efekt
krioskopowy. Ponadto substancje zanieczyszczone topią się w znacznie szerszym przedziale
temperatur: od kilku do nawet kilkudziesięciu stopni.
Dawniej przy pomiarze temperatury topnienia stosowano również ogrzewane palnikiem
łaźnie olejowe (np. aparat Thielego), podobnie jak przy opisywanym w podrozdz. IV.1
pomiarze temperatury wrzenia metodą Siwołobowa. Obecnie do tego celu służą elektryczne
urządzenia zwane kriometrami.
Najczęściej spotyka się trzy rodzaje konstrukcji. Aparaty Boëtiusa są rodzajem mikroskopu
z ogrzewanym stolikiem, w którego wnętrzu umieszczony jest dokładny termometr oraz
system oświetleniowy. Mały kryształ próbki (poniżej 1 mg) rozdrabnia się, umieszcza między
szkiełkami mikroskopowymi, kładzie na stoliku grzewczym aparatu i przykrywa specjalną
osłoną. Po włączeniu powolnego ogrzewania przez okular mikroskopu obserwuje się
jednocześnie podświetloną próbkę oraz termometr. W aparatach blokowych rozdrobnioną
próbkę wprowadza się do kapilary i umieszcza w specjalnym otworze wewnątrz miedzianego
lub aluminiowego bloku grzewczego, w bezpośrednim sąsiedztwie końcówki termometru.
Blok jest powoli ogrzewany elektrycznie, a zmiany zachodzące w próbce obserwuje się przez

oświetlane otwory zaopatrzone w szkła powiększające. Pomiar temperatury odbywa się za
pomocą termometru szklanego lub, coraz częściej, cyfrowego, a całe urządzenie jest
programowane i sterowane elektronicznie. Na podobnej zasadzie co aparaty blokowe działają
aparaty cieczowe, w których kapilara ze związkiem umieszczana jest w zbiorniku z olejem.
Ta wysokowrząca ciecz jest mieszana i ogrzewana elektrycznie, a urządzenie jest często
sterowane elektronicznie.
Ze względu na niższą cenę w powszechnym użyciu znajdują się aparaty blokowe. Pomiar
temperatury topnienia przy ich użyciu wymaga kilku kolejnych czynności:
Należy wybrać cienkościenną kapilarę o wewnętrznej średnicy ok. 0,5 mm i długości ok.
50 mm. Jeden z jej końców wprowadza się do płomienia małego palnika i cały czas obracając
w palcach kapilarę doprowadza się do zasklepienia otworu — trwa to zaledwie kilkanaście
sekund.
Kilkanaście miligramów dobrze wysuszonej próbki rozciera się starannie na szkiełku
zegarkowym. Otwartym końcem jednostronnie zatopionej i wystudzonej kapilary stuka się w
sproszkowany związek znajdujący się na twardym podłożu, umieszczając tym samym
preparat w kapilarze.
Następnie należy spowodować przesypanie się produktu w stronę zamkniętego końca
(„dna” kapilary). Czasami wystarczy chwycić kapilarę tuż przy zamkniętym końcu i postukać
jej dnem o twardą powierzchnię. Skuteczna metoda polega na wrzuceniu kapilary dnem do
dołu do długiej rury (ok. 1 m) o małej średnicy (ok. 2 cm) postawionej pionowo na twardym
podłożu. Spadając, kapilara odbija się kilkakrotnie od podłoża, a związek zazwyczaj
przesypuje się we właściwe miejsce.
Jeżeli w kapilarze znajduje się kilkumilimetrowa (optymalnie ok. 5 mm) warstwa
substancji, można przystąpić do zmierzenia jej temperatury topnienia. W przeciwnym razie
nabijanie kapilary trzeba powtórzyć.
Po włożeniu do kriometru próbkę ogrzewa się powoli, początkowo w tempie kilkunastu, a w
pobliżu temperatury topnienia 3–4 stopni na minutę, obserwując jednocześnie wzrastającą
temperaturę oraz zmiany w wyglądzie substancji. Zbyt szybkie ogrzewanie może
doprowadzić do dużych błędów w odczycie temperatury topnienia. Za początek topienia
przyjmuje się moment, gdy w kapilarze pojawiają się pierwsze kropelki, natomiast koniec
przemiany fazowej następuje, gdy powstaje warstwa cieczy z widocznym meniskiem.
Odczytany przedział temperatur podaje się jako temperaturę topnienia (tt.) substancji.
Precyzyjne określenie punktu topnienia jest utrudnione, gdy przemiana fazowa połączona jest
z rozkładem substancji (zwęglaniem, wydzielaniem się gazów) lub gdy związek sublimuje w
podwyższonej temperaturze. W tym ostatnim przypadku pomiar można próbować wykonać w
obustronnie zatopionej kapilarze. Zużyte kapilary należy umieścić w pojemniku: SZKŁO —
odpady.
Jeżeli nie wiadomo, jaka może być temperatura topnienia danego związku, należy wykonać
pomiar dwukrotnie: za pierwszym razem stosując cały czas większą szybkość ogrzewania, a
po rozpoznaniu przybliżonego zakresu topnienia pomiar powtórzyć zgodnie z wcześniejszymi
wskazówkami.
Pomiar temperatury topnienia jest też pomocny przy próbie identyfikacji związku.
Wykorzystuje się w tym celu wspomniany wcześniej efekt krioskopowy, dzięki któremu
można w prosty sposób sprawdzić, czy dwie podobne substancje o zbliżonych temperaturach
topnienia (np. analizowana próbka i wzorzec) są tym samym związkiem. Jednakowe ilości

obu substancji miesza się i dokładnie rozciera, a następnie oznacza temperaturę topnienia
otrzymanej mieszaniny. Obniżenie temperatury topnienia mieszaniny w stosunku do
substancji wyjściowych praktycznie wyklucza tożsamość porównywanych związków. Brak
obniżenia temperatury topnienia nie jest jednak niepodważalnym dowodem identyczności obu
substancji, gdyż znane są przypadki mieszanin charakteryzujących się temperaturą topnienia
taką samą lub nawet wyższą niż czyste substancje wchodzące w ich skład. Opisany powyżej
test nazywany jest często próbą mieszania.
V. Operacje rozdzielania i oczyszczania
V.1. Krystalizacja
W praktyce laboratoryjnej chemik często staje przed koniecznością oczyszczenia stałego
związku organicznego. Potrzeba taka istnieje, gdy
związek otrzymany w wyniku syntezy jest zanieczyszczony produktami ubocznymi i/lub
substratem,
produkt handlowy ma zbyt niski, w stosunku do potrzeb, stopień czystości,
podczas długiego przechowywania nastąpił częściowy rozkład związku w wyniku
utlenienia, hydrolizy itp.
Najczęściej stosowanym sposobem oczyszczania stałych związków organicznych jest proces
zwany krystalizacją. W tej metodzie wykorzystuje się różnice rozpuszczalności substancji
i zanieczyszczeń w odpowiednio dobranym rozpuszczalniku w wysokiej oraz niskiej
temperaturze (najczęściej w temperaturze wrzenia rozpuszczalnika oraz temperaturze
pokojowej lub niższej). W wyniku procesu krystalizacji można oddzielić od oczyszczanej
substancji związki, które są w danym rozpuszczalniku znacznie słabiej od niej rozpuszczalne
w wysokiej temperaturze (optymalnie: są praktycznie nierozpuszczalne), oraz te
zanieczyszczenia, które rozpuszczają się znacznie lepiej (optymalnie: całkowicie) w niskiej
temperaturze. W najprostszym ujęciu krystalizacja obejmuje pięć kolejnych etapów:
rozpuszczenie zanieczyszczonej substancji w odpowiednio dobranym rozpuszczalniku w
temperaturze wrzenia rozpuszczalnika;
szybkie przesączenie gorącego roztworu w celu oddzielenia nierozpuszczalnych w tych
warunkach zanieczyszczeń;
pozostawienie przesączu do oziębienia, co powoduje krystalizację oczyszczonego związku;
oddzielenie kryształów oczyszczonego związku od tzw. przesączu macierzystego (ługu
pokrystalizacyjnego, zawierającego dobrze rozpuszczalne zanieczyszczenia) i wysuszenie
związku;
kontrolę czystości związku.
W wyniku niewłaściwie przeprowadzonej krystalizacji mogą nastąpić duże straty
oczyszczanej substancji, a otrzymany związek może zawierać więcej zanieczyszczeń niż
wyjściowy preparat. Odpowiedni dobór rozpuszczalnika i prawidłowe przeprowadzenie
krystalizacji stanowią zatem jedną z podstawowych umiejętności każdego chemika. Należy
również pamiętać, że krystalizacja nie jest uniwersalną metodą oczyszczania substancji

stałych. Czasami lepsze efekty przynoszą inne techniki, takie jak destylacja pod
zmniejszonym ciśnieniem, sublimacja, chromatografia preparatywna, destylacja z parą wodną
czy ekstrakcja.
Przed przystąpieniem do krystalizacji należy wybrać rozpuszczalnik odpowiedni do
oczyszczania danej substancji. Jeżeli wiadomo dokładnie, jaki związek ma być oczyszczany,
wystarczy sprawdzić w literaturze chemicznej (np. w [23], [24], podręczniku preparatyki
organicznej lub innej literaturze źródłowej) rodzaj, a często również ilość rozpuszczalnika
zalecanego do krystalizacji tej substancji. Jednak, gdy mamy do czynienia ze związkiem o
nieznanej strukturze lub gdy literatura nie podaje potrzebnych informacji, należy
samodzielnie przeprowadzić dobór odpowiedniego rozpuszczalnika. Muszą być przy tym
spełnione następujące warunki:
Krystalizowana substancja nie może wchodzić w reakcję z rozpuszczalnikiem.
Rozpuszczalnik musi dobrze rozpuszczać krystalizowaną substancję w temperaturze
zbliżonej do temperatury wrzenia (optymalnie 5–25 g substancji w 100 g rozpuszczalnika) i
jednocześnie rozpuszczać ją tylko w niewielkim stopniu w temperaturze pokojowej lub
niższej (optymalnie mniej niż 2 g substancji w 100 g rozpuszczalnika).
Zanieczyszczenia powinny rozpuszczać się w tym rozpuszczalniku bardzo dobrze lub
rozpuszczać się tylko w niewielkim stopniu.
Rozpuszczalnik powinien sprzyjać tworzeniu się kryształów oczyszczanego związku, nie
zaś bezpostaciowego osadu (szkliwa).
Rozpuszczalnik powinien być łatwy do usunięcia z powierzchni kryształów (niska
temperatura wrzenia, mała lepkość).
Gdy dwa rozpuszczalniki w podobnym stopniu spełniają powyższe warunki, o ostatecznym
wyborze rozpuszczalnika decyduje bezpieczeństwo pracy (toksyczność, wybuchowość),
łatwość manipulacji oraz cena. Wiele substancji organicznych można krystalizować z wody.
Wybór wody jest zalecany, jeżeli tylko są spełnione cztery pierwsze warunki doboru
optymalnego rozpuszczalnika do krystalizacji.
Spośród tysięcy znanych obecnie związków ciekłych w temperaturze pokojowej do
krystalizacji wykorzystuje się w praktyce jedynie około trzydziestu. Najczęściej stosowane
rozpuszczalniki są podane w tabeli 3. Uszeregowano je według wzrastającej polarności,
podając temperatury wrzenia, zdolność mieszania się z wodą i uwagi dotyczące
bezpieczeństwa pracy.
W praktyce dobór rozpuszczalnika przeprowadza się na drodze eksperymentalnej. Polega ona
na badaniu rozpuszczalności niewielkiej ilości związku (zależnie od wprawy
eksperymentatora: 5–50 mg) w różnych rozpuszczalnikach, w temperaturze pokojowej
i temperaturze wrzenia. Zbyt dobra lub zbyt słaba rozpuszczalność powoduje wyeliminowanie
testowanego rozpuszczalnika. Na przykład, do probówki wsypuje się 50 mg związku i dodaje
1 cm
3
testowanego rozpuszczalnika. Jeżeli związek ulega rozpuszczeniu w temperaturze
pokojowej, rozpuszczalnik się odrzuca. Jeżeli mimo mieszania i wstrząsania substancja się nie
rozpuszcza, zawartość probówki ogrzewa się do wrzenia. Gdy związek nie ulega
rozpuszczeniu w podwyższonej temperaturze, dodaje się kolejne porcje rozpuszczalnika po
ok. 0,5 cm
3
. Jeśli substancja nie ulegnie rozpuszczeniu w 3 cm
3
wrzącego rozpuszczalnika,
taki rozpuszczalnik również należy wyeliminować. Jeżeli jednak substancja rozpuści się
całkowicie lub prawie całkowicie (zanieczyszczenia mogą być nierozpuszczalne!), probówkę
oziębia się i obserwuje proces krystalizacji. Spontaniczne formowanie się dobrze

wykształconych kryształów świadczy o właściwym doborze rozpuszczalnika. Na podstawie
takiej próby można oszacować niezbędną ilość rozpuszczalnika do przekrystalizowania całej
ilości związku wymagającego oczyszczenia.
W trakcie eksperymentu należy również zwrócić uwagę na pewne fakty, które mogą pomóc w
późniejszym prawidłowym przeprowadzeniu krystalizacji:
jakość powstających kryształów (np. czy związek nie tworzy oleju zamiast krystalizować,
czy należy zainicjować krystalizację przez dodatkowe oziębienie lub pocieranie bagietką
wewnętrznych ścian naczynia itp.);
zachowanie się zanieczyszczeń (czy pozostają nierozpuszczone, czy powodują silne
zabarwienie roztworu);
zachowanie się substancji podczas ogrzewania z rozpuszczalnikiem. (Ten problem dotyczy
substancji o temperaturze topnienia niższej niż temperatura wrzenia rozpuszczalnika. Podczas
ogrzewania substancja nierozpuszczalna w danym rozpuszczalniku ulega stopieniu i tworzy
wraz z zanieczyszczeniami małą kropelkę trudną do zauważenia. Można wówczas błędnie
przyjąć, że związek uległ rozpuszczeniu. Po ochłodzeniu substancja oczywiście krzepnie,
tworząc ciało stałe, ale nie nastąpiło jej pełne oczyszczenie przez krystalizację.)
Jeżeli znana jest struktura związku, w doborze rozpuszczalnika mogą nieco pomóc
następujące ogólne zasady:
Związki organiczne rozpuszczają się łatwiej w rozpuszczalnikach zbliżonych do nich pod
względem budowy oraz właściwości fizycznych i chemicznych. Związki polarne
rozpuszczają się zatem lepiej w rozpuszczalnikach polarnych niż niepolarnych.
W miarę przechodzenia do wyższych członów szeregu homologicznego dowolnej klasy
związków rozpuszczalność związku coraz bardziej zbliża się do rozpuszczalności
węglowodoru, z którego ten związek można wyprowadzić.
Często zachodzi konieczność przeprowadzania krystalizacji z mieszaniny rozpuszczalników.
Na przykład, gdy pewna substancja zbyt dobrze rozpuszcza się w etanolu, a zbyt słabo w
wodzie, dobre wyniki można uzyskać, stosując mieszaninę wody i etanolu w stosunku 1 : 1.
Naturalnie rozpuszczalniki dwuskładnikowe można wykorzystać jedynie wtedy, gdy składniki
są cieczami mieszającymi się ze sobą całkowicie (woda + etanol, woda + aceton, cykloheksan
+ toluen, aceton + toluen itp.).
Tabela 3. Najczęściej stosowane
rozpuszczalniki do krystalizacji,
uszeregowane według wzrastającej
polarności
Rozpuszczalnik
(nazwa zwyczajowa)
Temp.
wrz.
o
C
Mieszalność
z wodą
Uwagi
Cykloheksan
81
–
lotny, łatwopalny

Eter naftowy 40/60
(benzyna lekka)
40–60
–
b. lotny, szkodliwy, łatwopalny
Eter naftowy 60/90
(ligroina)
60–90
–
lotny, szkodliwy, łatwopalny
Tetrachlorometan
(tetrachlorek węgla, czterochlorek
węgla)
77
–
rakotwórczy
Toluen
111
–
szkodliwy
Eter dietylowy
35
–
b. lotny, niebezpieczeństwo
wybuchu
1,4-Dioksan
101
+
lotny, niebezpieczeństwo
wybuchu
Octan etylu
78
–
palny
Trichlorometan
(chloroform)
61
–
lotny, toksyczny
Dichlorometan
(chlorek metylenu)
41
–
b. lotny, toksyczny
Propan-2-on
(aceton)
56
+
palny, lotny
Propan-2-ol
(alkohol izopropylowy)
82
+
palny
Kwas octowy
118
+
ostry zapach, parzący, pary
duszące
Etanol
78
+
palny
Metanol
65
+
toksyczny, palny
Woda
100
+
stosować możliwie często
Przebieg krystalizacji
Przebieg krystalizacji
Po dobraniu najbardziej odpowiedniego rozpuszczalnika i oszacowaniu jego ilości potrzebnej
do oczyszczenia całej próbki, można przystąpić do właściwej krystalizacji. Poniżej krótko
omówione są kolejne etapy tej operacji. Jeżeli krystalizację prowadzi się z użyciem
rozpuszczalnika palnego, cały proces należy wykonywać z dala od źródeł otwartego
ognia.
A. Rozpuszczanie substancji we wrzącym rozpuszczalniku
Odważoną próbkę umieszcza się w kolbie okrągłodennej o pojemności dostosowanej do
przewidywanej ilości rozpuszczalnika (podrozdz. III.2, rys. 13a). Substancję należy wsypać
do naczynia przez szklany lub plastikowy lejek z szeroką nóżką lub, w ostateczności, przez

tutkę z gładkiego papieru, aby nie dopuścić do zabrudzenia szlifu kolby. Następnie dodaje się
przez lejek właściwą ilość rozpuszczalnika (ok. 2/3 ilości oszacowanej podczas doboru
rozpuszczalnika) i wrzuca kilka kamyczków wrzennych. W szyjce kolby mocuje się
chłodnicę zwrotną, pamiętając o pokryciu połączeń szlifowych odpowiednim smarem.
Zawartość kolby ogrzewa się do łagodnego wrzenia w płaszczu grzejnym. Gdy temperatura
wrzenia rozpuszczalnika nie przekracza 80
o
C, można zastosować łaźnię wodną. Jeżeli próbka
nie rozpuści się całkowicie, mimo kilkuminutowego ogrzewania do wrzenia, dodatkowe
porcje rozpuszczalnika można wprowadzać ostrożnie przez górny otwór chłodnicy zwrotnej.
Rozpuszczalnika dodaje się przez szeroki lejek, małymi porcjami, tak aby nie przerwać
wrzenia roztworu, gdyż w przeciwnym razie należałoby wprowadzić nowe kamyczki
wrzenne. Należy bacznie uważać, aby ciecz nie spływała po zewnętrznej ścianie chłodnicy i
nie trafiła przypadkiem do rozgrzanego płaszcza grzejnego!
Obserwując mieszaninę, trzeba pamiętać, że próbka może zawierać nierozpuszczalne
zanieczyszczenia, nie zawsze jest zatem możliwe uzyskanie klarownego roztworu mimo
wprowadzania kolejnych porcji rozpuszczalnika. Jeżeli krystalizowany związek jest
całkowicie rozpuszczony, a widoczny osad pochodzi od nierozpuszczalnych zanieczyszczeń,
nie należy dodawać już więcej rozpuszczalnika, gdyż utrudni to późniejsze wykrystalizowanie
oczyszczonego związku. Jeżeli próbka zawiera zanieczyszczenia, powodujące intensywne
zabarwienie całego roztworu, względnie zanieczyszczenia smoliste lub w postaci drobnej
zawiesiny, trudne do usunięcia przez zwykłe sączenie, należy do roztworu dodać węgla
aktywnego (odbarwiającego) i ogrzewać do wrzenia przez kilka minut. Węgiel aktywny
dzięki dobrze rozwiniętej, porowatej powierzchni silne adsorbuje związki barwne o dużych
cząsteczkach oraz zatrzymuje zawiesiny smół i silnie rozdrobnionych ciał stałych. W tym celu
można również zastosować ziemię okrzemkową. Środka odbarwiającego dodaje się w
niewielkiej ilości (do 2% masy próbki). Nie należy używać go w zbyt dużej ilości, gdyż może
adsorbować również krystalizowaną substancję oraz w niewielkim stopniu przechodzić przez
pory miękkiej bibuły filtracyjnej. Nie wolno dodawać stałych substancji do rozgrzanego
roztworu, gdyż może to spowodować gwałtowne wrzenie i wyrzucenie cieczy z naczynia!
Przed dodaniem węgla aktywnego roztwór należy zatem lekko ostudzić. Najwyższą
skuteczność węgiel aktywny wykazuje, gdy adsorpcja zanieczyszczeń jest przeprowadzana w
rozpuszczalnikach polarnych (np. w wodzie).
Podczas krystalizacji substancji z mieszaniny rozpuszczalników próbkę rozpuszcza się na
gorąco w niewielkiej ilości (ok. 10 cm
3
na każdy gram próbki) tego rozpuszczalnika, w
którym substancja rozpuszcza się dobrze (np. etanolu). W trakcie wrzenia powoli, małymi
porcjami wprowadza się rozpuszczalnik, w którym substancja rozpuszcza się słabo (np.
wodę), do momentu, gdy w kolbie pojawi się lekkie zmętnienie. Wówczas ponownie dodaje
się odrobinę pierwszego rozpuszczalnika (etanolu) do zaniku zmętnienia.
Jeżeli przeprowadza się krystalizację znanego, nieszkodliwego związku z wody,
rozpuszczanie substancji w wodzie można przeprowadzić w kolbie stożkowej, umieszczonej
na siatce ceramicznej na trójnogu i powoli ogrzewanej palnikiem. Aby można było prowadzić
ogrzewanie w takim zestawie, należy dodatkowo upewnić się, że związek nie jest lotny z parą
wodną. Lotność substancji z parą wodną można ocenić w trakcie dokonywania doboru
rozpuszczalnika do krystalizacji, na podstawie obserwacji skroplin powstających w górnej
części probówki. Jeżeli kondensujące krople są mętne, to związek jest lotny z parą wodną i
nie można ogrzewać go z wodą w otwartym naczyniu.
B. Sączenie gorącego roztworu

Wrzący roztwór należy możliwie szybko przesączyć, unikając ostudzenia. Przystępując do
tej operacji, na dłoń, którą będzie chwytana kolba, należy założyć rękawicę chroniącą przed
poparzeniem (z grubej skóry lub impregnowanej bawełny). Roztwór sączy się przez sączek
fałdowany umieszczony w dużym lejku stożkowym o krótkiej, szerokiej nóżce a przesącz
zbiera do kolby stożkowej z szeroką szyją bez szlifu lub, w przypadku krystalizacji z wody, w
zlewce (rysunek 14a). Lejek powinien być zawieszony w kółku umocowanym do statywu, a
jego nóżka wsunięta możliwie głęboko do szyjki odbieralnika. Istnieje kilka sposobów
pozwalających zapobiec szybkiemu stygnięciu roztworu i krystalizacji substancji w nóżce
lejka lub na sączku:
Najbezpieczniej jest używać dużych lejków i miękkiej bibuły oraz szybko przelewać
gorącą ciecz.
Przed przystąpieniem do sączenia lejek można ogrzać (np. w suszarce, w strumieniu
gorącego powietrza lub ostatecznie nad płaszczem grzejnym).
Jeśli zachodzi potrzeba przesączenia większej ilości gorącego roztworu, to podczas
sączenia lejek wraz sączkiem fałdowanym umieszcza się w odpowiednim płaszczu do lejków.
Najczęściej używa się miedzianych płaszczy wypełnionych wodą i ogrzewanych za pomocą
palnika (rys. 14b). Stosując taki płaszcz, należy pamiętać, aby wyłączyć palnik przed
rozpoczęciem sączenia.
Często zdarza się, że mimo opisanych powyżej zabiegów na sączku wydziela się tak duża
ilość kryształów, że sączenie staje się niemożliwe, a przemywanie niewielkimi porcjami
wrzącego rozpuszczalnika — nieefektywne. Wówczas należy cały sączek wraz z osadem
umieścić ponownie w kolbie, dodać nową porcję rozpuszczalnika, świeże kamyczki wrzenne i
doprowadzić zawartość kolby do wrzenia. Po kilku minutach otrzymany roztwór sączy się
przez nowy sączek fałdowany, a pozostały w kolbie zużyty sączek wygotowuje się z kolejną
porcją rozpuszczalnika. Wszystkie przesącze zbiera się do jednego naczynia.
W każdym przypadku po przesączeniu roztworu kolbę i sączek przemywa się dodatkową
porcją wrzącego rozpuszczalnika. Należy pamiętać, aby opróżnionej kolby kulistej nie
wkładać do gorącego płaszcza grzejnego! Podczas przelewania roztworu ostatnie jego krople
muszą być zbierane z krawędzi naczynia za pomocą bagietki i przenoszone na sączek, aby nie
dopuścić do ściekania roztworu po zewnętrznych ściankach kolby.
C. Krystalizacja próbki
Gorący przesącz pozostawia się w kolbie stożkowej (lub zlewce) zakrytej szkiełkiem
zegarkowym. Studzenie roztworu powinno się odbywać powoli, co zazwyczaj sprzyja
powstawaniu ładnie wykształconych kryształów. Zbyt szybkie oziębienie roztworu może
spowodować powstanie bezpostaciowego osadu, który zazwyczaj okluduje znaczną ilość
zanieczyszczeń wraz z roztworem. Po osiągnięciu temperatury pokojowej kolbę z przesączem
można oziębić dodatkowo w wodzie z lodem. Nie zawsze tworzenie kryształów następuje
szybko i zgodnie z oczekiwaniami. Najczęstszymi problemami jest brak kryształów oraz
wydzielanie się, zamiast kryształów, drugiej warstwy ciekłej, zwłaszcza w przypadku
substancji o niskich temperaturach topnienia. Brak kryształów może być spowodowany
użyciem zbyt dużej ilości rozpuszczalnika i otrzymaniem roztworu nienasyconego. Jedynym
wyjściem jest wówczas odparowanie na gorąco części rozpuszczalnika (uwaga:
rozpuszczalników organicznych nie wolno odparowywać w otwartych naczyniach; należy
oddestylować rozpuszczalnik, np. w wyparce — patrz podrozdz. V.3) i pozostawienie
roztworu do krystalizacji. Często zdarza się jednak, że otrzymuje się roztwory przesycone, z

których pierwsze kryształy wydzielają się z trudnością. Przyczyną mogą być niewielkie ilości
żywicowatych substancji, działających jak koloid ochronny na zarodki krystalizacyjne.
Krystalizację pozwalają zainicjować następujące metody:
Zaszczepienie roztworu niewielką ilością krystalicznej substancji. Metoda jest godna
polecenia, gdy przeprowadza się oczyszczanie znanej substancji, której próbka jest osiągalna
z innego źródła. W ostateczności można posłużyć się odrobiną zanieczyszczonej substancji.
Pocieranie wewnętrznych ścian naczynia szklaną bagietką. Mikroskopijne odłamki szkła i
rysy na szkle mogą działać jak zarodki krystalizacyjne.
Silne oziębienie mieszaniny np. w łaźni zawierającej lód z solą (temp. ok. –20
o
C) lub przez
wprowadzenie do wnętrza kolby stałego ditlenku węgla lub ciekłego azotu, i jednoczesne
pocieranie wewnętrznych ścian naczynia szklaną bagietką. Należy pamiętać, że silne
oziębienie z jednej strony zwiększa stopień przesycenia roztworu, z drugiej jednak zwiększa
jego lepkość, co może w efekcie czasem hamować krystalizację.
Powolne zatężanie roztworu (zwiększa się stopień przesycenia) lub przeciwnie —
stopniowe rozcieńczanie roztworu (zmniejsza się lepkość).
Dodanie niewielkiej ilości innego rozpuszczalnika, w którym substancja trudno się
rozpuszcza.
Pozostawienie roztworu na dłuższy czas (od kilku godzin do kilku tygodni) w łaźni
chłodzącej lub lodówce.
W trakcie krystalizacji związków o niskiej temperaturze topnienia, zwłaszcza niższej od
temperatury wrzenia użytego rozpuszczalnika, zamiast krystalicznego ciała stałego z
roztworów wydzielają się tzw. oleje. Aby zainicjować krystalizację warstwy oleistej, można
zastosować jedną z wyżej wymienionych metod lub intensywnie mieszać i rozcierać już
pierwsze krople oleju wydzielającego się podczas studzenia roztworu. Tworzące się z oleju
kryształy często okludują jednak rozpuszczone w nim zanieczyszczenia; lepiej jest zatem
powtórzyć krystalizację, stosując większą objętość rozpuszczalnika lub spróbować dobrać
inny rozpuszczalnik (np. o niższej temperaturze wrzenia).
Całkowite wydzielenie się kryształów z roztworu o stałej temperaturze następuje często już
po kilkudziesięciu minutach od chwili rozpoczęcia krystalizacji. Zdarza się też, że jest to
proces znacznie wolniejszy i trwa nawet kilka tygodni. Dłuższe przechowywanie roztworu
krystalizacyjnego w zamkniętym naczyniu i stałej temperaturze sprzyja również tworzeniu się
dobrze wykształconych kryształów. W praktyce rzadko można pozwolić sobie na długie
oczekiwanie i wytworzone kryształy oddziela się po kilkudziesięciu minutach lub kilku
godzinach.
D. Odsączanie i suszenie próbki
Wydzielone kryształy odsącza się pod zmniejszonym ciśnieniem, stosując zestaw
przedstawiony na rys. 14c, postępując dokładnie według opisu w podrozdz. III.5. Lejek
Büchnera (porcelanowy lub polipropylenowy) musi być dopasowany wielkością i kształtem
do ilości sączonego produktu oraz objętości roztworu i rodzaju rozpuszczalnika.
Pozostały w kolbie osad wypłukuje się niewielką ilością zimnego, czystego rozpuszczalnika,
przemywa nim produkt na lejku Büchnera, aby odmyć resztki przesączu z zawartymi w nim
zanieczyszczeniami i dokładnie odciska.

Przesącz zawarty w kolbce ssawkowej, czyli przesącz pokrystalizacyjny lub ług macierzysty,
może stać się, po zagęszczeniu, źródłem kolejnej porcji kryształów, zazwyczaj są one jednak
bardziej zanieczyszczone niż pierwsza frakcja kryształów. W przypadku niewielkiej ilości
pierwszej (głównej) porcji kryształów należy jednak zawsze podjąć próby wydobycia
kolejnych frakcji kryształów z ługu macierzystego. Najprostszy sposób polega na
odparowaniu większości rozpuszczalnika — do ok. 1/4 pierwotnej jego objętości (podrozdz.
V.3). Roztwory wodne zawierające substancje nielotne z parą wodną można odparować z
parownicy lub szerokiej zlewki umieszczonej na łaźni wodnej względnie pozostawić na kilka
dni w otwartym naczyniu (jeżeli związek jest nieszkodliwy i nielotny z parą wodną). Sposób
odparowania wody z roztworów zawierających substancję lotną z parą wodną zależy od
właściwości tej substancji, czasami (lecz nie zawsze!) możliwe jest zagęszczenie roztworu
pod zmniejszonym ciśnieniem.
Częściowo osuszone ciało stałe (nie powinno być lepkie ani maziste) suszy się na powietrzu
w temperaturze pokojowej, a gdy staje się sypkie, przenosi się do suszarki lub stawia pod
promiennikiem cieplnym (lampą do suszenia). Trzeba pamiętać o prowadzeniu suszenia
w temperaturze znacznie niższej od temperatury topnienia substancji oraz o innych
ograniczeniach związanych z suszeniem ciał stałych (podrozdz. III.6).
E. Ocena czystości, pomiar temperatury topnienia
Ostatnim etapem procesu krystalizacji jest sprawdzenie stopnia oczyszczenia otrzymanego
produktu. W przypadku oceny czystości ciał stałych, zwłaszcza o znanej strukturze, najłatwiej
jest przeprowadzić pomiar temperatury topnienia otrzymanej próbki. Dokładny opis
postępowania jest zamieszczony w podrozdz. IV.3. Jeżeli związek po krystalizacji topi się w
temperaturze zgodnej z danymi literaturowymi, w przedziale węższym niż 1
o
C, można
przyjąć, że krystalizacja została przeprowadzona poprawnie i otrzymano czysty produkt. Jeśli
krystalizowano związek o nieznanej strukturze lub nieopisany w literaturze chemicznej, to
„ostra” temperatura topnienia nie jest wystarczającym kryterium czystości preparatu. Dawniej
stosowanym kryterium czystości związku była tzw. krystalizacja do stałej temperatury
topnienia. Jeśli po kolejnych procesach krystalizacji zmierzona temperatura topnienia
produktu nie ulegała już podwyższeniu, można było przyjąć, że związek był czysty. Obecnie
jednorodność substancji oznacza się metodami instrumentalnymi — spektroskopowymi lub
chromatograficznymi.
W przypadku gdy związek topi się w szerokim zakresie (ponad 2
o
C) lub w temperaturze
odbiegającej więcej niż 2–3
o
C od wartości literaturowej, należy poddać go prawdopodobnie
dalszemu oczyszczaniu. Przed rozpoczęciem ponownej krystalizacji z innego rozpuszczalnika
warto jednak rozważyć inne przyczyny, które mogły wpłynąć na temperaturę topnienia, na
przykład:
Preparat mógł być niecałkowicie wysuszony. Należy spróbować go dokładniej wysuszyć.
Związek mógł wydzielić się jako bezpostaciowy osad (zakrzepnięty olej, szkliwo —
zamiast oczekiwanych kryształów) lub mógł zaokludować składniki ługu macierzystego, na
przykład podczas szybkiego ochładzania gorącego przesączu. Należy wówczas podjąć próbę
rekrystalizacji. Substancję rozpuszcza się na gorąco w rozpuszczalniku, z którego był
krystalizowany. Jeśli roztwór jest klarowny, nie ma konieczności sączenia go przez sączek
fałdowany, lecz pozostawia się go do powolnej krystalizacji.
Popełniono błędy podczas pomiaru temperatury topnienia (zbyt szybkie ogrzewanie,
grubościenna kapilara, niewłaściwe uchwycenie momentu przemiany fazowej, uszkodzony
termometr itp.). Warto ponownie zmierzyć temperaturę topnienia, najlepiej na innym
urządzeniu.
V.2. Sublimacja
Sublimacja to zjawisko przechodzenia ciał stałych w stan pary bez pośredniego przejścia
przez fazę ciekłą. Sytuację taką ilustruje następujący diagram fazowy, czyli wykres
obrazujący wzajemne relacje stanów skupienia danej substancji: ciała stałego A, cieczy B i
gazu C w zależności od ciśnienia i temperatury. Krzywe te spotykają się w punkcie
potrójnym, oznaczonym gwiazdką (rys. 24). Z wykresu tego wynika, że jeśli przy stałym
ciśnieniu p
1
podnosić się będzie temperaturę, to ciało stałe A najpierw stopi się przechodząc
w ciecz B, a następnie przejdzie w stan pary C. Natomiast przy niższym stałym ciśnieniu p
2
wzrost temperatury spowoduje przejście ciała stałego A bezpośrednio w stan pary.
Rysunek 24. Przykładowy diagram fazowy
Niektóre pierwiastki (np. jod) i substancje (np. zestalony ditlenek węgla, czyli tak zwany
suchy lód, naftalen, kwas benzoesowy czy związki organiczne zawierające fluor) sublimują
łatwo już pod normalnym ciśnieniem, ponieważ prężność ich par jest duża nawet poniżej
temperatury topnienia. W przypadku innych sublimacja zachodzi dopiero pod obniżonym
ciśnieniem. Sublimacja jest procesem odwracalnym, możliwe jest więc także zjawisko
odwrotne, czyli osadzanie się ciała stałego po oziębieniu gazu: resublimacja. Sublimacja
wykorzystywana jest do nanoszenia specjalnych barwników na różne powierzchnie (np. na
papier w kolorowych drukarkach, na tkaniny z kolorowymi nadrukami), do suszenia
substancji, a także oczyszczania niektórych substancji stałych. To ostatnie zastosowanie
ograniczone jest rodzajem zanieczyszczeń, które powinny być znacznie bardziej lub znacznie
mniej lotne od oczyszczanej substancji stałej.
Sublimację pod normalnym ciśnieniem najłatwiej jest przeprowadzić w porcelanowej
parownicy przykrytej krążkiem z bibuły filtracyjnej, w której wycięte są małe otwory, oraz
odwróconym lejkiem szklanym o tej samej średnicy co parownica (rys. 25b). Nóżkę lejka
zatyka się luźnym kłębkiem waty celulozowej lub szklanej. Substancję sublimowaną
umieszcza się w parownicy, którą ogrzewa się ostrożnie, najczęściej umieszczając ją na
statywie nad palnikiem. Należy tak ogrzewać, aby związek nie uległ stopieniu. Do sublimacji
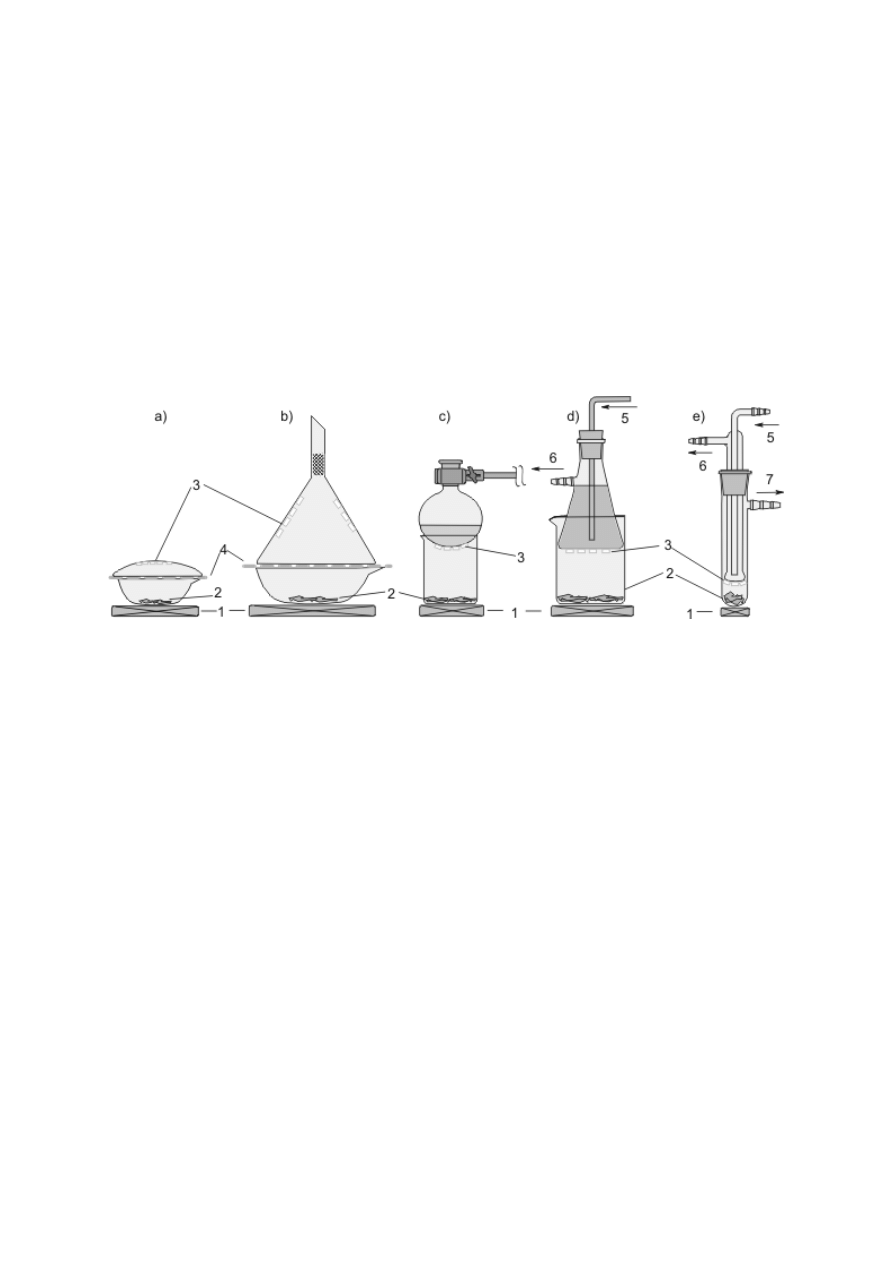
mniejszych ilości substancji lejek można zastąpić chłodzonym z zewnątrz szkiełkiem
zegarkowym (rys. 25a).
Pary substancji sublimowanej przedostają się przez otwory w bibule a następnie zestalają się
na zimnej powierzchni lejka. Krążek bibuły zapobiega opadaniu nadmiaru kryształów
z powrotem do parownicy. Tak oczyszczoną przez sublimację substancję łatwo jest zebrać
z powierzchni bibuły i wewnętrznej ścianki lejka lub szkiełka zegarkowego.
Sublimację można też przeprowadzić stosując ogrzewanie substancji w zlewce przykrytej
kolbą kulistą wypełnioną zimną wodą (rys. 25c). Substancja sublimowana osadza się
wówczas w postaci kryształów na ochłodzonej ściance kolby. W innej wersji nad dnem
zlewki można umieścić kolbę ssawkową, w której przepływa zimna woda (rys. 25d). Kolba
ssawkowa pełni wtedy rolę palca chłodzącego.
Rysunek 25. Różne wersje zestawów do sublimacji: 1 — źródło ciepła, 2 — związek
zanieczyszczony, 3 — sublimat, 4 — podziurkowana bibuła, 5 — wpływ wody, 6 — wypływ
wody, 7 — podłączenie do pompy próżniowej
Palec chłodzący z przepływającą przezeń zimną wodą jest głównym elementem aparatu do
sublimacji pod zmniejszonym ciśnieniem (rys. 25e). Nasadkę próżniową z palcem
chłodzącym łączy się przez odpowiedni szlif albo ze specjalnym tubusem, albo z kolbą
okrągłodenną, w których umieszcza się substancję sublimowaną. Kolbę lub tubus ogrzewa się
w łaźni olejowej o określonej temperaturze. Nasadkę łączy się przez kran z układem
próżniowym zawierającym manometr i pompę próżniową. Zarówno podwyższanie
temperatury, jak i obniżanie ciśnienia należy przeprowadzić wolno i ostrożnie, obserwując
równocześnie osadzanie się kryształów substancji sublimowanej na palcu chłodzącym. Taka
aparatura umożliwia nawet zbieranie kolejnych frakcji mieszaniny substancji sublimujących,
przy zachowaniu na przykład stałego ciśnienia i różnych temperatur sublimacji. Rozdzielenie
takie nie jest jednak nigdy zupełne.
Sublimacja ma tę przewagę nad innymi metodami oczyszczania substancji stałych, że
w przeciwieństwie do krystalizacji nie wymaga zastosowania rozpuszczalnika oraz że straty
w przypadku sublimacji są zwykle mniejsze niż w trakcie destylacji substancji stałej.
V.3. Destylacja

Destylacja jest jedną z najbardziej użytecznych metod rozdzielania wieloskładnikowych
mieszanin cieczy. Polega ona na przeprowadzeniu cieczy w stan pary, a następnie na
skropleniu par i zebraniu skroplonej cieczy (destylatu). Proces destylacji pozwala na
oddzielenie bardziej lotnego związku od mniej lotnych zanieczyszczeń, rozdzielenie
mieszaniny kilku związków oraz oznaczenie temperatury wrzenia. Podczas ogrzewania cieczy
prężność pary nad nią wzrasta aż do momentu, gdy staje się równa ciśnieniu
atmosferycznemu i rozpoczyna się wrzenie, czyli parowanie w całej objętości, obserwowane
jako pojawienie się wewnątrz cieczy pęcherzyków gazu. Temperatura wrzenia cieczy jest to
więc temperatura, w której prężność pary cieczy równa jest ciśnieniu zewnętrznemu
(atmosferycznemu). Substancje czyste chemicznie charakteryzują się praktycznie stałą
temperaturą wrzenia (w granicach 1–2 °C). Destylacja jest więc także najlepszą metodą
oznaczania temperatury wrzenia substancji, znacznie bardziej skuteczną niż opisana
wcześniej metoda Siwołobowa (podrozdz. IV.1). Czystość cieczy po destylacji można
sprawdzić różnymi metodami spektroskopowymi i fizykochemicznymi, a jedną z nich jest
pomiar współczynnika załamania światła (podrozdz. IV.2).
W praktyce laboratoryjnej stosuje się różnego rodzaju destylacje, przy czym najczęściej są
wykorzystywane ich następujące typy:
destylacja prosta,
destylacja frakcyjna (rektyfikacja),
destylacja pod zmniejszonym ciśnieniem (próżniowa) oraz jej wersja wykonywana przy
użyciu wyparki obrotowej (rotacyjnej),
destylacja z parą wodną.
Destylacja prosta
Do destylacji prostej wykonywanej pod ciśnieniem atmosferycznym, czyli około 1013 hPa
(760 mmHg), służą zestawy przedstawione na rys. 26a i b. W zestawie na rys. 26a
wykorzystana jest znacznie prostsza w użyciu chłodnica destylacyjna, łącząca w sobie funkcje
chłodnicy prostej, nasadki destylacyjnej i przedłużacza (zestaw zintegrowany).
Wielkość kolby destylacyjnej oraz całej aparatury jest uzależniona od ilości oczyszczanej
substancji, przy czym należy pamiętać, że kolba może być napełniona jedynie do 2/3 swojej
pojemności. Do kolby przymocowanej do statywu za pomocą łapy z mufą i umieszczonej w
łaźni wodnej lub płaszczu grzejnym dołącza się chłodnicę destylacyjną, pamiętając, aby
wszystkie połączenia szlifowe były uprzednio posmarowane specjalnym smarem do szlifów.
Jeżeli rozmiary szlifów kolby i chłodnicy są różne, to trzeba zastosować odpowiedni reduktor
szlifów. Chłodnicę przymocowuje się za pomocą łapy i mufy do statywu. Bardzo ważny jest
dobór właściwej chłodnicy. W przypadku cieczy o niskiej temperaturze wrzenia (np. eteru
dietylowego, tw. 35 °C lub chlorku metylenu, tw. 41 °C) powinna to być długa chłodnica z
płaszczem wodnym (np. chłodnica o długości 50 cm). Jeżeli ciecz ma wyższą temperaturę
wrzenia, to można zastosować nieco krótszą chłodnicę, także zaopatrzoną w płaszcz wodny.
Przy destylacji cieczy o temperaturze wrzenia wyższej niż 150 °C stosuje się długie
chłodnice powietrzne lub chłodnice wodne z zatrzymanym przepływem wody (woda stojąca
znajduje się wewnątrz płaszcza chłodzącego). Zastosowanie chłodnicy wodnej (z przepływem
wody) przy destylacji cieczy wysokowrzących może być przyczyną zniszczenia zestawu do
destylacji. Zimna woda chłodząca płaszcz w zetknięciu z powierzchnią szkła silnie
rozgrzanego parami cieczy wywoła duże naprężenia wewnątrz szklanej ścianki płaszcza i
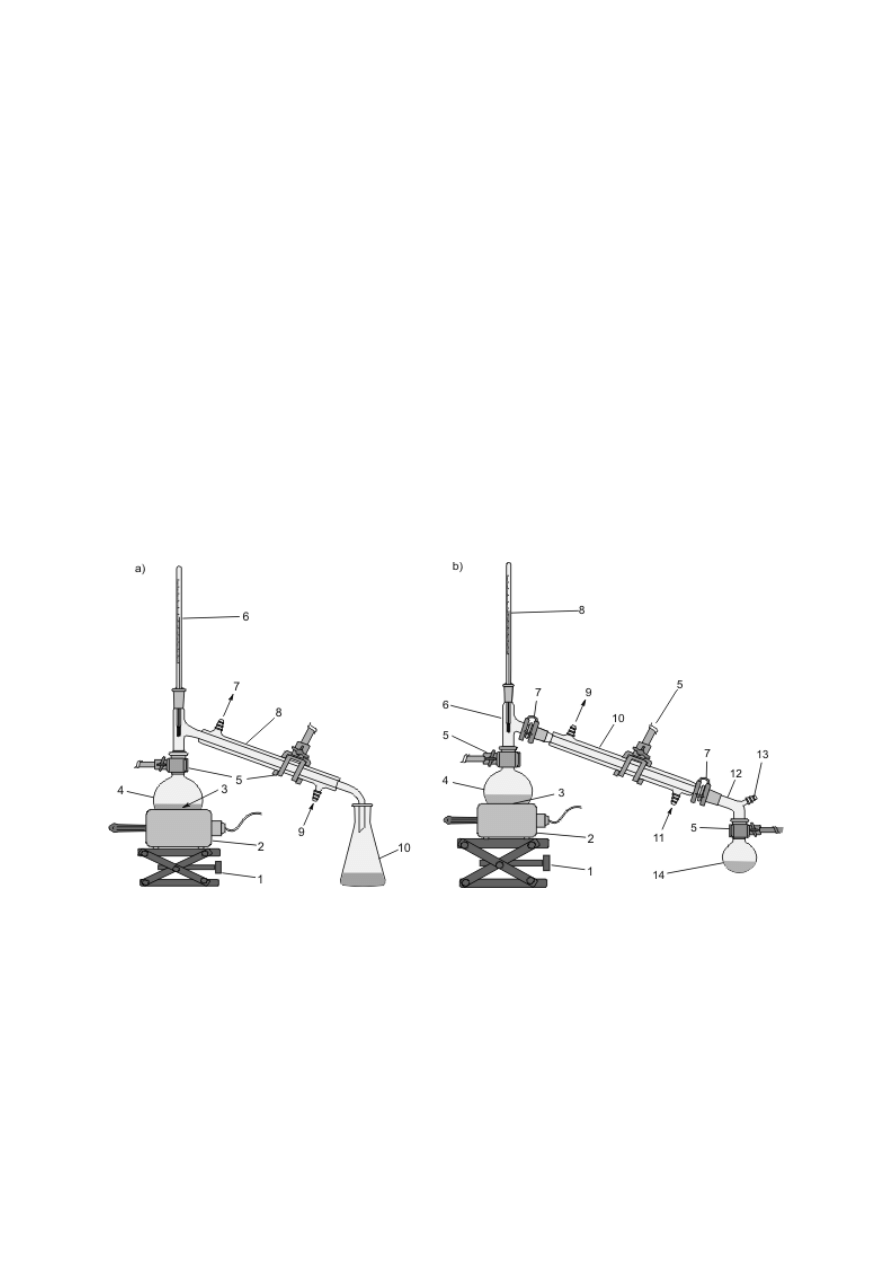
doprowadzi do jej pęknięcia. Chłodnicę wodną podłącza się do sieci wodociągowej w taki
sposób, aby woda wpływała niżej położonym, a wypływała do kanalizacji wyżej położonym
tubusem (zasada przeciwprądu). U wylotu chłodnicy umieszcza się odbieralnik, czyli
naczynie, w którym zbiera się destylat. Może nim być zlewka lub, lepiej, kolba stożkowa.
Odbieralnik przeznaczony do zbierania czystego destylatu powinien być uprzednio zważony,
co ułatwi obliczenie wydajności procesu.
Jeżeli nie ma do dyspozycji odpowiedniej chłodnicy destylacyjnej, to można zbudować
zestaw z chłodnicą prostą. Nie nadają się jednak do tego celu chłodnice kulowe,
wykorzystywane powszechnie jako chłodnice zwrotne. Do budowy zestawu destylacyjnego
oprócz chłodnicy potrzebna jest wówczas nasadka destylacyjna, służąca do połączenia kolby
destylacyjnej z chłodnicą i termometrem (rys. 26b). Wylot chłodnicy zaopatruje się w
przedłużacz kierujący destylat do odbieralnika. Przedłużacz może mieć kształt fajki lub
zgiętej rurki szklanej zaopatrzonej w dwa szlify oraz tubus boczny. Przy tym drugim typie
odbieralnikiem jest kolba okrągłodenna lub kolba stożkowa ze szlifem, zamocowana do
dolnego szlifu przedłużacza. Tubus pozostaje otwarty. Jeżeli zachodzi konieczność
zabezpieczenia destylatu przed wilgocią z powietrza, to do tubusa przyłącza się za pomocą
gumowego węża rurkę z chlorkiem wapnia lub innym środkiem suszącym (podrozdz. III.6).
Przy montażu takiego zestawu należy zwrócić uwagę na ostrożne łączenie nasadki
destylacyjnej i przedłużacza z chłodnicą. Wszystkie szlify muszą być ustawione dokładnie
w jednej linii prostej.
Rysunek 26. a) Zintegrowany zestaw do destylacji prostej pod ciśnieniem atmosferycznym: 1
— podnośnik, 2 — płaszcz grzejny, 3 — kamyczki wrzenne, 4 — kolba okrągłodenna, 5 —
łapy mocujące zestaw do statywu, 6 — termometr, 7 — odprowadzenie wody wężem do
zlewu, 8 — chłodnica destylacyjna, 9 — doprowadzenie wody wężem z kranu, 10 —
odbieralnik z destylatem; b) zestaw do destylacji prostej pod ciśnieniem atmosferycznym z
możliwością podłączenia rurki ze środkiem suszącym: 1 — podnośnik, 2 — płaszcz grzejny,
3 — kamyczki wrzenne, 4 — kolba okrągłodenna, 5 — łapy mocujące zestaw do statywu, 6
— nasadka destylacyjna, 7 — spinki trzymające szlify, 8 — termometr, 9 — odprowadzenie
wody wężem do zlewu, 10 — chłodnica, 11 — doprowadzenie wody wężem z kranu, 12 —
przedłużacz, 13 — tubus umożliwiający podłączenie np. rurki ze środkiem suszącym, 14 —
odbieralnik z destylatem

W każdym z omówionych zestawów do destylacji termometr umieszcza się w taki sposób,
aby zbiorniczek z rtęcią znajdował się nieco poniżej wlotu par do chłodnicy. Należy więc
dobrać termometr ze szlifem o odpowiedniej długości nóżki. Tylko ten sposób montażu
termometru umożliwi odczytanie prawdziwej temperatury wrzenia. Jeżeli nie ma możliwości
dobrania odpowiedniego termometru ze szlifem, to do zamontowania zwykłego termometru
stosuje się nagwintowany łącznik ze szlifem lub w ostateczności korek gumowy z dziurką.
Oczyszczaną ciecz oraz kamyczki wrzenne umieszcza się w kolbie destylacyjnej w ten
sposób, aby szlif nie został zabrudzony wlewaną cieczą. W poprawnie zmontowanym
zestawie termometr musi stać pionowo, a chłodnica być skierowana ukośnie ku dołowi.
Termometr montuje się na samym końcu. Często się zdarza, że wylot chłodnicy znajduje się
nisko nad powierzchnią blatu, co uniemożliwia podstawienie odbieralnika. Wówczas zestaw
trzeba zamontować odpowiednio wyżej, wykorzystując do tego celu podnośnik laboratoryjny.
W chwili rozpoczęcia ogrzewania kolby destylacyjnej termometr wskazuje temperaturę
otoczenia. Po pewnym czasie rozpoczyna się wrzenie cieczy i wtedy można zaobserwować
„pierścień” skraplającej się pary, podnoszący się w kolbie i nasadce destylacyjnej.
Temperatura zaczyna powoli wzrastać i mogą pojawić się pierwsze krople destylatu. Kolbę
trzeba ogrzewać w taki sposób, aby szybkość destylacji wynosiła 1–2 krople na sekundę.
Jeżeli mimo wrzenia cieczy w kolbie destylacyjnej nie obserwuje się tworzenia destylatu, to
prawdopodobnie pary cieczy są zbyt intensywnie wychładzane w nasadce destylacyjnej.
Wówczas należy owinąć nasadkę tkaniną izolującą przed stratami ciepła (np. tkaniną z
włókna szklanego). W pewnym momencie temperatura stabilizuje się w granicach 1–2 °C, co
świadczy o tym, że rozpoczęła się destylacja czystej frakcji. Przy destylacji cieczy o znanej
temperaturze wrzenia wszystko to, co zostało przedestylowane w temperaturze niższej
(przedgon) oraz w wyższej (pogon) zazwyczaj się odrzuca. Każda frakcja powstała w czasie
destylacji musi być zbierana do osobnego odbieralnika. Jeżeli ciecz destylowana nie jest
znana, to wszystkie frakcje trzeba zachować, a następnie poddać je analizie. Po zakończeniu
destylacji pierwszej czystej frakcji należy zwrócić uwagę na zawartość kolby destylacyjnej.
Jeżeli pozostało w niej jeszcze dużo cieczy, to zapewne wkrótce rozpocznie się destylacja
kolejnej frakcji. Przy dużej różnicy temperatur wrzenia między poszczególnymi frakcjami
zdarza się, że temperatura wskazywana przez termometr po zakończeniu destylacji pierwszej
frakcji obniża się zamiast rosnąć. Oznacza to, że w tym momencie do termometru nie
dochodzą jeszcze pary kolejnej frakcji, gdyż zbyt niska jest temperatura kolby destylacyjnej.
Dopiero wtedy, gdy zawartość kolby ogrzeje się do odpowiedniej temperatury, rozpocznie się
destylacja kolejnej frakcji. Destylacji nigdy nie wolno prowadzić „do sucha”. W kolbie
destylacyjnej zawsze musi pozostać pewna ilość cieczy. W przeciwnym razie może nastąpić
niekontrolowany wzrost temperatury kolby, jej pęknięcie, a w pewnych przypadkach
eksplozja rozgrzanych zanieczyszczeń. Po zakończeniu destylacji i ochłodzeniu kolby
destylacyjnej należy ją starannie wymyć. Pozostałości po destylacji w wyniku działania tlenu
i światła mogą ulegać polimeryzacji i później są bardzo trudne do usunięcia.
Zazwyczaj destylację prostą stosuje się do oczyszczania cieczy o temperaturze wrzenia
niższej od 200 °C, jakkolwiek wiele z nich destyluje bez rozkładu nawet w temperaturze 250
°C. Jeżeli substancja ciekła nie jest dostatecznie trwała termicznie lub ma jeszcze wyższą
temperaturę wrzenia, to do jej oczyszczania stosuje się destylację pod zmniejszonym
ciśnieniem.
Należy pamiętać, że temperatura wrzenia jednorodnej mieszaniny dwóch cieczy jest
zazwyczaj temperaturą pośrednią między temperaturami wrzenia czystych cieczy i zależy od
zawartości poszczególnych składników w mieszaninie. Para tworząca się podczas wrzenia

takiej mieszaniny również zawiera oba składniki, jednak zawartość w parze składnika
bardziej lotnego (o niższej temperaturze wrzenia) jest większa niż w ogrzewanej cieczy. W
żadnym jednak przypadku nie wolno zakładać, że w wyniku ogrzania do wrzenia mieszaniny
dwóch cieczy uda się odparować (oddestylować) wyłącznie składnik bardziej lotny w czystej
postaci. Jeżeli celem destylacji prostej ma być rozdzielenie dwóch cieczy, to dobrych
rezultatów można oczekiwać, gdy ich temperatury wrzenia różnią się o co najmniej 70–80 °C.
Jeżeli różnica temperatur wrzenia jest mniejsza, to praktycznie nie obserwuje się stałej
temperatury destylacji. Następuje więc ciągła zmiana składu destylatu i żadna z zebranych
frakcji nie jest czystą cieczą. W celu rozdzielenia cieczy o mniejszej różnicy temperatur
wrzenia stosuje się destylację frakcyjną (rektyfikację).
Istnieją mieszaniny substancji znacznie różniących się temperaturami wrzenia, których nie
można oddzielić od siebie mimo zastosowania najbardziej doskonałych technik
destylacyjnych. Są to mieszaniny azeotropowe, które destylują bez zmiany składu. Inaczej
mówiąc, faza ciekła i gazowa takiej mieszaniny mają taki sam skład. Z wieloma
mieszaninami azeotropowymi można spotkać się w codziennej praktyce laboratoryjnej. Do
najbardziej znanych należy mieszanina wody i etanolu znana pod nazwą spirytus lub alkohol
rektyfikowany (tw. wody = 100 °C, tw. etanolu = 78,3 °C, skład mieszaniny: 4,4% wody i
95,6% etanolu, tw. mieszaniny = 78,15 °C). Jest to mieszanina azeotropowa z minimum
temperatury wrzenia, czyli azeotrop dodatni. Innym przykładem jest mieszanina azeotropowa
wody i chlorowodoru, czyli kwas chlorowodorowy (tw. wody = 100 °C, tw. chlorowodoru = –
80 °C, skład mieszaniny 79,8% wody i 20,2% chlorowodoru, tw. mieszaniny = 108,6 °C). Jest
to mieszanina azeotropowa z maksimum temperatury wrzenia, czyli azeotrop ujemny).
Należy zwrócić uwagę na fakt, że dostępny w handlu stężony kwas chlorowodorowy o
stężeniu 36% produkowany jest przez dodatkowe nasycanie mieszaniny azeotropowej
gazowym chlorowodorem.
Zjawisko azeotropii jest bardzo rozpowszechnione; wiele związków organicznych tworzy
takie mieszaniny z wodą i jeżeli nie są one dostatecznie odwodnione przed destylacją, to
uzyskane temperatury wrzenia różnią się znacznie od oczekiwanych. Przykładem może być
otrzymywanie octanu benzylu — estru o przyjemnym kwiatowym zapachu, stosowanego w
przemyśle spożywczym i perfumeryjnym. Ostatnim etapem bardzo wydajnej syntezy jest
ekstrakcja i następnie suszenie fazy organicznej bezwodnym siarczanem(VI) magnezu.
Czysty ester powinien destylować w temperaturze 215 °C, zdarza się jednak, że temperatura
destylacji nie przekracza 100 °C, a mimo to w destylacie obecny jest oczekiwany ester.
Powodem tego zjawiska jest niedokładne osuszenie fazy organicznej, a octan benzylu tworzy
z wodą mieszaninę azeotropową o temperaturze wrzenia 99,6 °C, zawierającą aż 90,8%
wody.
Destylacja frakcyjna
Destylacja frakcyjna (rektyfikacja) stosowana jest wówczas, gdy istnieje konieczność
rozdzielenia substancji niewiele różniących się temperaturą wrzenia (mniej niż 70–80 °C).
Jeżeli natomiast różnica temperatur wrzenia wynosi mniej niż 10 °C, to nawet podczas
destylacji frakcyjnej nie należy oczekiwać dobrego rozdzielenia frakcji.
Zasadnicze elementy zestawu do destylacji frakcyjnej są takie same, jak w przypadku zestawu
do destylacji prostej. Dodatkowym elementem jest kolumna destylacyjna, pełniąca w tym
zestawie funkcję deflegmatora. Typowy zestaw do destylacji frakcyjnej przedstawiono na rys.
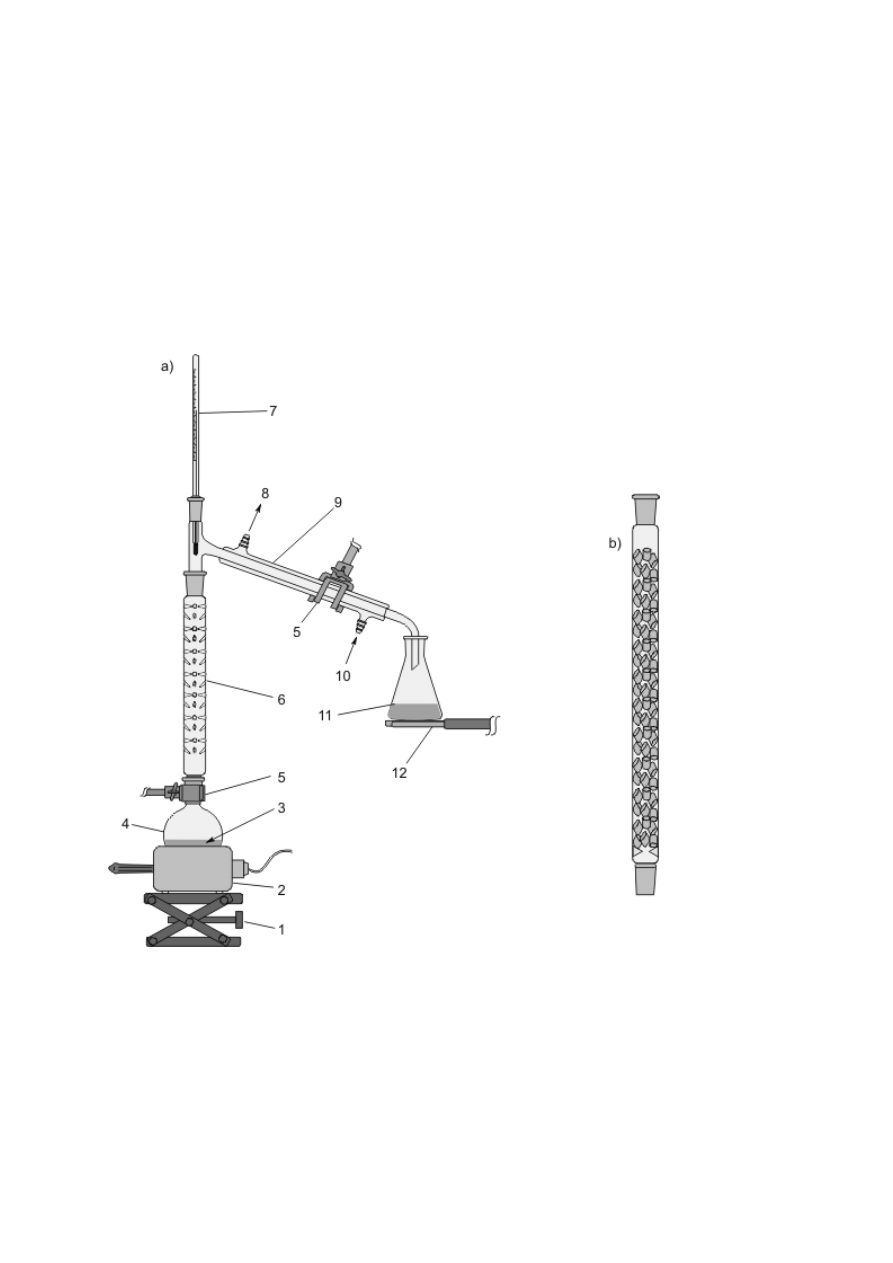
27a. Deflegmator montuje się pomiędzy kolbą i nasadką destylacyjną. Jest to długa pionowa
rura o znacznie rozwiniętej powierzchni wewnętrznej. Efekt ten uzyskuje się w różny sposób,
na przykład przez wypełnienie kolumny kształtkami szklanymi, ceramicznymi lub
metalowymi (kolumna Hempla na rys. 27b). Najprostszą i prawdopodobnie najczęściej
stosowaną jest kolumna Vigreux, czyli szklana rura z wgięciami skierowanymi do wewnątrz,
które zbierają ciecz ze ścian kolumny i skierowują ją do środka (element 6 na rys. 27a). W
czasie destylacji pary cieczy podążają ku górze kolumny, ulegając w tym czasie częściowemu
skropleniu. Utworzony kondensat spływa w dół kolumny i spotyka się z dążącymi ku górze
gorącymi parami. Następuje wymiana cieplna pomiędzy dwiema fazami, a pary wzbogacają
się w składnik bardziej lotny. W kolumnie zachodzi więc wielokrotny proces parowania i
skraplania. Po ustaleniu się równowagi następuje destylacja składnika najbardziej lotnego.
Rysunek 27. a) Zestaw do destylacji frakcyjnej pod ciśnieniem atmosferycznym: 1 —
podnośnik, 2 — płaszcz grzejny, 3 — kamyczki wrzenne, 4 — kolba okrągłodenna, 5 — łapy
mocujące zestaw do statywu, 6 — kolumna destylacyjna (deflegmator), 7 — termometr, 8 —
odprowadzenie wody wężem do zlewu, 9 — chłodnica destylacyjna, 10 — doprowadzenie
wody wężem z kranu, 11 — odbieralnik z destylatem, 12 — podstawka pod odbieralnik
zamocowana do statywu; b) kolumna Hempla z wypełnieniem ze szklanych pierścieni
Raschiga
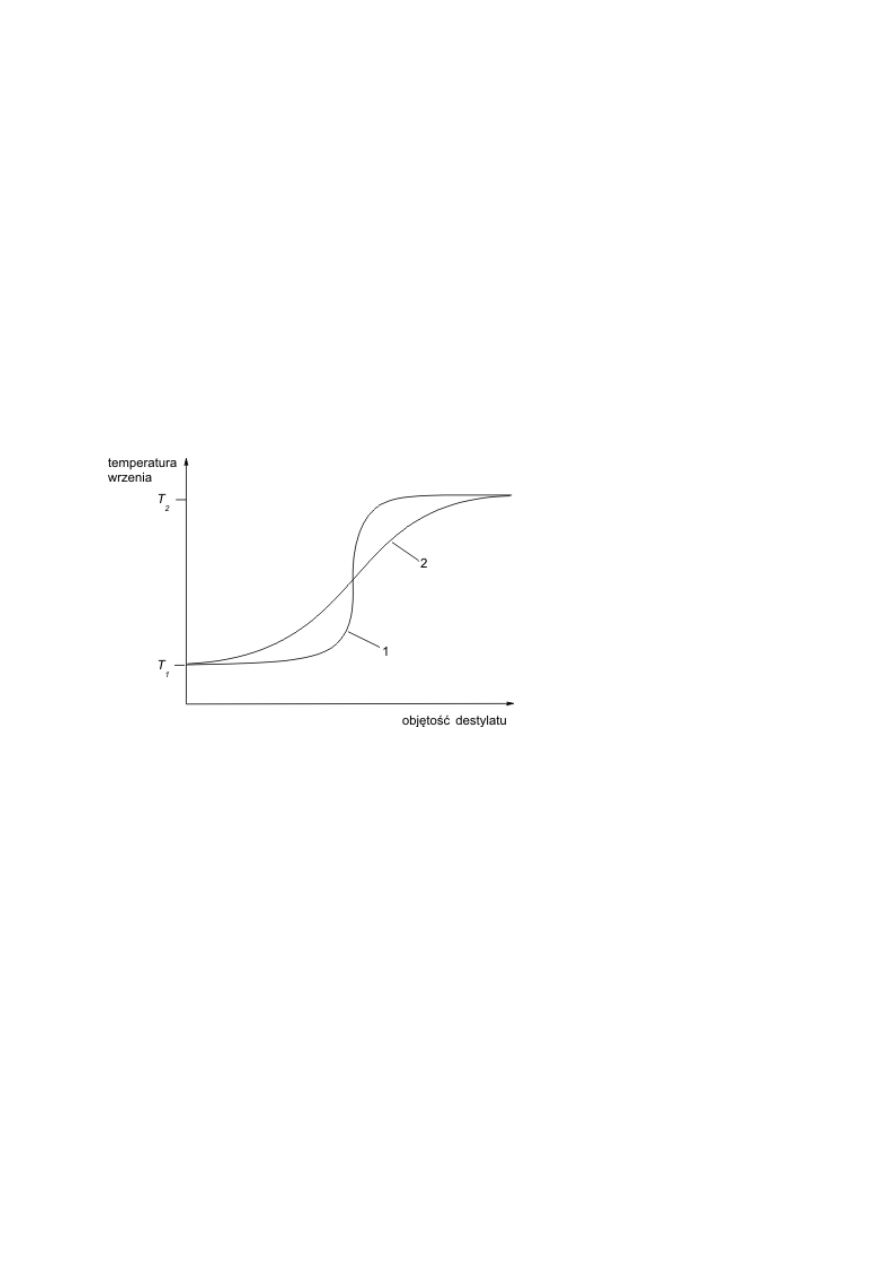
Po zakończeniu destylacji pierwszej frakcji zbiera się nieco frakcji mieszanej (jej ilość zależy
od różnicy temperatur wrzenia i sprawności kolumny), a następnie destyluje kolejna czysta
frakcja. Destylację należy prowadzić z szybkością jedna kropla destylatu na 2–3 sekundy,
zwracając uwagę, aby wewnątrz kolumny nie tworzyły się „korki” cieczy, podtrzymywane
przez wędrujące ku górze pary, czyli aby nie nastąpiło zalanie kolumny. Jeżeli destylacja jest
prowadzona zbyt szybko, to w kolumnie zachodzi bardzo niewiele aktów skraplania par i
efektywność pracy kolumny znacznie spada. W przypadku cieczy o wysokiej temperaturze
wrzenia wychładzanie par w kolumnie może być zbyt intensywne i ulegną one zupełnemu
skropleniu przed dojściem do nasadki. Wówczas należy owinąć kolumnę tkaniną izolującą
przed utratą ciepła lub, w skrajnych przypadkach, taśmą ogrzewaną elektrycznie.
Wykres na rysunku 28 przedstawia zależność temperatury wrzenia od objętości destylatu
podczas destylacji mieszaniny dwóch cieczy różniących się temperaturą wrzenia o około
30 °C. Krzywa 2 przedstawia przebieg destylacji prostej, a krzywa 1 — przebieg destylacji
frakcyjnej. Jak wynika z wykresu, w przypadku destylacji prostej praktycznie nie udało się
uzyskać czystej frakcji.
Rysunek 28. Zależność temperatury wrzenia od objętości destylatu podczas destylacji
mieszaniny: 1 — dla destylacji frakcyjnej, 2 — dla destylacji prostej, T
1
— temperatura
wrzenia pierwszego składnika, T
2
— temperatura wrzenia drugiego składnika
Destylacja pod zmniejszonym ciśnieniem
(próżniowa)
Destylacja pod zmniejszonym ciśnieniem służy do oczyszczania lub rozdzielania cieczy
o bardzo wysokich temperaturach wrzenia (ponad 200 °C) lub takich, które ulegają
znacznemu rozkładowi przed osiągnięciem temperatury wrzenia pod ciśnieniem
atmosferycznym. Typowy zestaw do destylacji pod zmniejszonym ciśnieniem przedstawiono
na rys. 29.
Zestaw taki różni się znacznie od zestawu do destylacji prostej. Stosuje się innego rodzaju
nasadkę destylacyjną — jest to nasadka Claisena (rys. 7e). Niekiedy nasadka wraz z chłodnicą
i przedłużaczem stanowią integralną całość noszącą nazwę „chłodnica Claisena”. W jednej
szyi nasadki, znajdującej się bezpośrednio nad kolbą destylacyjną, umieszcza się kapilarę.
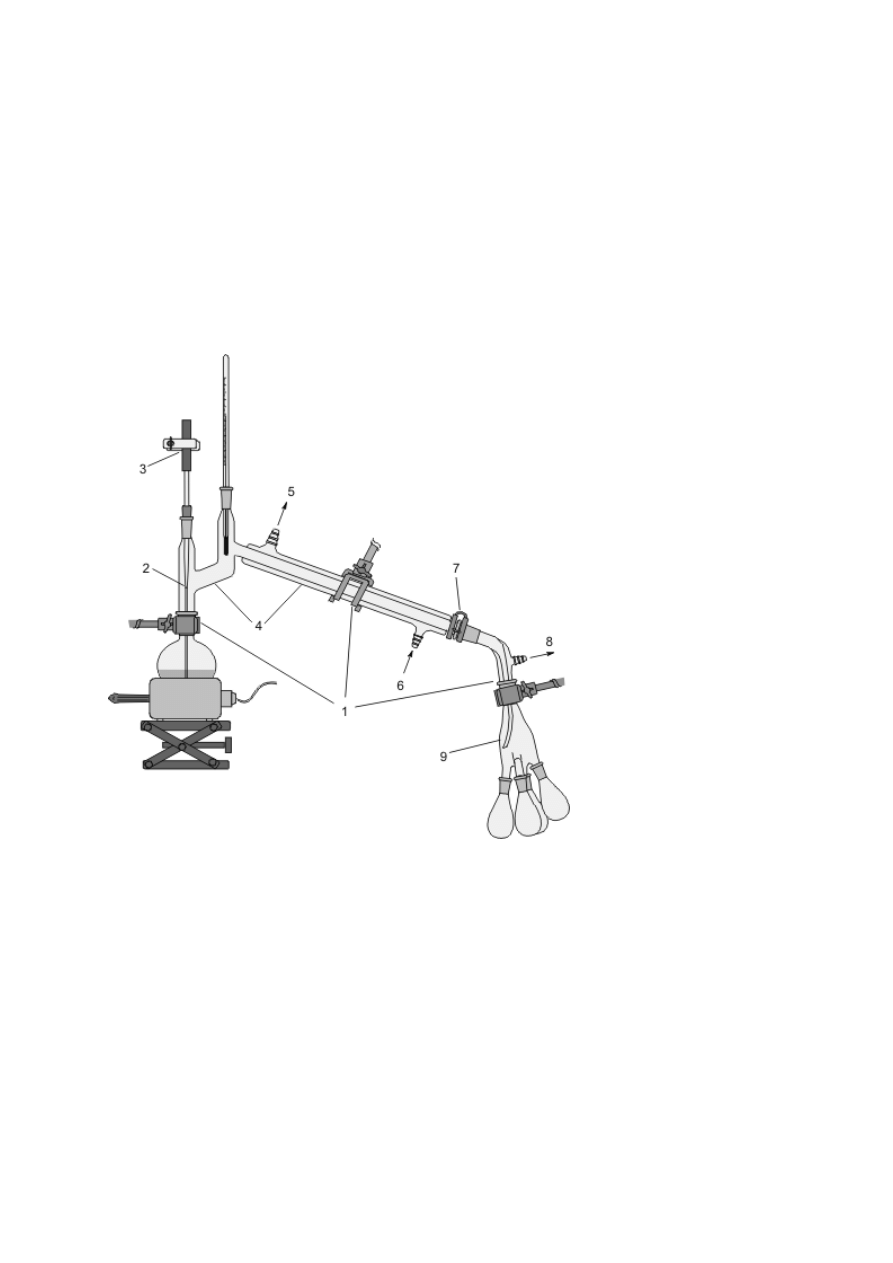
Jest to bardzo cienka rurka, której dolny koniec sięga niemal dna kolby destylacyjnej, a na
górny założony jest kawałek węża gumowego ze ściskaczem śrubowym. Zadaniem kapilary
jest doprowadzanie podczas destylacji strumienia pęcherzyków powietrza, zapobiegających
przegrzewaniu się cieczy. W drugiej szyi nasadki Claisena umieszcza się termometr.
Chłodnicę łączy się przez przedłużacz zaopatrzony w boczny tubus z odbieralnikiem, którym
przy pracy pod zmniejszonym ciśnieniem musi być kolba kulista. Znacznie wygodniejsze jest
stosowanie odbieralnika pozwalającego na zbieranie kolejno kilku różnych frakcji bez
wyłączania podciśnienia. Przez obrót wokół połączenia szlifowego można skierować strumień
destylatu do różnych naczyń (zazwyczaj małych kolbek okrągłodennych). Ten rodzaj
odbieralnika jest pospolicie zwany „krówką”, choć w żargonie chemicznym funkcjonują też
inne nazwy.
Rysunek 29. Zestaw do destylacji pod zmniejszonym ciśnieniem: 1 — łapy mocujące zestaw
do statywu, 2 — kapilara, 3 — zaciskacz regulujący przepływ strumienia powietrza, 4 —
chłodnica zespolona z nasadką Claisena, 5 — odprowadzenie wody wężem do zlewu, 6 —
doprowadzenie wody wężem z kranu, 7 — spinka do szlifów, 8 — wyjście do podłączenia
pompy próżniowej (przez wymrażacz par), 9 — system odbieralników (krówka)
Niewielką ilość cieczy (2–20 cm
3
) można bardzo wygodnie przedestylować pod
zmniejszonym ciśnieniem, posługując się kompaktowym zestawem przedstawionym na rys.
30. Główne różnice polegają na zastąpieniu klasycznej chłodnicy palcem chłodzącym oraz
wyeliminowaniu kłopotliwej w użyciu kapilary. Jednak aby nie następowało przegrzewanie
się cieczy, zawartość kolby musi być bardzo silnie mieszana. W zestawach w skali półmikro
stosuje się jednoczęściowe odbieralniki, działające podobnie jak omówione wcześniej
„krówki”. Poszczególne frakcje wydobywa się z osobnych nóżek za pomocą pipetki Pasteura.
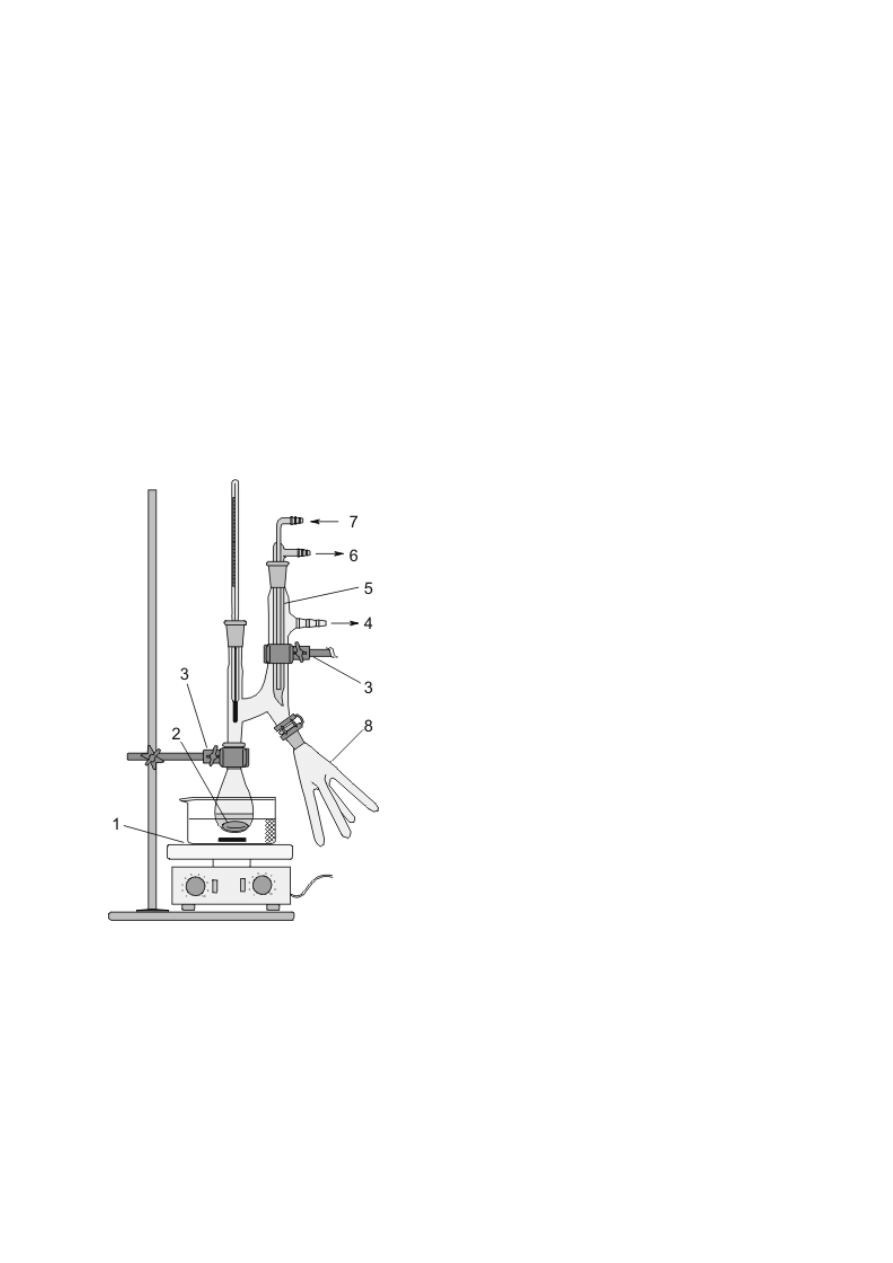
Obniżenie ciśnienia w układzie destylacyjnym uzyskuje się przy użyciu różnego rodzaju
pomp próżniowych. Najprostsza jest pompka wodna, za pomocą której można uzyskać
ciśnienie do 10 hPa. Należy zwrócić uwagę, że sprawność pompki wodnej zależy od
temperatury wody wodociągowej, a tym samym od pory roku. W lecie, gdy woda jest
cieplejsza, można uzyskać ciśnienie najwyżej 30 hPa. Ciśnienie 10 hPa, niezależnie od pory
roku, można osiągnąć za pomocą pompy membranowej. Jest ona ponadto znacznie bardziej
oszczędna — nie zużywa dużych ilości uzdatnionej do spożycia wody, a tylko nieco energii
elektrycznej. Do destylacji przy jeszcze niższym ciśnieniu (do około 0,1 hPa) stosuje się
rotacyjne pompy olejowe. Pompy próżniowe łączy się z zestawem do destylacji za pomocą
grubościennych węży gumowych Należy jednak pamiętać, że niedopuszczalne jest
bezpośrednie połączenie pompy z tubusem bocznym przedłużacza. Stosując pompkę wodną
lub pompę membranową, trzeba włączyć w zestaw kolbę bezpieczeństwa lub przynajmniej
pustą płuczkę wykonaną ze szkła o odpowiedniej grubości. Jest to zabezpieczenie przed
przerzuceniem wody z pompki wodnej do zestawu destylacyjnego lub na odwrót — przed
przerzuceniem destylatu do pompy. Jeżeli w zestawie nie ma specjalnej kolby bezpieczeństwa
z zaworem, to musi być zainstalowany między płuczką a pompą kran trójdrożny. Pozwala on
na łatwą regulację ciśnienia, a w razie awarii na szybkie zapowietrzenie układu.
Rysunek 30. Kompaktowy zestaw do destylacji pod zmniejszonym ciśnieniem: 1 —
mieszadło magnetyczne z łaźnią olejową, 2 — mieszalnik w kolbie destylacyjnej, 3 — łapy
mocujące zestaw do statywu, 4 — wyjście do podłączenia pompy próżniowej (przez
wymrażacz par), 5 — chłodnica palcowa (palec chłodzący), 6 — odprowadzenie wody
wężem do zlewu, 7 — doprowadzenie wody wężem z kranu, 8 — jednoczęściowy odbieralnik
do rozdzielania czterech frakcji
Niezbędną częścią zestawu do destylacji próżniowej jest manometr, który pozwala na pomiar
ciśnienia wewnątrz aparatury. Manometr powinien być połączony za pomocą trójnika
z kranem lub kranu trójdrożnego z wężem łączącym zestaw z pompą. Najbardziej popularne

są nadal manometry rtęciowe, których użycie wymaga ostrożności, gdyż zawierają znaczną
ilość toksycznej rtęci. Coraz częściej stosowane są znacznie bezpieczniejsze manometry
mechaniczne lub elektroniczne.
Jeżeli źródłem obniżonego ciśnienia ma być pompa olejowa, to dodatkowo trzeba zastosować
urządzenie pochłaniające nieskroplone pary destylatu. W przeciwnym razie będą one
dostawać się do pompy i rozcieńczać olej lub powodować jego chemiczną degradację. Traci
on wówczas swoje właściwości — pompa osiąga gorsze parametry i ulega korozji.
Urządzeniem do pochłaniania par może być odpowiedni zestaw pojemników z absorbentami
lub, w tańszej wersji, specjalna płuczka zanurzona w termosie z ciekłym azotem (tw. –196
°C). Musi być ona przyłączona do węża pomiędzy zestawem do destylacji a manometrem.
Ciekły azot powinien być nalewany do suchego termosu bezpośrednio przed
rozpoczęciem destylacji, a wkrótce po jej zakończeniu wylany na podłogę. Chodzi o to, że
w trakcie przechowywania ciekłego azotu w otwartym naczyniu skrapla się w nim tlen (tw. –
183 °C), który w kontakcie z substancjami organicznymi może powodować gwałtowną
eksplozję. Także długotrwałe wymrażanie w termosie z ciekłym azotem pary wodnej z
powietrza nie jest korzystne, bo wytworzona znaczna ilość lodu może zniszczyć delikatną
ściankę naczynia. Dobre wyniki daje zastosowanie płuczki chłodzonej mieszaniną suchego
lodu (czyli zestalonego ditlenku węgla) i acetonu (temperatura –78 °C).
Doboru właściwego rodzaju pompy próżniowej dokonuje się oczywiście na podstawie
przewidywanej temperatury wrzenia oczyszczanej cieczy. Jeżeli znana jest temperatura
wrzenia cieczy pod ciśnieniem normalnym lub zmniejszonym, to można dość łatwo
przewidzieć z pewnym przybliżeniem jej temperaturę wrzenia pod dowolnym ciśnieniem.
Służy do tego reguła empiryczna mówiąca, że obniżenie ciśnienia o połowę powoduje
obniżenie temperatury wrzenia o ok. 15 °C. Lepszym sposobem oszacowania temperatury
wrzenia znanej substancji pod określonym ciśnieniem jest skorzystanie ze specjalnego
nomogramu (rozdz. IX,
). W przypadku oczyszczania substancji nieznanej można
oszacować jej temperaturę wrzenia przez analogię do substancji znanych o podobnej budowie
cząsteczki.
Przed przystąpieniem do montażu zestawu należy dokładnie obejrzeć wszystkie części
szklane, a w przypadku jakichkolwiek uszkodzeń wymienić je na nowe. Nawet drobne
pęknięcie szkła może po włączeniu próżni spowodować gwałtowną implozję. Wszystkie
połączenia szlifowe powinny być posmarowane smarem do aparatury próżniowej.
Następnie krok po kroku montuje się aparaturę. Przy braku doświadczenia w destylacji
próżniowej praca powinna być kontrolowana przez osobę prowadzącą zajęcia. Wskazane
jest, aby osoby przebywające w pobliżu zestawu do destylacji próżniowej były zaopatrzone
oprócz okularów ochronnych także w maskę osłaniającą całą twarz. Zestaw powinien być
zmontowany za specjalną szybą ochronną. Po zmontowaniu zestawu uruchamia się pompę i
sprawdza szczelność wszystkich połączeń. Jeżeli zestaw jest sprawny, to należy wlać
ostrożnie przez lejek oczyszczaną ciecz do kolby destylacyjnej. Wszystkie substancje lotne
muszą być wcześniej dokładnie usunięte metodą destylacji prostej lub przy użyciu wyparki
rotacyjnej. Kolbę napełnia się najwyżej do połowy jej objętości, pamiętając, że nie wolno
ogrzewać substancji w kolbie destylacyjnej przed obniżeniem ciśnienia. Teraz już można
przystąpić do destylacji. Najpierw (przy zakręconym ściskaczu kapilary) uruchamia się
pompę i za pomocą kranów ustawia pożądane ciśnienie. Ściskacz przy kapilarze otwiera się
na tyle, aby przez ciecz przechodził łagodny strumień pęcherzyków powietrza. Płuczkę
umieszcza się w cieczy chłodzącej i rozpoczyna ogrzewanie kolby destylacyjnej. W czasie
destylacji należy odczytywać temperaturę i ciśnienie oraz pamiętać o skierowaniu kolejnych

frakcji do oddzielnych odbieralników. Zasady destylacji są takie same jak w przypadku
destylacji prostej. Po zakończeniu zbierania ostatniej frakcji lub zaobserwowaniu objawów
rozkładu cieczy (ciemnienie, dym itp.) należy wyłączyć ogrzewanie i odczekać, aż kolba
destylacyjna ostygnie. Wówczas należy wyjąć płuczkę z cieczy chłodzącej (jest to bardzo
istotne przy chłodzeniu ciekłym azotem), delikatnie otworzyć ściskacz przy kapilarze,
wprowadzić powietrze do układu przez otwarcie kranu i wyłączyć pompę. W pierwszej
kolejności odłącza się od zestawu kolbki z destylatem, a następnie demontuje wszystkie
części zestawu, pamiętając o starannym ich umyciu i usunięcie smaru ze szlifów.
Odmianą klasycznej wersji destylacji pod zmniejszonym ciśnieniem jest destylacja w
wyparce obrotowej (rotacyjnej). Służy ona najczęściej do szybkiego usuwania
rozpuszczalnika z roztworów związków organicznych (np. po ekstrakcji rozpuszczalnikiem
organicznym z fazy wodnej). Odparowanie przeprowadza się zazwyczaj pod zmniejszonym
ciśnieniem, uzyskanym za pomocą pompki wodnej lub pompy membranowej. Należy
pamiętać, aby pomiędzy pompą a wyparką była zainstalowana pusta płuczka zabezpieczająca.
Ten rodzaj destylacji nadaje się bardzo dobrze do usuwania cieczy o niezbyt wysokiej
temperaturze wrzenia. Źródłem ciepła jest łaźnia wodna z regulacją temperatury. W czasie
odparowywania cieczy kolba cały czas obraca się wzdłuż swojej osi, co zapobiega
przegrzewaniu się zawartości kolby (nie są konieczne kamyczki wrzenne) oraz zwiększa
powierzchnię parowania. Pary cieczy są ochładzane przez bardzo sprawną chłodnicę spiralną.
Mimo to w przypadku usuwania rozpuszczalników o bardzo niskiej temperaturze wrzenia
(np. eteru dietylowego lub chlorku metylenu) nie zaleca się stosować obniżonego ciśnienia.
Chodzi o to, aby uniknąć wciągania par rozpuszczalnika do pompy. Wówczas jednak należy
pamiętać, aby kran na wyparce pozostał otwarty.
Obsługa wyparki jest bardzo prosta. Kolbę destylacyjną nakłada się na szlif szklanej rury
wyparki i dokładnie przymocowuje za pomocą specjalnej spinki. Uruchamia się silnik i wtedy
kolba zaczyna wirować z zadaną prędkością. W razie potrzeby włącza się pompę i za pomocą
kranu umieszczonego na wyparce reguluje ciśnienie wewnątrz urządzenia. Po ustaleniu
odpowiedniego ciśnienia można ostrożnie zanurzyć kolbę w łaźni wodnej. Szybkość
destylacji kontroluje się przez dobór właściwej temperatury łaźni wodnej, głębokości
zanurzenia kolby destylacyjnej w łaźni oraz stopnia obniżenia ciśnienia. Należy uważać, aby
zawartość kolby nie była przerzucana do chłodnicy i odbieralnika. Po zakończeniu destylacji
trzeba w pierwszej kolejności otworzyć delikatnie kran na wyparce w celu likwidacji
podciśnienia. Następnie można wyłączyć pompę, zatrzymać silnik wyparki, wyłączyć dopływ
wody, unieść kolbę ponad poziom wody w łaźni wodnej i zdjąć kolbę ze szlifu rury wyparki.
Z odbieralnika należy usunąć destylat. Jeżeli w czasie destylacji wyparka uległa zabrudzeniu,
to trzeba ją wymyć, na przykład przez przedestylowanie większej ilości acetonu technicznego.
Chociaż obecnie dostępne są na rynku bardzo zróżnicowane modele wyparek, zasada ich
funkcjonowania pozostaje praktycznie taka sama. Najdroższe urządzenia tego typu
wyposażone są w cyfrowe wskazania prędkości obrotów, temperatury łaźni, temperatury par
w chłodnicy, ciśnienia wewnętrznego oraz pozwalają na ich sterowanie według zadanego
programu.
Destylacja z parą wodną
Destylacja z parą wodną jest bardzo dogodną metodą wyodrębniania substancji organicznej z
mieszanin poreakcyjnych oraz z surowców naturalnych. Nadaje się do oczyszczania cieczy
(także o wysokich temperaturach wrzenia) oraz ciał stałych o niezbyt wysokich temperaturach

topnienia. Oczyszczana tą drogą substancja nie może mieszać się z wodą, nie może z
wodą reagować oraz musi być lotna z parą wodną, czyli w temperaturze bliskiej 100 °C
wykazywać znaczną prężność pary (przynajmniej 10 hPa). Całkowita prężność pary
nasyconej nad mieszaniną niejednorodną (p
całk.
) wyraża się sumą prężności nasyconych par
poszczególnych składników (p
1
do p
n
). Jest to treść prawa Daltona, które w przypadku
destylacji z parą wodną można zapisać następująco:
p
całk.
= p
woda
+ p
2
+ p
3
+ ... + p
n
Mieszanina spełniająca warunki prawa Daltona osiągnie temperaturę wrzenia wtedy, gdy
suma prężności par składników osiągnie wartość ciśnienia zewnętrznego (atmosferycznego),
czyli temperatura wrzenia mieszaniny będzie zawsze niższa od temperatury wrzenia
najbardziej lotnego składnika. Zazwyczaj w przypadku destylacji z parą wodną najbardziej
lotnym składnikiem jest woda, a więc destylacja będzie odbywać się w temperaturze nieco
niższej niż 100 °C. Często podawanym w literaturze przykładem jest mieszanina wody i
bromobenzenu (tw. = 155 °C), która osiąga prężność pary równą ciśnieniu atmosferycznemu
(1013 hPa) w temperaturze 95,5 °C. Destylacja bromobenzenu z parą wodną zachodzi więc w
temperaturze o ok. 60 °C niższej niż destylacja czystej substancji.
Najprostszy test na sprawdzenie, czy substancja jest lotna z parą wodną, polega na
ogrzaniu do wrzenia niewielkiej ilości próbki z kilkoma cm
3
wody w zlewce przykrytej
szkiełkiem zegarkowym lub nawet w probówce. Jeśli skropliny osadzające się na szkiełku lub
w górnej części probówki są całkowicie klarowne, to substancja nie jest lotna z parą wodną.
Podstawowe elementy zestawu do destylacji z parą wodną są takie same jak w przypadku
destylacji prostej. Należy pamiętać o zastosowaniu bardzo wydajnej chłodnicy wodnej,
będącej w stanie skroplić duże ilości gorącej pary. W zestawie do destylacji z parą wodną nie
jest potrzebny termometr, bo temperatura wrzenia mieszaniny nie jest istotnym parametrem.
Zamiast termometru montuje się (za pomocą korka gumowego z dziurką) rurkę szklaną,
sięgającą niemal dna kolby destylacyjnej, przez którą będzie wprowadzana para wodna.
Opisany zestaw, pomimo że jest prosty w montażu i łatwo dostępny w laboratorium, ma
istotną wadę. Przy pionowym ustawieniu szyi kolby destylacyjnej występuje często tendencja
do przerzucania rozdzielanej mieszaniny do odbieralnika (rys. 31a). Aby temu zapobiec,
wykorzystuje się specjalną nasadkę destylacyjną, pozwalającą na ukośne zamontowanie kolby
destylacyjnej (rys. 7f). Nasadka destylacyjna musi być w tym przypadku dopasowana
wielkością do kolby destylacyjnej w ten sposób, aby rurka doprowadzająca parę sięgała
prawie dna kolby.
Źródłem pary wodnej jest kociołek wykonany z blachy miedzianej, ustawiony na trójnogu i
ogrzewany palnikiem gazowym. Kociołek napełnia się wodą (w miarę możliwości gorącą, np.
z termy elektrycznej) do ok. 3/4 objętości i zamyka korkiem gumowym z pionową rurą
szklaną zwaną rurą bezpieczeństwa. Kociołek łączy się z rurką doprowadzającą parę do kolby
destylacyjnej za pomocą węża gumowego dopiero wówczas, gdy pojawi się intensywny
strumień pary wodnej. Łączenie wykonuje się w rękawicach chroniących przed wysoką
temperaturą. Jeżeli oczyszczana substancja ma konsystencję stałą lub gęstej mazi, to do
kolby destylacyjnej należy dolać pewną ilość wody. Zaleca się, aby kolba destylacyjna była
dodatkowo ogrzewana płaszczem grzejnym. Wąż doprowadzający parę do zestawu powinien
być dołączony wówczas, gdy zawartość kolby jest dostatecznie ogrzana (blisko temperatury
wrzenia). Zapobiega to nadmiernemu skraplaniu się pary i gromadzeniu cieczy w kolbie
destylacyjnej. Szybkość destylacji należy dobrać tak, aby zawartość kolby destylacyjnej nie
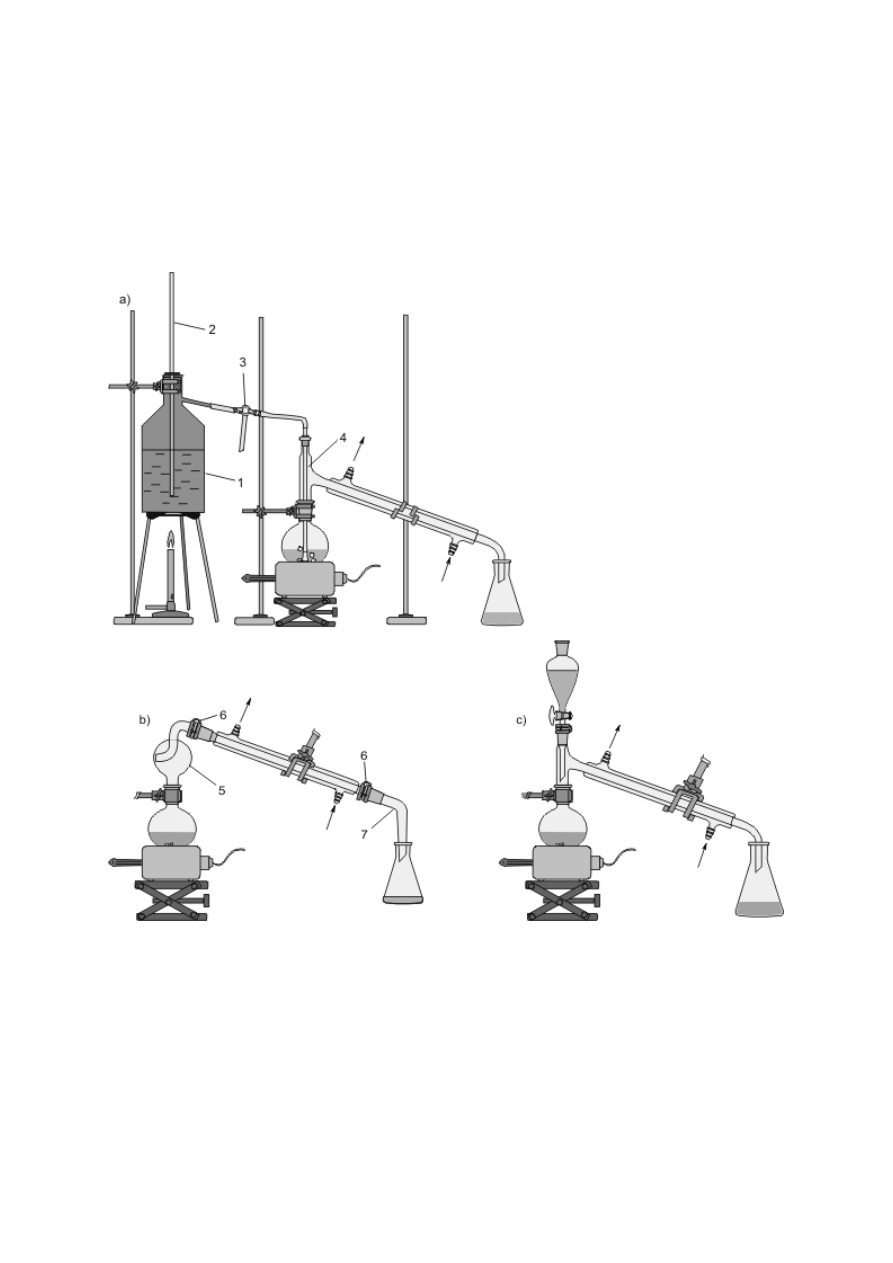
była przerzucana do chłodnicy, aby było możliwe całkowite skroplenie par w chłodnicy oraz
aby był zachowany w przybliżeniu stały poziom cieczy w kolbie destylacyjnej. W przypadku
destylacji substancji o temperaturze topnienia wyższej niż temperatura ściany chłodnicy,
często obserwuje się na niej krystalizację oczyszczanego związku. Jest to bardzo
niebezpieczne zjawisko, bo może doprowadzić do zablokowania przepływu destylatu przez
chłodnicę. Należy wówczas zakręcić dopływ wody chłodzącej i poczekać, aż substancja stopi
się i spłynie do odbieralnika. Wtedy należy ponownie włączyć dopływ wody do chłodnicy.
Rysunek 31. a) Zestaw do destylacji z parą wodną dużych ilości substancji: 1 — kociołek do
ogrzewania wody, 2 — rurka zabezpieczająca, 3 — kran trójdrożny, 4 — rurka
wprowadzająca parę wodną; b) zestaw do destylacji z parą wodną niewielkich ilości łatwo
lotnych związków z użyciem łapacza kropel: 5 — łapacz kropel, 6 — spinki do szlifów, 7 —
łącznik typu „fajka”; c) zestaw do destylacji z parą wodną z wykorzystaniem wkraplacza
Destylację prowadzi się tak długo, aż destylat stanie się klarowny. Najprościej można to
sprawdzić, zbierając 2–3 krople destylatu na szkiełko zegarkowe i obserwując ich wygląd na
ciemnym tle. Jeżeli ciecz jest zupełnie przezroczysta, to destylację można przerwać. W celu
zakończenia destylacji należy najpierw odłączyć wąż gumowy pomiędzy kociołkiem a

nasadką destylacyjną (korzystając oczywiście z rękawic ochronnych), co zapobiega
wciągnięciu zawartości kolby destylacyjnej do kociołka. Następnie, po ostygnięciu aparatury,
demontuje się ostrożnie pozostałe jej elementy. Oziębiony destylat poddaje się dalszej
obróbce w celu wydobycia związku organicznego. Jeżeli jest to substancja krystaliczna, to
można oddzielić ją od wody przez sączenie pod zmniejszonym ciśnieniem na lejku Büchnera.
W przypadku cieczy destylat poddaje się ekstrakcji odpowiednim rozpuszczalnikiem
organicznym.
Inny sposób postępowania, stosowany przy destylacji z parą wodną, polega na rezygnacji z
doprowadzania pary wodnej z zewnętrznego źródła. Przy tym rozwiązaniu należy umieścić
pewną ilość wody w kolbie destylacyjnej i ogrzewać ją płaszczem grzejnym. Stosowana jest
wówczas specjalna nasadka destylacyjna o kulistym kształcie, zwana „łapaczem kropel” (rys.
31b). Jest to metoda zalecana przy destylacji niewielkich ilości substancji o stosunkowo dużej
lotności. W innej wersji destylację taką można przeprowadzić w aparaturze do destylacji
prostej, umieszczając w niej zamiast termometru wkraplacz z wodą (rys. 31c). Poziom wody
w kolbie można zaznaczyć pisakiem, i prowadząc destylację z parą wodną, wkraplać wodę z
taką szybkością, aby poziom cieczy w kolbie pozostał stały.
V.4. Ekstrakcja
Ekstrakcja (z łaciny: extraho — wyciągam) jest to metoda wyodrębniania z mieszaniny ciał
stałych lub cieczy jakiegoś składnika za pomocą rozpuszczalnika tak dobranego, aby
rozpuszczał przede wszystkim żądany związek. Chemicy często stosują tę metodę do
otrzymywania związków naturalnych z materiału roślinnego (liści, kory itp.). Wszyscy
korzystamy z tej metody, na przykład, przy parzeniu kawy.
W syntezie organicznej produkt reakcji otrzymywany jest często wraz z innymi związkami w
postaci roztworu lub zawiesiny w wodzie. Podczas wytrząsania takiej mieszaniny
z niemieszającym się z wodą rozpuszczalnikiem produkt reakcji ulega ekstrakcji i może być
następnie odzyskany przez odparowanie rozpuszczalnika. Ekstrakcja związku z jednej fazy
ciekłej do drugiej jest procesem ustalania się równowagi zależnej od rozpuszczalności
związku w obu rozpuszczalnikach. Stosunek stężenia w jednym rozpuszczalniku do stężenia
w drugim nosi nazwę stałej podziału i jest wielkością stałą w danej temperaturze,
charakterystyczną dla danej substancji i określonej pary rozpuszczalników. Prawo to zwane
prawem Nernsta wyraża się następującym wzorem:
w którym c
A
i c
B
stanowią stężenia substancji w warstwach A i B, a K — stałą podziału.
Można przyjąć, że w przybliżeniu stała podziału jest równa stosunkowi rozpuszczalności
danej substancji w obu rozpuszczalnikach. Związki organiczne są zwykle lepiej rozpuszczalne
w rozpuszczalnikach organicznych niż w wodzie i dlatego mogą być ekstrahowane z
roztworów wodnych. Zgodnie z prawem Nernsta dużo bardziej korzystne jest
przeprowadzenie wielokrotnej ekstrakcji mniejszymi porcjami rozpuszczalnika niż jeden raz
dużą porcją. Jeśli do roztworu wodnego doda się elektrolitu, np. chlorku sodu, to
rozpuszczalność substancji organicznej maleje, inaczej mówiąc, substancja ulega wysoleniu.
Czynnik ten pomaga wyekstrahować związek organiczny.

Do ekstrakcji i rozdzielania warstw niemieszających się ze sobą cieczy używa się
rozdzielaczy gruszkowatych (rys. 6a, b, d). Rozdzielacz umieszcza się na dogodnej wysokości
w kółku na statywie. Przed użyciem zawsze należy sprawdzić, czy kran obraca się swobodnie.
Wszystkie szlifowane powierzchnie smaruje się bardzo cienką warstwą specjalnego smaru.
Rozdzielacza nie można napełniać więcej niż do ok. 3/4 wysokości. Należy sprawdzić, czy
dolny kran jest zamknięty, a następnie wlać, najlepiej przez lejek, roztwór wodny i pierwszą
część rozpuszczalnika. Podczas wytrząsania rozdzielacz trzyma się kranem do góry, przy
czym jedna dłoń zabezpiecza kran przed otwarciem lub wypadnięciem, a cała druga (a nie
tylko jeden palec) dociska korek. Od czasu do czasu, trzymając rozdzielacz nadal w tej
pozycji, otwiera się kran w celu wyrównania ciśnienia i usunięcia powietrza. Po krótkim
czasie ostrożnego wytrząsania i kilkakrotnym otwarciu kranu należy wytrząsać energicznie
przez kilka minut. Następnie rozdzielacz należy umieścić ponownie w kółku i pozostawić, aż
warstwy dokładnie się rozdzielą. Wówczas wyjmuje się korek i, otwierając kranik, wylewa
dolną warstwę do kolby stożkowej.
Niekiedy na granicy faz tworzy się trudna do rozdzielenia emulsja. Jest to spowodowane
podobną gęstością obu roztworów lub na przykład małym napięciem powierzchniowym
cieczy (zdarza się to w przypadku ekstrakcji roztworów alkalicznych). Emulsji tej można się
pozbyć przez:
zwiększenie gęstości warstwy wodnej, co można uzyskać przez dodanie soli kuchennej i
ponowne wytrząśnięcie,
mechaniczne rozbicie emulsji szklanym pręcikiem,
lekki ruch wirowy rozdzielacza,
pozostawienie roztworów na dłuższy czas w rozdzielaczu.
Do ekstrakcji roztworów wodnych używa się rozpuszczalników lżejszych od wody (czyli o
gęstości mniejszej niż 1 g/cm
3
, np. eteru dietylowego) lub cięższych od wody (czyli o gęstości
większej niż 1 g/cm
3
, np. chloroformu). W pierwszym przypadku po spuszczeniu warstwy
dolnej warstwę organiczną trzeba również wylać do osobnej kolby stożkowej. Następnie
warstwę wodną przenosi się ponownie do rozdzielacza i ekstrahuje nową porcją
rozpuszczalnika. W przypadku stosowania rozpuszczalnika cięższego od wody roztwór
wodny pozostaje w rozdzielaczu i może być wytrząsany z kolejnymi porcjami
rozpuszczalnika. W każdym przypadku należy się upewnić, czy warstwa wodna znajduje się
na górze, czy na dole. W tym celu należy zaznaczyć na rozdzielaczu granicę faz (np. pisakiem
do szkła), a następnie dodać nieco wody. Zwiększy się wówczas oczywiście objętość warstwy
wodnej.
Po ekstrakcji roztwór organiczny jest nasycony wodą i należy go osuszyć (podrozdz. III.6).
Roztwór pozostawia się nad środkiem suszącym ok. 15–20 min, mieszając od czasu do czasu.
Następnie odsącza się środek suszący przez fałdowany sączek i przemywa małą ilością
rozpuszczalnika (podrozdz. III.5). Z kolei usuwa się rozpuszczalnik, stosując wyparkę
obrotową (podrozdz. V.3), a pozostałość poddaje destylacji (podrozdz. V.3) lub krystalizacji
(podrozdz. V.1).
Opisany proces dotyczył pojedynczej, choć powtarzanej wielokrotnie, ekstrakcji typu
ciecz/ciecz. Proces ten można prowadzić również w sposób ciągły, a służą temu aparaty
zwane perkolatorami. Ich budowę i działanie opisano w podręcznikach preparatyki
organicznej.
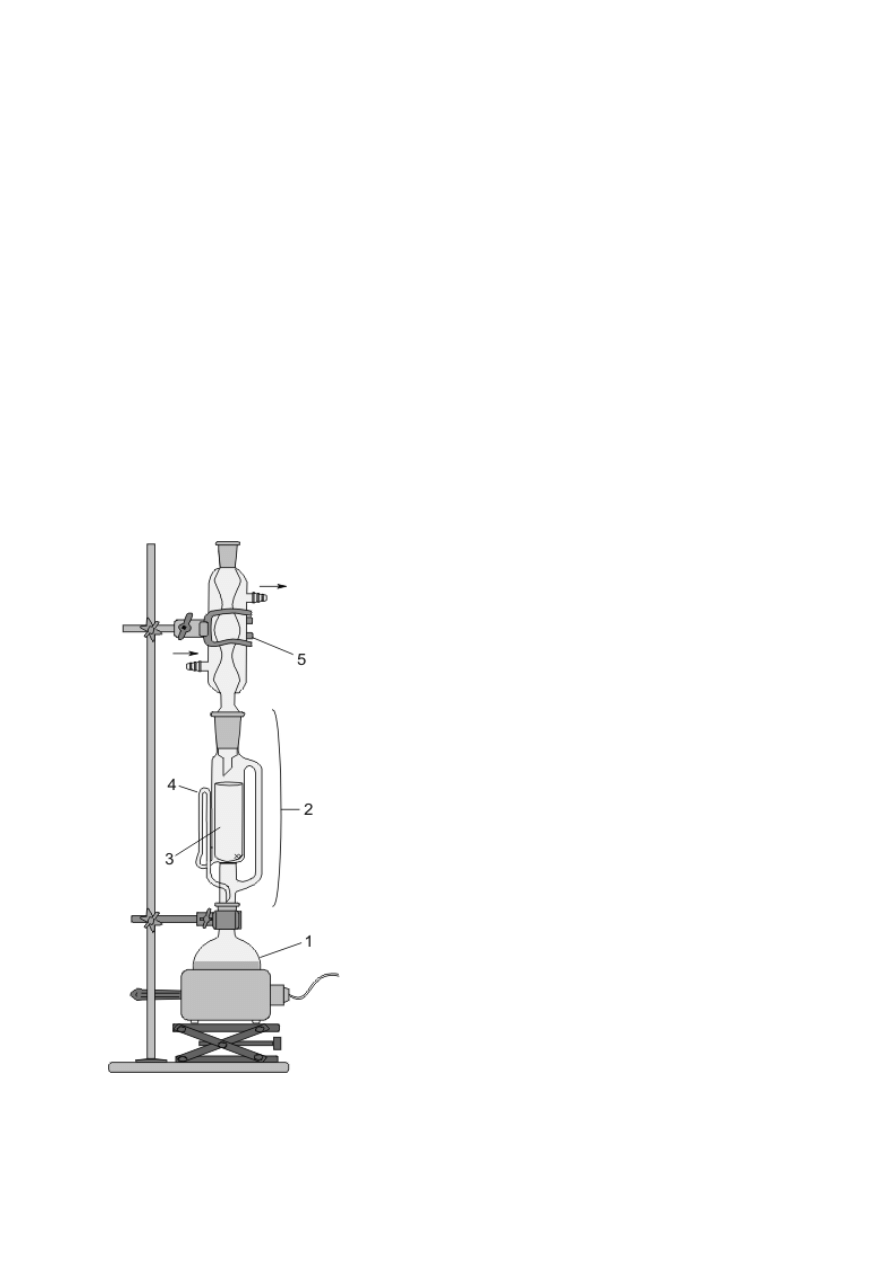
Innym rodzajem ekstrakcji jest ekstrakcja pojedyncza lub ciągła w układzie ciało stałe/ciecz.
Z pojedynczą, niekiedy powtarzaną wielokrotnie, ekstrakcją ciała stałego cieczą mamy do
czynienia, gdy na przykład rozdrobniony materiał roślinny traktujemy odpowiednio
dobranym rozpuszczalnikiem, w którym substancja ekstrahowana dobrze się rozpuszcza.
Dokładne wymieszanie obu faz może następować na zimno (tzw. macerowanie) lub na
gorąco, po czym rozdziela się obie fazy przez odsączenie ciała stałego, najczęściej na lejku
Büchnera. Następnie, aby odzyskać ekstrahowany materiał, należy z przesączu odparować
rozpuszczalnik, stosując wyparkę obrotową lub aparaturę do destylacji (podrozdz. V.3).
Ciągłą ekstrakcję ciała stałego cieczą prowadzi się w aparacie Soxhleta (rys. 32). Ten szklany
aparat składa się z dwóch części zespolonych szlifem. Górną część stanowi chłodnica
z płaszczem wodnym. W dolnej części aparatu umieszcza się specjalną gilzę z prasowanego
papieru filtracyjnego, a w niej rozdrobniony materiał do ekstrakcji. Dolną część aparatu łączy
się z kolbą okrągłodenną, wypełnioną do 3/4 rozpuszczalnikiem do ekstrakcji.
Rozpuszczalnik ten ogrzewa się do wrzenia, jego pary skraplają się w chłodnicy i spływają do
gilzy. Po jej wypełnieniu otrzymany roztwór substancji ekstrahowanej przepływa przez
syfonowy system rurek do kolby, skąd rozpuszczalnik ponownie odparowuje. W celu
wykonania efektywnej ekstrakcji proces ten powtarza się wielokrotnie. Po zakończeniu
ekstrakcji z roztworu otrzymanego w kolbie oddestylowuje się rozpuszczalnik, najwygodniej
w wyparce obrotowej.
Rysunek 32. a) Zestaw do ekstrakcji z aparatem Soxhleta: 1 — kolba z
rozpuszczalnikiem/ekstraktem, 2 — aparat Soxhleta, 3 — gilza z ekstrahowaną substancją, 4
— rurka syfonująca, 5 — chłodnica

Ekstrakcja jest jedną z częstych operacji stosowanych w laboratorium chemicznym.
Wykorzystując ją do rozdzielania skomplikowanych mieszanin, można się posłużyć kolejno
celowo dobranymi rozpuszczalnikami o zróżnicowanym charakterze chemicznym. Dzieje się
tak na przykład w przypadku wyodrębniania substancji naturalnych, ekstrahowanych
oddzielnie roztworem kwasów, roztworem zasadowym i neutralnymi rozpuszczalnikami.
Pamiętać jednak trzeba, że w przeciwieństwie do destylacji, czy nawet krystalizacji,
ekstrakcja jako metoda rozdzielania nie gwarantuje otrzymania czystych substancji. Pojawia
się więc konieczność sprawdzania czystości ekstraktów na przykład metodami
chromatograficznymi (podrozdz. V.5).
V.5. Chromatografia
Chromatografia zawdzięcza swe odkrycie rosyjskiemu chemikowi M. Cwietowi, który na
początku XX w. w Warszawie zajmował się badaniem pigmentów roślinnych. W czasie
przesączania roztworu zielonych barwników roślinnych przez pionową rurę wypełnioną
węglanem wapnia zaobserwował on tworzenie się szeregu pasm o barwie zielonej i żółtej,
odpowiadających poszczególnym składnikom mieszaniny. Szybki rozwój chromatografii
rozpoczął się od połowy XX wieku; powstały nowe rodzaje chromatografii, zaczęto
konstruować różnorodną aparaturę chromatograficzną.
W różnych metodach chromatograficznych wykorzystuje się ten sam efekt: poszczególne
składniki mieszaniny ulegają zróżnicowanemu podziałowi między dwie nie mieszające się
fazy. Jedna z tych faz jest nieruchoma (faza stacjonarna — porowaty adsorbent lub cienka
warstwa cieczy osadzona na obojętnym nośniku), druga zaś jest fazą ruchomą (gazem lub
cieczą). Składniki próbki poruszają się w układzie chromatograficznym wraz z fazą ruchomą,
przy czym szybkość ich migracji jest różna w zależności od powinowactwa poszczególnych
składników do fazy stacjonarnej i ruchomej.
W zależności od rodzaju fazy stacjonarnej związki mogą adsorbować się na jej powierzchni
(chromatografia adsorpcyjna) lub ulegać rozpuszczeniu w cienkiej błonce cieczy
pokrywającej nośnik (chromatografia podziałowa). Substancje silnie wiążące się z
adsorbentem poruszają się wolniej niż te, które wiążą się słabo i wykazują duże
powinowactwo do fazy ruchomej. Wielokrotnie powtarzający się dynamiczny proces podziału
składników między dwie fazy pozwala na lepsze rozdzielenie składników mieszaniny niż przy
zastosowaniu innych operacji jednostkowych (krystalizacji, destylacji i ekstrakcji).
Innym kryterium podziału chromatografii jest rodzaj zastosowanej fazy ruchomej. Jeśli fazą
ruchomą jest gaz, to mówimy o chromatografii gazowej (GC — ang. Gas Chromatography),
natomiast w przypadku ciekłej fazy ruchomej mówimy o chromatografii cieczowej. Do
najpopularniejszych technik chromatografii cieczowej należą: wysokosprawna
chromatografia cieczowa (HPLC — ang. High-Performance Liquid Chromatography),
chromatografia cienkowarstwowa (TLC — ang. Thin Layer Chromatography) i
chromatografia kolumnowa (CC — ang. Column Chromatography). Dwie ostatnie techniki ze
względu na swą prostotę, efektywność i brak wymagań aparaturowych są wykorzystywane
prawie w każdym laboratorium chemicznym. Chromatografia kolumnowa służy do
oczyszczania związków i preparatywnego rozdzielania mieszanin, a chromatografię
cienkowarstwową wykorzystuje się głównie do śledzenia przebiegu reakcji oraz identyfikacji
i sprawdzania czystości związków.

W chromatografii kolumnowej adsorbent zwilżony odpowiednim rozpuszczalnikiem
umieszcza się w pionowej, cylindrycznej kolumnie zaopatrzonej w kran. Po naniesieniu na
kolumnę badanej próbki w jak najmniejszej ilości rozpuszczalnika dolewa się od góry kolejne
porcje rozpuszczalnika. Powoduje to tzw. rozwijanie chromatogramu. Poszczególne
składniki mieszaniny docierają do wylotu kolumny w różnym czasie, co pozwala na
rozdzialenie mieszaniny na czyste składniki.
Natomiast w chromatografii cienkowarstwowej roztwory badanych substancji nanosi się na
płytki pokryte warstwą adsorbenta, które następnie umieszcza się w komorze
chromatograficznej zawierającej odpowiedni rozpuszczalnik (eluent). Siły kapilarne
powodują przesuwanie się eluenta po powierzchni płytki. Wraz z eluentem wędrują z różnymi
szybkościami poszczególne składniki mieszaniny, tworząc po zakończeniu procesu układ
plamek, których liczba zależy od liczby związków wchodzących w skład mieszaniny.
Materiały stosowane w chromatografiach
TLC i CC
Faza stacjonarna
Wszystkie adsorbenty stosowane w chromatografii adsorpcyjnej muszą spełniać kilka
podstawowych warunków. Adsorbent nie może reagować ani z substancjami rozdzielanymi
(wyjątek stanowi chromatografia jonowymienna) ani z rozpuszczalnikiem. Powinien być
nierozpuszczalny w stosowanych rozpuszczalnikach, a wielkość jego ziaren powinna
zapewnić odpowiednią szybkość filtracji. Adsorbenty dzielą się na polarne i niepolarne. Do
polarnych należą: żel krzemionkowy, tlenek glinu oraz odpowiednio spreparowane tlenki,
siarczany, węglany i fosforany metali alkalicznych. Najczęściej stosowane, żel krzemionkowy
i tlenek glinu, to drobno sproszkowane, bezbarwne ciała stałe o silnie porowatej strukturze.
Średnica ziaren sorbentów stosowanych w TLC waha się od 2 do 60 μm, natomiast w
chromatografii kolumnowej stosuje się ziarna większe, od 60 do 200 μm.
Atomy tlenu znajdujące się na powierzchni żelu krzemionkowego występują w postaci grup
hydroksylowych, co nadaje temu nośnikowi właściwości bardzo słabego kwasu i powoduje
jego wysoką polarność. Cząsteczki związku chemicznego wprowadzonego na powierzchnię
żelu ulegają tam procesowi adsorpcji. Związki niepolarne wiążą się z powierzchnią żelu tylko
siłami dyspersyjnymi oraz indukowanymi oddziaływaniami dipolowymi. Cząsteczki
zawierające ugrupowania polarne, np. C–O, C=O, O–H, N–H, N=O, C–Cl, tworzą silne
wiązania dipolowe, a nawet wodorowe. Im bardziej zatem polarna jest cząsteczka, tym silniej
adsorbuje się na powierzchni żelu krzemionkowego. Podobnie zachodzi adsorpcja na
powierzchni tlenku glinu.
Efektywność adsorpcji zależy również od sposobu przygotowania fazy stacjonarnej. Dodatek
wody, której cząsteczki niezwykle silnie wiążą się z powierzchnią nośnika i blokują dostęp do
niej cząsteczkom organicznym, powoduje znaczne obniżenie aktywności fazy stacjonarnej.
Dlatego spotyka się różne rodzaje tego samego adsorbenta, różniące się stopniem
aktywności. Postać adsorbenta o najwyższej aktywności jest określana cyfrą I, mniej aktywne
zaś postaci, zawierające coraz większe ilości wody, cyframi od II do V (aktywność wg
Brockmanna).

Do adsorbentów niepolarnych zalicza się m.in. węgiel aktywny, sacharozę, skrobię oraz
specjalnie modyfikowany żel krzemionkowy, którego polarne grupy hydroksylowe zostały
podstawione grupami alkilosililowymi. Hydrofobowa powierzchnia tych adsorbentów
utrudnia wiązanie się z nimi cząsteczek polarnych.
Każdy z opisanych wyżej adsorbentów może zawierać związany trwale wskaźnik
fluorescencyjny (F
254
). Adsorbent taki naświetlany promieniowaniem UV o długości 254 nm
wykazuje intensywną zieloną fluorescencję, która jest wygaszana przez zaadsorbowany
związek organiczny (obserwuje się to w postaci ciemnych plam na zielonym tle).
Faza ruchoma
Fazę ruchomą (eluent) w chromatografii cienkowarstwowej i kolumnowej stanowią
rozpuszczalniki organiczne lub ich mieszaniny. Jest oczywiste, że stosowany rozpuszczalnik
nie może reagować z analizowaną substancją, powinien rozpuszczać ją choć w niewielkim
stopniu, musi być względnie łatwy do usunięcia, bezpieczny i niezbyt drogi.
Przepływ eluenta wzdłuż fazy stacjonarnej zachodzi pod wpływem sił grawitacji
(w chromatografii kolumnowej) lub sił kapilarnych (w chromatografii TLC). Przemieszczanie
się rozpuszczalnika przez warstwę fazy stacjonarnej, na której została zaadsorbowana próbka,
powoduje wymywanie składników próbki z podłoża i ich przemieszczanie w kierunku ruchu
eluenta. Skuteczność eluowania zależy od dwóch podstawowych czynników:
„powinowactwa” substancji do fazy stacjonarnej,
„powinowactwa” eluenta do fazy stacjonarnej.
Zaadsorbowana substancja może zostać wyparta z adsorbenta przez rozpuszczalnik tylko
wtedy, gdy wykazuje on większe od niej powinowactwo do tego adsorbenta.
W przypadku adsorbentów polarnych, największym „powinowactwem” do nośnika cechują
się związki o polarnych cząsteczkach. Zarówno zatem adsorpcja związku, jak również
zdolność rozpuszczalnika do eluowania substancji zaadsorbowanej zależy głównie od ich
polarności (momentu dipolowego cząsteczki, przenikalności elektrycznej) i tendencji do
tworzenia wiązań wodorowych. Rozpuszczalniki niepolarne mogą wymyć z powierzchni
polarnego adsorbenta jedynie niepolarne związki, natomiast rozpuszczalniki polarne, o dużym
„powinowactwie” do nośnika, eluują skutecznie zarówno związki polarne, jak i niepolarne.
Stosowane w chromatografii rozpuszczalniki uszeregowane zgodnie ze wzrostem zdolności
wymywania zaadsorbowanych związków polarnych z polarnego nośnika tworzą szereg
eluotropowy (tabela 4). Kolejność rozpuszczalników w tym szeregu jest skorelowana z ich
polarnością, może się jednak w pewnym stopniu zmieniać, zależnie od rozpatrywanego
adsorbenta (nieco inna jest dla żelu krzemionkowego niż dla np. tlenku glinu).
Znajomość szeregu eluotropowego pozwala odpowiednio dobrać rozpuszczalniki i kolejność
ich użycia w celu rozdzielenia mieszanin związków różniących się polarnością. Na przykład,
jeżeli mieszanina składająca się z węglowodoru i estru zostanie naniesiona na żel
krzemionkowy, to bardziej polarny ester silniej adsorbuje się na powierzchni adsorbenta niż
niepolarny węglowodór. Wymywanie rozpoczyna się od rozpuszczalnika o możliwie małej
polarności (np. cykloheksanu); spowoduje to wyeluowanie jedynie niepolarnego
węglowodoru. Natomiast, aby wymyć ester, należy zmienić rozpuszczalnik na bardziej

polarny (np. chloroform). Często użycie czystych rozpuszczalników nie daje spodziewanych
efektów, stosuje się wówczas mieszaniny dwóch lub więcej rozpuszczalników.
Należy zdawać sobie sprawę, że w przypadku użycia adsorbenta niepolarnego, zależności się
odwracają: „powinowactwo” do nośnika związków polarnych jest dużo mniejsze niż
związków niepolarnych. Wymywanie rozpoczyna się zatem rozpuszczalnikiem polarnym,
który powoduje w pierwszej kolejności eluowanie związków najbardziej polarnych.
Tabela 4. Szereg eluotropowy wybranych
rozpuszczalników dla żelu krzemionkowego
Eter naftowy
Cykloheksan
Tetrachlorometan (tetrachlorek węgla)
Toluen
Dichlorometan (chlorek metylenu)
Trichlorometan (chloroform)
Eter dietylowy
Octan etylu
Propanon (aceton)
Propan-2-ol (alkohol izopropylowy)
Etanol
Metanol
Woda
Wzrost polarności
Chromatografia cienkowarstwowa (TLC)
Chromatografia cienkowarstwowa jest szybką metodą pozwalającą orientacyjnie określić, ile
związków wchodzi w skład badanej mieszaniny, jak również dokonać ich identyfikacji, jeżeli
dysponuje się odpowiednimi wzorcami. Dzięki doborowi odpowiedniego adsorbenta i układu
rozwijającego daje ona możliwość rozdzielenia dowolnej mieszaniny związków
organicznych. Zaletami tej metody są: krótki czas rozwijania, minimalne zużycie materiałów
oraz prostota wykonania. Wszystko to powoduje, że jest ona najczęściej używaną metodą
wstępnej identyfikacji związków organicznych, kontroli ich czystości, pozwala śledzić postęp

prowadzonej reakcji oraz dobierać rozpuszczalniki do rozdzielenia mieszanin metodą
chromatografii kolumnowej. Istnieje również możliwość wykorzystania tej metody w wersji
preparatywnej do rozdzielenia niewielkich (miligramowych) porcji mieszanin.
Sposób postępowania w analitycznej chromatografii cienkowarstwowej jest prosty. Polega on
na naniesieniu na płytkę z odpowiednim adsorbentem analizowanej substancji oraz substancji
wzorcowych, rozwinięciu chromatogramu odpowiednim rozpuszczalnikiem, a następnie
wywołaniu chromatogramu.
Obecnie zrezygnowano z metody ręcznego (obarczonego licznymi błędami) pokrywania
płytek na rzecz wykorzystania gotowych, dostępnych w handlu. Adsorbent jest naniesiony na
arkusze folii aluminiowej lub poliestrowej, które można łatwo przycinać do żądanych
rozmiarów np. za pomocą nożyka do tapet.
Przed naniesieniem próbek należy ostrożnie zaznaczyć ołówkiem (nie długopisem!) tzw. linię
startu w odległości ok. 1 cm od dolnego brzegu płytki oraz linię mety w odległości zazwyczaj
0,5 cm od górnej krawędzi płytki. Na tak przygotowane płytki wprowadza się próbki
substancji badanych oraz wzorcowych, przygotowane przez rozpuszczenie tych substancji w
możliwie lotnych rozpuszczalnikach. Bardzo cienką kapilarę zanurza się w roztworze, po
wyjęciu przykłada się koniec kapilary najpierw do bibuły, a następnie dotyka bardzo
delikatnie warstwy adsorbenta. Należy to robić bardzo ostrożnie, aby nie naruszyć
powierzchni adsorbenta. Pracować należy w okularach, gdyż może się zdarzyć, że kapilara
pęknie i jej fragmenty mogą trafić do oczu.
Między punktami nanoszenia poszczególnych substancji powinna być zachowana odległość
ok. 1 cm, a skrajne plamki startowe powinny znajdować się w odległości nie mniejszej niż 1
cm od brzegu płytki. Należy się starać, aby naniesione plamki miały możliwie najmniejszą
średnicę. Gdy analizowane roztwory są silnie rozcieńczone, próbkę nanosi się na to samo
miejsce kilkakrotnie. Po każdym naniesieniu roztworów na miejsce startu płytkę suszy się
w strumieniu ciepłego powietrza (np. suszarką do włosów) w celu usunięcia rozpuszczalnika.
Niedokładne wysuszenie płytek powoduje rozmywanie się plamek w czasie rozwijania
chromatogramu. Opisane czynności należy wykonywać delikatnie, w żadnym razie nie można
dotykać powierzchni adsorbenta palcami (rys. 33a).
Tak przygotowaną płytkę umieszcza się ostrożnie za pomocą pęsetki w komorze
chromatograficznej, której wielkość uzależniona jest od wymiarów płytek. Niewielkie płytki
można umieścić na przykład w naczynkach wagowych o odpowiedniej wysokości lub
słoiczkach szklanych z zakrętkami. Do komory wlewa się eluent na wysokość 0,5 cm.
Komora winna być wysycona parami eluenta, gdyż skraca to czas rozwijania i korzystnie
wpływa na wyniki rozdzielenia. Szybkie wysycenie komory parami eluenta można osiągnąć
przez wyłożenie ścianek komory bibułą filtracyjną zwilżoną eluentem. Po włożeniu płytki do
komory nie wolno nią poruszać do momentu zakończenia rozwijania chromatogramu.
Chromatogram rozwija się w wyniku wędrówki cieczy w górę płytki na skutek działania sił
kapilarnych warstwy adsorbenta (rys. 33b). Gdy czoło eluenta osiągnie linię mety, płytkę
wyjmuje się z komory i dokładnie suszy.
Odczytanie wyników jest sprawą prostą, gdy analizowane składniki są barwne. Jeśli
analizowane substancje są bezbarwne, chromatogram należy wywołać. W tym celu płytkę
spryskuje się roztworem związku dającego reakcję barwną ze składnikami analizowanej
mieszaniny, np. roztworem ninhydryny przy analizie aminokwasów. Uniwersalnym
wskaźnikiem jest alkoholowy roztwór kwasu siarkowego. Po jego użyciu należy ogrzać
płytkę do temperatury ok. 200
o
C, co powoduje zwęglenie związków organicznych i
pojawienie się brunatnych plam. Inną metodą wywołania chromatogramu jest umieszczenie
płytki w komorze z kilkoma małymi kryształami jodu, którego pary tworzą barwne połączenia
z większością związków organicznych. Specyficzną metodą wizualizacji chromatogramu jest
obserwacja płytki w świetle UV. Obserwację prowadzi się przez specjalną szybę lub w
okularach ochronnych, co zapobiega dostawaniu się odbitego, szkodliwego promieniowania
UV do wnętrza gałki ocznej. Niezależnie od zastosowanej techniki położenie wszystkich
wywołanych plamek należy niezwłocznie zaznaczyć ołówkiem, mogą one bowiem po
niedługim czasie zniknąć.
Rysunek 33. Kolejne etapy wykonywania chromatogramu cienkowarstwowego: a)
nanoszenie roztworów substancji za pomocą kapilary (naniesiono dwa roztwory wzorcowe
związku A oraz B oraz analizowaną mieszaninę X), b) rozwijanie chromatogramu w komorze
chromatograficznej, c) analiza chromatogramu: 1 — linia startu, 2 — plamka wzorca B, 3 —
plamki analizowanej mieszaniny X, 4 — plamka wzorca A, 5 — linia mety
Cechą charakterystyczną każdej substancji identyfikowanej metodą chromatografii
cienkowarstwowej jest współczynnik opóźnienia R
F
. Jest to stosunek odległości przebytej
przez plamkę (np. długość d
A
dla plamki związku A na rys. 33c) do odległości przebytej przez
rozpuszczalnik od linii startu do linii mety (np. d
rozp
), czyli R
F
= d
A
/d
rozp
. Z definicji wynika,
że wartości R
F
muszą zawierać się w przedziale od 0 do 1. Jeżeli R
F
= 0, to substancja jest
całkowicie adsorbowana i pozostaje na starcie, natomiast R
f
= 1 oznacza, że substancja jest
bardzo słabo adsorbowana, rozpuszcza się świetnie w fazie ruchomej i porusza się z czołem
rozpuszczalnika. W praktyce analitycznej należy unikać takich skrajności. Parametr R
F
powinien mieć dla danej substancji wartość stałą w tych samych warunkach doświadczalnych.
Jednak w praktyce rzadko udaje się dokładnie odtworzyć wartości R
F
. Nawet gdy zastosuje
się taki sam adsorbent i eluent, nie można uniknąć różnic, których przyczyną mogą być:
stopień aktywacji płytek (zależy od czasu i sposobu przechowywania),
drobne fluktuacje w składzie rozpuszczalnika zwłaszcza przy stosowaniu mieszanin,
ilość wilgoci w nanoszonym roztworze,
stopień nasycenia komory parami rozpuszczalnika.
Porównanie wartości R
F
substancji badanej i wzorca stanowi podstawę do jej identyfikacji
pod warunkiem, że chromatogram był wykonywany w identycznych warunkach (np. na tej
samej płytce). Oczywiście, zgodność wartości R
F
może być czasami przypadkowa. Jeżeli
jednak dwie porównywane substancje w identycznych warunkach (na tej samej płytce) dają
plamki o różnych wartościach względnych przesunięć, to należy wnioskować, że są to różne

związki. Na przykład analiza chromatogramu analizowanej substancji X na rys. 33c pozwala
stwierdzić, że X jest mieszaniną przynajmniej dwóch składników, przy czym jednym z nich
jest prawdopodobnie związek A, a drugim nie jest żaden z wzorcowych związków A i B.
Chromatografia kolumnowa (CC)
Chromatografia kolumnowa jest najstarszą techniką chromatograficzną. Kolumnę
chromatograficzną stanowi rura szklana o stosunku średnicy do długości od 1:15 do 1:60. U
dołu kolumny znajduje się zabezpieczenie, mające na celu utrzymanie adsorbenta w
kolumnie. Może to być spiek szklany, ale wystarczający jest kłębek waty. Kolumna
zakończona jest kranem do regulacji szybkości wypływu rozpuszczalnika (rys. 34).
Warunkiem uzyskania dobrych wyników w chromatografii jest jednorodne wypełnienie
kolumny adsorbentem. W nierówno wypełnionej kolumnie tworzą się szczeliny i kanaliki, co
prowadzi do znacznych deformacji pasm. Istnieją dwa sposoby napełniania kolumn: na mokro
i na sucho.
Napełnianie na mokro prowadzi się w następujący sposób: w zlewce przygotowuje się papkę
z adsorbenta i na ogół tego rozpuszczalnika, który będzie później użyty do rozwijania
chromatogramu. Zawartość zlewki miesza się bardzo dokładnie, aby usunąć wszystkie
pęcherzyki powietrza. Otrzymaną zawiesinę wlewa się porcjami przez lejek z długą nóżką do
czystej i suchej kolumny umocowanej na statywie pod wyciągiem. Po odczekaniu aż
adsorbent osiądzie, otwiera się kran i spuszcza nadmiar rozpuszczalnika, pozostawiając
jednak w kolumnie kilkumilimetrową warstwę eluenta nad powierzchnią adsorbenta. Nie
wolno dopuścić do zapowietrzenia kolumny!
W przypadku napełniania na sucho do kolumny wsypuje się małymi porcjami suchy
sproszkowany adsorbent. Kolumnę ostukuje się przy tym bardzo delikatnie drewnianym lub
gumowym młoteczkiem. Następnie nalewa się rozpuszczalnik, aby uzyskać całkowite
zwilżenie adsorbenta. Bezbłędne napełnienie kolumny adsorbentem na sucho, bez
pozostawienia pęcherzyków powietrza jest trudne i sposób ten jest rzadko stosowany.
Do wypełnionej kolumny wsypuje się 5-milimetrową warstwę piasku, aby zabezpieczyć
powierzchnię adsorbenta przed naruszeniem podczas nanoszenia substancji, a następnie
bardzo ostrożnie za pomocą pipetki Pasteura wprowadza roztwór przeznaczony do
rozdzielania. Pożądane jest możliwie duże stężenie roztworu, aby uzyskać zwarte pasmo u
góry kolumny. Rozpuszczalnik powinien być jak najmniej polarny. Roztwór ten wlewa się po
ściance kolumny, a następnie otwiera kran u dołu. Gdy badana substancja zostanie
zaadsorbowana w górnej części kolumny, wprowadza się dalsze porcje rozpuszczalnika,
uważając, aby nie dopuścić do wysuszenia kolumny. Nad warstwą adsorbenta musi się
zawsze znajdować niewielka ilość eluenta. W przypadku gdy rozdziela się substancje o dużej
różnicy polarności, zdarza się, że po wymyciu substancji mniej polarnej, kolejne frakcje
wymywane są bardzo wolno. Wówczas można przyspieszyć proces przez stopniowe
zwiększanie polarności eluenta (patrz szereg eluotropowy).
Jeśli analizowane substancje są barwne, obserwacja powstawania pasm jest prosta
i poszczególne frakcje wypływające z kolumny zbiera się do oddzielnych naczyń. Natomiast,
gdy rozdzielane substancje są bezbarwne stosuje się czasami naświetlanie kolumny lampą
kwarcową. Można wówczas zauważyć strefy o wyraźnej fluorescencji oraz ciemne pasma w
miejscach, gdzie światło uległo absorpcji i tym kierować się przy odbiorze frakcji. Można
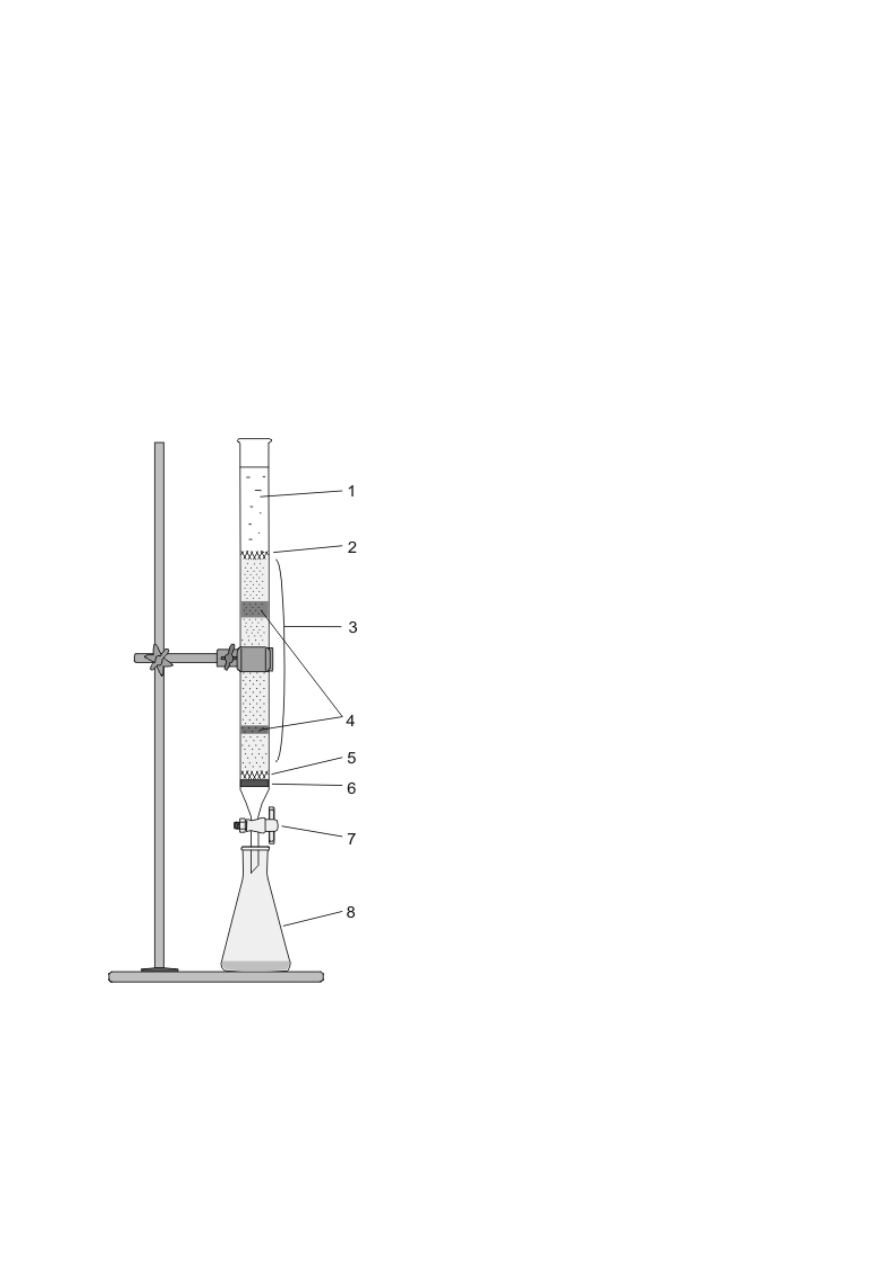
również zbierać eluat porcjami o określonej objętości do ponumerowanych probówek.
Zawartość poszczególnych probówek bada się metodą chromatografii cienkowarstwowej i
łączy frakcje zawierające ten sam składnik. Po odparowaniu rozpuszczalnika uzyskuje się
rozdzielone substancje.
Rozpuszczalniki stosowane w chromatografii kolumnowej są zazwyczaj cieczami lotnymi,
często toksycznymi. Dlatego też chromatografię kolumnową należy wykonywać pod
sprawnie działającym wyciągiem. Ważnym problemem jest bezpieczne oczyszczenie
kolumny po zakończeniu chromatografowania. Nośnik nasączony eluentem na ogół nie daje
się łatwo usunąć. Należy więc przyłączyć wylot kolumny na kilka minut do pompki wodnej,
aby przechodzący strumień powietrza wysuszył nośnik. Wówczas można go już zazwyczaj
wysypać z kolumny. Gdyby ta próba się nie powiodła, należy podłączyć kolumnę wężem do
kranu i delikatnym strumieniem wody wypłukać zawartość kolumny do dużej zlewki.
Kolumnę należy następnie wymyć acetonem i wysuszyć, przepuszczając przez nią strumień
powietrza.
Rysunek 34. Kolumna chromatograficzna: 1 — rozpuszczalnik (eluent), 2 — warstwa
czystego piasku, 3 — nośnik chromatograficzny, 4 — warstwy poszczególnych składników
spływające z różną szybkością, 5 — warstwa waty, 6 — płytka ze szkła porowatego, 7 —
kranik, 8 — odbieralnik

VI. Ustalanie składu pierwiastkowego
związków organicznych
Analiza jakościowa związków organicznych stanowi osobną część ćwiczeń laboratoryjnych z
chemii organicznej i wymaga korzystania ze specjalnego podręcznika. Ale już w trakcie
wykonywania syntez może okazać się konieczne przeprowadzenie prób na obecność
pierwiastków w otrzymanym produkcie. Wyobraźmy sobie sytuację, że w wyniku
przeprowadzonej syntezy otrzymaliśmy pewien surowy produkt, ale nie mamy pewności, że
jest to produkt oczekiwany, czy też może jeden z zanieczyszczonych substratów, bo reakcja
nie zaszła. Jeżeli znamy skład pierwiastkowy oczekiwanego produktu i potrafimy skutecznie
wykryć te pierwiastki, to możemy w krótkim czasie ocenić, czy warto zajmować się
oczyszczaniem tej substancji i jej dalszą identyfikacją (np. metodami spektroskopowymi), czy
też powtórzyć syntezę w zmienionych warunkach.
Pierwszą próbą, którą chemik organik wykonuje, mając do czynienia z nieznaną substancją,
jest spalanie niewielkiej ilości próbki badanego związku na szpatułce w płomieniu palnika.
Już sama palność związku z wytworzeniem produktów gazowych (głównie CO
2
i pary
wodnej) świadczy o jego organicznej naturze. Natomiast po spaleniu związków
nieorganicznych pozostaje popiół tlenków. Należy pamiętać, że sole związków organicznych
również pozostawiają na szpatułce osad, świadczący o obecności metalu w związku.
Dalszy krok w analizie związku organicznego stanowi oznaczenie zawartości pierwiastków
najbardziej popularnych w związkach organicznych obok węgla i wodoru, a więc fluorowców
(chloru, bromu lub jodu), azotu i siarki. Fluorowce najprościej można wykryć w tzw. próbie
Beilsteina, czyli spalaniu niewielkiej próbki badanego związku na siatce miedzianej. Związki
organiczne zawierające fluorowce ogrzewane z tlenkiem miedzi(II) tworzą, z wyjątkiem
fluoru lotne halogenki miedzi, które barwią płomień na zielono lub niebieskozielono. Fluorek
miedzi(II) również się tworzy w takiej próbie, nie jest jednak lotny. Zabarwienie płomienia
wykazują także inne związki (między innymi tiomocznik, cyjanki, tiocyjaniany, niektóre
pochodne pirydyny i puryny), ma więc ona znaczenie jedynie orientacyjne. Należy pamiętać,
że w próbie Beilsteina mogą się tworzyć bardzo toksyczne i odporne temperaturowo
związki z grupy halogenopochodnych dibenzodioksyn lub dibenzofuranów; próbę, jako
bardzo czułą, należy więc wykonać dla niewielkiej ilości badanej substancji pod
sprawnie działającym wyciągiem!
W próbie Beilsteina nie można jednak wykryć rodzaju fluorowca. Aby to ustalić, należy
dokonać rozkładu badanej substancji w procesie stapiania próbki z metalicznym sodem, co
pozwala także na wykrycie azotu i siarki (rys. 35). Wszystkie te pierwiastki zawarte w
związku organicznym w trakcie jego degradacji termicznej z udziałem sodu zostają
przeprowadzone w związki jonowe z sodem, których wykrycie za pomocą czułych reakcji z
odczynnikami nieorganicznymi nie jest skomplikowane.
Rysunek 35. Mineralizacja próbki organicznej podczas stapiania z sodem

UWAGA: W trakcie wykonywania poniższych prób pracuje się z odczynnikami
toksycznymi i żrącymi. Obowiązują rękawice ochronne i praca pod wyciągiem!
Szczególną ostrożność należy zachować podczas pracy z metalicznym sodem!
Próba Beilsteina na fluorowce
Na wyprażoną w płomieniu siatkę miedzianą wprowadza się odrobinę badanej substancji, po
czym ogrzewa się siatkę w płomieniu palnika. Zielononiebieskie zabarwienie płomienia
wskazuje na obecność fluorowca.
Stapianie z sodem
Do szklanej mikroprobówki wprowadza się mały kawałek metalicznego sodu, a następnie
odrobinę badanej substancji (kilka kryształów lub kropli). Fiolkę trzymaną przy pomocy
metalowymi szczypcami wprowadza się do płomienia palnika i bardzo ostrożnie ogrzewa aż
do stopienia sodu, a następnie ogrzewa się mocno jej dno do czerwonego żaru i gorącą fiolkę
wrzuca do parowniczki z 5–10 cm
3
wody destylowanej. (Uwaga: nadmiar użytej wody
zmniejsza stężenie badanych jonów w roztworze, co może prowadzić do błędnych wyników
analizy.) Jeśli fiolka nie pęknie, rozbija się ją szklanym pręcikiem. Płyn z parowniczki się
sączy, a bezbarwny przesącz wykorzystuje do dalszych prób. Jeśli przesącz jest żółty lub
brunatny, stapianie należy powtórzyć, gdyż rozkład związku nie był całkowity. W przypadku
szczególnie lotnych związków można dodać do fiolki niewielką ilość cukru przed stopieniem
substancji z sodem.
Próba na siarkę
Do około 1 cm
3
badanego alkalicznego przesączu dodaje się 4–5 kropli 0,1-procentowego
wodnego, świeżo przygotowanego roztworu pentacyjanonitrozylożelazianu(III) sodu
(nitroprusydku sodu) Na
2
[Fe(CN)
5
NO]. Krótkotrwałe pojawienie się intensywnego,
ciemnopurpurowego zabarwienia związku kompleksowego Na
4
[Fe(CN)
5
NOS] wskazuje na
zawartość siarki w badanej próbce.
Próba Lassaigne'a na azot
Około 3 cm
3
silnie alkalicznego przesączu ogrzewa się do wrzenia z małym kryształem
siarczanu(VI) żelaza(II). Początkowo strąca się wodorotlenek żelaza(II), który w trakcie
ogrzewania na powietrzu ulega częściowo utlenieniu do związków żelaza(III) Po ochłodzeniu
dodaje się do zawiesiny rozcieńczonego kwasu siarkowego(VI) do odczynu kwasowego
(kontrola papierkiem wskaźnikowym). Jeśli substancja zawierała azot, występuje
zielononiebieskie zabarwienie roztworu, a po dłuższym odstaniu wydziela się osad związku
kompleksowego heksacyjanożelazianu(II) żelaza(III) (błękitu pruskiego) Fe
4
[Fe(CN)
6
]
3
.
Próba na fluorowce
Jeśli badana substancja nie zawierała ani azotu, ani siarki, to 2 cm
3
alkalicznego przesączu
zakwasza się rozcieńczonym kwasem azotowym(V) i dodaje się kilka kropli 1-procentowego
roztworu azotanu(V) srebra. Powstanie białego lub żółtego osadu trudno rozpuszczalnego
halogenku srebra wskazuje na zawartość chloru, bromu lub jodu. Jeśli substancja zawiera azot
lub siarkę, to 2 cm
3
badanego przesączu po zakwaszeniu rozcieńczonym kwasem
azotowym(V) ogrzewa się do wrzenia przez kilka minut — do odpędzenia siarkowodoru lub

cyjanowodoru, które z azotanem srebra również tworzą trudno rozpuszczalne związki. Po
zagęszczeniu do połowy objętości i uzupełnieniu wodą destylowaną do pierwotnej objętości
wykonuje się próbę z azotanem(V) srebra.
Ustalanie rodzaju fluorowca
Otrzymany w próbie na fluorowce osad halogenku srebra, po zdekantowaniu płynu, zadaje się
niewielką objętością stężonego roztworu amoniaku. Rozpuszczenie się osadu AgX wskazuje
na obecność chloru. Jeśli osad jest jasnożółty i rozpuszcza się tylko częściowo, oznacza to, że
substancja zawiera brom. Osad żółty i zupełnie nierozpuszczalny w wodzie amoniakalnej
wskazuje na zawartość jodu. W praktyce zdarza się, że zamiast wyraźnego osadu pojawia się
niewielka ilość rozproszonego koloidalnego osadu, wtedy objętość dodawanego amoniaku
powinna być znikoma, a rozpoznanie fluorowca jest szczególnie trudne.
Rodzaj obecnego w próbce fluorowca można też ustalić, stosując reakcje redoks. Opiera się to
na fakcie, że bardziej aktywny chlor wypiera brom i jod z ich związków. W celu
przeprowadzenia takiej próby 1–2 cm
3
roztworu po stopieniu z sodem zakwasza się
rozcieńczonym kwasem siarkowym, ochładza i dodaje 1 cm
3
chloroformu. Następnie
przygotowuje się 5-procentowy roztwór kwasu chlorowodorowego z dodatkiem kilku
kryształów KMnO
4
. Stanowi on ekwiwalent wody chlorowej [jony chlorkowe zostają
utlenione manganianem(VII) potasu do wolnego chloru]. Roztwór ten dodaje się kroplami do
badanej próbki przy energicznym mieszaniu. Warstwa organiczna może pozostać bezbarwna,
co wskazuje na obecność chloru w próbce, może przyjąć barwę brunatną lub
czerwonobrunatną, co wskazuje na obecność bromu w próbce, lub może przyjąć barwę
fioletową, co wskazuje na obecność jodu w próbce. Ta ostania barwa zanika po dodaniu
nadmiaru wody chlorowej, gdyż dalsze utlenianie jodu daje bezbarwny jodan.
VII. Komentarz do organizacji ćwiczeń
laboratoryjnych w ZChO WCh UJ
W ramach ćwiczeń laboratoryjnych odbywanych w Zakładzie Chemii Organicznej Wydziału
Chemii UJ każdy student chemii musi wykonać:
zadania wstępne (krystalizację oraz chromatografię cienkowarstwową i kolumnową);
określoną liczbę preparatów ułożonych w zestawy;
zadanie specjalne;
zadanie asystenckie (o jego wykonaniu decyduje prowadzący ćwiczenia);
zadanie z klasycznej analizy jakościowej;
zadanie z analizy jakościowej z załączonym kompletem widm.
Przed rozpoczęciem zadań syntetycznych student losuje zestaw syntez (preparatów).
Zestawy syntez są ułożone w sposób nieprzypadkowy: wykonanie każdego z nich wymaga
przeprowadzenia wszystkich operacji jednostkowych, które są zawarte w programie kursu
podstawowego. W skład poszczególnych zestawów wchodzą eksperymenty o zróżnicowanym
stopniu trudności, w każdym przypadku przewiduje się realizację syntez zarówno jedno-, jak i
wieloetapowych. Syntezy obejmują możliwie różne typy reakcji chemicznych (patrz: podział
preparatów), co zmusza do stopniowego powtarzania kolejnych partii materiału

teoretycznego. Załącznik 9 (rozdz. IX) przedstawia jeden z zestawów zadań syntetycznych,
które były wykonywane w roku akademickim 2006/2007.
Instrukcje do wykonania ćwiczeń wstępnych oraz opisy otrzymywania poszczególnych
preparatów są zamieszczone w uzupełniającej części skryptu (na stronie www lub płycie CD
dołączonej do książkowego wydania skryptu), w dziale „instrukcje do ćwiczeń dla studentów
chemii”. Zostały one podzielone na kilka grup, według typów przeprowadzanych reakcji:
I. Addycja do wiązań podwójnych C=C (I.1–I.11);
III. Substytucja elektrofilowa w układach aromatycznych:
a) nitrowanie (III.a.1–III.a.10),
b) sulfonowanie (III.b.1–III.b.6) ,
c) chlorowcowanie (III.c.1–III.c.7),
d) alkilowanie, acylowanie i formylowanie (III.d.1–III.d.4),
e) reakcje sprzęgania soli diazoniowych (III.e.1–III.e.7);
IV. Substytucja nukleofilowa i rodnikowa w układach aromatycznych (IV.1–IV.10);
V. Reakcje aldehydów i ketonów (V.1–V.13);
VI. Reakcje redoks:
a) utlenianie (VI.a.1–VI.a.8),
b) redukcja (VI.b.1–VI.b.11);
VII. Synteza związków heterocyklicznych i reakcje przegrupowania (VII.1–VII.25).
W instrukcji do każdego ćwiczenia są zamieszczone hiperlinki, pozwalające szybko zapoznać
się z najważniejszymi informacjami o stosowanych związkach chemicznych — kliknięcie
tytułu ćwiczenia lub każdej z pozycji spisu odczynników otwiera zbiór zawierający krótki
wyciąg z karty charakterystyki danej substancji.
Zadanie specjalne jest przydzielane ze zbioru tego typu zadań, który każdego roku ulega
pewnym zmianom. W ramach zadania specjalnego syntetyzuje się substancje o ciekawych
właściwościach (luminol, barwniki solwatochromowe, indygo itp. — przepisy na
wykonywanie tych preparatów nie są zawarte w tym skrypcie), wykonuje pewne dodatkowe
eksperymenty z wykorzystaniem otrzymanego związku, rozwiązuje problemy
spektroskopowe itp. Zadanie specjalne wymaga ponadto samodzielnego znalezienia danych
dotyczących toksyczności używanych związków. Zadanie asystenckie jest przydzielane
opcjonalnie. Może być połączone z zadaniem specjalnym, może też to być jakiś preparat z
bazy przepisów lub też synteza według przepisu dostarczonego przez asystenta.
Do każdego zestawu preparatów jest przygotowana tabela dla asystenta, w której
odnotowuje on zakończenie przez studenta kolejnych etapów ćwiczeń, a także wydajności
otrzymanych preparatów. Bardzo istotne znaczenie ma ostatnia kolumna tej tabeli, w której są
zamieszczone wydajności akceptowalne, czyli takie, w ramach których wykonanie preparatu
może być zaliczone. Wydajności akceptowalne są ustalone na podstawie wyników
wydajności zgromadzonych przez kilka lat w Zakładzie Chemii Organicznej Wydziału
Chemii UJ, uzyskiwanych przez studentów, a nie przez doświadczonych chemików. Jeżeli
wydajność syntezy osiągnięta przez studenta znacznie odbiega od wydajności akceptowalnej,

to zadanie musi być powtórzone lub jest ocenione na ocenę niedostateczną. Wydajności
akceptowalne są danymi poufnymi tylko do wiadomości asystenta.
Studenci tych kierunków studiów, na których chemia organiczna jest nauką pomocniczą
(np. biologii lub kierunków pokrewnych), wykonują:
zadania typu analitycznego (wybrane spośród ćwiczeń oznaczonych symbolami od A.1.1
do A.6.4);
zadania syntetyczne (wybrane spośród ćwiczeń oznaczonych symbolami od S.1.1 do S.4.4).
Opis wykonywania poszczególnych ćwiczeń jest zamieszczony w uzupełniającej części
skryptu, w dziale „instrukcje do ćwiczeń dla studentów biologii i kierunków pokrewnych”.
Również w tym przypadku kliknięcie nazwy związku powoduje otworzenie się zbioru
zawierającego krótką charakterystykę danej substancji chemicznej.
Przed przystąpieniem do wykonywania ćwiczeń praktycznych każdy student musi zapoznać
się z zasadami bezpiecznej pracy w laboratorium chemicznym oraz zaznajomić się z
podstawowym wyposażeniem pracowni chemii organicznej (rozdziały I i II zasadniczej części
skryptu). Właściwe przygotowanie się do wykonania każdego ćwiczenia obejmuje:
dokładne przestudiowanie instrukcji do ćwiczenia (w przypadku studentów biologii i
kierunków pokrewnych również wstępów do poszczególnych rozdziałów i podrozdziałów);
sprawdzenie w dostępnych źródłach informacji o właściwościach używanych i
otrzymywanych substancji chemicznych (np. na podstawie charakterystyk substancji
niebezpiecznych);
zaznajomienie się ze stosowanym w danym ćwiczeniu szkłem laboratoryjnym,
urządzeniami, technikami oraz operacjami laboratoryjnymi (na podstawie informacji
zawartych w rozdz. III, IV i V zasadniczej części skryptu oraz zdjęć zamieszczonych w części
uzupełniającej);
poznanie podstaw teoretycznych przeprowadzanej reakcji, przygotowanie
szczegółowego mechanizmu tej przemiany, określenie możliwych produktów ubocznych
itp. (np. na podstawie informacji zawartych w pozycjach [4–13] spisu literatury w rozdz.
VIII);
zrozumienie przyczyn i celu wykonywania poszczególnych czynności oraz roli wszystkich
stosowanych odczynników i urządzeń;
ocenę ryzyka wykonywanych eksperymentów, a w szczególności analizę procedur pod
kątem stwarzanych przez nie potencjalnych zagrożeń, zaproponowanie sposobów
zmniejszenia ryzyka (np. wybór odpowiedniego miejsca pracy, użycie właściwych środków
ochrony osobistej) i określenie sposobu postępowania z powstającymi odpadami;
racjonalne rozplanowanie kolejności wykonywania poszczególnych czynności podczas
trwania zajęć laboratoryjnych.
Niektóre zadania wymagają wykonania wielu operacji jednostkowych. Bardzo pomocne w
zrozumieniu celowości poszczególnych czynności jest narysowanie schematu postępowania z
substratami, produktami głównymi i ubocznymi oraz odpadami. Przykładowy schemat
postępowania przedstawia Załącznik 8 w rozdz. IX. Taki diagram powinien być
przygotowany przed rozpoczęciem pracy laboratoryjnej i przedstawiony do zaakceptowania
asystentowi, a następnie może być dołączony do sprawozdania.

Wszyscy studenci po zakończeniu każdego z zadań muszą napisać sprawozdanie. Wzory
sprawozdań z zadań typu analitycznego dla studentów kierunków, na których chemia jest
nauką pomocniczą, zamieszczone są na końcu każdego przepisu, natomiast wzór
sprawozdania z zadań syntetycznych i wskazówki co do jego przygotowania przedstawiono w
Załączniku 7 w rozdz. IX.
Preparaty oddaje się asystentowi zazwyczaj łącznie ze sprawozdaniem. Substancje ciekłe
powinny być umieszczone w szklanej fiolce lub buteleczce, ciała stałe zaś w pudełeczku
wykonanym z gładkiego papieru techniką origami. Schemat wykonania takiego prostego
pudełka przedstawiono w Załączniku 6 w rozdz. IX. Fiolki i pudełka podpisuje się czytelnie
nazwiskiem studenta oraz nazwą preparatu i jego numerem. Należy pamiętać, że ocenie
podlega nie tylko wydajność wykonanej syntezy, ale również jakość preparatu! Oprócz
standardowych sposobów oceny czystości (chromatografia cienkowarstwowa, pomiar
temperatury topnienia lub wrzenia, oznaczenie współczynnika załamania światła) otrzymany
związek można porównać z preparatami wzorcowymi. Niska jakość uzyskanego produktu
może być podstawą do niezaliczenia ćwiczenia.
VIII. Bibliografia
Chemia organiczna
[1] H. Hart, L.E. Craine, D.J. Hart, Chemia organiczna. Krótki kurs, PZWL, Warszawa
1999.
[2] D.J. Hart, H. Hart, Chemia organiczna. Repetytorium i rozwiązanie zadań, PZWL,
Warszawa 2000.
[3] E. Białecka-Florjańczyk, J. Włostowska, Chemia organiczna, WNT, Warszawa 2003.
[4] J. McMurry, Chemia organiczna, PWN, Warszawa 2005.
[5] S. McMurry, Chemia organiczna. Rozwiązania problemów, PWN, Warszawa 2005.
[6] R.T. Morrison, R.N. Boyd, Chemia organiczna, PWN, Warszawa 1998.
[7] J. March, Chemia organiczna. Reakcje, mechanizmy, budowa, WNT, Warszawa
1975.
[8] J. March, Advanced Organic Chemistry. Reactions, Mechanisms, and Structure, John
Wiley & Sons, New York, Chichester, Brisbane, Toronto, Singapore 1992.
[9] L.G. Wade, Jr, Organic Chemistry, Prentice Hall, Upper Saddle River, New Jersey
1999.
[10] J. Clayden, N. Greeves, S. Warren, P. Wothers, Organic Chemistry, Oxford
University Press, Oxford 2000.
Preparatyka organiczna
[11] A.I. Vogel, Preparatyka organiczna, WNT, Warszawa 1984.
[12] Preparatyka i elementy syntezy organicznej, (praca zbiorowa, red. J.T. Wróbel),
PWN, Warszawa 1983.
[13] Preparatyka organiczna, (praca zbiorowa, tłum. z języka niemieckiego, red. B.
Bochwic), PWN Warszawa 1975.
[14] Organikum, Organisch-chemisches Grundpraktikum, (praca zbiorowa), VEB
Deutscher Verlag der Wissenschaften, Berlin 1986.
[15] H. Hart, L.E. Craine, Organic Chemistry, A Short Course, Laboratory Manual,
Houghton Mifflin Company, Boston 1991.
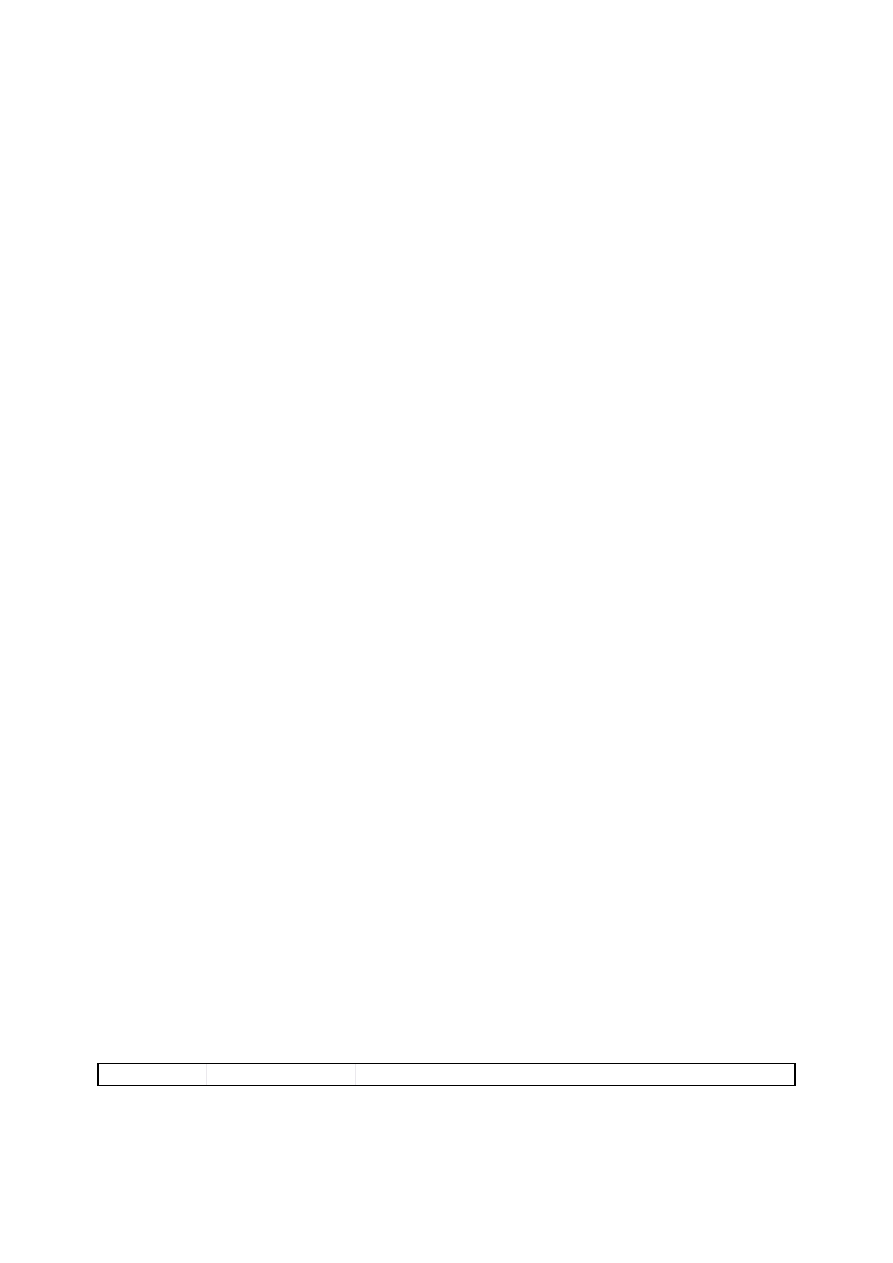
[16] L.F. Tietze, W. Eicher, Reaktionen und Synthesen im organisch-chemischen
Praktikum und Forschungslaboratorium, Georg Thieme Verlag, Stuttgart, New York 1991.
[17] J.W. Lehman, Operational Organic Chemistry, A Problem-Solving Approach to the
Laboratory Course, Prentice Hall, Upper Saddle River, New Jersey 1999.
Bezpieczeństwo i technika laboratoryjna
[18] G. Kupryszewski, Podstawowe zasady bezpieczeństwa pracy w laboratorium
chemicznym, Wydawnictwo Gdańskie, Gdańsk 1999.
[19] Laboratorium chemii organicznej, techniki pracy i przepisy bhp, (praca zbiorowa,
red. P. Kowalski), WNT, Warszawa 2004.
Analiza jakościowa związków organicznych
[20] Z. Jerzmanowska, Analiza jakościowa związków organicznych, PZWL, Warszawa
1951.
[21] J. Woliński, J. Terpiński, Organiczna analiza jakościowa, PWN, Warszawa 1973.
[22] R. Walczyna, J. Sokołowski, G. Kupryszewki, Analiza związków organicznych,
Wydawnictwo Uniwersytetu Gdańskiego, Gdańsk 1996.
Dane fizykochemiczne
[23] Poradnik fizykochemiczny, (praca zbiorowa), WNT, Warszawa 1974.
[24] W. Mizerski, Tablice chemiczne, Wydawnictwo Adamantan, Warszawa 2004.
IX. Załączniki
Załącznik 1B. Nowe piktogramy i hasła
ostrzegawcze.
Załącznik 1B. Nowe piktogramy i hasła ostrzegawcze.
Załącznik 1. Objasnienia piktogramów
identyfikujących zagrożenia stwarzane
przez substancje niebezpieczne
1A: System klasyfikacji i oznakowań dozwolony do 2015 roku
Znak
Symbol
Opis zagrożeń

T+
bardzo toksyczna
stanowi poważne zagrożenie dla zdrowia po
wprowadzeniu do organizmu niewielkich ilości
(np. po spożyciu DL
50
< 25 mg/kg masy ciała)
*
T
toksyczna
stanowi zagrożenie dla zdrowia po wprowadzeniu do
organizmu (np. po spożyciu DL
50
25–200 mg/kg masy
ciała)
*
lub powoduje nieodwracalne zmiany przy
długotrwałej ekspozycji
Xn
szkodliwa
stanowi zagrożenie dla zdrowia po wprowadzeniu do
organizmu większej ilości (np. po spożyciu DL
50
200–
2000 mg/kg masy ciała)
*
lub może powodować
nieodwracalne zmiany przy długotrwałej ekspozycji
Xi
drażniąca
powoduje podrażnienie oczu, skóry lub dróg
oddechowych;
może wywołać reakcję alergiczną
F+
skrajnie
łatwopalna
substancja o temperaturze wrzenia <35
o
C i temperaturze
samozapłonu <0
o
C lub zapalająca się w kontakcie z
powietrzem
F
wysoce
łatwopalna
substancja, która nie jest skrajnie łatwopalna, lecz ma
temperaturę zapłonu poniżej 21
o
C lub może ulec
zapłonowi w kontakcie z wodą lub wilgocią z powietrza
O
utleniająca
może powodować lub podtrzymywać pożar w kontakcie z
materiałami łatwopalnymi
C
żrąca
substancja może spowodować trwałe uszkodzenie tkanek
żywych organizmów oraz sprzętu laboratoryjnego w
wyniku krótkotrwałego kontaktu
N
niebezpieczna
dla środowiska
substancja, która stanowi bezpośrednie lub pośrednie,
długotrwałe zagrożenie dla życia organizmów we
wszystkich częściach środowiska biosfery
E
wybuchowa
substancja może wybuchać pod wpływem temperatury,
tarcia, uderzenia itp.
*
DL
50
— łac. dosis letalis 50% — dawka powodująca zgon 50% osobników badanej
populacji (ang. LD
50
— lethal dose).
1B: System klasyfikacji i oznakowań obowiązujący od 2011 roku
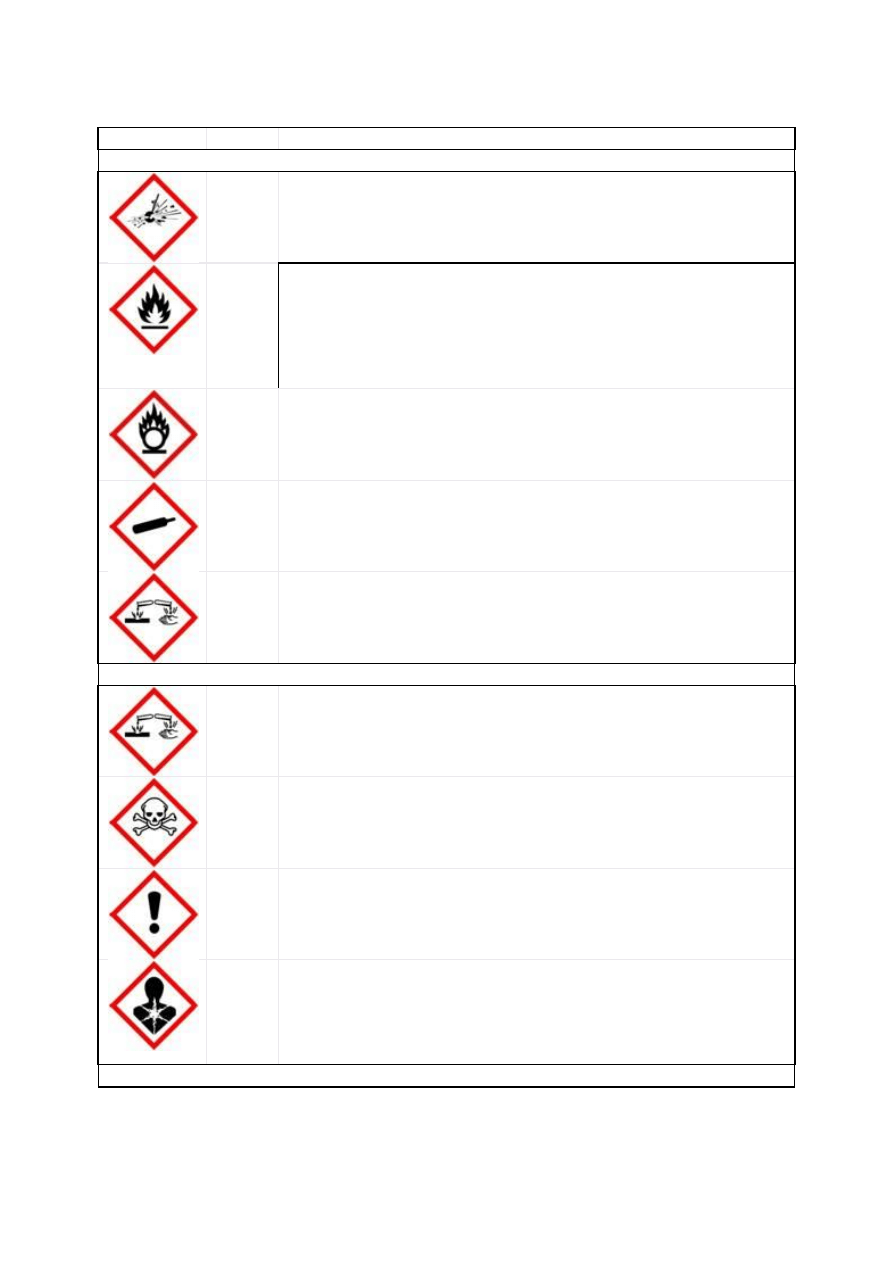
Znak
Symbol Opis zagrożeń
zagrożenia fizykochemiczne
GHS01 - związki wybuchowe lub samo reaktywne
GHS02 - łatwopalne (ciecze/gazy/aerozole/ciała stałe)
- samonagrzewające się, samoreaktywne
- piroforyczne (stałe/ciekłe)
- nadtlenki organiczne
- substancje/mieszaniny które w kontakcie z wodą uwalniają palne
gazy
GHS03 - utleniające (gazy/ciecze/ciała stałe)
GHS04 - gazy pod ciśnieniem (sprężone)
GHS05 - powodujące korozje metali
zagrożenia dla zdrowia
GHS05 - wykazujące działanie żrące na skórę kat. 1A, 1B, 1C
- ryzyko poważnego uszkodzenia oczu
GHS06 - toksyczność ostra kategorii 1, 2 lub 3
GHS07 - toksyczność ostra kat. 4 (dawniej: substancja szkodliwa)
- działanie drażniące na skórę lub oczy
- działanie uczulające na skórę
GHS08 - działanie rakotwórcze, mutagenne lub działanie na rozrodczość
(tzw. CMR)
- toksyczne działanie na narządy krytyczne przy narażeniu
jednorazowym lub przewlekłym
- działanie uczulające na układ oddechowy
zagrożenia dla środowiska
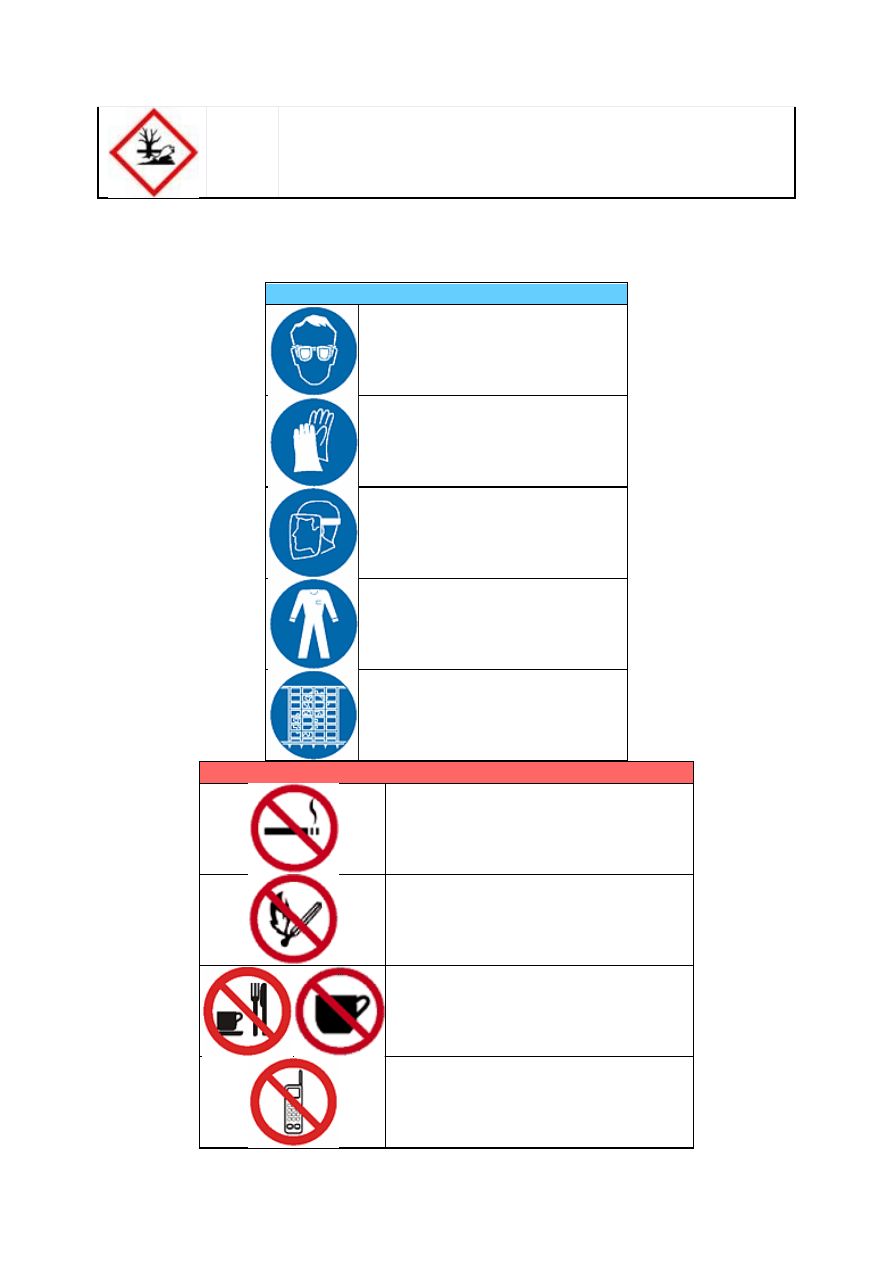
GHS09 - niebezpieczne dla środowiska, w szczególności stwarzające
zagrożenie dla środowiska wodnego
- stwarzające zagrożenie dla warstwy ozonowej
Załącznik 2. Znaki BHP
Znaki nakazu
Nakaz stosowania ochrony oczu
Nakaz stosowania ochrony rąk
Nakaz stosowania ochrony twarzy
Nakaz stosowania odzieży ochronnej
Nakaz stosowania osłony
Znaki zakazu
Zakaz palenia papierosów
Zakaz używania otwartego ognia
Zakaz spożywania posiłków
Zakaz używania telefonów komórkowych
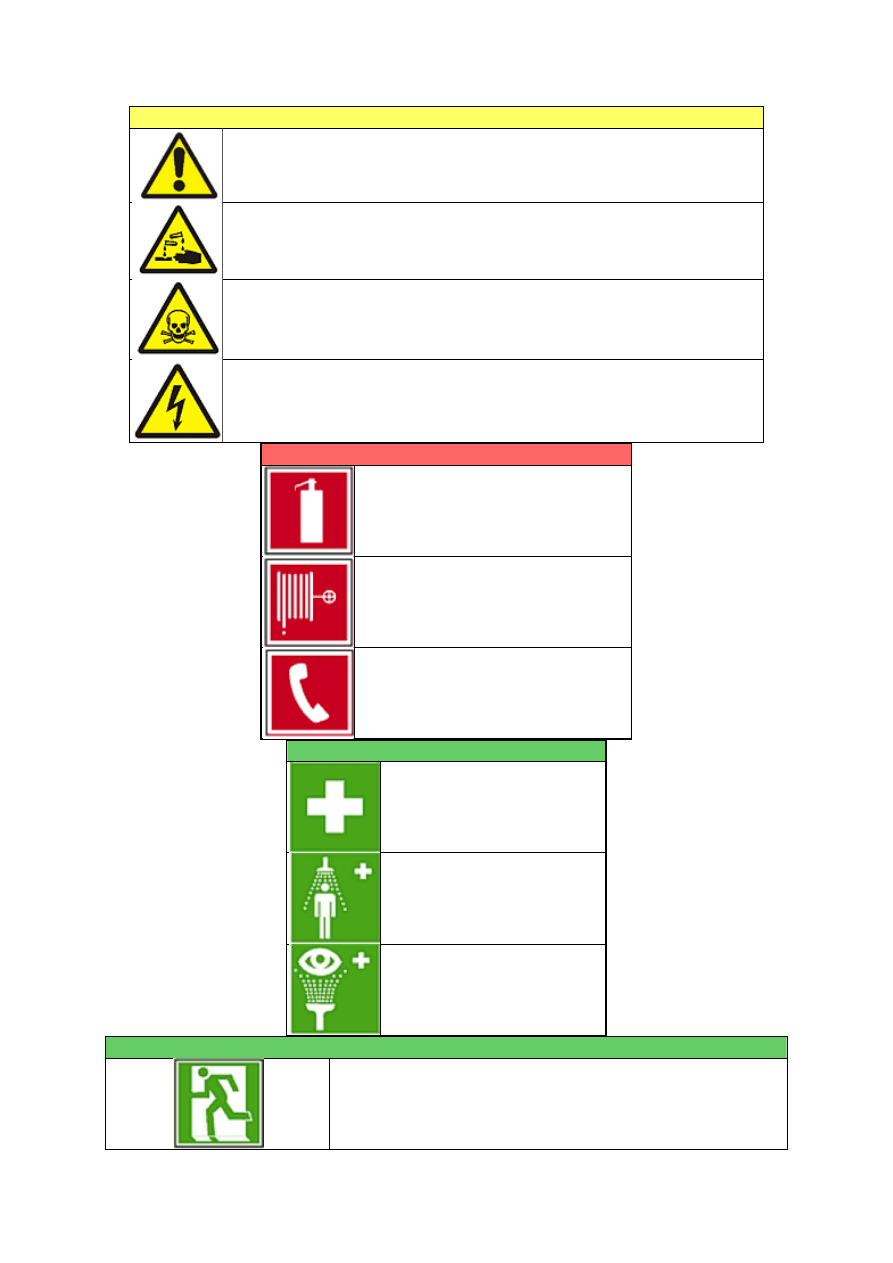
Znaki ostrzegawcze
Ogólny znak ostrzegawczy (ostrzeżenie, ryzyko niebezpieczeństwa)
Ostrzeżenie przed substancjami żrącymi
Ostrzeżenie przed niebezpieczeństwem zatrucia substancjami toksycznymi
Ostrzeżenie przed porażeniem prądem elektrycznym
Znaki ochrony przeciwpożarowej
Gaśnica
Hydrant wewnętrzny
Telefon do użycia w stanie zagrożenia
Znaki informacyjne
Apteczka pierwszej pomocy
Prysznic bezpieczeństwa
Prysznic do przemywania oczu
Znaki ewakuacyjne
Drzwi ewakuacyjne
Kierunek drogi ewakuacyjnej
Wyjście ewakuacyjne
Kierunek do wyjścia drogi ewakuacyjnej
Kierunek do wyjścia drogi ewakuacyjnej schodami w górę (w
dół)
Pchać (ciągnąć), aby otworzyć
Załącznik 3. Zwroty określające zagrożenia
oraz środki bezpieczeństwa (R i S oraz H i
P)
3A. ZWROTY R oraz ZWROTY S (dopuszczone do użytku do 2015 roku):
ZAGROŻENIA — ZWROTY R:
UWAGA: Na przykład zapis „R26-35” (pojawiający się w kartach charakterystyk
substancji chemicznej) oznacza, że związku (preparatu) dotyczą zwroty R26 oraz R35, a
nie wszystkie zwroty od R26 do R35!
R1 Produkt wybuchowy w stanie suchym.
R2 Zagrożenie wybuchem wskutek uderzenia, tarcia, kontaktu z ogniem lub innymi
źródłami zapłonu.
R3 Skrajne zagrożenie wybuchem wskutek uderzenia, tarcia, kontaktu z ogniem lub
innymi źródłami zapłonu.
R4 Tworzy łatwo wybuchające związki metaliczne.
R5 Ogrzanie grozi wybuchem.
R6 Produkt wybuchowy z dostępem i bez dostępu powietrza.
R7 Może spowodować pożar.
R8 Kontakt z materiałami zapalnymi może spowodować pożar.
R9 Grozi wybuchem po zmieszaniu z materiałem zapalnym.

R10 Produkt łatwo palny.
R11 Produkt wysoce łatwo palny.
R12 Produkt skrajnie łatwo palny.
R14 Reaguje gwałtownie z wodą.
R15 W kontakcie z wodą uwalnia skrajnie łatwopalne gazy.
R16 Produkt wybuchowy po zmieszaniu z substancjami utleniającymi.
R17 Samorzutnie zapala się w powietrzu.
R18 Podczas stosowania mogą powstawać łatwopalne lub wybuchowe mieszaniny par
z powietrzem.
R19 Może tworzyć wybuchowe nadtlenki.
R20 Działa szkodliwie przez drogi oddechowe.
R21 Działa szkodliwie w kontakcie ze skórą.
R22 Działa szkodliwie po połknięciu.
R23 Działa toksycznie przez drogi oddechowe.
R24 Działa toksycznie w kontakcie ze skórą.
R25 Działa toksycznie po połknięciu.
R26 Działa bardzo toksycznie przez drogi oddechowe.
R27 Działa bardzo toksycznie w kontakcie ze skórą.
R28 Działa bardzo toksycznie po połknięciu.
R29 W kontakcie z wodą uwalnia toksyczne gazy.
R30 Podczas stosowania może stać się wysoce łatwo palny.
R31 W kontakcie z kwasami uwalnia toksyczne gazy.
R32 W kontakcie z kwasami uwalnia bardzo toksyczne gazy.
R33 Niebezpieczeństwo kumulacji w organizmie.
R34 Powoduje oparzenia.
R35 Powoduje poważne oparzenia.
R36 Działa drażniąco na oczy.
R37 Działa drażniąco na drogi oddechowe.
R38 Działa drażniąco na skórę.
R39 Zagraża powstaniem bardzo poważnych nieodwracalnych zmian w stanie zdrowia.
R40 Ograniczone dowody działania rakotwórczego.
R41 Ryzyko poważnego uszkodzenia oczu.
R42 Może powodować uczulenie w następstwie narażenia drogą oddechową.
R43 Może powodować uczulenie w kontakcie ze skórą.
R44 Zagrożenie wybuchem po ogrzaniu w zamkniętym pojemniku.
R45 Może powodować raka.
R46 Może powodować dziedziczne wady genetyczne.
R48 Stwarza poważne zagrożenie zdrowia w następstwie długotrwałego narażenia.
R49 Może powodować raka w następstwie narażenia drogą oddechową.
R50 Działa bardzo toksycznie na organizmy wodne.
R51 Działa toksycznie na organizmy wodne.

R52 Działa szkodliwie na organizmy wodne.
R53 Może powodować długo utrzymujące się niekorzystne zmiany w środowisku
wodnym.
R54 Działa toksycznie na rośliny.
R55 Działa toksycznie na zwierzęta.
R56 Działa toksycznie na organizmy glebowe.
R57 Działa toksycznie na pszczoły.
R58 Może powodować długo utrzymujące się niekorzystne zmiany w środowisku.
R59 Stwarza zagrożenie dla warstwy ozonowej.
R60 Może upośledzać płodność.
R61 Może działać szkodliwie na dziecko w łonie matki.
R62 Możliwe ryzyko upośledzenia płodności.
R63 Możliwe ryzyko szkodliwego działania na dziecko w łonie matki.
R64 Może oddziaływać szkodliwie na dzieci karmione piersią.
R65 Działa szkodliwie; może powodować uszkodzenie płuc w przypadku połknięcia.
R66 Powtarzające się narażenie może powodować wysuszanie lub pękanie skóry.
R67 Pary mogą wywoływać uczucie senności i zawroty głowy.
R68 Możliwe ryzyko powstania nieodwracalnych zmian w stanie zdrowia.
Łączone zwroty R:
R14/15
Reaguje gwałtownie z wodą, uwalniając skrajnie łatwo palne gazy.
R15/29
W kontakcie z wodą uwalnia skrajnie łatwo palne, toksyczne gazy.
R20/21
Działa szkodliwie przez drogi oddechowe i w kontakcie ze skórą.
R20/22
Działa szkodliwie przez drogi oddechowe i po połknięciu.
R20/21/22
Działa szkodliwie przez drogi oddechowe, w kontakcie ze skórą i po
połknięciu.
R21/22
Działa szkodliwie w kontakcie ze skórą i po połknięciu.
R23/24
Działa toksycznie przez drogi oddechowe i w kontakcie ze skórą.
R23/25
Działa toksycznie przez drogi oddechowe i po połknięciu.
R23/24/25
Działa toksycznie przez drogi oddechowe, w kontakcie ze skórą i po
połknięciu.
R24/25
Działa toksycznie w kontakcie ze skórą i po połknięciu.
R26/27
Działa bardzo toksycznie przez drogi oddechowe i w kontakcie ze skórą.
R26/28
Działa bardzo toksycznie przez drogi oddechowe i po połknięciu.
R26/27/28
Działa bardzo toksycznie przez drogi oddechowe, w kontakcie ze skórą i
po połknięciu.
R27/28
Działa bardzo toksycznie w kontakcie ze skórą i po połknięciu.
R36/37
Działa drażniąco na oczy i drogi oddechowe.
R36/38
Działa drażniąco na oczy i skórę.
R36/37/38
Działa drażniąco na oczy, drogi oddechowe i skórę.
R37/38
Działa drażniąco na drogi oddechowe i skórę.
R39/23
Działa toksycznie przez drogi oddechowe; zagraża powstaniem bardzo
poważnych, nieodwracalnych zmian w stanie zdrowia.
R39/24
Działa toksycznie w kontakcie ze skórą; zagraża powstaniem bardzo
poważnych, nieodwracalnych zmian w stanie zdrowia.

R39/25
Działa toksycznie po połknięciu; zagraża powstaniem bardzo poważnych,
nieodwracalnych zmian w stanie zdrowia.
R39/23/24
Działa toksycznie przez drogi oddechowe i w kontakcie ze skórą; zagraża
powstaniem bardzo poważnych, nieodwracalnych zmian w stanie
zdrowia.
R39/23/25
Działa toksycznie przez drogi oddechowe i po połknięciu; zagraża
powstaniem bardzo poważnych, nieodwracalnych zmian w stanie
zdrowia.
R39/24/25
Działa toksycznie w kontakcie ze skórą i po połknięciu; zagraża
powstaniem bardzo poważnych nieodwracalnych zmian w stanie zdrowia.
R39/23/24/25
Działa toksycznie przez drogi oddechowe, w kontakcie ze skórą i po
połknięciu; zagraża powstaniem bardzo poważnych nieodwracalnych
zmian w stanie zdrowia.
R39/26
Działa bardzo toksycznie przez drogi oddechowe; zagraża powstaniem
bardzo poważnych nieodwracalnych zmian w stanie zdrowia.
R39/27
Działa bardzo toksycznie w kontakcie ze skórą; zagraża powstaniem
bardzo poważnych nieodwracalnych zmian w stanie zdrowia.
R39/28
Działa bardzo toksycznie po połknięciu; zagraża powstaniem bardzo
poważnych nieodwracalnych zmian w stanie zdrowia.
R39/26/27
Działa bardzo toksycznie przez drogi oddechowe i w kontakcie ze skórą;
zagraża powstaniem bardzo poważnych nieodwracalnych zmian w stanie
zdrowia.
R39/26/28
Działa bardzo toksycznie przez drogi oddechowe i po połknięciu; zagraża
powstaniem bardzo poważnych, nieodwracalnych zmian w stanie
zdrowia.
R39/27/28
Działa bardzo toksycznie w kontakcie ze skórą i po połknięciu; zagraża
powstaniem bardzo poważnych, nieodwracalnych zmian w stanie
zdrowia.
R39/26/27/28
Działa bardzo toksycznie przez drogi oddechowe, w kontakcie ze skórą i
po połknięciu; zagraża powstaniem bardzo poważnych nieodwracalnych
zmian w stanie zdrowia.
R42/43
Może powodować uczulenie w następstwie narażenia drogą oddechową i
w kontakcie ze skórą.
R48/20
Działa szkodliwie przez drogi oddechowe; stwarza poważne zagrożenie
zdrowia w następstwie długotrwałego narażenia.
R48/21
Działa szkodliwie w kontakcie ze skórą; stwarza poważne zagrożenie
zdrowia w następstwie długotrwałego narażenia.
R48/22
Działa szkodliwie po połknięciu; stwarza poważne zagrożenie zdrowia w
następstwie długotrwałego narażenia.
R48/20/21
Działa szkodliwie przez drogi oddechowe i w kontakcie ze skórą; stwarza
poważne zagrożenie zdrowia w następstwie długotrwałego narażenia.
R48/20/22
Działa szkodliwie przez drogi oddechowe i po połknięciu; stwarza
poważne zagrożenie zdrowia w następstwie długotrwałego narażenia.
R48/21/22
Działa szkodliwie w kontakcie ze skórą i po połknięciu; stwarza poważne
zagrożenie zdrowia w następstwie długotrwałego narażenia.
R48/20/21/22
Działa szkodliwie przez drogi oddechowe, w kontakcie ze skórą i po
połknięciu; stwarza poważne zagrożenie zdrowia w następstwie
długotrwałego narażenia.
R48/23
Działa toksycznie przez drogi oddechowe; stwarza poważne zagrożenie
zdrowia w następstwie długotrwałego narażenia.

R48/24
Działa toksycznie w kontakcie ze skórą; stwarza poważne zagrożenie
zdrowia w następstwie długotrwałego narażenia.
R48/25
Działa toksycznie po połknięciu; stwarza poważne zagrożenie zdrowia w
następstwie długotrwałego narażenia.
R48/23/24
Działa toksycznie przez drogi oddechowe i w kontakcie ze skórą; stwarza
poważne zagrożenie zdrowia w następstwie długotrwałego narażenia.
R48/23/25
Działa toksycznie przez drogi oddechowe i po połknięciu; stwarza
poważne zagrożenie zdrowia w następstwie długotrwałego narażenia.
R48/24/25
Działa toksycznie w kontakcie ze skórą i po połknięciu; stwarza poważne
zagrożenie zdrowia w następstwie długotrwałego narażenia.
R48/23/24/25
Działa toksycznie przez drogi oddechowe, w kontakcie ze skórą i po
połknięciu; stwarza poważne zagrożenie zdrowia w następstwie
długotrwałego narażenia.
R50/53
Działa bardzo toksycznie na organizmy wodne; może powodować długo
utrzymujące się niekorzystne zmiany w środowisku wodnym.
R51/53
Działa toksycznie na organizmy wodne; może powodować długo
utrzymujące się niekorzystne zmiany w środowisku wodnym.
R52/53
Działa szkodliwie na organizmy wodne; może powodować długo
utrzymujące się niekorzystne zmiany w środowisku wodnym.
R68/20
Działa szkodliwie przez drogi oddechowe; możliwe ryzyko powstania
nieodwracalnych zmian w stanie zdrowia.
R68/21
Działa szkodliwie w kontakcie ze skórą; możliwe ryzyko powstania
nieodwracalnych zmian w stanie zdrowia.
R68/22
Działa szkodliwie po połknięciu; możliwe ryzyko powstania
nieodwracalnych zmian w stanie zdrowia.
R68/20/21
Działa szkodliwie przez drogi oddechowe i w kontakcie ze skórą;
możliwe ryzyko powstania nieodwracalnych zmian w stanie zdrowia.
R68/20/22
Działa szkodliwie przez drogi oddechowe i po połknięciu; możliwe
ryzyko powstania nieodwracalnych zmian w stanie zdrowia.
R68/21/22
Działa szkodliwie w kontakcie ze skórą i po połknięciu; możliwe ryzyko
powstania nieodwracalnych zmian w stanie zdrowia.
R68/20/21/22
Działa szkodliwie przez drogi oddechowe, w kontakcie ze skórą i po
połknięciu; możliwe ryzyko powstania nieodwracalnych zmian w stanie
zdrowia.
PRAWIDŁOWE POSTĘPOWANIE — ZWROTY S:
UWAGA: Na przykład zapis „S26-36” (pojawiający się w kartach charakterystyk
substancji chemicznej) oznacza, że związku (preparatu) dotyczą zwroty S26 oraz S36, a
nie wszystkie zwroty od S26 do S36!
S1 Przechowywać pod zamknięciem.
S2 Chronić przed dziećmi.
S3 Przechowywać w chłodnym miejscu.
S4 Nie przechowywać w pomieszczeniach mieszkalnych.
S5 Przechowywać w ... (cieczy wskazanej przez producenta).
S6 Przechowywać w atmosferze ... (obojętnego gazu wskazanego przez producenta).
S7 Przechowywać pojemnik szczelnie zamknięty.
S8 Przechowywać pojemnik w suchym pomieszczeniu.

S9 Przechowywać pojemnik w miejscu dobrze wentylowanym.
S12 Nie przechowywać pojemnika szczelnie zamkniętego.
S13 Nie przechowywać razem z żywnością, napojami i paszami dla zwierząt.
S14 Nie przechowywać razem z ... (materiałami określonymi przez producenta).
S15 Przechowywać z dala od źródeł ciepła.
S16 Nie przechowywać w pobliżu źródeł zapłonu — nie palić tytoniu.
S17 Nie przechowywać razem z materiałami zapalnymi.
S18 Zachować ostrożność w trakcie otwierania i manipulacji z pojemnikiem.
S20 Nie jeść i nie pić podczas stosowania produktu.
S21 Nie palić tytoniu podczas stosowania produktu.
S22 Nie wdychać pyłu.
S23 Nie wdychać gazu/dymu/pary/rozpylonej cieczy (rodzaj określi producent).
S24 Unikać zanieczyszczenia skóry.
S25 Unikać zanieczyszczenia oczu.
S26
Zanieczyszczone oczy przemyć natychmiast dużą ilością wody i zasięgnąć porady
lekarza.
S27 Natychmiast zdjąć całą zanieczyszczoną odzież.
S28
Zanieczyszczoną skórę natychmiast przemyć dużą ilością ... (cieczy określonej
przez producenta).
S29 Nie wprowadzać do kanalizacji.
S30 Nigdy nie dodawać wody do tego produktu.
S33 Zastosować środki ostrożności zapobiegające wyładowaniom elektrostatycznym.
S35 Usuwać produkt i jego opakowanie w sposób bezpieczny.
S36 Nosić odpowiednią odzież ochronną.
S37 Nosić odpowiednie rękawice ochronne.
S38
W przypadku niedostatecznej wentylacji stosować odpowiednie indywidualne
środki ochrony dróg oddechowych.
S39 Nosić okulary lub ochronę twarzy.
S40
Czyścić podłogę i wszystkie inne obiekty zanieczyszczone tym produktem ...
(środkiem wskazanym przez producenta).
S41 Nie wdychać dymów powstających w wyniku pożaru lub wybuchu.
S42
Podczas fumigacji/rozpylania/natryskiwania stosować odpowiednie środki ochrony
dróg oddechowych (rodzaj określi producent).
S43
W przypadku pożaru używać ... (podać rodzaj sprzętu przeciwpożarowego). Nigdy
nie używać wody.
S45
W przypadku awarii lub jeżeli źle się poczujesz, niezwłocznie zasięgnij porady
lekarza — jeżeli to możliwe, pokaż etykietę.
S46
W razie połknięcia niezwłocznie zasięgnij porady lekarza — pokaż opakowanie lub
etykietę.
S47 Przechowywać w temperaturze nieprzekraczającej ... °C (określi producent).
S48 Przechowywać produkt zwilżony ... (właściwy materiał określi producent).
S49 Przechowywać wyłącznie w oryginalnym opakowaniu.
S50 Nie mieszać z ... (określi producent).
S51 Stosować wyłącznie w dobrze wentylowanych pomieszczeniach.
S52 Nie zaleca się nanoszenia na duże płaszczyzny wewnątrz pomieszczeń.
S53 Unikać narażenia — przed użyciem zapoznać się z instrukcją.
S56
Zużyty produkt oraz opakowanie dostarczyć na składowisko odpadów
niebezpiecznych.
S57 Używać odpowiednich pojemników zapobiegających skażeniu środowiska.
S59 Przestrzegać wskazówek producenta lub dostawcy dotyczących odzysku lub

wtórnego wykorzystania.
S60 Produkt i opakowanie usuwać jako odpad niebezpieczny.
S61
Unikać zrzutów do środowiska. Postępować zgodnie z instrukcją lub kartą
charakterystyki.
S62
W razie połknięcia nie wywoływać wymiotów: niezwłocznie zasięgnąć porady
lekarza i pokazać opakowanie lub etykietę.
S63
W przypadku zatrucia drogą oddechową wyprowadzić lub wynieść
poszkodowanego na świeże powietrze i zapewnić warunki do odpoczynku.
S64
W przypadku połknięcia wypłukać usta wodą — nigdy nie stosować u osób
nieprzytomnych.
Łączone zwroty S:
S1/2
Przechowywać pod zamknięciem i chronić przed dziećmi.
S3/7
Przechowywać pojemnik szczelnie zamknięty w chłodnym miejscu.
S3/9/14
Przechowywać w chłodnym, dobrze wentylowanym miejscu, z dala od ...
(materiału wskazanego przez producenta).
S3/9/14/49
Przechowywać wyłącznie w oryginalnym opakowaniu, w chłodnym, dobrze
wentylowanym miejscu; nie przechowywać razem z ... (materiałami
wskazanymi przez producenta).
S3/9/49
Przechowywać wyłącznie w oryginalnym opakowaniu w chłodnym, dobrze
wentylowanym miejscu.
S3/14
Przechowywać w chłodnym miejscu; nie przechowywać razem z ...
(materiałami wskazanymi przez producenta).
S7/8
Przechowywać pojemnik szczelnie zamknięty w suchym pomieszczeniu.
S7/9
Przechowywać pojemnik szczelnie zamknięty w miejscu dobrze
wentylowanym.
S7/47
Przechowywać pojemnik szczelnie zamknięty w temperaturze
nieprzekraczającej ... °C (określi producent).
S20/21
Nie jeść i nie pić oraz nie palić tytoniu podczas stosowania produktu.
S24/25
Unikać zanieczyszczenia skóry i oczu.
S27/28
W przypadku zanieczyszczenia skóry natychmiast zdjąć całą
zanieczyszczoną odzież i przemyć zanieczyszczoną skórę dużą ilością ...
(rodzaj cieczy określi producent).
S29/35
Nie wprowadzać do kanalizacji, a produkt i opakowanie usuwać w sposób
bezpieczny.
S29/56
Nie wprowadzać do kanalizacji, a zużyty produkt i opakowanie dostarczyć
na składowisko odpadów niebezpiecznych.
S36/37
Nosić odpowiednią odzież ochronną i odpowiednie rękawice ochronne.
S36/37/39
Nosić odpowiednią odzież ochronną, odpowiednie rękawice ochronne i
okulary lub ochronę twarzy.
S36/39
Nosić odpowiednią odzież ochronną i okulary lub ochronę twarzy.
S37/39
Nosić odpowiednie rękawice ochronne i okulary lub ochronę twarzy.
S47/49
Przechowywać wyłącznie w oryginalnym opakowaniu w temperaturze
nieprzekraczającej ... °C (określi producent).
3B. ZWROTY H oraz ZWROTY P (obowiązujące od 2011 roku):
Zwroty H:

H 200 materiały wybuchowe niestabilne
H 201
materiał wybuchowy; zagrożenie wybuchem masowym
H 202
materiał wybuchowy; poważne zagrożenie rozrzutem
H 203
materiał wybuchowy; zagrożenie pożarem, wybuchem lub rozrzutem
H 204
zagrożenie pożarem lub rozrzutem
H 205
może wybuchać masowo w przypadku pożaru
H 220
skrajnie łatwopalny gaz
H 221
gaz łatwopalny
H 222
skrajnie łatwopalny aerozol
H 223
aerozol łatwopalny
H 224
skrajnie łatwopalna ciecz i pary
H 225
wysoce łatwopalna ciecz i pary
H 226
łatwopalna ciecz i pary
H 228
substancja stała łatwopalna
H 240
ogrzanie grozi wybuchem
H 241
ogrzanie może spowodować pożar lub wybuch
H 242
ogrzanie może spowodować pożar
H 250
zapala się samorzutnie w przypadku wystawienia na działanie powietrza
H 251
substancja samonagrzewająca się; może się zapalić
H 252
substancja samonagrzewająca się w dużych ilościach; może się zapalić
H 260
w kontakcie z wodą uwalnia łatwopalne gazy, które mogą ulec samozapaleniu
H 261
w kontakcie z wodą uwalnia łatwopalne gazy
H 270
może spowodować lub intensyfikować pożar; utleniacz
H 271
może spowodować pożar lub wybuch; silny utleniacz
H 272
może intensyfikować pożar; utleniacz
H 280
zawiera gaz pod ciśnieniem; ogrzanie grozi wybuchem
H 281
zawiera schłodzony gaz; może spowodować oparzenia kriogeniczne lub
obrażenia
H 290
może powodować korozję metali
H 300
połknięcie grozi śmiercią
H 301
działa toksycznie po połknięciu
H 302
działa szkodliwie po połknięciu
H 304
połknięcie i dostanie się przez drogi oddechowe może grozić śmiercią
H 310
grozi śmiercią w kontakcie ze skórą
H 311
działa toksycznie w kontakcie ze skórą
H 312
działa szkodliwie w kontakcie ze skórą
H 314
powoduje poważne oparzenia skóry oraz uszkodzenia oczu
H 315
działa drażniąco na skórę
H 317
może powodować reakcję alergiczną skóry
H 318
powoduje poważne uszkodzenie oczu
H 319
działa drażniąco na oczy
H 330
wdychanie grozi śmiercią
H 331
działa toksycznie w następstwie wdychania
H 332
działa szkodliwie w następstwie wdychania
H 334
może powodować objawy alergii lub astmy lub trudności w oddychaniu, w
następstwie wdychania
H 335
może powodować podrażnienie dróg oddechowych
H 336
może wywoływać uczucie senności lub zawroty głowy
H 340
może powodować wady genetyczne
H 341
podejrzewa się, że powoduje wady genetyczne

H 350
może powodować raka
H 351
podejrzewa się, że powoduje raka
H 360
może działać szkodliwie na płodność lub na dziecko w łonie matki
H 361
podejrzewa się, że działa szkodliwie na płodność lub na dziecko w łonie matki
H 362
może działać szkodliwie na dzieci karmione piersią
H 370
powoduje uszkodzenie narządów .........
H 371
może powodować uszkodzenie narządów ...........
H 372
powoduje uszkodzenie narządów ........., poprzez długotrwałe lub powtarzane
narażenie
H 373
może powodować uszkodzenie narządów .........., poprzez długotrwałe lub
powtarzane narażenie
H 400
działa bardzo toksycznie na organizmy wodne
H 410
działa bardzo toksycznie na organizmy wodne, powodując długotrwałe skutki
H 411
działa toksycznie na organizmy wodne, powodując długotrwałe skutki
H 412
działa szkodliwie na organizmy wodne, powodując długotrwałe skutki
H 413
może powodować długotrwałe szkodliwe skutki dla organizmów wodnych
Dla krajów Unii Europejskiej ponadto:
EUH 001
produkt wybuchowy w stanie suchym
EUH 006 produkt wybuchowy z dostępem lub bez dostępu powietrza
EUH 014 reaguje gwałtownie z wodą
EUH 018 podczas stosowania mogą powstawać łatwopalne lub wybuchowe
mieszaniny par z powietrzem
EUH 019 może tworzyć wybuchowe nadtlenki
EUH 044 zagrożenie wybuchem po ogrzaniu w zamkniętym pojemniku
EUH 029 w kontakcie z wodą uwalnia toksyczne gazy
EUH 031 w kontakcie z kwasami uwalnia toksyczne gazy
EUH 032 w kontakcie z kwasami uwalnia bardzo toksyczne gazy
EUH 066 powtarzające się narażenie może powodować wysuszanie lub pękanie
skóry
EUH 070 działa toksycznie w kontakcie z oczami
EUH 071 działa drażniąco na drogi oddechowe
EUH 059 stwarza zagrożenie dla warstwy ozonowej
EUH 201 zawiera ołów; nie należy stosować na powierzchniach, które mogą być
gryzione lub ssane przez dzieci
EUH 201A Uwaga! Zawiera ołów.
EUH 202 Cyjanoakrylany. Niebezpieczeństwo. Skleja skórę i powieki w ciągu kilku
sekund. Chronić przed dziećmi.
EUH 203 Zawiera chrom(VI). Może powodować wystąpienie reakcji alergicznej.
EUH 204 Zawiera izocyjaniany. Może powodować wystąpienie reakcji alergicznej.
EUH 205 Zawiera składniki epoksydowe. Może powodować wystąpienie reakcji
alergicznej.
EUH 206 Uwaga! Nie stosować razem z innymi produktami. Może wydzielać
niebezpieczne gazy (chlor).
EUH 207 Uwaga! Zawiera kadm. Podczas stosowania wydziela niebezpieczne pary.
Zapoznaj się z instrukcją dostarczoną przez producenta. Przestrzegaj
instrukcji bezpiecznego stosowania.
EUH 208 Zawiera ............ . Może powodować wystąpienie reakcji alergicznej.
EUH 209 Podczas stosowania może przekształcić się w substancję wysoce
łatwopalną.
EUH 209A Podczas stosowania może przekształcić się w substancję łatwopalną.

EUH 210 Karta charakterystyki dostępna na żądanie.
EUH 401 W celu uniknięcia zagrożeń dla zdrowia ludzi i środowiska, należy
postępować zgodnie z instrukcją użycia.
Zwroty P:
P101
W razie konieczności zasięgnięcia porady lekarza należy pokazać
pojemnik lub etykietę.
P102
Chronić przed dziećmi.
P103
Przed użyciem przeczytać etykietę.
P201
Przed użyciem zapoznać się ze specjalnymi środkami ostrożności.
P202
Nie używać przed zapoznaniem się i zrozumieniem wszystkich
środków bezpieczeństwa.
P210
Przechowywać z dala od źródeł ciepła/iskrzenia/otwartego
ognia/gorących powierzchni. Palenie wzbronione.
P211
Nie rozpylać nad otwartym ogniem lub innym źródłem zapłonu.
P220
Trzymać/przechowywać z dala od odzieży/.../materiałów
zapalnych.
P221
Zastosować wszelkie środki ostrożności w celu uniknięcia
mieszania z innymi materiałami zapalnymi ...
P222
Nie dopuszczać do kontaktu z powietrzem.
P223
Chronić przed wszelkim kontaktem z wodą z powodu gwałtownej
reakcji i możliwości wystąpienia błyskawicznego pożaru.
P230
Przechowywać produkt zwilżony....
P231
Używać w atmosferze obojętnego gazu.
P232
Chronić przed wilgocią.
P233
Przechowywać pojemnik szczelnie zamknięty
P234
Przechowywać wyłącznie w oryginalnym pojemniku.
P235
Przechowywać w chłodnym miejscu.
P240
Uziemić/połączyć pojemnik i sprzęt odbiorczy.
P241
Używać elektrycznego/wentylującego/oświetleniowego/.../.
przeciwwybuchowego sprzętu
P242
Używać wyłącznie nieiskrzących narzędzi.
P243
Przedsięwziąć środki ostrożności zapobiegające statycznemu
rozładowaniu.
P244
Chronić zawory redukcyjne przed tłuszczem i olejem.
P250
Nie poddawać szlifowaniu/wstrząsom/.../tarciu.
P251
Pojemnik pod ciśnieniem. Nie przekłuwać ani nie spalać, nawet po
zużyciu.
P260
Nie wdychać pyłu/dymu/gazu/mgły/par/rozpylonej cieczy.
P261
Unikać wdychania pyłu/dymu/gazu/mgły/par/rozpylonej cieczy.
P262
Nie wprowadzać do oczu, na skórę lub na odzież
P263
Unikać kontaktu w czasie ciąży/karmienia piersią.
P264
Dokładnie umyć ... po użyciu.
P270
Nie jeść, nie pić i nie palić podczas używania produktu.
P271
Stosować wyłącznie na zewnątrz lub w dobrze wentylowanym
pomieszczeniu
P272
Zanieczyszczonej odzieży ochronnej nie wynosić poza miejsce
pracy.
P273
Unikać uwolnienia do środowiska.

P280
Stosować rękawice ochronne/ odzież ochronną/ ochronę oczu
/ochronę twarzy.
P281
Stosować wymagane środki ochrony indywidualnej.
P282
Nosić rękawice izolujące od zimna/maski na twarz/ochronę oczu.
P283
Nosić odzież ognioodporną/płomienioodporną/opóźniającą
zapalenie.
P284
Stosować indywidualne środki ochrony dróg oddechowych.
P285
W przypadku niedostatecznej wentylacji stosować indywidualne
środki ochrony dróg oddechowych.
P231 + P232
Używać w atmosferze obojętnego gazu Chronić przed wilgocią.
P235 + P410
Przechowywać w chłodnym miejscu. Chronić przed światłem
słonecznym.
P301
W PRZYPADKU POŁKNIĘCIA:
P302
W PRZYPADKU KONTAKTU ZE SKÓRĄ:
P303
W PRZYPADKU KONTAKTU ZE SKÓRĄ (lub z włosami):
P304
W PRZYPADKU DOSTANIA SIĘ DO DRÓG
ODDECHOWYCH:
P305
W PRZYPADKU DOSTANIA SIĘ DO OCZU:
P306
W PRZYPADKU KONTAKTU Z ODZIEŻĄ:
P307
W PRZYPADKU narażenia:
P308
Z PRZYPADKU narażenia lub styczności:
P309
W PRZYPADKU narażenia lub złego samopoczucia:
P310
Natychmiast skontaktować się z OŚRODKIEM ZATRUĆ lub
lekarzem.
P311
Skontaktować się z OŚRODKIEM ZATRUĆ lub lekarzem.
P312
W przypadku złego samopoczucia skontaktować się z
OŚRODKIEM ZATRUĆ lub z lekarzem.
P313
Zasięgnąć porady/zgłosić się pod opiekę lekarza.
P314
W przypadku złego samopoczucia zasięgnąć porady/zgłosić się
pod opiekę lekarza.
P315
Natychmiast zasięgnąć porady/zgłosić się pod opiekę lekarza.
P320
Pilnie zastosować określone leczenie (patrz ... na etykiecie).
P321
Zastosować określone leczenie (patrz ... na etykiecie).
P322
Środki szczególne (patrz ... na etykiecie).
P330
Wypłukać usta.
P331
NIE wywoływać wymiotów.
P332
W przypadku wystąpienia podrażnienia skóry:
P333
W przypadku wystąpienia podrażnienia skóry lub wysypki:
P334
Zanurzyć w zimnej wodzie/owinąć mokrym bandażem.
P335
Nie związaną pozostałość strzepnąć ze skóry.
P336
Rozmrozić oszronione obszary letnią wodą. Nie trzeć
oszronionego obszaru.
P337
W przypadku utrzymywania się działania drażniącego na oczy:
P338
Wyjąć soczewki kontaktowe, jeżeli są i można je łatwo usunąć.
Nadal płukać.
P340
Wyprowadzić lub wynieść poszkodowanego na świeże powietrze i
zapewnić warunki do odpoczynku w pozycji umożliwiającej
swobodne oddychanie.
P341
W przypadku trudności z oddychaniem, wyprowadzić lub wynieść
poszkodowanego na świeże powietrze i zapewnić warunki do

odpoczynku w pozycji umożliwiającej swobodne oddychanie.
P342
W przypadku wystąpienia objawów ze strony układu
oddechowego:
P350
Delikatnie umyć dużą ilością wody z mydłem.
P351
Ostrożnie płukać wodą przez kilka minut.
P352
Umyć dużą ilością wody z mydłem.
P353
Spłukać skórę pod strumieniem wody/prysznicem.
P360
Natychmiast spłukać zanieczyszczoną odzież i skórę dużą ilością
wody przed zdjęciem odzieży.
P361
Natychmiast usunąć/zdjąć całą zanieczyszczoną odzież.
P362
Zanieczyszczoną odzież zdjąć i wyprać przed ponownym użyciem.
P363
Wyprać zanieczyszczoną odzież przed ponownym użyciem.
P370
W przypadku pożaru:
P371
W przypadku poważnego pożaru i dużych ilości:
P372
Ryzyko wybuchu w razie pożaru.
P373
NIE gasić pożaru, jeżeli ogień dosięgnie materiały wybuchowe
P374
Gasić pożar z rozsądnej odległości z zachowaniem zwykłych
środków ostrożności.
P375
Z powodu ryzyka wybuchu gasić pożar z odległości.
P376
Jeżeli jest to bezpieczne zahamować wyciek.
P377
W przypadku płonięcia wyciekającego gazu: Nie gasić, jeżeli nie
można bezpiecznie zahamować wycieku.
P378
Użyć ... do gaszenia.
P380
Ewakuować teren.
P381
Wyeliminować wszystkie źródła zapłonu, jeżeli jest to bezpieczne.
P390
Usunąć wyciek, aby zapobiec szkodom materialnym.
P391
Zebrać wyciek.
P301 + P310
W PRZYPADKU POŁKNIĘCIA: Natychmiast skontaktować się z
OŚRODKIEM ZATRUĆ lub z lekarzem.
P301 + P312
W PRZYPADKU POŁKNIĘCIA: W przypadku złego
samopoczucia skontaktować się z OŚRODKIEM ZATRUĆ lub z
lekarzem.
P301 + P330 + P331
W PRZYPADKU POŁKNIĘCIA: wypłukać usta. NIE wywoływać
wymiotów.
P302 + P334
W PRZYPADKU KONTAKTU ZE SKÓRĄ: Zanurzyć w zimnej
wodzie/owinąć mokrym bandażem.
P302 + P350
W PRZYPADKU DOSTANIA SIĘ NA SKÓRĘ: Delikatnie umyć
dużą iloścą wody z mydłem.
P302 + P352
W PRZYPADKU KONTAKTU ZE SKÓRĄ: Umyć dużą ilością
wody z mydłem.
P303 + P361 + P353
W PRZYPADKU KONTATKU ZE SKÓRĄ (lub z włosami):
Natychmiast usunąć/zdjąć całą zanieczyszczoną odzież. Spłukać
skórę pod strumieniem wody/prysznicem.
P304 + P340
W PRZYPADKU DOSTANIA SIĘ DO DRÓG
ODDECHOWYCH: wyprowadzić lub wynieść poszkodowanego
na świeże powietrze i zapewnić warunki do odpoczynku w pozycji
umożliwiającej swobodne oddychanie.
P304 + P341
W PRZYPADKU DOSTANIA SIĘ DO DRÓG
ODDECHOWYCH: W przypadku trudności z oddychaniem,
wyprowadzić lub wynieść poszkodowanego na świeże powietrze i

zapewnić warunki do odpoczynku w pozycji umożliwiającej
swobodne oddychanie.
P305 + P351 + P338
W PRZYPADKU DOSTANIA SIĘ DO OCZU: Ostrożnie płukać
wodą przez kilka minut. Wyjąć soczewki kontaktowe, jeżeli są i
można je łatwo usunąć. Nadal płukać.
P306 + P360
W PRZYPADKU KONTAKTU Z ODZIEŻĄ: natychmiast
spłukać zanieczyszczoną odzież i skórę dużą ilością wody przed
zdjęciem odzieży.
P307 + P311
W przypadku narażenia: Skontaktować się z OŚRODKIEM
ZATRUĆ lub z lekarzem.
P308 + P313
W przypadku narażenia lub styczności: Zasięgnąć porady/zgłosić
się pod opiekę lekarza.
P309 + P311
W przypadku narażenia lub złego samopoczucia: Skontaktować się
z OŚRODKIEM ZATRUĆ lub z lekarzem.
P332 + P313
W przypadku wystąpienia podrażnienia skóry: Zasięgnąć
porady/zgłosić się pod opiekę lekarza.
P333 + P313
W przypadku wystąpienia podrażnienia skóry lub wysypki:
Zasięgnąć porady/zgłosić się pod opiekę lekarza.
P335 + P334
Nie związaną pozostałość strzepnąć ze skóry. Zanurzyć w zimnej
wodzie/owinąć mokrym bandażem.
P337 + P313
W przypadku utrzymywania się działania drażniącego na oczy:
Zasięgnąć porady/zgłosić się pod opiekę lekarza.
P342 + P311
W przypadku wystąpienia objawów ze strony układu
oddechowego: Skontaktować się z OŚRODKIEM ZATRUĆ lub z
lekarzem.
P370 + P376
W przypadku pożaru: Jeżeli jest to bezpieczne zahamować wyciek.
P370 + P378
W przypadku pożaru: Użyć ... do gaszenia.
P370 + P380
W przypadku pożaru: Ewakuować teren.
P370 + P380 + P375
W przypadku pożaru: Ewakuować teren. Z powodu ryzyka
wybuchu gasić pożar z odległości.
P371 + P380 + P375
W przypadku poważnego pożaru i dużych ilości: Ewakuować
teren. Z powodu ryzyka wybuchu gasić pożar z odległości.
P401
Przechowywać ...
P402
Przechowywać w suchym miejscu.
P403
Przechowywać w dobrze wentylowanym miejscu.
P404
Przechowywać w zamkniętym pojemniku.
P405
Przechowywać pod zamknięciem.
P406
Przechowywać w pojemniku odpornym na korozję /... o odpornej
powłoce wewnętrznej.
P407
Zachować szczelinę powietrzną pomiędzy stosami/paletami.
P410
Chronić przed światłem słonecznym.
P411
Przechowywać w temperaturze nieprzekraczającej ...
o
C/...
o
F.
P412
Nie wystawiać na działanie temperatury przekraczającej 50
o
C/122
o
F.
P413
Przechowywać luzem masy przekraczające ... kg/... funtów w
temperaturze nieprzekraczającej ...
o
C/...
o
F.
P420
Przechowywać z dala od innych materiałów.
P422
Zawartość przechowywać w ...
P402 + P404
Przechowywać w suchym miejscu. Przechowywać w zamkniętym
pojemniku.

P403 + P233
Przechowywać w dobrze wentylowanym miejscu. Przechowywać
pojemnik szczelnie zamknięty.
P403 + P235
Przechowywać w dobrze wentylowanym miejscu. Przechowywać
w chłodnym miejscu.
P410 + P403
Chronić przed światłem słonecznym. Przechowywać w dobrze
wentylowanym miejscu.
P410 + P412
Chronić przed światłem słonecznym. Nie wystawiać na działanie
temperatury przekraczającej 50
o
C/122
o
F.
P411 + P235
Przechowywać w temperaturze nieprzekraczającej ...
o
C/...
o
F.
Przechowywać w chłodnym miejscu.
P501
Zawartość/pojemnik usuwać do ...
Załącznik 4. Nomogram określający
zależność pomiędzy ciśnieniem a
temperaturą wrzenia
Sposób użycia:
1. Aby określić temperaturę wrzenia cieczy pod normalnym ciśnieniem (760 mmHg)
znając jej temperaturę wrzenia pod ciśnieniem zmniejszonym p (np. 80 °C przy 10
mmHg), należy poprowadzić linię prostą łączącą zmierzoną temperaturę wrzenia
(80 °C; SKALA A) z wartością ciśnienia p zanotowaną podczas destylacji (10 mmHg;
SKALA C). Punkt przecięcia ze SKALĄ B określa temperaturę wrzenia pod
ciśnieniem normalnym (ok. 200 °C).
2. Bardzo podobnie postępuje się, gdy znana jest temperatura wrzenia pod ciśnieniem
normalnym (np. 300 °C; SKALA B) i ciśnienie p panujące w zestawie destylacyjnym
(np. 2,0 mmHg; SKALA C). Po połączeniu linią prostą odpowiednich punktów na
SKALI B i na SKALI C, punkt przecięcia tej prostej ze SKALĄ A wskaże
przybliżoną wartość oczekiwanej temperatury wrzenia (ok. 130 °C).
Załącznik 5. Dane fizykochemiczne
wybranych rozpuszczalników oraz
roztworów amoniaku i kwasów
nieorganicznych
Wybrane rozpuszczalniki
Rozpuszczalnik
(nazwa zwyczajowa)
Temp. wrzenia
*
o
C
Rozpuszczalność
w wodzie
*
g w 100 g
Gęstość
*
g/cm
3
Symbole
zagrożeń
Cykloheksan
81
< 0,1
0,78
F
Dichlorometan
(chlorek metylenu)
41
1,3
1,33
Xn
1,4-Dioksan
101
∞
1,03
F
Etanol
78
∞
0,79
F
Eter dietylowy
35
6
0,71
F+, Xn
Eter naftowy 40/60
(benzyna lekka)
40–60
< 0,1
ok. 0,65 F, Xn
Eter naftowy 60/90
(ligroina)
60–90
< 0,1
ok. 0,70 F, Xn
Kwas octowy
118
∞
1,05
C
Metanol
65
∞
0,79
F, T
Octan etylu
78
8,1
0,9
F, Xi
Propan-2-ol
(alkohol izopropylowy)
82
∞
0,78
F, Xi
Propan-2-on
(aceton)
56
∞
0,79
F, Xi
Tetrachlorometan
(tetrachlorek węgla)
77
< 0,1
1,59
T, N
Toluen
111
< 0,1
0,87
F, Xn
Trichlorometan
(chloroform)
61
0,8
1,49
Xn
Roztwory amoniaku i kwasów nieorganicznych
Nazwa systematyczna
(zwyczajowa)
Stężenie procentowe
%
Stężenie molowe
mol/dm3
Gęstość*
g/cm
3
Symbole
zagrożeń
Kwas azotowy(V) stęż.
65
14,4
1,39
C
Kwas azotowy(V) dym.
70
15,8
1,41
C
Kwas chlorowodorowy stęż.
(kwas solny)
36
11,7
1,18
C
Kwas fosforowy(V) stęż.
(kwas ortofosforowy)
85
14,8
1,7
C
Kwas siarkowy(VI) stęż.
95
17,8
1,83
C
98
18,4
1,84
C
Woda amoniakalna stęż.
25
13,4
0,91
C, N
Załącznik 6. Schemat wykonania
pudełeczka z papieru techniką origami
(Rysunki i opisy na podstawie książki:
F. Temko, „Origami. Pudełka z papieru”,
, za zgodą Wydawnictwa.)
Pudełko można wykonać z dowolnej prostokątnej kartki papieru. Rozmiar kartki jest
uzależniony od ilości substancji. Najczęściej wykorzystuje się papier formatu A5 lub A6.
Papier nie powinien być pokryty zbyt ciemnym drukiem, bo wówczas nie jest widoczny
podpis na pudełku. Pudełeczek nie wolno wykonywać z bibuły filtracyjnej.
Krok 1. Złóż kartkę papieru na pół wzdłuż dłuższego boku, a następnie rozłóż papier na
płasko:
Krok 2. Przyłóż obie krawędzie do środkowego zgięcia, a następnie rozłóż papier na płasko:
Krok 3. Złóż kartkę papieru na pół wzdłuż krótszego boku, a następnie rozłóż papier na
płasko:
Krok 4. Przyłóż obie krótsze krawędzie do zgięcia w środku. Nie rozkładaj papieru:
Krok 5. Zagnij wszystkie cztery narożniki do najbliższych zgięć poprzecznych:
Krok 6. Wywiń obie wystające krawędzie w środku papieru na zagięte narożniki. Zaznacz
ostro te linie zagięcia, gdyż one trzymają konstrukcję pudełka:
Krok 7. Rozciągnij konstrukcję zgodnie z narysowanymi strzałkami:
Krok 8. Pudełeczko prawie gotowe. Należy tylko dokładnie „zaprasować” krawędzie pudełka
oraz podpisać go nazwiskiem i nazwą oraz numerem preparatu. Podpis można zrobić dużo
łatwiej w Kroku 6, gdy konstrukcja jest jeszcze płaska.
Załącznik 7. Wzór sprawozdania z syntezy
związku organicznego
Imię i nazwisko osoby wykonującej
ćwiczenie
Data wykonania ćwiczenia
A) Numer i tytuł ćwiczenia
np. II.33. Synteza benzoesanu fenylu
B) Schemat reakcji
C) Mechanizm reakcji
Jest to przykład reakcji estryfikacji, w której następuje atak nukleofilowy anionu
fenolanowego na karbonylowy atom węgla w chlorku benzoilu. Reakcja jest prowadzona w
warunkach katalizy przeniesienia międzyfazowego (PTC). Katalizatorem jest chlorek
benzylotrietyloamoniowy (TEBA).

Anion fenolanowy jest przenoszony do fazy organicznej (CH
2
Cl
2
) w postaci pary jonowej z
kationem benzylotrietyloamoniowym. W fazie organicznej zachodzi reakcja substytucji
nukleofilowej, a następnie kation organiczny w postaci soli z anionem chlorkowym jest
przenoszony do fazy wodnej. Ester pozostaje w fazie organicznej.
D) Opis w punktach kolejnych etapów syntezy
Sprawozdanie należy pisać zwięźle w czasie przeszłym podając własne obserwacje i uwagi.
Nie należy przepisywać instrukcji do ćwiczenia. W przypadku bardziej skomplikowanych
przepisów pomocne jest narysowanie schematu obrazującego kolejne etapy postępowania z
produktem oraz powstającymi odpadami (tzw. „drzewko” w Załączniku 8).
1. W kolbie stożkowej o poj. 100 cm
3
umieszczono 20 cm
3
wody, do której dodano
kolejno 1,5 g wodorotlenku sodu, 3,5 g fenolu oraz 0,1 g TEBA.
2. Zawartość kolby mieszano aż do uzyskania klarownego roztworu.
3. Do uzyskanego roztworu dodano 3,0 cm
3
chlorku benzoilu w 20 cm
3
chlorku
metylenu.
4. Dwufazową mieszaninę mieszano intensywnie mieszadłem magnetycznym przez 1
godzinę.
5. Mieszaninę umieszczono w rozdzielaczu i oddzielono warstwę organiczną (dolną).
Przemyto ją wodnym roztworem NaOH, a następnie 20 cm
3
wody.
6. Warstwę organiczną wysuszono bezwodnym siarczanem(VI) magnezu, następnie
przesączono ją przez sączek fałdowany do uprzednio starowanej kolby okrągłodennej
i odpędzono chlorek metylenu na wyparce.
7. Surowy ester (ok. 4,5 g) oczyszczono przez krystalizację z ok. 35 cm
3
etanolu i
wysuszono go dokładnie na powietrzu.
8. Otrzymano 3,44 g produktu barwy kremowej o tt. 67–68
o
C.
9. Na polecenie asystenta sprawdzono czystość produktu metodą chromatografii
cienkowarstwowej (CHCl
3
/ SiO
2
)
Chromatogram należy wkleić do sprawozdania i opatrzyć go krótkim komentarzem.
E) Porównanie zmierzonej temperatury topnienia z wartością podaną w przepisie
zmierzona tt. 67–68
o
C; literaturowa tt. 69
o
C

W przypadku gdy różnica pomiędzy tymi wartościami jest dość znaczna (>5
o
C), należy ten
fakt skomentować lub powtórzyć krystalizację związku ewentualnie poddać go dalszemu
suszeniu.
F) Obliczenie wydajności reakcji
Do reakcji wzięto:
3,50 g fenolu (masa cząsteczkowa = 94,1)
3,00 cm
3
chlorku benzoilu (masa cząsteczkowa = 140,6)
W reakcji otrzymano:
3,44 g benzoesanu fenylu (masa cząsteczkowa = 198,2)
Przy użyciu nadmiaru jednego z substratów wydajność oblicza się zawsze względem
substratu będącego w niedomiarze, ponieważ jego ilość warunkuje maksymalną teoretyczną
wydajność reakcji. W tym celu należy wyrazić użyte ilości substratów w molach i porównać je
ze sobą.
Liczba moli cząsteczek fenolu = 3,50 g / 94,1 g/mol = 0,037 mola
Liczba moli cząsteczek chlorku benzoilu:
gęstość chlorku benzoilu d = 1,21 g/cm
3
masa chlorku benzoilu = 3,00 cm
3
· 1,21 g/cm
3
= 3,63 g
liczba moli cząsteczek chlorku benzoilu = 3,63 g / 140,61 g/mol = 0,026 mola
Wydajność reakcji liczymy więc względem chlorku benzoilu.
Przy wydajności 100% powinno się otrzymać
140,6 g chlorku benzoilu ----------------- 198,2 g benzoesanu fenylu
3,63 g ---------------------------------------- x g
x = (3,63 g · 198,2 g) / 140,6 g = 5,12 g benzoesanu fenylu
Wydajność praktyczna wynosi zatem:
5,12 g benzoesanu fenylu ------------- 100%
3,44 g -------------------------------------- x%
x = (3,44 g / 5,12 g) · 100% = 67,2%
W przypadku obniżonej wydajności należy próbować uzasadnić logicznie ten fakt, a w
przypadku zbyt niskiej wydajności powtórzyć syntezę, unikając uprzednio popełnionych
błędów. Także zbyt wysoka wydajność reakcji sugeruje, że produkt nie jest dostatecznie
wysuszony lub oczyszczony.
G) Komentarz

Otrzymano benzoesan fenylu z dobrą wydajnością 67,2%. Pewne straty produktu
zaobserwowano podczas krystalizacji z etanolu, bo użyto nieco zbyt dużo rozpuszczalnika.
Barwa związku nieco odbiega od podanej w przepisie, ale produkt jest dostatecznie czysty, co
potwierdza oznaczona temperatura topnienia (różnica zaledwie 2
o
C w stosunku do wartości
literaturowej) oraz załączony chromatogram (na płytce chromatograficznej widoczna jest po
naświetleniu promieniowaniem UV jedna wyraźna plamka).
H) Przykład obliczenia wydajności syntezy wieloetapowej
Dla syntezy wieloetapowej oblicza się najpierw wydajność każdego etapu w sposób podany
powyżej. Wydajność syntezy wieloetapowej jest iloczynem wydajności poszczególnych etapów
syntezy. Sposób obliczeń obrazuje następujący schemat:
wyd. produktu B = 50%
wyd. syntezy dwuetapowej (produkt C) = 50% · 40% = 20%
wyd. syntezy trójetapowej (produkt D) = 50% · 40% · 70% = 14%
Załącznik 8. Schemat rozdzielenia
mieszaniny otrzymanej podczas syntezy
związku organicznego
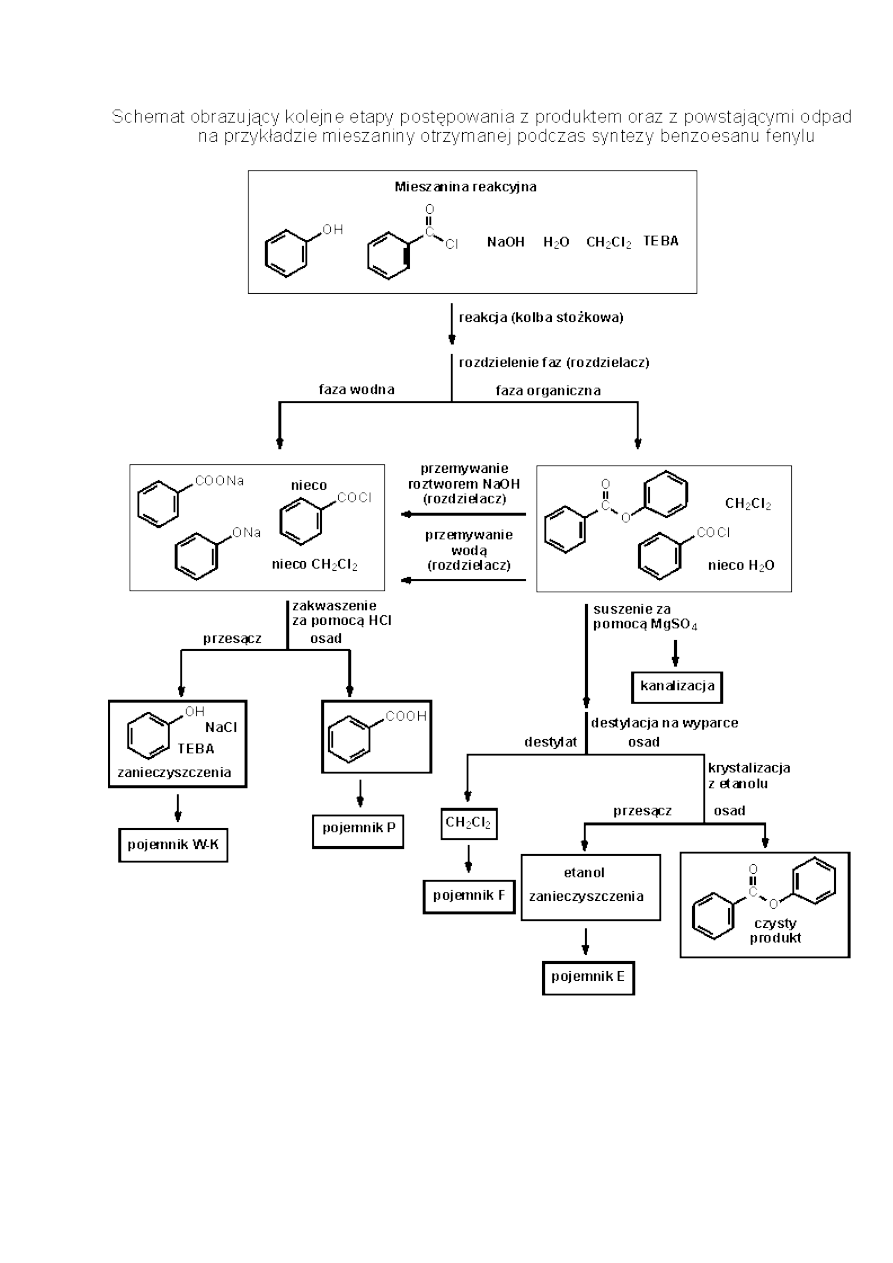
Załącznik 9. Przykładowy zestaw
preparatów do wykonania podczas
pracownii chemii organicznej dla studentów
chemii
III.b.3
III.e.6 T
V.11 D
s
, W
I.4 R
IV.6
VI.a.7
II.18
III.c.7
II.27 E,W
VI.b.6 E, D
p
VII.18
III.a.4
Zadanie specjalne
(obowiązkowe)
Zadanie asystenckie (decyduje prowadzący
ćwiczenia)
ciągi reakcji:
1) III.b.3 → III.e.6
2) V.11 → I.4
5) II.18 → III.c.7 → II.27
7) VII.18 → III.a.4
syntezy jednoetapowe:
3) IV.6
4) VI.a.7
6) VI.b.6
Objaśnienia skrótów:
E — ekstrakcja
D
s
— destylacja substratu

D
p
— destylacja produktu
R — dobór rozpuszczalnika do krystalizacji
W — destylacja z parą wodną
T — chromatografia cienkowarstwowa
Wyszukiwarka
Podobne podstrony:
ćwiczenia laboratoryjne nr 2
!Program ćwiczeń laboratoryjnych 2012id 602
Program ćwiczeń laboratoryjnych
Ćwiczenie laboratoryjne nr 6 materiały
Ćwiczenia laboratoryjne PBiI (1) - konspekt, Studia INiB, Podstawy bibliotekoznawstwa i informacji n
SPRAWOZDANIE Z ĆWICZEŃ LABORATORYJNYCII
ĆWICZENIA LABORATORYJNE Z CHEMII grafik
Instrukcja do ćwiczenia laboratoryjnego PDH
chromatografia - ćwiczenie laboratoryjne, Studia Zip, Semestr 1, Chemia
CLAB 6-1 2008-2009, Tematy ćwiczeń laboratoryjnych z Języka Programowania
26, wstep, ĆWICZENIE LABORATORYJNE NR 26.
TiSP - dok, Lab. TiSP - Wykaz ćwiczeń, LABORATORIUM
Grafika rastrowa 2, Opis ćwiczenia2, Ćwiczenia laboratoryjne
SPRAWOZDANIE Z ĆWICZEŃ LABORATORYJNYC III
IBN Pytania na ćwiczenia laboratoryjne z chemii
ZAGADNIENIA NA EGZAMIN Z MECHANIKI TECHNICZNEJ II DLA SEMESTRU III, sem III, +Mechanika Techniczna I
4SPRAWOZDANIE DO CWICZENIA LABORATORYJNEGO Z FIZYKI BUDOWLI
więcej podobnych podstron