
Genetyka w praktyce klinicznej
Rozdział 3
Genetyka w praktyce klinicznej
Niezwykły wzrost liczby chorób uwarunkowa-
nych mutacjami genowymi wynika z zastoso-
wania nowych technologii, szeroko omówio-
nych w poprzednich rozdziałach. Liczba chorób
genetycznie uwarunkowanych i loci skatalogo-
wanych w bazie Mendelian Inheritance in Man
wzrosła z około 1500 w 1964 roku do ponad 15
tysięcy obecnie. Od 1995 roku powyższy katalog
dostępny jest w wersji elektronicznej, jako On-
line Mendelian Inheritance in Man (OMIM),
na stronie: http://www.ncbi.nlm.nih.gov/si-
tes/entrez?db=OMIM. W poniższym rozdziale
przedstawiono wybrane choroby o znanym lub
prawdopodobnym podłożu genetycznym oraz
opisano technologie stosowane w celu ich roz-
poznania i w próbach leczenia.
CHOROBY GENETYCZNIE
UWARUNKOWANE
Choroby dziedziczne uwarunkowane są sze-
rokim spektrum defektów genetycznych, od
zmian pojedynczych nukleotydów (niedo-
krwistość sierpowatokrwinkowa) po utratę
lub obecność dodatkowego całego chromo-
somu (zespół Downa). Najpowszechniej wy-
stępujące choroby genetyczne u człowieka
są uwarunkowane poligenowo i wynikają
z obecności wielu mutacji lub wariantów ge-
nowych w różnych loci. Spośród nich każda
zmiana wywiera niewielki, lecz addytyw-
ny, wpływ na ujawnienie się choroby. Wiek,
w którym pojawiają się pierwsze objawy cho-
roby oraz jej ekspresja kliniczna, zależą także
od wpływu czynników środowiskowych, stąd
określenie choroby wieloczynnikowe.
ABERRACJE CHROMOSOMOWE
Zagadnienia ogólne
Wiele nieprawidłowości (aberracji) chromo-
somowych wynika z utraty całego chromo-
somu (zespół Turnera) [ryc. 3.1], obecności
dodatkowego chromosomu (zespół Downa)
[ryc. 3.2] lub podobnych zmian, lecz doty-
czących fragmentów chromosomów, np. de-
lecje, duplikacje, inwersje, translokacje [patrz
rozdz. 2, str. 67]. Obserwowane w przebiegu
tych chorób problemy medyczne i rozwojo-
we uwarunkowane są dysfunkcją jednego lub
wielu ważnych genów.
Aberracje chromosomowe występują często
i stanowią prawie połowę przyczyn wczesnych
poronień. Stwierdza się je również u 1 na
200 żywo urodzonych noworodków. Badania
genetyczne wykonywane u nosicieli aberracji
chromosomowych umożliwiają często lo-
kalizację genów determinujących określone
choroby, a także poznanie patogenezy chorób
występujących w populacji ogólnej, u osób
z prawidłowym garniturem chromosomo-
wym. Dalej przedstawiono przykłady niektó-
rych częstszych aberracji chromosomowych.

Genetyka medyczna
100
Rozpoznanie
Aberracje chromosomowe obejmujące ob-
szar większy od 5 milionów par zasad (Mpz)
widoczne są po zastosowaniu technik analizy
prążkowej chromosomów. Wzrastająca liczba
identyfi kowanych obecnie mniejszych niepra-
widłowości, niewidocznych w standardowym
badaniu kariotypu, wynika z wprowadzenia
technik cytogenetyki molekularnej, takich
jak fl uorescencyjna hybrydyzacja in situ
(FISH) [patrz str. 74] czy porównawcza hy-
brydyzacja genomowa (CGH) [patrz str. 75].
Zespół Turnera i zespół Downa
W zespole Turnera dochodzi do utraty, fragmen-
tu lub całego, jednego z dwóch chromosomów
X w komórce. Powyższa aberracja chromosomo-
wa występuje często w zygocie bezpośrednio
po zapłodnieniu. Zespół Turnera stwierdza się
u 15% płodów poronionych samoistnie. Głów-
ne objawy choroby to niskorosłość oraz przed-
wczesne wygasanie czynności jajników.
Osoby z zespołem Downa mają dodatkowy
chromosom 21. Charakterystyczne cechy klinicz-
ne to dysmorfi a twarzoczaszki, wrodzona wada
serca oraz niepełnosprawność intelektualna.
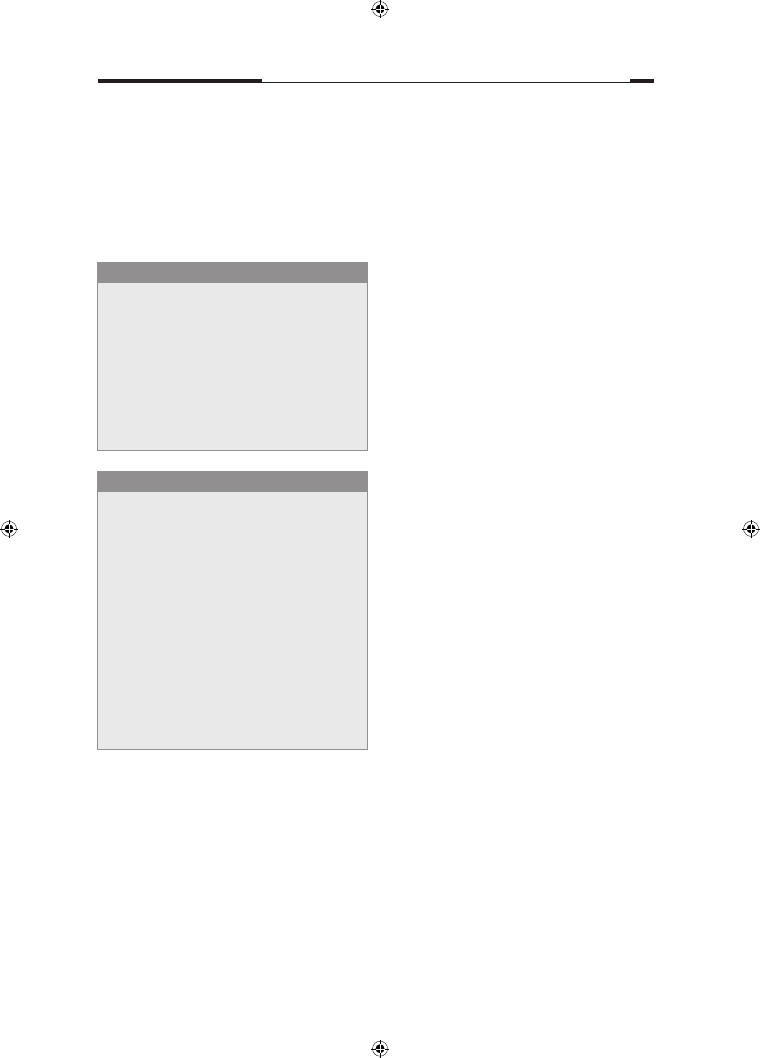
Rycina 3.1 Kariotyp w zespole
Turnera (45,X). Badanie wykona-
no w Genetics Laboratories, Ad-
denbrooke’s Hospital, Cambridge.
Poszczególne chromosomy iden-
tyfi kowane są metodami prążko-
wymi. Sposób klasyfi kacji chro-
mosomów uwzględnia zarówno
ich wielkość, jak i kształt (grupy
A–G).
Rycina 3.2 Kariotyp w zespole
Downa (trisomia chromosomu 21
pary).
Analiza kariotypu
Chromosomy płciowe
Chromosomy płciowe
Genetyka w praktyce klinicznej
101
Zespół Downa
Charakterystyka kliniczna
Zespół Downa najczęściej uwarunkowany
jest trisomią chromosomu 21 pary. Wystę-
puje z częstością około 1/1000 noworodków
żywo urodzonych. Wiążą się z nim różne
problemy kliniczne, m.in. wrodzone wady
serca, niedosłuch, niedoczynność tarczycy,
niepełnosprawność intelektualna oraz choro-
ba Alzheimera. W ostatnich dziesięcioleciach
średnia długość życia osób z zespołem Dow-
na wzrosła i obecnie wynosi około 50 lat.
Geny i ich produkty
Na chromosomie 21 zmapowano ponad 300
genów. Potrójna dawka niektórych z nich,
a w konsekwencji nadmierna ekspresja, przy-
czynia się do ujawnienia fenotypu zespołu
Downa. Na przykład za wystąpienie wcze-
snej postaci choroby Alzheimera, stosunko-
wo częstej w przebiegu zespołu, odpowiada
prawdopodobnie gen białka prekursorowe-
go amyloidu APP, zlokalizowany w regionie
21q21. Akumulacja amyloidu w tkankach jest
typowym objawem, który stwierdza się w ba-
daniach neuropatologicznych osób z choro-
bą Alzheimera. Obecność trzech kopii genu
APP u pacjentów z zespołem Downa skutkuje
nadmierną produkcją tego białka.
Powyższy związek genotypowo-fenotypo-
wy umożliwił identyfi kację genu APP jako
genu-kandydata w chorobie Alzheimera. Mu-
tacje punktowe w genie APP zidentyfi kowa-
no także w części przypadków występowania
rodzinnego tej choroby w populacji ogólnej
[patrz str. 147].
Rozpoznanie
Potwierdzenie diagnozy możliwe jest w stan-
dardowym badaniu kariotypu, a także przy
zastosowaniu metody inter- lub metafazowe-
go FISH.
Technika FISH umożliwia identyfi kację tzw.
zespołów mikrodelecyjnych, uwarunkowa-
nych utratą fragmentu DNA mniejszego niż
5 Mpz. Mikrodelecje powstają w sąsiedztwie
wysoce powtarzalnych sekwencji DNA, tzw.
duplikonów, które predysponują chromoso-
my do nieallelicznej rekombinacji w trakcie
podziału komórki.
Duplikony
Sekwencje o niskiej powtarzalności (ang. low-
-copy repeats) występują powszechnie w geno-
mie, przy czym specyfi czna sekwencja dupli-
konu różni się w zależności od lokalizacji chro-
mosomowej. Duplikony odgrywają istotną rolę
w procesie homologicznej rekombinacji chro-
mosomów. Poprzez zwiększenie prawdopodo-
bieństwa utraty fl ankowanej przez nie sekwencji
DNA prowadzą także do powstawania zespołów
mikrodelecyjnych.
FISH subtelomerowy
FISH subtelomerowy umożliwia identyfi kację
bardzo małych aberracji w pobliżu końców chro-
mosomów (telomerów), które nie są widoczne
przy użyciu standardowych metod prążkowych.
Zastosowanie tej techniki doprowadziło do roz-
poznania nieznanych dotychczas zespołów mi-
krodelecji i mikroduplikacji, w których obrazie
występuje niepełnosprawność intelektualna ze
współistnieniem lub bez wad rozwojowych.
Fluorescencyjna hybrydyzacja in situ poszerza
możliwości diagnostyczne w przypadkach wad
rozwojowych i niepełnosprawności intelektual-
nej oraz ułatwia rozpoznawanie i leczenie naby-
tych aberracji chromosomowych w niektórych
nowotworach [patrz tekst o nowotworach w tym
rozdziale].
Postępowanie terapeutyczne
Postępowanie terapeutyczne w aberracjach
chromosomowych obejmuje przede wszyst-
kim działania wspierające i edukację rodzin
osób chorych. Identyfi kacja zmienionych
w wyniku rearanżacji chromosomowych ge-
nów umożliwiła poznanie genetycznego pa-
tomechanizmu niektórych chorób występu-
jących w populacji ogólnej.

Genetyka medyczna
102
Postępowanie terapeutyczne
Opublikowano szereg medycznych algorytmów
postępowania, które w znaczący sposób popra-
wiają standard opieki nad chorymi z zespołem
Downa. Prowadzone są także badania nad le-
kami, które mogłyby zapobiec wystąpieniu lub
zmniejszyć skutki choroby Alzheimera.
Zespół Turnera
Charakterystyka kliniczna
Zespół Turnera powstaje w wyniku częściowej
lub całkowitej monosomii chromosomu X u osób
płci żeńskiej. Występuje z częstością około
1/2500 noworodków żywo urodzonych. Najczę-
ściej manifestuje się niskorosłością oraz przed-
wczesnym wygasaniem czynności jajników, choć
w jego przebiegu mogą ujawnić się inne proble-
my medyczne, m.in. nieprawidłowości układu
limfatycznego czy wrodzona wada serca. Osoby
z zespołem Turnera zwykle nie wykazują niepeł-
nosprawności intelektualnej, niemniej mają spe-
cyfi czne trudności w nauce.
Geny i ich produkty
Na chromosomie X znajduje się ponad 1000
genów. Utrata tych, które zlokalizowane są na
krótkim ramieniu, determinuje fenotyp zespo-
łu Turnera. Brak kopii genu SHOX w regionie
pseudoautosomalnym Xp22.33 jest prawdopo-
dobnie odpowiedzialny za niskorosłość [patrz
Homeobox]. Mutacje w tym genie u osób
z prawidłowym kariotypem są przyczyną
idiopatycznej niskorosłości oraz, stosunkowo
rzadkiej, łagodnej dysplazji szkieletowej (ze-
spół Leriego-Weilla), wykazującej podobień-
stwo fenotypowe do zespołu Turnera.
Rozpoznanie
Zwykle potwierdzenie zespołu Turnera uzy-
skuje się w klasycznym badaniu kariotypu,
choć ze względu na wysoką częstość występo-
wania mozaikowości należy przeanalizować
większą niż zwykle liczbę metafaz. Czasem
stosuje się także techniki inter- lub metafazo-
wego FISH.
Postępowanie terapeutyczne
Opublikowano standardy rozpoznania zespo-
łu Turnera i postępowania terapeutycznego.
Obejmują one zastosowanie hormonu wzro-
stu i estrogenów. Dawka oraz okres stosowa-
nia tych leków są przedmiotem sporów.
Homeobox
Homeobox (kaseta homeo) to krótki fragment
DNA, kodujący 60-aminokwasowy polipeptyd,
którego sekwencja jest identyczna we wszyst-
kich zawierających go genach (geny homeo-
tyczne). Białka, w których skład wchodzi ten po-
lipeptyd, są kluczowymi regulatorami rozwoju
embrionalnego.
Zespół Williamsa-Beurena
Charakterystyka kliniczna
Zespół Williamsa-Beurena, jeden z najlepiej po-
znanych zespołów mikrodelecyjnych, spowo-
dowany jest ubytkiem około 1,5 Mpz w regionie
7q11.23. Główne objawy kliniczne to: zwężenia
naczyń krwionośnych, niskorosłość, hiperkal-
cemia, niepełnosprawność intelektualna w stop-
niu lekkim oraz charakterystyczne cechy osobo-
wości. Zespół niemal zawsze występuje de novo.
Geny i ich produkty
Wystąpienie objawów zespołu Williamsa-Beu-
rena spowodowane jest delecją około 25 genów.
Niektórym z nich przyporządkowano wystę-
powanie poszczególnych cech fenotypowych.
Wiele prac potwierdza rolę delecji genu elastyny
(ELN) w powstawaniu zmian naczyniowych.
Późniejsze badania wykazały, że także nie-
które rodzaje zwężeń naczyń niezwiązane
z zespołem Williamsa-Beurena spowodowa-
Genetyka w praktyce klinicznej
103
ne są mutacjami punktowymi lub delecjami
w genie ELN i wynikającą z tego haploinsufi -
cjencją produktu genu elastyny.
Rozpoznanie
Mikrodelecja wywołująca zespół Williamsa-
-Beurena jest niewidoczna w standardowym
badaniu kariotypu. Metodą z wyboru jest za-
tem FISH metafazowy z użyciem sondy iden-
tyfi kującej gen elastyny.
Postępowanie terapeutyczne
Dostępne są standardy zarówno rozpoznania
tego zespołu, jak i postępowania z osobami
chorymi. Opisano metody leczenia i zapo-
biegania niektórym problemom zdrowotnym
oraz wynikającym z cech osobowości.
pełniąc istotną rolę w powstaniu struktur
twarzy, szyi i klatki piersiowej. Zaburzenie
procesu migracji neuronalnej obserwowa-
ne w zespole mikrodelecji 22q11 skutkuje
wystąpieniem anomalii twarzoczaszki, im-
munologicznych oraz sercowo-naczynio-
wych, m.in.: cech dysmorfi i twarzy, wady
wrodzonej serca (np. tetralogii Fallota lub
hipoplazji łuku aorty), hipokalcemii związa-
nej z niedorozwojem przytarczyc, niedobo-
rów immunologicznych oraz strukturalnych
i funkcjonalnych nieprawidłowości podnie-
bienia miękkiego. Wielu chorych wykazuje
ponadto cechy niepełnosprawności intelek-
tualnej w stopniu lekkim, a około 25% ma
zaburzenia psychiatryczne, m.in. schizofre-
nię lub depresję.
Haploinsufi cjencja białka
Haploinsufi cjencja oznacza, że zmiana ilości pro-
duktu danego genu prowadzi do wystąpienia
określonego fenotypu. W przypadku utraty jed-
nej funkcjonalnej kopii genu obecność jedynie
50% produktu białkowego doprowadzi do ujaw-
nienia efektu fenotypowego.
Tetralogia Fallota
Tetralogia Fallota jest złożoną wadą serca obej-
mującą cztery nieprawidłowości anatomiczne:
ubytek w przegrodzie międzykomorowej, zwę-
żenie drogi odpływu krwi z prawej komory do
płuc, kompensacyjny przerost prawej komory
i prawostronne przemieszczenie aorty. Częstą
przyczyną tej wady jest zaburzenie migracji ko-
mórek grzebienia nerwowego w kierunku serca
w czasie życia płodowego.
Zespół mikrodelecji 22q11
(zespół DiGeorge’a lub zespół
podniebienno-sercowo-twarzowy)
Charakterystyka kliniczna
Zespół DiGeorge’a oraz zespół podniebienno-
-sercowo-twarzowy, które niegdyś uznawane
były za różne jednostki chorobowe, są innymi
nazwami tego samego zespołu mikrodelecyj-
nego. Jest to najczęstszy zespół mikrodelecyjny,
stwierdzany u 1/4000 osób, zwykle uwarunko-
wany obecnością delecji około 3 Mpz w regionie
22q11.
Komórki grzebienia nerwowego rozwija-
jącego się zarodka, powstałe z brzegów cewy
nerwowej, migrują w różne okolice ciała,
Około 90% przypadków zespołu występuje
de novo. Pozostałe są wynikiem przekazania
mikrodelecji 22q11 przez rodzica, czasem
uprzednio niezdiagnozowanego. Prawdo-
podobieństwo wystąpienia zespołu u potom-
stwa osoby chorej wynosi 50% zgodnie z au-
tosomalnym dominującym trybem uwarun-
kowania choroby.
Geny i ich produkty
Kumulacyjny efekt utraty około 30 genów
w regionie 22q11 odpowiada za wystąpienie
objawów zespołu. Utrata kopii genu UFD1L
(kodującego białko szlaku ubikwityno-protea-
somowego, odpowiedzialnego za degradację
białek komórkowych) lub genu COMT (kodu-
jącego enzym katecholo-O-metylotransfera-

Genetyka medyczna
104
zę) prawdopodobnie predysponuje do ujaw-
nienia zaburzeń psychiatrycznych.
Identyfi kacja genu(-ów) odpowiedzialne-
go(-ych) za patogenezę powyższych objawów
może mieć istotne znaczenie dla populacji
ogólnej, jako że mutacje lub inne zmiany
w obrębie tych genów są przypuszczalnie
w dużej mierze odpowiedzialne za pojawie-
nie się choroby psychicznej u osób bez delecji
w obrębie chromosomu 22 pary.
Rozpoznanie
Standardowe badanie cytogenetyczne umożli-
wia identyfi kację delecji 22q11 jedynie u mniej
niż 10% chorych. Najczulszym i szeroko dostęp-
nym testem diagnostycznym jest FISH z sondą
zawierającą sekwencje DNA z regionu 22q11.
Postępowanie terapeutyczne
Standardowe postępowanie obejmuje zwal-
czanie problemów medycznych i psychia-
trycznych, a także profi laktykę lub minimali-
zowanie zaburzeń neurorozwojowych.
CHOROBY MONOGENOWE
(MENDLOWSKIE)
Zagadnienia ogólne
Znamy mutacje genowe odpowiedzialne za
wystąpienie tysięcy chorób monogenowych.
Zmutowane geny zostały zidentyfi kowane
przy użyciu różnych technik biologii mole-
kularnej, takich jak klonowanie miejsc zła-
mań w translokacjach chromosomowych,
analiza sprzężeń, analiza genów-kandydatów
oraz badanie genów na modelach zwierzę-
cych. Zsekwencjonowanie genomu ludzkiego
w ramach projektu HUGO sprawiło, że iden-
tyfi kacja patogennych zmian w genach, które
odpowiadają za choroby monogenowe, stała
się niemal codziennością.
Rozpoznanie
Stosowane techniki diagnostyczne różnią
się w zależności od rodzaju defektu odpo-
wiedzialnego za wystąpienie choroby. Wiele
mutacji punktowych identyfi kuje się tech-
niką PCR i przez sekwencjonowanie. Testy
diagnostyczne dla chorób monogenowych
stają się powszechnie dostępne. Laboratoria
kierują swoje oferty nawet do lekarzy podsta-
wowej opieki zdrowotnej. Mimo to w wielu
zespołach jednogenowych (np. zespół Ehler-
sa-Danlosa lub zespół Marfana) zasadnicze
znaczenie ma nadal rozpoznanie kliniczne ze
względu na nieznane podłoże genetyczne lub
zbyt złożoną diagnostykę molekularną.
Podstawą diagnostyki wszystkich chorób
genetycznie uwarunkowanych powinno być
badanie kliniczne i analiza rodowodu. W ra-
mach analizy rodowodu należy uwzględnić:
objawy kliniczne stwierdzane u krewnych
chorego, ewentualne pokrewieństwo rodzi-
ców, powtarzające się poronienia, występo-
wanie niepełnosprawności intelektualnej oraz
wad rozwojowych.
Postępowanie terapeutyczne
Postępowanie terapeutyczne jest zależne od
rodzaju i specyfi ki stwierdzanego zaburze-
nia. W chorobach uwarunkowanych muta-
cjami genów kodujących białka strukturalne
(np. w zespole Marfana lub wrodzonej łam-
liwości kości) ogranicza się ono głównie do
zapobiegania lub minimalizowania powi-
kłań. W przypadku defektów enzymatycz-
nych (wrodzonych zaburzeń metabolizmu,
np. fenyloketonurii lub rodzinnej hipercho-
lesterolemii) stosuje się dietę restrykcyjną
i/lub substytucję enzymatyczną, która stała
się szeroko dostępna dzięki innowacyjnej
technologii produkcji rekombinowanych
enzymów.
Wielkie nadzieje wiązane są z terapią geno-
wą. Choć dotychczasowe próby nie przynio-
sły oczekiwanych rezultatów, z pewnością jest
to obiecująca technologia.

Genetyka w praktyce klinicznej
105
CHOROBY AUTOSOMALNE
DOMINUJĄCE
Achondroplazja
Charakterystyka kliniczna
Achondroplazja to najczęstsza genetycznie
uwarunkowana choroba układu szkieletowe-
go. Występuje z częstością 1/20 000–1/40 000
żywo urodzonych noworodków. Główne ob-
jawy kliniczne to niskorosłość (średni wzrost
osoby dorosłej ok. 120 cm), skrócenie kończyn,
nadmierna lordoza lędźwiowa i wielkogłowie
(powiększenie obwodu głowy) z wydatnym
czołem. W okresie niemowlęcym stwierdza się
hipotonię, a także zwiększone ryzyko wystąpie-
nia bezdechu, będącego wynikiem ucisku na
pień mózgu lub zwężenia dróg oddechowych.
Wśród najczęstszych powikłań wieku dorosłe-
go wymienia się otyłość oraz objawy z ucisku
rdzenia kręgowego lub nerwów rdzeniowych,
wynikające ze zwężenia kanału kręgowego
(stenoza kanału kręgowego). Rozwój intelek-
tualny jest zwykle prawidłowy.
W ponad 80% przypadków wywiad rodzin-
ny jest ujemny, a chorobę powoduje mutacja
de novo na chromosomie 4, odziedziczonym
od ojca. Istnieje związek pomiędzy częstszym
występowaniem achondroplazji a zaawanso-
wanym wiekiem ojca.
Geny i ich produkty
Achondroplazja jest zawsze wywołana muta-
cją w genie receptora czynnika wzrostu fi bro-
blastów (FGFR3, 4p16), który koduje białko
o tej samej nazwie. Osoby chore są heterozy-
gotycznymi nosicielami mutacji. Homozy-
gotyczność to niskorosłość letalna. Prawdo-
podobieństwo jej wystąpienia u potomstwa
dwóch osób chorych wynosi 25%.
Białko FGFR3 jest receptorem kinazy tyrozy-
nowej, który wiąże białka czynników wzrostu
fi broblastów. W warunkach prawidłowych zwią-
zanie liganda aktywuje receptor, inicjując kaska-
dy sygnałowe, istotne dla wzrostu i różnicowania
komórek. Mutacja odpowiedzialna za wystąpie-
nie achondroplazji skutkuje konstytutywną (nie-
zależną od liganda) aktywacją receptora FGFR3.
Szereg innych mutacji w genie FGFR3 powodu-
je wystąpienie dysplazji szkieletowych, przy czym
im większa konstytutywna aktywacja receptora,
tym większe nasilenie objawów choroby.
Rozpoznanie
Rozpoznanie kliniczne ustalane jest na podsta-
wie charakterystycznych cech klinicznych i ob-
jawów radiologicznych. Dostępna jest też diag-
nostyka molekularna. Praktycznie u wszystkich
chorych z achondroplazją stwierdza się taką
samą mutację genową w pozycji 1138. Prawdo-
podobnie jest to najczęściej ulegający mutacjom
nukleotyd w genomie człowieka.
Postępowanie terapeutyczne
Stosuje się chirurgiczną korekcję występujących
w przebiegu choroby powikłań, np. zwężenia ka-
nału kręgowego. Stymulację wzrostu uzyskuje się
poprzez terapię hormonem wzrostu. Można też
wykonać ortopedyczne wydłużanie kończyn.
Zespół policystycznych nerek typu
dorosłych
Charakterystyka kliniczna
Zespół policystycznych nerek typu dorosłych
jest najczęstszą dziedziczną chorobą nerek.
Skróty używane w nazwach genów
Skrócone nazwy genów pisze się kursywą. Gen LDLR
(gen receptora lipoprotein o niskiej gęstości) kodu-
je białko receptorowe o tej samej nazwie. Mutacje
w LDLR wywołują rodzinną hipercholesterolemię.
W skróconych nazwach genów mysich pierwsza lite-
ra jest wielka, pozostałe małe, np. Ldlr. Locus chromo-
somowe umieszcza się w nawiasie, np. LDLR (19p13).

Genetyka medyczna
106
Występuje u 1/500–1/1000 osób. Dziedziczy
się w sposób autosomalny dominujący. Nie-
wydolność nerek jest wynikiem postępującego
zwyrodnienia torbielowatego. Torbiele mogą
tworzyć się także w innych narządach, m.in.
w wątrobie. Często spotyka się również ano-
malie pozanerkowe: tętniaki, uchyłkowatość,
przepukliny i wady zastawkowe serca. Wcze-
snym objawem jest nadciśnienie tętnicze.
Geny i ich produkty
Około 85% przypadków zespołu wynika z mu-
tacji w genie PKD1 (16p13.3), który koduje
policystynę 1. Znaczenie tego białka nie jest
znane. Wiadomo, że stanowi receptor błono-
wy rzęsek nabłonka nerkowego i występuje
w kompleksie z innymi białkami. Większość
mutacji PKD1 to mutacje typu utraty funkcji.
Znaczną część pozostałych przypadków
zespołu policystycznych nerek wywołuje mu-
tacja w genie PKD2 (4q13-23) kodującym po-
licystynę 2. Białko to jest częścią napięciowo-
zależnego kanału wapniowego i jednocześnie
homologiem strukturalnym policystyny 1.
Wykazano interakcję policystyny 2 z policy-
styną 1 na poziomie komórkowym, szczegól-
nie w obrębie rzęsek nabłonka nerkowego.
Początek i przebieg choroby są szybsze i bar-
dziej dramatyczne u pacjentów z mutacją w ge-
nie PKD1 w porównaniu z nosicielami mutacji
PKD2.
Rozpoznanie
Podstawą rozpoznania klinicznego jest
stwierdzenie charakterystycznych torbieli w ba-
daniu ultrasonografi cznym nerek. Liczba tor-
bieli wzrasta z czasem.
Dotychczas zidentyfi kowano prawie 100
mutacji w genach PKD1 i PKD2. Komercyj-
nie dostępne testy diagnostyczne pozwalają
potwierdzić chorobę na poziomie molekular-
nym u 75% chorych z mutacją w genie PKD1.
Czułość badania jest jeszcze większa w przy-
padkach uwarunkowanych mutacjami w ge-
nie PKD2.
Ponad 75% sekwencji genu PKD1 zdupliko-
wane jest w innej lokalizacji – na chromosomie
16. Badanie w kierunku mutacji w tym genie
wymaga więc szczególnej uwagi, aby zidentyfi -
kować defekt w genie PKD1, a nie w jego kopii.
Postępowanie terapeutyczne
Bliższe poznanie ubytku czynnościowego, któ-
ry prowadzi do rozwoju tego zespołu, pozwoli
zapewne w przyszłości na opracowanie metod
leczenia. Niemal u połowy pacjentów do 50.
roku życia rozwija się niewydolność nerek.
Wymagają oni dializoterapii lub przeszczepu.
Choroba Charcot-Marie-Tooth
typu 1 i 2 (dziedziczne neuropatie
czuciowo-ruchowe, HMSN)
Charakterystyka kliniczna
Grupa chorób Charcot-Marie-Tooth (CMT, od
nazwisk lekarzy francuskich żyjących w XIX
wieku) charakteryzuje się w badaniu neurolo-
gicznym: postępującym osłabieniem siły mię-
śniowej, zanikami dystalnych mięśni kończyn,
osłabieniem głębokich odruchów ścięgni-
stych, osłabieniem czucia w obrębie kończyn
oraz obecnością stopy wydrążonej (pes ca-
vus). Choroby te występują z częstością oko-
ło 1/2500 osób. CMT typu 1 i 2 dziedziczą się
w sposób autosomalny dominujący. Początek
choroby przypada zwykle pomiędzy okresem
dzieciństwa a 30. rokiem życia. Na podstawie
wyników badań elektroneurografi cznych, mo-
lekularnych i analizy sposobu uwarunkowania
wyróżnia się liczne podtypy CMT.
CMT1A jest najczęstszym podtypem CMT.
Spowodowana jest obecnością duplikacji
wielkości 1,5 Mpz w obrębie chromosomu 17
(17p11), która skutkuje m.in. obecnością trzech
kopii genu PMP22 kodującego obwodowe biał-
ko mieliny 22. Stwierdza się zwolnienie prze-
wodnictwa nerwowego. CMT1A jest zatem
przykładem choroby demielinizacyjnej. Zanik

Genetyka w praktyce klinicznej
107
mięśni łydek i stóp skutkuje osłabieniem mięśni
kostki i obustronnym opadaniem stóp. Kończy-
ny dolne przyjmują charakterystyczny kształt,
przypominający odwróconą butelkę szampana.
Mimo to chorzy zwykle chodzą samodzielnie.
Przewidywana długość życia w CMT1A nie od-
biega od normy dla populacji ogólnej.
CMT1B, o podobnym do CMT1A obrazie kli-
nicznym, spowodowana jest obecnością mutacji
w genie MPZ kodującym białko mielinowe zero.
Fenotyp CMT2 jest łagodniejszy od obser-
wowanego w CMT1, a wyniki badań szybko-
ści przewodzenia w nerwach obwodowych są
prawidłowe. Opisano wiele podtypów CMT2
i zidentyfi kowano siedem genów, w których
mutacje odpowiedzialne są za wystąpienie tej
choroby.
Geny i ich produkty
CMT1A jest zespołem mikroduplikacyjnym.
Podobnie jak w zespołach mikrodelecyjnych
[patrz str. 101], homologiczne sekwencje DNA
(duplikony) fl ankujące region krytyczny na
chromosomie 17 predysponują do nierówne-
go crossing-over w trakcie mejozy. Efektem jest
m.in. duplikacja genu PMP22. Jego dodatkowa
kopia powoduje zwiększenie poziomu specy-
fi cznego mRNA, co prowadzi, w nieznanym
mechanizmie, do uszkodzenia mieliny nerwów
obwodowych i zwyrodnienia aksonalnego. Dal-
sze zwiększenie liczby kopii PMP22 zarówno
u ludzi, jak i u myszy, skutkuje cięższym efektem
fenotypowym. Poważniejszy obraz kliniczny
CMT1A stwierdza się także u nosicieli mutacji
punktowych w genie PMP22, których patogen-
ność wynika prawdopodobnie ze zmniejszenia
aktywności lub eliminacji prawidłowego białka
eksprymowanego z allelu dzikiego [patrz str.
111, „Mutacje dominujące negatywne”]. Co
ciekawe, o ile duplikacja genu PMP22 powo-
duje fenotyp CMT, jego delecja odpowiada za
ujawnienie innej choroby, dziedzicznej neuro-
patii z nadwrażliwością na ucisk (HNPP), któ-
rą charakteryzuje obecność porażeń nerwów
obwodowych, np. strzałkowego z opadaniem
stopy, po stosunkowo niewielkim urazie.
Opis mechanizmów, które wywołują CMT,
a wynikają z mutacji w innych genach, prze-
kracza zakres tej książki.
Rozpoznanie
Podejrzenie kliniczne CMT powinno zostać
zweryfi kowane badaniem elektroneurogra-
fi cznym.
Identyfi
kację duplikacji genu PMP22
umożliwiają technika FISH i metoda Sou-
therna – obie powszechnie dostępne. Rzadkie
przypadki CMT1A uwarunkowane mutacja-
mi punktowymi w genie PMP22 wykrywa
się poprzez sekwencjonowanie całego genu
w poszukiwaniu defektu molekularnego.
Analiza mutacji w innych genach, które rów-
nież wywołują CMT, możliwa jest jedynie
w wybranych laboratoriach.
Prawdopodobnie odkryte zostaną także
kolejne geny, których mutacje skutkują wy-
stąpieniem choroby. Opisano bowiem rodzi-
ny dotknięte CMT, ale niebędące nosicielami
mutacji w znanych dotychczas genach.
Postępowanie terapeutyczne
Leczenie CMT jest objawowe. Ze względu na
autosomalny dominujący tryb uwarunkowania
większości podtypów wskazane jest badanie neu-
rologiczne i molekularne krewnych pierwszego
stopnia osób chorych. Wyniki badań na mode-
lach zwierzęcych z mutacją lub duplikacją genu
PMP22 mogą w przyszłości pomóc w poznaniu
mechanizmu procesu demielinizacyjnego.
Dodatkową metodą leczenia, zapobiegającą
obumieraniu aksonów, może być podawanie
czynników neurotrofi cznych.
Rodzinna hipercholesterolemia
Charakterystyka kliniczna
Rodzinna hipercholesterolemia (FH, ang. fami-
lial hypercholesterolaemia) jest najczęstszą cho-

Genetyka medyczna
108
robą monogenową z grupy zaburzeń przemiany
lipidów. Zwykle spowodowana jest obecnością
mutacji w genie LDLR kodującym białko recep-
torowe lipoprotein niskiej gęstości (LDL). Około
1 na 500 osób jest heterozygotą FH, nosicielem
mutacji w jednym allelu genu LDLR. Wiąże się
z tym podwyższone stężenie cholesterolu LDL
w surowicy krwi i wyższe od populacyjnego
ryzyko wczesnego wystąpienia choroby niedo-
krwiennej serca. Homozygoty FH (około 1 na
milion osób) charakteryzuje bardzo wysokie
stężenie LDL w surowicy krwi oraz oporna na
leczenie miażdżyca naczyń, pojawiająca się już
w dzieciństwie. FH jest uwarunkowana w spo-
sób autosomalny dominujący. Obecność dwóch
kopii zmutowanego genu skutkuje cięższym ob-
razem klinicznym niż tylko jednej.
Geny i ich produkty
Wolny cholesterol nie jest rozpuszczalny w wo-
dzie, dlatego we krwi przenoszony jest z udzia-
łem nośników. Z jednym z nich tworzy tzw.
kompleks LDL. Większość tak dostarczonego
do tkanek cholesterolu wykorzystywana jest
do budowy błon komórkowych i syntezy ste-
roli. Kompleks LDL jest rozpoznawany przez
układ receptorowy LDLR, po czym choleste-
rol zostaje wchłonięty w procesie receptoro-
wo swoistej endocytozy.
Receptor dla LDL jest białkiem przezbłono-
wym, składającym się z szeregu funkcjonal-
nych podjednostek, z których każda odgrywa
istotną rolę w wiązaniu cholesterolu. Jego de-
fekty skutkują podwyższonym stężeniem tej
lipoproteiny w surowicy krwi, co prowadzi do
rozwoju miażdżycy. Dotychczas zidentyfi ko-
wano kilkaset różnych mutacji w genie LDLR
(19p13), prowadzących do utraty lub obniżenia
funkcji receptora (np. poprzez utratę zdolności
jego syntezy lub zaburzenie transportu cząste-
czek receptora na powierzchnię komórki).
Rozpoznanie
Podstawową rolę w rozpoznaniu choroby od-
grywają: dokładna analiza rodowodu, także
ewentualnych przypadków podwyższonych
wartości cholesterolu u innych członków ro-
dziny, obecność lub brak żółtaków oraz wynik
lipidogramu na czczo. Stężenie całkowitego
cholesterolu między 9 a 13 mmol/l występuje
u heterozygot, zaś 18–31 mmol/l u homozy-
got FH, przy czym najwyższe odchylenie od
normy osiąga cholesterol LDL.
Z uwagi na dużą liczbę różnych mutacji
w genie LDLR, z których większość identyfi -
kowana jest jedynie w pojedynczych rodzi-
nach, analizę molekularną wykonuje się w wa-
runkach klinicznych rzadko.
Postępowanie terapeutyczne
Farmakoterapia heterozygot FH obejmuje
podawanie inhibitorów reduktazy HMGCoA
(statyny) i/lub żywic jonowymiennych, które
zapobiegają wchłanianiu egzogennego chole-
sterolu z przewodu pokarmowego. Efekt ta-
kiej terapii u homozygot jest niewielki. Cho-
rzy ci uzyskują znaczną poprawę kliniczną
dopiero po transplantacji wątroby. Obecnie
prowadzone są badania nad zastosowaniem
terapii genowej z użyciem prawidłowej kopii
genu LDLR.
Dziedziczna telangiektazja
krwotoczna
Charakterystyka kliniczna
Dziedziczna telangiektazja krwotoczna
(HHT, ang. hereditary haemorrhagic telan-
giectasia), znana także jako choroba Rendu-
-Oslera-Webera, jest zaburzeniem procesu
waskulogenezy. Częstość jej występowania
szacuje się na około 1/10 000 osób. Głów-
ne objawy choroby nasilają się z wiekiem
i zawsze ujawniają się przed 40. rokiem ży-
cia. Zaliczamy do nich rozszerzenia naczyń
krwionośnych w obrębie skóry i błon ślu-
zowych (telangiektazje – niewielkie anasto-
mozy tętniczo-żylne), które predysponują

Genetyka w praktyce klinicznej
109
do nawracających krwawień z nosa (obser-
wowanych już w dzieciństwie) i przewodu
pokarmowego. Często występują też duże
malformacje tętniczo-żylne, zwykle w płu-
cach, wątrobie i mózgu. Wady naczyniowe
w obrębie płuc, w wyniku mieszania się krwi
tętniczej z żylną, mogą prowadzić do wystą-
pienia objawów nietolerancji wysiłku i sini-
cy. Nierzadko są także przyczyną krwotoku
z płuc oraz ropnia lub udaru mózgu. Mal-
formacje naczyń mózgowych powodują bóle
głowy, przemijające napady niedokrwienne
(TIA), udar oraz krwotok mózgowy.
Geny i ich produkty
Chorobę Rendu-Oslera-Webera warunku-
ją mutacje w dwóch genach. Za większość
przypadków odpowiedzialne są defekty
w genie endogliny (ENG, 9q34.1), które po-
wodują tworzenie wadliwego, skróconego
produktu białkowego. Ilość prawidłowej en-
dogliny jest wtedy niewystarczająca [patrz
„Haploinsufi cjencja białka”, str. 103]. Feno-
typ choroby znakomicie odzwierciedla jej
mysi model (myszy z knock-outem genu).
Endoglina jest białkiem błonowym zlokali-
zowanym w endotelium naczyniowym, gdzie
wiąże się z transformującym czynnikiem
wzrostu β (TGF-β). Uważa się, iż obniżenie
jej aktywności zakłóca w nieznanym mecha-
nizmie funkcjonowanie kaskady sygnałowej
TGF-β, co przyczynia się do rozwoju patolo-
gii naczyniowych.
Mutacje w innym genie kodującym recep-
tor aktywiny A (ACVRL1, 12q11-14) są od-
powiedzialne za 20% przypadków choroby.
Produktem genu ACVRL1 jest białko homo-
logiczne do TGF-β, którego wpływ na rozwój
HHT również nie został poznany.
Rozpoznanie
Rozpoznanie kliniczne choroby Rendu-
-Oslera-Webera opiera się na stwierdzeniu
trzech z następujących objawów: telangiek-
tazje, nawracające krwotoki z nosa, obciążo-
ny wywiad rodzinny, malformacje tętniczo-
żylne.
Diagnostyka molekularna polega na po-
szukiwaniu mutacji w genach ENG i ACVRL1
z wykorzystaniem metody sekwencjonowa-
nia fragmentów kodujących genów, wspoma-
ganej techniką ilościowego PCR, szczególnie
przydatną w identyfi kacji delecji.
Postępowanie terapeutyczne
W przypadku nawracających krwawień sto-
suje się suplementację preparatami żelaza,
a w poważnych przypadkach – przetoczenia
krwi. Nieprawidłowe połączenia tętniczo-
-żylne zamykane są techniką embolizacji.
Stworzenie modelu zwierzęcego choroby
(myszy z knock-outem genu) może przyczy-
nić się do lepszego poznania jej patomechani-
zmu oraz przyspieszyć rozwój nowych metod
terapeutycznych.
Zespół Marfana
Charakterystyka kliniczna
Zespół Marfana występuje z częstością 1/5000
osób. Stwierdza się zaburzenia układu mięśnio-
wo-szkieletowego: względną wysokorosłość
z nieproporcjonalnie długimi kończynami
i palcami (w porównaniu ze zdrowymi krew-
nymi), skoliozę (skrzywienie kręgosłupa) oraz
deformacje mostka. Objawy ze strony układu
sercowo-naczyniowego mogą być względnie
łagodne: wypadanie płatka zastawki mitralnej;
lub zagrażające życiu: poszerzenie łuku aorty,
tętniaki (miejscowe osłabienie ściany naczynia
z jego odcinkowym poszerzeniem, z lub bez
rozwarstwienia ściany naczynia). Występują
też różnego rodzaju defekty oka, od krótko-
wzroczności po podwichnięcie soczewki.
W około 75% przypadków cechy kliniczne
zespołu Marfana stwierdza się także u jedne-
go z rodziców pacjenta. Pozostałe przypadki
uwarunkowane są mutacjami de novo.
Wyszukiwarka
Podobne podstrony:
226 Example 1 Mix)
226 235
226 Example 1 Image Check)
(LABORATORIUM 022 Sterownik GE 226 Fanuc 90 30)
226 Zasada odzyskiwania danych)
226 Rodzaje notatek
Prawo spółdzielcze, ART 226 PrSpółdz, 2001
226 Example 1 FAT Search)
226 Where look for skipped sectors)
786
226
226
dz u 09 226 1817 rodzaje dok zazadac zamaw od wykonawcy
1 786 skiero leczenie przerz, pobrane
226
226 i 227, Uczelnia, Administracja publiczna, Jan Boć 'Administracja publiczna'
SHSBC 226 INSTRUCTORS' BUGBEAR
KPRM. 226, WSZYSTKO O ENERGII I ENERGETYCE, ENERGETYKA, KOPYDŁOWSKI
więcej podobnych podstron