
ZADANIE 1. DEFINICJE.
1. Katalizator- , substancja, której obecność w układzie reagentów wywołuje zmianę
szybkości reakcji chemicznej. Bierze udział w zachodzących reakcjach, jednakże tak,
że pozostaje po ich zajściu w niezmienionej ilości i postaci chemicznej, w związku z
czym nie pojawia się w sumarycznych równaniach opisywanych reakcji.
2. Aktywność katalizatora Ak- określa się jako różnicę między szybkościami reakcji
chemicznej zachodzącej w obecności katalizatora, vk, i bez niego, vh:
Ak = vk – vh
Szybkość reakcji bez katalizatora jest znikomo mała w porównaniu z reakcją
katalizowaną stąd miarą aktywności katalizatora jest wprost szybkość reakcji vk.
Aktywność katalizatora ma sens tylko w odniesieniu do układu katalizator-reagenty,
ma ona sens tylko w odniesieniu do danego typu reakcji.
3. Selektywność katalizatora- definiuje się jako stosunek ilości cpi jednego z kilku
możliwych produktów (Pi) reakcji do całkowitej ilości produktów Scpi:
X ==> P1 + P2 + P3 + ...+Pi
4. Stopień konwersji- stosunek ilości (wybranego) substratu, która uległa przemianie w
produkty reakcji chemicznej, do początkowej ilości tego substratu. Stopień
przereagowania równy jest ilorazowi chwilowej (aktualnej) wartości liczby postępu
reakcji chemicznej i maksymalnej osiągalnej dla wybranego reagenta wartości tej
liczby. Stopień przereagowania zawiera się w przedziale od 0 (przed rozpoczęciem
reakcji) do wartości bliskich 1 (dla tzw. reakcji praktycznie nieodwracalnych).
5. Stopień zaadsorbowanej substancji-
c
k
c
x
x
θ
p
k
p
x
x
o
o
lub
. (2)
gdzie
- stopień pokrycia powierzchni (ułamek miejsc zajętych przez cząsteczki
zaadsorbowane), x
o
– liczba moli substancji zaadsorbowanej przez jednostkową masę
adsorbentu w stanie pełnego obsadzenia jego powierzchni warstwą jednocząsteczkową
(wszystkie centra aktywne zajęte), x – liczba moli substancji zaadsorbowanej przez
jednostkową masę adsorbentu z fazy gazowej o równowagowym ciśnieniu cząstkowym p lub
stężeniu molowym c, k – stała zależna od temperatury.

6. Nośnik – jego zadaniem jest zwiększenie powierzchni fazy aktywnej i zwiększenie
wytrzymałości mechanicznej i termicznej katalizatora. Może być źródłem centrów
aktywnych innego typu niż faza aktywna, albo modyfikować jej strukturę
elektronową. Przykłady nośników to SiO2, γ-Al2O3, α-Al2O3, Cr2O3, MgO, CaO.
7. Faza aktywna – jest źródłem centrów aktywnych jednego bądź różnych typów, na
których powstają kompleksy przejściowe. Przykłady fazy aktywnej to Fe, Ni, Pt, Cu
Pd, Ag, Ni O, ZnO.
8. Promotory – modyfikują strukturę fizyczna i elektronową substancji aktywnej,
hamują jej niekorzystne przemiany fazowe i ułatwiają regenerację. Przykłady
promotorów to ZrO2, HCl, MgO, K2O, pierwiastki ziem rzadkich.
9. Szybkość reakcji chemicznej- szybkość przybywania lub ubywania reagenta w
wyniku przebiegu reakcji chemicznej.
Szybkość reakcji chemicznej definiuje się jako zmianę liczby moli składnika
odniesienia w czasie.
Najczęściej spotykane równania:
= ±
= ±
= ±
gdzie
± - znak minus odnosi się do substratu, znak plus do produktu,
A - składnik odniesienia (najczęściej substrat),
- liczba moli składnika odniesienia,
V - objętość przestrzeni reakcyjnej,
m - masa katalizatora,
S - powierzchnia kontaktu międzyfazowego.
Ilościowo szybkość reakcji określa się jako zmianę molowego stężenia substratu
lub produktu w jednostce czasu.
10.
Stała szybkości reakcji – współczynnik proporcjonalności k w równaniu
kinetycznym reakcji chemicznej
szybkość reakcji = k[A]
n
[B]
m
[C]
p
[D]
q
...
Zależność stałej szybkości reakcji od temperatury opisuje równanie Arrheniusa:
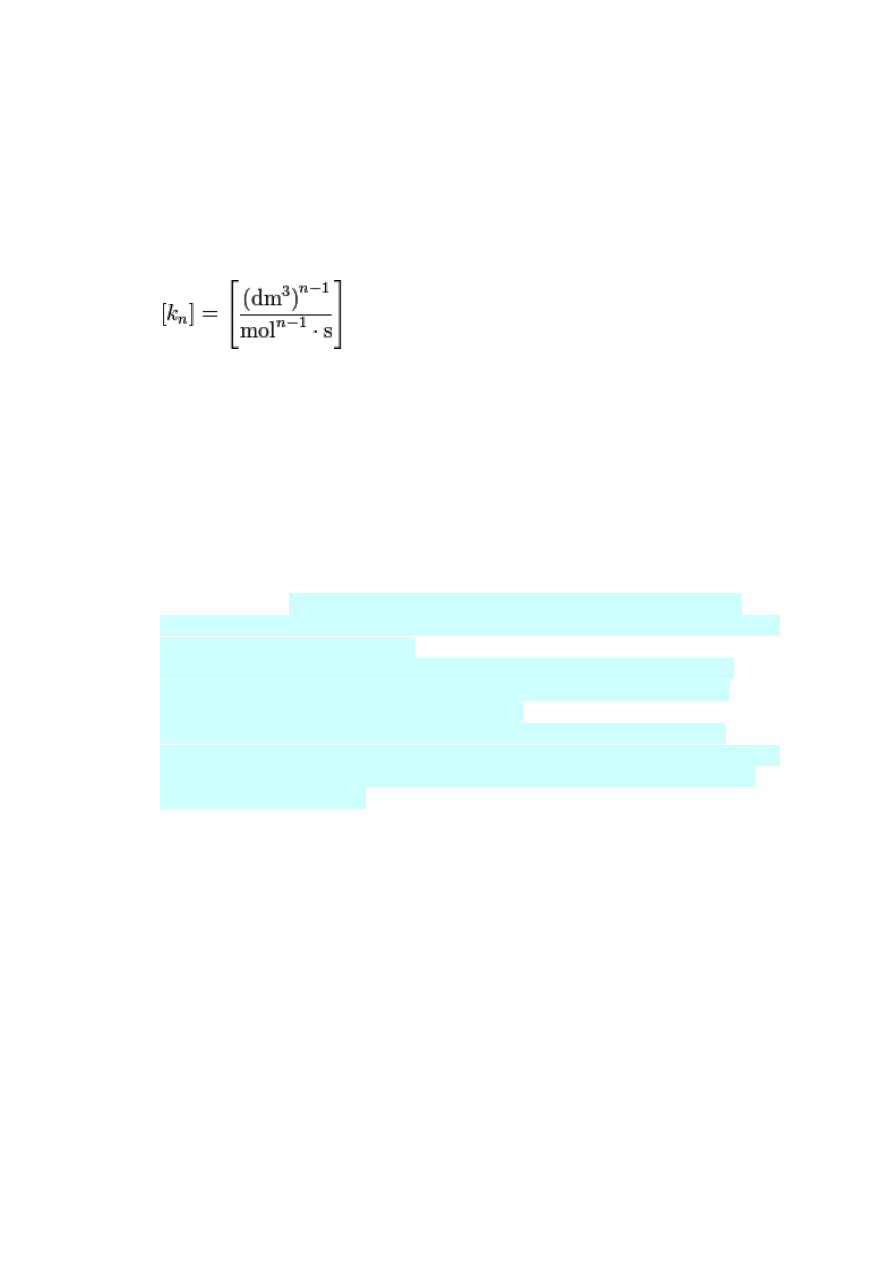
gdzie A – czynnik przedwykładniczy Arrheniusa, E
a
– energia
aktywacji Arrheniusa, R – stała gazowa, T – temperatura w kelwinach
Jednostka stałej szybkości reakcji zależna jest od rzędowości reakcji
k dla reakcji I-rzędowej [1/s]
dla reakcji II-rzędowej [dm^3/mol *s]
dla reakcji III-rzędowej [ (dm3)^2/mol^2 *s]
11. Rząd reakcji (rzędowość reakcji) – suma wykładników potęg w równaniu
kinetycznym reakcji chemicznej w postaci jednomianu potęgowego.
r = k[A]
0
= k – reakcja zerowego rzędu
r = k[A]
1
= k[A] – reakcja pierwszego rzędu
r = k[A]
2
r = k[A][B] – reakcja drugiego rzędu; pierwszego względem składnika A i pierwszego
względem składnika B
r = k[A]
1
– reakcja rzędu ułamkowego (postać równania kinetycznego sugeruje
złożony mechanizm).
Cząsteczkowość-definiuje liczba cząsteczek biorących udział w najwolniejszym
stadium reakcji. Cząsteczkowość jest zwykle równa rzędowości reakcji, natomiast nie
jest słuszne stwierdzenie odwrotne.
Cząsteczkowość i rząd reakcji wyznacza się tylko eksperymentalnie, nie można
obliczyć ich teoretycznie.Na ogół rząd reakcji jak i cząsteczkowość są z reguły
małymi liczbami nie przekraczającymi wartości 3.
Zagadnienie sprowadza się do tego, że równoczesne zderzenia większej liczby
cząsteczek są mało prawdopodobne a na sumaryczną szybkość reakcji wpływa przede
wszystkim najpowolniejszy etap pośredni bedący przemianą elementarną i dlatego
rząd reakcji jest małą liczbą.
ZADANIE 2. PODSTAWOWE WŁAŚCIWOŚCI KATALIZATORA ORAZ
WIELKOŚCI CHARAKTERYZUJĄCE DZIAŁANIE KATALIZATORA.
Podstawowe właściwości katalizatorów:
Aktywność (miara szybkości reakcji)
Selektywność (preferencja jednego kierunku przemiany)
Stabilność (żywotność katalizatora w czasie jego eksploatacji)
Zakres temperaturowy efektywnego działania katalizatora
Wielkości charakteryzujące działanie katalizatora:
Do oceny działania katalizatora w danej reakcji służą dwie wielkości: aktywność i
selektywność.

Aktywność jest miarą działania przyspieszającego daną reakcję chemiczną, a
selektywność miarą
działania ukierunkowującego. Aktywność katalizatora Ak określa się jako różnicę
między szybkością
reakcji chemicznej zachodzącej w obecności katalizatora vk i szybkością
reakcji bez udziału katalizatora vh.
Ak = vk – vh
Ponieważ zwykle vh<<vk jako miarę aktywności przyjmuje się szybkość reakcji w
obecności katalizatora. Inną, często stosowaną, miarą aktywności katalizatora jest
określenie konwersji α, czyli stopnia przereagowania substratów.
α=c0-c/c0 *100%
gdzie c jest stężeniem substratu, a c0 to początkowe stężenie substratu (gdy reakcja
jeszcze nie biegnie). Selektywność katalizatora Si definiuje się jako stosunek ilości
Cpi jednego z kilku możliwych produktów reakcji (Pi) reakcji do całkowitej ilości
produktów (suma Cpi po i)
Si=Cpi/suma Cpi po i
Zarówno aktywność jak i selektywność odnoszą się do działania katalizatora w danej
reakcji.
ZADANIE 3. ETAPY PRZYGOTOWANIA KATALIZATORA.
1. ROZTWÓR PREKURSORÓW SKŁADNIKÓW.
2. STRĄCANIE LUB WSPÓŁSTRĄCANIE.
3. FILTRACJA, WIROWANIE, DEKANTACJA.
4. SUSZENIE OSADU.
5. MIELENIE, PRZESIEWANIE.
6. TABLETKOWANIE, WYTŁACZANIE, GRANULOWANIE.
7. SUSZENIE, KALCYNACJA.
8. NANOSZENIE SKŁADNIKÓW AKTYWNYCH (PRZEZ METODĘ
WYTRĄCANIA, ADSORPCJI, WYMIANE JONOWĄ, IMPREGNACJA
MOKRA, IMPREGNACJA SUCHA).
9. WPROWADZANIE PROMOTORÓW.
10. AKTYWACJA (UTLENIANIE/REDUKCJA).

ZADANIE 4. METODY FIZYKOCHEMICZNE BADAŃ KATALIZATORÓW.
METODA BET- badanie powierzchni katalizatorów tlenkowych.
RENTGENOWSKA DYFRAKTOMETRIA PROSZKOWA- wyznaczanie
średnic wielkości krystalitów.
METODA RIETVELDA- ilościowa analiza fazowa.
ELEKTRONOWA MIKROSKOPIA SKANINGOWA I MIKROANALIZA
RENTGENOWSKA.
SPEKTROSKOPIA FOTOELEKTRONÓW WZBUDZONYCH
PROMIENIOWANIEM RENTGENOWSKIM.
ELEKTRONOWA MIKROSKOPIA TRANSMISYJNA.
SPEKTROSKOPIA IR- badanie właściwości kwasowych powierzchni ciał
stałych.
ZJAWISKO RAMANOWSKIEGO ROZPRASZANIA ŚWIATŁA.
NMR.
SPEKTROSKOPIA ELEKTRONOWEGO REZONANSU
PARAMAGNETYCZNEGO.
SPEKTROSKOPIA MOSSBAUEROWSKA.
TECHNIKI TEMPERATUROWO-PROGRAMOWANE.
BADANIE AKTYWNOŚCI KATALIZATORÓW W REAKTORACH
KATALITYCZNYCH.
SPEKTROMETR MAS.
ZADANIE 5. METODA WYTWARZANIA KATALIZATORÓW.
Metoda zol-żel polega na utworzeniu zolu (postaci koloidalnej), a następnie związaniu
jego składników w postaci żelu i kserożelu (wysuszonego żelu). Kserożele poddaje się
kalcynacji w celu uzyskania jednorodnych drobnokrystalicznych tlenków.
Prekursorami składników katalizatora/nośnika są w metodzie zol-żel związki
ulegające kontrolowanej hydrolizie i kondensacji, zwykle alkoholany metali i
niemetali. Rozpuszczalność alkoholanów (alkoksylanów) w wodzie jest ograniczona,
dlatego zwykle wymagane jest dodatkowo stosowanie rozpuszczalnika organicznego.
Stosując powyższą metodę otrzymuje się katalizatory (nośniki), w których składniki
sąwymieszane homogenicznie, jednakże wadą jest wysoki koszt stosowanych w
syntezie odczynników.
Lub inne: metoda strącania (współstrącania) homogenicznego (tj. metoda
mocznikowa, metoda spaleniowa – mocznikowa, metoda spaleniowa –
cytrynianowa), metoda pechiniego, metoda z dodatkiem surfaktantu lub
polimeru*.

*Metoda z dodatkiem surfaktantu lub polimeru- surfaktanty lub polimery
mogąstanowić istotny składnik mieszaniny podczas syntezy metodą
strącania lub zol-żel, zmieniający właściwości otrzymywanych osadów. Poprzez
wpływanie na procesy olacji i oksolacji związki organiczne mogą modyfikować
proces wzrostu krystalitów i co za tym idzie, właściwości teksturalne otrzymywanych
katalizatorów. Dodatkowa porowatość może zostać wygenerowana także na etapie
kalcynacji, podczas rozkładu części organicznej otrzymanego prekursora.
ZADANIE 6. NANOSZENIE FAZY AKTYWNEJ KATALIZATORA PRZEZ
IMPREGNACJĘ.
Fazę aktywną katalizatora można nanosić kilkoma metodami. Jedną z nich jest
impregnacja sucha zwana również impregnacją pierwszej wilgotności. Metoda ta
polega na bezpośrednim nanoszeniu składników aktywnych, poprzez wypełnienie
porów katalizatora roztworem soli o odpowiednim stężeniu. Chcąc uzyskać założoną
zawartość procentową metalu aktywnego w próbce niezbędne jest przygotowanie
roztworu soli o odpowiednim stężeniu. Przed wprowadzeniem fazy aktywnej należy
wyznaczyć pojemność sorpcyjną nośnika. Najczęściej dokonuje się tego poprzez
miareczkowanie materiału wodą destylowaną, otrzymując wartośćchłonności nośnika
w cm3/g. W trakcie tej czynności należy ściśle przestrzegać zasady, że objętość
wprowadzanego roztworu soli powinna być dokładnie równa, bądź tylko o kilka
procent większa od porowatości nośnika. Zaimpregnowany materiał należy wysuszyć
w celu usunięcia rozpuszczalnika. Parametry suszenia należy dobierać w taki sposób,
aby zapobiec rekrystalizacji fazy aktywnej. Przy wolnym suszeniu większość fazy
aktywnej gromadzi się na dnie porów, a w przypadku suszenia szybkiego ze względu
na gradient temperatur faza aktywna gromadzi się na zewnątrz porów. Po wysuszeniu
materiału należy go kalcynować w celu usunięcia reszt kwasowych pochodzących z
użytych soli (np. azotany, octany, szczawiany).
ZADANIE 7. CENTRA AKTYWNE NA POWIERZCHNI KATALIZATORA.
Powierzchnia katalizatora nie jest jednorodna. W reakcji katalitycznej aktywność
wykazuje nie cała powierzchnia katalizatora, lecz tylko jej niewielka część. Są to
atomy, jony lub grupy atomów i jonów, które tworzą wiązanie (wiązania) z
cząsteczkami reagującej substancji w procesie powstawania kompleksu aktywnego.
Rolę centrów aktywnych mogą pełnić atomy na narożach i krawędziach płaszczyzn
krystalograficznych lub defekty. Ze względu na charakter chemiczny centra można
podzielić na kwasowe i zasadowe (typu Brønsteda lub Lewisa) oraz centra redoksowe.
Obecność w reagentach nawet niewielkich ilości substancji, które z centrami
aktywnymi tworzą bardzo trwałe połączenia powoduje szybki spadek aktywności
katalizatora, tzw. zatruwanie.
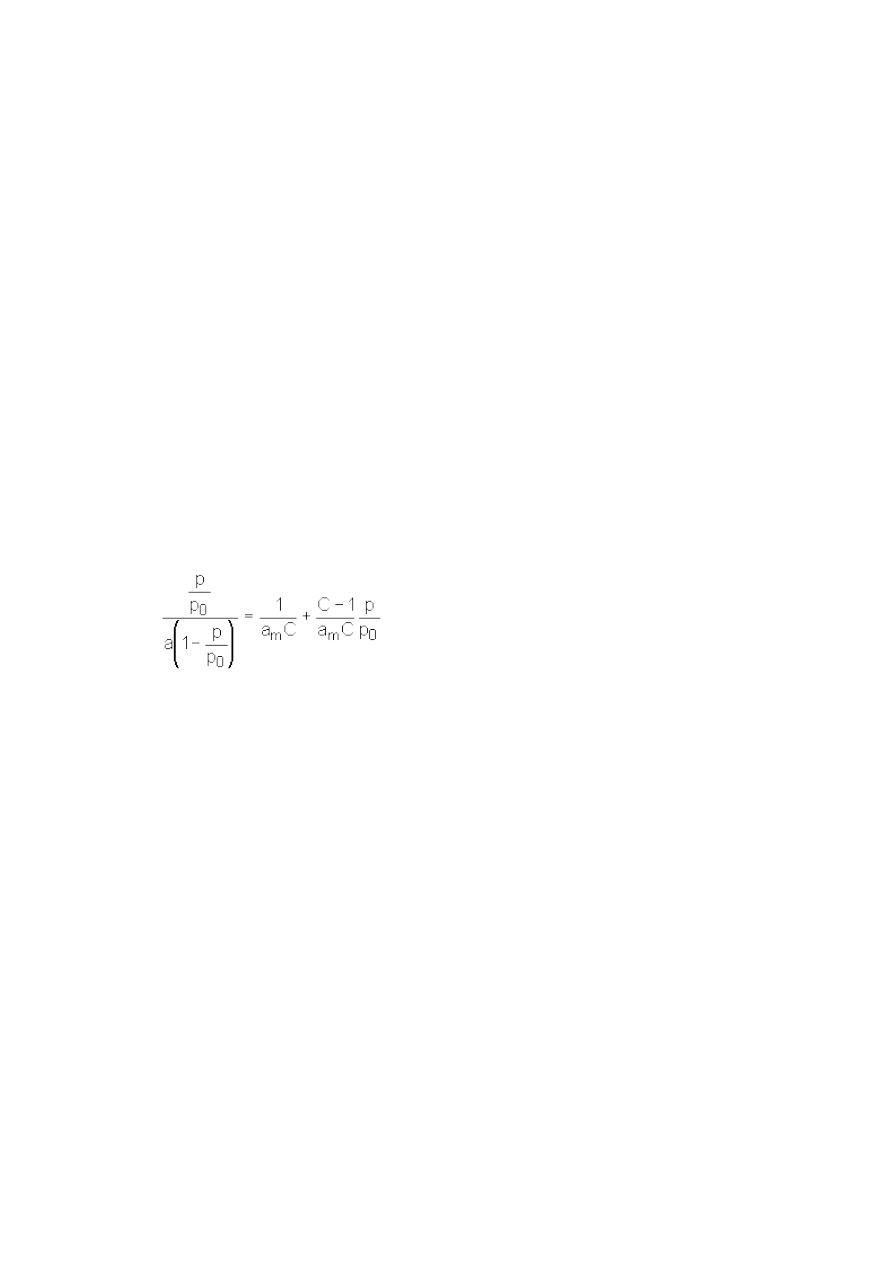
ZADANIE 8. IZOTERMY ADSORPCJI. RÓWNANIE BET, RÓWNANIE
DUBININA-RADUSZKIEWICZA.
Izoterma adsorpcji to zależność ilości zaadsorbowanej substancji od stężenia lub
ciśnienia adsorbatu przy ustalonej temperaturze.
Typ I - charakterystyczny dla adsorbentów mikroporowatych nazywany izotermą
Langmuira;
Typ II - (najczęściej spotykany) i III-ci (bardzo rzadko występujący)
charakterystyczny dla adsorbentów mikroporowatych;
Typ IV - (rozpowszechniony) i V (rzadko występujący) – charakterystyczny dla
mezoporowatych adsorbentów;
Typ VI - adsorpcja wielowarstwowa na powierzchni jednorodnej.
Równanie adsorpcji BET, izoterma BET,
równanie izotermywielowarstwowej adsorpcji pary jednoskładnikowej Brunauera,
Emmetta i Tellera, równanie postaci:
gdzie: p - ciśnienie zewnętrzne pary, p
0
- prężność pary nasyconej, a - całkowita ilość
zaadsorbowanej pary, a
m
- ilość adsorbatu (pary) pokrywającego powierzchnię
adsorbentu monowarstwą, C - stała zależna od ciepła adsorpcji pierwszej warstwy i
ciepła kondensacji.
Stałe a
m
i C mogą być wyznaczone doświadczalnie. Gdy C>>1 i p<<p
0
, równanie
adsorpcji BET przechodzi w izotermę adsorpcji Langmuira. Równanie adsorpcji BET
odnosi się do zjawiska adsorpcji par, któremu, przy wysokich ciśnieniach,
towarzyszy kondensacja par. Znajduje zastosowanie podczas doświadczalnego
wyznaczania powierzchni adsorbentów.
Izoterma DA, czyli izoterma Dubinina i Astachowa opisuje adsorpcję w
niewielkich porach, tzw. mikroporach, o średnicach porównywalnych z rozmiarami
cząsteczki adsorbatu (< 2nm wgIUPAC). Adsorpcja w małych porach jest znacznie
silniejsza niż na takiej samej chemicznie powierzchni płaskiej (większa ilość atomów
adsorbatu oddziałuje z bliska z adsorbatem) i najczęściej jest opisywana za pomocą
równania Dubinina-Raduszkiewicza (izoterma DR), izotermy Dubinina-Astachowa -
uogólnienia izotermy DR i Freundlicha - lub równań pochodnych.
gdzie:
- pojemność adsorpcyjna mikroporów,
- ciśnienie przy którym wszystkie mikropory są zapełnione (z reguły niższe niż
ciśnienie pary nasyconej, p
s
),
- stała związana z rozmiarem porów,
- stała związana z typem i rozkładem porów,
T - temperatura bezwzględna.
Dla n=2 z izotermy DA otrzymujemy izotermę DR, a dla n=1 izotermę Freundlicha.
Równanie izotermy DA może być uważane za jedno z rozwiązań całkowego
równania Stoeckliego, które pozwala na opisanie adsorpcji na mikroporach o
zróżnicowanej strukturze i rozmiarze.
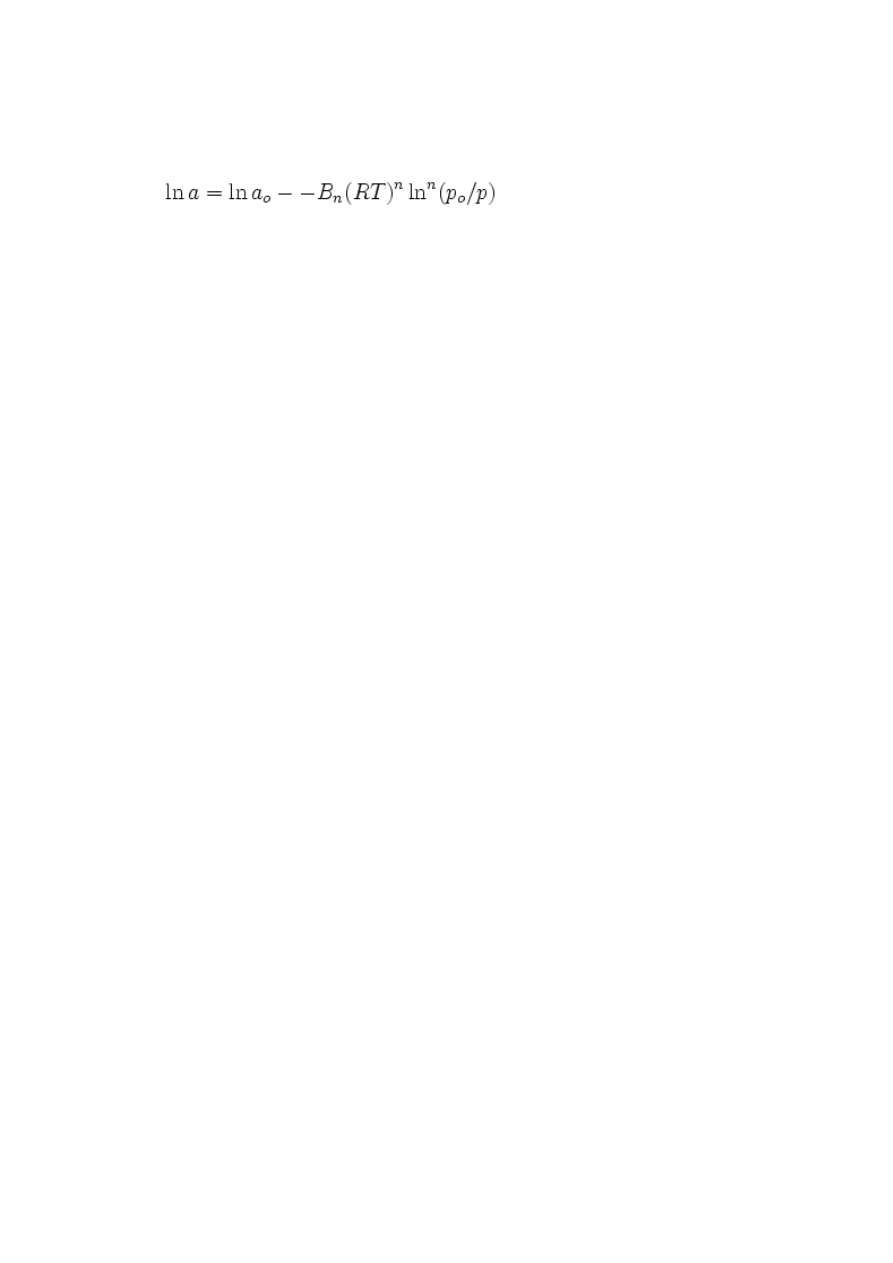
Równanie DA wykorzystuje się często w postaci logarytmicznej (log
10
lub ln), w
której dane doświadczalne zgodne z modelem powinny układać się wzdłuż linii
prostej ln(a)=f[ln
n
(p
o
/p)]:
Niestety stosowanie tej zależności do wyznaczania parametrów równania wymaga
uprzedniego wyznaczenia wartości parametrów n i p
o
. Można je jednak stosować
również w celu zademonstrowania zgodności danych doświadczalnych z modelem po
uprzednim dopasowaniu parametrów np. metodą najmniejszych kwadratów.
ZADANIE 9. WŁASCIWOŚCI TEKSTURALNE ADSORBENTÓW I
METODY ICH WYZNACZANIA.
Pełną informację o teksturze badanych materiałów adsorpcyjnych można uzyskać w
oparciu o następujące parametry: powierzchnia właściwa (powierzchnia przypadająca
na jednostkę masy adsorbentu wyrażana w m2/g); kształt, objętość i dystrybucja
porów (porozymetria).
Określenie tekstury adsorbentów można uzyskać na podstawie wielu metod.
Najczęściej korzysta się z tzw. standardowej metody analizy izoterm adsorpcji w
niskotemperaturowej sorpcji azotu (77 K) w oparciu o liniową formę równania
izotermy BET (metoda BET). Znając wielkość pojemności adsorpcyjnej
odpowiadającej utworzeniu monowarstwy (am) można wyznaczyć powierzchnię
właściwą (Sm) korzystając z równania:
Sm = am N ωm [m2/g]
gdzie: am - pojemność adsorpcyjna (wielkość adsorpcji odpowiadająca zapełnieniu
monowarstwywyrażana w cm3/g lub mol/g);
ωm - powierzchnia siadania (powierzchnia zajmowana przez cząsteczkę adsorbatu w
monowarstwie (np. dla azotu wynosi 0.16 nm2);
N - liczba Avogadro.
Czasami do wyznaczenia powierzchni właściwej stosuje się tzw. jednopunktową
metodę BET(metoda pkt B). Z wykresu izotermy adsorpcji odczytuje się wielkość
adsorpcji (am) tylko dla jednej wartości ciśnienia (najczęściej p/po= 0.2) przyjmując
założenie, że przy tej wartości ciśnienia względnego powierzchnia adsorbentu
pokrywa się monowarstwą. Inna metoda zwana metodą t (Lipsena i de Boera) zakłada,
że statystyczna grubość warstwy adsorpcyjnej „t’’ zależy od ciśnienia względnego
p/po (modele matematyczne Halseya, Harkinsa-Jury). Zmodyfikowaną metodą t jest
opracowana przez Singa metoda α, gdzie współczynnik α określa stosunek ilości
zaadsorbowanego azotu na badanym adsorbencie do ilości zaadsorbowanego azotu na
nieporowatym odnośniku dla ciśnienia względnego p/po.

ZADANIE 10. OBSZARY ZASTOSOWAŃ XRD, INTERPRETACJA
WYNIKÓW XRD.
Obszary zastosowań XRD:
Krystalografia rentgenowska
• układ krystalograficzny i klasę dyfrakcyjną,
• parametry komórki elementarnej,
• typ sieci Bravais’a i grupę symetrii przestrzennej,
• pozycje atomów w komórce elementarnej.
Rentgenowska analiza fazowa
• identyfikacja faz krystalicznych,
• skład fazowy próbek krystalicznych,
• rozróżnienie faz stałych amorficznych od krystalicznych
Interpretacja wyników XRD:
Pozycja refleksów (Identyfikacja substancji krystalicznej analiza fazowa
jakościowa i ilościowa)
Grupa symetrii przestrzennej.
Parametry komórki elementarnej.
Naprężenia wewnętrzne (jednorodne) - ciśnienie
Intensywność (j.w)
Rozmieszczenie jonów w komórce elementarnej
Tekstura.
Ilość materiału w substancjach wielofazowych.
Szerokość połówkowa
Naprężenia wewnętrzne (niejednorodne)
Wielkość krystalitów

Wyszukiwarka
Podobne podstrony:
Kataliza pytania lab
pytania z katalizy, Poda˙ wymiar sta˙ych szybko˙ci reakcji w r˙wnaniu szybko˙ci:
Mechanika Semest I pytania egz
prelekcja ZUM z pytaniami
pytania przykladowe exam zaoczne(1)
pytania nowe komplet
Pytania egzaminacyjneIM
Przeciwciała katalityczne u zdrowych ludzi i pacjentów z
EGZAMIN PKM2 pytania2011
Podstawy Teorii Okretow Pytania nr 4 (20) id 368475
haran egzamin opracowane pytania
NAI A2 pytaniaKontrolne
OU pytania id 342624 Nieznany
więcej podobnych podstron