
Charakterystyka Produktu Leczniczego, Ulotka dla
Pacjenta i opakowania produktów leczniczych w świetle
istniejących i powstających regulacji prawnych.
Anna Wachnik-Święcicka
Kierownik Działu Oceny Druków Informacyjnych
Urząd Rejestracji Produktów Leczniczych,
Wyrobów Medycznych i Produktów Biobójczych
12 października 2010

CHARAKTERYSTYKA PRODUKTU LECZNICZEGO
Podstawy prawne
➤ Wymagana dla każdego produktu leczniczego
Dyrektywa 2001/83/EC
w sprawie wspólnotowego kodeksu odnoszącego się
do produktów leczniczych stosowanych
u ludzi
Dyrektywa 2004/27/EC
zmieniająca Dyrektywę
2001/83/EC
Rozporządzenie (EC) 726/2004
w sprawie procedur wydawania pozwoleń
dla produktów leczniczych
Ustawa Prawo Farmaceutyczne
z 20 września 2001 r. ze zmianami

Wytyczne unijne
Forma:
szczegóły techniczne
CMD(h)
Annotated
QRD Template
for MR/DC
Procedure
Treść: szczegółowe zasady przedstawienia
informacji w ChPL
Notice to Applicants
(NTA):
A Guideline on Summary of Product
Characteristics,
October 2005
December 2007 (Draft)

Zalecenia ogólne
Zawarta w module 1.3 wspólnego dokumentu
technicznego. Uzgadniana w procesie rejestracji i
może być
zmieniona tylko za zgodą
władz
rejestracyjnych.
Stanowi integralną
część
pozwolenia na dopuszczenie
do obrotu.
Przeznaczona dla fachowego personelu medycznego i
stanowi podstawę
wiedzy o tym, jak stosować
produkt leczniczy bezpiecznie i skutecznie.

Zalecenia ogólne
Przygotowana z uwzględnieniem dodatkowych
wytycznych:
−
wytyczne dotyczące substancji pomocniczych
−
szczegółowe wytyczne dotyczące niektórych produktów
leczniczych, np.
antybiotyków
benzodiazepin
szczepionek
białek pegylowanych
produktów otrzymywanych z osocza
Stanowi podstawę
do przygotowania Ulotki dla pacjenta.

Zalecenia ogólne
Nie zawiera opisów sposobu leczenia poszczególnych
przypadków chorób -
jednak szczegóły związane z
leczeniem danym produktem oraz skutki jego działania.
Nie zawiera ogólnego opisu procedur medycznych -
opisuje szczegóły dotyczące sposobu stosowania
danego leku.
W każdym punkcie spójny opis zagadnień, zgodnie z
tytułem punktu. Jeśli dana informacja odnosi się
do
kilku punktów, zamieszcza się
stosowne odnośniki
.

Zalecenia ogólne
Oddzielna dla każdej postaci farmaceutycznej i mocy.
Ulotki mogą
być
wspólne dla kilku mocy, jeśli mają
identyczne wskazania i dawkowanie, sposób
stosowania, drogę
podania, przeciwwskazania,
ostrzeżenia i środki ostrożności oraz działania
niepożądane.
W razie konieczności można zamieścić
informacje o
innych dostępnych mocach lub postaciach, jeśli jest to
ważne dla całego schematu dawkowania.

Zasady przedstawiania informacji
Używać
jasnego i zwięzłego języka.
Używać
konsekwentnej terminologii medycznej
zaleca się
stosowanie terminologii MedDRA, zwłaszcza w punktach 4.3, 4.4 i
4.8.
Unikać
powtarzania informacji.
Najpierw podawać
informacje dotyczące całej populacji, a potem informacje
istotne dla szczególnych grup pacjentów.
Charakterystyka jest informacją
o konkretnym produkcie leczniczym -
nie należy
odwoływać
się
do innych produktów leczniczych z danej grupy (np. pisząc „jak
inne leki z tej samej grupy ...”), chyba że istnieją
oficjalne zalecenia dotyczące
ostrzeżeń
w danej grupie produktów.
Korzystać
z praktycznych zaleceń
zawartych w dokumentach grupy EMEA-
QRD.

Wytyczne EMEA-QRD
Wzorce druków
(Product
Information
Templates) do procedury centralnej
oraz MRP, DCP i procedury odwoławczej
-
annotated
template
(wzorce z objaśnieniami, wersja angielska)
-
clean
templates
(wzorce bez objaśnień
we wszystkich językach EU)
Dodatki
-
Appendix I
zapisy do punktu 4.6. Ciąża i laktacja
-
Appendix II
MedDRA
-
Appendix III
warunki przechowywania (do punktu 6.4)
-
Appendix IV
sposoby zapisu okresu ważności i numeru serii
Konwencja zapisu we wzorcach
tekst
tekst
obligatoryjny (np. tytuły punktów)
{tekst}
informację
w nawiasie należy uzupełnić
<tekst 1>
<tekst 2>
teksty do wyboru
[tekst]
objaśnienia


Części Wytycznych KE
Wytyczne unijne składają
się
z 3 części:
W rozdziale 1 wytyczne opisują
to, co w Polsce znajduje
się
w załącznikach do rozporządzenia o oznakowaniu
produktu leczniczego i treści ulotki
W rozdziale 2 wytyczne odnoszą
się
do wymagań
oznakowania produktu leczniczego związanych z
populacją
niewidomych i słabowidzących
W rozdziale 3 wytyczne dają
wskazówki do badania
czytelności ulotki, co w Polsce opisuje rozporządzenie o
badaniu czytelności.

12
ROZPORZĄDZENIE
MINISTRA ZDROWIA
z dnia 20 lutego 2009 r.
w sprawie wymagań
dotyczących oznakowania
opakowań
produktu leczniczego
i treści ulotki
Na podstawie art. 26 ust. 2 ustawy z dnia 6
września 2001 r. -
Prawo farmaceutyczne (Dz. U. z
2008 r. Nr 45, poz. 271 i Nr 227, poz. 1505)
zarządza się, co następuje:

13
§
1. Rozporządzenie określa wymagania
dotyczące oznakowania opakowań
produktu
leczniczego i treści ulotki oraz zakres
dostępności treści ulotki w formie właściwej
dla osób niewidomych i słabowidzących.

14
§
2. Na opakowaniu zewnętrznym, a jeżeli produkt ten
nie ma opakowania zewnętrznego -
na opakowaniu
bezpośrednim, umieszcza się
następujące informacje w
systemie Braille’a:
1) nazwę
produktu leczniczego;
2) moc produktu leczniczego, jeżeli
produkt jest dostępny w kilku mocach;
3) postać
farmaceutyczną, jeżeli produkt jest
dostępny w kilku postaciach.

15
§
5. 1. Do każdego opakowania produktu
leczniczego dołącza się
ulotkę, chyba że
opakowanie zewnętrzne lub opakowanie
bezpośrednie oznakowano zgodnie z
wymaganiami określonymi w §
6.
2. Pełna treść
ulotki jest udostępniana w formie
właściwej dla osób niewidomych i
słabowidzących.

16
§
7. 1. Na opakowaniu zewnętrznym i w ulotce mogą
być
zamieszczone symbole lub piktogramy, mające na
celu podanie w przystępniejszej formie niektórych
informacji określonych w §
3 ust. 1 i w §
6, lub
dodatkowe informacje dotyczące produktu
leczniczego, zgodne z danymi zawartymi w
Charakterystyce Produktu Leczniczego, jeżeli są
użyteczne do celów promocji zdrowia i nie zawierają
elementów reklamy. Umieszczone znaki graficzne
muszą
być
zatwierdzone w procesie
dopuszczenia do
obrotu.
2.
Wszystkie dane umieszczone na opakowaniu
zewnętrznym i opakowaniu bezpośrednim muszą
być
czytelne, zrozumiałe i nieusuwalne.

17
§
15. Informacje zamieszczone na opakowaniach należy
przedstawiać
czytelnie, zrozumiale dla użytkownika i
w sposób uniemożliwiający ich usunięcie bez
zniszczenia tego opakowania.
§
16. 1. Wymagania dotyczące sposobu sporządzania
oznakowania opakowań
określa załącznik nr 1 do
rozporządzenia.
2. Wymagania dotyczące sposobu sporządzania ulotki
określa załącznik nr 2 do rozporządzenia.

18
§
18. Treść
ulotki nie może zawierać
elementów promocyjnych
ani podawać
właściwości czy wskazań
do stosowania, które nie
są
zawarte w Charakterystyce Produktu Leczniczego, oraz nie
może zachęcać
do nieuzasadnionego wskazaniami stosowania
produktu leczniczego.

19
ROZPORZĄDZENIE MINISTRA ZDROWIA
z dnia 18 grudnia 2002 r.
w sprawie dokonywania zmian w pozwoleniu i
dokumentacji dotyczącej wprowadzenia do obrotu
produktu
leczniczego
§
6.
1. Zmiany inne niż
określone w §
3 i 4, a dotyczące
opakowań
i ulotek i niezwiązane ze zmianami w
Charakterystyce Produktu Leczniczego, zgłasza się
Prezesowi Urzędu Rejestracji Produktów Leczniczych,
Wyrobów Medycznych i Produktów Biobójczych.
2. Jeżeli Prezes Urzędu, o którym mowa w ust. 1, w ciągu 90
dni od dnia zgłoszenia nie wniesie sprzeciwu, to
proponowane zmiany uważa się
za przyjęte.

ROZPORZĄDZENIE MINISTRA ZDROWIA
z dnia 14 listopada
2008
r.
w sprawie
kryteriów zaliczenia produktu leczniczego
do poszczególnych kategorii dostępności
Rp
Rpw
Lz
Rpz
OTC

21
1.
NOWE KATEGORIE DOSTĘPNOŚCI
•
Produkt leczniczy wydawany z przepisu lekarza -
Rp
•
Produkt leczniczy wydawany z przepisu lekarza, zawierający
środki odurzające
lub substancje psychotropowe, określone
w odrębnych przepisach -
Rpw
•
Produkt leczniczy stosowany wyłącznie w lecznictwie
zamkniętym -
Lz
•
Produkt leczniczy wydawany z przepisu lekarza do
zastrzeżonego stosowania -
Rpz
•
Produkt leczniczy wydawany bez przepisu lekarza -
OTC”

22
Proponujemy pozostawienie czytelnych dla pacjenta i
krótszych opisów poszczególnych kategorii:
•
Rp
-
Produkt leczniczy wydawany z przepisu lekarza
•
Rpw
-
Produkt leczniczy wydawany z przepisu lekarza
•
Lz
-
Lek stosowany wyłącznie w lecznictwie zamkniętym
•
Rpz -
Produkt leczniczy wydawany z przepisu lekarza
•
OTC –
Produkt leczniczy
wydawany bez recepty

23
Zwolnienie z konieczności oznakowania
Braille’m
–
projekt rozporządzenia
Obowiązek umieszczania nazwy produktu leczniczego w systemie
Braille’a
nie dotyczy:
1) produktu
leczniczego,
który jest przeznaczony do podawania
wyłącznie przez lekarza, lekarza
dentystę, felczera, pielęgniarkę,
położną
lub ratownika medycznego;
2) produktu
leczniczego
przeznaczonego do specjalnych celów
żywieniowych;
3)
produktu leczniczego, którego opakowanie zewnętrzne, a w
przypadku jego braku, opakowanie bezpośrednie nie przekracza
objętości 10 ml;
4) produktu
leczniczego,
którego
zawartość
opakowania nie jest większa
niż
zalecana dawka dobowa;
5) produktu
leczniczego
roślinnego w postaci farmaceutycznej zioła do
zaparzania.

OCENA RAPORTU Z BADANIA
CZYTELNOŚCI ULOTKI
25
Artyku
Artyku
ł
ł
10 ust. 9
10 ust. 9
Minister w
Minister w
ł
ł
a
a
ś
ś
ciwy do spraw zdrowia okre
ciwy do spraw zdrowia okre
ś
ś
li,
li,
w drodze rozporz
w drodze rozporz
ą
ą
dzenia, spos
dzenia, spos
ó
ó
b badania
b badania
czytelno
czytelno
ś
ś
ci ulotki oraz kryteria dla raportu z
ci ulotki oraz kryteria dla raportu z
tego badania, w szczeg
tego badania, w szczeg
ó
ó
lno
lno
ś
ś
ci z
ci z
uwzgl
uwzgl
ę
ę
dnieniem
dnieniem
wytycznych Wsp
wytycznych Wsp
ó
ó
lnoty
lnoty
Europejskiej
Europejskiej
.
.
26
ROZPORZĄDZENIE
MINISTRA ZDROWIA
z dnia 26 kwietnia 2010 r.
w sprawie badania czytelności ulotki
Na podstawie art. 10 ust. 9 ustawy z dnia 6 września
2001 r. -
Prawo farmaceutyczne (Dz. U. z 2008 r. Nr
45, poz. 271 i Nr 227, poz.1505) zarządza się, co
następuje:
1) Minister Zdrowia kieruje działem administracji
rządowej -
zdrowie, na podstawie §
1 ust. 2
rozporządzenia Prezesa Rady Ministrów z dnia 16
listopada 2007 r. w sprawie szczegółowego zakresu
działania Ministra Zdrowia (Dz. U. Nr 216, poz.
1607).
Rozporządzenie bez załącznika
Zawiera 9 paragrafów
§
5. Wynik badania czytelności zatwierdzonej
ulotki w całości lub części, może być
wykorzystany przez podmiot odpowiedzialny
do potwierdzenia czytelności ulotki innego
produktu leczniczego.


Zawartość
raportu z badania czytelności
1.
opis produktu leczniczego;
2.
informacje dotyczące badania czytelności ulotki, w tym w
szczególności:
a)
opis sposobu przeprowadzenia badania,
b)
wyjaśnienie sposobu doboru uczestników badania,
c)
informację, w jakim języku przeprowadzono badanie;
d)
stwierdzenie, iż
przed badaniem zdefiniowano istotne
informacje dotyczące bezpieczeństwa stosowania produktu
leczniczego, w tym:
- najważniejsze przeciwwskazania,
- działania niepożądane,
- ostrzeżenia,
-
wskazania,
-
sposób stosowania produktu leczniczego;

Zawartość
raportu z badania czytelności
3.
kwestionariusz z instrukcją
dla przeprowadzającego badanie i
wypełnionym formularzem wyników obserwacji w jaki sposób
uczestnik badania posługiwał
się
ulotką
szukając odpowiednich
informacji;
4.
omówienie wyników badania, w szczególności w zakresie
zaistniałych problemów, a także omówienie dokonanych
modyfikacji w poszczególnych częściach ulotki;
5.
wnioski końcowe;
6.
ulotkę
poddawaną
badaniu i ulotkę
z wprowadzonymi
modyfikacjami na skutek przeprowadzonego badania.

§
8. Raport z badania czytelności ulotki
podlega zatwierdzeniu, jeżeli na podstawie
danych wymienionych w §
7., można
stwierdzić, że użytkownik, do którego
ulotka jest skierowana, zrozumie istotne
informacje zawarte w ulotce oraz będzie w
stanie prawidłowo stosować
produkt
leczniczy.

32

33
Porównanie metod oceny raportów z testów czytelności
w EMEA i MHRA
•
EMEA
Rekrutacja
Kwestionariusz
Czas trwania testu
Procedury testu
Przeprowadzenie wywiadu
Ocena wyników testu
Użycie punktowej skali oceny
odpowiedzi
Zapis i dokumentacja danych
Ocena wartości diagnostycznej pytań
Ocena wyglądu ulotki
Ocena jakości diagnostycznej testu
Wnioski
•
MHRA
Ocena wyglądu ulotki
Styl
Czcionka
Wygląd
Tytuły
Symbole i piktogramy
Treść
ulotki
Zgodność
z ChPL
Porządek zgodny z rozporządzeniem
Tekst podany w formie osobowej w
stronie czynnej
Ocena raportu z testu czytelności
Kluczowe informacje dot. bezpieczeństwa
stosowania
……

OCENA RAPORTU Z BADANIA
(według wytycznych EMEA)
1.
Sprawdzenie czy prawidłowo dobrano populację
badaną
Metoda rekrutacji, skład populacji pod względem
płci, wieku, edukacji, doświadczenia z użyciem
produktu leczniczego
Czy badana populacja jest z grupy ryzyka,
opiekunów czy jest nieświadoma choroby
Czy wiadomo jaka liczba pacjentów została
włączona do badania i czy była wystarczająca

2.
Ocena kwestionariusza
Czy liczba pytań
jest wystarczająca (15 do 20)
Czy pytania dotyczą
ważnych części ulotki
Czy są
pytania dotyczące wyglądu ogólnego ulotki
Czy bezpieczeństwo stosowania leku jest
sprawdzone wystarczającą
liczbą
pytań
Czy pytania są
otwarte, czy zawierają
gotowe
odpowiedzi

3.
Czas trwania testu
Czy czas na odpowiedź
jest wystarczający
Czy raport zawiera informacje dotyczące
długości wywiadu i czy był
to
wystarczający czas na jego
przeprowadzenie

4.
Procedury testu
Czy przeprowadzono 2 rundy badania
Czy ostatecznie ulotka została
przetestowana na minimum 20 pacjentach
Czy wprowadzono zmiany zwiększające
czytelność
Czy użyto scenariusza zmuszającego do
demonstracji

5.
Przeprowadzenie wywiadu
Czy osoba przeprowadzająca wywiad została
przeszkolona
Czy pacjenci mogli pokazywać
w ulotce miejsce
gdzie znajduje się
odpowiedź
na pytanie
Czy pacjenci byli proszeni o podanie
odpowiedzi własnymi słowami
Czy wywiad był
filmowany lub nagrywany

6.
Czy ocena wyników testu została
przeprowadzona prawidłowo
Jakość
oceny odpowiedzi
Czy w badaniu określono oddzielnie
łatwość
znalezienia informacji i
zrozumienie informacji

7.
Czy użyto punktowej skali oceny
odpowiedzi
1 –
brak odpowiedzi
2 –
zła odpowiedź
3 –
odpowiedź
niekompletna
4 –
odpowiedź
niejednoznaczna
5 –
odpowiedź
prawidłowa

8.
Czy dane z testu są
prawidłowo zapisane i
udokumentowane
W jaki sposób
Czy odpowiedzi zostały przekształcone
według skali punktowej

9.
Ocena jakości testu
Czy metodologia badania jest oparta na
wytycznych dotyczących czytelności
Czy w przypadku każdego z pytań
90%
uczestników
(18 z 20) prawidłowo
zlokalizowało
informację
a z tych 90%
następne 90% uczestników
(16,2 z 18)
informację
zrozumiało

10.
Ocena wyglądu ulotki
Czy wykorzystano zasady projektowania
ulotki z wytycznych dotyczących
czytelności
Czy język ulotki jest przyjazny dla
pacjenta
Czy odpowiedzi na pytania były łatwo
lokalizowane przez badanych
Czy wykresy, rysunki są
wystarczająco
zrozumiałe

Leaflet A

Leaflet B

Analysing search time
Overall search time taken
0
50
100
150
200
250
300
350
400
450
500
Q0
Q1
Q2
Q3
Q4
Q5
Q6
Q7
Q8
Q9
Q10
Q11
Q12
Question
A1
A2
A3
A4
A5
A6
A7
A8
A9
A10
Overall search time taken
0
50
100
150
200
250
300
350
400
450
500
Q0
Q1
Q2
Q3
Q4
Q5
Q6
Q7
Q8
Q9
Q10
Q11
Q12
Question
B1
B2
B3
B4
B5
B6
B7
B8
B9
B10

11.
Ocena jakości diagnostycznej testu
Czy znaleziono słabe strony ulotki
Czy słabe strony zostały prawidłowo
poprawione
Czy wyniki odnoszą
się
do odpowiedniej rundy
testu
Czy przeprowadzający test prawidłowo wyjaśnia
trudności ze zrozumieniem tekstu (zły tytuł,
podwójne zaprzeczenie, terminologia zbyt
trudna)

Czy przeprowadzono 2 rundy
Czy poprawiono tekst odnoszący się
do pytań
ze
słabym wynikiem zrozumienia (<80%)
Czy wyraźnie określono jakie były podstawy
nowelizacji tekstu (na podstawie jakich obserwacji
dokonano zmiany tekstu)
Czy zaznaczono jakie obserwacje nie zostały
wzięte pod uwagę
i dlaczego
Czy nowelizacje tekstu zostały przetestowane i
dowiodły poprawy czytelności

FINAL REPORT
FINAL REPORT
Analysis of Responses
Percentage of answers found and understood
0
20
40
60
80
100
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
Question numbers
Very Easy
Easy
Some difficulty
Lots of difficulty
Understanding
80% Benchmark

12.
Czy cel badania czytelności został
osiągnięty
Czy wnioski z badania są
prawidłowe
Czy metodologia badania była prawidłowa
Czy ulotka została prawidłowo
zbudowana

PODSUMOWANIE
Ocena raportu musi określić, czy oceniono
czytelność
ulotki
Czy zlokalizowano problemy ze
zrozumieniem tekstu i prawidłowym
użyciem leku
Czy wystarczająco opisano zmiany
wprowadzone w celu zwiększenia
czytelności

Doświadczenia wybranych krajów UE w zakresie
oceny raportów z badania czytelności ulotek
Wielka Brytania –
MHRA
Akceptacja ulotki nie zależy wyłącznie od
wyników prezentowanych w raporcie z badania
czytelności
Można zaakceptować
ulotkę, a w ocenie napisać,
że test czytelności nie może stanowić
podstawy do
pomostowania jego wyników na inną
ulotkę

GRAFIKA OPAKOWAŃ
PRODUKTÓW LECZNICZYCH

Wizerunek dziecka/dzieci, np. na
opakowaniach leków przeznaczonych do
stosowania u dzieci.

Ilustracja roślin, zwierząt, np. na
opakowaniach leków
przeciwalergicznych.

Ilustracja określonych roślin i (lub) owoców
na opakowaniach leków o danym smaku, np.
liść
mięty, wiśnia, na wpół
obrany banan.
Fragmenty układów i narządów, np.
układu pokarmowego, wątroby, górnych
dróg oddechowych.

Personifikacja postaci farmaceutycznych,
np. tabletki

Zamieszczanie haseł, tj. skróconych
wskazań
lub objawów, na które lek działa,
na opakowaniach leków bez recepty.

produkt leczniczy
≠
produkt spożywczy


WZGLĘDY BEZPIECZEŃSTWA!
OPAKOWANIE PRODUKTU
LECZNICZEGO NIE MOŻE BYĆ
ATRAKCYJNE DLA DZIECI!

symbole umieszczane na opakowaniu
zewnętrznym mogą, zgodnie z §
7
Rozporządzenia Ministra Zdrowia z dnia 20
lutego 2009 r. w sprawie wymagań
dotyczących
oznakowania opakowań
produktu leczniczego i
treści ulotki, w przystępniejszej formie podawać
niektóre informacje określone w § 3 ust 1 i § 6,
jeżeli są
użyteczne do celów promocji zdrowia i
nie zawierają
elementów reklamy;

na opakowaniu zewnętrznym mogą
być
zamieszczone dodatkowe informacje
dotyczące produktu leczniczego, zgodne z
danymi zawartymi w Charakterystyce
Produktu Leczniczego, jeżeli są
użyteczne
do celów promocji zdrowia i nie zawierają
elementów reklamy;

przykłady symboli lub piktogramów,
które mogą
być
zamieszczane na
opakowaniach są
podane w Załączniku 1.
do Rozporządzenia Ministra Zdrowia z
dnia 20 lutego 2009 r. w sprawie
wymagań
dotyczących oznakowania
opakowań
produktu leczniczego i treści
ulotki w punkcie 10;

grafika oznakowania opakowań
produktu
leczniczego o kilku mocach w celu
ułatwienia rozróżnienia mocy i
zmniejszenia ryzyka pomyłki powinna
odróżniać
kolorami poszczególne moce;

na projektach graficznych oznakowań
opakowań
produktów leczniczych nie należy
umieszczać
grafik, takich jak: wizerunki
zabawek, baloników, tabliczek czekolady itp.,
które mogłyby spowodować, że tak oznakowany
lek zostanie potraktowany przez użytkownika,
jako, np. środek spożywczy. Wyżej wymienione
symbole, powodując możliwość
pomyłki,
promocji zdrowia nie służą;

rysunek cytryny lub inny element tego typu,
odzwierciedlający smak produktu, może być
umieszczony, jeśli składnik produktu
warunkujący ten smak jest pochodzenia
naturalnego i rysunek taki nie stanowi
głównego elementu graficznego opakowania,
nie powoduje zmniejszenia czcionki w tekście
oznakowania ani nie jest przedstawiony w
sposób wskazujący na jego reklamowy cel;

wizerunki dzieci, jako informacja, że lek jest
przeznaczony dla dzieci, mogą
być
umieszczane, jeśli
nie stanowią
głównego elementu graficznego
opakowania, nie powodują
zmniejszenia czcionki w
tekście oznakowania ani nie są
przedstawione w sposób
wskazujący na ich reklamowy cel;

kolory tła opakowania i kolory napisów
powinny w odpowiednim stopniu kontrastować
ze sobą, tak aby napisy były czytelne i nie ginęły
w kolorach tła, gdyż
zgodnie z §
15
Rozporządzenia Ministra Zdrowia z dnia 20
lutego 2009 r. w sprawie wymagań
dotyczących
oznakowania opakowań
produktu leczniczego i
treści ulotki, informacje zamieszczone na
opakowaniach należy przedstawiać
czytelnie i
zrozumiale dla użytkownika;

Safety Agencies
3. National Safety Patients Agency (NPSA)
Design for safety. Labelling and Packaging Guidelines for
Injectable
Medicines. NPSA, London June 2007.
Pages 20 and 21
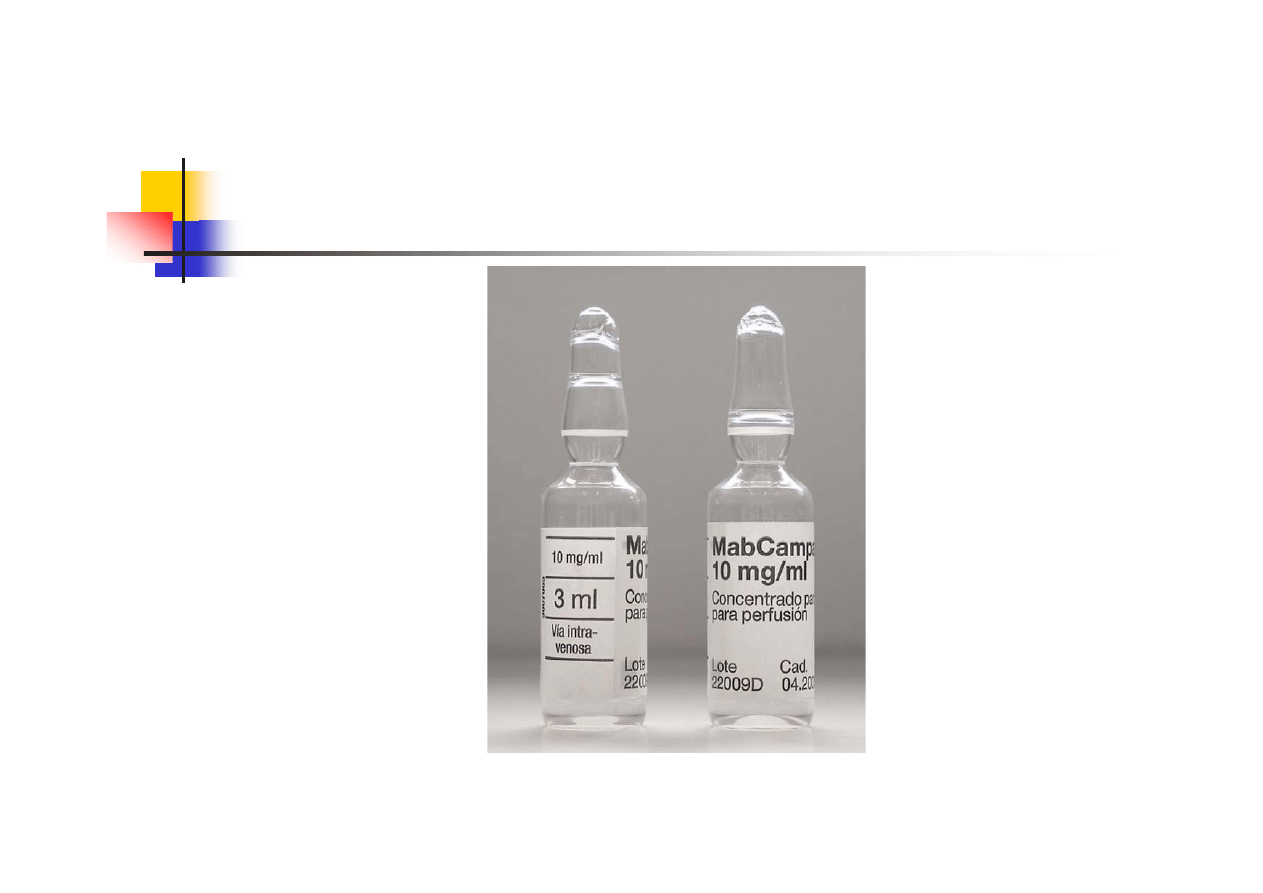
“Issue: It can be misleading if the strength of a medicine is
expressed in quantity/unit volume e.g. mg/ml. This strength
can be confused as the total concentration in the ampoule,
vial or syringe. Recommendation: It should be expressed as
the total quantity / total volume (x mg/y ml)”.



Dziękuję
za uwagę!
Document Outline
- Charakterystyka Produktu Leczniczego, Ulotka dla Pacjenta i opakowania produktów leczniczych w świetle istniejących i powstających regulacji prawnych.
- CHARAKTERYSTYKA PRODUKTU LECZNICZEGO Podstawy prawne
- Slajd numer 3
- Zalecenia ogólne
- Zalecenia ogólne
- Zalecenia ogólne
- Zalecenia ogólne
- Zasady przedstawiania informacji
- Wytyczne EMEA-QRD
- http://ec.europa.eu/enterprise/pharmaceuticals/eudralex/vol-2/c/2009_01_12_readability_guideline_final.pdf
- Części Wytycznych KE
- Slajd numer 12
- Slajd numer 13
- Slajd numer 14
- Slajd numer 15
- Slajd numer 16
- Slajd numer 17
- Slajd numer 18
- Slajd numer 19
- Slajd numer 20
- Slajd numer 21
- Slajd numer 22
- Zwolnienie z konieczności oznakowania Braille’m – projekt rozporządzenia
- Slajd numer 24
- Slajd numer 25
- Slajd numer 26
- Slajd numer 27
- Slajd numer 28
- Zawartość raportu z badania czytelności
- Zawartość raportu z badania czytelności
- Slajd numer 31
- Slajd numer 32
- Porównanie metod oceny raportów z testów czytelności w EMEA i MHRA
- OCENA RAPORTU Z BADANIA (według wytycznych EMEA)
- Slajd numer 35
- Slajd numer 36
- Slajd numer 37
- Slajd numer 38
- Slajd numer 39
- Slajd numer 40
- Slajd numer 41
- Slajd numer 42
- Slajd numer 43
- Slajd numer 44
- Slajd numer 45
- Analysing search time
- Slajd numer 47
- Slajd numer 48
- FINAL REPORT
- Slajd numer 50
- Slajd numer 51
- Doświadczenia wybranych krajów UE w zakresie oceny raportów z badania czytelności ulotek
- Slajd numer 53
- Slajd numer 54
- Slajd numer 55
- Slajd numer 56
- Slajd numer 57
- Slajd numer 58
- Slajd numer 59
- Slajd numer 60
- Slajd numer 61
- Slajd numer 62
- Slajd numer 63
- Slajd numer 64
- Slajd numer 65
- Slajd numer 66
- Slajd numer 67
- Slajd numer 68
- Slajd numer 69
- Slajd numer 70
- Slajd numer 71
- Slajd numer 72
- Slajd numer 73
- Slajd numer 74
- Slajd numer 75
- Slajd numer 76
Wyszukiwarka
Podobne podstrony:
anx 130011 pl Charakterystyka produktu leczniczego
cyclodynon ulotka dla pacjenta
Pobierz charakterystykę produktu leczniczego w formacie
sinupret tabletki ulotka dla pacjenta
sinupret tabletki ulotka dla pacjenta
wymagania jakościowe dla produktów leczniczych
Szczegółowy sposób przedstawiania dokumentacji dla produktów leczniczych weterynaryjnych
Reklama produktów leczniczych
PRODUKTY DOZWOLONE I PRZECIWWSKAZANE DLA KARMIĄCYCH, pliki zamawiane, edukacja
02 Charakteryzowanie produkcji Nieznany (2)
02 Charakteryzowanie produkcji roślinnej i zwierzęcejid 3593
Charakterystyka nowej podstawy programowej dla przedszkoli i klas początkowych, Współczesne koncepcj
Rozp MZ w sprawie określenia grup produktów leczniczych oraz wymagań dotyczących dokumentacji wynik
Porównanie projektów wdrożenia opieki farmaceutycznej dla pacjentów z astmą
poradnik neurorehabilitacji dla pacjentów wcm
Zalecenia dla pacjentów po zabiegu augmentacji dziąsła
Rozp MZ w sprawie sposobu ustalania i uiszczania opłat związanych z dopuszczeniem do obrotu produkt
ROŽLINNE PRODUKTY LECZNICZE wstŕp
więcej podobnych podstron