
1
Wydział Inżynierii Materiałowej i Ceramiki
Akademii Górniczo-Hutniczej w Krakowie
Materiały pomocnicze do zajęć laboratoryjnych z przedmiotu
„CHEMIA ORGANICZNA”
Kraków 2010

2
SPIS TREŚCI
Str.
1. Zasady pracy w laboratorium chemii organicznej ................................... 3
2. Wykaz ćwiczeń laboratoryjnych .............................................................. 4
3. Szkło i sprzęt laboratoryjny używany na zajęciach .................................. 5
4. Instrukcje do ćwiczeń
Ćwiczenie 1 ............................................................................................... 12
Ćwiczenie 2 ............................................................................................... 26
Ćwiczenie 3 ............................................................................................... 34
Ćwiczenie 4 ............................................................................................... 40
Ćwiczenie 5 ............................................................................................... 46

Zasady pracy w laboratorium chemii organicznej
3
1. Zasady pracy w laboratorium chemii organicznej
Podczas pracy w laboratorium chemii organicznej - jak w każdym laboratorium
chemicznym - należy zachować ostrożność. Specyfika zajęć laboratoryjnych
z „Chemii organicznej” polega jednak na tym, że wymagają one stosowania
substancji organicznych, które często są palne i niebezpieczne dla zdrowia. Dlatego
w laboratorium chemii organicznej należy:
1. Zawsze nosić odzież ochronną, tj. fartuch, rękawice, okulary ochronne,
a długie włosy na czas pracy w laboratorium związywać z tyłu głowy.
2. Niebezpieczne ciecze, takie, jak: lotne, palne, toksyczne rozpuszczalniki
organiczne, stężone kwasy nieorganiczne, przelewać pod sprawnie
działającym wyciągiem.
3. Odmierzone pod wyciągiem ciecze przenosić do swojego miejsca pracy
w kolbach zamkniętych odpowiednimi korkami.
4. Ogrzewanie rozpuszczalników organicznych prowadzić zawsze pod
chłodnicą zwrotną.
5. Zużyte lub niepotrzebne rozpuszczalniki i substancje organiczne umieszczać
w specjalnie przeznaczonych na nie naczyniach lub pojemnikach.
6. Jakiekolwiek wątpliwości związane z wykonaniem ćwiczenia wyjaśniać
u prowadzącego zajęcia.
7. Po zakończeniu ćwiczenia umyć szkło, wytrzeć stół laboratoryjny, ustawić
wszystko na właściwym miejscu, wyłączyć przewody elektryczne z sieci
i sprawdzić, czy wszystkie krany wodociągowe i kurki gazowe są dokładnie
zamknięte. Umyć ręce.
W laboratorium chemii organicznej nie wolno:
1. Przebywać, uruchamiać sprzętów/aparatury, rozpoczynać eksperymentów
bez zgody prowadzącego zajęcia.
2. Pozostawiać mieszaniny reakcyjnej bez nadzoru.
3. Spożywać pokarmów, pić napojów, palić tytoniu.
4. Badać smaku związków chemicznych, a badając ich zapach należy zachować
jak największą ostrożność – w żadnym wypadku nie wolno wdychać par
związku.
5. Pipetować ustami.
6. Dotykać substancji chemicznych rękami – do tego celu używa się
specjalnych łopatek lub łyżeczek.
7. Włączać palników gazowych, jeżeli w ich pobliżu znajdują się substancje
palne.
8. Palić palników gazowych bez potrzeby – palniki należy wyłączać
bezpośrednio po ich użyciu.

Wykaz ćwiczeń laboratoryjnych
4
2. Wykaz ćwiczeń laboratoryjnych
Ćwiczenie 1: DESTYLACJA
Ćwiczenie 2: KRYSTALIZACJA
Ćwiczenie 3: EKSTRAKCJA
Ćwiczenie 4: REAKCJE ZWIĄZKÓW ORGANICZNYCH: PODSTAWIENIE
ELEKTROFILOWE W ZWIĄZKACH AROMATYCZNYCH –
SULFONOWANIE p-KSYLENU
Ćwiczenie 5: REAKCJE ZWIĄZKÓW ORGANICZNYCH: ESTRYFIKACJA
– OTRZYMYWANIE OCTANU n-BUTYLU
lub OCTANU IZOAMYLU

Szkło i sprzęt laboratoryjny używany na zajęciach
5
3.
Szkło i sprzęt laboratoryjny używany na zajęciach
1
1
Opracowanie: dr inż. Edyta Stochmal
Zlewki
Kolbki stożkowe (erlenmayerki)
ze szlifem, korki szklane ze szlifem
Kolbki okrągłodenne i kolbka
gruszkowa ze szlifem
Cylindry miarowe
Kolba ssawkowa

Szkło i sprzęt laboratoryjny używany na zajęciach
6
Chłodnica Liebiega
Nasadki do destylacji,
nasadka do gazu
Termometr z regulowanym
szlifem
Kolumna frakcjonująca,
(kolumna rektyfikacyjna,
deflegmator) wypełniona
pierścieniami Raschiga
Bagietka, szpatułka

Szkło i sprzęt laboratoryjny używany na zajęciach
7
Lejek ze spiekiem szklanym,
lejek zwykły
Rozdzielacz gruszkowy
Łapa, łącznik,
kółko do rozdzielacza
Statyw
Nasadka azeotropowa

Szkło i sprzęt laboratoryjny używany na zajęciach
8
Podnośniki
Czasza grzejna (płaszcz grzejny)
z regulatorem mocy
Aparat do pomiaru temperatury
topnienia – typ SMP3
Kapilarki do pomiaru
temperatury topnienia
Aparat do pomiaru temperatury
topnienia – typ SMP10

Szkło i sprzęt laboratoryjny używany na zajęciach
9
Zestaw aparatury do destylacji
prostej pod ciśnieniem
atmosferycznym
Zestaw aparatury do destylacji
frakcyjnej
Zestaw do sączenia pod
zmniejszonym ciśnieniem
Zestawy laboratoryjne

Szkło i sprzęt laboratoryjny używany na zajęciach
10
Zestaw aparatury do ogrzewania
pod chłodnicą zwrotną
Zestaw do prowadzenia ekstrakcji
periodycznej w układzie ciecz-ciecz
Zestaw aparatury do
prowadzenia estryfikacji

11
4. Instrukcje do ćwiczeń

Ćwiczenie 1: Destylacja
12
Ć
wiczenie 1:
D E S T Y L A C J A
2
Cel ćwiczenia: Opanowanie dwu podstawowych technik destylacji – destylacji różniczkowej
i rektyfikacji okresowej w kolumnie z wypełnieniem.
1. WSTĘP
Jednym z najważniejszych etapów przygotowujących do pracy ze związkami chemicznymi jest
ich oczyszczenie. Szczególną uwagę należy zwrócić na substancje w stanie ciekłym, bowiem większość
syntez chemicznych prowadzi się w takim środowisku. Destylacja w różnych jej odmianach jest
najczęściej stosowaną metodą rozdzielania i oczyszczania ciekłych substancji. Destylacja polega na
przeprowadzeniu cieczy w stan pary w temperaturze wrzenia, a następnie skropleniu powstałej pary.
Skropliny noszą nazwę destylatu.
W zależności od właściwości fizykochemicznych mieszanin stosuje się różnego rodzaju warianty
destylacji. Są to destylacje proste - równowagowa, różniczkowa, z parą wodną, pod zmniejszonym
ciśnieniem, azeotropowa, molekularna - i cały szereg destylacji frakcjonowanej (rektyfikacji) - jak
rektyfikacja w kolumnach półkowych, w kolumnach z wypełnieniem – zarówno jako okresowe bądź
ciągłe.
W ćwiczeniu prowadzi się destylację różniczkową i rektyfikację okresową w kolumnie
z wypełnieniem. Porównując ze sobą przebiegi tych procesów będzie można stwierdzić, jakie różnice
dzielą te dwie metody. Aby wyjaśnić, na czym polega istota destylacji przedyskutujemy na wstępie, jak
zachowują się ciecze podczas ogrzewania pod stałym ciśnieniem i podczas izotermicznej (w stałej
temperaturze) zmiany ciśnienia, bo takie właśnie działania są podstawą destylacji.
Substancje czyste (jednoskładnikowe).
Ciecze takie wrą przy zachowaniu stałego ciśnienia w stałej temperaturze, mimo ich ogrzewania.
Dopiero po całkowitym odparowaniu temperatura pary będzie wzrastać, gdy nadal będziemy dostarczać
ciepła z zewnątrz. Natomiast gdyby ciecz umieścić w naczyniu zamkniętym ruchomym szczelnym
tłokiem, a następnie tłok nieco podnieść, to ciśnienie w naczyniu nie zmieni się, bo w „wolne” miejsce
odparuje kolejna porcja cieczy. Tak będzie się działo dopóty, dopóki cała ciecz nie przejdzie w stan pary.
Potem ciśnienie w naczyniu będzie maleć w miarę podnoszenia tłoka.
Mieszaniny
Zachowanie się cieczy będącej mieszaniną dwu lub więcej ciekłych składników w obydwu
przypadkach będzie odmienne od zachowania się czystych substancji. I tak ogrzewanie wrzącego
roztworu (poza kilkoma szczególnymi wyjątkami) będzie powodować stały wzrost temperatury wrzącej
cieczy. Natomiast podniesienie tłoka nad roztworem umieszczonym w naczyniu spowoduje obniżenie
ciśnienia par w naczyniu. Właściwe wykorzystanie tych odmiennych zachowań roztworów i substancji
czystych pozwala rozdzielić mieszaninę na składniki lub uwolnić od zanieczyszczeń, co prowadzi do
procesu destylacji.
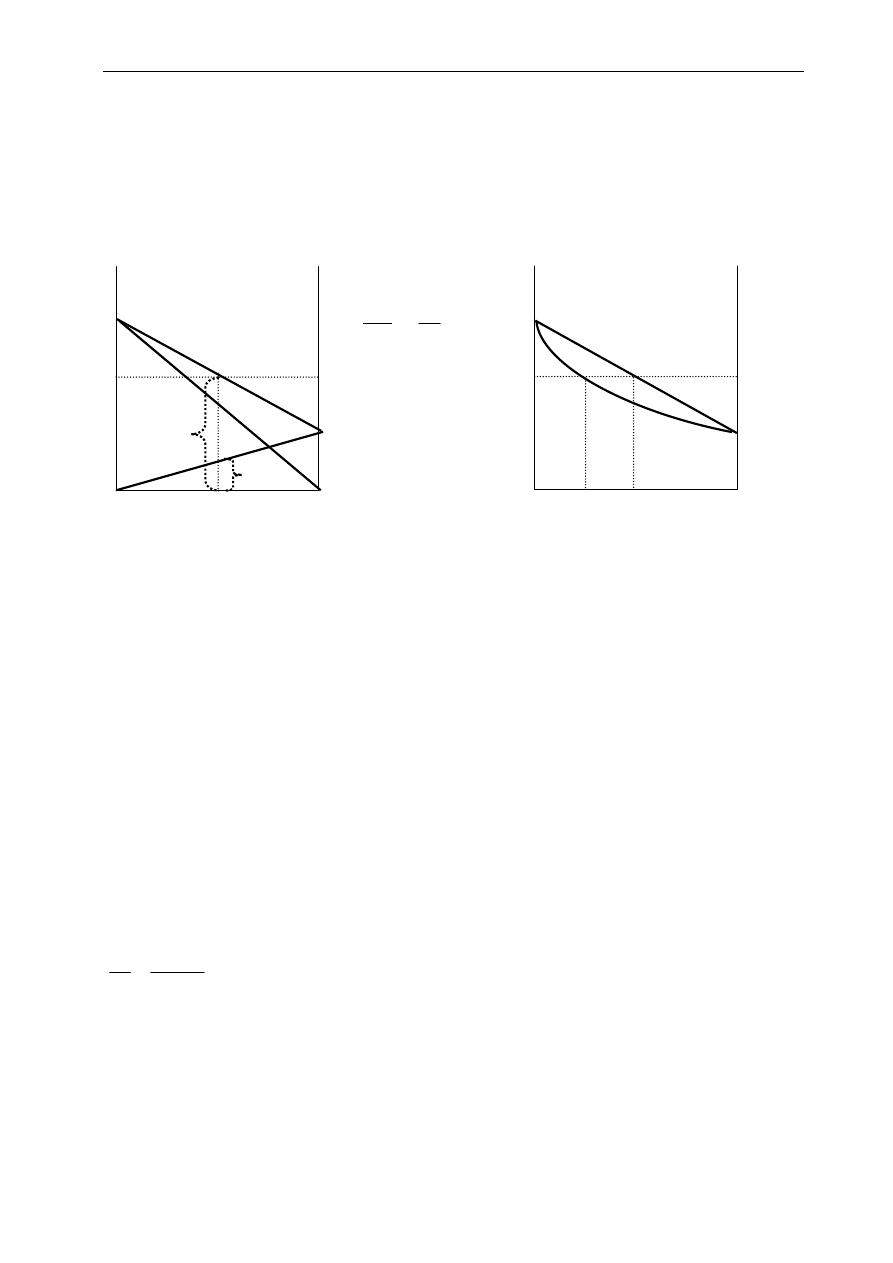
2. DIAGRAMY FAZOWE
Do opisu zachowania się cieczy będącej mieszaniną dwu (lub większej liczby) składników
ciekłych podczas zmiany ciśnienia lub temperatury służą diagramy fazowe. Rozróżniamy dwa rodzaje
takich diagramów:
a) podający zależność prężności pary nasyconej od składów cieczy i pary przy stałej temperaturze;
b) podający zależność temperatury wrzenia od składów cieczy i pary pod stałym ciśnieniem
(nawiązujący do codziennej praktyki gotowania w kuchni)
2
Opracował dr Antoni Szumiło-Kulczycki; opracowanie graficzne: dr hab. Magdalena Hasik.
Poszerzone ramy instrukcji podyktowane są faktem, że kurs chemii fizycznej, będącej podstawą destylacji, jest
wykładany w późniejszym terminie
Ćwiczenie 1: Destylacja
13
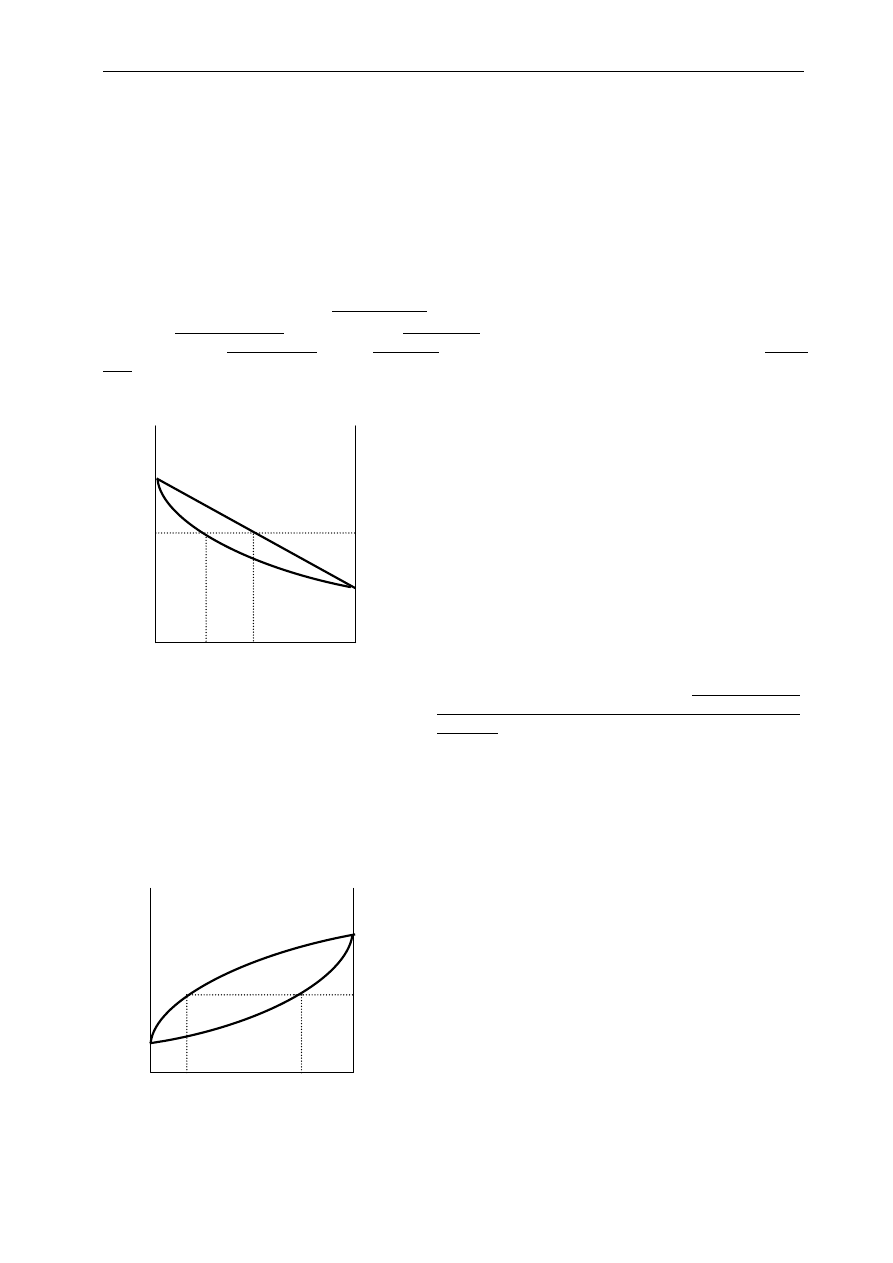
Ad a) Diagram fazowy: prężność pary – skład (temperatura stała)
Diagram taki przedstawiono na Rys. 2.1. Osi odciętych odpowiada skład (stężenie), a osi
rzędnych prężność pary układu. Oś odciętych zawiera informacje zarówno o stężeniu składników w
roztworze, jak i w parze. Stężenie składnika B - tak w roztworze, jak i w parze nasyconej nad nim - w
postaci ułamka molowego, lub wagowego składnika B są zaznaczone na osi odciętych
i zmieniają się od
0 do 1 idąc z
lewa do prawa. (Można też na tej osi odłożyć wartości stężeń składnika A, ale wówczas po
prawej stronie – na końcu – będzie 0 a po lewej – na początku osi 1). Na osi rzędnych odłożono prężności
pary, w szczególności na lewej osi odpowiadającej czystemu składnikowi A zaznaczona jest prężność
pary nasyconej czystego składnika A -
0
A
P
, a na prawej odpowiadającej czystemu składnikowi B prężność
pary nasyconej składnika B -
0
B
P
. Krzywa górna jest wykresem prężności pary nasyconej nad roztworem
w funkcji składu roztworu i nazywana jest linią cieczy, bo wiąże prężność pary nasyconej ze składem
cieczy. Natomiast krzywa dolna, zwaną linią pary przedstawia prężność pary nasyconej w funkcji składu
pary . Konstrukcję tego diagramu dla roztworów doskonałych omówiono w 7 rozdziale uzupełniającym:
ANEKS I.
Rys.2.1. Diagram fazowy prężność pary -
skład
diagramu fazowego, prowadząc prostą równoległą do osi odciętych na wysokości tego ciśnienia P
1
.
Punkty przecięcia tej prostej z krzywymi cieczy i pary mają współrzędne „x-owe” odpowiadające
składowi cieczy (x
B
) i pary (y
B
), patrz Rys. 2.1.
Ad b) Diagram fazowy temperatura wrzenia – skład (ciśnienie stałe)
x
B
y
B
P
B
0
T=const
P
A
0
P
1
para
ciecz
p
p
Ułamek molowy składnika B
0
1
Z linią równoległą do osi odciętych poprowadzoną na
wysokości odpowiadającej całkowitej prężności
1
P
związane są dwa szczególne punkty o współrzędnych
na osi odciętych wynoszących
B
B
y
x
i
, które podają
odpowiednio składy roztworu i pary nad nim. Punkty
należące do obszaru leżącego powyżej krzywej górnej
przedstawiają stany, w których nasz układ może
wystąpić tylko w postaci fazy ciekłej, a punkty
poniżej dolnej krzywej charakteryzują stany, w
których nasz układ występuje tylko jako faza gazowa -
para. Natomiast każdy punkt znajdujący się w
obszarze
pomiędzy
tymi
dwiema
krzywymi
reprezentuje
sytuację,
w
której
występują
równocześnie dwie fazy - ciekła i gazowa -
pozostające ze sobą w równowadze. Składy tych faz
zależą tylko od ciśnienia całkowitego panującego w
układzie. Np. pod ciśnieniem P
1
, układ złożony jest z
cieczy i pary o składach, które możemy odczytać z
T
B
0
para
ciecz
p=const
T
A
0
x
B
y
B
T
T
Rys.2.2. Diagram fazowy
temperatura- skład
0
1
T
1
Oś odciętych ma identyczne, co poprzednio, oznaczenia. Na
osi rzędnych odłożono temperatury -
0
0
i
B
A
T
T
temperatury
wrzenia czystych składników pod zadanym ciśnieniem
(zazwyczaj
atmosferycznym).
W
odróżnieniu
od
poprzedniego diagramu, górna krzywa jest krzywą pary, a
dolna krzywą cieczy (rys.2.2). Punkty leżące na górnej
krzywej odpowiadają stanom w których para zaczyna się
skraplać, a punkty z dolnej krzywej odpowiadają stanom, w
których ciecz zaczyna wrzeć i pojawia się pierwsza porcja
pary. Linia równoległa do osi odciętych na wysokości
1
T
przecina krzywe górną i dolną w punktach o współrzędnych
na osi odciętych
B
x
i
B
y
. Z tego diagramu odczytujemy, że
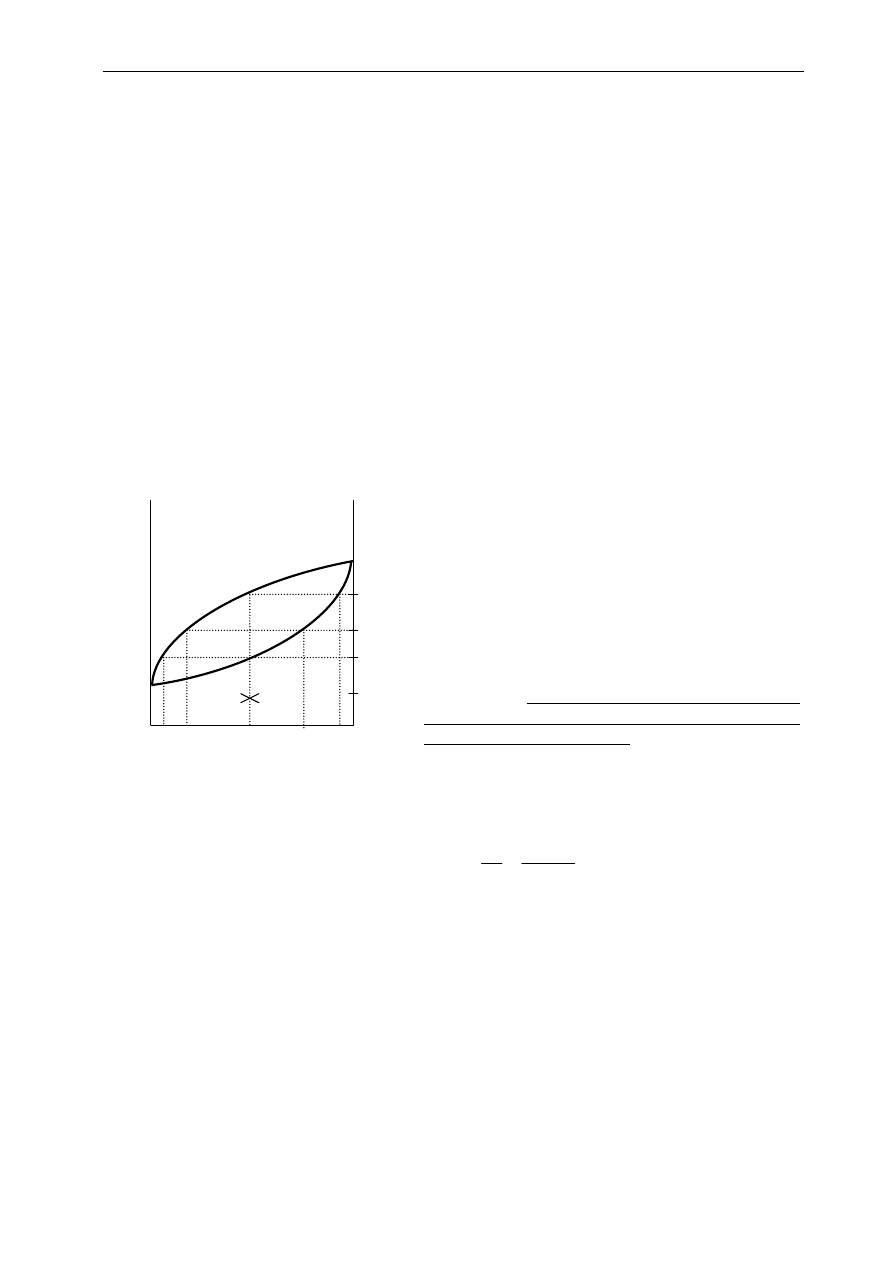
Ćwiczenie 1: Destylacja
14
w temperaturze
1
T
skład wrzącej cieczy wynosi
B
x
, a skład pary nad nią
B
y
.Powyżej górnej krzywej
układ występuje jedynie w postaci pary, a poniżej krzywej dolnej jedynie w postaci ciekłej. Punkty
pomiędzy tymi krzywymi odpowiadają obecności dwu faz: pary i wrzącego roztworu w równowadze.
3. ZACHOWANIE SIĘ ROZTWORÓW PODCZAS OGRZEWANIA.
Aby zrozumieć procesy rozdzielania składników roztworu drogą destylacji omówimy zachowanie
się układu podczas jego ogrzewania, posługując się diagramem fazowym temperatura – skład.
Rozpatrzymy trzy przypadki.
Przypadek 1
Zakładamy, że ciecz znajduje się w naczyniu zamkniętym ruchomym tłokiem (to pozwala
utrzymać stałe ciśnienie w naczyniu). Ciecz o temperaturze początkowej
0
T
i składzie
0
x
(stężenie
składnika B - ułamek molowy względnie wagowy) jest systematycznie ogrzewana. (Rys.3.1). W punkcie
p
T
ciecz zaczyna wrzeć i pojawia się pierwsza porcja pary o stężeniu składnika B równym
0
y
(jak to
wynika z diagramu).
w równowadze wyraża się następującym wzorem:
s
s
c
p
y
x
x
x
n
n
−
−
=
0
0
. Parę zebraną nad cieczą możemy
odprowadzić z naczynia i ewentualnie skroplić. W ten sposób można otrzymać dwie ciecze - jedną w
kolbie - bogatszą w składnik B, a drugą w destylacie - w niego uboższą, w porównaniu ze składem cieczy
wyjściowej. Ten proces jest podstawą destylacji równowagowej. W praktyce jest ona realizowana w
procesie ciągłym, polegającym na tym, że ciecz jest stale uzupełniana w miarę odbierania destylatu (Rys.
3.2).
Gdyby jednak nie odbierać pary znad cieczy, a cieczy nie uzupełniać i dalej ogrzewać, to dojdzie
się w końcu do momentu, w którym znika ostatnia porcja cieczy. Zajdzie to w temperaturze
k
T
,wyznaczonej przez rzędną przecięcia linii składu początkowego cieczy z krzywą pary (Rys. 3.1).
W układzie jest tylko sama para o składzie
0
x
takim, jaki miał nasz układ na początku. Dalsze
ogrzewanie będzie jedynie podnosić temperaturę pary. Wniosek z tej dyskusji jest oczywisty - całkowite
odparowanie cieczy nie zmieni jej składu w stosunku do składu początkowego cieczy.
Rys. 3.1. Odparowywanie roztworu
T
T
T
A
0
para
ciecz
p=const
T
B
0
x
0
y
0
y
S
x
S
y
K
T
0
T
P
T
S
T
K
Ułamek molowy składnika B
0
1
Ponieważ
0
y
<
0
x
to zawartość składnika B (trudniej
lotnego) w parze jest mniejsza niż w cieczy; ciecz
zubożała silniej w składnik bardziej lotny A – o niższej
temp wrzenia
0
A
T
, a przez to wzbogaciła się nieco w
składnik mniej lotny B. Aby podtrzymać wrzenie układ
należy stale ogrzewać z tym, że temperatura wrzenia
będzie się stopniowo podnosić. Z cieczy będzie
odparowywać mieszanina składników A i B z przewagą
lotniejszego składnika A. I tak, np. w temperaturze T
s
wrząca ciecz będzie zawierać składnik B o stężeniu x
s
,
a para nad nią składnik B o stężeniu y
s
Należy
podkreślić, że składy cieczy i pary nad nią zależą
jedynie od temperatury roztworu, a nie od składu
początkowego
mieszaniny
.
Skład
początkowy
mieszaniny ma wpływ jedynie na proporcje ilościowe
(molowe,
masowe)
cieczy
do
pary
w
danej
temperaturze. Zgodnie z regułą dźwigni (patrz ANEKS
II) liczba moli pary do liczby moli cieczy będącej z nią

Ćwiczenie 1: Destylacja
15
Rys. 3.2. Praktyczny sposób realizacji destylacji równowagowej w warunkach laboratoryjnych.
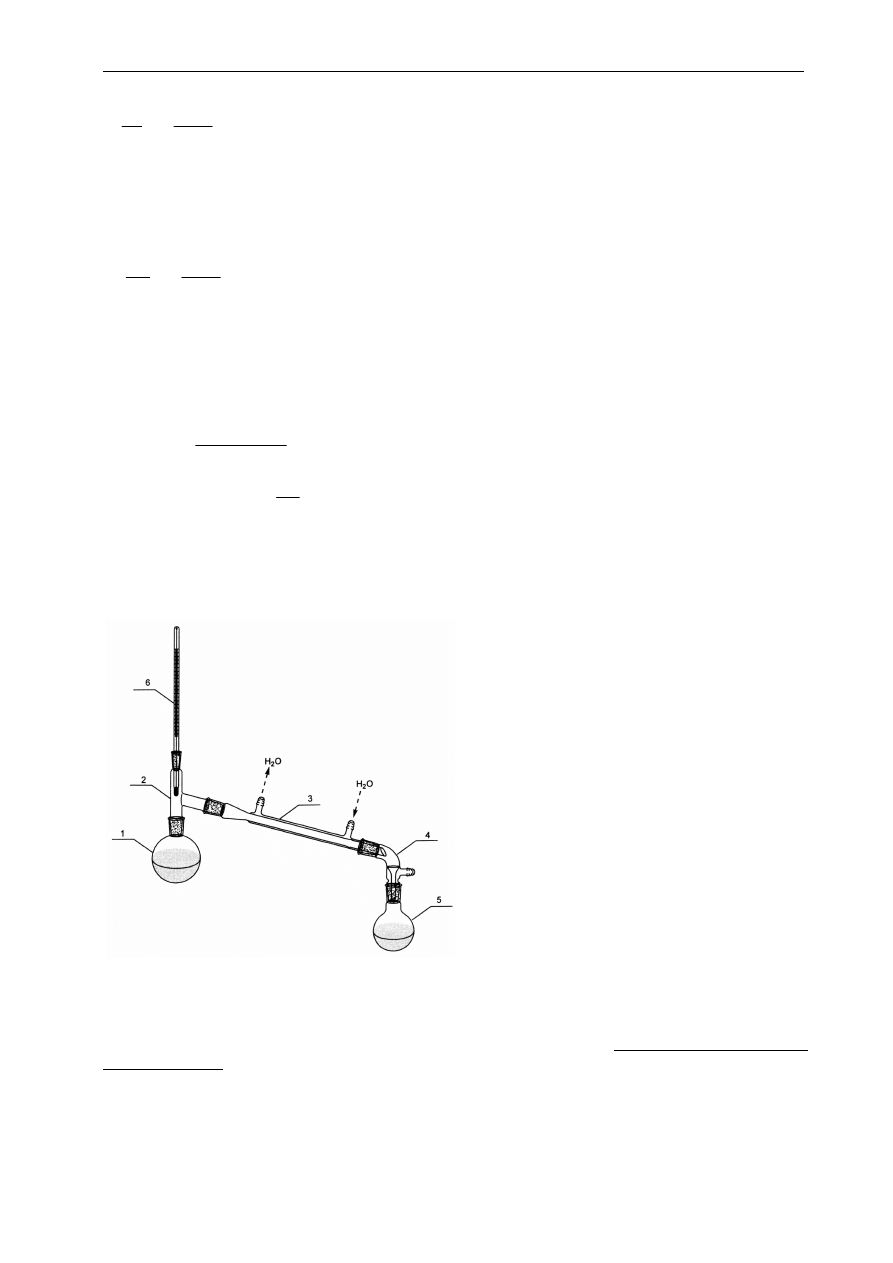
Przypadek 2
Zakładamy, że roztwór o początkowym stężeniu składnika mniej lotnego B
0
x
znajduje się w
naczyniu, z którego można momentalnie odprowadzać wytworzoną parę. Ciecz tę stopniowo ogrzewamy
od temperaturze początkowej
0
T
do końcowej
k
T
(patrz Rys.3.3 ).
Rys.3.3 Destylacja różniczkowa
wyprowadzić z bilansu molowego (masowego) jednego ze składników układu. Jeżeli oznaczymy przez:
n – liczbę moli substancji ciekłej
dn – liczbę moli cieczy która przeszła w stan pary
x – aktualny skład cieczy (ułamek molowy składnika wyżej wrzącego B)
y – aktualny stan powstającej pary (ułamek molowy składnika B)
dx - zmianę stężenia cieczy wskutek oddestylowania
to bilans moli liczony na składnik B dany jest równością:
0
)
)(
(
=
+
−
−
−
ydn
nx
dx
x
dn
n
(3.1)
Przyjmując, że iloczyn dwu nieskończenie małych wielkości
(dn)(dx) jest równy zeru otrzymamy po
ugrupowaniu wyrażeń równanie różniczkowe:
x
y
dx
n
dn
−
=
(3.2)
które po scałkowaniu przybierze postać:
1. kolba destylacyjna
2. nasadka destylacyjna
3. chłodnica Liebiega
4. przedłużacz
5. odbieralnik
6. rozdzielacz, z którego
dodawana jest ciecz tak, aby
w czasie procesu destylacji
zachować jej stałą objętość
w kolbie destylacyjnej
1
4
3
5
6
2
para
ciecz
p=const
T
A
0
T
B
0
x
0
y
0
T
0
T
P
y
K
x
K
T
K
T
T
Ułamek molowy składnika B
0
1
W chwili, gdy dojdziemy do temperatury
p
T
ciecz zaczyna
wrzeć, a powstałą parę o składzie
0
y
natychmiast
odprowadzamy w postaci skroplin.
Para jest wzbogacona w składnik lotniejszy A, ciecz
w składnik wyżej wrzący B. Aby podtrzymać wrzenie
temperatura cieczy musi się podnieść. Powstająca nowa
porcja pary o jeszcze większej zawartości lotniejszego
składnika A znowu jest usunięta z układu, a ciecz coraz
bardziej zatęża się w składnik B. Ponieważ ten proces
można traktować jako ciąg następujących po sobie
infinitezymalnych (nieskończenie małych) ubytków cieczy
na korzyść pary i infinitezymalnych zmiany stężeń
roztworu,
nazywamy
go
destylacją
różniczkową.
W temperaturze
k
T
skład roztworu osiąga wartość x
k
,
a będącej w równowadze z nim pary wartość y
k
.Formalnie
można go ująć równaniem różniczkowym, które łatwo
Ćwiczenie 1: Destylacja
16
∫
−
=
k
x
x
k
x
y
dx
n
n
0
0
ln
(3.3)
gdzie indeksy
0
i
k
opisują stan początkowy i końcowy procesu;
zatem:
k
n
n
i
0
to liczby moli cieczy na początku i na końcu destylacji;
k
x
x
i
0
- ułamki molowe składnika B w cieczy przed i po zakończeniu destylacji.
Jeżeli operować ułamkami wagowymi zamiast ułamkami molowymi to równanie (3.3) przyjmie postać :
∫
−
=
k
x
x
k
x
y
dx
m
m
0
0
ln
(3.4)
a m
k
i m
0
to masy cieczy w kolbie przed i po zakończeniu destylacji.
Znając zależność stężenia składnika w parze od jego stężenia w roztworze, tzn. funkcję y(x) można tę
całkę obliczyć i w ten sposób powiązać ilość odparowanej cieczy z jej końcowym składem, a stąd stężenie
składników w destylacie. Dla roztworów doskonałych zależność pomiędzy składem pary nasyconej y -
ułamkiem molowym składnika wyżej wrzącego B – a składem roztworu x – ułamkiem molowym
składnika wyżej wrzącego wyraża się wzorem:
1
)
1
(
+
⋅
−
⋅
=
x
x
y
α
α
(3.5)
gdzie współczynnik
0
0
A
B
P
P
=
α
(3.6)
Wzór (3.5), słuszny też dla ułamków wagowych, zostanie wyprowadzony w rozdziale 7 (ANEKS III,
patrz wzór (7.7). Ten wariant destylacji jest powszechnie stosowany w praktyce w sytuacji dużych
rozpiętości temperatur wrzenia czystych składników. Rysunek 3.4 przedstawia zestaw laboratoryjny do
prowadzenia takiej destylacji.
Rys.3.4. Destylacja pod ciśnieniem atmosferycznym
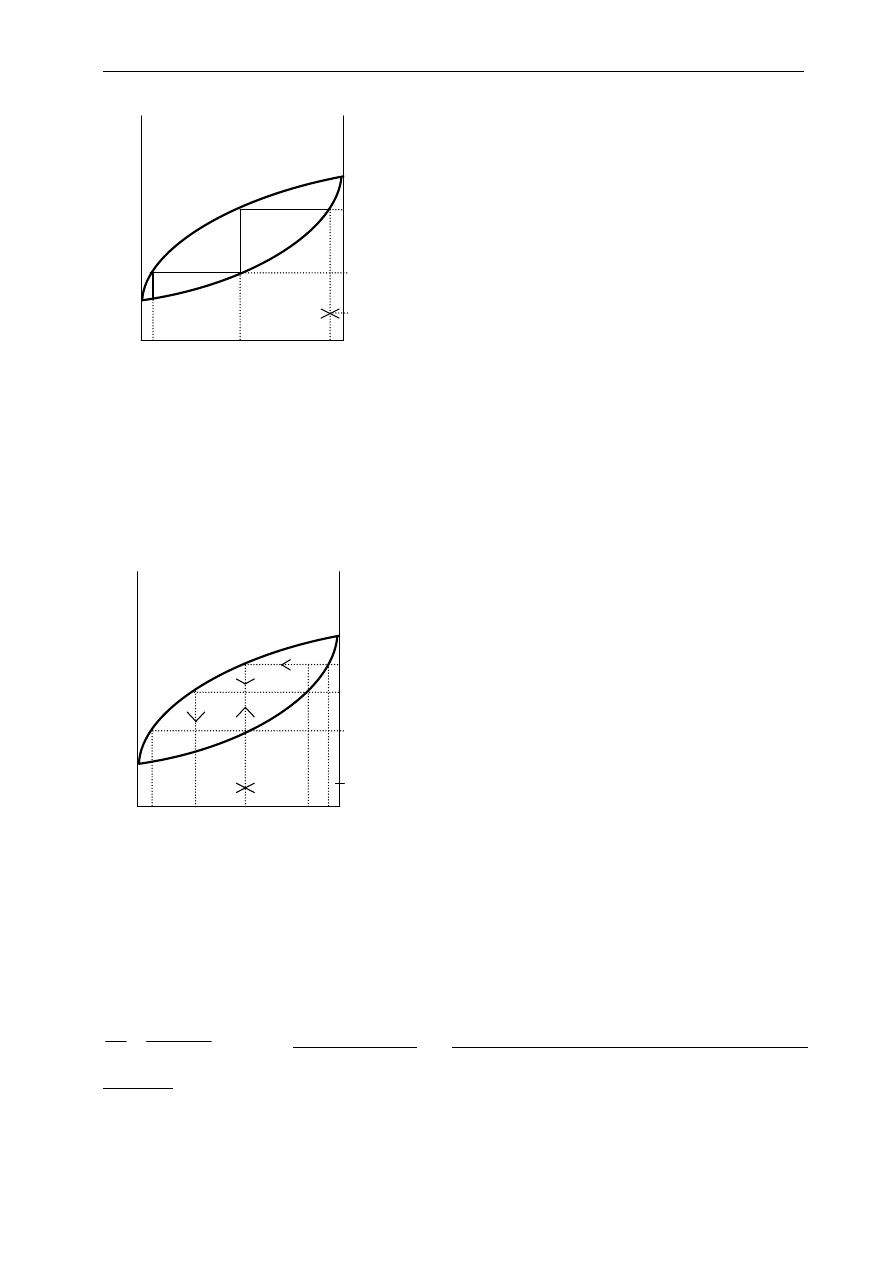
Przypadek 3
Rozpatrzymy wyidealizowany przypadek destylacji, podczas której skraplamy pierwszą porcję
odparowanej pary. Przyjmujemy, że badany roztwór jest złożony z cieczy A i B, których prężności par
nasyconych wynoszą
)
(
i
)
(
0
0
T
P
T
P
B
A
i ciecz A jest bardziej lotna od cieczy B, tzn.
)
(
)
(
0
0
T
P
T
P
B
A
>
.
Zawartość początkowa składnika mniej lotnego B jest równa
1
x
, a temperatura początkowa roztworu
wynosi
0
T
. Roztwór ogrzewamy do temperatury
1
T
, w której pojawia się pierwsza porcja pary o zawar-
1. kolba destylacyjna
2. nasadka destylacyjna
3. chłodnica Liebiega z wodnym
płaszczem chłodzącym
4. przedłużacz
5. odbieralnik
6. termometr
Ćwiczenie 1: Destylacja
17
4. REKTYFIKACJA (DESTYLACJA FRAKCJONOWANA)
Rektyfikacją nazywamy proces rozdzielania cieczy w czasie którego przeciwprądowo (płynąc
w przeciwnych kierunkach) stykają się strumienie cieczy i pary i równocześnie zachodzi pomiędzy nimi
.
ss
x
(znowu bogatszy w składnik B) i parę o składzie
ss
y
zbliżonym do składu początkowego cieczy
0
x
, w
proporcjach moli pary do moli cieczy wynikających z reguły dźwigni. Natomiast parę otrzymaną w
pierwszym etapie o składzie
s
y
skraplamy i oddestylowujemy w temperaturze
ss
T
−
uzyskując parę o
składzie
ss
y
−
i roztwór o składzie
ss
x
−
, zbliżonym do składu początkowego roztworu
0
x
w proporcjach
ss
s
s
ss
c
p
y
y
y
x
n
n
−
−
−
−
=
. Teraz ten roztwór o składzie
ss
x
−
łączymy z destylatem, otrzymanym wcześniej z pary
o składzie
ss
y
i ponownie ogrzewamy do temperatury
s
T
. Powtarzając tę procedurę na coraz to nowszych
frakcjach cieczy
,...,
ss
x
i pary
,...,
ss
y
−
uzyskanych w kolejnych etapach tego procesu zwielokrotniamy
wydajność destylacji. Ten proces nazwiemy pseudorektyfikacją.
para
ciecz
p=const
T
A
0
T
B
0
T
T
Rys. 4.1. Pseudorektyfikacja
0
1
x
0
x
s
x
ss
y
s
y
-ss
T
0
T
-ss
T
ss
T
s
y
ss
x
-ss
wymiana masy i ciepła. Końcowy produkt rektyfikacji,
otrzymany
przez
skroplenie
pary
jest
nazywany
rektyfikatem
lub
destylatem,
a
pozostałość
ciekłą
nazywamy cieczą wyczerpaną. Aby w prosty sposób
przybliżyć opis tego procesu przeanalizujemy następujący
przykład. Początkowo prowadzimy proces w sposób
opisany w punkcie pt. Przypadek 1. Po ogrzaniu roztworu o
temperaturze
0
T
i składzie
0
x
(patrz Rys. 4.1) do
temperatury
s
T
otrzymujemy parę o składzie
s
y
i nowy
roztwór
o
składzie
s
x
(bogatszy
w
składnik
B
w porównaniu ze składem roztworu przed destylacją)
w proporcjach moli pary do moli cieczy wynikających
z reguły dźwigni.
Frakcję gazową (parę) oddzielamy od roztworu.
Nieprzedestylowaną ciecz o składzie
s
x
ogrzewamy do
temperatury
ss
T
otrzymując z niej roztwór o składzie
Rys.3.5. Pierwowzór destylacji
frakcjonowanej (rektyfikacji)
para
ciecz
p=const
T
A
0
T
B
0
T
T
Ułamek molowy składnika B
0
1
x
1
y
1
y
2
T
2
T
1
T
0
tości składnika B równej
1
y
. Parę tę skraplamy w
temperaturze
2
T
, otrzymując ciecz o takim samym
składzie, co para. Ta ciecz wrząc w
2
T
daje parę o
zawartości y
2
składnika mniej lotnego B. Zabieg
ten powtarzamy
wielokrotnie, dochodząc w
granicy do czystego składnika A. Mankamentem
tego procesu są znikome ilości odparowywanych
porcji fazy lotnej. Obrazuje to Rys. 3.5. Ten sposób
destylacji jest pierwowzorem rektyfikacji, ale z
uwagi na wspomnianą wadę, nie ma praktycznego
zastosowania. Praktyczny wariant rektyfikacji
przedstawiono w następnym rozdziale.
Ilościową zależność pomiędzy składem
destylatu,
składem
roztworu,
a
krotnością
skraplania można łatwo otrzymać dla układu
będącego roztworem doskonałym. Wyprowadzimy
ją później w ANEKSIE III.
Ćwiczenie 1: Destylacja
18
Najbardziej efektywnym wariantem tej skomplikowanej czynnościowo procedury jest ustawienie
pionowo nad kolbą z roztworem kolumny, np. wypełnionej pierścieniami szklanymi, w której procesy
destylacji, skraplania, zlewania cieczy o podobnych składach zachodzą automatycznie. Najwyższa
temperatura panuje w kolbie destylacyjnej, a w miarę oddalania się, wzdłuż kolumny, od kolby
temperatura stopniowo spada i najniższa temperatura jest u szczytu kolumny. Typowy zestaw aparatury
stosowanej w warunkach laboratoryjnych do prowadzenia procesu rektyfikacji przedstawiono Rys. 4.2.
Pary ogrzewanego do wrzenia w kolbie roztworu, wędrując w górę kolumny oziębiają się, ulegają
skraplaniu i zawracają do kolby. Pary roztworu o najniższej temperaturze wrzenia (najbardziej
wzbogacone w lotniejszy składnik) wędrują najwyżej, pary mniej lotne (o mniejszej zawartości składnika
lotniejszego) wykraplają się po drodze w kolumnie. Przy odpowiednio dobranym tempie grzania
dochodzimy do sytuacji, w której u wierzchołka kolumny pary składu o najniższej temperaturze wrzenia
wnikają do chłodnicy i w niej dopiero ulegają skropleniu, natomiast pary o innym składzie, o wyższej
temperaturze wrzenia, wskutek chłodzenia kolumny przez otoczenie, ulegają skropleniu wcześniej, zanim
dojdą do szczytu kolumny. Wędrówka par w górę kolumny odpowiada, na rysunku 3.5 „schodzeniu”
układu po schodkach od temperatury odpowiadającej wrzeniu roztworu w kolbie do temperatury wrzenia
czystego składnika (tego o niższej temperaturze wrzenia).
Rys. 4.2. Zestaw aparatury laboratoryjnej do rektyfikacji
Odparowana frakcja, po pewnym czasie wyczerpie się i jeżeli nie podniesiemy tempa grzania, to
destylacja ustanie (Wówczas pary wykonują cyrkulację - wznoszą się wzdłuż kolumny, ale zanim dojdą
do chłodnicy skroplą się i zawrócą do kolby). Dopiero zwiększenie tempa grzania spowoduje skraplanie w
chłodnicy, po opuszczeniu kolumny, frakcji o wyższej temperaturze wrzenia. W taki sposób regulując
tempem grzania roztworu możemy odbierać frakcje o różnych, coraz to wyższych temperaturach wrzenia.
Podniesienie temperatury kolby wymagane jest z tego powodu, że roztwór w kolbie zatężany w składnik
mniej lotny (na diagramie fazowym odpowiada to przesuwaniu się składu roztworu w prawo) wrze w
coraz to wyższej temperaturze. Jest to destylacja frakcjonowana czyli rektyfikacja.
Trzeba jednak pamiętać, że zbyt gwałtowne grzanie może sprowadzić rektyfikację do zwykłej
destylacji, której zdolność rozdzielania składników jest znacznie gorsza. Jest to związane z tym, że w
kolumnie nie zdąży ustalić się odpowiedni gradient temperatury dla wykraplania poszczególnych
składników, a skraplać się będą frakcje o mieszanych składach.
Jednak nie zawsze celem rektyfikacji jest rozdział roztworu na czyste składniki, ale wykraplanie
frakcji będących mieszaninami składników o różnych zakresach temperatur wrzenia, jak to ma miejsce w
rafineriach. W ten sposób z ropy naftowej otrzymujemy eter naftowy (t. wrz.40-90
0
C), benzynę lekką
t.wrz.90- 120
0
C) , ligroinę czyli benzynę ciężką t. wrz. 120 – 200
0
C), naftę (t.wrz.170-270
0
C) itd.
Ważną charakterystyką kolumn rektyfikacyjnych jest liczba półek teoretycznych jaka mieści się
w kolumnie. Jest to pojęcie uniwersalne i charakteryzuje każdy rodzaj kolumny. Półką teoretyczną
1. kolba destylacyjna
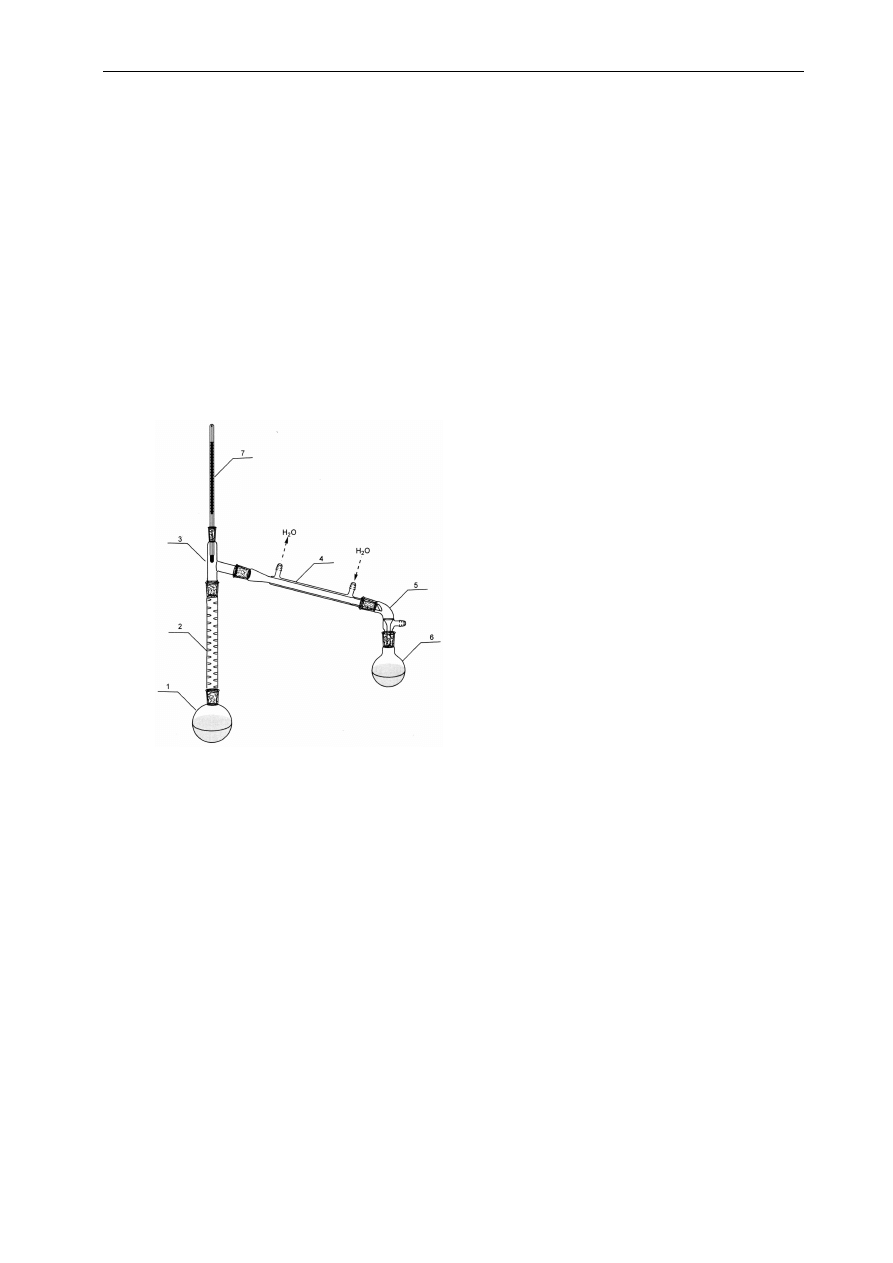
2. kolumna destylacyjna
3. nasadka destylacyjna
4. chłodnica Liebiega z wodnym
płaszczem chłodzącym
5. przedłużacz
6. odbieralnik
7. termometr

Ćwiczenie 1: Destylacja
19
nazywamy wyidealizowane miejsce w kolumnie rektyfikacyjnej, z którego odparowująca para, jest w
równowadze termodynamicznej ze spływająca z niego cieczą. Liczba półek teoretycznych zależy od
rodzaju destylowanej cieczy, a także od sposobu prowadzenia rektyfikacji, tzn. czy destylat w całości
odprowadzamy z układu (zerowy powrót), czy po części czy też w całości zawracamy górą (powrót
pełny). Liczbę półek teoretycznych kolumny rektyfikacyjnej można oszacować w oparciu o wzór (7.10).
Często zbiornik jest traktowany jako pierwsza półka kolumny i wówczas obliczoną wartość n należy
powiększyć o 1. Inne aspekty rektyfikacji oraz destylację z parą wodną i destylację próżniową omówiono
w ANEKSIE IV, V i VI
5. WYKONANIE ĆWICZENIA
Zestaw laboratoryjny
1. Kolba okrągło-denna ze szlifem na 50 cm
3
2. Chłodnica Liebiega
3. Nasadka destylacyjna (do umieszczenia termometru)
4. Przedłużacz (łączy chłodnicę z odbieralnikiem)
5. Kolumna rektyfikacyjna z pierścieniami Raschiga
6. Lejek, odbieralniki (erlenmayerki) (3)
7. Płaszcz grzejny z regulatorem prądu,
8.Termometr, kamyki wrzenne (6-9), lub sita molekularne (3)
5.1. Destylacja czystego związku chemicznego; wyznaczanie temperatury wrzenia
Do kolby odmierzyć 10-15 cm
3
czystego związku chemicznego wziętego od prowadzącego
ć
wiczenia, a następnie wsypać do niej 2-3 sztuki kamyków wrzennych. Podłączyć chłodnicę przez
nasadkę, a do wolnego wylotu nasadki wstawić termometr. Przy wylocie chłodnicy ustawić erlenmayerkę
odbierającą destylat. Pod kolbą ustawić płaszcz grzejny podłączony do sieci. Puścić niewielkim
strumieniem z kranu zimną wodę do chłodnicy i nie zamykać kranu aż do zakończenia destylacji.
Zapisywać temperaturę co minutę. Zestaw aparatury pokazano
na rys. 3.4 i stronicy 9 – rysunek górny.
5.2. Destylacja roztworu złożonego z dwu cieczy
Do kolby na 50 cm
3
odmierzyć 25 cm
3
mieszaniny (1:1 objętościowo) dwóch ciekłych składników
wziętej od prowadzącego ćwiczenia. Powtórzyć czynności opisane w poprzednim ćwiczeniu.
5.3. Rektyfikacja
Do kolby na 50 cm
3
odmierzyć 25 cm
3
mieszaniny z p.5.2, wrzucić 2-3 sztuki kamyków
wrzennych. Na kolbę nałożyć kolumnę rektyfikacyjną i przyłączyć do niej chłodnicę. U góry kolumny
wstawić termometr, a na wylocie chłodnicy erlenmayerkę do odbioru destylatu. Przepuszczać przez
chłodnicę zimną wodę i włączyć grzanie. Regulator mocy na początku ustawić na wysoki poziom, a gdy
temperatura będzie bliska temperatury wrzenia składnika niżej wrzącego, skręcić na niższy poziom.
Odpowiednio dobrane tempo grzania pozwoli rozdzielić badany układ na frakcje różniące się między sobą
temperaturą wrzenia. Po oddestylowaniu pierwszej frakcji temperatura układu „stoi w miejscu” i nie
obserwuje się dalszego odparowywania cieczy. Należy nieco podnieść poziom grzania pokrętłem
regulatora. Po chwilowym oddestylowaniu frakcji pośredniej – temperatura wrzenia wzrasta - następuje
stabilizacja temperatury i oddestylowuje frakcja druga do drugiego odbieralnika. (W zależności od stopnia
złożoności układu destylowanego prowadzi się proces w podobny sposób aż do wyczerpania cieczy w
kolbie – uzyskania czystego składnika wysoko wrzącego.) Gdy rozpocznie się skraplanie pary należy
notować temperaturę co minutę. Zestaw aparatury pokazano na rys. 4.2
i stronicy 9 – rysunek prawy.
6. OPRACOWANIE WYNIKÓW
1. Sporządzić na jednym rysunku wykresy zależności temperatury wrzenia od czasu dla trzech
wykonanych sposobów destylacji. Na podstawie wyznaczonych temperatur wrzenia składników
Ćwiczenie 1: Destylacja
20
mieszaniny rozdzielonych metodą rektyfikacji (p.5.3) określić, jakie związki chemiczne tworzą
badaną mieszaninę.
2. W oparciu o dane wyjściowe dla przeprowadzonej destylacji różniczkowej mieszaniny cieczy i
informację , że destylację zakończono w momencie gdy stężenie składnika trudniej lotnego w kolbie
wyniosło x
k
= 0.8 [ułamek wagowy] przeprowadzić odpowiednie obliczenia do wypełnienia jednego z
zestawów (A lub B) tabel. Potrzebne do obliczeń dane są dostępne w tablicach u prowadzącego
ć
wiczenie. Wyniki przedstawić wg. jednego z zamieszczonych schematów zestawu:
Zestaw A Jeżeli liczono całkę, w której zmiennymi były ułamki molowe [wzory (3.3) i (3.5)]
)
ln(
0
n
n
k
liczba moli cieczy w kolbie
przed destylacją n
0
liczba moli cieczy w kolbie po
destylacji n
k
W kolbie przed destyl
W kolbie po destyl.
W destylacie
uł. molowy składnika
wyżej wrzącego (B)
uł. wagowy składnika
wyżej wrzącego (B)
Zestaw B Jeżeli liczono całkę, w której zmiennymi były ułamki wagowe [wzory (3.4) i (3.5)]
)
ln(
0
m
m
k
masa cieczy w kolbie przed
destylacją m
0
masa cieczy w kolbie po
destylacji m
k
W kolbie przed destyl
W kolbie po destyl.
W destylacie
uł. molowy składnika
wyżej wrzącego (B)
uł. wagowy składnika
wyżej wrzącego (B)
7. ANEKS
I. Diagram fazowy: prężność pary – skład (temperatura stała)
Diagram taki łatwo skonstruować znając zależność prężności par nasyconych poszczególnych
składników (ciśnień cząstkowych) od stężenia tych składników w roztworze. Najczęściej stężenia podaje
się w postaci ułamka molowego (lub ułamka wagowego) jednego ze składników układu. Np. dla
roztworów doskonałych – zachowujących się zgodnie z prawem Raoulta - ciśnienia cząstkowe
składników A i B spełniają zależności:
A
A
A
x
P
p
0
=
oraz
B
B
B
x
P
p
0
=
gdzie:
B
A
x
x
i
to odpowiednio ułamki molowe składników A i B w roztworze, a temperatura układu jest
ustalona. Ułamki molowe zdefiniowane są w sposób następujący:
B
A
A
A
n
n
n
x
+
=
;
B
A
B
B
n
n
n
x
+
=
, gdzie: n
A
, n
B
to odpowiednio liczby moli składników A i B
w roztworze. Na Rys.7.1 ciśnienia cząstkowe p
A
i p
B
w funkcji składu roztworu przedstawione są liniami
prostymi wychodzącymi z naroży rysunku (punkty 0 i 1). Całkowitą prężność pary nasyconej nad
roztworem w funkcji jego składu otrzymuje się sumując przyczynki od jej składników:
B
A
p
p
P
+
=
i
na Rys.7.1 jest to linia prosta łącząca punkty
0
0
i
B
A
P
P
. Linia ta stanowi też jedną z krzywych - krzywą
cieczy (górną ) diagramu fazowego z Rys.7.2 Natomiast krzywą przedstawiającą prężność pary nasyconej
w funkcji składu pary - krzywą pary (dolną na tym diagramie) wyznacza się z Rys.7.1 korzystając z tego,
Ćwiczenie 1: Destylacja
21
ż
e ułamek molowy składnika w parze równy jest stosunkowi jego ciśnienia cząstkowego do prężności
całkowitej pary, tzn.:
P
p
y
B
B
/
=
oraz
B
A
A
y
p
y
−
=
=
1
P
/
. Na przykład, na Rys. 7.1 widać, że nad
roztworem o składzie
2
/
1
=
B
x
istnieje para nasycona, której prężność wynosi P
1
= 4 (w jednostkach
umownych), podczas gdy ciśnienie cząstkowe składnika B, p
B
= 1 (w tych samych jednostkach). To
oznacza, że w tej parze ułamek molowy składnika B wynosi
4
/
1
/
1
=
=
P
p
y
B
B
. Zatem punktowi o
rzędnej odpowiadającej wartości P
1
należy na osi odciętych rysunku 7.2 nadać wartość
1
/ P
p
B
= 1/4.
Rys. 7.1. Zależność prężności pary
od składu nad roztworem doskonałym
II. Reguła dźwigni.
Reguła ta pozwala w oparciu o diagramy fazowe typu temperatura - skład lub ciśnienie – skład
określić proporcje (stosunki liczby moli lub stosunki mas) pary i cieczy na podstawie początkowego
składu cieczy lub pary w wybranej temperaturze w pierwszym przypadku lub ciśnieniu w drugim.
Wyprowadzenie wzoru opiera się o bilans molowy (lub masowy) całości oraz jednego ze składników.
Niech skład początkowy cieczy wynosi np. w ułamkach molowych
0
x
(patrz Rys.3.1)
.
W temperaturze
s
T
ustala się równowaga pomiędzy wrzącym roztworem którego skład wynosi
s
x
, a parą o składzie
s
y
.
Dla całości mamy następujący bilans moli:
c
p
n
n
n
+
=
(7.1)
Dla jednego ze składników , np. składnika B mamy bilans:
s
p
s
c
y
n
x
n
x
n
⋅
+
⋅
=
⋅
0
(7.2)
gdzie:
n
= liczba całkowita moli w układzie
c
n
- liczba moli składników A i B w cieczy, a
p
n
- liczba moli składników A i B w parze
Po odpowiednim ugrupowaniu wyrażeń otrzymujemy równanie:
)
(
)
(
0
0
s
p
s
c
y
x
n
x
x
n
−
=
−
(7.3)
stąd ostatecznie:
0
0
x
x
y
x
n
n
s
s
p
c
−
−
=
(7.4)
III. Wielokrotne skraplanie pary roztworów doskonałych
Prężności par składników B i A nad wrzącym roztworem w temperaturze
1
T
wynoszą:
1
1
0
)
(
x
T
P
P
B
B
⋅
=
(7.5)
)
1
(
)
(
1
1
0
x
T
P
P
A
A
−
⋅
=
(7.6)
Rys. 7.2. Diagram fazowy prężność pary-skład
P
A
0
p
B
P=p
A
+p
B
p
A
P
B
0
T=const
P
1
x
B
=1/2
p
B
=1
P
1
=4
p
B
P
1
=
1
4
p
p
0
1
x
B
=1/2
y
B
=1/4
P
B
0
T=const
P
A
0
P
1
para
ciecz
p
p
0
1
Ćwiczenie 1: Destylacja
22
stąd
1
]
1
)
/
[(
)
/
(
)
1
(
1
0
0
1
0
0
1
0
1
0
1
0
1
+
−
=
−
⋅
+
⋅
⋅
=
+
=
x
P
P
x
P
P
x
P
x
P
x
P
P
P
P
y
A
B
A
B
A
B
B
B
A
B
(7.7)
oraz:
)
1
(
)
(
)
(
1
1
1
1
0
1
0
1
1
x
x
T
P
T
P
P
P
y
y
A
B
A
B
−
⋅
=
=
−
(7.8)
Jeżeli destylat o składzie
1
y
przeprowadzimy w parę o składzie
2
y
, a z jej skroplin z kolei parę o
składzie
3
y
etc. (jak na Rys. 3.5), a stosunki prężności par nasyconych czystych składników nie zmieniają
się wyraźnie z temperaturą, co w zdarza się dość często, to wzór wiążący skład pary po n - krotnej
operacji
n
y
, z początkowym składem cieczy
1
x
ma postać:
)
1
(
,...,
)
1
(
1
1
1
0
0
1
1
0
0
x
x
P
P
y
y
P
P
y
y
n
A
B
n
n
A
B
n
n
−
⋅
=
=
−
⋅
=
−
−
−
. (7.9)
Należy pamiętać, że wzór (7.8) obowiązuje jedynie w przypadku procesów izotermicznych. Jednakże w
przypadku słabej zależności wyrażenia
0
0
/
A
B
P
P
od temperatury stosuje się go również do opisu procesów
izobarycznych (bo takie zachodzą podczas destylacji czy rektyfikacji). Wówczas korzystamy wprost z
wartości
0
0
/
A
B
P
P
dla konkretnej temperatury lub ze średniej geometrycznej tych wartości z kilku
temperatur, gdy zachodzi zauważalna zmienność ilorazu prężności par. Na podstawie równania (7.9)
można określić wymaganą krotność skraplania, jeżeli znamy skład początkowy roztworu i wiemy jaką
zawartość składników ma mieć końcowy destylat:
1
0
0
1
1
ln
)
1
(
1
ln
−
⋅
−
⋅
−
=
A
B
n
n
P
P
x
x
y
y
n
(7.10)
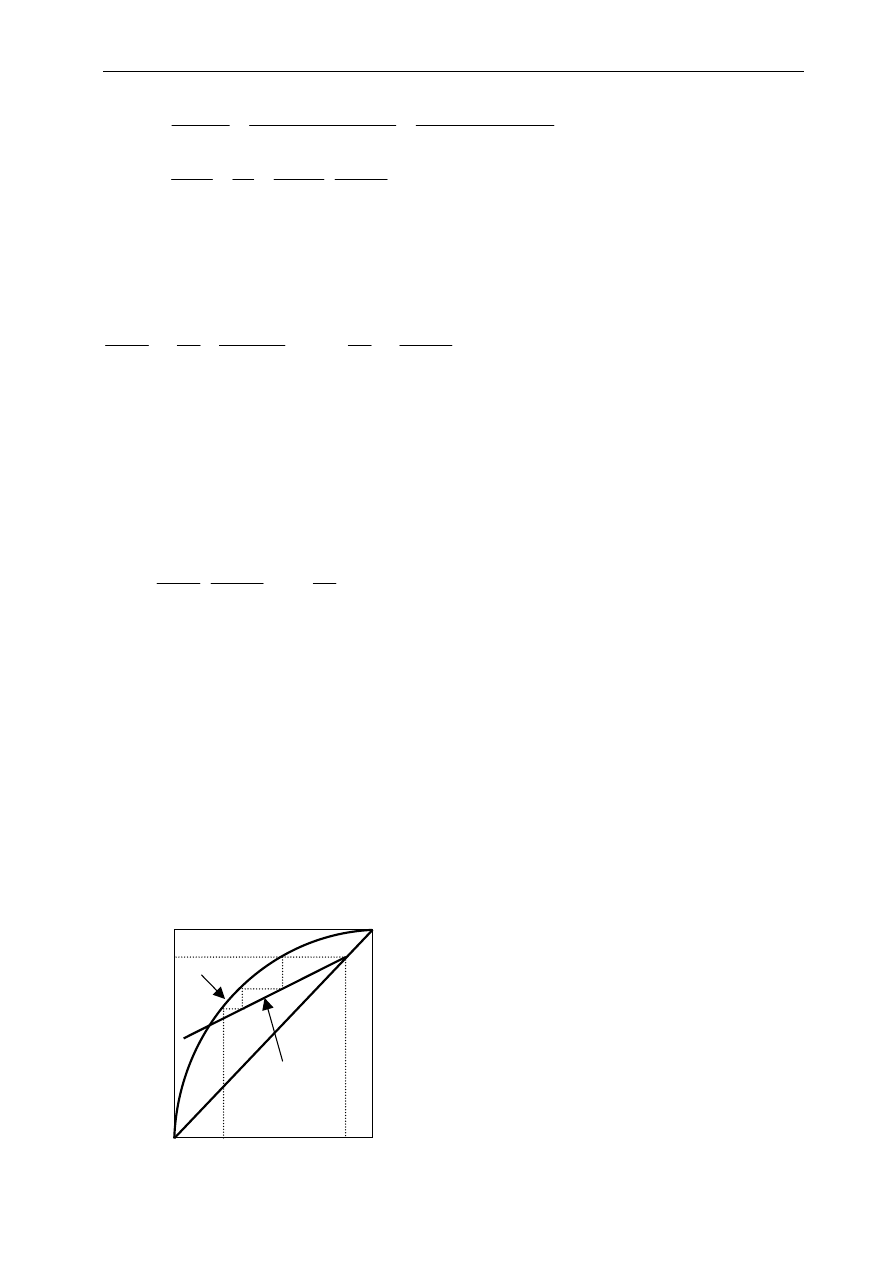
IV. Rektyfikacja
Istnieje szereg praktycznych rozwiązań rektyfikacji. Zasadnicze różnice dotyczą rodzaju kolumny
rektyfikacyjnej, sposobu podawania orosienia czyli części destylatu, która jest zawracana od góry do
kolumny na kolumnę oraz dostarczania surowca. Orosienie może być całkowite, gdy nie odbieramy
produktu destylacji, a w całości zawracamy go na kolumnę, częściowe, lub zerowe, gdy cały destylat
odprowadzamy poza kolumnę. Może być ono stałe w czasie rektyfikacji lub zmieniające się w taki
sposób, aby odbierany destylat miał stały skład. Kolumny stosowane na skalę przemysłową różnią się od
tych, na których pracuje się w laboratorium. Różnice występują w gabarytach, w materiale wypełniającym
kolumnę, w sposobie doprowadzenia roztworu do destylacji.
Kolumny rektyfikacyjne mogą być z wypełnieniem bądź półkowe. Wypełnieniem jest materiał o
rozwiniętej powierzchni, co zapewnia wzmożony kontakt pary wznoszącej się z dolnych partii kolumny z
cieczą spływającą z jej górnych partii. Materiałem wypełniającym kolumnę są najczęściej pierścienie
Raschiga (w kształcie walców), pierścienie Lessinga z przegrodą, wirująca wstęga etc. Pierścienie mogą
być wykonane ze szkła, ceramiki, metalu lub tworzywa sztucznego, a wstęga z metalu.
Rys.7.3. Graficzny obraz rektyfikacji
x
0
1
y
0
1
x
D
y
D
=x
D
Linia
operacyjna
Linia
równowagi
x
W
W
Przebieg rektyfikacji wygodnie jest zaprezentować
na wykresach podających równowagowe składy pary
i cieczy - krzywa równowagowa - oraz składy pary i cieczy
w warunkach panujących w kolumnie rektyfikacyjnej -
linia operacyjna (patrz Rys. 7.3). Krzywa równowagi to
inny obraz diagramu fazowego. Na osi rzędnych
odkładamy stężenia (ułamki molowe) pary, a na osi
odciętych stężenia cieczy będącej z nią w równowadze.
Taką krzywą równowagi można wykreślić dla procesu
izotermicznego (z diagramu fazowego ciśnienie- skład),
bądź dla procesu izobarycznego (z diagramu fazowego
temperatura- skład). Na rys.7.3 jest to krzywa w kształcie
łuku. Linię operacyjną kreślimy w oparciu o proste bilanse
„materiałowe” obowiązujące w każdym miejscu kolumny.

Ćwiczenie 1: Destylacja
23
Jeżeli G oznacza całkowitą ilość odparowanej cieczy zebranej u szczytu kolumny o składzie y, D
część odprowadzoną na zewnątrz jako destylat o składzie x
D
, a R resztę o składzie x jako zawróconą od
góry do kolumny, to mamy:
D
R
G
+
=
(7.11)
D
x
D
x
R
y
G
⋅
+
⋅
=
⋅
(7.12)
Po podzieleniu stronami otrzymujemy:
D
D
x
x
x
D
R
D
x
D
R
R
y
1
1
1
+
+
+
=
+
+
+
=
ψ
ψ
ψ
(7.13)
D
R
=
ψ
określany jest mianem stopnia deflegmacji
Jest to równanie prostej - linii operacyjnej - wiążącej skład cieczy i pary w dowolnym miejscu kolumny
rektyfikacyjnej. Linia operacyjna nachylona jest do osi odciętych pod kątem
α,
spełniającym
zależność:
1
tg
+
=
ψ
ψ
α
i bierze początek w punkcie o współrzędnych (x
D
, y
D
= x
D
). Im więcej destylatu
zawracamy do kolumny, tzn. im większa jest wartość stopnia deflegmacji
ψ
, tym linia operacyjna staje
się bardziej stroma. W granicznym przypadku pełnego powrotu
∞
=
ψ
linia operacyjna pokrywa się z
przekątną. Zwiększanie stopnia deflegmacji poprawia zdolność rozdzielczą kolumny. Jest to istotne w
przypadku rozdzielania cieczy o zbliżonych temperaturach wrzenia.
Ponieważ skład cieczy w zbiorniku ulega w czasie rektyfikacji ciągłej zmianie z uwagi na
zmniejszanie się stężenia składnika bardziej lotnego od początkowej wartości x
F
do założonej końcowej
x
W
. to rektyfikację prowadzi się też na inne sposoby. Mianowicie, albo utrzymywać skład destylatu na
stałym poziomie (x
D
= const) regulując ciągle ilością zawracanej części R, bądź utrzymywać stałą wartość
stopnia deflegmacji, za to ze zmieniającym się składem odbieranego destylatu. Z rysunku 4.1 możemy
łatwo odczytać liczbę półek teoretycznych potrzebnych do uzyskania rektyfikatu o składzie x
D
i cieczy
wyczerpanej o składzie x
W
. Liczba ta równa jest liczbie schodków przeprowadzonych pomiędzy krzywą
równowagi , a linią operacyjną (biegnącą poniżej punktu W), rozpoczynając od położenia x
D
, a kończąc
na x
W
. Zbiornik z roztworem jest tu potraktowany jako pierwsza pólka teoretyczna. W przypadku pełnego
powrotu linia operacyjna pokrywa się z przekątną diagramu przedstawionego na rys. 7.3 i liczę półek
teoretycznych można obliczyć kierując się procedurą opisaną wyżej (patrz wzór (7.10).
V. Destylacja z parą wodną
Bardzo ważną w chemii organicznej odmianą destylacji jest destylacja z parą wodną. Stosuje się
ją do oczyszczania związków organicznych nierozpuszczalnych w wodzie, w szczególności gdy te
związki są nietrwałe w wyższych temperaturach. Układ złożony z wody i nie mieszającej się z nią cieczy
(organicznej) będzie wrzał pod ciśnieniem atmosferycznym zawsze poniżej 100
0
C, co zazwyczaj
zapobiega rozkładowi związku. Co więcej, temperatura wrzenia będzie utrzymywała się na stałym
poziomie, do wyczerpania jednego ze składników (wody lub oczyszczanego związku). O zachowaniu się
mieszaniny cieczy podobnym do zachowania się czystych składników (stałość temperatury wrzenia)
wspomniano już we wcześniejszych partiach materiału. Prężność pary nasyconej nad układem nie
mieszających się cieczy: woda-związek organiczny, (w skrócie A i B) jest równa sumie prężności par
nasyconych czystych składników w danej temperaturze
0
0
B
A
P
P
P
+
=
i nie zależy od składu mieszaniny.
Obrazuje to Rys.7.4 Ogrzewana ciecz o składzie
0
x
(patrz rys.7.5) wrze pod ciśnieniem, np.
atmosferycznym w temperaturze
E
T
, podczas gdy jej czyste składniki wrą odpowiednio w temperaturach
0
B
0
T
i
A
T
.W temperaturze
E
T
obok dwu wcześniej powstałych faz ciekłych pojawia się faza gazowa, czyli
mieszanina par A i B o składzie wyznaczonym z diagramu w punkcie
P
/
P
0
B
=
=
E
E
x
y
. Pomimo
ogrzewania temperatura wrzenia utrzymuje się na jednej wartości (jak to ma miejsce dla czystych
substancji), a z cieczy odparowują obydwa składniki w proporcjach spełniających zależność:
Ćwiczenie 1: Destylacja
24
0
0
A
B
A
B
P
P
n
n
=
(7.14)
gdzie:
B
A
n
n
i
są liczbami moli składników A i B odparowywanymi jednocześnie z mieszaniny.
Rys. 7.6 przedstawia zestaw aparatury laboratoryjnej stosowanej do destylacji z parą wodną.
VI. Destylacja próżniowa
Destylację roztworu w niższej temperaturze, niż by to wynikało z jego zachowania się w
warunkach normalnych, można przeprowadzić też w inny sposób niż opisany wyżej. Temperatura wrzenia
to temperatura w której prężność pary nasyconej nad cieczą osiąga wartość równą ciśnieniu
zewnętrznemu. Przez zmianę ciśnienia zewnętrznego możemy wpływać na temperaturę wrzenia cieczy.
Wychodząc z jednego z podstawowych równań chemii fizycznej – równania Clausiusa – Clapeyrona
wiążącego prężność pary nasyconej dowolnego czystego składnika z temperaturą:
2
0
)
(
))
(ln(
RT
H
T
d
p
d
∆
=
(7.15)
gdzie:
0
p
prężność pary nasyconej nad cieczą
Rys. 7.4. Zależność prężności pary od
składu dla cieczy nie mieszających się
Rys. 7.5. Zależność temperatury wrzenia od
składu dla cieczy nie mieszających się
Rys. 7.6. Zestaw aparatury laboratoryjnej stosowanej do destylacji z parą wodną.
1. kolba destylacyjna
2. nasadka do destylacji z parą
wodną
3. chłodnica Liebiega z wodnym
płaszczem chłodzącym
4. przedłużacz
5. odbieralnik
6. kociołek
7. wskaźnik poziomu wody
8. rurka bezpieczeństwa
P
B
0
T=const
P
A
0
P= P
A
0
+ P
B
0
p
p
Ułamek molowy składnika B
0
1
T
B
0
p=const
T
A
0
T
E
T
T
E
X
E
X
0
Ułamek molowy składnika B
0
1

Ćwiczenie 1: Destylacja
25
T temperatura (maksymalna wartość nie może przekroczyć temperatury krytycznej)
∆
H entalpia parowania cieczy
R stała gazowa
po scałkowaniu otrzymujemy zależność:
)
1
1
(
)
ln(
2
1
1
2
T
T
R
H
p
p
−
∆
=
(7.16)
gdzie: T
1
temperatura wrzenia dowolnego składnika pod ciśnieniem p
1
(np. atmosferycznym)
T
2
temperatura wrzenia tego składnika pod ciśnieniem p
2
.
Po prostym przekształceniu równania (7.16) dostajemy:
( )
)
)
(
ln(
1
1
2
0
2
1
0
1
1
2
T
p
T
p
H
R
T
T
∆
+
=
(7.17)
Z tego wzoru łatwo wydedukować, że z relacji
0
1
0
2
p
p
<
wynika zależność T
2
< T
1
Zatem im niższe będzie ciśnienie zewnętrzne tym niższa będzie temperatura wrzenia cieczy. Ta
zależność w sposób oczywisty przenosi się na każdą mieszaninę składników, tzn. na dowolny roztwór.
Ten sposób destylacji jest bardzo często stosowany w chemii organicznej i nosi nazwę destylacji
próżniowej lub destylacji pod obniżonym ciśnieniem.
Literatura
1. K. Gumiński „Chemia fizyczna”, PWN, Warszawa 1973
2. Z. Ziołkowski „Destylacja i rektyfikacja w przemyśle chemicznym”, WNT, Warszawa 1978
3.
„Laboratorium
chemii
organicznej”,
red.
P.
Kowalski,
WNT,
Warszawa
2004

Ć
wiczenie 2: Krystalizacja
26
Ć
wiczenie 2:
K R Y S T A L I Z A C J A
3
Cele ćwiczenia: 1) Zapoznanie się z techniką krystalizacji.
2) Zapoznanie się z metodyką pomiaru temperatury topnienia jako testu czystości
substancji krystalicznej.
3) Przeprowadzenie krystalizacji dwóch zanieczyszczonych związków organicznych .
1. WSTĘP
Związki chemiczne w stanie krystalicznym otrzymane drogą reakcji chemicznych, są z reguły
zanieczyszczone bądź substratami, bądź produktami ubocznymi, bądź jednym i drugim. Przed ich
dalszym wykorzystaniem należy je oczyścić. Najczęściej stosowaną metodą oczyszczania substancji
stałych jest krystalizacja. Krystalizacja polega na rozpuszczeniu substancji w odpowiednim
rozpuszczalniku i wytrąceniu z roztworu kryształów związku chemicznego, który dzięki temu zabiegowi
jest pozbawiony substancji zanieczyszczających. Jeżeli nie znamy pochodzenia związku, to przed jego
użyciem należy sprawdzić, czy nie jest on zanieczyszczony. Tę wydawałoby się trudną sprawę, bo często
nie wiemy dokładnie jakimi związkami chemicznymi są zanieczyszczenia, można rozwiązać w bardzo
prosty sposób, odwołując się do podstawowych praw chemii fizycznej. Substancje czyste topią się
w stałej temperaturze, substancje zanieczyszczone nie mają stałej temperatury topnienia
(z wyjątkiem składu eutektycznego – patrz DODATEK I) i zanim przejdą całkowicie w fazę ciekłą
temperatura topnienia będzie stopniowo wzrastać.
Rozpuszczanie ciała stałego w rozpuszczalniku (kluczowy proces w krystalizacji) jest tym samym
procesem fizycznym, co topnienie dwu różnych ciał stałych. Różnica polega jedynie na tym, że
w procesie rozpuszczania jeden ze składników (rozpuszczalnik) w warunkach pokojowych jest już
w stanie ciekłym, podczas gdy przy topnieniu oba składniki są ciałami stałymi, najczęściej o różnych
temperaturach topnienia. Aby ten układ, złożony z dwu faz stałych, przeprowadzić w stan ciekły, należy
go podgrzać. W przypadku rozpuszczania substancja stała może rozpuścić się na zimno
w danym rozpuszczalniku, bądź dopiero po podgrzaniu.
Tak samo pełna analogia występuje pomiędzy wytrącaniem osadu z roztworu i krzepnięciem
stopionej mieszaniny. Z uwagi na podobieństwo procesów topnienia do rozpuszczania, jak
i krzepnięcia do wytrącania można je w spójny sposób przedstawić posługując się jednym diagramem
fazowym temperatura- skład. W rozdziałach, pt. DODATEK I, II zamieszczono krótkie omówienie tych
zagadnień.
2. CZYNNOŚCI PODCZAS PROCESU OCZYSZCZANIA SUBSTANCJI METODĄ
KRYSTALIZACJI
W procesie oczyszczania substancji krystalicznych od zanieczyszczeń metodą krystalizacji
wykorzystuje się różnicę rozpuszczalności substancji i zanieczyszczeń w odpowiednio dobranym
rozpuszczalniku. Substancja oczyszczana nie może rozpuszczać się już na zimno w rozpuszczalniku, bo
wytrącić ją z roztworu będzie niezwykle trudno. Właściwym rozpuszczalnikiem do krystalizacji jest taki
rozpuszczalnik, w którym substancja rozpuszcza się dopiero na gorąco w temperaturze wrzenia
rozpuszczalnika, a krystalizuje (wytrąca z niego) po ochłodzeniu. Ponadto w rozpuszczalniku stosowanym
do krystalizacji zanieczyszczenia nie powinny się wcale rozpuszczać, rozpuszczać w bardzo niewielkim
stopniu albo przeciwnie – rozpuszczać się bardzo dobrze. Ponieważ do krystalizacji stosujemy roztwory
stężone, to w przypadku złej rozpuszczalności zanieczyszczeń oziębianie roztworu spowoduje przesycenie
go względem zanieczyszczeń, a konsekwencji wypadanie zanieczyszczeń wraz z substancją oczyszczaną.
Lepszym wariantem jest stosowanie rozpuszczalników, w których zanieczyszczenia rozpuszczają się
znacznie lepiej od substancji oczyszczanej, bo wówczas zanieczyszczenia przejdą do ługu
pokrystalicznego, a krystalizować będzie substancja oczyszczana. Rozpuszczalnik nie może ponadto
wchodzić w reakcję z oczyszczanym związkiem chemicznym. Przed rozpoczęciem całej procedury
oczyszczania należy zmierzyć temperaturę topnienia substancji, w celu sprawdzenia jej czystości
i zważyć. Temperaturę topnienia mierzy się w specjalnym aparacie, o czym będzie mowa w rozdziale 2.6.
3
Opracował dr Antoni Szumiło-Kulczycki
Ć
wiczenie 2: Krystalizacja
27
Warto tu wspomnieć, że metodę opartą na różnicy rozpuszczalności składników stosuje się również, do
rozdzielania mieszaniny stałych związków chemicznych.
Proces oczyszczania przez krystalizację składa się z następujących etapów:
1
.
rozpuszczenie oczyszczanej substancji na gorąco w odpowiednim rozpuszczalniku;
2. odsączenie roztworu od zanieczyszczeń mechanicznych;
3. wytrącenie z przesączu kryształów oczyszczanej substancji;
4. oddzielenie kryształów od roztworu;
5. suszenie kryształów.
Po wykonaniu krystalizacji również należy zmierzyć temperaturę topnienia uzyskanych kryształów po
to, żeby sprawdzić jakość oczyszczenia. W niektórych przypadkach otrzymanie związku o zadawalającym
stopniu czystości wymaga wielokrotnego przeprowadzenia krystalizacji. Na końcu wysuszony związek
chemiczny ważymy – co da nam informację o stopniu zanieczyszczenia próbki.
2.1 Rozpuszczanie
Oczyszczaną substancję umieszczamy w kolbie i zalewamy wcześniej dobranym rozpuszczalnikiem.
Jeśli krystalizację prowadzimy z rozpuszczalnika organicznego, to rozpuszczanie wykonujemy
ogrzewając zanieczyszczoną substancję wraz z rozpuszczalnikiem w kolbie zaopatrzonej w chłodnicę
zwrotną. Zestaw odpowiedniej aparatury przedstawiono na Rys. 2.1. Jeżeli rozpuszczalnikiem wybranym
do krystalizacji jest woda, to można ogrzewać
substancję w kolbie z wodą bez specjalnych osłon.
krystalizację dodaje się do kolby rozpuszczalnik porcjami (wlewając od góry przez chłodnicę zwrotną) aż
do momentu, w którym cała substancja znajdująca się w kolbie rozpuści się we wrzącym rozpuszczalniku.
2.2 Odsączanie
Po rozpuszczeniu substancji przesączamy gorący roztwór na lejku z sączkiem karbowanym do zlewki
lub kolbki stożkowej. Pozostałość na sączku stanowi zanieczyszczenie i jest niepotrzebnym odpadem.
Rys. 2.1. Zestaw aparatury
do ogrzewania pod chłodnicą
zwrotną
Warto
zwrócić
uwagę,
ż
e
chłodnica
zwrotna
pełni,
w pewnym sensie, rolę dynamicznego korka – przez schładzanie
gorące pary rozpuszczalnika zawracają w postaci skroplin do kolby,
co zapobiega ulatnianiu się rozpuszczalnika na laboratorium.
Zatkanie kolby zwykłym korkiem, w celu zapobieżenia temu
ulatnianiu, prowadziłoby do wzrostu ciśnienia par w kolbie, a w
konsekwencji
do
rozsadzenia
kolby
i
sprowadzenia
niebezpieczeństwa na osoby wykonujące ćwiczenia.
Istotną sprawą w procesie krystalizacji jest dobór odpowiedniej
ilości używanego rozpuszczalnika. Ze względu na fakt, że
oczyszczany związek może rozpuszczać się w pewnym stopniu w
rozpuszczalniku stosowanym do krystalizacji również na zimno,
należy stosować możliwie najmniejszą ilość rozpuszczalnika (to
pozwoli zminimalizować straty oczyszczanego związku). W
praktyce przy doborze ilości rozpuszczalnika do krystalizacji
najlepiej jest kierować się danymi dotyczącymi rozpuszczalności
oczyszczanego związku w danym rozpuszczalniku w różnych
temperaturach (o ile takie są dostępne).
Krystalizację prowadzi się tak, aby uzyskać roztwór nasycony
w temperaturze wrzenia rozpuszczalnika. Jeśli rozpuszczalność
związku w różnych temperaturach nie jest znana, to wykonując
Ć
wiczenie 2: Krystalizacja
28
2.3 Wytrącanie
Przesącz chłodzimy, np. w kąpieli z zimnej wody, w celu wypadnięcia osadu – kryształów naszej
substancji. Im wolniej krystalizacja zachodzi, tym wykształcają się dorodniejsze kryształki. Zbyt szybkie
wytrącenie osadu nie jest też korzystne, gdyż może prowadzić do zaokludowania w nim zanieczyszczeń.
Pełniejsze omówienie powyższych procedur znajduje się w rozdziałach DODATEK I, II.
2.4 Oddzielanie
2.5 Suszenie
Wilgotne kryształki rozprowadzamy na bibule
i czekamy, aż resztki rozpuszczalnika wyparują
z substancji. Jeżeli związek topi się, bądź sublimuje w niskich temperaturach, to nie należy go suszyć w
suszarce. Substancję wysuszoną ważymy.
2.6 Pomiar temperatury topnienia substancji
Pomiar temperatury topnienia wykonuje się w specjalnych aparatach. Niewielką ilość próbki
umieszcza się w kapilarze i ogrzewa, obserwując zmiany preparatu w trakcie wzrostu temperatury.
Pojawienie się w kapilarze pierwszych porcji cieczy to początek topnienia (należy zapisać temperaturę), a
moment, w którym cała próbka przechodzi w stan ciekły to koniec procesu topnienia (należy zapisać
temperaturę). Jak wspomniano wcześniej, wykonuje się pomiar temperatury topnienia substancji przed
i po krystalizacji.
Przy prawidłowym wykonaniu oczyszczania substancji powinniśmy zarejestrować stałą
temperaturę topnienia związku. Brak stałości temperatury topnienia świadczy, że nasze postępowanie było
obarczone niedokładnością. Formalne ujęcie procesu topnienia znajduje się w rozdziale DODATEK I, II
3. WYKONANIE ĆWICZENIA
Szkło laboratoryjne, aparatura, materiały: Odczynniki
1. Płaszcz grzejny 1. Kwas salicylowy zanieczyszczony
2. Podnośnik 2. Acetanilid zanieczyszczony
2. Łapy (2), łączniki (2) 3. Toluen
3. Zlewki 50 ml (2), zlewka 250 ml, zlewka 100 ml
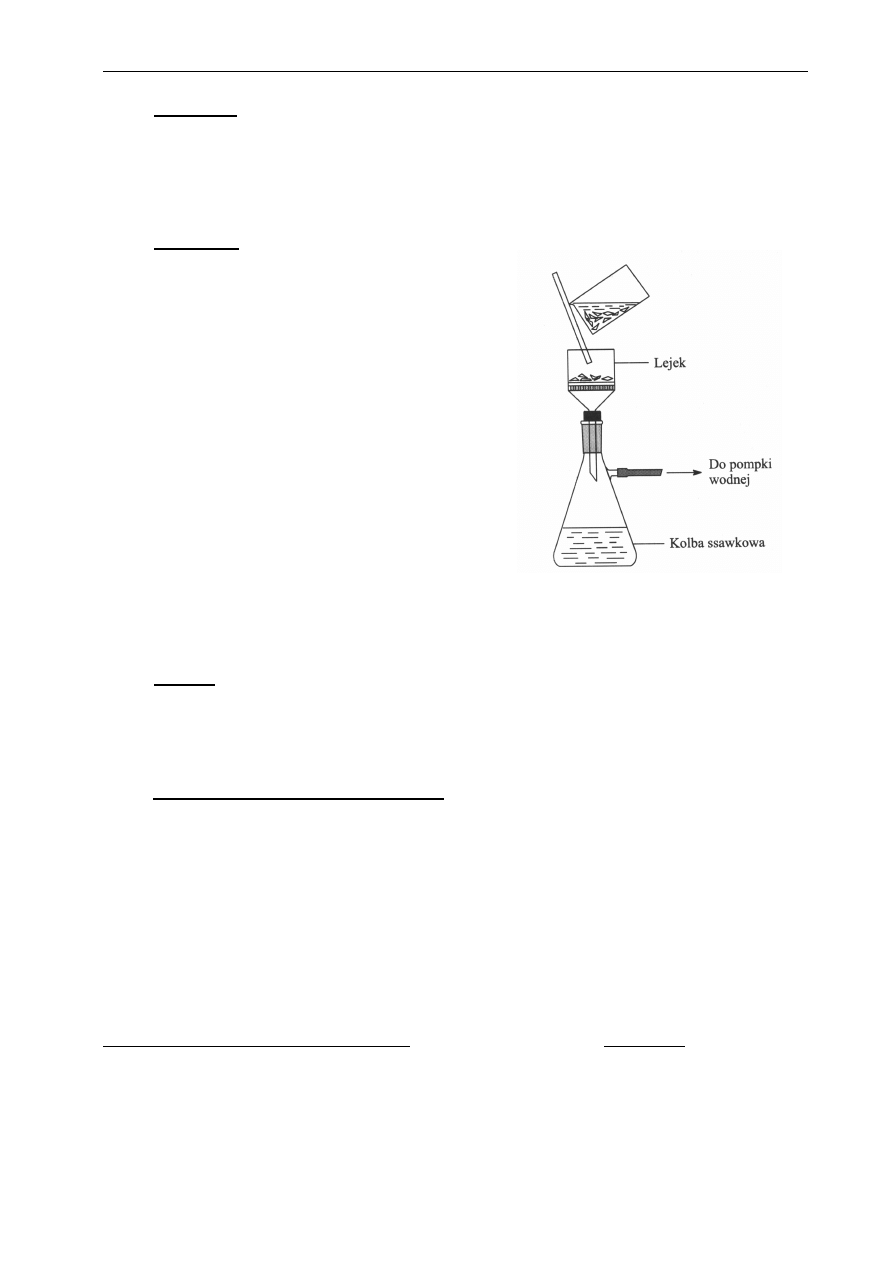
Rys. 2.2. Zestaw aparatury do sączenia pod
zmniejszonym ciśnieniem
Oddzielenie wykrystalizowanej subs-
tancji od cieczy prowadzi się na lejku
Büchnera wyścielonym bibułą filtracyjną
i
osadzonym
w
kolbie
ssawkowej
podłączonej do pompki wodnej. Zamiast
lejka Büchnera można użyć lejka z
wtopionym
spiekiem
szklanym
o
odpowiedniej porowatości. Zestaw aparatury
stosowanej do sączenia pod zmniejszonym
ciśnieniem pokazano na Rys. 2.2.
Pompka wodna służy do wytwarzania
podciśnienia w kolbie ssawkowej, co
znacznie
przyspiesza
odfiltrowanie
przesączu. (Drobne kryształki filtruje się
znacznie wolniej od dużych.). Krótkie
omówienie zasady działania i rysunek
przedstawiający różne konstrukcje pompki
wodnej (Rys. 4.3) podano w DODATKU
III.

Ć
wiczenie 2: Krystalizacja
29
4. Lejek zwykły, lejek ze spiekiem
5. Kolba ssawkowa
6. Kolba okrągło-denna 50 ml
7. Chłodnica
8. Cylindry miarowe (menzurki) 25 ml i 50 ml
9. Pręcik szklany, bagietka metalowa
10. Kapilarki (2 + 2)
11. Bibuła miękka, bibuła twarda
12. Szalka Petriego
13. Kamyki wrzenne (2) lub sita molekularne (1)
Ć
wiczenie polega na oczyszczeniu drogą krystalizacji dwóch zanieczyszczonych substancji. Jedną
krystalizację prowadzi się z wody (kwas salicylowy zanieczyszczony mocznikiem, drugą – z
rozpuszczalnika organicznego (acetanilid zanieczyszczony naftalenem).
1. Przed przystąpieniem do krystalizacji należy zmierzyć temperatury topnienia (zakres)
2.
Zważyć próbki (po koło 0.8 g) związków otrzymanych od prowadzącego ćwiczenia – kwasu
salicylowego zanieczyszczonego mocznikiem i acetanilidu zanieczyszczonego naftalenem
3.
Próbkę, kwasu salicylowego, który rozpuszcza się w wodzie przenieść do zlewki na 50 ml i zalać 45
ml wody destylowanej i ogrzewać płaszczem grzejnym do całkowitego jej rozpuszczenia.
4.
Na lejku z sączkiem karbowanym odsączyć gorący roztwór od zanieczyszczeń do erlenmayerki.
5.
Przesącz odstawić do wystudzenia i wytrącenia kryształków oczyszczanego związku.
6.
Po krystalizacji całość przenieść na lejek ze spiekiem nałożony na kolbkę ssawkową podłączoną do
pompki wodnej. Przyciskając mocno lejek do kolbki (nie naciskać otwartą dłonią !) otworzyć kran z
wodą przy pompce wodnej na średni strumień .
7.
Po odessaniu roztworu znad osadu należy osad jeszcze przemyć niewielką ilością zimnej wody, przy
włączonej pompce wodnej
8.
Po dokładnym odsączeniu kryształów zamknąć wodę od pompki, zdjąć lejek z kolbki i kryształy
suszyć na powietrzu rozpostarte na świeżej bibule.
9.
Zmierzyć temperaturę topnienia oczyszczonego związku i zważyć go.
Próbkę acetanilidu , który rozpuszcza się w rozpuszczalniku organiczym:
1’ Po sprawdzeniu stopnia jej czystości na aparacie do pomiaru temperatury topnienia,
2’. Przenieść do kolbki okrągło-dennej i zalać pod dygestorium 23 ml toluenu
3’. Zestawić aparaturę do ogrzewania cieczy z chłodnica zwrotną (wg schematu pokazanego na
Rys.2.1). Po rozpuszczeniu substancji postępować w sposób opisany wyżej, z tym że chłodzenie
przesączu prowadzić pod dygestorium.
Temperatury plateau dla zanieczyszczonego acetanilidu 90
0
C, a dla kwasu salicylowego 110
0
C
Temperatury plateau dla oczyszczonych związków 105
0
C i 150
0
C odpowiednio.
3.1. Opracowanie wyników
1. Na podstawie wyników ważenia substancji zanieczyszczonych i po oczyszczeniu obliczyć procent
wagowy zanieczyszczeń w wyjściowych substancjach.
2. W oparciu o scałkowaną postać równania (4.1) – por. DODATEK II:
∫
∫
∆
=
0
2
1
))
(ln(
T
T
Xz
k
T
dT
R
H
X
d
gdzie: X
z
– szukany ułamek molowy substancji zanieczyszczanej, tj. głównego składnika
mieszaniny
T
0
- zmierzona temperatura topnienia oczyszczonego związku
T
k
- zmierzona końcowa temperatura topnienia substancji zanieczyszczonej
H
∆
- entalpia topnienia czystego związku:
Ć
wiczenie 2: Krystalizacja
30
H
∆
dla kwasu salicylowego: 24,60 [kJ/mol]
H
∆
dla acetanilidu : 21,65 [kJ/mol]
obliczyć ułamek molowy zanieczyszczanego związku (w obydwu badanych układach),
a następnie przeliczyć ten wynik na % wagowy zanieczyszczenia (potrzebne masy molowe
policzyć w oparciu o wzory chemiczne związków zawartych w mieszaninach).
3. Wyniki zestawić w tabeli wg poniższego wzoru:
Masa
związku
zanieczysz-
czonego [g]
Masa
związku po
oczyszcze-
niu [g]
Zakres temp
topnienia
związku za-
nieczyszczo-
nego [
0
C]
Zakres temp
topnienia
zwiazku po
oczyszczeniu
[
0
C]
Zanieczysz-
czenie
związku ob-
liczone
wg
p.1 [% wag]
Zanieczysz-
czenie
związku ob-
liczone
wg
p.2 [% wag]
Kwas
salicylowy
Acetanilid
4. Skomentować otrzymane wyniki.
4. DODATEK
I. Diagram fazowy temperatura - skład.
Zachowanie się związków krystalicznych podczas ogrzewania lub chłodzenia wygodnie jest
przedstawić na diagramie fazowym temperatura – skład, przy ustalonym ciśnieniu. Rysunek 4.1
przedstawia bardzo często spotykany przypadek zachowania się mieszaniny dwu związków chemicznych
A i B .
Punkty leżące poniżej linii równoległej do osi odciętych na wysokości punktu E reprezentują układy
dwufazowe – jedną fazą jest krystaliczny związek A, a drugą krystaliczny związek B. Sama linia
poprowadzona na wysokości punktu E, równolegle do osi odciętych (linia aET
E
) reprezentuje stany, w
których w równowadze są trzy fazy – kryształy A, kryształy B i roztwór o składzie x
E
. Ten wyróżniony
skład nosi nazwę składu eutektycznego.
T
B
0
p=const
T
A
0
T
E
T
T
E
X
E
X
0
Ułamek molowy składnika B
A
B
T
m
T
k
T
0
a
Rys. 4.1. Diagram fazowy temperatura-skład
układu dwuskładnikowego z eutektykiem
Na osi rzędnych odłożona jest temperatura, a na osi
odciętych skład zarówno mieszaniny stałej, jak i
roztworu w postaci ułamka molowego (wagowego)
składnika B. Na osi rzędnych po lewej stronie
zaznaczono
temperaturę
topnienia
czystego
związku chemicznego A (
0
A
T
), a na osi rzędnych
po prawej stronie temperaturę topnienia czystego
związku chemicznego B(
0
B
T
).Krzywe łączące
punkt
0
A
T
z
punktem
E
oraz
punkt
0
B
T
z punktem E przedstawiają stany, w których
rozpoczyna się krystalizacja roztworu. Punkty
leżące powyżej tych krzywych reprezentują
jednofazowe stany ciekłe A-B, różniące się
temperaturą i składem. Punkty pod tymi krzywymi,
ale powyżej linii aET
E
reprezentują stany, w
których istnieją dwie fazy w równowadze - faza
stała i roztwór A - B. Dla punktów na lewo od E
fazą stałą są kryształy związku A, na prawo -
kryształy związku B.

Ć
wiczenie 2: Krystalizacja
31
II. Zachowanie się ciał stałych pod wpływem temperatury
a) czyste składniki
Czysty składnik A znajdujący się w temperaturze T
0
po ogrzaniu będzie się topił w
temperaturze
0
A
T
. Dopóki cała substancja nie stopi się temperatura, mimo ogrzewania, będzie stała w
miejscu. Gdy całość przejdzie w ciecz, to ogrzewanie będzie powodować podnoszenie temperatury (jak to
ma miejsce, np. podczas grzania wody- otrzymanej z lodu- na herbatę). W analogiczny sposób będzie
zachowywać się czysta substancja B z tym, że temperatura topnienia będzie dla niej inna. Idąc teraz w
kierunku przeciwnym – ochładzając stopiony składnik A o początkowej temperaturze T
k
jego temperatura
będzie spadać do wartości
0
A
T
. Od tego momentu, aż do całkowitego skrzepnięcia cieczy ochładzanie nie
spowoduje zmiany temperatury układu. Po całkowitym zniknięciu fazy ciekłej temperatura kryształów
substancji A, przy dalszym chłodzeniu będzie spadać. Podobny obraz przedstawia ochładzanie czystego
składnika B.
b) mieszaniny
Zupełnie inaczej od czystych składników krystalicznych będzie zachowywać się układ złożony z
dwu substancji krystalicznych - mieszaniny kryształów A i B. Rysunek 4.1 przedstawia sytuację, w której
kryształy obydwu substancji wykazują całkowity brak mieszalności w stanie stałym, ale pełną
mieszalność w stanie ciekłym.
Przyjmijmy, dla skoncentrowania uwagi, że wyjściowy skład mieszaniny będzie gdzieś w
przedziale na lewo od punktu x
E
. (Wnioski płynące z przeprowadzonej niżej dyskusji można będzie łatwo
przenieść na sytuację w której skład mieszaniny jest na prawo od x
E
, z tą różnicą, że składnik B będzie
teraz pełnił rolę składnika A, np. wytrącał się przy chłodzeniu roztworu zamiast składnika A).
b1) topnienie
Mieszanina kryształów o składzie x
0
i temperaturze początkowej T
0
, (Rys. 4.1) po ogrzaniu,
zacznie się topić w temperaturze T
E
. Dopóki w układzie będą kryształy substancji B, dopóty powstający
podczas tego procesu roztwór, będzie wykazywał niezmienny skład x
E
. Dopiero po całkowitym stopieniu
kryształów B, dalsze ogrzewanie będzie powodować topnienie reszty kryształów A, wzrost temperatury
układu i zmianę składu roztworu na korzyść składnika A. Skład roztworu zależy tylko od temperatury
układu i jest wyznaczony rzędną punktu przecięcia linii temperatury (równoległej do osi odciętych) z
krzywą T
0
E. Proporcje ilościowe fazy stałej do fazy ciekłej znajdujemy z reguły dźwigni (poznanej już
wcześniej) i one zależą od temperatury, ale też od składu wyjściowego mieszaniny. Po osiągnięciu przez
układ temperatury T
m
otrzymamy układ jednofazowy – ciecz o składzie wyjściowym x
0
jaki miała
mieszanina kryształów. Dalsze ogrzewanie roztworu będzie powodować systematyczny wzrost
temperatury. Tak więc, w odróżnieniu od czystych substancji krystalicznych temperatura topnienia
mieszaniny ciał stałych zmienia się w pewnym przedziale. Początek topnienia jest zawsze w
temperaturze T
E
, niezależnie od składu początkowego mieszaniny. Powyższe sentencje są podstawą
weryfikacji czystości próbki stałej.
b2) krystalizacja
Schładzanie roztworu o składzie x
0
i początkowej temperaturze T
n
, spowoduje, że dopiero w
temperaturze T
m
, niższej od
0
A
T
rozpocznie się krystalizacja, zaczną wypadać kryształy substancji A. W
miarę schładzania temperatura krystalizacji będzie spadać, a roztwór będzie zatężać się w związek
chemiczny B. W temperaturze T
E
z roztworu zaczną wypadać obydwa składniki. Końcowy skład
mieszaniny kryształów będzie taki sam, jaki miał roztwór na początku, x
0
. Odmiennie niż to wykazują
czyste substancje, mieszaniny krystalizują w pewnym przedziale temperaturowym. Dalsze chłodzenie
mieszaniny stałych A i B będzie powodować obniżanie temperatury układu.
Szczególną pozycję w tej analizie zajmuje skład eutektyczny. Mieszanina o składzie eutektycznym
x
E
, topi się jak i krystalizuje w stałej, najniższej temperaturze T
E
, a podczas przemiany fazowej skład
roztworu (przy krystalizacji), czy skład powstającego roztworu (przy topnieniu) nie ulegają zmianie.

Ć
wiczenie 2: Krystalizacja
32
Relację pomiędzy początkową temperaturą krzepnięcia, a składem próbki dla układów
dwuskładnikowych o całkowitej rozpuszczalności w stanie ciekłym i zupełnym braku mieszalności w
stanie stałym (Rys. 4.1) można łatwo wyprowadzić bazując na podstawowych zależnościach
termodynamicznych. Ponieważ problem ten jest omawiany w każdym podręczniku chemii fizycznej nie
będziemy tej zależności wyprowadzać, a podamy tylko finalną postać:
2
T
H
dT
))
X
(ln(
d
R
∆
=
(4.1)
Gdzie: R - stała gazowa
T - temperatura
X - ułamek molowy substancji powyżej składu eutektycznego
H
∆
- entalpia topnienia tej substancji
Zależność przedstawiona wzorem (4.1) jest spełniona również dla przypadków niewielkich udziałów
drugiego składnika w mieszaninie, a więc substancji zanieczyszczonej innym związkiem. Wówczas
H
∆
oraz X będą dotyczyć substancji zanieczyszczanej. Mieszaniny kryształów tworzące jednorodną fazę
stałą - rozwór stały mają diagram fazowy inny (w kształcie „rybki”), ale wniosek dotyczący płynności
temperatury topnienia jest spełniony i tym przypadku.
Ilościowy opis procesów topnienia i krystalizacji oparty jest na poznanym już przy destylacji
różniczkowej bilansie masy (moli) wybranego składnika - wiążącym zmianę składu roztworu ze zmianą
jego masy (liczby moli) zachodzącej w trakcie procesu.
III Zasada działania pompki wodnej
Pompka wodna jest niesłychanie prostym urządzeniem, służącym do wytwarzania podciśnienia. Jest
ono przykładem wykorzystania prostych praw fizyki przepływów cieczy. Podstawowym prawem,
któremu podlegają przepływy cieczy jest prawo Bernoulliego. Podczas przepływu cieczy działają dwa
rodzaje ciśnienia:
a) ciśnienie statyczne p - działające prostopadle do kierunku przepływu (patrz Rys. 4.2)
b) ciśnienie dynamiczne
2
2
v
⋅
ρ
- działające w kierunku przepływu cieczy.
Dla
płynów
nieściśliwych
prawo
zachowania
energii
można
zapisać
w
następujący
sposób:
const
v
p
v
p
=
⋅
+
=
⋅
+
2
2
2
2
2
2
1
1
ρ
ρ
(4.2)
Gdzie: p
1
, p
2
ciśnienia statyczne w rurach o przekrojach większym i mniejszym
odpowiednio
ρ
gęstość cieczy
2
1
, v
v
prędkości liniowe w rurach o przekrojach większym i mniejszym
odpowiednio.
Prawo Bernoulliego głosi, że podczas przepływu stacjonarnego suma ciśnienia statycznego
i dynamicznego jest wielkością stałą, równą ciśnieniu hydrostatycznemu cieczy pozostającej
w spoczynku.
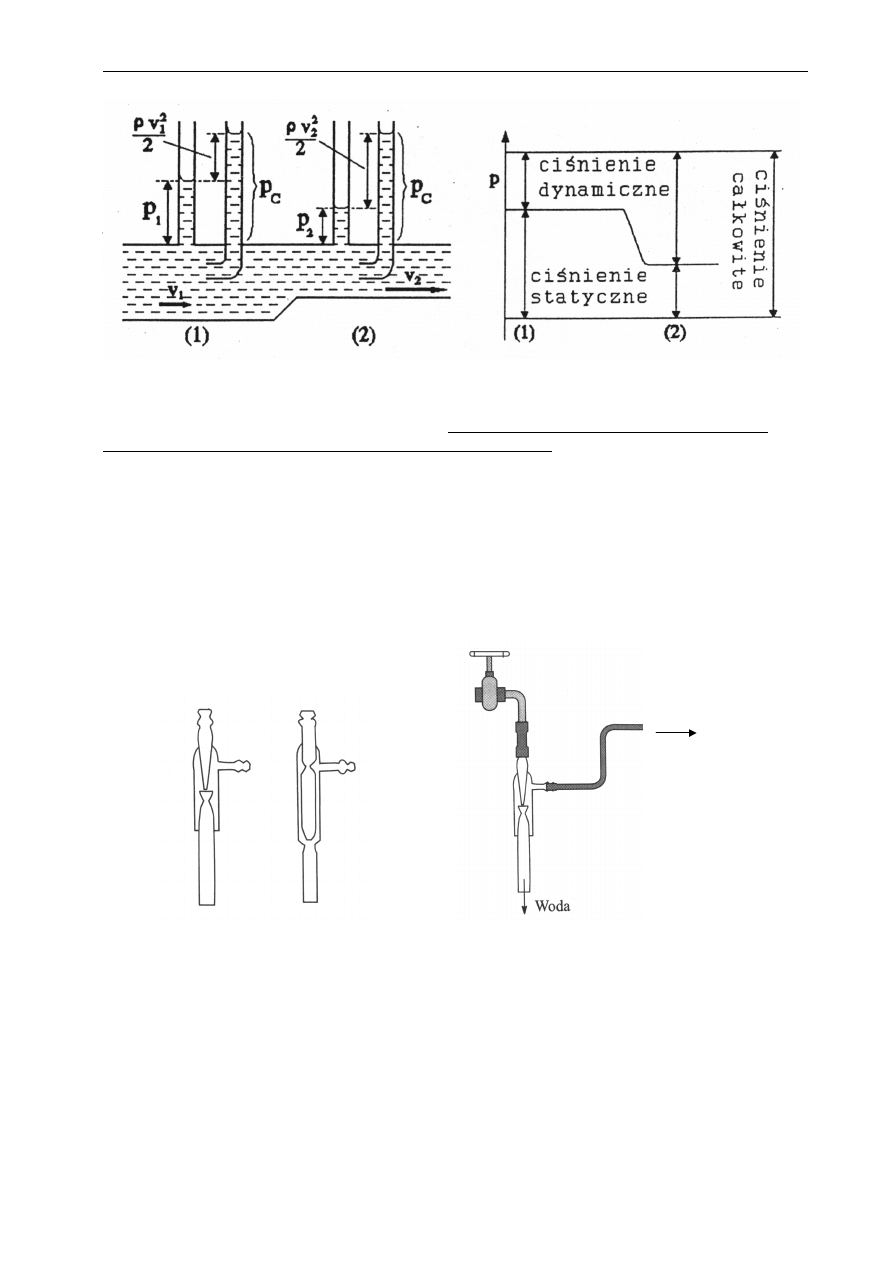
Ć
wiczenie 2: Krystalizacja
33
Jeżeli ciecz nieściśliwa przepływa z rury o większej średnicy do rury o mniejszej średnicy, to jej
prędkość w rurze o mniejszej średnicy musi odpowiednio wzrosnąć, aby spełnić prawo zachowania masy.
Zwiększenie prędkości przepływu spowoduje zwiększenie wartości drugiego członu (ciśnienia
dynamicznego) we wzorze (4.2), ale pociągnie obniżenie wartości członu pierwszego (ciśnienia
statycznego). W konsekwencji, przy przepływie cieczy z rury o większej średnicy do rury o mniejszej
ś
rednicy, ciśnienie statyczne - prostopadłe do kierunku przepływu – spadnie; przy dużych prędkościach
nawet do bardzo małych wartości w porównaniu z ciśnieniem atmosferycznym. Zasada ta została
wykorzystana w konstrukcji pompki wodnej (patrz Rys. 4.3). Wygenerowane przez zwężającą się rurkę
obniżone ciśnienie (statyczne –prostopadłe do kierunku przepływu) pozwala zasysać ośrodek z otoczenia
np. powietrze w erlenmayerce przykrytej szczelnie lejkiem Büchnera wyłożonym bibułą filtracyjną.
Wytworzone tak podciśnienie powoduje odsysanie cieczy znajdującej się na lejku.
Literatura
A. Vogel „Preparatyka organiczna”,WNT Warszawa 1984
K.Gumiński „Chemia fizyczna”, PWN Warszawa 1973
Rys. 4.2. Rozkład ciśnień przy przepływie płynów przez różne przekroje.
Do aparatury
Rys. 4.3. Pompki wodne: a) różne konstrukcje; b) sposób podłączenia do wody i aparatury
a)
b)

Ćwiczenie 3: Ekstrakcja
34
Ć
wiczenie 3:
E K S T R A K C J A
4
Cele ćwiczenia: 1) Zapoznanie się z techniką ekstrakcji.
2) Przeprowadzenie ekstrakcji w układzie ciecz-ciecz: ekstrakcja kwasu benzoesowego
z roztworu w toluenie za pomocą wodnego roztworu wodorotlenku sodowego
1. Podstawowe informacje dotyczące procesu ekstrakcji
Ekstrakcja to metoda wydzielania składnika (lub składników) z mieszaniny związków chemicznych
przez jego (ich) rozpuszczenie w odpowiednio dobranym rozpuszczalniku. Po dodaniu wybranego
rozpuszczalnika do mieszaniny tworzy się układ dwufazowy. Jedną fazę stanowi roztwór ekstrahowanego
składnika (lub składników) mieszaniny w rozpuszczalniku użytym do ekstrakcji, drugą – mieszanina
pozostała po ekstrakcji.
Mieszaniny poddawane procesowi ekstrakcji mogą być w fazie ciekłej (najczęściej w roztworze
wodnym lub w rozpuszczalniku organicznym) – wówczas ekstrakcja przebiega w układzie dwóch nie
mieszających się cieczy. Jest to ekstrakcja ciecz-ciecz, nazywana często ekstrakcją właściwą lub po prostu
ekstrakcją. Mieszaniny mogą też być ciałami stałymi – wtedy ekstrakcja przebiega w układzie ciało stałe-
ciecz. Taką ekstrakcję nazywa się ługowaniem.
Z techniką ekstrakcji wiążą się następujące terminy:
•
substancja ekstrahowana – to związek chemiczny wyodrębniany z mieszaniny metodą
ekstrakcji;
•
ekstrahent – to rozpuszczalnik używany do ekstrakcji;
•
ekstrakt – to roztwór substancji ekstrahowanej w ekstrahencie uzyskiwany po ekstrakcji;
•
rafinat – to pozostałość po ekstrakcji.
W procesie ekstrakcji wykorzystuje się różną rozpuszczalność składników mieszaniny
w ekstrahencie. Decydujący wpływ na skuteczność rozdziału związków chemicznych tą metodą ma więc
właściwy dobór rozpuszczalnika do ekstrakcji. Dobry rozpuszczalnik do ekstrakcji powinien być:
•
selektywny, tzn. dobrze rozpuszczać składnik (składniki) wyodrębniane z mieszaniny, a nie
rozpuszczać pozostałych jej składników;
•
obojętny chemicznie, tj. nie reagować ze składnikami mieszaniny (o ile nie jest to
zamierzone);
•
łatwy do usunięcia z ekstraktu, a więc najlepiej lotny, bo wtedy można go oddestylować.
Należy także dążyć do tego, aby rozpuszczalnik wybrany do ekstrakcji był niepalny, nietoksyczny,
łatwo dostępny oraz tani. W przypadku ekstrakcji ciecz-ciecz wskazane jest ponadto, żeby ekstrahent
i faza ciekła poddawana ekstrakcji różniły się znacznie gęstością, żeby nie tworzyły emulsji i były
całkowicie niemieszalne. Wszystko to ułatwia rozdział ekstraktu i rafinatu, tj. dwóch faz ciekłych
uzyskiwanych po ekstrakcji.
Chociaż istotą procesu ekstrakcji jest zjawisko fizyczne, czyli rozpuszczanie związków chemicznych
w ekstrahencie, to jednak czasami celowo prowadzi się ekstrakcję tak, że w jej trakcje przebiega reakcja
chemiczna. W przypadku mieszanin związków organicznych ma to miejsce wtedy, gdy zawierają one
substancje o właściwościach kwasowych i/lub zasadowych. Ekstrakcja związków kwasowych (kwasów
karboksylowych, fenoli) roztworem wodnym zasady (NaOH, NaHCO
3
) powoduje, że zachodzi reakcja
zobojętniania. W jej wyniku powstaje sól, która – jako związek jonowy – jest rozpuszczalna w wodzie,
a więc przechodzi do warstwy wodnej. Jeśli jest to konieczne, można zakwasić powstały ekstrakt i w ten
sposób wydzielić z niego kwas. Związki zasadowe (aminy) najczęściej ekstrahuje się rozcieńczonym
kwasem solnym. Działanie zasadą (zwykle NaHCO
3
) na warstwę wodną, w której rozpuszczone są
utworzone sole (chlorowodorki amin), pozwala wyodrębnić z niej zasadę organiczną.
Ekstrakcja znajduje szerokie zastosowanie zarówno w praktyce laboratoryjnej, jak i przemysłowej.
Wszyscy mamy z nią do czynienia na co dzień, gdy parzymy herbatę lub kawę. W syntezie organicznej
ekstrakcja jest często pierwszą metodą używaną do obróbki mieszaniny poreakcyjnej, zawierającej zwykle
4
Opracowanie: dr hab. Magdalena Hasik
Ćwiczenie 3: Ekstrakcja
35
wiele związków chemicznych. Stosuje się ją w celu wyodrębnienia pożądanego produktu lub w celu
oddzielenia od niego ubocznych produktów reakcji i/lub nie przereagowanych substratów. W pierwszym
przypadku ekstrakt jest roztworem zawierającym cenny związek, w drugim – roztworem związków, które
można traktować jako jego zanieczyszczenia. Zawsze jednak otrzymanie czystej substancji z ekstraktu
wymaga przeprowadzenia dodatkowych operacji: usunięcia rozpuszczalnika (np. przez oddestylowanie)
i oczyszczenia wyekstrahowanego związku (przez destylację, jeśli jest to substancja ciekła lub przez
krystalizację, jeśli jest to ciało stałe).
1.1. Ekstrakcja w układzie ciecz-ciecz
Taką ekstrakcję prowadzi się w układzie dwóch faz ciekłych, z których jedną często jest roztwór
wodny, a drugą – nie mieszający się z wodą rozpuszczalnik organiczny taki, jak: chlorek metylenu,
chloroform, toluen lub eter dietylowy (eter jest niebezpieczny, bo łatwopalny!).
Ekstrakcja ciecz-ciecz może być procesem okresowym, czyli periodycznym lub ciągłym.
W warunkach laboratoryjnych ekstrakcję periodyczną prowadzi się w rozdzielaczu gruszkowym.
Periodyczna ekstrakcja ciecz-ciecz przebiegająca w układzie, w którym nie zachodzi reakcja
chemiczna, jest procesem równowagowym. Zgodnie z prawem podziału Nernsta
5
, w stanie równowagi
ekstrahowany związek dzieli się między obie fazy ciekłe w określonej proporcji. Stosunek
równowagowych ilości związku w każdej z faz nosi nazwę współczynnika podziału:
B
A
C
C
K
=
(1)
gdzie: K- współczynnik podziału
C
A
– równowagowe stężenie ekstrahowanego związku w jednej z faz ciekłych
C
B
– równowagowe stężenie ekstrahowanego związku w drugiej fazie ciekłej
Współczynnik podziału zależy od temperatury. W danej temperaturze jest to wielkość
charakterystyczna dla układu trójskładnikowego, jakim jest związek ekstrahowany i para
rozpuszczalników stosowanych w ekstrakcji. W przybliżeniu wartość współczynnika podziału
wyznaczona jest przez stosunek rozpuszczalności związku w obu rozpuszczalnikach w danej
temperaturze. Gdy rozpuszczalność ekstrahowanego związku w obu rozpuszczalnikach jest jednakowa, to
K = 1. Jeśli przyjmie się, że we wzorze (1) C
A
oznacza równowagowe stężenie
ekstrahowanego związku
w ekstrahencie, C
B
– w rafinacie
,
a substancja ekstrahowana lepiej rozpuszcza się w ekstrahencie
niż
w rozpuszczalniku, w którym znajduje się mieszanina poddawana ekstrakcji, to K
>
1. Ekstrakcję ciecz-
5
Prawo podziału Nernsta jest spełnione tylko w rozpuszczalnikach, w których związek rozpuszczony nie ulega
asocjacji ani dysocjacji. W praktyce ma to miejsce w roztworach rozcieńczonych.
Rozdzielacz umieszcza się w metalowym kółku
przymocowanym do statywu (Rys. 1)
i napełnia cieczą
poddawaną ekstrakcji. Następnie dodaje się ekstrahent,
zakrywa rozdzielacz korkiem i miesza (wytrząsa) jego
zawartość. Co pewien czas należy odpowietrzać
rozdzielacz przez odwrócenie go na chwilę nóżką do góry
i otwarcie kranu. Po wymieszaniu zawartości, rozdzielacz
znów umieszcza się na statywie i pozostawia do
rozdzielenia faz. Ekstrakt zlewa się do przygotowanego
wcześniej naczynia (kolbki stożkowej lub zlewki),
a mieszaninę pozostałą po pierwszej ekstrakcji wytrząsa
się w rozdzielaczu z kolejną porcją rozpuszczalnika
w celu wyodrębnienia większej ilości ekstrahowanego
związku. W ten sposób ekstrakcję prowadzi się
wielokrotnie aż do momentu, gdy z mieszaniny zostanie
wyodrębniona zadowalająca ilość związku.
Rys. 1. Zestaw laboratoryjny do prowadzenia
ekstrakcji periodycznej.
Rozdzielacz
Kolbka
stożkowa
Statyw
Kółko

Ćwiczenie 3: Ekstrakcja
36
ciecz korzystnie jest prowadzić w układach o jak najwyższej wartości K między ekstrahent a fazę rafinatu,
gdyż wtedy jest ona szybka i skuteczna.
Ilość substancji ekstrahowanej pozostałej w rafinacie po wielokrotnie przeprowadzonej ekstrakcji
można obliczyć ze wzoru (2). Wzór ten łatwo jest wyprowadzić (Warto spróbować to zrobić! Można
wzorować się na wyprowadzeniu przedstawionym w pozycji literatury oznaczonej w spisie numerem [1]),
rozpatrując zawartości substancji ekstrahowanej w każdej z faz ciekłych po kolejnych ekstrakcjach
i pamiętając, że ich stosunek jest równy współczynnikowi podziału.
n
A
B
B
n
V
K
V
V
W
W
⋅
+
=
0
(2)
gdzie: n - liczba przeprowadzonych ekstrakcji;
W
0
– początkowa ilość ekstrahowanego składnika w cieczy poddawanej ekstrakcji;
W
n
– ilość składnika w rafinacie po przeprowadzeniu n-ekstrakcji;
K - współczynnik podziału substancji między fazę ekstrahenta, a fazę rafinatu;
V
A
– objętość ekstrahenta używanego w kolejnych ekstrakcjach;
V
B
– objętość cieczy poddawanej ekstrakcji.
Na podstawie wzoru (2) można stwierdzić, jak należy postąpić, jeśli dysponuje się ograniczoną ilością
rozpuszczalnika przeznaczonego do ekstrakcji. Lepiej jest wówczas podzielić rozpuszczalnik na mniejsze
porcje i przeprowadzić ekstrakcję periodyczną więcej razy niż używać większych porcji rozpuszczalnika
i prowadzić ekstrakcję mniej razy. Pierwszy sposób pozwala bowiem na wyodrębnienie z mieszaniny
większej ilości ekstrahowanej substancji (gdy iloczyn V
A
•
n we wzorze (2) ma stałą wartość, to W
n
maleje,
jeśli V
A
jest małe, a n duże).
W przypadku małej wartości współczynnika podziału substancji ekstrahowanej między ekstrahent
a drugą fazę ciekłą (fazę rafinatu)
,
ekstrakcja periodyczna musi być prowadzona wiele razy. Taki proces
jest długotrwały, uciążliwy i nieekonomiczny, gdyż wymaga użycia dużej ilości ekstrahenta, a kolejne
porcje ekstraktu to roztwory bardzo rozcieńczone. Wtedy lepiej jest zastosować ekstrakcję ciągłą,
w której ekstrahent znajduje się w obiegu zamkniętym. Ze względu na stosowanie stałej ilości
rozpuszczalnika, ekstrakt w czasie takiej ekstrakcji ulega zatężeniu, co ułatwia późniejsze wydzielenie
z niego wyekstrahowanego związku. Istnieją specjalne zestawy aparatury laboratoryjnej do prowadzenia
ekstrakcji ciągłej ciecz-ciecz. Ich opis znajduje się w pozycjach literatury oznaczonych w spisie numerami
[1, 2].
1.2. Ekstrakcja w układzie ciało stałe-ciecz (ługowanie)
Ekstrakcja w układzie ciało stałe-ciecz, podobnie jak ekstrakcja ciecz-ciecz, może być procesem
periodycznym lub ciągłym.
Periodyczną ekstrakcję ciało stałe-ciecz można prowadzić w temperaturze otoczenia (jest to tzw.
„ekstrakcja na zimno”) lub podwyższonej (tzw. „ekstrakcja na gorąco”). Gdy proces przebiega
w temperaturze pokojowej rozdrobnioną stałą mieszaninę umieszczoną w odpowiednim naczyniu (kolbie,
zlewce) zalewa się ekstrahentem, miesza powstałą zawiesinę i odsącza ekstrakt (najlepiej pod
zmniejszonym ciśnieniem). Do mieszaniny pozostałej po pierwszej ekstrakcji dodaje się następną porcję
rozpuszczalnika i oddziela kolejny ekstrakt. W ten sposób proces ekstrakcji prowadzi się wielokrotnie,
ekstrakty łączy, a następnie wydziela z nich wyekstrahowaną substancję. W przypadku ekstrakcji na
gorąco - jeśli ekstrahentem jest rozpuszczalnik organiczny - stałą mieszaninę ogrzewa się
z rozpuszczalnikiem pod chłodnicą zwrotną, sączy na ciepło, ekstrakt pozostawia, a mieszaninę poddaje
kolejnej ekstrakcji w podwyższonej temperaturze. Ogrzewanie poprawia skuteczność ekstrakcji, gdyż
rozpuszczalność większości związków chemicznych wzrasta wraz ze wzrostem temperatury. Niezależnie
jednak od tego, w jakiej temperaturze jest prowadzona, periodyczna ekstrakcja ciało stałe-ciecz jest
procesem kłopotliwym. Konieczne jest stosowanie dużych ilości ekstrahenta, a także wielokrotne sączenie
mieszaniny. Dlatego ługowanie najczęściej prowadzi się w sposób ciągły.
Typowym zestawem aparatury laboratoryjnej do prowadzenia ekstrakcji ciągłej ciało stałe-ciecz jest
aparat Soxhleta pokazany na Rys. 2.
Ćwiczenie 3: Ekstrakcja
37
2. Opis ćwiczenia
W ćwiczeniu wyodrębnia się kwas benzoesowy z jego roztworu w toluenie przez ekstrakcję wodnym
roztworem wodorotlenku sodowego. Jest to więc ekstrakcja w układzie ciecz-ciecz, w trakcie której
zachodzi reakcja zobojętniania kwasu:
COOH
NaOH
COONa
O
H
2
+
+
Jeżeli zastosujemy odpowiednio duży nadmiar molowy wodorotlenku sodowego w stosunku do liczby
moli kwasu benzoesowego znajdującego się w toluenie, to jedna ekstrakcja wystarcza, żeby cały kwas
zawarty w warstwie organicznej utworzył sól i w tej postaci przeszedł do fazy wodnej. Z warstwy wodnej
po zakwaszeniu wytrąca się kwas benzoesowy, który oczyszcza się przez krystalizację z wody. Toluen po
ekstrakcji „odzyskuje się” przez destylację prostą prowadzoną pod ciśnieniem atmosferycznym.
Warto wspomnieć, że kwas benzoesowy jest produktem utlenienia toluenu. Reakcję jego
otrzymywania można przedstawić schematycznie następującym równaniem:
CH
3
O
COOH
Ekstrakcja prowadzona w ćwiczeniu stanowi jeden z końcowych etapów syntezy kwasu
benzoesowego, polegający na oddzieleniu produktu reakcji (kwasu benzoesowego) od nie
przereagowanego substratu (toluenu). Surowy produkt, tj. kwas benzoesowy otrzymany po ekstrakcji,
poddaje się następnie krystalizacji, a toluen – po oczyszczeniu przez destylację - można wykorzystać jako
substrat w syntezie następnej partii kwasu. Tak więc wszystkie eksperymenty wykonywane w trakcie
ć
wiczenia (ekstrakcja, krystalizacja kwasu benzoesowego, destylacja toluenu) stanowią elementy obróbki
mieszaniny poreakcyjnej po utlenieniu toluenu do kwasu benzoesowego.
Aparat składa się z trzech elementów szklanych: kolby
okrągłodennej (C), części środkowej (B) oraz chłodnicy
zwrotnej (D). Przed ekstrakcją kolbę napełnia się
rozpuszczalnikiem, a w części środkowej aparatu, w specjalnej
porowatej gilzie (A) umieszcza się stałą mieszaninę poddawaną
ekstrakcji. Gilza wykonana jest z twardej bibuły filtracyjnej lub
masy papierowej. Ługowanie w aparacie Soxhleta prowadzi się
w ten sposób, że rozpuszczalnik w kolbie C ogrzewa się do
wrzenia. Jego opary przez ramię E części środkowej wędrują do
góry i są skraplane w chłodnicy zwrotnej D. Już jako ciecz,
rozpuszczalnik z chłodnicy opada do gilzy A z mieszaniną,
wypłukując z niej ekstrahowany związek. W ten sposób
powstaje więc ekstrakt. Kiedy poziom ekstraktu zrówna się
z wysokością syfonu F, opada on do kolby C. W kolbie
rozpuszczalnik z ekstraktu ulega odparowaniu, w postaci pary
wędruje ramieniem E do chłodnicy D, skrapla się i opadając do
gilzy z mieszaniną tworzy następną partię ekstraktu, który znów
- po osiągnięciu odpowiedniej wysokości - spływa do kolby.
Dzięki tym kolejnym, powtarzającym się wielokrotnie, etapom
ekstrakcji prowadzonej w aparacie Soxhleta, stężenie
ekstrahowanej substancji w ekstrakcie jest coraz wyższe.
Pewną niedogodnością stosowania aparatu Soxhleta jest jednak
długotrwałe ogrzewanie ekstraktu, które może spowodować
rozkład substancji o niskiej trwałości termicznej. Fakt ten
należy zawsze brać pod uwagę, planując ekstrakcję w aparacie
Soxhleta, zwłaszcza w przypadku, gdy ekstrakt zawiera
związek (lub związki), na których nam zależy.
Rys. 2. Aparat Soxhleta (opis
elementów znajduje się w tekście).

Ćwiczenie 3: Ekstrakcja
38
2.1. Wykonanie ćwiczenia
Uwaga: Ćwiczenie należy wykonywać w rękawicach ochronnych.
Odczynniki: Szkło laboratoryjne, aparatura, materiały:
1. Roztwór kwasu benzoesowego w toluenie 1.Rozdzielacz gruszkowy o pojemności 100 cm
3
2. Roztwór wodny NaOH, 5% wag. 2. Kolbki stożkowe o poj. 50 cm
3
3. Woda destylowana
3. Lejek/sączek karbowany
4. Kwas solny stężony 4. Zestaw do krystalizacji z wody
5. Siarczan magnezu bezwodny 5. Zestaw do destylacji prostej
6. Zestaw do sączenia pod zmniejszonym
ciśnieniem
7. Czasza grzejna z regulatorem mocy
8. Statywy, łączniki, łapy, kółko na rozdzielacz
Ć
wiczenie należy przeprowadzić zgodnie z następującym opisem:
1. Na statywie w odpowiednim kółku umieścić rozdzielacz jak pokazano na Rys. 1.
2. Po sprawdzeniu, że kran rozdzielacza jest nasmarowany i zabezpieczony przed wypadaniem
gumką-recepturką, nalać do rozdzielacza przez lejek 25 cm
3
roztworu kwasu benzoesowego
w toluenie (kran rozdzielacza powinien być zamknięty!).
3. Do roztworu toluenowego znajdującego się w rozdzielaczu dodać 25 cm
3
wodnego roztworu
NaOH.
4. Po zamknięciu rozdzielacza korkiem (korek powinien być nasmarowany smarem) wytrząsać jego
zawartość – najpierw powoli i często odpowietrzając, a po odpowietrzeniu energicznie.
5. Po wymieszaniu zawartości, rozdzielacz umieścić na statywie i pozostawić do rozdzielenia faz.
6. Gdy fazy rozwarstwią się, warstwę dolną (wodną) zlać do zlewki. Warstwę górną (toluenową)
pozostawić w rozdzielaczu i zakryć korkiem.
7. Warstwę wodną pod działającym wyciągiem zakwasić stężonym kwasem solnym. Kwas należy
dodawać pipetką aż do uzyskania odczynu kwaśnego roztworu (test odczynu wykonać za pomocą
papierka uniwersalnego). Wytrącony osad kwasu benzoesowego przesączyć pod zmniejszonym
ciśnieniem i przekrystalizować z wody (zanotować objętość wody użytej do krystalizacji).
Pozostawić do wysuszenia do następnych zajęć. Wysuszony kwas benzoesowy zważyć i zmierzyć
jego temperaturę topnienia.
8. Warstwę toluenową pozostałą w rozdzielaczu przemyć dwukrotnie wodą destylowaną, używając
za każdym razem po 25 cm
3
wody. Takie przemywanie przeprowadza się w rozdzielaczu – jest to
ekstrakcja warstwy toluenowej za pomocą wody. Po przemyciu, warstwę toluenową zlać do
kolbki stożkowej, dodać do niej środka suszącego (bezwodnego siarczanu magnezu), zakryć
kolbkę korkiem i odstawić na 10-15 minut. Po tym czasie pod działającym wyciągiem odsączyć
ś
rodek suszący przez sączek karbowany. Przesączony toluen przedestylować metodą destylacji
prostej pod ciśnieniem atmosferycznym. Zapisać temperaturę wrzenia toluenu.
2.2. Opracowanie wyników
1. Na podstawie ilości wyodrębnionego kwasu benzoesowego obliczyć jego stężenie molowe
w roztworze użytym do ekstrakcji.
2. Porównując liczbę moli kwasu benzoesowego znajdującego się w roztworze użytym do ekstrakcji
oraz liczbę moli wodorotlenku sodowego w ekstrahencie (gęstość roztworu wodnego NaOH
o stężeniu 5 % wag. wynosi 1,05 g/cm
3
) stwierdzić, jak duży nadmiar zasady został użyty
do ekstrakcji. Nadmiar należy wyrazić jako:
a)
różnicę ilości NaOH użytego do ekstrakcji i ilości koniecznej do zobojętnienia kwasu
benzoesowego znajdującego się w roztworze użytym do ekstrakcji, podaną zarówno
w molach, jak i mililitrach 5% roztworu wodnego NaOH;
b)
proporcję liczby moli NaOH użytego do ekstrakcji i liczby moli NaOH koniecznej do
zobojętnienia kwasu benzoesowego znajdującego się w roztworze użytym do ekstrakcji.

Ćwiczenie 3: Ekstrakcja
39
Taka proporcja pozwoli zorientować się, ilokrotny nadmiar NaOH (w stosunku do
stechiometrycznego) był użyty do ekstrakcji.
3. Problemy do rozwiązania
1. Proszę zaproponować dwa warianty ekstrakcji, która pozwoli rozdzielić kwas 1-naftoesowy
od aniliny. Oba związki rozpuszczone są w eterze dietylowym.
2. 100 cm
3
roztworu wodnego zawiera po 1 gramie związków A i B. Współczynniki podziału między
chlorek metylenu (CH
2
Cl
2
) i wodę w temperaturze 20
0
C wynoszą: dla związku A: 0,1, dla związku B:
5. Proszę policzyć, ile każdego z tych związków (w gramach i w procentach w stosunku do
wyjściowej ilości) uda się wyodrębnić z roztworu przez ekstrakcję za pomocą 100 cm
3
CH
2
Cl
2
, jeśli
ekstrakcję przeprowadzi się
a) jeden raz używając całej ilości CH
2
Cl
2
;
b) dwa razy używając za każdym razem po 50 cm
3
CH
2
Cl
2
;
c) cztery razy używając za każdym razem po 25 cm
3
CH
2
Cl
2
.
Proszę skomentować otrzymane wyniki porównując, w jaki sposób liczba przeprowadzonych
ekstrakcji mniejszymi porcjami rozpuszczalnika wpływa na jej skuteczność w przypadku związku
o
małej
wartości
współczynnika
podziału
oraz
związku
o
dużej
wartości
współczynnika podziału między CH
2
Cl
2
i wodę.
4. Sprawozdanie
Sprawozdanie z ćwiczenia powinno zawierać:
1. Cele ćwiczenia.
2. Opis wykonanych czynności.
3. Wyniki i ich opracowanie. Opracowanie wyników powinno uwzględniać zarówno wskazówki
zawarte w opisie wykonania ćwiczenia (punkt 2.1 instrukcji), jak i polecenia z punktu 2.2
instrukcji.
4. Rozwiązania problemów z punktu 3 instrukcji.
5. Wnioski.
Literatura
[1] A. Vogel, „Preparatyka organiczna, WNT, Warszawa 1984, str. 104-112
[2]
„Laboratorium chemii organicznej”, red. P. Kowalski, WNT, Warszawa 2004, str. 102-108
Ćwiczenie 4: Reakcje podstawienia elektrofilowego w związkach aromatycznych
40
Ć
wiczenie 4:
REAKCJE ZWI
Ą
ZKÓW ORGANICZNYCH:
PODSTAWIENIE ELEKTROFILOWE
W ZWI
Ą
ZKACH AROMATYCZNYCH
6
Sulfonowanie p-ksylenu
Cele ćwiczenia: 1) Utrwalenie wiedzy dotyczącej reakcji i reagentów organicznych, a także budowy
i właściwości chemicznych związków aromatycznych.
2) Przeprowadzenie sulfonowania p-ksylenu jako przykładowej reakcji podstawienia
elektrofilowego w pierścieniu aromatycznym.
1. Reakcje i reagenty organiczne
Jak wiadomo, chemia organiczna jest nauką zajmującą się badaniami związków węgla. Czasami
można spotkać się również z określeniem, że chemia organiczna to chemia związków kowalencyjnych.
Atomy węgla w cząsteczkach związków organicznych połączone są bowiem z innymi atomami
wiązaniami kowalencyjnymi. Wiązania te mogą być pojedyncze (nasycone) lub wielokrotne
(nienasycone), bardziej lub mniej spolaryzowane. Krotność, a także polaryzacja wiązań kowalencyjnych
decydują o tym, jakim reakcjom ulegają cząsteczki związków organicznych i z jakimi reagentami reagują
najłatwiej.
Najbardziej charakterystycznymi reakcjami nienasyconych związków organicznych są reakcje
addycji, czyli przyłączania atomów lub grup atomów do wiązania wielokrotnego. W trakcie addycji
następuje rozpad wiązania wielokrotnego, a jej produktem jest związek, w którym występuje wiązanie
o niższej krotności. Tak więc, jeśli substrat zawierał wiązanie potrójne, to w produkcie (najczęściej
przejściowym) jest w jego miejscu wiązanie podwójne. W wyniku addycji do wiązania podwójnego
natomiast powstaje związek o wiązaniu pojedynczym. W chemii organicznej reakcje addycji oznacza się
symbolem A. Ogólny przebieg takich reakcji pokazano schematycznie na Rys. 1A na przykładzie
wiązania podwójnego węgiel-węgiel. Należy jednak pamiętać, że jest to reakcja typowa także dla
połączeń atomów węgla z innymi atomami, np. grupy karbonylowej (>C=O) lub nitrylowej (
−
C
≡
N).
Najbardziej charakterystycznymi reakcjami nasyconych związków organicznych są reakcje
substytucji, czyli podstawienia atomów lub grup atomów w cząsteczce. Takie reakcje oznacza się
symbolem S. Związki nasycone mogą także ulegać reakcjom eliminacji, oznaczanym symbolem E.
Eliminacja jest przeciwieństwem addycji: w jej wyniku ze związków nasyconych powstają nienasycone.
Substytucję i eliminację pokazano schematycznie na Rys. 1B i 1C. Osobną grupę stanowią związki
aromatyczne, dla których – mimo ich niensyconego charakteru – najbardziej typowe są reakcje
substytucji. Przyczyną tego faktu jest ogromna trwałość pierścienia aromatycznego.
6
Opracowanie: dr hab. Magdalena Hasik
C
Z
Y +
C
Y
+
Z
C
C
Z
Y
+ ZY
C
C
+ ZY
C
C
Z
Y
C
C
Rys. 1. Reakcje organiczne: A) addycja, B) substytucja, C) eliminacja.
A)
B)
C)
Ćwiczenie 4: Reakcje podstawienia elektrofilowego w związkach aromatycznych
41
Addycja, substytucja i eliminacja to główne typy reakcji organicznych. Każda z nich może przebiegać
według mechanizmu wolnorodnikowego lub polarnego. Reakcje wolnorodnikowe zapoczątkowywane są
przez wolne rodniki, tj. reagenty zawierające niesparowane elektrony. Do reakcji polarnych natomiast
zalicza się procesy elektrofilowe i nukleofilowe. Te pierwsze inicjowane są przez elektrofile, czyli
reagenty charakteryzujące się niedoborem elektronów (kationy lub kwasy Lewisa). Te drugie
wywoływane są przez nukleofile, tj. reagenty zawierające nadmiar elektronów (aniony lub zasady
Lewisa). Mechanizm reakcji oznacza się za pomocą dolnego indeksu: R – reakcja wolnorodnikowa, E-
reakcja elektrofilowa, N- reakcja nukleofilowa. I tak, oznaczenie S
R
odnosi się do reakcji substytucji
wolnorodnikowej, A
N
- addycji nukleofilowej, A
E
- addycji elektrofilowej, itd.
O mechanizmie reakcji często decyduje polaryzacja wiązań kowalencyjnych występujących
w cząsteczkach związków organicznych. Warto jednak pamiętać, że istotny wpływ na mechanizm ma
również środowisko reakcji.
2. Związki aromatyczne i ich znaczenie
Związki aromatyczne to klasa związków organicznych, wykazujących szereg wspólnych cech budowy
oraz wspólne właściwości chemiczne. Wszystkie związki aromatyczne mają budowę pierścieniową, czyli
cykliczną. Ich cząsteczki są płaskie, ale zawierają rozbudowany w przestrzeni układ zdelokalizowanych
wiązań
π
. Dzięki tym wiązaniom pierścienie aromatyczne są bardzo trwałe i trudno ulegają rozerwaniu.
Dlatego najbardziej charakterystycznymi reakcjami tej klasy związków są nie naruszające układu
cyklicznego reakcje podstawienia atomu lub grupy atomów związanych z pierścieniem aromatycznym.
Istnieją karbocykliczne (nazywane inaczej homocyklicznymi) i heterocykliczne związki aromatyczne.
Szkielet pierścienia tych pierwszych tworzą wyłącznie atomy węgla. W pierścieniach związków
heterocyklicznych – oprócz atomów węgla
– znajdują się atomy innych pierwiastków, nazywane
heteroatomami. Mogą nimi być atomy tlenu, azotu, siarki. Najważniejszymi przedstawicielami
karbocyklicznych związków aromatycznych są benzen i jego analogi. Heterocykliczne związki
aromatyczne to na przykład: furan, pirol, tiofen, pirydyna. Wzory strukturalne wybranych związków
aromatycznych przedstawiono na Rys. 2.
Rys. 2. Wzory strukturalne niektórych związków aromatycznych.
Każdy, nawet najbardziej podstawowy, kurs chemii organicznej zawiera informacje na temat
związków aromatycznych. Rozdziały poświęcone tym związkom znajdują się we wszystkich
podręcznikach chemii organicznej. Nie wydaje się więc celowe powtarzanie w obecnym opracowaniu
wiadomości, które każdy student może znaleźć w swoich notatkach z wykładów lub w dowolnym
podręczniku chemii organicznej. Warto natomiast zastanowić się, dlaczego związki aromatyczne są tak
ważne, że nie sposób ich pominąć w żadnym kursie chemii organicznej. Odpowiedź jest prosta: układy
aromatyczne to bardzo rozpowszechnione elementy budowy związków organicznych. Wchodzą
w skład cząsteczek substancji o dużym znaczeniu praktycznym, takich jak: leki, środki ochrony roślin,
barwniki, substancje zapachowe, przyprawy i konserwanty używane w przemyśle spożywczym, polimery.
W Tabeli 1 podano przykłady znanych, szeroko stosowanych związków zawierających
w cząsteczkach pierścienie benzenowe.
N
N
N
Benzen Naftalen Antracen Pirydyna Pirymidyna
O
S
Furan Tiofen Pirol
N
H
Ćwiczenie 4: Reakcje podstawienia elektrofilowego w związkach aromatycznych
42
Tabela 1. Wybrane związki zawierające w cząsteczkach pierścienie benzenowe i ich zastosowania.
Wzór strukturalny związku
Nazwa
Zastosowanie
O
H
N
S
H
COOH
CH
3
CH
3
H
HN
C
O
CH
2
Penicylina G
(penicylina benzylowa)
Antybiotyk przeciwbakteryjny
odkryty
przez
Aleksandra
Fleminga
w
1928
roku
i stosowany w lecznictwie od
początku lat 1940-tych do
chwili obecnej.
COOH
O
C
O
CH
3
Kwas acetylosalicylowy
(aspiryna, polopiryna)
Lek o działaniu przeciwbó-
lowym, przeciwgorączkowym
i przeciwzapalnym. Hamuje
także tworzenie się skrzepów
krwi.
N
H O
C
CH
3
H
O
N-(4-hydroksyfenylo)acetamid
(paracetamol)
Lek o działaniu przeciwbólo-
wym i przeciwgorączkowym.
O
H
N
N
O
H
Indygo
Niebieski barwnik używany do
barwienia tkanin, m.in. tkaniny
dżinsowej.
CHO
H O
CH
3
O
4-hydroksy-3-metoksybenzaldehyd
(wanilina)
Jeden ze składników aroma-
tycznych wanilii. Syntetyczną
wanilinę
stosuje
się
jako
aromat w przemyśle spożyw-
czym.
CH C
O
OCH
3
N
H
C
O
CH
CH
2
HOOC
H
2
N
Aspartam (nutrasweet)
Substancja słodząca o symbolu
E951 dodawana do soków
owocowych
oraz
napojów
gazowanych typu „light”.
COOH
Kwas benzoesowy
Zarówno sam kwas, oznaczany
symbolem E210, jak i jego
sole: sodowa (E211), potasowa
(E212) i wapniowa (E213) to
konserwanty
dodawane
do
majonezów, przetworów wa-
rzywnych, margaryn i napojów
gazowanych.
CH CH
2
n
Polistyren
Polimer. W postaci spienionej
(styropian)
stosowany
jako
materiał
opakowaniowy,
a
także jako materiał izolacyjny
w budownictwie.
C
O
OCH
2
CH
2
O
C
O
n
Poli(tereftalan etylenu)
Polimer używany masowo do
produkcji butelek na napoje
(butelki PET) oraz włókien
poliestrowych,
stosowanych
np. do wytwarzania tkaniny
polar.
Ćwiczenie 4: Reakcje podstawienia elektrofilowego w związkach aromatycznych
43
3. Podstawienie elektrofilowe w związkach aromatycznych
Ze
względu
na
swoją
budowę
elektronową
związki
aromatyczne
są
nukleofilami
i łatwo reagują z czynnikami charakteryzującymi się niedoborem elektronów, tj. elektrofilami.
Najbardziej charakterystyczną reakcją związków aromatycznych jest reakcja podstawienia
elektrofilowego w pierścieniu aromatycznym, oznaczana w skrócie symbolem S
E
. Ogólny mechanizm tej
reakcji na przykładzie benzenu przedstawia schematycznie Rys. 3. W pierwszym etapie (Rys. 3a) reagent
elektrofilowy (Y
+
) atakuje pierścień aromatyczny, w wyniku czego tworzy się karbokation
o zdelokalizowanym ładunku (trzeba umieć narysować struktury rezonansowe tego karbokationu!).
Następnie, wskutek reakcji karbokationu z nukleofilem (Z
-
) odtwarza się układ aromatyczny i powstaje
związek zawierający atom lub grupę atomów Y (Rys. 3b). Sumarycznie, każda reakcja S
E
prowadzi do
podstawienia atomu (lub atomów) wodoru w pierścieniu aromatycznym innym atomem lub grupami
atomów, co pokazano na Rys. 3c.
YZ
HZ
Y
+
+
Podstawniki znajdujące się w pierścieniu aromatycznym wywierają wpływ na przebieg reakcji S
E
.
Od nich też zależy miejsce wprowadzenia następnego podstawnika do pierścienia.
Reakcjami podstawienia elektrofilowego związków aromatycznych są: sulfonowanie, nitrowanie,
chlorowanie, bromowanie, alkilowanie, acylowanie. Szczegółowe mechanizmy tych reakcji można
znaleźć w podręcznikach lub w notatkach z wykładów.
4. Opis ćwiczenia
W ćwiczeniu prowadzi się reakcję sulfonowania p-ksylenu, przebiegającą zgodnie z następującym
równaniem:
CH
3
H
2
SO
4
O
H
2
H
3
C
CH
3
H
3
C
SO
3
H
+
+
Sulfonowany p-ksylen w temperaturze pokojowej jest ciałem stałym i po oziębieniu mieszaniny
reakcyjnej wytrąca się z niej w postaci osadu, który należy oczyścić przez krystalizację z wody.
Y
Y
H
+
+
+
Y
H
Z
HZ
Y
+
+
-
+
a)
b)
c)
Rys. 3. Reakcja podstawienia elektrofilowego w pierścieniu aromatycznym:
a) pierwszy etap, b) drugi etap, c) równanie sumaryczne.
∆
T

Ćwiczenie 4: Reakcje podstawienia elektrofilowego w związkach aromatycznych
44
4.1. Wykonanie ćwiczenia
Uwaga: Ćwiczenie należy wykonywać w rękawicach ochronnych.
Odczynniki: Szkło laboratoryjne, aparatura, materiały:
1. p-Ksylen 1. Kolba okrągłodenna ze szlifem o pojemności 50 cm
3
2. Kwas siarkowy stężony 2. Chłodnica zwrotna
3. Woda destylowana
3. Zlewki o pojemności 50 i 250 cm
3
4. Zestaw do krystalizacji z wody
5.
Zestaw do sączenia pod zmniejszonym
ciśnieniem
6. Pipety z nasadkami do zasysania cieczy
7. Czasza grzejna z regulatorem mocy
8. Statyw, łącznik, łapa
Ć
wiczenie należy przeprowadzić zgodnie z następującym opisem:
1. Do kolby okrągłodennej wlać pipetą 3 cm
3
p-ksylenu. Następnie (inną pipetą) dodać do kolby
5 cm
3
stężonego kwasu siarkowego. Obie czynności należy przeprowadzić pod sprawnie
działającym wyciągiem.
2. Kolbę z zawartością umieścić w łapie na statywie. Zamontować chłodnicę zwrotną. Tak
przygotowany układ reakcyjny umieścić w zlewce o pojemności 250 cm
3
, napełnionej wodą
i wstawionej do czaszy grzejnej. Zlewka z wodą pełni rolę łaźni, zapobiegającej przegrzaniu
mieszaniny reakcyjnej.
3. Odkręcić doprowadzenie wody do chłodnicy i rozpocząć ogrzewanie układu reakcyjnego.
Ogrzewanie powinno być łagodne tak, żeby temperatura wody w zlewce stanowiącej łaźnię
wynosiła 70 – 80
0
C (kontrola temperatury nie jest konieczna). W trakcie ogrzewania należy co
pewien czas ostrożnie mieszać zawartość kolby ruchem wirowym.
4. Obserwować zmiany mieszaniny reakcyjnej: po pewnym czasie (30-45 minut) z powierzchni
kwasu siarkowego znika warstwa p-ksylenu, a zawartość kolby staje się jednorodną, jasnożółtą
mieszaniną. Wtedy reakcja jest zakończona i należy wyłączyć ogrzewanie.
5. Po ochłodzeniu do temperatury pokojowej, zawartość kolby wylać do zlewki o pojemności
50 cm
3
, a następnie dodać do niej ostrożnie, mieszając bagietką, 3 cm
3
wody destylowanej
i oziębić w naczyniu z zimną wodą. Czynności te należy wykonać pod działającym wyciągiem.
6. Wytrącony krystaliczny osad sulfonowanego p-ksylenu odsączyć pod zmniejszonym ciśnieniem
na lejku ze spiekiem szklanym (lejka nie należy wykładać bibułą filtracyjną!) i oczyścić przez
krystalizację z wody (ok. 3 cm
3
) i pozostawić do wysuszenia w eksykatorze.
7. Po wysuszeniu, produkt sulfonowania p-ksylenu należy zważyć i zmierzyć jego temperaturę
topnienia.
5. Opracowanie wyników i problemy do rozwiązania
1. Obliczyć wydajność przeprowadzonej syntezy, wyrażając ją w procentach wydajności teoretycznej.
Wydajność teoretyczna to ilość produktu, jaką uzyskałoby się, gdyby reakcja przebiegła do końca
zgodnie z jej równaniem. W obliczeniach wydajności teoretycznej należy uwzględniać ilości
reagentów używane w syntezie. Dane potrzebne do obliczeń: gęstość p-ksylenu wynosi 0,86 g/cm
3
,
jego masa molowa równa jest 106,16 g/mol, masa molowa sulfonowanego p-ksylenu wynosi 186,16
g/mol.
2. Napisać mechanizm reakcji sulfonowania p-ksylenu. Wyjaśnić, dlaczego produktem tej reakcji jest
związek o budowie pokazanej w równaniu z punktu 4 instrukcji. Proszę też nazwać sulfonowany
p-ksylen zgodnie z zasadami nomenklatury systematycznej.
3. Napisać reakcję sulfonowania toluenu. Który związek ulega łatwiej tej reakcji: p-ksylen czy toluen?
Proszę uzasadnić odpowiedź.

Ćwiczenie 4: Reakcje podstawienia elektrofilowego w związkach aromatycznych
45
6. Sprawozdanie
Sprawozdanie z ćwiczenia powinno zawierać:
6. Cele ćwiczenia.
7. Opis wykonanych czynności.
8. Wyniki i ich opracowanie zgodne z punktem 4.2 instrukcji.
9. Wnioski dotyczące przeprowadzonych eksperymentów.
Literatura
Przygotowując się do ćwiczenia i opracowując jego wyniki można korzystać z notatek z wykładów
oraz z dowolnych podręczników chemii organicznej, w których opisano budowę i właściwości chemiczne
związków aromatycznych. Spośród podręczników specjalnie zalecane są dwa podane niżej.
[1] R. T. Morrison, R. T. Boyd, „Chemia organiczna”, PWN, Warszawa 1985, str. 380-418.
[2] J. McMurry, „Chemia organiczna”, PWN, Warszawa 2006, str. 499-553.
Ćwiczenie 5: Estryfikacja
46
REAKCJE ZWI
Ą
ZKÓW ORGANICZNYCH: ESTRYFIKACJA
7
Otrzymywanie octanu n-butylu lub octanu izoamylu
8
Cele ćwiczenia:1) Utrwalenie wiedzy dotyczącej kwasów karboksylowych i ich pochodnych .
2) Przeprowadzenie syntezy octanu n-butylu, w której wykorzystuje się poznane wcześniej
techniki laboratoryjne
1. Budowa, znaczenie, właściwości fizyczne i chemiczne estrów
Estry to pochodne kwasów organicznych (karboksylowych) lub nieorganicznych, w których
cząsteczkach zamiast atomów wodoru grup hydroksylowych znajdują się grupy alkilowe lub arylowe,
najczęściej pochodzące z alkoholi. Kwasy wielowodorotlenowe mogą tworzyć mono-, di-, triestry, itd.,
podczas gdy w przypadku kwasów jednowodorotlenowych możliwe jest wyłącznie powstawanie
monoestrów. Ogólne wzory strukturalne estrów kwasów karboksylowych oraz wybranych kwasów
nieorganicznych pokazano na Rys. 1.
Estry są związkami szeroko rozpowszechnionymi w przyrodzie. Tłuszcze, czyli źródło energii oraz
materiał zapasowy w organizmach zwierzęcych i roślinnych, są estrami wyższych kwasów
karboksylowych (tj. kwasów o długich łańcuchach węglowych) i gliceryny (propan-1,2,3-triolu). Estry
różnych kwasów karboksylowych oraz alkoholi zawierających długie łańcuchy węglowe to woski,
tworzące na powierzchniach owoców, liści i futer zwierząt warstwy ochronne, zapobiegające nadmiernej
utracie wody lub nadmiernemu zwilżaniu wodą. Fosfolipidy, podstawowy składnik budulcowy błon
komórkowych, to estry kwasu fosforowego i różnych alkoholi. Wiele estrów kwasów karboksylowych ma
charakterystyczny, przyjemny zapach. Wraz z innymi związkami, wchodzą one w skład mieszanin
nadających charakterystyczną
woń niektórym owocom i kwiatom. Nazwy oraz wzory strukturalne takich
estrów zestawiono w Tabeli 1.
Estry kwasów karboksylowych mają także duże znaczenie techniczne. Stosowane są jako substancje
zapachowe w perfumach i aromatach spożywczych. Używa się ich także jako rozpuszczalników farb
i lakierów. Rozpuszczalnik nitro na przykład to mieszanina octanów propylu, butylu i pentylu. Ważnymi
estrami kwasów karboksylowych są także kwas acetylosalicylowy i poli(tereftalan etylenu). Pierwszy
z tych związków to aspiryna (polopiryna), lek o działaniu przeciwgorączkowym, przeciwbólowym
i przeciwzakrzepowym, stosowany już od ponad 100 lat. Drugi związek to polimer (poliester). Ogólnie
7
Opracowanie: dr hab. Magdalena Hasik
8
O tym, jaka synteza zostanie przeprowadzona, decyduje prowadzący ćwiczenie
Rys. 1. Wzory ogólne estrów: a) kwasu karboksylowego, b) kwasu azotowego(V), c) kwasu siarkowego(VI),
d) kwasu fosforowego(V). R, R”, R’’’ oznaczają grupę alkilową lub arylową (aromatyczną).
W cząsteczkach estrów grupy te mogą być różne lub jednakowe.
R'O P OH
OH
O
R'O P OH
OR"
O
R'O P OR"'
OR"
O
d)
monoester
diester
triester
a)
b)
c)
R' C
O
OR"
R'O S OH
O
O
R'O S OR"
O
O
R'O N
O
O
monoester
diester
Ćwiczenie 5: Estryfikacja
47
przyjętym skrótem jego nazwy jest PET. Z PET wytwarza się butelki na napoje, a w postaci włókien
stosuje się go do produkcji tkanin (elana, polar). Wzory strukturalne kwasu acetylosalicylowego oraz
poli(tereftalanu etylenu) przedstawiono w instrukcji do ćwiczenia laboratoryjnego poświęconego
reakcjom związków aromatycznych (ćwiczenie 4).
Tabela 1. Nazwy i wzory estrów kwasów karboksylowych wchodzących w skład mieszanin
związków odpowiedzialnych za zapach wybranych owoców.
Nazwa estru
Wzór strukturalny
Zapach
Maślan butylu
CH
3
(CH
2
)
2
C
O
O(CH
2
)
3
CH
3
ananasów
Mrówczan izobutylu
H C
O
OCH
2
CH(CH
3
)
2
malin
Octan amylu
CH
3
C
O
O(CH
2
)
4
CH
3
jabłek, bananów
Octan izoamylu
CH
3
C
O
O(CH
2
)
2
CH(CH
3
)
2
gruszek, bananów
Octan oktylu
CH
3
C
O
O(CH
2
)
7
CH
3
pomarańczy
Estry kwasów karboksylowych o krótkich i średnich łańcuchach węglowych są cieczami,
charakteryzującymi się niższymi temperaturami wrzenia i mniejszą rozpuszczalnością w wodzie niż
kwasy karboksylowe o porównywalnej masie molowej. Można to stwierdzić na przykładzie kwasu
butanowego i dwóch estrów, które są jego izomerami: octanu etylu i propanianu metylu (Tabela 2).
Różnice te tłumaczy się różnicami w zdolności do tworzenia wiązań wodorowych (Rys. 2).
W cząsteczkach kwasów karboksylowych znajdują się zarówno atomy tlenu, jak i wodoru. Cząsteczki te
mogą więc łączyć się wiązaniami wodorowymi między sobą, co decyduje o wysokich temperaturach
wrzenia kwasów. Z cząsteczkami wody natomiast kwasy karboksylowe tworzą wiązania wodorowe
dwóch rodzajów: takie, w których są donorami atomów wodoru i takie, w których pełnią rolę akceptorów
atomów wodoru. Jest to przyczyną zwiększonej rozpuszczalności kwasów w wodzie. W odróżnieniu od
kwasów karboksylowych, cząsteczki estrów nie tworzą wiązań wodorowych między sobą, gdyż nie
zawierają atomów wodoru. Z cząsteczkami wody natomiast estry mogą łączyć się wyłącznie jako
akceptory wodoru. Dlatego estry są związkami bardziej lotnymi i bardziej hydrofobowymi od kwasów
karboksylowych o porównywalnej masie molowej.
Tabela 2. Właściwości fizyczne kwasu butanowego i dwóch estrów, będących jego izomerami (masa
molowa wszystkich związków wynosi 88 g/mol).
Nazwa związku
Temperatura wrzenia
[
0
C]
Rozpuszczalność w wodzie
Kwas butanowy (masłowy)
164
wysoka
Octan etylu
77
ś
rednia
(8,3 g/100 ml H
2
O w 20
0
C)
Propanian metylu
80
niska
(5,0 g/100 ml H
2
O w 20
0
C)

Ćwiczenie 5: Estryfikacja
48
Najważniejszą reakcją chemiczną estrów jest hydroliza. Prowadzona w środowisku kwaśnym jest
procesem odwracalnym i przeciwnym do estryfikacji (por. punkt 2). Hydroliza zachodząca w obecności
zasady to reakcja nieodwracalna, nazywana zmydlaniem estrów. Jednym z jej produktów jest sól kwasu
karboksylowego. W wyniku zmydlania tłuszczów w środowisku NaOH lub KOH powstają odpowiednio:
sole sodowe lub potasowe wyższych kwasów tłuszczowych, czyli mydła.
Estry ulegają także reakcjom uwodornienia, w wyniku których tworzą się alkohole pierwszorzędowe,
reakcjom z odczynnikami Grignarda, prowadzącym do otrzymania alkoholi trzeciorzędowych oraz
reakcjom z amoniakiem, których produktami są amidy. Dokładniejszy opis tych reakcji znajduje się
w pozycji oznaczonej [1] w wykazie literatury zamieszczonym na końcu instrukcji.
2. Reakcja estryfikacji
Istnieje kilka metod otrzymywania estrów kwasów karboksylowych [1, 2]. Najważniejszymi z nich są
reakcje kwasów karboksylowych, ich bezwodników lub chlorków z alkoholami, nazywane ogólnie
reakcjami alkoholizy.
Bezpośrednią syntezę estru z kwasu karboksylowego i alkoholu nazywa się estryfikacją. Jest to
przykład reakcji kondensacji, tzn. takiej, w wyniku której powstają: związek zawierający w swojej
cząsteczce fragmenty cząsteczek substratów oraz prosty związek nieorganiczny, np. woda. Ogólne
równanie reakcji estryfikacji kwasu karboksylowego przedstawiono na Rys. 3.
R' C
O
OH
R''OH
H
R' C
O
OR''
O
H
2
+
+
+
Do zapoczątkowania estryfikacji konieczna jest obecność w środowisku reakcji mocnego kwasu
protonowego. Jest to związane z mechanizmem reakcji (Rys. 4), której pierwszy etap polega na
protonowaniu atomu tlenu grupy karbonylowej. Do utworzonego w ten sposób karbokationu przyłącza się
następnie cząsteczka alkoholu, który – ze względu na obecność niewiążących par elektronowych na
atomie tlenu – jest nukleofilem. W końcowym etapie reakcji od przejściowego kationu odłącza się jon
hydroniowy (H
3
O
+
) i powstaje cząsteczka estru. Estryfikacja przebiega więc według mechanizmu addycji-
eliminacji. Warto zwrócić uwagę, że atom tlenu w cząsteczce estru pochodzi z cząsteczki alkoholu.
R'
C
O
OH
H
H
R''
O
H
R''
O
R'
C
O
OH
H
H
O
R' C
O
OH
H
H
3
O
R' C
O
OH
H
R''
R' C
O
O
R''
+
+
+
+
+
-
+
R C
O
O H
R
C
O
O
H
R C
O
O H
H
O
O H
H
H
R' C
O
O R''
H
O
O H
H
H
Rys. 2. Wiązania wodorowe tworzone przez cząsteczki : a) kwasu karboksylowego,
b) kwasu karboksylowego z cząsteczkami wody, c) estru z cząsteczkami wody.
a)
b)
c)
Rys. 3. Ogólne równanie reakcji estryfikacji kwasu karboksylowego
Rys. 4. Mechanizm reakcji estryfikacji kwasu karboksylowego
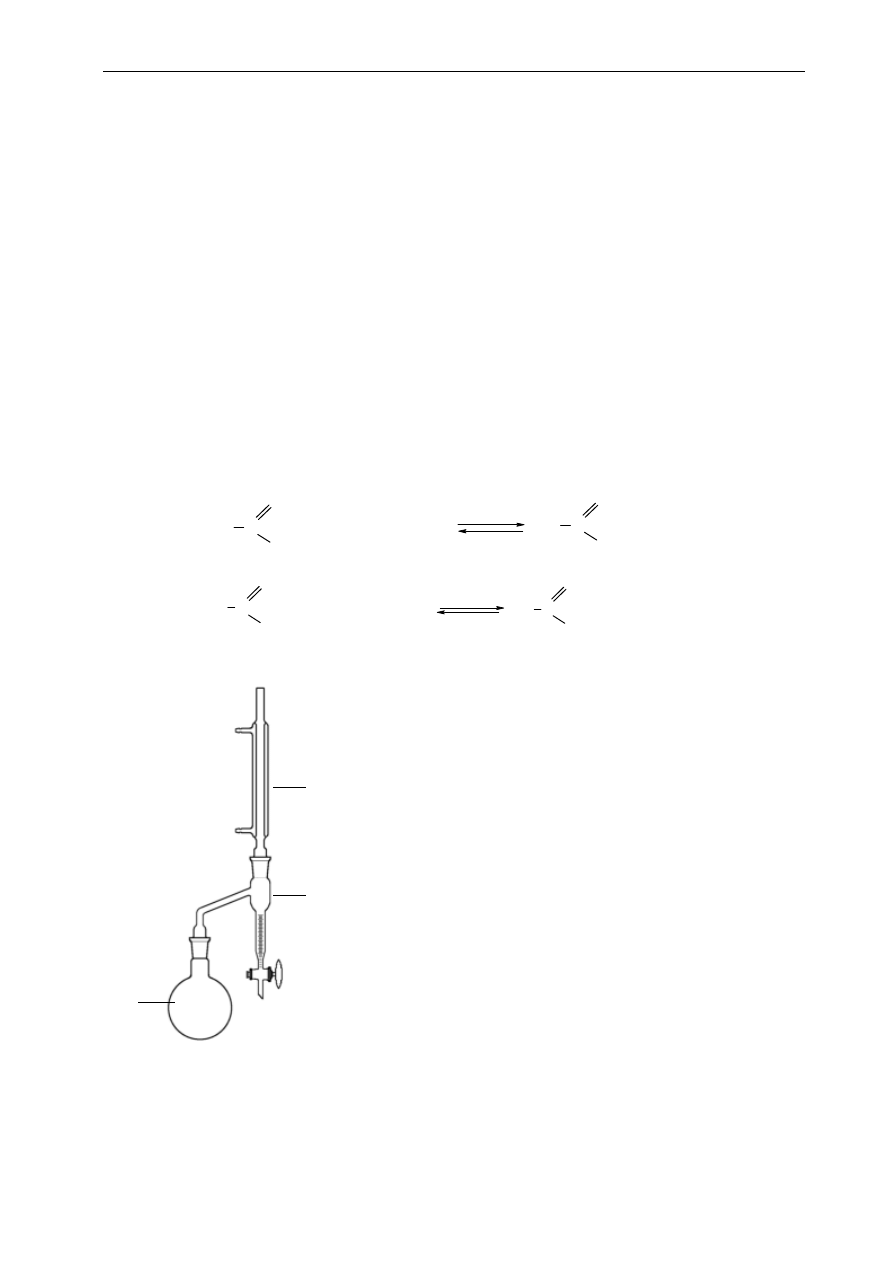
Ćwiczenie 5: Estryfikacja
49
Estryfikacja jest procesem odwracalnym. Oznacza to, że zawsze dochodzi do stanu równowagi,
w którym proporcje produktów i substratów obecnych w układzie są stałe, zależne wyłącznie od
temperatury i określone przez stałą równowagi reakcji. W praktyce istotne jest, żeby równowagę
estryfikacji przesunąć w prawo, to znaczy w kierunku tworzenia estru. Można to osiągnąć, stosując
nadmiar jednego z substratów (zwykle tańszego) lub usuwając ze środowiska reakcji jeden z jej
produktów (najczęściej wodę). Wodę można odprowadzać w postaci azeotropu. Azeotrop jest ciekłą
mieszaniną dwu lub większej liczby składników wrzącą w stałej temperaturze, czyli tak, jak substancje
czyste.
Związków wchodzących w skład azeotropu nie można więc rozdzielić
metodą destylacji. W celu
usunięcia wody powstającej w czasie estryfikacji do środowiska reakcji dodaje się związek tworzący
z wodą mieszaninę azeotropową, czyli tak zwany składnik azeotropujący. Może nim być: chloroform,
czterochlorek węgla, toluen. Mieszanina azeotropowa składnika azeotropującego z wodą jest
oddestylowywana z układu w miarę postępu reakcji. Jeżeli jeden z reagentów sam tworzy azeotrop
z wodą, to dodawanie składnika azeotropującego do układu reakcyjnego nie jest konieczne.
3. Opis ćwiczenia
W ćwiczeniu prowadzi się syntezę octanu n-butylu lub octanu izoamylu z kwasu octowego
i odpowiedniego alkoholu (butan-1-olu lub 3-metylobutan-1-olu, czyli alkoholu izoamylowego)
w obecności stężonego kwasu siarkowego jako katalizatora.
Synteza octanu n-butylu przebiega zgodnie
z równaniem reakcji (1), a octanu izoamylu – zgodnie z równaniem (2):
CH
3
C
O
OH
CH
3
(CH
2
)
2
CH
2
OH
H
2
SO
4
CH
3
C
O
O(CH
2
)
3
CH
3
O
H
2
+
+
CH
3
C
O
OH
(CH
3
)
2
CH(CH
2
)
2
OH
H
2
SO
4
CH
3
C
O
O(CH
2
)
2
CH(CH
3
)
2
O
H
2
+
+
.
Zestaw aparatury do syntezy estrów przedstawiono
na
Rys. 5. Składa się on z kolby okrągłodennej ze szlifem (A),
nasadki azeotropowej (B) oraz chłodnicy zwrotnej (C).
Reakcja
estryfikacji
zachodzi
podczas
ogrzewania
substratów znajdujących się w kolbie A. Produkt uboczny,
woda, tworzy z alkoholami azeotrop, którego opary
dochodzą przez ukośne ramię nasadki azeotropowej B do
chłodnicy zwrotnej C. Tam ulegają skropleniu, a skropliny
opadają do pionowego ramienia nasadki azeotropowej
(z kranem), gdzie rozdzielają się na dwie fazy: znajdującą
się na dole wodę i stanowiącą warstwę górną fazę
organiczną. Rozdzielenie składników azeotropu w nasadce
jest możliwe, gdyż skład mieszanin azeotropowych silnie
zależy od temperatury. Górna warstwa częściowo zawraca
do układu reakcyjnego, a dolną można co pewien czas
wylewać
do
przygotowanego
naczynia
(cylindra
miarowego). Reakcję prowadzi się tak długo, aż woda
przestanie opadać z chłodnicy do nasadki. Ilość
wydzielonej
wody
pozwoli
określić
stopień
przereagowania substratów.
Syntezowane estry to ciecze wrzące pod ciśnieniem
atmosferycznym w temperaturze 126
o
C (octan n-butylu)
i 142
o
C (octan izoamylu). Otrzymane po syntezie surowe
produkty, po przemyciu odpowiednimi roztworami,
oczyszcza się metodą destylacji prostej
Rys. 5. Zestaw aparatury
do estryfikacji
B
A
C
(1)
(2)

Ćwiczenie 5: Estryfikacja
50
3.1 Wykonanie syntezy octanu n-butylu
Uwaga: Ćwiczenie należy wykonywać w rękawicach ochronnych.
Odczynniki: Szkło laboratoryjne, aparatura, materiały:
1. Butan-1-ol 1.Kolba okrągłodenna ze szlifem o pojemności 100 cm
3
2. Kwas octowy lodowaty 2. Nasadka azeotropowa
3. Kwas siarkowy stężony
3. Chłodnica zwrotna
3. Wodorowęglan sodu, nasycony r-r wodny
4. Cylinder miarowy o pojemności 25 cm
3
4.
Siarczan magnezu bezwodny
5. Rozdzielacz gruszkowy o pojemności 100 cm
3
5. Woda destylowana
6. Lejek z sączkiem karbowanym
7
. Zestaw do destylacji prostej
8. Czasza grzejna z regulatorem mocy
9. Podnośnik laboratoryjny
10. Statywy, łączniki, łapy, kółko na rozdzielacz
Ć
wiczenie należy przeprowadzić zgodnie z następującym opisem:
1. W kolbie okrągłodennej umieścić kolejno: 20 cm
3
butan-1-olu, 26 cm
3
kwasu octowego lodowatego,
0,5 cm
3
stężonego kwasu siarkowego i 2 sztuki kamyków wrzennych. Wszystkie te czynności należy
przeprowadzić pod działającym wyciągiem, zachowując szczególną ostrożność przy nalewaniu
stężonego kwasu siarkowego. Następnie kolbę zakryć korkiem i przenieść na swoje miejsce pracy.
2. Kolbę z zawartością umieścić w łapie na statywie. Ustawić pod nią na podnośniku czaszę grzejną, po
czym zamontować nasadkę azeotropową i chłodnicę zwrotną w sposób pokazany na Rys. 5.
3. Odkręcić kran doprowadzający zimną wodę do chłodnicy i rozpocząć ogrzewanie mieszaniny
reakcyjnej. Reakcję prowadzić w temperaturze wrzenia do czasu, gdy w nasadce azeotropowej
przestanie zbierać się woda (30 – 45 minut).
4. Po zakończeniu reakcji wyłączyć ogrzewanie, odstawić czaszę grzejną na bok. Zlać wodę z nasadki
azeotropowej do cylindra miarowego i zapisać jej ilość. Zawartość kolby ostudzić do temperatury
pokojowej i przelać przez lejek do rozdzielacza napełnionego 30 cm
3
wody. Dodać warstwę
organiczną z nasadki azeotropowej. Wymieszać zawartość rozdzielacza i pozostawić do rozdzielenia
faz.
5. Warstwę dolną (wodną) wylać z rozdzielacza do zlewki lub kolbki stożkowej i odrzucić. Pozostałą
w rozdzielaczu warstwę górną (organiczną), zawierającą octan n-butylu, przemyć kolejno: 25 cm
3
wody, 15 cm
3
nasyconego roztworu wodorowęglanu sodu i 20 cm
3
wody. Zachować ostrożność
zwłaszcza przy przemywaniu roztworem NaHCO
3
, w trakcie którego wydziela się CO
2
. (Takie
przemywanie wykonuje się w rozdzielaczu – jest to po prostu kolejno przeprowadzana ekstrakcja
warstwy organicznej wodą lub roztworem NaHCO
3
).
6. Przemytą warstwę organiczną wylać z rozdzielacza do kolbki stożkowej o pojemności 25 cm
3
lub 50
cm
3
(w zależności od ilości produktu),
nasypać do niej niewielką ilość środka suszącego
(bezwodnego siarczanu magnezu) i pozostawić zakrytą korkiem na około 15 minut.
7. W czasie, gdy surowy produkt reakcji pozostaje nad środkiem suszącym, przygotować zestaw do
destylacji prostej. Kolbą destylacyjną powinna być kolba o pojemności 25 cm
3
lub 50 cm
3
,
a odbieralnikem przeznaczonym na octan n-butylu – zważona kolbka stożkowa o pojemności
25 cm
3
(pustą kolbkę należy zważyc z korkiem!). Trzeba też przygotować odbieralnik na przedgon,
tj. frakcję substancji o niższej temperaturze wrzenia niż octan n-butylu.
8. Po wysuszeniu, surowy octan n-butylu przesączyć przez lejek z sączkiem karbowanym do kolby
destylacyjnej. Przeprowadzić destylację (pamiętać o dodaniu kamyków wrzennych do kolby
destylacyjnej), zbierając przedgon (zapisać zakres jego temperatury wrzenia) oraz octan n-butylu o
temperaturze wrzenia 125-126
0
C.
9. Po zakończeniu destylacji otrzymany ester zważyć w zakorkowanej kolbie. Obliczyć masę produktu
przeprowadzonej syntezy.
S
Y
N
T
E
Z
A
O
C
T
A
N
U
n
-B
U
T
Y
L
U

Ćwiczenie 5: Estryfikacja
51
3.2. Wykonanie syntezy octanu izoamylu
Uwaga: Ćwiczenie należy wykonywać w rękawicach ochronnych.
Odczynniki: Szkło laboratoryjne, aparatura, materiały:
1. 3-metylobutan-1-ol (alkohol izoamylowy) 1.Kolba okrągłodenna ze szlifem o pojemności 100 cm
3
2. Kwas octowy lodowaty 2. Nasadka azeotropowa
3. Kwas siarkowy stężony
3. Chłodnica zwrotna
4. Wodorowęglan sodu, 5 % r-r wodny
4. Cylinder miarowy o pojemności 25 cm
3
5.
Siarczan magnezu bezwodny
5. Rozdzielacz gruszkowy o pojemności 100 cm
3
6. Chlorek sodu, nasycony r-r wodny
6. Lejek z sączkiem karbowanym
7. Woda destylowana
7
. Zestaw do destylacji prostej
8.Czasza grzejna z regulatorem mocy
9. Podnośnik laboratoryjny
10. Statywy, łączniki, łapy, kółko na rozdzielacz
Ć
wiczenie należy przeprowadzić zgodnie z następującym opisem:
1. W kolbie okrągłodennej umieścić kolejno: 25 cm
3
alkoholu izoamylowego, 15,8 cm
3
lodowatego
kwasu octowego, 2 krople stężonego kwasu siarkowego i 2 sztuki kamyków wrzennych. Wszystkie te
czynności należy przeprowadzić pod działającym wyciągiem, zachowując szczególną ostrożność przy
nalewaniu stężonego kwasu siarkowego. Następnie kolbę zakryć korkiem i przenieść na swoje
miejsce pracy.
2. Kolbę z zawartością umieścić w łapie na statywie. Ustawić pod nią na podnośniku czaszę grzejną, po
czym zamontować nasadkę azeotropową i chłodnicę zwrotną w sposób pokazany na Rys. 5.
3. Odkręcić kran doprowadzający zimną wodę do chłodnicy i rozpocząć ogrzewanie mieszaniny
reakcyjnej. Reakcję prowadzić w temperaturze wrzenia do czasu, gdy w nasadce azeotropowej
przestanie zbierać się woda (około 20 minut).
4. Po zakończeniu reakcji wyłączyć ogrzewanie, odstawić czaszę grzejną na bok. Zlać wodę z nasadki
azeotropowej do cylindra miarowego i zapisać jej ilość. Zawartość kolby ostudzić do temperatury
pokojowej i przelać przez lejek do rozdzielacza. Dodać warstwę organiczną i wodę z nasadki
azeotropowej oraz 15 cm
3
wody destylowanej. Wymieszać zawartość rozdzielacza i pozostawić do
rozdzielenia faz.
5. Warstwę dolną (wodną) wylać z rozdzielacza do zlewki lub kolbki stożkowej i odrzucić. Pozostałą
w rozdzielaczu warstwę górną (organiczną), zawierającą octan izoamylu, przemyć dwukrotnie
porcjami po 15 cm
3
wody, potem 25 cm
3
5 % roztworu wodorowęglanu sodu, a na koniec 25 cm
3
nasyconego roztworu chlorku sodu. Zachować ostrożność zwłaszcza przy przemywaniu roztworem
NaHCO
3
,
w trakcie którego wydziela się CO
2
. (Takie przemywanie wykonuje się w rozdzielaczu – jest to po
prostu kolejno przeprowadzana ekstrakcja warstwy organicznej wodą lub odpowiednimi roztworami).
6. Przemytą warstwę organiczną wylać z rozdzielacza do kolbki stożkowej o pojemności 25 cm
3
lub 50
cm
3
(w zależności od ilości produktu),
nasypać do niej niewielką ilość środka suszącego (bezwodnego
siarczanu magnezu) i pozostawić zakrytą korkiem na około 15 minut.
7. W czasie, gdy surowy produkt reakcji pozostaje nad środkiem suszącym, przygotować zestaw do
destylacji prostej. Kolbą destylacyjną powinna być kolba o pojemności 25 cm
3
lub 50 cm
3
,
a odbieralnikem przeznaczonym na octan izoamylu – zważona kolbka stożkowa o pojemności
25 cm
3
(pustą kolbkę należy zważyć z korkiem!). Trzeba też przygotować odbieralnik na przedgon,
tj. frakcję substancji o niższej temperaturze wrzenia niż octan izoamylu.
8. Po wysuszeniu, surowy octan izoamylu przesączyć przez lejek z sączkiem karbowanym do kolby
destylacyjnej. Przeprowadzić destylację (pamiętać o dodaniu kamyków wrzennych do kolby
destylacyjnej), zbierając przedgon (zapisać zakres jego temperatury wrzenia) oraz octan izoamylu
o temperaturze wrzenia 142
0
C.
9. Po zakończeniu destylacji otrzymany ester zważyć w zakorkowanej kolbie. Obliczyć masę produktu
przeprowadzonej syntezy.
S
Y
N
T
E
Z
A
O
C
T
A
N
U
I
Z
O
A
M
Y
L
U

Ćwiczenie 5: Estryfikacja
52
4. Opracowanie wyników i problemy do rozwiązania
1. Na podstawie masy otrzymanego estru obliczyć wydajność przeprowadzonej reakcji, wyrażając ją
w procentach wydajności teoretycznej. Dane potrzebne do obliczeń: gęstość butan-1-olu wynosi
0,811 g/cm
3
, masa molowa butan-1-olu: 74,12 g/mol, gęstość alkoholu izoamylowego wynosi
0,815 g/cm
3
, masa molowa alkoholu izoamylowego: 88,15 g/mol, gęstość lodowatego kwasu
octowego: 1,05 g/cm
3
, masa molowa kwasu octowego: 60,05 g/mol, masa molowa octanu n-
butylu: 116,16 g/mol, masa molowa octanu izoamylu: 130,19 g/mol. Obliczyć też wydajność
estryfikacji na podstawie ilości wody wydzielonej w trakcie reakcji.
2. Proszę napisać reakcję zasadowej hydrolizy octanu n-butylu lub octanu izoamylu i nazwać jej
produkty.
3. Estry to pochodne kwasów karboksylowych. Podane niżej wzory przedstawiają różne pochodne
kwasu octowego. Proszę napisać, jakie to są pochodne i podać ich nazwy systematyczne.
5. Sprawozdanie
Sprawozdanie z ćwiczenia powinno zawierać:
1. Cele ćwiczenia.
2. Opis wykonanych czynności.
3. Wyniki i ich opracowanie. Opracowanie wyników powinno uwzględniać polecenia z punktu 4
instrukcji.
4. Rozwiązania problemów z punktu 4 instrukcji.
5. Wnioski.
Literatura
[1] J. McMurry, „Chemia organiczna”, PWN, Warszawa 2006, rozdział 21 (pochodne kwasów
karboksylowych).
[2]
R. T. Morrison, R. T. Boyd, „Chemia organiczna”, PWN, Warszawa 1985, rozdział 18.16,
str. 692-694 (synteza estrów z kwasów karboksylowych) i rozdział 20 (pochodne kwasów
karboksylowych).
CH
3
C
O
Cl
CH
3
C
O
NHCH
3
CH
3
C
O
O
CH
3
C
O
a)
b)
c)
d)
CH
3
C
O
NH
2
Wyszukiwarka
Podobne podstrony:
instrukcja laboratoryjna id 216 Nieznany
Instrukcja laboratoryjna PR4G
Copy of Cw3 instrukcja laboratoryjna INF
Instrukcja laboratorium ETP ćw 2 2012
Operacje na rekordach i przetwarzanie plików Instrukcja laboratoryjna
Instrukcja laboratorium z ochrony środowiska, Górnictwo i Geologia AGH, ochrona środowiska
Instrukcja laboratorium 20 21
instrukcja laboratoryjna dla makiety zd537
MATERIALY INFORMATYKA, OPIS CW LAB INFORMATYKA, CD instrukcji laborat. z fizyki
Instrukcja laboratorium ETP ćw 1 2012
instrukcja laboratoryjna id 216 Nieznany
instrukcja laboratoryjna
Bluetooth Instrukcja laboratoryjna Teoria
instrukcja - HYDROLIZA SOLI, Inżynieria środowiska, inż, Semestr II, Chemia ogólna, laboratorium
więcej podobnych podstron