Mutacje
założycielskie
Pewne mutacje genów, często będące przyczyną
poważnych chorób, pozwalają prześledzić
na przestrzeni tysięcy lat migracje
i rozwój ludzkich populacji
Dennis Drayna
34
ŚWIAT NAUKI
LISTOPAD 2005

Dwaj mężczyźni w średnim wieku, miesz-
kający w Stanach Zjednoczonych tysiące
kilometrów od siebie, nigdy w życiu się
nie spotkali. Mają jednak pewną wspólną
cechę: tak wydajnie przyswajają żelazo, że
ta pozorna, zdawałoby się, korzyść może
w rzeczywistości obrócić się przeciwko
nim, grożąc uszkodzeniem wielu narzą-
dów, a nawet śmiercią. Stan ten nazy-
wany jest hemochromatozą dziedziczną
i powoduje go mutacja w specyficznym
genie, przekazana potomstwu przez oboje
rodziców. Jest ona błędem, który dawno
temu pojawił się w sekwencji nukleotydo-
wej DNA u jednego mieszkańca Europy.
Od tamtego czasu mutacja rozprzestrze-
niła się wśród potomków tego człowie-
ka, których tylko w USA żyje obecnie
około 22 mln. Są wśród nich również
dwaj wspomniani mężczyźni, którzy za-
pewne byliby zaskoczeni, słysząc o swoim
„pokrewieństwie”. Wszyscy oni mają co
najmniej jedną kopię zmutowanego genu.
Początek ich populacji dał wspólny odle-
gły przodek, a jego genetyczna spuścizna
nazywana jest mutacją założycielską.
Genetycy znaleźli już tysiące mutacji
odpowiedzialnych za choroby ludzi, jed-
nak mutacje założycielskie są szczególną
grupą. Ofiary wielu chorób genetycznych
umierają młodo, nie osiągnąwszy dojrza-
łości płciowej, co ogranicza przekazywa-
nie błędnych sekwencji przyszłym poko-
leniom. Jednak mutacje założycielskie
często oszczędzają swych nosicieli i prze-
chodzą na ich potomstwo. W rezultacie
niektóre choroby uwarunkowane tymi błę-
dami upowszechniają się, jak wspomnia-
na hemochromatoza dziedziczna, anemia
sierpowata czy mukowiscydoza. Dlacze-
go ewolucja utrwaliła pozornie szkodliwe
mutacje, zamiast się ich pozbyć? Poniżej
przedstawię ten mechanizm, którym kie-
rowała się przyroda.
LISTOPAD 2005 ŚWIAT NAUKI
35

Naukowcy badający mutacje będące
przyczyną chorób dziedzicznych mają
nadzieję, że znajdą prosty sposób okre-
ślania, kto może być nimi zagrożony i w
jakim stopniu, oraz że wymyślą metody
zapobiegania im i ich leczenia [ramka
na stronie 39
]. Odkrycie, że mutacje zało-
życielskie można uznać za pozostawio-
ne w czasie ślady ludzkości, bo pozwala-
ją one antropologom śledzić przeszłość
ludzkich populacji i ich wędrówek w
świecie – to niezwykły „produkt ubocz-
ny” tych wysiłków.
Wyjątkowe mutacje
ZROZUMIENIE
NIEZWYK
ŁEJ
ROLI
mutacji
założycielskich oraz odczytanie infor-
macji, które one niosą, wymaga krót-
kiego wprowadzenia. Mutacje powsta-
ją w wyniku przypadkowych zmian w
DNA. Większość tego typu uszkodzeń
jest oczywiście naprawiana albo dziec-
ko z ich powodu umiera tuż po narodze-
niu i dlatego nie dziedziczą ich kolej-
ne pokolenia. Jednak pewne mutacje,
te, które powstają w komórkach linii
zarodkowej, są przekazywane dalej,
niekiedy z bardzo poważnymi konse-
kwencjami zdrowotnymi dla potom-
stwa, które je otrzymuje. Mutacje w
różnych ludzkich genach powodują
ponad tysiąc chorób.
Mutacje założycielskie pasują do
kategorii mutacji linii zarodkowej, ale
są nietypowe. Chorobami dziedzicz-
nymi zwykle rządzą dwie ogólne zasa-
dy. Przede wszystkim różne mutacje
w jednym genie mogą być przyczyną
tej samej choroby. W rezultacie w róż-
nych rodzinach, w których występuje
ta sama choroba, zwykle odpowiadają
za nią odmienne mutacje. Na przykład
hemofilia, będąca zaburzeniem krzep-
nięcia krwi, jest spowodowana muta-
cją w genie kodującym czynnik VIII
– składnik układu odpowiedzialnego
za powstawanie skrzepu. Okazuje się,
że każdy przypadek hemofilii związany
jest z wyraźną pojedynczą mutacją w
sekwencji DNA tego genu, ale występu-
jącą w różnych jego miejscach – bada-
cze znaleźli ich setki.
W kilku jednak przypadkach chorobę
może powodować ciągle ta sama muta-
cja. Mutacje na ogół powstają w dwojaki
sposób – w tzw. gorących miejscach lub
właśnie jako mutacja założycielska. Gorą-
ce miejsce to wrażliwy punkt genomu
– para zasad, będąca pojedynczym ele-
mentem sekwencji DNA, w którym szcze-
gólnie łatwo powstają błędy. I tak achon-
droplazja, częsta forma karłowatości,
zwykle pojawia się jako wynik mutacji
w pozycji 1138 w genie FGFR3, znajdu-
jącym się na krótkim ramieniu ludzkiego
chromosomu 4. Osoby z mutacjami, któ-
re powstały w gorących miejscach, zwy-
kle nie są ze sobą spokrewnione, dlatego
różnice w ich DNA są takie same jak w
przypadku innych obcych sobie ludzi.
Mutacje założycielskie, które są
przekazywane w niezmienionej for-
mie od pokoleń, różnią się całkowicie
od mutacji powstających w gorących
miejscach. W każdym przypadku zwią-
zanym z mutacją założycielską zmie-
niona sekwencja znajduje się w więk-
szym fragmencie DNA takim samym,
jaki miał jej protoplasta. (Badacze opi-
sują ten fenomen jako „identyczny z
pochodzenia”). Ów charakterystyczny
fragment DNA –-kompletny zapis znaj-
dującej się w nim informacji genetycz-
nej, charakterystycznej dla wszystkich
potomków danego przodka-założyciela
– nazywa się haplotypem. Co więcej,
analiza geograficznego występowania
takiego haplotypu pozwala na odnale-
zienie miejsca powstania mutacji zało-
życielskiej i śledzenie migracji ludzi.
Wiek mutacji założycielskiej można
określić na podstawie długości haploty-
pu, który z upływem czasu staje się coraz
krótszy [ramka na stronie 38]. Wyjścio-
wym haplotypem założyciela jest w rze-
czywistości cały chromosom zawiera-
jący mutację. Potomek otrzymuje go od
swojego protoplasty wraz ze „zdrowym”
chromosomem (bez mutacji), pochodzą-
cym od jego partnera. Dwa takie chro-
mosomy, po jednym od każdego z rodzi-
ców (tzw. chromosomy homologiczne),
ulegają rekombinacji, przypadkowo
wymieniając między sobą pewne frag-
menty DNA, podobnie jak mieszają się
karty dwóch talii podczas tasowania.
Po jednej rundzie rekombinacji muta-
cja nadal pozostanie wewnątrz bardzo
długiego odcinka DNA pochodzącego od
założyciela, tak jak po jednym przełoże-
niu i tasowaniu oznaczonej karcie ciągle
będą towarzyszyć karty znajdujące się
obok niej w konkretnym miejscu wyjścio-
wej talii. Każda nowa runda przełożenia i
tasowania sprawi jednak, że coraz mniej
będzie w sąsiedztwie kart, które otaczały
ją na początku. Podobnie haplotyp, który
zawiera zmutowany gen, po każdej kolej-
nej rekombinacji będzie krótszy.
Młoda mutacja założycielska, powiedz-
my, taka, która powstała kilkaset lat
temu, u ludzi żyjących obecnie powinna
zatem znajdować się w środku długiego
haplotypu. Z kolei bardzo stara mutacja
założycielska, na przykład sprzed kilku-
dziesięciu tysięcy lat, tkwi zapewne w
bardzo krótkim haplotypie.
Gen, którego uszkodzenie prowadzi
do rozwoju hemochromatozy, jest tylko
jednym z wielu w kolekcji poznanych
mutacji założycielskich. Dobrze zbada-
liśmy już wiele innych mutacji wystę-
pujących u Europejczyków oraz kilka
nowszych, występujących w populacji
północnoamerykańskich Indian, Azjatów
i Afrykanów [ramka na stronie 40]. Zdu-
miewające jest to, jak bardzo się one roz-
powszechniły – są setki, a może nawet
tysiące razy częstsze niż typowe mutacje
powodujące choroby. Większość mutacji
odpowiedzialnych za choroby pojawia
się w genomie z częstością raz na kilka
tysięcy lub milionów narodzin, jednak
mutacje założycielskie mogą dotyczyć
nawet kilku procent populacji.
Ta anomalia – czyż ewolucja nie powin-
na pozbyć się tych szkodliwych genów,
zamiast je preferować – jest cennym
tropem, prowadzącym do wyjaśnienia,
dlaczego mutacje założycielskie utrzyma-
36
ŚWIAT NAUKI
LISTOPAD 2005
SLIM FILMS (
popr
zednie str
ony
)
n
Mutacje założycielskie to szczególna grupa mutacji genetycznych we fragmentach DNA,
które są identyczne u osób je posiadających. Wszyscy ludzie z mutacją założycielską
mają wspólnego przodka-założyciela, u którego ta zmiana w DNA pojawiła się
po raz pierwszy.
n
Mierząc długość odcinka DNA, który zawiera mutację założycielską, i ustalając,
kto obecnie ją ma, naukowcy mogą w przybliżeniu oszacować datę pierwszego
jej pojawienia się i obecny stopień jej rozprzestrzenienia. Dane te dostarczają informacji
o migracjach poszczególnych grup ludzi w przeszłości.
n
W miarę jak odrębne populacje mieszają się, zmiany w sekwencjach DNA powodujące
choroby, kojarzone obecnie z konkretną grupą etniczną, będą wykrywane bardziej
przypadkowo. W przyszłości medycyna powinna sięgać do analiz DNA, by określić
ryzyko choroby zagrażającej danej społeczności.
Przegląd /
Zapisane w sekwencji
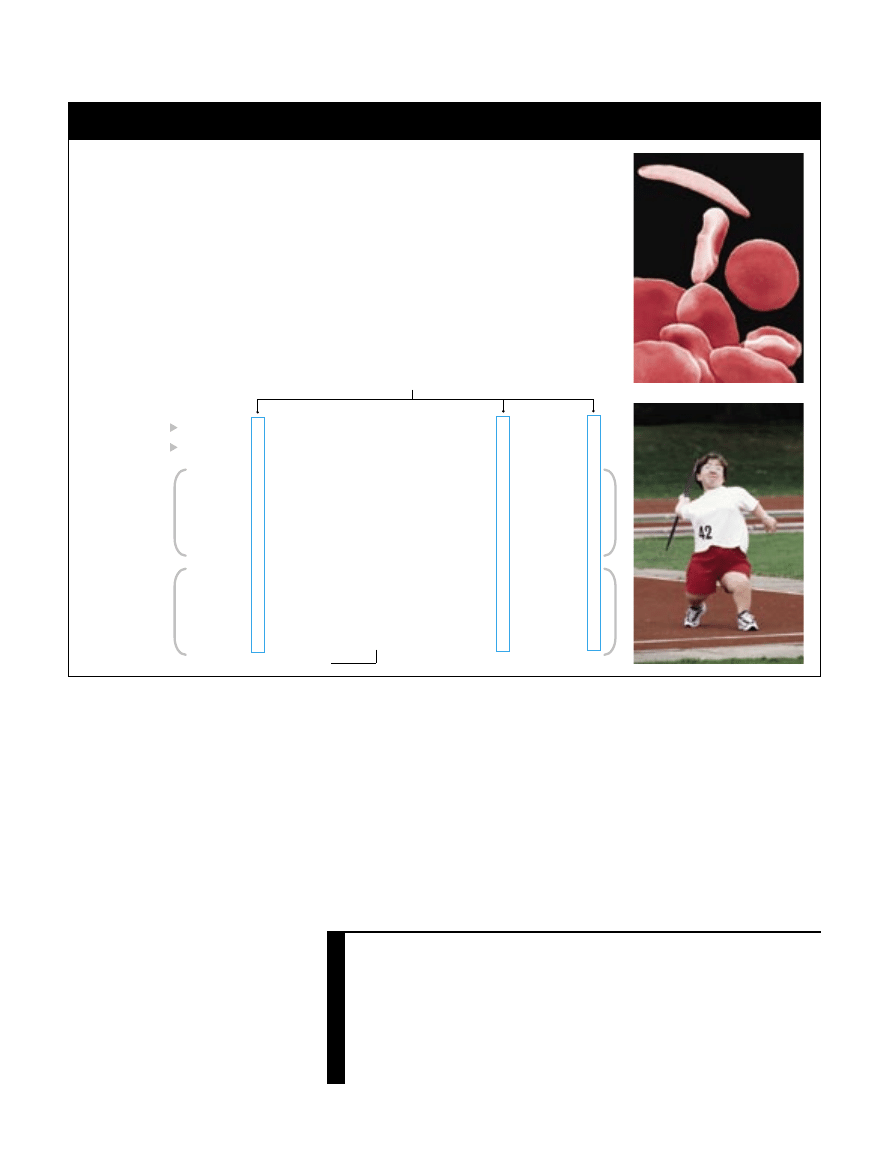
ły się w populacji i rozprzestrzeniły po
całym świecie.
Odpowiedź nie powinna być dla
nikogo zaskoczeniem: w pewnych oko-
licznościach mutacje założycielskie są
korzystne. Większość z nich jest rece-
sywna – jedynie osoba, której genom
zawiera dwie kopie tej samej wersji
zmienionego genu, po jednej od każde-
go z rodziców, zachoruje. Znacznie więk-
szy procent ludzi mających tylko jedną
kopię tak zmienionego genu będzie jego
nosicielami. Przekażą gen swoim dzie-
ciom, ale sami nie doświadczą żadnych
objawów choroby. Co więcej, pojedyn-
cza kopia genu z mutacją założycielską
w pewnych warunkach daje jego nosicie-
lowi przewagę w walce o przetrwanie.
Na przykład nosiciele mutacji odpo-
wiedzialnej za dziedziczną hemochroma-
tozę są zabezpieczeni przed niedoborem
żelaza powodującym anemię (co w prze-
szłości było częstym zagrożeniem życia),
gdyż białko kodowane przez zmutowany
gen sprawia, że dana osoba przyswaja
żelazo znacznie efektywniej, niż gdyby
miała dwie prawidłowe kopie tego genu.
Dzięki temu nosiciele tej mutacji mają
przewagę nad resztą populacji, gdy die-
ta jest uboga w żelazo.
Prawdopodobnie najlepiej pozna-
nym przykładem takiej „dwuznacznej”
mutacji jest mutacja odpowiedzialna
za anemię sierpowatą. Pojawiała się
ona wielokrotnie w Afryce i na Bliskim
Wschodzie, w rejonach wyniszczanych
przez malarię. Pojedyncza kopia zmuto-
wanego genu ułatwia nosicielowi prze-
życie infekcji pierwotniakiem wywołu-
jącym malarię (Plasmodium). Jednak
dwie kopie skazują go na cierpienie i
przedwczesną śmierć. Obecnie mutację
przyczyniającą się do rozwoju anemii
sierpowatej można odnaleźć w pięciu
różnych haplotypach. Wynika z tego, że
pojawiła się ona niezależnie pięć razy,
u pięciu różnych założycieli. (Choć
zwykle anemia sierpowata jest powo-
dowana przez mutację założycielską,
w niektórych przypadkach może być
LISTOPAD 2005 ŚWIAT NAUKI
37
ALISON KENDALL (
ilustracja
); GOP
AL MUR
TI
Photo R
esear
cher
s, Inc.
(
górne zdjęcie
); WELLCOME PHOTO LIBRARY (
dolne zdjęcie
)
DENNIS DRAYNA uzyskał licencjat w 1975 roku w University of Wisconsin-Madison, a w
1981 doktoryzował się w Harvard University. Staż podoktorski odbył w Howard Hughes
Medical Institute w University of Utah, a następnie przez 14 lat pracował w firmie bio-
technologicznej w San Francisco Bay Area, w której zidentyfikował u ludzi wiele genów
będących przyczyną zaburzeń sercowo-naczyniowych i metabolicznych. W 1996 roku
przeszedł do National Institutes of Health, gdzie obecnie jest szefem sekcji zajmującej się
głuchotą i innymi zaburzeniami komunikowania się. Jego początkowe zainteresowania
badawcze dotyczyły genetyki zaburzeń komunikacji – pracował w ośmiu krajach na czte-
rech kontynentach, poszukując tam rodzin z tego typu zaburzeniami. W czasie wolnym
lubi uprawiać wspinaczkę górską w równie odległych miejscach.
O-AUTORZE
ODLEGŁY PRZODEK KONTRA NOWICJUSZE
Jeśli grupa pacjentów cierpiących na tę samą chorobę ma identyczną mutację w jakimś punkcie
genomu, to jak lekarze mają odróżnić, czy jest to mutacja założycielska, czy też powstała w tzw.
gorącym miejscu? Otóż jest na to sposób: należy zbadać sekwencję otaczającą rejon mutacji.
Przyjmijmy, że u wszystkich pacjentów zmiana w sekwencji DNA polegała na zastąpieniu
tyminy (T) adeniną (A) (czerwony, poniżej). Jeśli A byłoby mutacją założycielską, to otaczająca
je sekwencja u wszystkich pacjentów powinna być taka sama – pacjenci odziedziczyliby pełną
sekwencję od tego samego odległego przodka. Jednak jeśli A było mutacją w gorącym miejscu,
pojawiającą się spontanicznie we wrażliwym na błędy odcinku DNA, sekwencja otaczająca
mutację powinna wykazywać także inne różnice (żółty) w miejscach, gdzie DNA ma tendencję
do mutowania bez wywoływania choroby.
Przyczyną anemii sierpowatej, wywoływanej przez zniekształcone erytrocyty (zdjęcie na górze),
jest zwykle mutacja założycielska. Z kolei achondroplazję, pewną formę karłowatości (zdjęcie
na dole), wywołują mutacje pojawiające się w gorącym punkcie genomu.
G AT T C A C A G G T C T C T AT C C G A AT C G AT T C C A T
G AT T C A C A G G T C T C
A
AT C C G A AT C G AT T C C A T
G AT T C A C A G G T C T C
A
AT C C G A AT C G AT T C C A T
G AT T C A C A G G T C T C
A
AT C C G A AT C G AT T C C A T
G AT T C A C A G G T C T C
A
AT C C G A AT C G AT T C C A T
G AT T C A C A G G T C T C
A
AT C C G A AT C G AT T C C A T
G AT T C
T
C A G G T C T C
A
AT C C G A AT C
C
AT T C C A
G
G AT T C A C A G G T C T C
A
AT C C G A AT C
C
AT T C C A
G
G AT T C
T
C A G G T C T C
A
AT C C G A AT C G AT T C C A T
G AT T C A C A G G T C T C
A
AT C C G A AT C
C
AT T C C A T
Niezmieniona
sekwencja
ZmiennoÊç typowa dla populacji
Mutacja
Chromosomy
z mutacjà
za∏o˝ycielskà
Chromosomy
z mutacjà
w goràcych
miejscach
Mutacja

Za∏o˝yciel
Miejsce mutacji
Mutacja
Rekombinacja
Przeniesienie na nast´pne
pokolenie
Przeniesienie na nast´pne
pokolenie
Mutacja
Rekombinacja przez
wiele pokoleƒ
Potomek
nosiciel
Wnuk
nosiciel
Nosiciel
obecnie
Mutacja
Normalny
chromosom
Chromosom
z mutacjà
także wynikiem spontanicznej mutacji
w obszarze genomu, szczególnie wrażli-
wym na powstawanie błędów).
Częstość mutacji założycielskich w
populacji jest regulowana przez dwie
przeciwstawne siły – z jednej strony ktoś
z dwiema kopiami zmutowanego genu
prawdopodobnie umrze, zanim wyda na
świat potomstwo, z drugiej – z pojedynczą
kopią ma większe szanse na przeżycie niż
osoby niemające żadnej. Prowadzi to do
wytworzenia tzw. doboru stabilizującego,
kiedy korzystny efekt zwiększa częstość
pojawiania się zmutowanego genu, a
szkodliwy tłumi jego występowanie. Ewo-
lucja daje i zabiera, dlatego mimo upływu
czasu geny utrzymują się w populacji na
względnie stałym poziomie.
Naukowcy nadal nie wiedzą, jaką prze-
wagę nad innymi dają niektóre mutacje
założycielskie swoim nosicielom, jednak
stała obecność takich zmutowanych
genów przemawia za istnieniem konkret-
nych korzyści. Na przykład najnowsze
odkrycie może wyjaśnić utrzymywanie
się czynnika V Leiden (mutacji w genie
kodującym czynnik V), który jest odpo-
wiedzialny za współdziałanie kolejnych
składników układu krzepnięcia krwi. Ta
mutacja założycielska, obecna u-4% Euro-
pejczyków, prowadzi do rozwoju zakrze-
picy, objawiającej się nieprawidłowym
krzepnięciem krwi. W 2003 roku Bryce
A. Kerlin wraz ze współpracownikami z
Blood Center of Southeast Wisconsin i
Medical College of Wisconsin wykazali,
że nosiciele tej mutacji są odporni na bak-
teryjną infekcję krwi, zwaną posocznicą
(inaczej sepsą). W czasach, gdy nie zna-
no jeszcze antybiotyków, była ona ogrom-
nym zagrożeniem życia, zresztą tragiczne
przypadki notuje się także dziś.
Geny rozrzucone po świecie
JU
Ż
NA
D
ŁUGO
PRZED
POJAWIENIEM
SI
Ę
nowoczesnych środków transportu muta-
cje założycielskie pokonywały olbrzymie
odległości. Niekiedy „podróż” taka obej-
mowała dziesiątki, a nawet setki, poko-
leń. Anemia sierpowata przedostała
się z Afryki na zachód, do Ameryki, na
statkach niewolniczych oraz na północ,
do Europy. Ustalono, że mutacja zało-
życielska powodująca głuchotę, która
występuje często w genie GJB2, od
chwili pojawienia się w starożytności
na Bliskim Wschodzie przebyła dwie
drogi: jedną wzdłuż wybrzeża Morza
Śródziemnego do Włoch i Hiszpanii, a
drugą dolinami Renu i Dunaju na północ
38
ŚWIAT NAUKI
LISTOPAD 2005
ALLISON KENDALL
KRÓTSZY Z WIEKIEM
Wyjątkowo łatwo rozpoznawalny obszar chromosomu – haplotyp – który otacza
mutację założycielską, jest krótszy w każdym nowym pokoleniu dzięki temu, że
fragmenty chromosomów homologicznych mieszają się podczas procesu zwanego
rekombinacją. W poniższym przykładzie żółty chromosom jest miejscem powsta-
nia mutacji założycielskiej w genomie założyciela, a odpowiadający mu niebieski u
drugiego rodzica jest niezmieniony. Gdy założyciel wytwarza plemniki lub komórki
jajowe, między chromosomami zachodzi rekombinacja, polegająca na wzajemnej
wymianie odpowiednich fragmentów. Potomstwo dziedziczy jedynie nowe, wymie-
szane chromosomy, które zawierają mutację i pozostałą część haplotypu założyciela
(żółty obszar). Taki proces powtarza się we wszystkich pokoleniach i prowadzi do
skracania haplotypu.

Europy. Mutacja założycielska w genie
ABCA4
, powodująca ślepotę, pojawiła
się najprawdopodobniej w Szwecji oko-
ło 2700 lat temu i rozprzestrzeniła na
południe i zachód Europy.
Jednak najbardziej spektakularnym
przykładem takiej migracji jest prawdo-
podobnie genetyczna zmienność nasze-
go zmysłu smaku. Około 75% ludności
świata substancja zwana fenylotiokar-
bamidem (PTC) wydaje się okropnie
gorzka, jednak pozostałe 25% żadnej
goryczy w niej nie wyczuwa. Wraz ze
współpracownikami z National Institu-
tes of Health i innych instytucji odkryli-
śmy niedawno, że za powstanie wersji
genu kodującej receptor nierozpoznają-
cy PTC jest odpowiedzialna kombina-
cja trzech różnych zmian. Praktycznie
wszyscy, którzy nie czują smaku PTC,
są potomkami jednego człowieka, w
którego genie po raz pierwszy pojawi-
ły się takie zmiany. (Zmysł odczuwania
gorzkiego smaku istnieje po to, by chro-
nić nas przed połknięciem toksycznych
substancji pochodzenia roślinnego, ale
jakie korzyści miałby nieść gen uniemoż-
liwiający odczuwanie danego smaku?
Podejrzewamy, że wersja genu „pozba-
wiona” umiejętności czucia smaku kodu-
je tak zmieniony receptor dla PTC, że
może on rozpoznawać inne, jeszcze nie-
zidentyfikowane trujące związki).
Mutacja powodująca brak danego
smaku tkwi w niezmiernie krótkim,
rodowym fragmencie DNA. U niektó-
rych nosicieli jest to ledwie 30 tys. par
zasad, co oznacza, że mutacja założy-
cielska jest szczególnie stara – praw-
dopodobnie ma ponad 100 tys. lat.
Badania przeprowadzone w zeszłym
roku, którymi objęto cały świat, wyka-
zały że na obszarze subsaharyjskiej
Afryki istnieje siedem różnych wersji
genu związanego z PTC. Jednak tylko
podstawowa jego wersja, umożliwiają-
ca odczuwanie smaku PTC, i główny
wariant, pozbawiający tego smaku
odnajdowane są z dużą częstością
poza afrykańską populacją. Z pozosta-
łych pięciu wariantów jeden występuje
sporadycznie w nieafrykańskich popu-
lacjach, ale nigdy u rdzennych miesz-
kańców Nowego Świata, Indian, pod-
czas gdy pozostałe cztery spotyka się
wyłącznie w Afryce.
Mutacja uniemożliwiająca rozpozna-
wanie PTC jako gorzkiej substancji do-
starcza wielu danych o wczesnych
migracjach naszych przodków. Jej obec-
ne rozmieszczenie i częstość występowa-
nia wspiera archeologiczne i antropolo-
giczne dowody na to, że populacja ludzi
współczesnych pierwotnie żyła w Afry-
ce i przed mniej więcej 75 tys. lat jakaś
mała jej grupa rozprzestrzeniła się na
pozostałe pięć kontynentów. Jest to tzw.
hipoteza „pożegnania z Afryką”, zakła-
dająca, że człowiek nowoczesny anato-
micznie powstał w jednym miejscu na
tym kontynencie. Wszystkie obecnie ist-
niejące populacje pochodzą od tej jed-
nej. Dodatkowo, oprócz potwierdzenia
wcześniejszych przypuszczeń, wariant
genu, który nie pozwala na rozpozna-
wanie smaku PTC, ułatwia odpowiedź
na jedno z najbardziej kontrowersyj-
nych pytań współczesnej antropologii:
czy przodek Homo sapiens, zasiedlając
kulę ziemską, krzyżował się z bardziej
archaicznymi hominidami żyjącymi
w Europie i Azji?
LISTOPAD 2005 ŚWIAT NAUKI
39
LARRY WILLIAMS
Corbis
Wczoraj geny, jutro leki
Zdolność do identyfikowania mutacji założycielskich zapewne znajdzie praktyczne
zastosowanie w medycynie. Wiedza o takich mutacjach może na przykład pomóc
lekarzom w ustaleniu, którzy pacjenci powinni zostać poddani testom na daną choro-
bę. Obecnie, by określić ryzyko wystąpienia niektórych chorób i zaplanować dalsze
postępowanie, lekarze mogą jedynie odwołać się do danych dotyczących pochodzenia
etnicznego pacjenta. I tak większość przypadków anemii sierpowatej stwierdza się
u osób mających afrykańskie korzenie. Jednak w miarę jak ludność świata mie-
sza się coraz bardziej, trudniej określić geograficzne miejsce pojawienia się konkretnego
rodu czy ustalić tożsamość etniczną jakiejś osoby. Pochodzenie etniczne przestaje być
dobrą wskazówką diagnostyczną, dlatego lekarze, próbując określić ryzyko choroby
lub przyczyny objawów obserwowanych u pacjenta, będą bardziej polegać na indywi-
dualnych testach DNA. Znajdowanie mutacji założycielskich obecnie, gdy populacje
ludzkie pozostają genetycznie zróżnicowane, pomoże w przyszłości zidentyfikować
specyficzne geny odpowiedzialne za liczne schorzenia.
Rzeczywiście, znane mutacje założycielskie można traktować jako szczególny przy-
padek znacznie większej grupy wariantów w naszym DNA, które są przyczyną chorób.
Choć nie wiemy jeszcze, jak wiele ich istnieje, takie warianty są prawdopodobnie bar-
dzo stare. Jak usiłowaliśmy to pokazać w artykule, takie kojarzone z chorobami warian-
ty były prawdopodobnie korzystne dla ludzi żyjących w dawnych czasach i dlatego są
obecnie częste w populacji. Jednak spotkanie naszych starych genów, pochodzących
z odległych czasów, z nowoczesnym środowiskiem i stylem życia może prowadzić do
chorób, które z czasem się upowszechnią.
Badania genetyczne zyskają na randze w praktyce medycznej, ponieważ te liczne
warianty mutacji prawdopodobnie predysponują nas do zapadania na różne choroby,
nie tylko rzadkie, dziedziczne. Przykładem takich genetycznych wariantów mogły być
te, które pomogły nam syntetyzować cholesterol, jednak teraz wpływają na podwyż-
szenie jego poziomu, lub te, które umożliwiły oszczędną gospodarkę solami, a obec-
nie prowadzą do wrażliwości na sól i podwyższonego ciśnienia krwi. Rozpoznanie
specyficznych profili genetycznych związanych z częstymi dziś zaburzeniami może
oznaczać, że genetyka przechodzi od marginesowej specjalności medycznej, tropiącej
rzadkie i mało znane dolegliwości, do kluczowej w zapobieganiu, diagnozowaniu i
monitorowaniu chorób.
ANALIZOWANIE PRZYNALEŻNOŚCI ETNICZNEJ jest obecnie szybkim sposobem stosowa-
nym przez lekarzy do ustalenia ryzyka niektórych schorzeń. W miarę jak DNA współcze-
snego człowieka coraz bardziej się miesza, ta sama sekwencja DNA będzie informować
lekarza o predyspozycjach pacjenta do pewnych chorób.

Owe archaiczne hominidy mogły
z dużą dozą prawdopodobieństwa
mieć własny wariant genu kodującego
receptor rozpoznający PTC, wyselek-
cjonowany w odpowiedzi na toksyny
występujące w lokalnej florze. Jeśli te
hominidy miały potomstwo z osobnika-
mi należącymi do H. sapiens, to odna-
leźlibyśmy różne warianty tego genu w
populacji europejskiej, zachodnioazja-
tyckiej i południowo-zachodnioazjatyc-
kiej. Jednak uderza ich brak. Zatem
analiza mutacji założycielskich u ludzi
żyjących współcześnie dowodzi, że
przed dziesiątkami tysięcy lat, podczas
afrykańskiego exodusu, nie doszło do
skutecznego krzyżowania H. sapiens z
innymi gatunkami ludzi.
W poszukiwaniu protoplasty
JE
ŚLI
BLI
ŻEJ
PRZYJRZYMY
SI
Ę
haplotypowi
powodującemu dziedziczną hemo-
chromatozę, zrozumiemy, że analiza
genetyczna obecnej populacji i zapisy
historyczne mogą dostarczyć nowych
informacji na temat przyczyn i przebie-
gu tej szczególnej choroby. W latach
osiemdziesiątych, zanim zidentyfiko-
wano gen odpowiedzialny za tę przy-
padłość, genetycy zauważyli, że prawie
każdy człowiek z objawami hemochro-
matozy ma na chromosomie 6 prak-
tycznie identyczny fragment sekwen-
cji DNA. Było to szokujące odkrycie,
ponieważ większość chorych nie była
ze sobą spokrewniona i spodziewano
się raczej jakichś przypadkowych róż-
nic w dowolnym miejscu wspomnia-
nej sekwencji. Ten niezwykły odcinek
DNA uświadomił naukowcom, jak bar-
dzo prawdopodobne jest, że wszystkie
osoby cierpiące na dziedziczną hemo-
chromatozę mają wspólnego, dawno
zmarłego przodka oraz że gen odpowie-
dzialny za rozwój tej choroby najpew-
niej tkwi wewnątrz tego odcinka.
Gdy w latach dziewięćdziesiątych
nasza grupa badawcza zajmowała się tą
hipotezą, przeprowadziliśmy u 101 cho-
rych na hemochromatozę dokładną ana-
lizę genów, które udało się nam znaleźć
w odpowiednim regionie chromoso-
mu 6. Jednocześnie analizowaliśmy
DNA 64 zdrowych osób z grupy kontrol-
nej. U większości pacjentów stwierdzili-
śmy taką samą długą sekwencję – kilka
milionów par zasad – ale u niektórych
całkowitą zgodność wykazywał znacznie
krótszy fragment. Gdy przyjrzeliśmy mu
się bliżej, odkryliśmy, że rejon ten za-
wiera 16 genów. 13 z nich kodowało
histony, które są białkami oddziału-
jącymi z DNA i odpowiadają za jego
upakowanie w zwartą, widoczną pod
mikroskopem w czasie podziału komór-
ki strukturę przypominającą kształtem
serdelek. Histony i kodujące je geny są
praktycznie takie same u wszystkich, dla-
tego uznaliśmy za nieprawdopodobne,
by to one były odpowiedzialne za hemo-
chromatozę. Zajęliśmy się wobec tego
pozostałymi trzema genami.
Dwa z nich były identyczne u osób
cierpiących na hemochromatozę i nie
różniły się niczym od genów ochotni-
ków z grupy kontrolnej. W trzecim
genie, obecnie określanym jako HFE,
odkryliśmy mutację, która występuje
u ludzi chorych, ale wyraźnie brakuje
jej u tych, którzy nie mają kłopotów
z nadmiernym przyswajaniem żela-
za. Oznaczało to, że znaleźliśmy gen
zawierający-mutację założycielską, któ-
ra przyczynia się do rozwoju hemochro-
matozy dziedzicznej.
Nasze odkrycie dotyczące muta-
cji założycielskiej odpowiedzialnej
za hemochromatozę sprowokowało
natychmiast kilka pytań. Kto był zało-
życielem? Kiedy i gdzie żyła ta osoba?
Poszukiwanie odpowiedzi na te
pytania stało się możliwe dopiero dzię-
ki połączeniu sił genetyków oraz antro-
pologów i historyków. Nasze badania
wykazały, że dziedziczna hemochro-
matoza występuje w całej Europie,
jednak zdecydowanie częściej na pół-
nocy kontynentu. Co więcej, mutacja
założycielska jest obecna praktycznie u
wszystkich pacjentów pochodzących z
północy, ale u mniej niż dwóch trzecich
ze wschodu i południa. Oznacza to, że
pozostałe 30% populacji albo ma jakąś
40
ŚWIAT NAUKI
LISTOPAD 2005
ALISON KENDALL
DOBÓR STABILIZUJĄCY sprawia, że poten-
cjalnie szkodliwy gen utrzymuje się w puli
genowej. W rejonach, gdzie plagą jest rozno-
szona przez komary malaria, posiadanie poje-
dynczej kopii zmutowanego genu hemoglobiny
działa ochronnie – osoby z taką mutacją czę-
ściej przeżywają zakażenie. Jednak ci, którzy
odziedziczyli dwie kopie zmutowanego genu,
chorują na anemię sierpowatą i żyją znacznie
krócej. Konkurencyjne mechanizmy prowadzą
do osiągnięcia w populacji stabilnego poziomu
mutacji powodujących anemię sierpowatą.
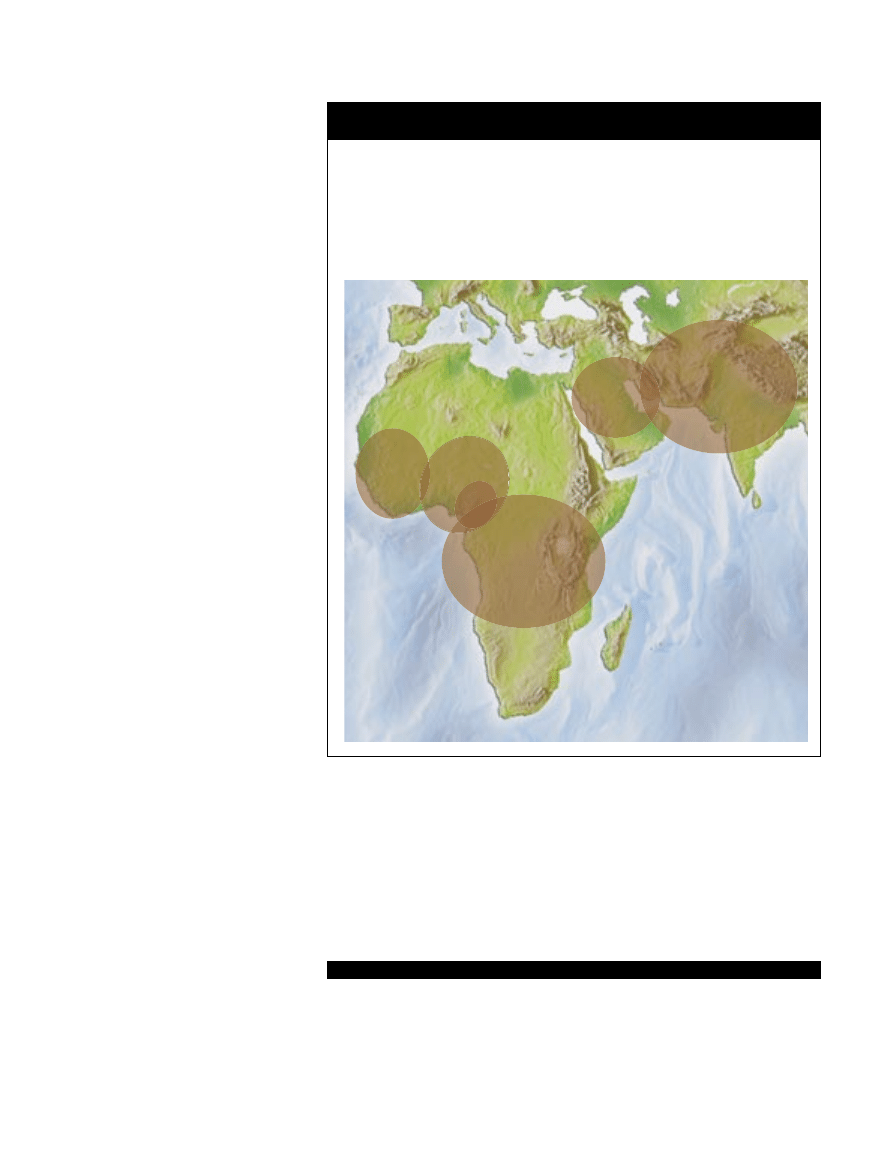
Interesujące mutacje założycielskie
Zmutowany gen Choroba
Miejsce powstania mutacji
Migracja
Potencjalna korzyść z jednej kopii
HFE
Hemochromatoza
Daleka północno-zachodnia Europa
Przez południową i wschodnią Europę Ochrona przed niedokrwistością
CFTR
Mukowiscydoza
Południowo-wschodnia Europa
Przez zachodnią
Odporność na biegunkę
i Bliski Wschód
i północną Europę
HbS
Anemia sierpowata Afryka i Bliski Wschód
Do Nowego Świata
Ochrona przed malarią
FV Leiden
Zakrzepica
Zachodnia Europa
Po całym świecie
Ochrona przed posocznicą
ALDH2
Toksyczność
Daleka wschodnia Azja
Przez północną i zachodnią Azję
Ochrona przed alkoholizmem
alkoholu
i prawdopodobnie zapaleniem wątroby typu B
LCT
Tolerancja laktozy
Azja
Przez zachodnią i północną Eurazję
Umożliwia spożywanie mleka
udomowionych zwierząt
GJB2
Głuchota
Bliski Wschód
Przez zachodnią i północną Europę
Nieznana
inną mutację w HFE, albo cierpi na cał-
kiem inny rodzaj zaburzeń związanych
z wchłanianiem żelaza.
Dokładniejsza analiza genetyczna
próbek pobranych od ludzi z północno-
zachodniej Europy ujawniła, że najczę-
ściej mutacja założycielska występuje
w Irlandii, zachodniej Wielkiej Brytanii
oraz po drugiej stronie Kanału La Man-
che, we francuskiej Bretanii. Taki wzór
rozmieszczenia tej mutacji niemal ideal-
nie pokrywa się z obecnym rozmieszcze-
niem ludności pochodzenia celtyckiego.
Celtowie zamieszkiwali Europę Środ-
kową ponad dwa tysiące lat temu. Część
z nich została wyparta na północny
zachód w wyniku ekspansji imperium
rzymskiego, podczas gdy inni wymie-
szali-się z mieszkańcami południowej
Europy i pozostali w pierwotnym miej-
scu. Czy wobec tego mutacja założyciel-
ska odpowiedzialna za hemochromato-
zę pojawiła się w Europie Środkowej,
a następnie przenieśli ją migrujący nosi-
ciele? A może pojawiła się na północy?
Odpowiedź przyniosły dodatkowe bada-
nia sekwencji DNA otaczającej miejsce
mutacji na chromosomie 6.
Współczesny halotyp jest dość długi,
można zatem przypuszczać, że mutacja
założycielska jest całkiem młoda, pojawi-
ła się prawdopodobnie zaledwie od 60 do
70 pokoleń temu, a więc przed około 800
laty. Tak wczesna data sugeruje, że założy-
ciel żył w Europie Środkowej, a mutacja
rozprzestrzeniła się na północ i zachód
razem z jego potomkami wypieranymi
przez Rzymian. Jednak imperium rzym-
skie upadło przed rokiem 800, jest więc
bardzo prawdopodobne, że nasza muta-
cja założycielska pojawiła się w północ-
no-zachodniej Europie, a dopiero potem
rozprzestrzeniła na południe i wschód
razem z potomkami założyciela.
Antropolodzy, w szczególności Luigi
Cavalli-Sforza, już wcześniej badali
inne rodzaje wariantów DNA, aby śle-
dzić migrację populacji. Jednak mutacje
założycielskie wprowadzają obecnie do
badań DNA nową jakość: wyskalowanie
długości haplotypu pozwala na określe-
nie daty powstania mutacji, a dzięki osza-
cowaniu częstości haplotypu w populacji
można mierzyć geograficzne rozprze-
strzenianie się potomków założyciela.
Każdy z nas nosi biochemiczne
świadectwo potwierdzające, że wszy-
scy ludzie są tak naprawdę członkami
jednej rodziny, powiązanymi ze sobą
wspólnym dziedzictwem naszego geno-
mu. Poza potwierdzeniem hipotezy
„pożegnania z Afryką” analiza muta-
cji założycielskich ujawniła wspólne
pochodzenie różnych innych pozornie
niespokrewnionych ze sobą grup – na
przykład najnowsze badania prowadzo-
ne przez Davida B. Goldsteina z Duke
University wykazały niespodziewany
genetyczny związek między Celtami i
Baskami. Dalsze badania mutacji zało-
życielskich i otaczających je haploty-
pów bez wątpienia pokażą więcej gene-
tycznych relacji, co pozwoli na nowy
wgląd w kwestię, skąd pochodzimy i w
jaki sposób znaleźliśmy się w miejscu,
w którym obecnie żyjemy. Takie badania
ujawnią także zaskakujące pokrewień-
stwa, co zapewne przyczyni się do głęb-
szego zrozumienia wspólnych korzeni
drzewa genealogicznego człowieka.
n
LISTOPAD 2005 ŚWIAT NAUKI
41
ALISON KENDALL
NIEZALEŻNE POCHODZENIE
Wszyscy ludzie chorzy na anemię sierpowatą mają taką samą mutację. Prawdopo-
dobnie jednak ta zmiana w sekwencji DNA powstała w pięciu różnych haplotypach,
co świadczy, że mutacja ta pojawiała się w przeszłości niezależnie pięć razy. Chorzy
mogą mieć haplotyp charakterystyczny dla obszaru Senegalu, Beninu, Bantu, krajów
arabskich i Indii oraz niedawno odkryty haplotyp typowy dla rejonu Kamerunu. 8%
czarnoskórych Amerykanów jest nosicielami co najmniej jednej kopii zmutowanego
genu, przyczyniającego się do rozwoju anemii sierpowatej.
Pożegnania z Afryką. Ian Tattersall; Świat Nauki, VI/1997.
Natural Selection and Molecular Evolution in PTC, a Bitter-Taste Receptor Gene. S.
Wooding, U.-K. Kim, M. J. Bamshad, J. Larsen, L. B. Jorde i D. Drayna; American Jour-
nal of Human Genetics, tom 74, nr 4, s. 637-646; 2004.
The Great Human Diasporas: The History of Diversity and Evolution. Luigi Cavalli-Sforza;
Addison-Wesley, 1995.
Omówienie International Haplotype Map Project na stronie National Human Genome
Research Institute: www.genome.gov/10001688
JEŚLI CHCESZ WIEDZIEĆ WIĘCEJ
Senegal
Benin
Kamerun
Bantu
Kraje arabskie i Indie
Wyszukiwarka
Podobne podstrony:
PREZ metody wykrywania mutacji
Mutacje chromosomowe strukturalne
Materiał genetyczny, mutacje, systemy naprawy DNA, test Amesa
dodatkowy artykul 2
ARTYKUL
jak zalozyc fundacje id 377540 Nieznany
laboratorium artykul 2010 01 28 Nieznany
Fizjologia snu Artykul
energoefekt artykul transmisja danych GPRS NiS[1]
Komunikacja interpersonalna Artykul 4 id 243558
artykul profilaktyka cz2 id 695 Nieznany (2)
jak zalozyc dzialalnosc
kryteria oceny podręczników artykuł
Artykul (2015 International Jou Nieznany
więcej podobnych podstron