
UNIWERSYTET JAGIELLOŃSKI
WYDZIAŁ CHEMII
I ROK SUM: ANALITYKA ŚRODOWISKOWA
Edyta Racławska
REAKCJA NEFA
Tarnów 2008

2
1. WSTĘP
3
2. MECHANIZM
4
3. MOŻLIWOŚCI I OGRANICZENIA
6
3.1. Z
WIĄZKI NITROWE I SOLE NITRONOWE
6
3.2. R
EAKCJE BOCZNE
,
KOMPLIKUJĄCE REAKCJĘ
N
EFA
9
3.3. Z
MODYFIKOWANE REAKCJE
N
EFA
18
3.4. P
OKREWNE REAKCJE ZWIĄZKÓW NITROWYCH PROWADZĄCE DO PRODUKTÓW REAKCJI
N
EFA
26
3.5. U
ŻYTECZNOŚĆ SYNTETYCZNA
32

3
1.
Wstęp
Reakcja Nefa jest zazwyczaj definiowana, jako przemiana pierwszorzędowych lub
drugorzędowych nitroalkanów w odpowiednie związki karbonylowe. Reakcja ta została
opisana przez szwajcarskiego chemika J. U. Nefa w 1894 roku na dwóch przykładach.
Przekształcenie grupy nitrowej w grupę karbonylową stało się ważnym narzędziem
syntezy z powodu łatwości przygotowania podstawionych związków nitrowych przez
kondensację nitroalkanów z aldehydami (reakcja Henry'ego), sprzężoną addycję
nitroalkanów do elektrofilowych alkenów lub przez alkilację węglową dianionów
pierwszorzędowych nitroparafin. Reakcja Nefa jest jednym z najlepszych przykładów
reakcji z inwersją polarności
1
, w której pierwotny anion związku nitrowego pełni rolę
równoważnego anionu acylowego.
W pracy tej zostanie omówiona reakcja Nefa oraz zostaną omówione modyfikacje
pierwotnego procesu, które zwiększają różnorodność związków, które są użyteczne, jako
substraty. Te modyfikacje będą rozważane według ogólnego typu mechanizmu reakcji oraz
będą opisane według otrzymywania odpowiedniego reagenta.
1
Inwersja polarności w chemii organicznej jest chemiczną modyfikacją grupy funkcyjnej w celu zmiany
(odwrócenia) polarności tej grupy.
CH
3
CHNO
2
- Na
+
H
+
CH
3
CHO
(70%)
(CH
3
)
2
CNO
2
-
Na
+
H
+
(CH
3
)
2
C=O
(67%)

4
2.
Mechanizm
Mechanizm reakcji Nefa został rozlegle przebadany. Początkowa konwersja związku
nitrowego w sól nitronową
2
jest realizowana za pomocą zasady. Jednakże, kluczowym
krokiem reakcji jest zakwaszenie tego związku pośredniego do otrzymania związku
karbonylowego i nieorganicznych produktów ubocznych (reakcja 1).
Reakcja ta jest zależna od pH. Dodatkowo mogą występować reakcje uboczne (tabela A).
Wydajność (%)
pH
(CH
3
)
2
CHNO
2
(CH
3
)
2
C=O
(CH
3
)
2
C=NOH (CH
3
)
2
C(NO)NO
2
5.4
(100)
(0)
(0)
(0)
5.0
(85)
(8)
(8)
(0)
4.3
(44)
(20)
(19)
(15)
3.1
(10)
(30)
(30)
(29)
2.0
(0)
(39)
(32)
(29)
1.5
(0)
(49)
(28)
(22)
1.2
(0)
(80)
(12)
(7)
0.5
(0)
(100)
(0)
(0)
Tabela A. Zależność rozdziału produktu w zakwaszeniu soli 2-nitropropanu w 21
o
od pH
Słabo kwaśne warunki reakcji faworyzują regenerację związku nitrowego, podczas gdy
reakcja Nefa zachodzi w silnie kwaśnym środowisku. Oksymy i pseudonitrole (związki
nitrowe z grupą nitrozową w pozycji α) uzyskuje się w pośrednim zakresie kwasowości.
Dla reakcji Nefa zostało zaproponowanych kilka mechanizmów. Kinetyczne analizy
oraz fakt, że dodatkowa woda w rozpuszczalniku wodno-alkoholowym spowalnia reakcję,
doprowadziły do wniosku, że reakcja może zachodzić według dwóch mechanizmów.
2
Z ang. nitronate salt
zasada
RR
1
C=NO
2
-
RR
1
CHNO
2
H
+
RR
1
C=O +1/2 N
2
O + 1/2 H
2
O
Reakcja 1.
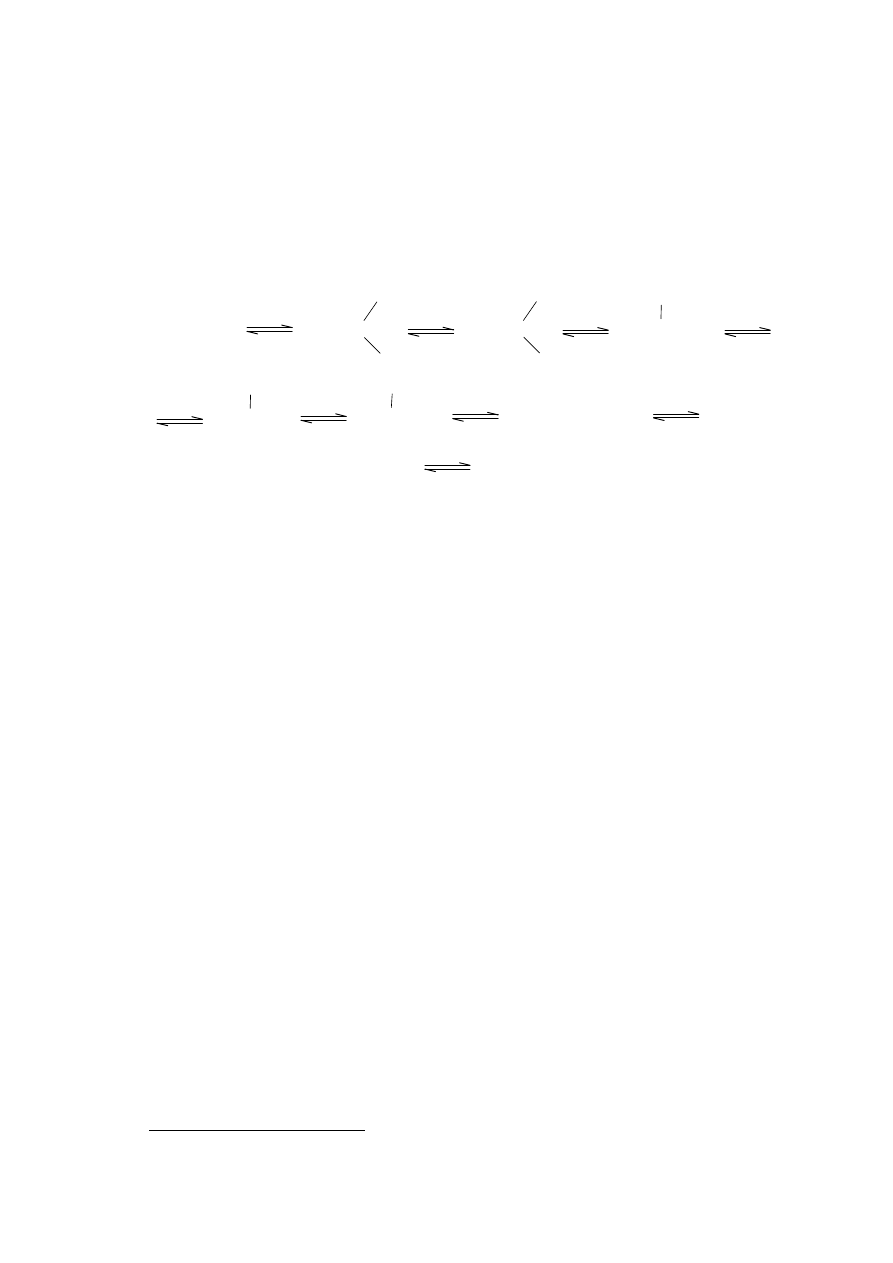
5
Różnicę między tymi mechanizmami stanowi moment utraty wody. Podstawowym etapem
jest stopniowa protonacja soli nitronowej na każdym tlenie, a następnie przyłączenie wody
i rozkład otrzymanego związku pośredniego (schemat 1). Inny opis twierdzi, że kwas
nitronowy
3
nie jest związkiem pośrednim w trakcie protonacji soli nitronowej. Niemniej
jednak reakcja jest czuła zarówno na pH jak i na stężenie wody.
W rezultacie, dodanie kwasu do soli nitronowej faworyzuje regenerację związku nitrowego
w reakcji konkurującej z reakcją Nefa, podczas gdy dodanie soli nitronowej do mocnego
kwasu faworyzuje reakcję Nefa. Mechanizm jasno pokazuje, że należy się spodziewać
reakcji pobocznych w pewnych układach z powodu tworzenia się tlenku azotu(I).
3
Z ang. nitric acid
RR
1
C=NO
2
-
H
+
RR
1
C=N
O
OH
-
+
H
+
RR
1
C=N
OH
OH
+
-H
+
H
2
O,
RR
1
CN(OH)
2
OH
-H
2
O
-H
2
O
RR
1
CN=O
OH
H
+
RR
1
CN=OH
OH
+
RR
1
C=OH + HNO
-H
+
+
RR
1
C=O
2HNO
H
2
O + N
2
O
Schemat 1.

6
3.
Możliwości i ograniczenia
3.1.
Związki nitrowe i sole nitronowe
Związki nitrowe są łatwo dostępne i służą, jako idealne syntetyczne związki pośrednie.
Najbardziej powszechna metoda ich otrzymywania polega na wypieraniu grupy
opuszczającej przez jon azotanu(III).
Większość pierwszorzędowych i drugorzędowych halogenków reaguje z azotanem(III)
sodu w aprotonowym rozpuszczalniku, takim jak sulfotlenek dimetylu (DMSO) lub
dimetyloformamid (DMF) dając związki nitrowe w użytecznej wydajności. W innej
metodzie, stabilizowane karboaniony mogą być nitrowane przy użyciu estrów nitrowych
(RONO
2
). Dodatkowo enole octanowe mogą być nitrowane przez azotan acetylu. A zatem
α
-nitroketony, α-nitroestry i α-nitrosulfony mogą być łatwo syntetyzowane. Trzecią
metodą jest utlenianie pierwszorzędowych amin za pomocą nadmanganianu potasu, kwasu
m-chloroperoksybenzoesowego (MCPBA), ozonu lub dimetylooksiranu. Ta metoda, jest
użyteczna do spreparowania praktycznie każdego związku nitrowego – nawet
czwartorzędowych pochodnych. Takiej możliwości nie daje, reakcja wypierania halogenku
w procesie S
N
2. Oksymy również mogą być utlenione za pomocą peroksykwasów.
Ewentualnie, oksymy mogą być bromowane do powstania związku nitrowego
podstawionego bromem w pozycji α, który może być utleniony za pomocą kwasu
azotowego(V) i nadtlenku wodoru. Redukcyjne usunięcie bromu jest wartościową drogą
przygotowania drugorzędowych nitroparafin. Drugorzędowe nitroparafiny mogą być
również otrzymane z dobrą wydajnością, przez alkilację dianionów pierwszorzędowych
związków nitrowych. Inna metoda używa kwasu chlorowego(I) do chlorowania oksymów
z wydajnością rzędu 71-93%. Produkty następnie są redukowane za pomocą magnezu,
cynku lub wodoru osadzonego na palladzie do otrzymania drugorzędowych związków
nitrowych z wydajnością 77-95%.
Wiele reakcji związków nitrowych odzwierciedla równowagę z kwasami nitronowymi
1 (zwanymi również związkami acy-nitrowymi lub izonitrowymi).
RX + NO
2
-
RNO
2
+ X
-
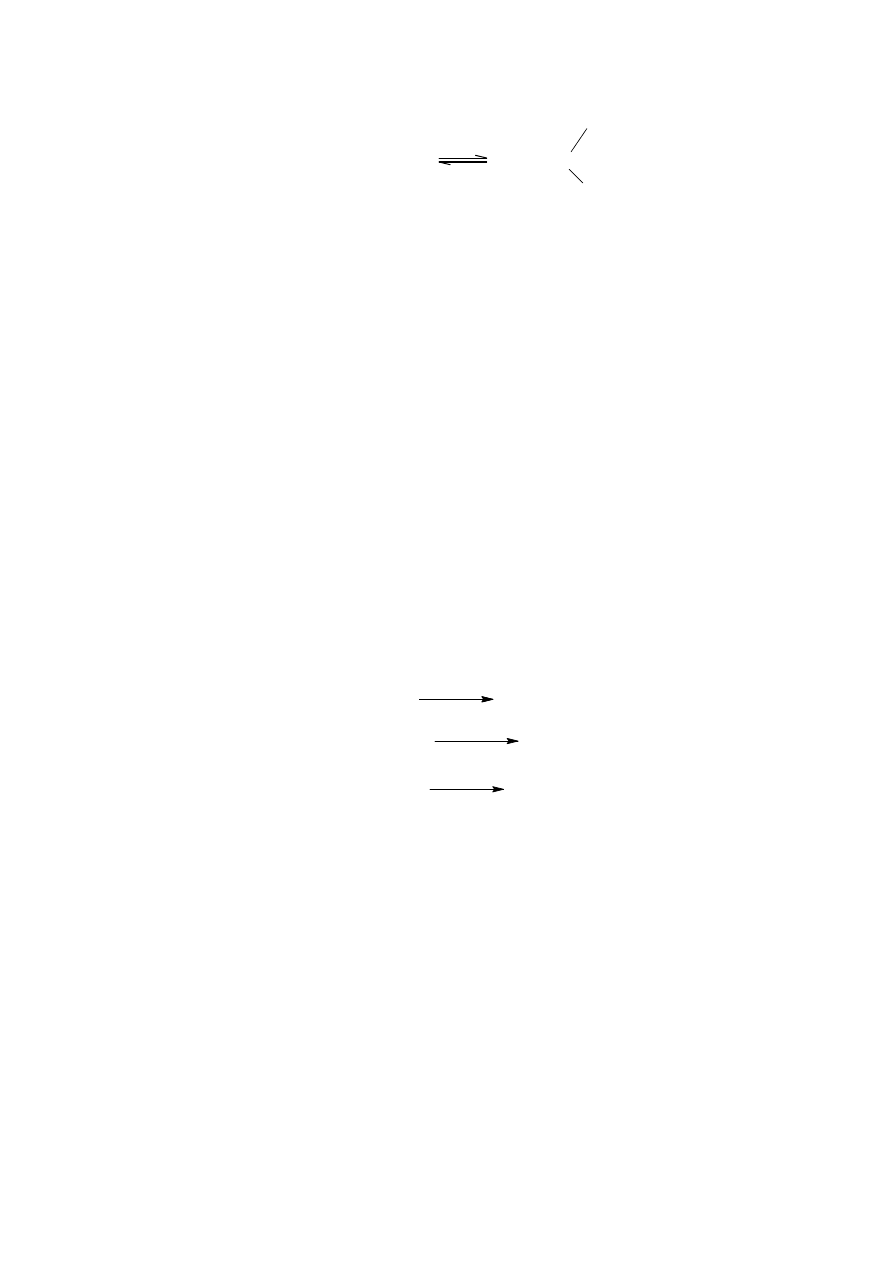
7
Kwasy te przypominają bardziej formę enolową ketonów – są bardziej kwasowe niż
związki nitrowe (2-5 jednostek pK
a
), a równowaga leży bardziej po stronie izomeru nitro.
Typowe wartości stałej równowagi K
eq
wynoszą od 10
-5
do 10
-7
.
Sole nitronowe są otrzymywane przez traktowanie związków nitrowych wodnymi
alkaliami. Można użyć również rozpuszczalników mieszających się z wodą, takich jak
dioksan, tetrahydrofuran (THF) lub alkohole, szczególnie kiedy związki nitrowe mają
ograniczoną rozpuszczalność w wodzie. Mocniejsze zasady są używane w nośnikach
aprotycznych. Dla zilustrowania, sole nitronowe pierwszorzędowych związków nitrowych
mogą być deprotonowane przez użycie n-butylolitu, jako zasady w aprotycznym
rozpuszczalniku jak np. tetrahydrofuran.
Wiele podstawionych związków nitrowych ulega reakcji Nefa. Zalicza się do nich γ-
nitroketony, γ-nitroalkohole, γ-nitroestry i γ-nitronitryle. Wszystkie z nich są dostępne
dzięki reakcji Michaela.
α
-Ketoaldehydy otrzymuje się z α-nitroketonów. α-Hydroksyaldehydy i α-hydroksyketony
są izolowane w kondensacji związków nitrowych z aldehydami, a następnie z reakcji Nefa.
Aldozy są często używane w reakcjach kondensacji z nitroparafinami w celu otrzymania
wysokofunkcyjnych związków nitrowych, które ulegają reakcji Nefa. α-Acetamidoaldozy
również mogą być otrzymywane na tej drodze. Inne polifunkcyjne związki, które ulegają
reakcji Nefa zawierają azydo-
β
-laktam 2.
RR
1
CHNO
2
RR
1
CH=N
O
-
OH
+
1
R
1
CH
2
NO
2
+ R
2
CH=CHY
R
1
CH(NO
2
)CHR
2
CH2Y
R
3
CHNO
2
-
+ R
2
CH=CHCOR
1
1. addycja
2. redukcja
R
3
CH(NO
2
)CHR
2
CH2CHOHR
1
R
1
CH
2
Y + R
2
CH=C(NO
2
)R
3
R
1
CH(Y)CHR
2
CH(NO
2
)R
3
8
Reakcja Nefa została użyta w kluczowym etapie przeniesienia 1,2 grupy karbonylowej.
Tak, więc keton jest nitrowany w pozycji alfa za pomocą estru nitrowego, a grupa
karbonylowa jest redukowana za pomocą borowodorku sodu. Utrata wody, a po niej
sprzężona redukcja z użyciem borowodorku sodu daje związek nitrowy, który jest
następnie poddawany reakcji Nefa. Niestety, ponieważ redukcja pewnych nitroolefin nie
jest kompletna, redukcyjny proces Nefa, w którym używa się cynku jest niezbędny, żeby
otrzymać przyzwoite rezultaty.
Pewne aromatyczne związki nitrowe ulegają addycji nukleofili do otrzymania anionów
soli nitronowej, które mogą być protonowane albo do związku nitrowego albo do produktu
reakcji Nefa.
Na przykład, addycja reagenta Grignara do 9-nitroantracenu, po której następuje działanie
zbuforowanego kwasu octowego, daje izomer cis. Podobne rezultaty otrzymano z
nitronaftalenem. Proces Nefa jest obserwowany również w reakcji o-nitrobenzonitrylu z
cyjankiem sodu, gdzie 2,6-dicyjanofenol jest otrzymywany z 60 - 70% wydajnością.
N
N
3
(CH
2
)
2
NO
2
O
CH
2
C
6
H
6
(OCH
3
)
2
-2, 4
1. NaOCH
3
2. H
2
SO
4
, CH
3
OH
N
N
3
CH
2
CH(OCH
3
)
2
O
CH
2
C
6
H
6
(OCH
3
)
2
-2, 4
2
(95%)
R
1
COCH
2
R
R
1
COCHRNO
2
NaBH
4
R
1
CHOHCHRNO
2
R
1
CH=CRNO
2
R
1
CH
2
CHRNO
2
R
1
CH
2
COR
O
2
N
1. RMgX
2. CH
3
CO
2
H, CH
3
CO
2
K
NO
2
R
(100%)
9
3.2.
Reakcje boczne, komplikujące reakcję Nefa
Sole nitronowe są reaktywne wobec elektrofili z powodu delokalizacji ładunku
ujemnego, a to często prowadzi do komplikacji. Addycja protonu do atomu węgla alfa
regeneruje związek nitrowy, podczas gdy pożądany proces Nefa wymaga protonacji na
jednym z atomów tlenu do otrzymania kwasu azotowego. Bardziej stabilne sole nitronowe
dążą do przemiany w związek nitrowy po zakwaszeniu. Ta regeneracja nitroparafin jest
jedyną reakcją, jeżeli zostaną użyte łagodne kwasy, zdolne do niszczenia kwasu
azotowego(III) (takie jak chlorowodorek hydroksyloaminy lub mocznik i kwas octowy).
Inny problem pojawia się na skutek tego, że jon azotanowy(III) jest dobrą grupą
odchodzącą. Stanowi to problem szczególnie wtedy, gdy grupa nitrowa jest w położeniu
beta do grupy zakwaszanej, jak na przykład grupa karbonylowa. Kiedy jest to reakcja
boczna dla reakcji Nefa, może mieć użyteczność syntetyczną.
Jak wspomniano powyżej, kwasy mogą powodować reakcje inne niż proces Nefa,
chociaż produkty mogą być identyczne. Istnieje nawet raport, gdzie opisana jest
bezpośrednia konwersja związku nitrowego w keton dzięki użyciu kwasu. Tak więc 2-
nitrooktan daje 2-oktanon po długotrwałym przepływie z kwasem hydrochlorowym w
układzie heterogenicznym. Przemiana po blisko dwóch tygodniach ogrzewania zachodzi
tylko w 35%. Działanie mocnych kwasów na związki nitrowe zostały odkryte przez
Meyera 11 lat przed tym, jak Nef zapisał swoje pierwsze obserwacje. W reakcji Meyera,
pierwszorzędowe związki nitrowe są przekształcane w kwasy karboksylowe przez
traktowanie ich kwasem hydrochlorowym lub kwasem siarkowym(VI).
NO
2
CN
NaCN
DMSO, 100
o
CN
OH
CN
(75%)
RCH
2
NO
2
H
3
O
+
RCO
2
H
10
W procesie pojawiają się kwasy hydroksamowe 3 jako związki pośrednie, które można
zazwyczaj łatwo wyizolować przez uniknięcie ogrzania.
Mechanizm reakcji Meyera pokazano na przykładzie reakcji 2.
Gruntowne przebadanie kinetyki wskazuje, że reakcja biegnie w maksymalnym tempie,
kiedy pH jest mniejsze niż pH wymagane do protonacji wszystkich neutralnych kwasów
nitronowych. To sugeruje, że konkurencyjna reakcja ma miejsce, gdy zachodzi O-
protonacja kwasu nitronowego, po której następuje utrata wody i protonu, w wyniku czego
tworzy się tlenek azotu. Tlenek azotu jest wyłapywany w trakcie 1,3-dipolarnej
cykloaddycji do alkenów i alkinów.
Kilka raportów wskazuje na to, że sole nitronowe pierwszorzędowych związków
nitrowych, po zakwaszeniu dają kwasy karboksylowe. Chociaż może to się wydawać
"nienormalną" reakcją Nefa, niewątpliwie reakcja Meyera jest prawdziwą ścieżką, z
powodu zastosowania mocnego kwasu i energicznych warunków. Bezpośrednie
zakwaszenie 1-fenylonitroetanu i 1-fenylonitropropanu w obecności azotanu(III) potasu
daje odpowiednio acetofenon oraz propiofenon.
Nitroalkany czasem ulegają kondensacji między sobą, po wystawieniu na działanie
zasady. Jak można przewidywać, proces ten jest poważnym problemem, kiedy używamy
małych, nierozbudowanych związków nitrowych, takich jak nitrometan, który pod
wpływem jonu wodorotlenowego łatwo formuje jon (
-
O
2
N=CHCH=NO
-
). Dodatkowo
małe, pierwszorzędowe nitroparafiny (nitroetan, 1-nitropropan i 1-nitrobutan) ulegają
trimeryzacji do izoksazoli, w obecności nawet słabych zasad, takich jak trietyloamina lub
RCH
2
NO
2
H
3
O
+
RCHHOH
O
RC
NOH
OH
3
RCH
2
NO
2
H
+
C
H
2
N
+
OH
O
-
-H
2
O
RC
N
+
O
-
H
2
O
3
H
2
O
ogrzew.
RCO
2
H + NH
2
OH
H
2
O
ogrzew.
Reakcja 2.
11
węglan potasu. Przemiana ta biegnie przez aldehyd, który ulega kondensacji z
nitroalkanami.
Inną reakcją, która komplikuje proces Nefa, jest tworzenie pseudonitroli 4 z
drugorzędowych związków nitrowych i kwasów 5 z pierwszorzędowych związków
nitrowych. Te produkty są faworyzowane w reakcji powolnej addycji soli nitronowych do
kwasów.
Ponieważ kwas azotowy(III) jest powodem nitrozowania, addycja dobrego związku,
który usunie kwas azotowy(III), takiego jak mocznik, zapobiega temu problemowi.
Pewne polifunkcyjne molekuły mogą prowadzić do niepożądanych produktów, z
powodu interakcji sąsiadujących grup lub utraty właściwości pewnych grup pod wpływem
warunków prowadzonej reakcji. Niecałkowita reakcja Nefa związku dinitrowego 6 lub
kwasu nitrowego 7 prowadzi głównie do produktów heterocyklicznych, przypuszczalnie
przez odpowiednie kwasy nitronowe.
3 RCH
2
NO
2
K
2
CO
3
H
2
O, ogrzew.
N
O
R
R
R
RR
1
CHNO
2
1. NaOH
2. H
+
RR
1
C(NO)NO
2
R
1
=H
RC(NO
2
)=NOH
4
5
CH
2
NO
2
CH
2
NO
2
Na
2
CO
3
ogrzew.
O
OH
O
CH
2
NO
2
CO
2
H
NH
3
, H
2
O
ogrzew.
O
O
NOH
6
7
(50%)
(67%)
12
Nitrolakton 8 ulega zarówno otwarciu pierścienia jak i reakcji Nefa i daje nieoczekiwany
acetal.
Utrata jonu azotanu(III) przez wewnątrzmolekularne wyparcie zachodzi szybciej niż
zakwaszenie i komplikuje reakcję α-nitrotoluenu z nitrostilbenem.
Mocny kwas użyty w procesie Nefa powoduje dehydratację związku 9 zamiast reakcji
Nefa, i prowadzi do częściowego odwodnienia związku nitrowego 10, co powoduje
powstanie produktów ubocznych.
O
CH
2
NO
2
O
1. NaOCH
3
2. H
2
SO
4
, CH
3
OH
O
OCH
3
CH
2
CO
2
CH
3
8
(98%)
C
6
H
5
CH
2
NO
2
+ C
6
H
5
C(NO
2
)=CHC
6
H
5
NaOCH
3
ogrzew.
O
N
+
H
5
C
6
O
-
C
6
H
5
C
6
H
5
(82%)
S
OH
NO
2
OH
1. zasada
2. HCl
S
OH
NO
2
+
S
NO
2
OH
9
(65%)
(35%)
13
2-Nitro-1-butanol daje mieszaninę zarówno produktu Nefa (1-hydroksy-2-butanon) i 2-
nitro-1-butenu jak i pewną ilość oksymu. Te współzawodniczące reakcje są zależne od pH.
Proces Nefa jest faworyzowany w wysoko kwaśnym środowisku (najlepsze jest pH
wynoszące ok 1.1).
γ
-Nitroketony
pochodzące
z
addycji
β
-ketoestrów
do
nitroolefin
ulegają
wewnątrzmolekularnym reakcjom w obecności alkoholanu sodu lub wodorotlenku potasu
oraz niecałkowita reakcja Nefa z kwasami do otrzymania furanów 11, 12, lub 13.
N
CH
3
H
O
2
N
N
O
OH
OCH
3
1. KOH, CH
3
OH
2. H
2
SO
4
N
CH
3
H
O
N
O
OH
OCH
3
+
N
CH
3
H
O
N
O
OCH
3
(28%)
(46%)
10
CH
3
CHOCH
2
CO
2
R + R
1
CH=C(NO
2
)R
2
zasada
O
CO
2
R
R
1
R
2
O
CO
2
R
R
1
R
2
O
CO
2
R
N
RO
2
C
O
R
1
R
2
11
12
13
14
R
R
1
R
2
C
2
H
5
C
6
H
5
C
6
H
5
(72%)
−
−
C
2
H
5
4-ClC
6
H
4
C
6
H
5
(3%)
(14%)
−
CH
3
2,4-Cl
2
C
6
H
3
CH
3
−
−
(70%)
W przeciwieństwie, obecność sąsiadującej grupy karboksylowej powoduje przyspieszenie
reakcji Nefa z kwasem 4-nitrowalerianowym, być może przez intramolekularną
protonację.
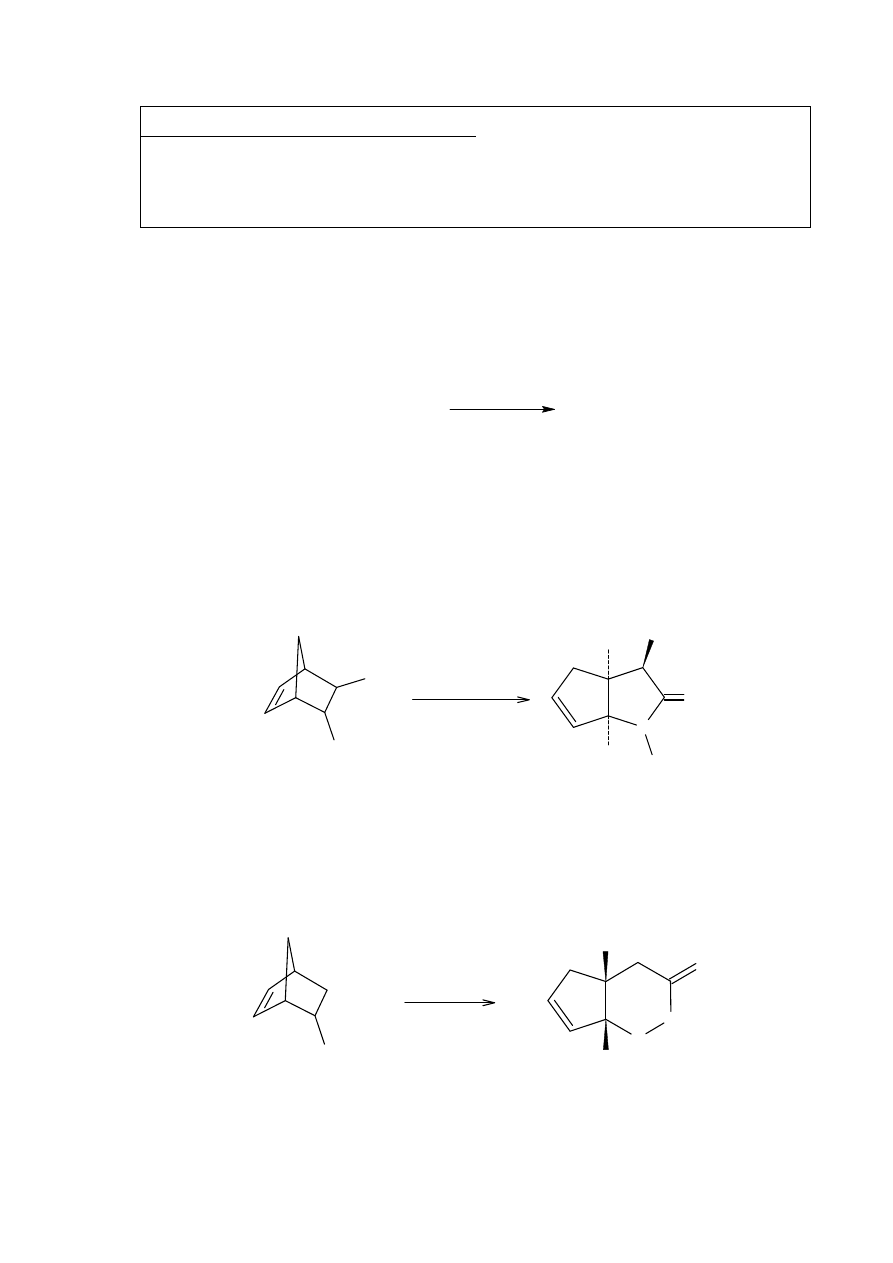
Kiedy substraty mają skłonność do formowania karbokationów, w warunkach reakcji
Nefa zdarza się również przestawienie; na przykład, substraty zawierające szkielet
bicyklo[2.2.1]heptanu.
Próba
przeprowadzenia
reakcji
Nefa
z
5-nitro-6-
fenylobicyklo[2.2.1]hept-2-enem zakończyła się niepowodzeniem. Struktura produktu
przestawienia została później pokazana, jako N-hydroksylaktam 14.
Podobna reakcja zachodzi, kiedy grupa fenylowa jest przestawiona z grupą metylową.
Produkty przestawienia z bardzo podobnych związków takich jak 15 są strukturalnie różne
od siebie.
CH
3
CH(NO
2
)(CH
2
)
2
CO
2
H
NaOH, H
2
O
CH
3
CO(CH
2
)
2
CO
2
H
NO
2
C
6
H
5
1. KOH, CH
3
OH
2. HCl
N
H
H
OH
O
C
6
H
5
14 (40%)
NO
2
1. NaOH
2. HCl
H
H
NH
O
O
15
(42%)
15
Oba produkty powstają przez otwarcie pierścienia związku acy-nitrowego, dzięki czemu
trzymuje się tlenek azotu a następnie kwas hydroksamowy, który może formować
pierścień albo przez atak na tlen albo przez atak na azot.
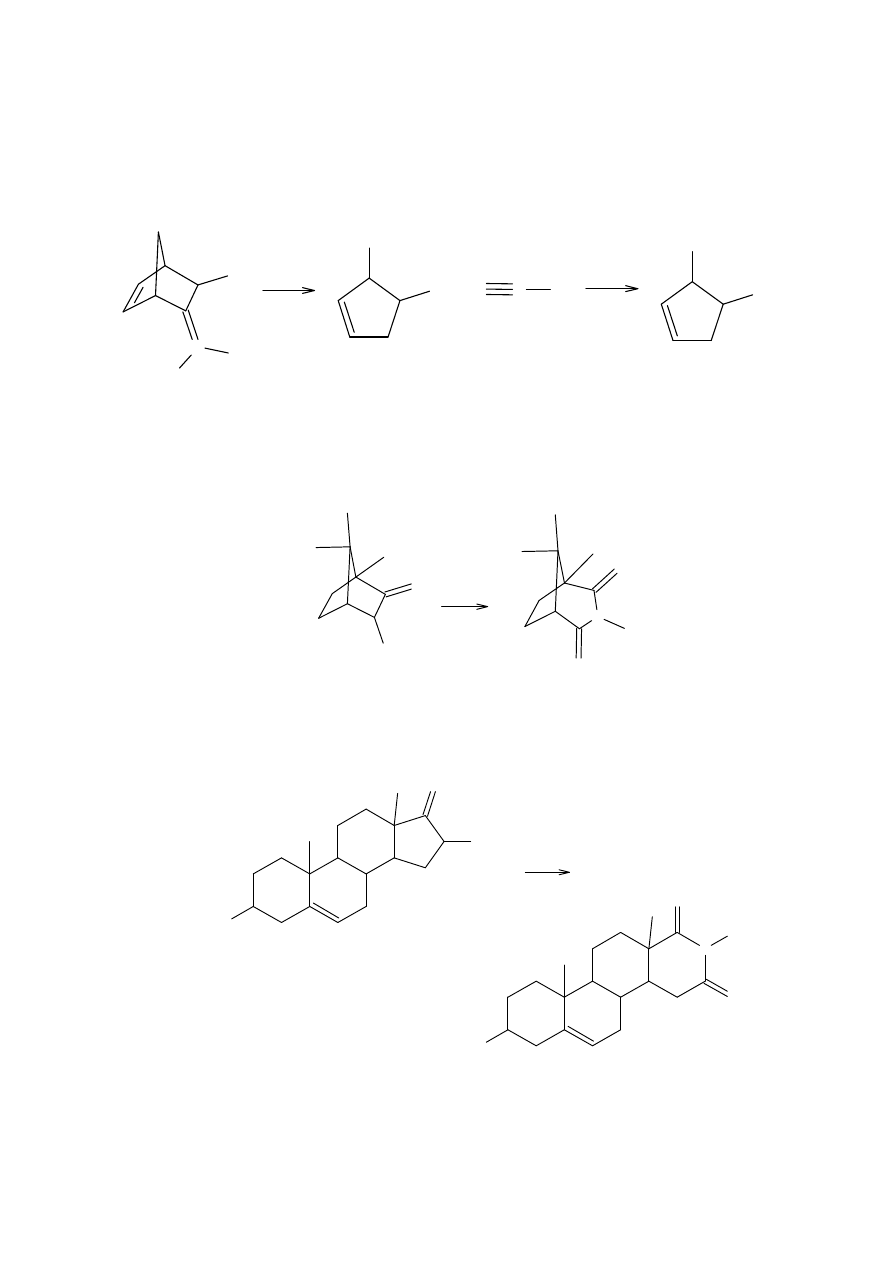
α
-Nitrokamfora jak również odpowiednia sól nitronowa, pod działaniem kwasu
hydrochlorowego, w podobnym mechanizmie dają N-hydroksyimid.
Podobna reakcja zachodzi z nitrosteroidem 16.
Wiele cyklicznych α-nitroketonów ulega temu typowi przestawienia w warunkach
kwaśnych. Jednakże, działanie nukleofili takich jak woda czy alkohole w warunkach albo
O
NO
2
HCl
N
O
OH
O
(94%)
O
H
O
NO
2
HCl
O
H
N
O
OH
O
(75%)
16
N
+
O
H
OH
R
OH
CH(R)C
N
+
O
-
OH
CH(R)CONHOH
16
kwaśnych albo zasadowych daje "otwarty" kwas nitrowy lub ester na drodze wstecznej
reakcji aldolowej.
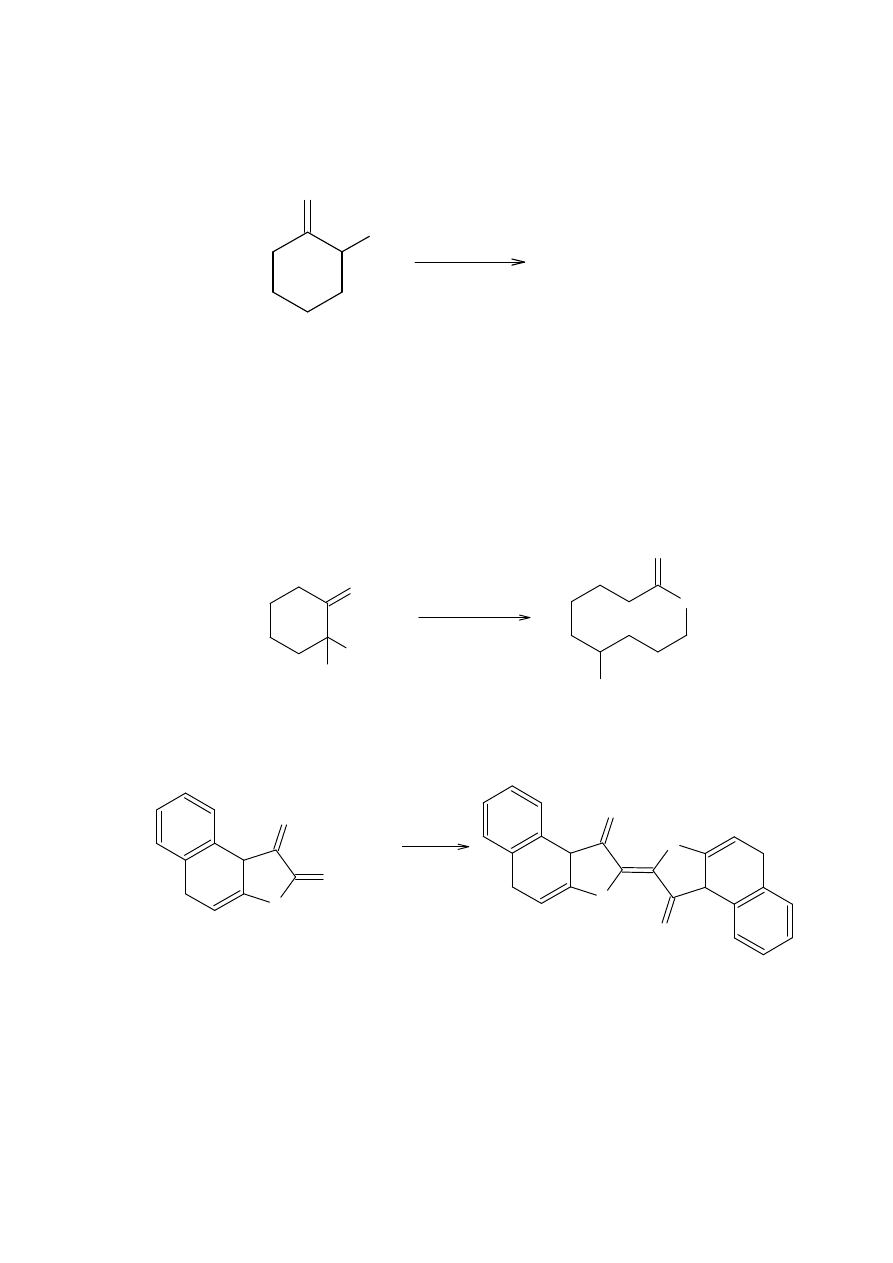
Interesującą modyfikacją, jest wewnątrzcząsteczkowy wariant tej reakcji, który może być
użyty do otrzymania makrocyklicznych związków nitrowych. Na przykład, 10-członowy
pierścień
nitrolaktonu
jest
otrzymywany
przez
reakcję
podstawionego
nitrocykloheksanonu 17 z katalityczną ilością wodorku sodu w gorącym 1,2-
dimetoksyetanie. Taki cykliczny związek nitrowy może być następnie poddany reakcji
Nefa do otrzymania ketolaktonu lub keto dikwasu.
Sól α-nitroketonu 18 dimeryzuje, kiedy jest poddana działaniu kwasu.
Niektóre produkty reakcji Nefa mają skłonność do ulegania epimeryzacji. Ester
nitrowy jest otrzymywany prze cykloaddycję 1-nitrocykloheksenu do cykloheksenu. Ester
ten jest czuły na utratę aktywności optycznej pod zwykłymi warunkami reakcji Nefa z
kwasem siarkowym i wodą. Kiedy reakcja jest prowadzona w obecności glikolu
O
NO
2
NaHCO
3
H
2
O
O
2
N(CH
2
)
5
CO
2
H
(85%)
O
(CH
2
)
3
OH
NO
2
kat. NaH
CH
3
O(CH
2
)
2
OCH
3
,
ogrzew
O
O
NO
2
(93%)
17
O
O
NO
2
-
Na
+
H
3
O
+
ogrzew.
O
O
O
O
18
17
etylenowego w 0
o
, nie obserwuje się żadnej epimeryzacji w wyizolowanym
hydroksyketalu.
Pewne związki nitrowe nie są wstanie przereagować w warunkach reakcji Nefa. Na
przykład, niezmienioną mieszaninę nitrodeoksyinozytolu 19 można odzyskać z
niecałkowicie przebiegającej reakcji Nefa z użyciem wodorotlenku baru a następnie kwasu
siarkowego.
Wiele fluorowanych związków nitrowych również nie ulega reakcji Nefa. Możliwe jest, że
reakcje te nie zachodzą, ponieważ aniony soli nitronowej nie są całkowicie formowane.
Użycie mocniejszych zasad lub zmodyfikowanie warunków reakcji Nefa mogłoby być
pomocne. W innych układach reakcja Nefa nie zachodzi, ponieważ odpowiednie sole
nitronowe są wysoko stabilizowane i mają skłonność do protonowania raczej węgla niż
tlenu. Przykładami mogą być związki nitrowe 20 i związki heterocykliczne 21. Benzylowy
związek nitrowy 22 również nie daje użytecznych ilości produktów reakcji Nefa, możliwe,
ż
e z podobnych powodów.
O
N
+
H
H
O
-
H
H
2
SO
4
HO(CH
2
)
2
OH, 0
O
O
O
H
OH
(88%)
OH
OH
OH
OH
OH
OH
19
O
H
R
NO
2
N
NC
CN
C
H
3
CH
2
NO
2
CH
3
OCH
3
OCH
3
CH(NO
2
)R
CH
2
CH=CH
2
OCH
3
20
21
22
R=H, CH
3
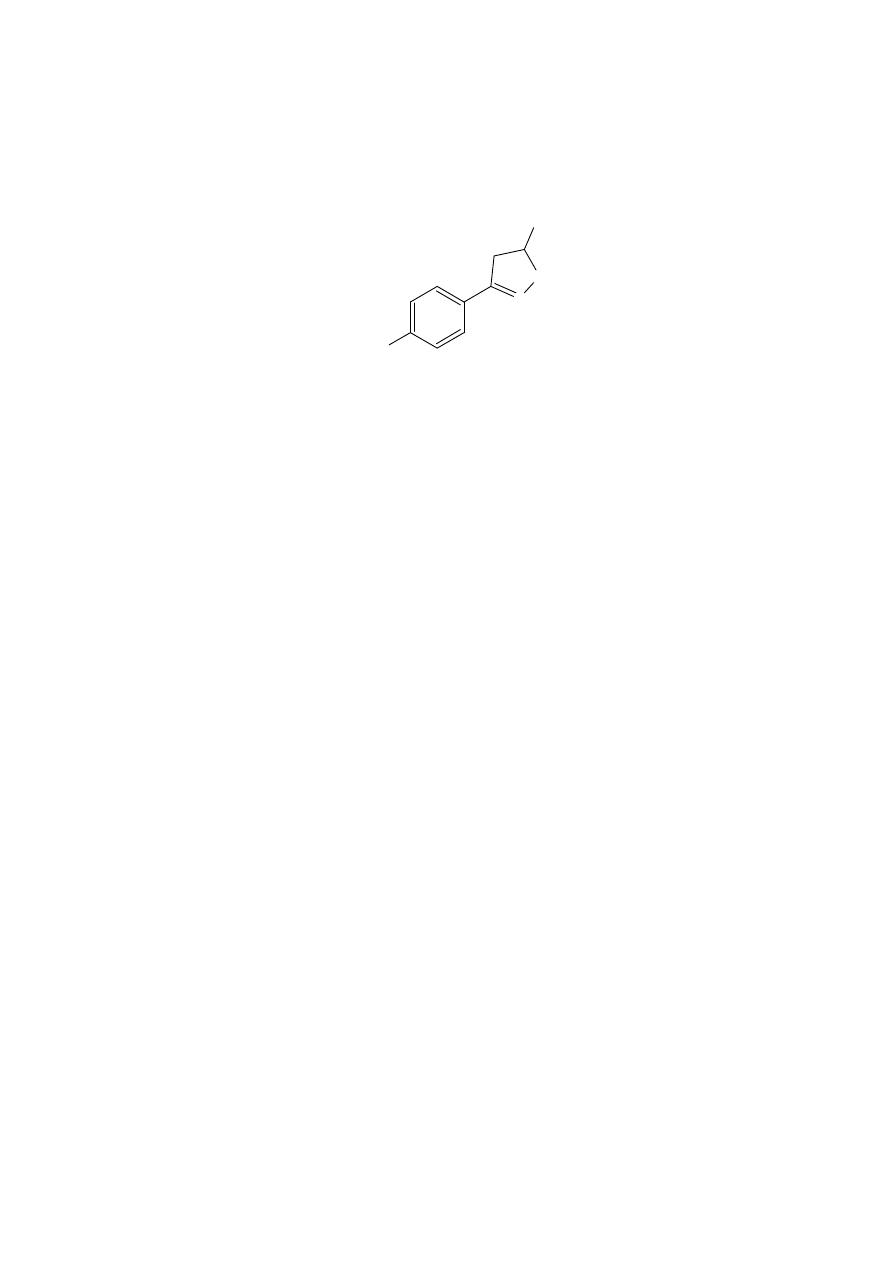
18
Innymi układami w których reakcja Nefa nie zachodzi są związki nitrowe 23, w których
tylko początkowy materiał jest odzyskiwany po działaniu etanolanu sodu w -15
o
a
następnie dodaniu kwasu siarkowego.
Użycie bromu zamiast kwasu siarkowego daje związek α-bromonitrowy, i pokazuje, że jest
formowana sól nitronowa. Rezultaty wydają się wykluczać eliminację do nitroolefin,
chociaż zakwaszenie nitroolefin może dać 23 przez sprzężoną addycję pośredniego
oksymowego nitroalkenu. Przez bromowanie nitroolefin można otrzymać związek z
bromem podstawionym w pozycji alfa.
3.3.
Zmodyfikowane reakcje Nefa
Z kilku powodów znaczne wysiłki zostały włożone w modyfikację warunków reakcji
Nefa. Po pierwsze, pewne związki mają skłonność do ulegania reakcjom bocznym bądź nie
ulegają reakcji Nefa w ogóle, o czym wspomniano w poprzednim rozdziale. Po drugie,
użycie najpierw zasady a następnie kwasu, tak jak w tradycyjnej reakcji Nefa, jest nie do
pogodzenia jeśli mamy do czynienia ze związkami z wieloma grupami funkcyjnymi.
Dlatego też, zakres reakcji Nefa został znacznie poszerzony przez użycie
zmodyfikowanych metod do osiągnięcia tej przemiany. Wiele modyfikacji wykorzystuje
reagenty utleniające bądź redukujące. Te metody zostaną omówione.
Odczynniki utleniające. Liczne czynniki realizują przemianę Nefa na drodze
utleniania. Omówiono je poniżej w kolejności ich odkrycia.
Nadmanganian potasu. Jedna z modyfikacji reakcji Nefa, odkryta około 1900 roku,
używa nadmanganianu potasu do rozszczepienia soli nitronowych różnorodnych
N
O
R
CH
2
NO
2
23
R=H, NO
2
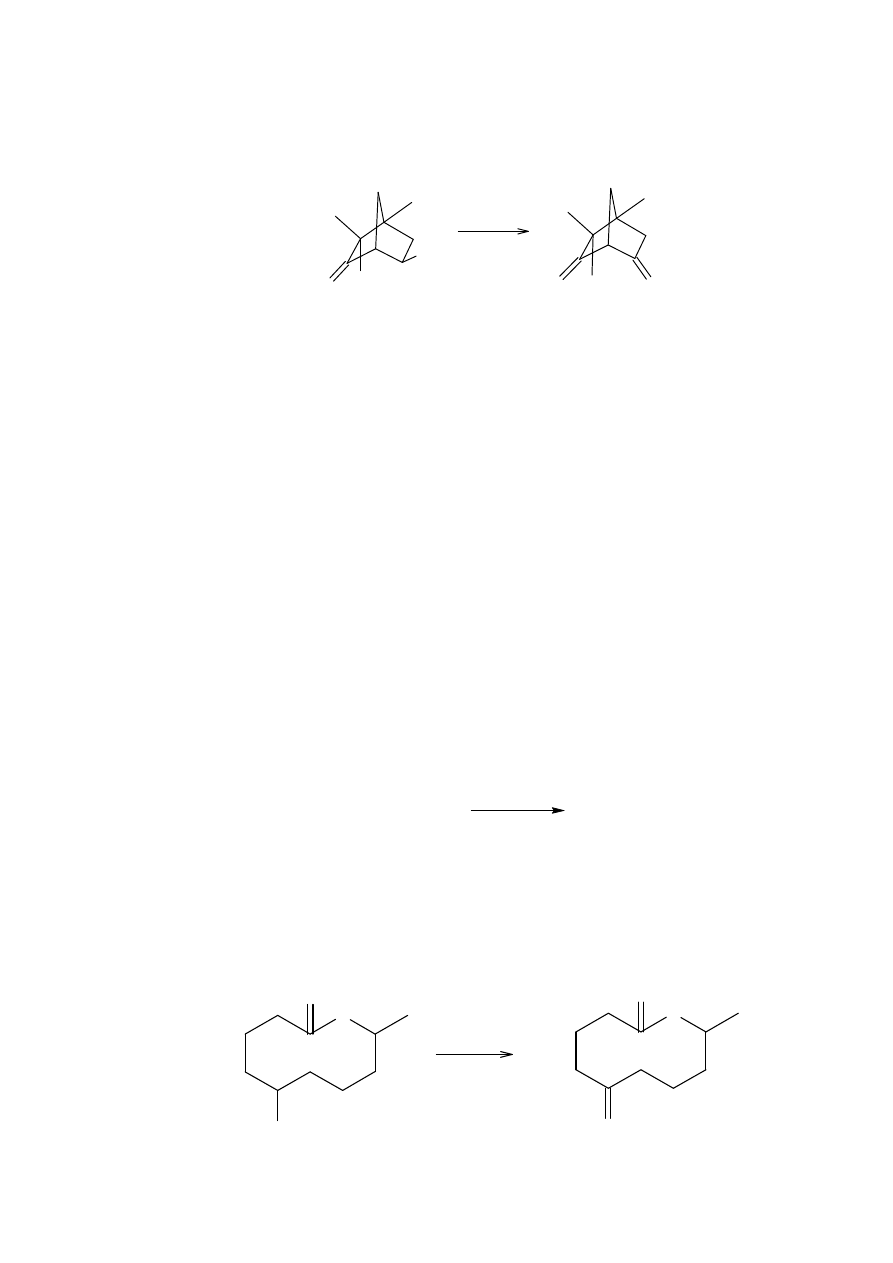
19
związków. Kiedy stosowano go do prostych związków nitrowych lub nienasyconych
bicyklicznych związków nitrowych jak 24, zakres wydajności wynosił 12-100%.
Znamienne jest, że utlenienie soli nitronowych jest szybsze niż rozszczepienie
podwójnego wiązania alkenów. Reakcja ta była ponownie badana w 1962 roku. Odkryto
wtedy, że zachodzi ona z wyższą wydajnością niż normalna reakcja Nefa. Dodatkowo,
przez uniknięcie nadmiaru nadmanganianu potasu można wyizolować aldehydy.
Kwasy karboksylowe są otrzymywane z pierwszorzędowych nitroparafin, kiedy użyje
się nadmiar reagenta. Reakcja jest zazwyczaj przeprowadzana w środowisku buforu z
siarczanem ołowiu lub solą boranu. Analizy kinetyki sugerują, że kluczowym etapem jest
atak jonu nadmanganianu na podwójne wiązanie C=N soli nitronowej.
Oryginalna procedura wymaga użycia wodorotlenku potasu do utworzenia soli
nitronowej, ale czasami prowadzi to do niekonsekwentnych rezultatów. Problem ten może
zostać ominięty przez użycie wodorku sodu w alkoholu tert-butylowym i pentanie. W tych
warunkach addycja uwodnionego nadmanganianu potasu prowadzi do wyizolowania z 59-
95% wydajnością aldehydu, takiego jak 25.
Użycie metanolanu litu a następnie nadmanganianu potasu daje z 95% wydajnością
ketolakton 95. Analogiczny ketolaktam można otrzymać w tej samej ogólnej procedurze.
NO
2
O
KMnO
4
24
(88%)
O
NO
2
O
O
O
O
1. LiOCH
3
2. KMnO
4
26
(95%)
t-C
4
H
9
O
2
CC(CH
3
)
2
C(CH
3
)
2
CH
2
NO
2
NaOC
4
H
9
-t
KMnO
4
, 0
O
t-C
4
H
9
O
2
CC(CH
3
)
2
C(CH
3
)
2
CHO
25
(91%)
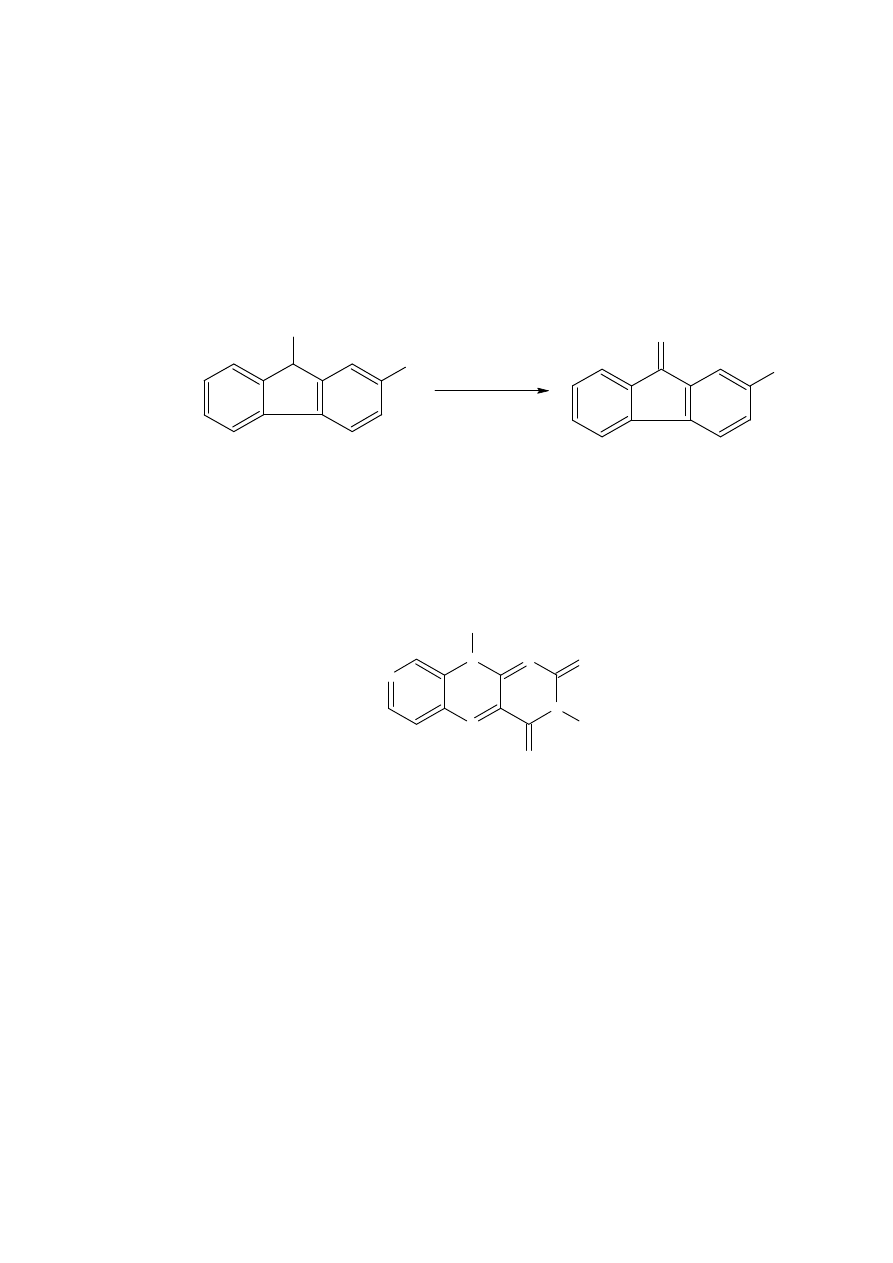
20
Nadmanganian cetylotrimetyloazaniowy w chlorku metylenu przekształca liczne
związki nitrowe w aldehydy i ketony w temperaturze pokojowej z dobrą wydajnością. Na
przykład, kamfora jest izolowana z 65% wydajnością, a heptanal z 71% wydajnością.
Tlen i ozon. Przekształcenie związku nitrowego 27 w odpowiedni keton przez użycie
etanolanu potasu i następnie działanie powietrza, z bliżej nieokreśloną wydajnością zostało
opisane 50 lat temu.
Ten typ reakcji był badany 20 lat później i odkryto, że przedstawia on autooksydację.
A zatem 2-nitropropan jest przekształcany w aceton i jon azotanowy(III) przez działanie
wodorotlenkiem sodu i powietrzem. Odkryto również, że reakcję to katalizuje 8-azaflawin
28.
Okazuje się, że utlenienie soli nitronowych cząsteczkowym tlenem nie ma zbyt dużej
użyteczności w syntezie, chyba, że zostanie wynaleziony jakiś niedrogi katalizator.
Chlorek żelaza przyspiesza formowanie acetonu, ale zakres i możliwość zastosowania
syntetycznego nie zostały zbadane.
Singletowy tlen również przekształca sole nitronowe w aldehydy i ketony. A zatem
napromieniowanie podstawowego roztworu czterech różnych związków nitrowych w
NO
2
Br
KOC
2
H
5
powietrze, kilka dni
O
Br
27
N
N
N
N
N
CH
3
O
CH
3
O
28
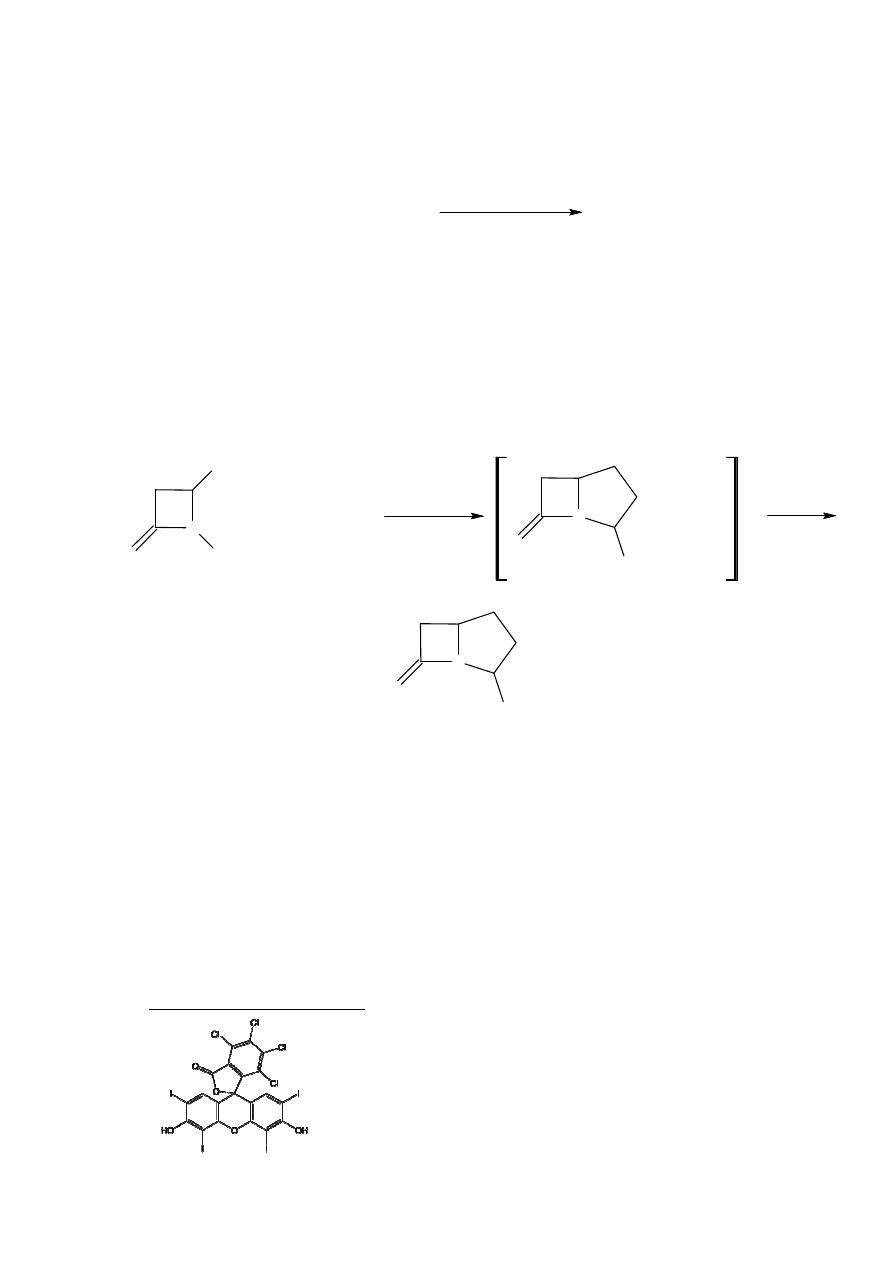
obecności tlenu oraz "Ró
67% wydajnością. Grupa ta zawiera nitroalkeny takie jak zwi
Ozon również realizuje reakcje podobne do reakcji Nefa. Zwi
deprotonowane metanolanem sodu w metanolu a nast
78
o
. Badania z siarczkiem dimetylu dały zwi
88%. Aldehydy również
ozonolizę soli nitronowej
Kwas m-chloroperoksybenzoesowy.
drugorzędowych związków nitrowych, zasad (np. 1,8
DBU), i chlorosilanów
chloroperoksybenzoesowym (MCPBA) do ut
Podstawione związki nitrowe, b
nitroolefin.
4
CH
3
CH(NO
2
)(CH
29
N
(CH
2
)
2
CH=C(NO
Si(CH
3
)
2
C
4
H
9
-t
O
"Róży Bengalskiej"
4
daje odpowiednie związki karbonylowe z 49
. Grupa ta zawiera nitroalkeny takie jak związek 29
realizuje reakcje podobne do reakcji Nefa. Związki nitrowe mog
deprotonowane metanolanem sodu w metanolu a następnie poddane
. Badania z siarczkiem dimetylu dały związki karbonylowe z wydajno
88%. Aldehydy również można otrzymać bez trudu. Tioestry mogą by
soli nitronowej wygenerowanej przez sprzężoną addycję.
chloroperoksybenzoesowy. Nitronany trialkilosilylu
ą
zków nitrowych, zasad (np. 1,8-diazabicyklo[5.4.0]undec
ów. Te estry soli nitronowych. reaguj
chloroperoksybenzoesowym (MCPBA) do utworzenia ketonów z 70
zki nitrowe, będące substratami mogą być przygotowane z odpowiednich
)(CH
2
)
2
CH=CH
2
NaOH, CH
3
OH, 0
O
Rose Bengal, O
2
, hv
CH
3
CO(CH
CH=C(NO
2
)SC
6
H
5
(n-C
4
H
9
)
4
N
+
F
-
-55
O
N
O
C(SC
N
O
COSC
6
H
5
21
ą
zki karbonylowe z 49-
29.
realizuje reakcje podobne do reakcji Nefa. Związki nitrowe mogą być
pnie poddane działaniu ozonem w -
zki karbonylowe z wydajnością rzędu 65-
bez trudu. Tioestry mogą być otrzymane przez
Nitronany trialkilosilylu są tworzone z
diazabicyklo[5.4.0]undec-2-en,
reagują z kwasem m-
worzenia ketonów z 70-99% wydajnością.
β
-
przygotowane z odpowiednich
CO(CH
2
)
2
CH=CH
2
(66%)
C(SC
6
H
5
)=NO
2
+ -
O
3

22
Nadtlenek wodoru. Inną zmodyfikowaną reakcją Nefa, w której używa się delikatnych
warunków reakcji, jest ta z użyciem nadtlenku wodoru. Związek nitrowy jest mieszany w
temperaturze pokojowej z 30% nadtlenkiem wodoru i węglanem potasu w metanolu, a
następnie poddany zakwaszeniu za pomocą rozcieńczonego kwasu hydrochlorowego.
Wydajności zarówno otrzymanych aldehydów jak i ketonów wynoszą 76-96%. Na
przykład, heksanal jest izolowany z 80% wydajnością, podczas gdy cykloheksanol jest
otrzymywany z 88% wydajnością. Kombinacja łagodnych warunków z wysokimi
wydajnościami sprawia, że jest to bardzo atrakcyjna alternatywa dla reakcji Nefa. Liczne
inne grupy funkcyjne mogą przetrwać w tych warunkach, chociaż to nie jest potwierdzone.
Odczynniki redukujące. Niewielka ilość reagentów przekształca związki nitrowe w
odpowiednie aldehydy i ketony na drodze procesu redukcji. Pomimo to, jest to ważne
rozszerzenie reakcji Nefa. Poniżej omówiono kilka najbardziej ważnych metod.
Trichlorek tytanu. Najbardziej szeroka redukcyjna metoda Nefa używa świeżo
przygotowanego uwodnionego trichlorku tytanu. Reaktywność tego odczynnika wymaga
modyfikacji w obojętnej atmosferze. Odczynnik redukcyjny może być przechowywany
nad cynkiem dla dłuższego okresu czasu przydatności. Niestety uwodniony trichlorek
tytanu ma mocno kwaśny odczyn (pH<1), dlatego estry mogą ulegać hydrolizie a
podwójne wiązanie węgiel-węgiel może ulegać izomeryzacji.
2-Metylo-2-nitropropan jest rozszczepiany do acetonu gorącym trichlorkiem tytanu.
Użycie buforu octanu amonu lub octanu sodu pozwala na zajście reakcji w pH równym ok.
5-6, oraz pozwala na przeżycie grup funkcyjnych. W tych warunkach, reakcja zachodzi
pomyślnie nawet z układami mającymi skłonność do katalizowanego kwasami
przestawienia, tak jak np. związek 31.
C
6
H
5
CH
2
OCH(CH
3
)CH(CH
3
)NO
2
1. DBU, (CH
3
)
3
SiCl
2. MCPBA
C
6
H
5
CH
2
OCH(CH
3
)COCH
3
(91%)
C
2
H
5
CH(NO
2
)(CH
2
)
2
COCH
3
TiCl
3
, H
2
O
CH
3
O(CH
2
)
2
OCH
3
, 25
O
C
2
H
5
CO(CH
2
)
2
COCH
3
(85%)
23
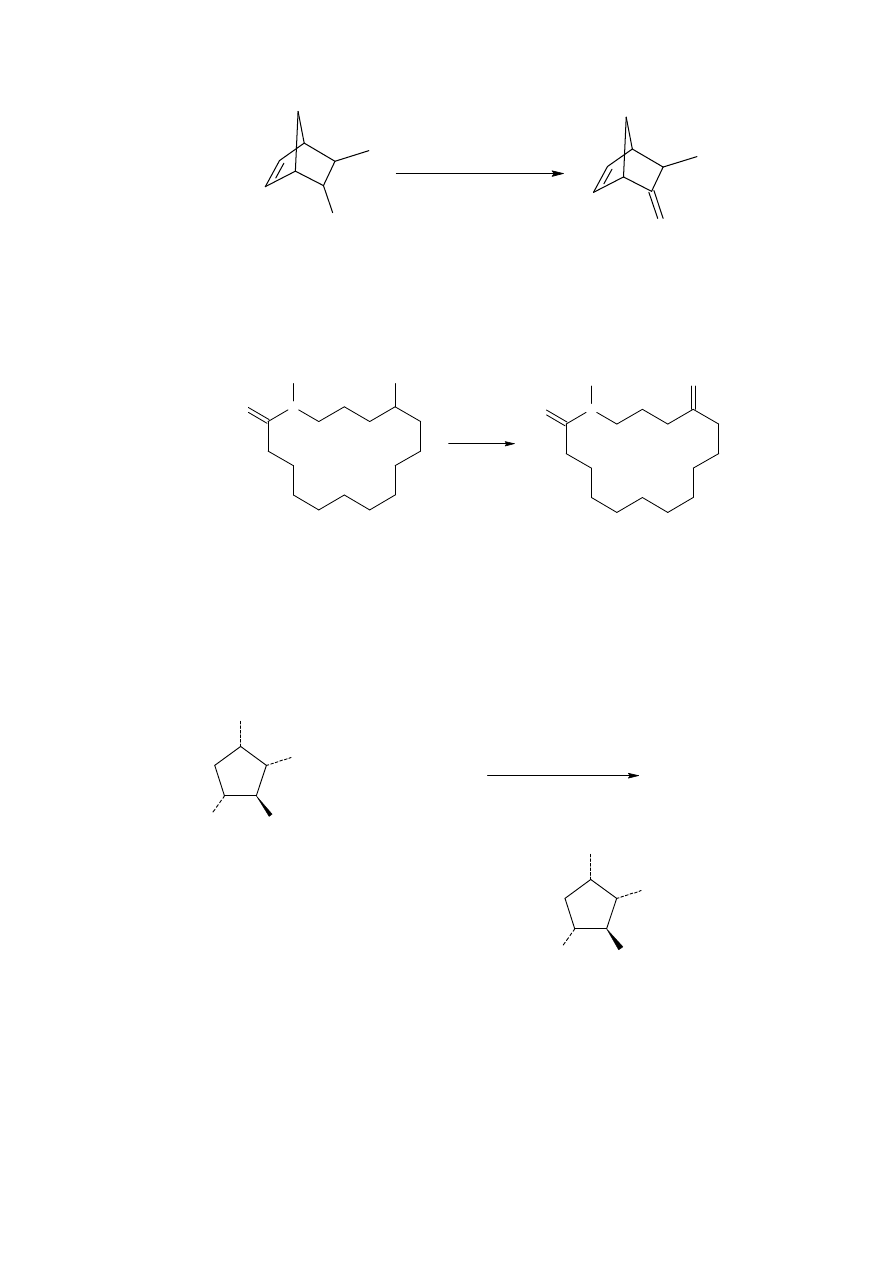
Aldehydy mogą być otrzymywane z nitrosteroidów, które nie ulegają konwencjonalnej
reakcji Nefa. Ta metoda również nie zawodzi w pewnych przypadkach, w których nie
udaje się metoda Nefa oparta na utlenieniu, tak jak związek 32.
Związki zawierające kilka grup funkcyjnych również ulegają pożądanej reakcji (reakcja
4). Przykład ten pokazuje że anion soli nitronowej jest często formowany przed addycją
zbuforowanej soli tytanu, chociaż istnieją przykłady kiedy sól nitronowa nie jest tworzona
przed addycją.
Użytecznym zastosowaniem syntetycznym reakcji Nefa jest generacja związków 1,4-
dikarbonylowych z α,
β
-nienasyconych prekursorów karbonylowych przez związek
nitrowy 33.
NO
2
O
1. NaOCH
3
, CH
3
OH
2. TiCl
3
, NH
4
O
2
CCH
3
31
(61%)
N
O
CH
2
C
6
H
5
NO
2
N
O
CH
2
C
6
H
5
O
TiCl
3
NaO
2
CCH
3
32
(63%)
OH
CH
2
CH(NO
2
)(CH
2
)
4
CO
2
CH
3
CH=CHCH(OR)C
5
H
11
-n
RO
R = t-C
4
H
9
Si(CH
3
)
2
1. NaOCH
3
, CH
3
OH
2. TiCl
3
, NH
4
O
2
CCH
3
, H
2
O
OH
CH
2
CO(CH
2
)
4
CO
2
CH
3
CH=CHCH(OR)C
5
H
11
-n
RO
(70%)
Reakcja 4.
24
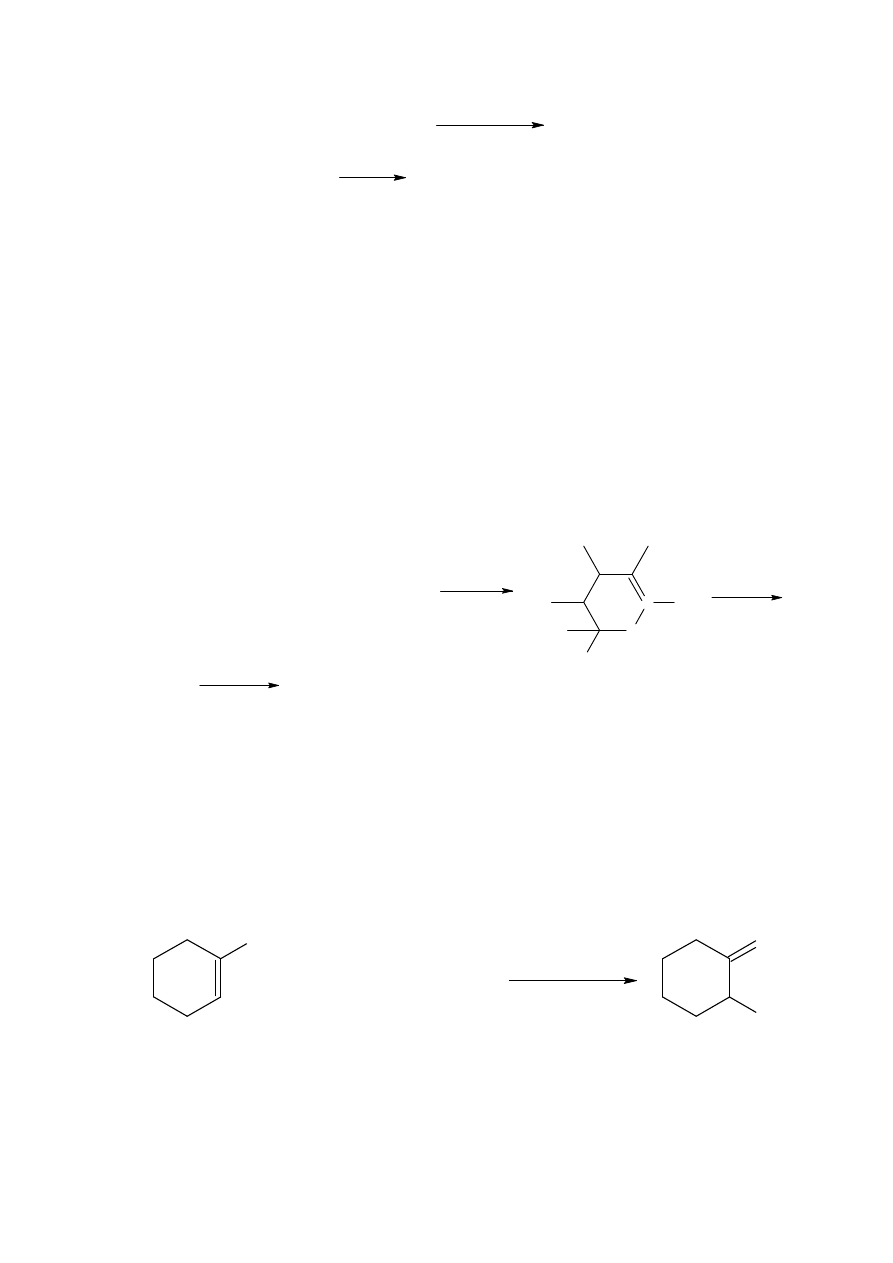
Chlorek tytanu jest używany w etapie zmodyfikowanej reakcji Nefa w tej sekwencji. Na
przykład, 1-nitropropan reaguje z ketonem metylowo-winylowym w obecności
diizopropyloaminy do otrzymania 5-nitro-2-heptanonu z 55% wydajnością. Traktowanie
otrzymanego związku trichlorkiem tytanu daje 2,5-heptanodion z 85% wydajnością.
Alternatywną drogą otrzymania związku 33 przez użycie kwasu Lewisa jako
katalizatora reakcji eteru enolowo sillilowego z nitroolefinami jest nawet bardziej dogodną
procedurą syntetyczną, ponieważ jest to często jednoetapowa reakcja. Często używane
kwasy Lewisa to tetrachlorek tytatu i chlorek cyny; chlorek glinu używany jest
okazjonalnie. W reakcji 5, związek O-silylowy 34 jest związkiem pośrednim.
Związek 34 został wyizolowany, przebadany spektroskopowo i oczyszczony kiedy jako
katalizatora użyto dichloroizopropoksytytanu. Hydroliza 34 do związku 1,4-
dikarbonylowego jest prosta i zachodzi ilościowo kiedy użyte zostają tetrachlorek tytanu,
chlorek cyny lub chlorek glinu. Zmiana setereoselektywności w nitro ketonie 33 jest
obserwowana
w
pewnych
układach
kiedy
jako
katalizatora
używa
się
dichloroizopropoksytytanu.
R
1
CH
2
NO
2
+ R
2
CH=CR
3
COR
4
R
1
CH(NO
2
)CHR
2
CHR
3
COR
4
TiCl
3
R
1
COCHR
2
CHR
3
COR
4
33
R
3
CH=CR
4
OSi(CH
3
)
3
+ R
2
CH=CR
1
NO
2
O
N
+
(H
3
C)
3
SiO
O
-
R
1
R
2
R
3
R
4
R
4
COCHR
3
CHR
2
COR
1
34
Reakcja 5.
OSi(CH
3
)
3
+
CH
2
=C(NO
2
)C
2
H
5
1. TiCl
4
, CH
2
Cl
2
2. H
3
O
+
CH
2
COC
2
H
5
O
(76%)
25
Chlorek wanadu(II). Proste ketony i aldehydy mogą być otrzymywane z 24-71%
wydajnością przez mieszanie związków nitrowych z chlorkiem wanadu(II), uwodnionym
kwasem hydrochlorowym i dimetyloformamidem. pH reakcji jest tak niskie, że grupy
wrażliwe na kwasy nie przeżywają. W rzeczywistości, oktanal jest otrzymywany z 1-
nitrooktanu tylko z 24% wydajnością z powodu konkurującej reakcji aldolowej.
Chlorek chromu(II). Związki nitrowe są przekształcane przez chlorek chromu(II) i
wodny kwas hydrochlorowy w gorącym metanolu w odpowiednie aldehydy i ketony z 32-
77% wydajnością i izolowane jako 2,4-dinitrofenylohydrazony. Ponieważ ten odczynnik
redukuje nitrobenzeny i sulfotlenki, inne dające się zredukować grupy funkcyjne nie mogą
być obecne. Oksymy są otrzymywane przez połączenie steroidowych związków nitrowych
z chlorkiem chromu(II) z krótkim okresem przepływu lub w temperaturze pokojowej
(reakcja 6). Chlorek chromu(II) jest niestabilny i musi być generowany in situ jak
pokazano w reakcji 6.
Inne odczynniki.
Azotan(III) sodu/ azotany(III) alkilu. Odczynnik złożony z azotanu(III) sodu i z estru
azotanu(III) w sulfotlenku dimetylu jest użyteczny, ponieważ dzięki niemu można uniknąć
mocnych kwasów i zasad. Związki nitrowe są deprotonowane przez azotan(III) sodu a
anion soli nitronowej jest nitrozowany przez azotan(III) alkilu. Otrzymywane wydajności
w temperaturze pokojowej wynoszą 67-90%. Ketony, amidy i pierścienie aromatyczne
przeżywają tą reakcję. Kwasy karboksylowe są otrzymywane z pierwszorzędowych
związków nitrowych, podczas gdy ketony są otrzymywane z drugorzędowych związków
nitrowych.
CH
3
CO
2
Cl
NO
2
C
8
H
17
1. Na
2
Cr
2
O
7
, Zn
(CrCl
2
)
2. CH
3
OH, HCl
przeplyw, 5 min
CH
3
CO
2
OH
3
C
NOH
C
8
H
17
Reakcja 6.

26
3.4.
Pokrewne reakcje związków nitrowych prowadzące do
produktów reakcji Nefa.
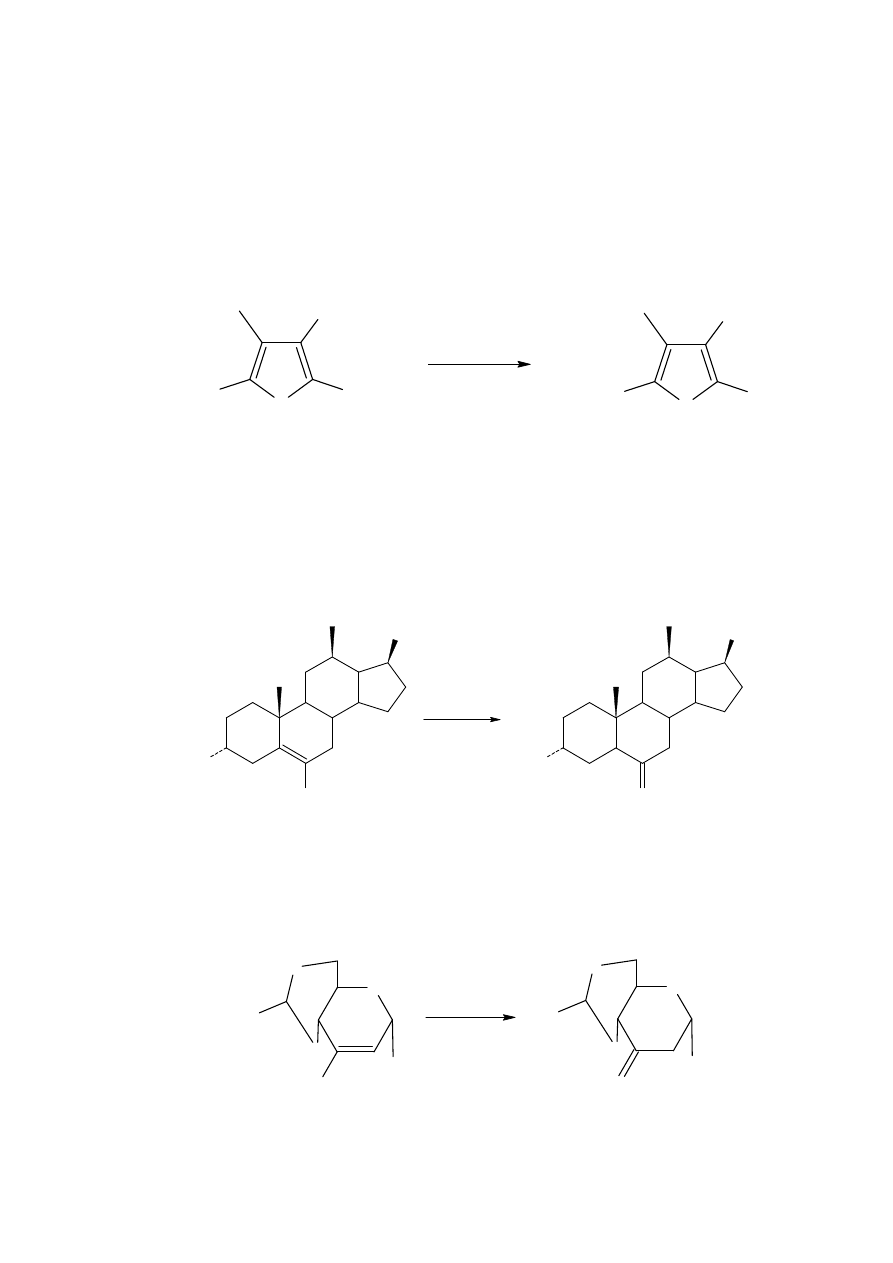
Alkilacja i acylacja związków nitrowych. Aniony soli nitronowej reagują z
elektrofilami albo przez tlen albo przez węgiel. Protonowanie prowadzi albo do regeneracji
związku nitrowego albo do reakcji Nefa. Alkilacja lub acylacja normalnie prowadzi do
produktów O-alkilowych (estru kwasu azotowego(III)) albo do O-acylowych (bezwodnik
kwasu azotowego(III)). Estry kwasu azotowego(III) są otrzymywane najbardziej
efektywnie przez alkilację soli nitronowych z solą oksoniową. Są szybko przekształcane w
związki karbonylowe przez uwodnione kwasu. Bezwodniki kwasu azotowego(III)
generalnie są niestabilne, a te utworzone z pierwszorzędowych związków nitrowych dają
tlenki azotu, które mogą być wyłapywane przez acetylenodikarboksylan dimetylu.
Alkilowanie związków nitrowych, a następnie hydroliza daje związki karbonylowe. Na
przykład, sól nitronowa 36 daje z 85% wydajnością odpowiednie diketony po działaniu na
niego siarczanem dimetylu i hydrolizie.
Sól dinitronowa 37 daje odpowiedni trion z 55% wydajnością.
Reakcje nitroolefin. Sprzężone nitroolefiny używane jako akceptory wielu nukleofili
dostarczają wielu użytecznych substratów dla reakcji Nefa, o czym wspomniano we
CH
3
CO(CH
2
)
2
CH(NO
2
)CH
3
NaNO
2
n-C
3
H
7
ONO
(CH
3
)
2
SO, 25
O
CH
3
CO(CH
2
)
2
COCH
3
(76%)
C
6
H
5
COCH=CHC(CH
3
)=NO
2
K
1. (CH
3
O)
2
SO
2
2. H
3
O
+
C
6
H
5
COCH=CHCOCH
3
36
(85%)
O
KO
2
N=C(H
3
C)HC
CHC(CH
3
)=NO
2
K
1. (CH
3
O)
2
SO
2
2. C
2
H
5
OH, ogrzew.
3. H
+
O
CH
3
COCH
CHCOCH
3
37
27
wcześniejszych rozdziałach. Jednakże istnieją inne reakcje, w których nitroolefiny są
przekształcane bezpośrednio w produkty reakcji Nefa.
Nitroolefiny mogą być redukowane przez metale do otrzymania albo oksymów albo
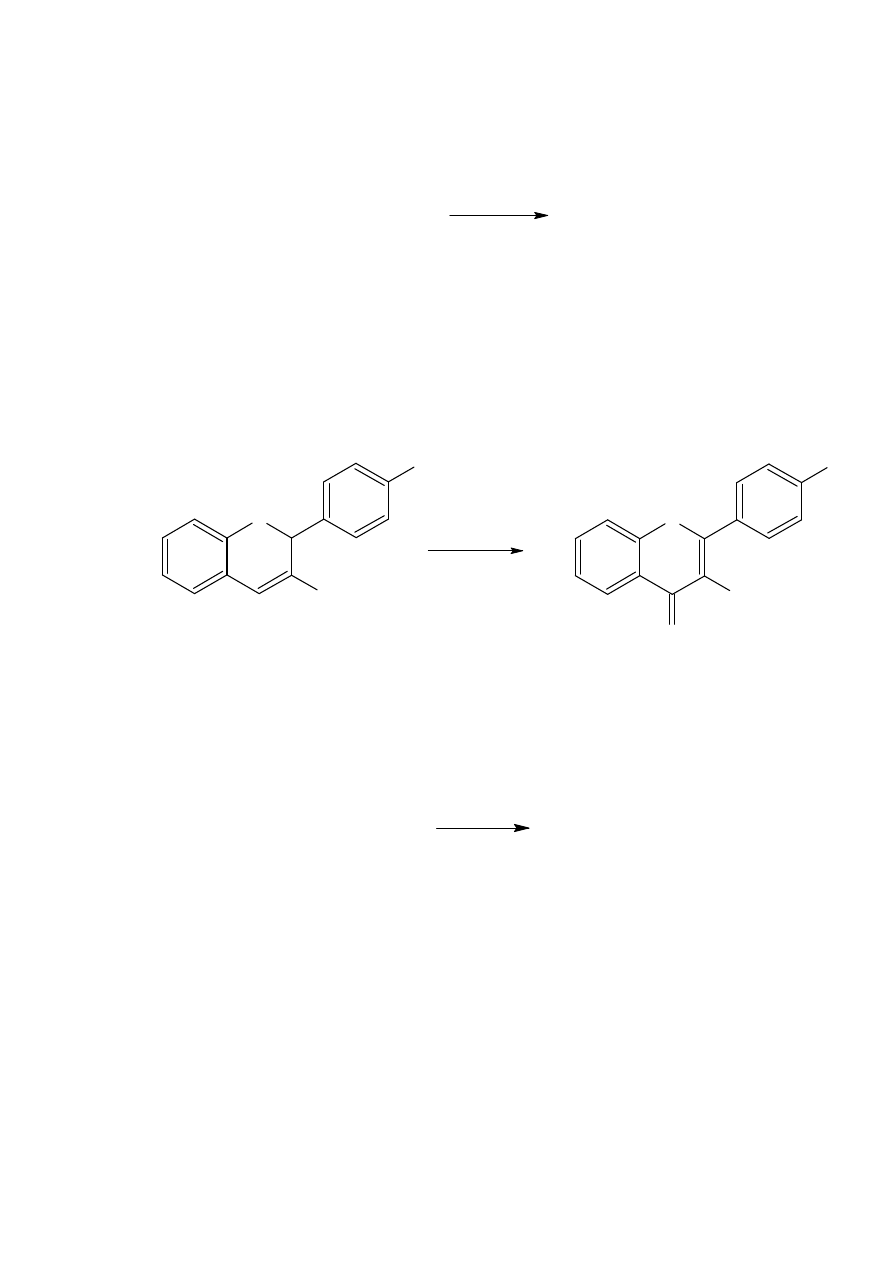
ketonów. Na przykład amalgamat glinu przekształca polifunkcyjną cząsteczkę 38 w oksym
39.
Do tej konwersji używa się również cynku w kwasie octowym. Na przykład, chlorek 6-
nitrocholesterylu daje ketosteroid 40 z 79-93% wydajnością kiedy reakcja zachodzi w tych
warunkach.
Przestawienie może się zdarzać, jednakże, kilka metylopiranozydów zawierających grupy
nitroolefinowe produkuje odpowiednie oksymy w wysokiej wydajności, co pokazuje że
grupa acetylowa przeżywa te warunki.
Cl
NO
2
C
8
H
17
Cl
O
C
8
H
17
Zn, HOAc
40
(79-93%)
O
O
H
5
C
6
O
O
2
N
OCH
3
O
O
H
5
C
6
O
HON
OCH
3
Zn, HOAc
(88%)
N
H
CH=CHNO
2
CO
2
C
2
H
5
C
2
H
5
O
2
C
N
H
CH
2
CH=NOH
CO
2
C
2
H
5
C
2
H
5
O
2
C
Al(Hg)
38
39
(50%)
28
ś
elazo i chlorek żelaza(III) reagują z nitrostyrenami w kwasie hydrochlorowym do
otrzymania ketonów szerokiej różnorodności układach.
Chlorek chromu(II) również tworzy ketony z 52-81% wydajnością z nitroolefin. Jest to
przeciwieństwo do wcześniejszych opisów ich przemiany w α-hydroksyketony, która była
początkowo proponowana jako metoda powstawania nitrozoalkenów. Chlorek chromu(II)
może być użyty do otrzymania α-diketonów przez ten sam tym związku pośredniego.
Pewne acykliczne
β
-arylo-α,
β
-nienasycone nitroolefiny w reakcji z chlorkiem chromu(II)
dają nasycone oksymy.
Chlorek cyny w alkoholach lub tiolach przemienia związki nitrowe w odpowiednie α-
alkoksy lub α-alkilotioketony. Na przykład, odczynnik ten powoduje przemianę
nitrocykloheksenu w etanolu w 2-etoksycykloheksanon z 79% wydajnością.
Istnieją również inne redukujące odczynniki dające różnorodne rezultaty. Borowodorek
sodu daje nasycone związki nitrowe. Borowodorek sodu/boran pozwala na otrzymanie
hydroksyloaminy przez redukcję pośrednich soli nitronowych. Tri-sec-butyloborowodorek
litu przemienia nitroalkeny w ketony po hydrolizie katalizowanej kwasem z 80-83%
wydajnością. Odwrotna addycja wodorku litu glinu do terminalnych nitroalkenów daje
aldiminy. Połączenie fosforanu(I) sodu i niklu Raney'a redukuje nitroolefiny do ketonu z
4-(CH
3
CONH)C
6
H
4
CH=C(NO
2
)CH
3
Fe, FeCl
3
HCl
4-(CH
3
CONH)C
6
H
4
CH
2
COCH
3
(68%)
ArCH=C(R)NO
2
ArCH
2
C(R=NOH
CrCl
2
O
NO
2
R
O
OH
R
O
CrCl
2
, HCl
THF
(60-65%)

29
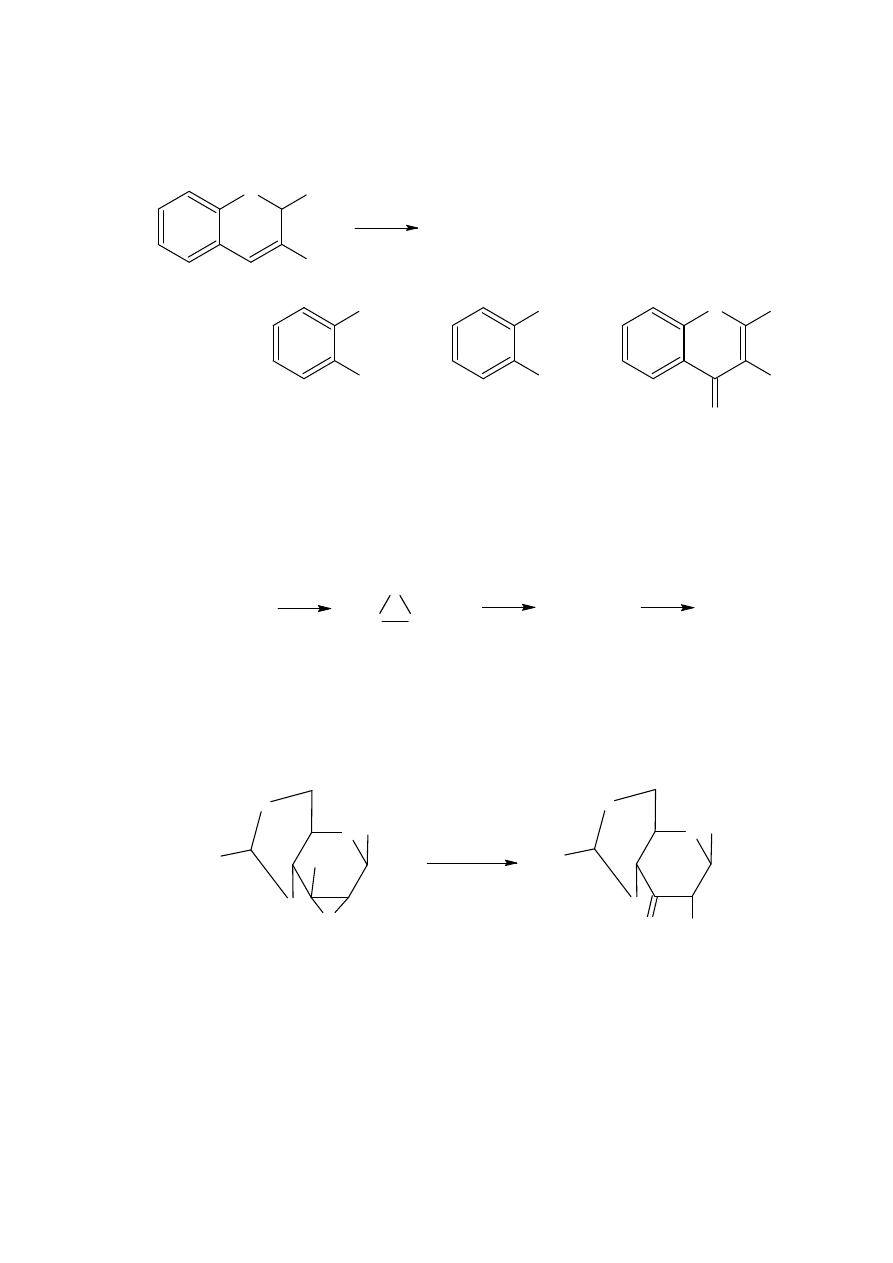
52-92% wydajnością bez wpływu na inne grupy funkcyjne takie jak estrowa czy
aromatyczna grupa nitrowa. Fosforan(I) sodu i pallad przemieniają nitroolefiny w oksymy.
Pewne nitroolefiny są przekształcane w α-chlorooksymy z dobrą wydajnością przez
działanie gazowym chlorkiem wodoru w eterze. Wodorek tributylocyny redukuje
nitroalkeny do estrów nitronowych tributylocyny, które reagują z kwasem m-
chloroperoksybezoesowym lub ozonem do aldehydów lub ketonów.
Wolnorodnikowa addycja nitroolefin do podstawionych triopirydyn w obecności
azobis(izobutyronitrylu) (AIBN) daje addukty które są rozkładane za pomocą trichlorku
tytanu do otrzymania ketonów lub kwasów z dobrą wydajnością.
Nitrohydrazony takie jak 41 dają acylohydrazyny po działaniu kwasem hydrochlorowym a
następnie uwodnioną pirydyną.
4-BrC
6
H
4
CH=C(NO
2
)CH
3
4-BrC
6
H
4
CH
2
COCH
3
4-BrC
6
H
4
CH
2
C(CH
3
)=NOH
NaH
2
PO
2
RaNi, pH 5
C
2
H
5
OH
NaH
2
PO
2
Pd/C, H
2
O, THF
(77%)
(44%)
4-CH
3
OC
6
H
4
CH=C(NO
2
)CH
3
4-CH
3
OC
6
H
4
CH
2
COCH
3
1. (n-C
4
H
9
)
3
SnH
2. MCPBA
(90%)
R
1
CH=C(NO
2
)R
2
N
RCH(R
1
)C(NO
2
)(R
2
)S
AIBN
RCO
2
N
S
C
6
H
5
C(NO
2
)=NNHC
6
H
5
C
6
H
5
CONHNHC
6
H
5
1. HCl
2. Py, H
2
O
41
30
Ponadtlenek potasu produkuje flawonole z niską wydajnością z nitroalkenów. Znaczącymi
produktami są kwasy salicylowe i kwasy benzoesowe.
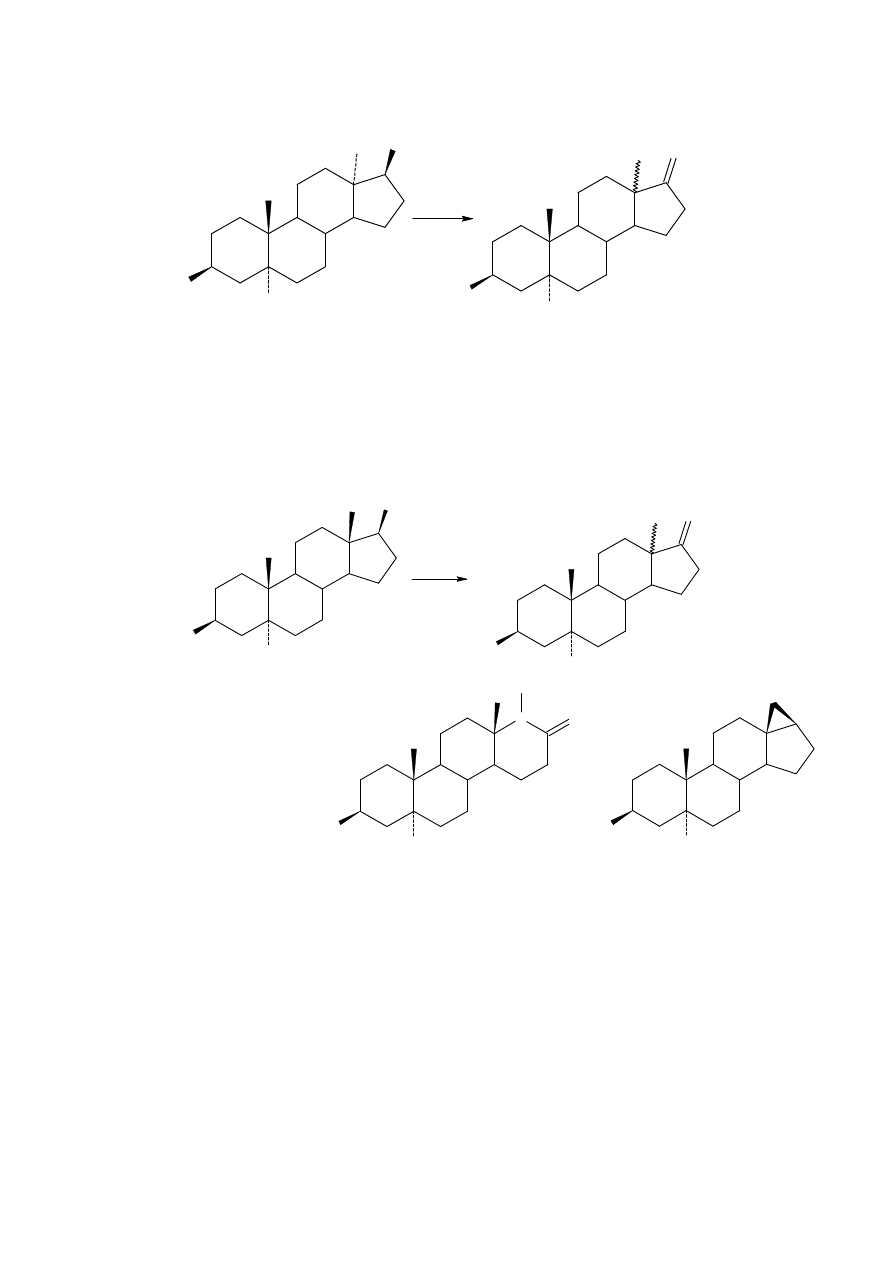
Reakcje nitroepoksydów. Sprzężone nitroolefiny mogą być przekształcone w
odpowiednie nitroepoksydy za pomocą nadtlenku wodoru.
Redukcja za pomocą wodorku litu glinu lub borowodorku sodu daje alkohol oczekiwany w
redukcji produktu Nefa. Otwarcie epoksydu za pomocą nukleofila innego niż wodorek daje
α
-podstawione ketony.
Fotoliza. Istnieje kilka raportów o fotolizie związków nitrowych w wyniku które
powstają produkty Nefa. Kilka nitrosteroidów ulega fotolizie w obecności zasad takich jak
etanolan sodu w etanolu do otrzymania ketonów i kwasów hydroksamowych jako
znaczące produkty.
O
NO
2
C
6
H
5
OH
COOH
OH
COOH
O
NO
2
C
6
H
5
O
+
+
KO
2
DMSO
(60-70%)
(10-15%)
RCH=C(NO
2
)R
1
RCH
O
C(NO
2
)R
1
(RCH
2
COR
1
)
RCH
2
CHOHR
1
H
2
O
2
OH
-
H
-
H
-
O
O
O
O
NO
2
OCH
3
H
5
C
6
O
O
O
OCH
3
H
5
C
6
O
Br
LiBr
THF, ogrzew.
(75%)
31
ś
aden keton nie jest otrzymywany w trakcie fotolizy alkoholu izopropylowego czy eteru
dietylowego. Odpowiedni związek nitrowy z grupą 13
β
metylową daje 11% ketonu, 55%
kwasu hydroksamowy i 7% cyklopropanu.
Jest zaskakujące, że napromieniowanie tego nitrosteroidu z metanolanem sodu w metanolu
daje 78% kwasu hydroksamowego i tylko 1% ketonu.
CH
3
CO
2
H
NO
2
O
H
H
O
O
H
H
N
OH
O
O
H
H
+
+
hv
NaOC
2
H
5
C
2
H
5
OH
(11%)
(55%)
(7%)
CH
3
CO
2
H
NO
2
O
H
H
O
+
hv
NaOC
2
H
5
C
2
H
5
OH
(55%)
(22%)

32
3.5.
Użyteczność syntetyczna
Powszechne występowanie grupy karbonylowej w cząsteczkach organicznych sprawia,
ż
e reakcja Nefa jest ważna w syntezach organicznych. Grupa nitrowa jest najbardziej
powszechnie wprowadzana jako nukleofil – albo jako anion soli nitronowej albo jako jon
azotanowy(III). Reakcja Nefa pozwala anionowi soli nitronowej na przemianę w anion
acylowy o ogromnej użyteczności – szczególnie do reakcji sprzężonej addycji.
Pomimo, że wiele aldehydów i ketonów, łącznie z tymi które są wrażliwe, jak te
zawierające
β
-laktam, mogą być otrzymane w reakcji Nefa, przeprowadzenie procesu
sprawia kilka problemów. Tradycyjnie, reakcja jest prowadzana w wodnym nośniku, ale
związki nitrowe o dużej masie cząsteczkowej nie ulegają jej dobrze. Użycie organicznych
rozpuszczalników mieszających się z wodą, pozwala w dużej mierze ominąć ten problem.
Bardziej poważnym problemem jest surowość warunków reakcji, szczególnie pH. W
rezultacie liczne związki polifunkcyjne ulegają reakcjom bocznym. Zmodyfikowane
reakcje Nefa, unikają tego problemu przez użycie łagodniejszych warunków. Najbardziej
znaczącymi modyfikacjami, są te z użyciem nadmanganianu potasu, ozonu i trichlorku
tytanu.
Nie jest łatwo uogólnić które metody pracują najlepiej z nowymi związkami
nitrowymi, ponieważ ich wydajność wydaje się być silnie zależna od substratów. To
znaczy, jedna metoda jest lepsza do pewnych związków nitrowych, podczas gdy inna
będzie sprawdzała się lepiej z innym rodzajem związków nitrowych.
Jasnym jest, że związki nitrowe o niższej masie molekularnej mogą być przekształcane
w związki karbonylowe na wielu drogach. Nieobecność innych grup funkcyjnych
rozszerza wybór metod, które mogą być użyte, chociaż tradycyjna reakcja Nefa może być
wśród nich najlepsza. Na przykład, nitrocykloheksan daje cykloheksanon z 85-97%
wydajnością kiedy jest traktowany najpierw zasadą a potem kwasem. Przy użyciu
nadmanganianu potasu z uwodnionym wodorotlenkiem transformacja ta zachodzi
ilościowo, ale wydajność spada do 93% kiedy jako rozpuszczalnika używa się metanolu.
Inne modyfikacje reakcji Nefa są tylko trochę mniej efektywne.
Im bardziej wzrasta złożoność substratu, tym wybór wykonalnej metody gwałtownie
spada. Grupy estrowa i acetalowa rzadko przeżywają zarówno zwykłą reakcję Nefa jak i

33
przy użyciu niezbuforowanego trichlorku tytanu. Takie wrażliwe na kwasy związki
najlepiej reagują z nadmanganianem, zbuforowanym trichlorkiem tytanu lub ozonem.
Ostatnia metoda nie może być użyta z nienasyconymi układami bądź acetalami jeżeli ilość
ozonu nie jest starannie kontrolowana.
Wyszukiwarka
Podobne podstrony:
procesy fotochemiczne i reakcje Nieznany
Chromatografia Cieczowa, Ochrona Środowiska, Sprawozdania z Chemii Analitycznej Środowiska
Praca socjalna w srodowisku lok Nieznany
Program, Ochrona Środowiska, Chemia analityczne środowiska
Chromatografia cieczowa 2, Ochrona Środowiska, Sprawozdania z Chemii Analitycznej Środowiska
Funkcje Analityczne Notatki do Nieznany
Mechanika analityczna id 290740 Nieznany
Ochrona srodowiska syllabus Nieznany
korki chemia materialy reakcje Nieznany
geometria analityczna zadani am Nieznany
Automation Studio Srodowisko id Nieznany (2)
Inspekcja ochrony srodowiska id Nieznany
Drobnoustroje a srodowisko zywn Nieznany
gazowka, Ochrona Środowiska, Sprawozdania z Chemii Analitycznej Środowiska
POPRAWA STANU SRODOWISKA W POLS Nieznany
Chemia analityczna TCh zaoczne Nieznany
Geometria analityczna AK, 2011 Nieznany
Refraktometria, Chemia Analityka środowiska, żywności i leków PWR
więcej podobnych podstron