
Ćwiczenie nr 1F
Temat :
Badanie struktury ciał stałych.
Cel ćwiczenia:
Celem
ćwiczenia jest poznanie budowy strukturalnej ciał stałych oraz
wybranych metod badania tej struktury.
1. WIADOMOŚCI PODSTAWOWE.
1.1 Struktura ciała stałego - wstęp.
Pojęcie struktury ciała stałego obejmuje:
1) charakterystykę uporządkowania atomowego, tzn. struktury krystalograficznej,
rodzaju i gęstości defektów punktowych, dyslokacji i defektów powierzchniowych
(granic ziaren);
2) opis wielkości, kształtu i jednorodności, ewentualnie uprzywilejowanej orientacji
ziaren (krystalitów) w układach polikrystalicznych;
3) rodzaj, liczbę i rozmieszczenie faz w układach wieloskładnikowych;
4) opis makrostruktury - tj. niejednorodności i defektów, odnoszących się do większych
obszarów materiału;
3)charakterystykę naprężeń własnych.
Każda materia może występować w jednym z trzech stanów skupienia: gazowym, ciekłym
bądź stałym. Zarówno w stanie gazowym jak i ciekłym atomy mają swobodę poruszania
się ograniczoną ściankami zbiornika lub zderzeniami z innymi atomami. Może to
powodować lokalne fluktuacje gęstości. W przeciwieństwie do tego ciało stałe
charakteryzuje się stałością, tzn.:
- ma określony kształt (atomy oscylują wokół położeń równowagi, tworzących
niezmienną strukturę); wyjątkowo zdarzające się przeskoki na puste miejsca po innym
atomie, nie mają w zasadzie wpływu na kształt ciała stałego,
- wykazuje sprężystą sztywność ze względu na odkształcenia skręcające.
Położenia równowagi są konsekwencja istnienia wiązań międzyatomowych,
odpowiedzialnych za spójność ciał stałych.
Struktura
ciał stałych może być rozmaita w zależności od warunków, w jakich się
tworzy:
1) w wyniku zestalania się cieczy (metale i materiały topione);
2) w procesach wytwórczych kompozytów;
3) w wyniku syntezy związków węgla i wodoru ( w przypadku tworzyw sztucznych).
Proces kondensacji do stanu stałego może przebiegać dwojako:
a) jako krystalizacja - tworzy się struktura krystaliczna;
b) jako zestalanie się cieczy wskutek dostatecznie szybkiego zwiększania lepkości przy
obniżaniu temperatury - tworzy się struktura amorficzna.
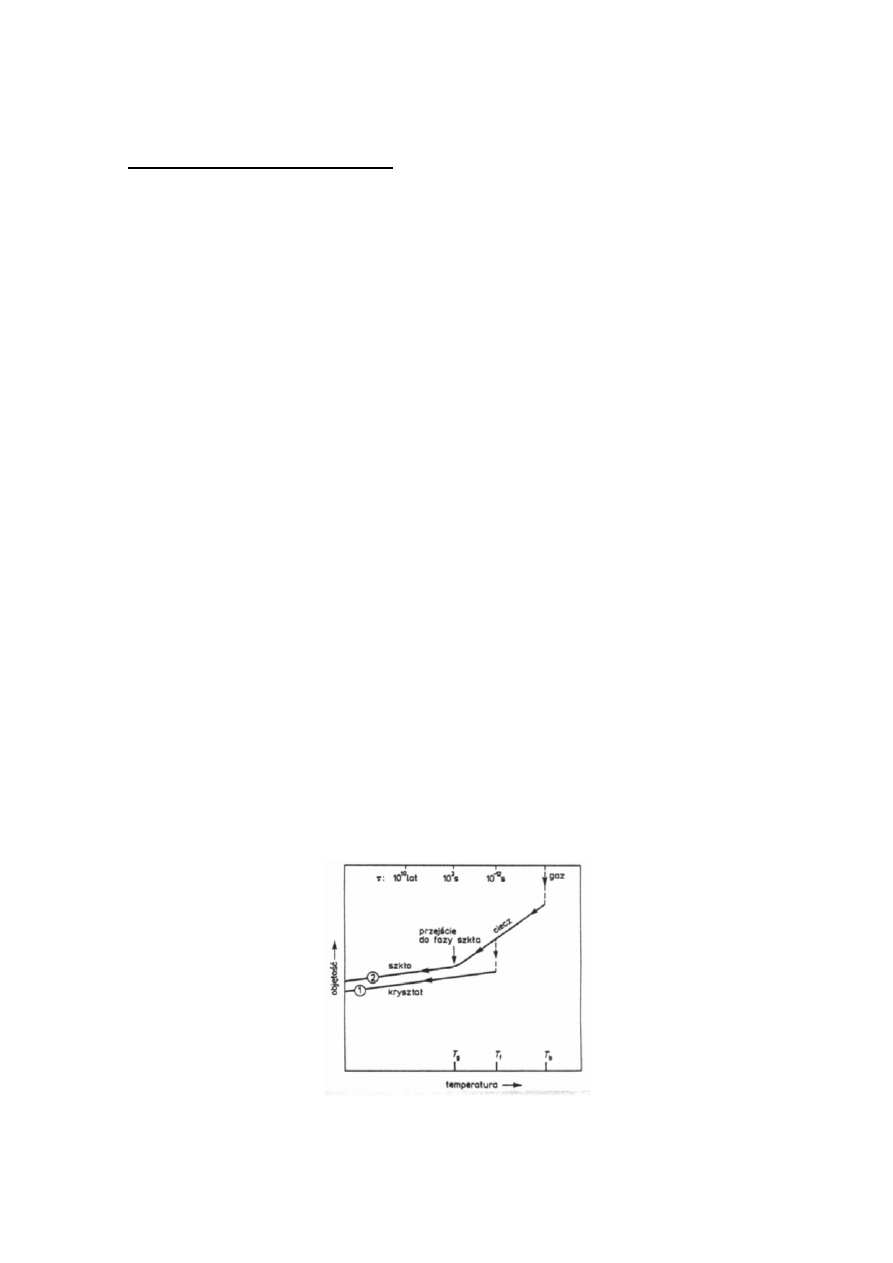
Na rysunku 1. przedstawiono przebieg zmiany objętości substancji w funkcji temperatury
dla tych dwu procesów.
Rys. 1. Zmiany objętości V w funkcji temperatury podczas różnych procesów
kondensacji do stanu stałego. T
b
oznacza temperaturę wrzenia.
Podczas ochładzania (od prawej strony rysunku 1. do lewej) następuje ciągła zmiana
objętości. Nachylenie krzywej określa wspólczynnik rozszerzalności cieplnej przy stałym
ciśnieniu. Gdy temperatura staje się dostatecznie niska następuje przejście ciecz- ciało
stałe, charakteryzujące się znacznie mniejszym współczynnikiem rozszerzalności cieolnej.
W temperaturze T
f
(rys.1) zachodzi nieciągłe przejście do stanu krystalicznego,
charakteryzującego się niższą energią. Następuje wtedy nagłe zmniejszenie objętości do
objętości kryształu. Natomiast, jeżeli stan ciekły istnieje do pewnej temperatury T
g
(rys.1)
- to w wąskim zakresie temperatur w pobliżu tej temperatury następuje ciągłe przejście do
fazy szkła. Obecnie niemal wszystkie substancje można otrzymać w postaci ciała
amorficznego, jeżeli zostaną dostatecznie szybko i dostatecznie silnie ochłodzone.
2.2 Porównanie ciał amorficznych i krystalicznych.
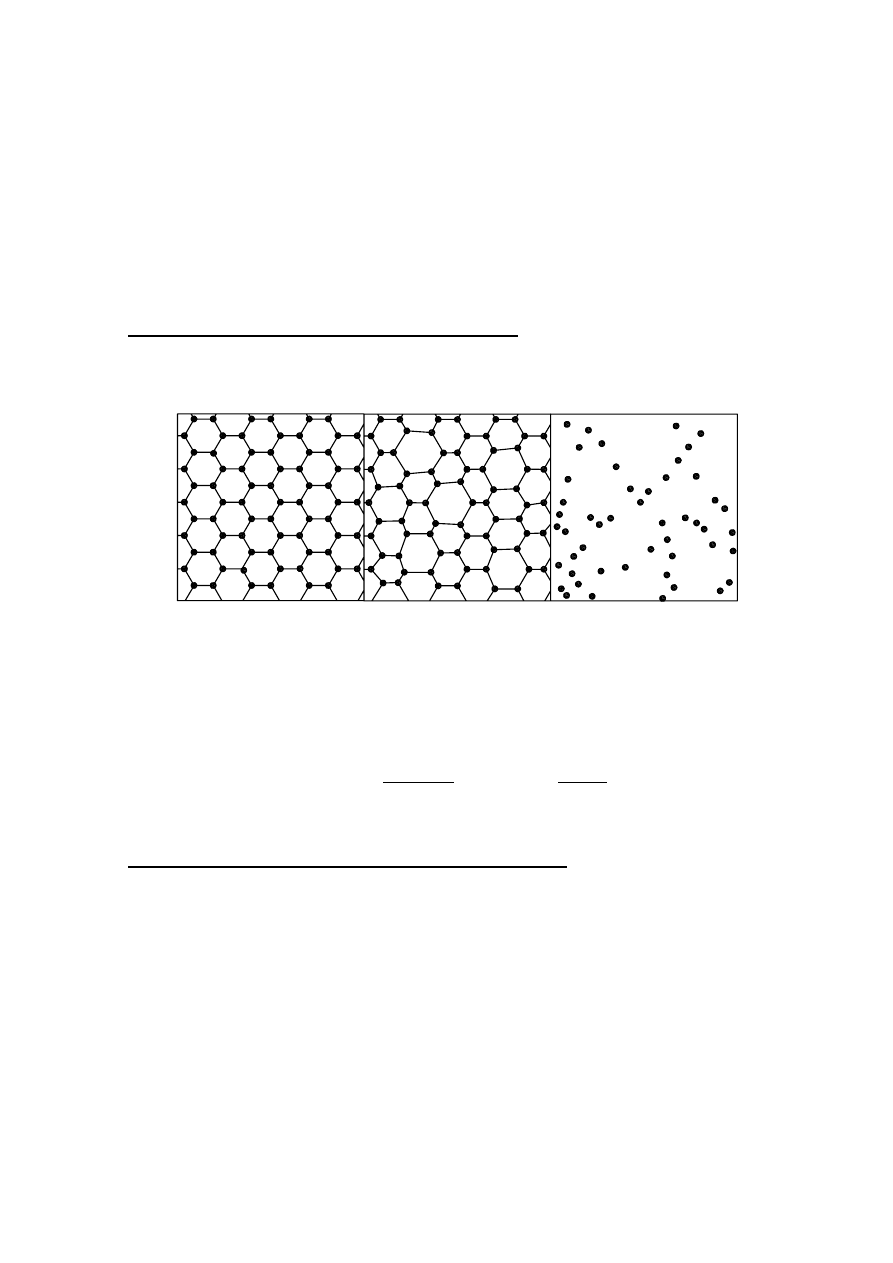
Najistotniejszą cechą odróżniającą strukturę atomową ciała amorficznego od ciała
krystalicznego jest brak uporządkowania dalekiego zasięgu (rysunek 2).
Rys. 2. Szkic położenia atomów w ciele a) krystalicznym, b) amorficznym, c)
gazowym.
W szkłach podobnie jak w kryształach istnieje wysoki stopień uporządkowania bliskiego
zasięgu (tzn.: struktura w skali atomowej jest nieprzypadkowa dla odległości rzędu kilku
odległości międzyatomowych od danego atomu, każdy atom ma trzech najbliższych
sąsiadów, kąty wiązań są niemal równe). W przypadku ciała krystalicznego odległości
między najbliższymi sąsiadami są dokładnie równe, a nie prawie równe jak w przypadku
szkieł. Uporządkowanie to zarówno w ciałach krystalicznych jak i amorficznych jest
konsekwencją istnienia wiązań chemicznych odpowiedzialnych za spójność ciał stałych.
2.3. Rodzaje wiązań międzyatomowych w ciałach stałych.
Struktura ciał stałych jest konsekwencją dzałania sił międzyatomowych i
międzycząsteczkowych. Dzielą się one na:
1)
siły przyciągające:
a)
między dodatnimi jonami i elektronami - w wiazaniu metalicznym (w metalach
) bądź kowaletnym ( w półprzewodnikach i polimerach);
b)
między jonami dodatnimi i ujemnymi - w wiązaniach jonowych ( w solach
metali, dielektrykach);
c)
oddziaływania słabe międzycząsteczkowe - w wiązaniach Van der Waalsa;
2)
siły odpychania - między jonami tego samego znaku.
Siły odpychania utrzymują jony w określonych odległościach od siebie, natomiast siły
przyciągania zapewniają spójność ciała stałego. Kryształ bądź ciało amorficzne jako całość
pozostaje elektrycznie obojętny. Odległości między atomami w siatce
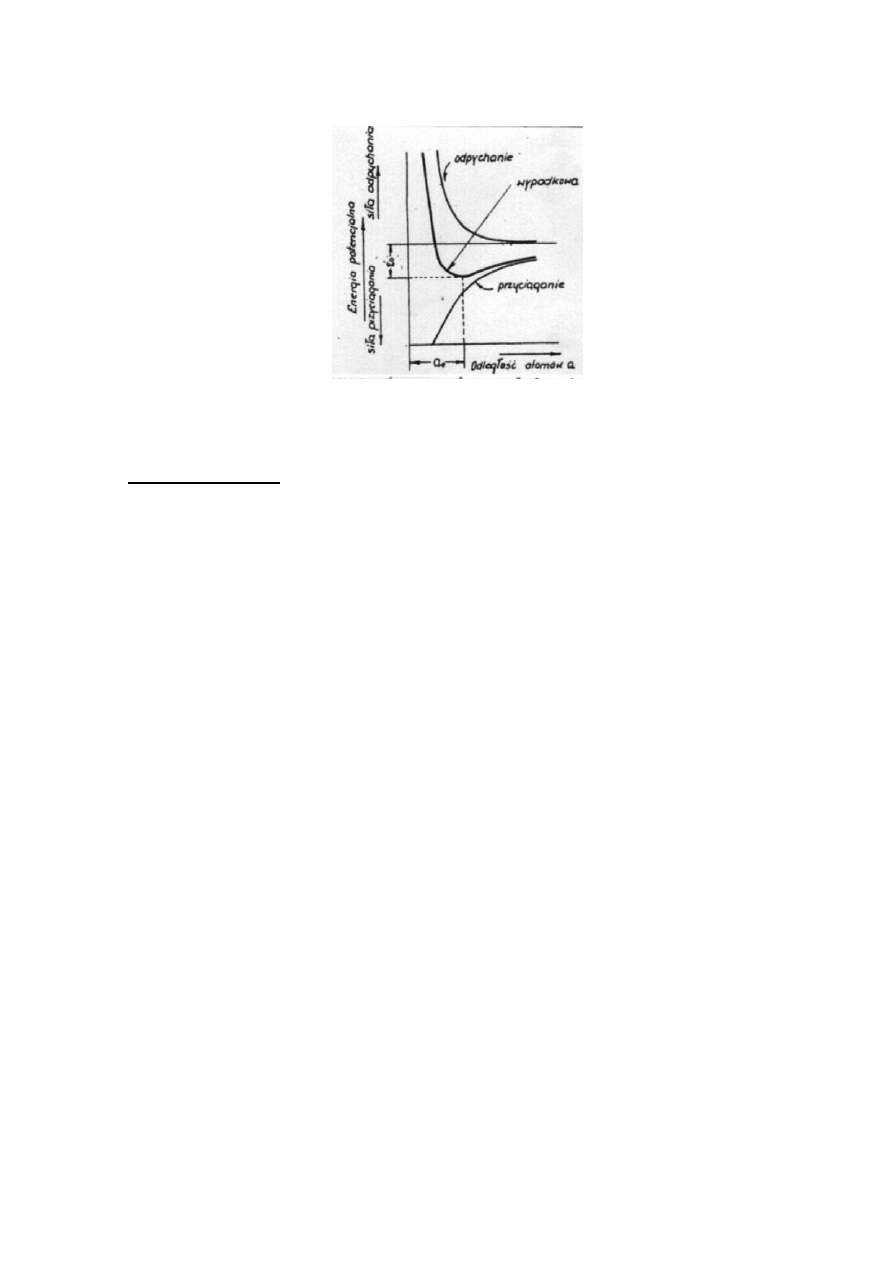
krystalicznej są cechą charakterystyczna danego pierwiastka bądź związku chemicznego i
wynikają z równowagi poprzednio wymienionych sił.
Rys. 3. Siły pomiędzy dwoma atomami w zależności od ich odległości, oraz energia
potencjalna w sieci przestrzennej.
2.4. Monokryształy.
Najmniejsze, powtarzające się ugrupowanie atomów w przestrzeni nazywa się komórką
elementarną sieci krystalicznej, a wielkości, określające długości jej krawędzi nazywają sie
parametrami sieci. Odległości między atomami w siatce krystalicznej są cechą
charakterystyczna danego pierwiastka bądź związku chemicznego i wynikają z równowagi
poprzednio wymienionych sił. Trójwymiarowy rozkład węzłów sieci krystalicznej, w
którym każdy węzeł ma identyczne otoczenie nazywa się siecią przestrzenną. Na rysunku
4. przedstawiono 14 podstawowych sieci krystalicznych.
Ciało stałe nie wykazujące w swej budowie wewnętrznej zakłóceń w żadnym kierunku
nazywa się kryształem doskonałym. Budowa olbrzymiej wiekszości kryształów
naturalnych i otrzymywanych sztucznie odbiega znacznie od modelu doskonałego,
wykazując szereg defektów - takie kryształy nazywa sie kryształami rzeczywistymi. Tylko
niektóre minerały mają budowę zbliżoną do kryształu doskonałego (np. niektóre okazy
diamentu, kwarcu i kalcytu). W ostatnich czasach są wytwarzane zarówno w laboratoriach
naukowych jak i na skalę przemysłową monokryształy, które ze względu na bardzo
znikomą liczbę defektów można nazwać kryształami prawie doskonałymi. Dotyczy to
głównie takich kryształów półprzewodnikowych, jak krzem, german i arsenek galu.
W procesie powstawania struktury kryształu można wyróżnić dwa etapy:
1) zarodkowanie, czyli tworzenie się w cieczy grup atomów w położeniach, chwilowo
odpowiadajacych położeniu i odległościom międzyatomowym, występujących w
krysztale;
2) wzrost kryształów.
W teorii krystalizacji rozróżnia się głównie dwa rodzaje zarodkowania:
a) zarodkowanie homogeniczne - dotyczące wyłącznie metalu idealnie czystego,
pozbawionego w stanie ciekłym wszelkich stałych wtrąceń i zanieczyszczeń oraz
krystalizującego w masie, nie ograniczonej ścianami materialnymi;
b) zarodkowanie heterogeniczne - w którym aktywnymi ośrodkami krystalizacji są
cząstki zanieczyszczeń, domieszek lub podłoże (np. ściany wnęki formy).
Natomiast w teorii wzrostu kryształów rozważa się:
a) mechanizm dołączania się atomów z cieczy do powierzchni międzyfazowej;
b) warunki cieplne, umożliwiające postęp krystalizacji ( tzn. odprowadzanie ciepła
krystalizacji do otoczenia, czyli zmniejszenie energii swobodnej układu, warunkujące
postęp krystalizacji).
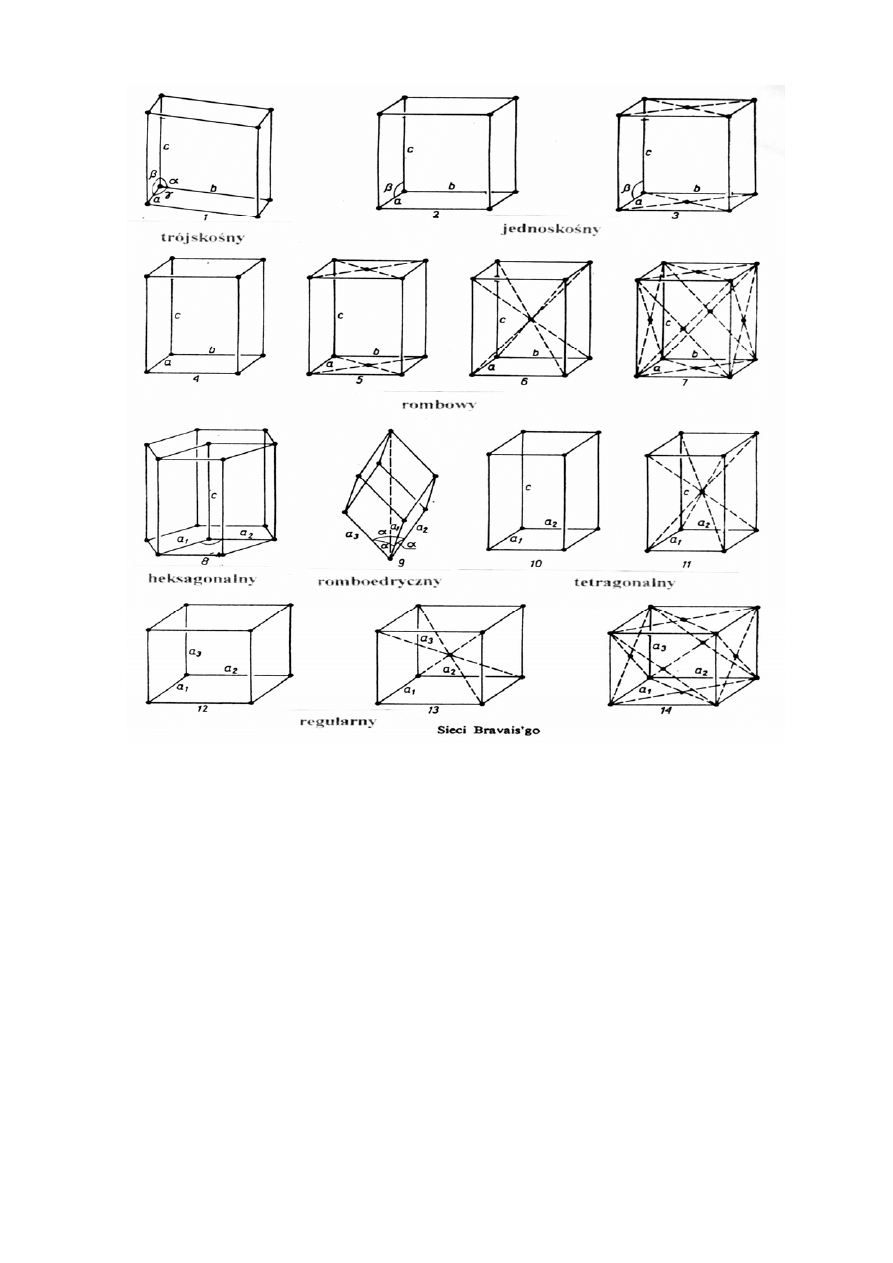
Rys. 5. Typy sieci krystalicznych.
Cechą charakterystyczną sieci metali jest duża gestość upakowania struktury. Kryształy
jonowe natomiast składają się z dużych anionów, tworzących główny szkielet, między
węzłami którego tkwią małe kationy. Na przykład w strukturze chlorku cezu CsCl jony
chloru tworzą regularną sieć prymitywną, a jony cezu znajdują się w lukach sześciennych.
W strukturze soli kuchennej NaCl duże aniony tworzą szkielet regularny ściennie
centrowany (RSC), a kationy znajdują się w lukach oktaedrycznych i mają również
uporządkowanie RSC (rysunek 5).
W kryształach o wiązaniach kowalencyjnych (np. Ge, Si, Sb) liczbę wiązań, jaką dany
atom tworzy ze swoimi sąsiadami określa reguła "8-N". Oznacza to, że dla uzupełnienia
oktetu elektronów, potrzebych do zapewnienia atomowi stabilności, pierwiastek, mający N
elektronów wartościowości otrzymuje 8-N dodatkowych elektronów od takiej samej
liczby, sąsiadujących z nim , atomów, z którymi te elektrony dzieli (rysunek 6).
Powstające struktury są twarde ( diament, Si, Ge - pierwiastki IV- wartościowe) bądź
łupliwe (As, Sb, Bi - V- wartościowe; sieć regularna prawie ściennie centrowana ale z
warstwami o różnej gęstości kowalencyjnie zwązanych atomów, równoległych do
płaszczyzny oktaedrycznej).
Niektóre pierwiastki ( Fe, Ti, Co, C, S, Sn) mają kilka odmian alotropowych, czyli
różniących się budową sieci przestrzennej.
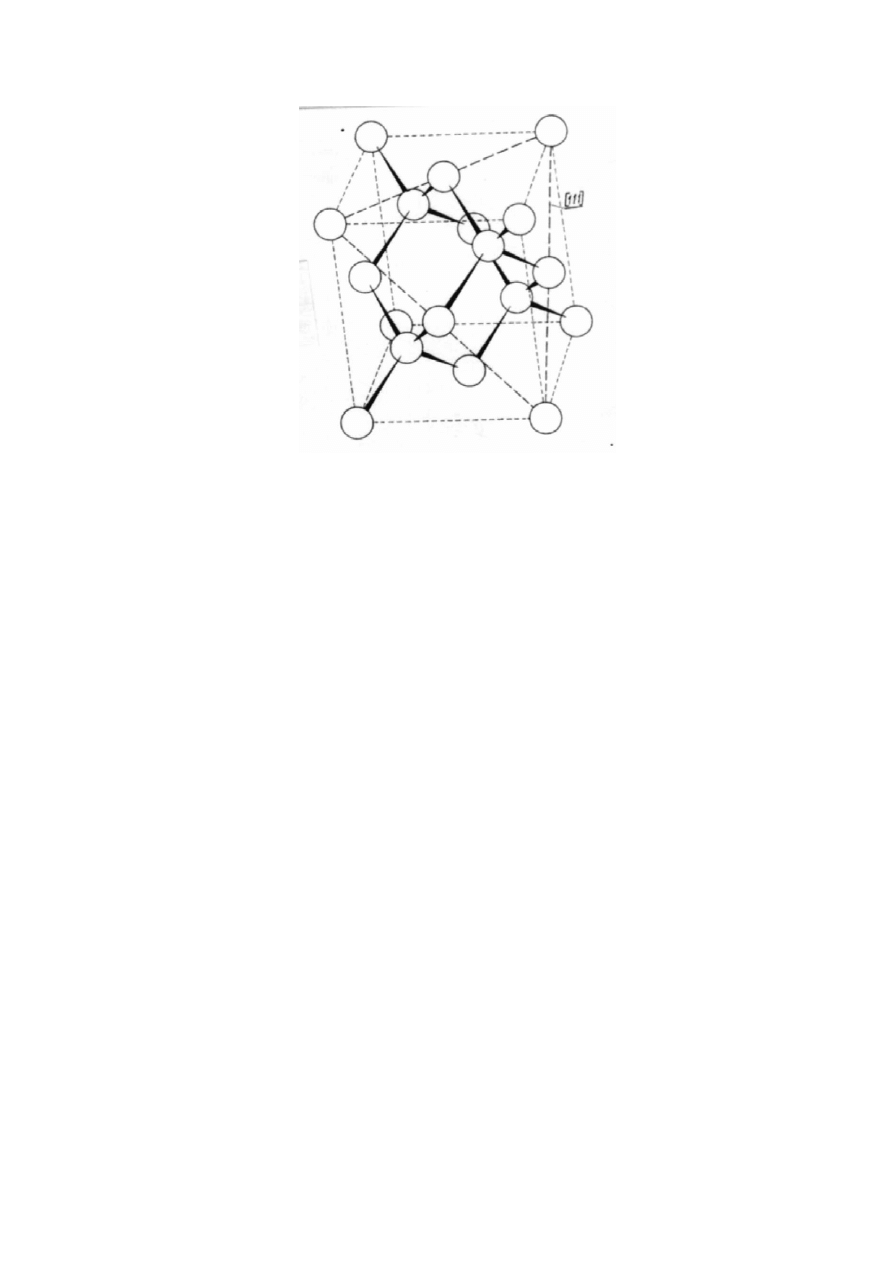
Rys. 6. Struktura RSC diamentu.
Wyhodowanie niezaburzonej struktury krystalicznej dowolnej z omówionych
poprzednio typów sieci wiąże się z dużymi trudnościami (np. metoda wyciągania
kryształów Czochralskiego, metoda Bridgmana- Stockbargera, metoda wędrującej strefy
stopionej). Niekiedy w technice (np. półprzewodnikowej) lub dla badań naukowych
konieczne jest wytworzenie takich monokryształów. Ich rozmiary mogą osiągać wielkości
nawet kilkunastu centymetrów. W ostatnich latach wyhodowano też kryształy metali w
postaci cienkich kryształów nitkowych o dużym stopniu doskonałości (tzw. wiskery).
Nową strukturą osiagniętą w praktyce w latach 90 jest struktura fullerenu (rysunek 7).
Tworzą ją wieloatomowe cząsteczki węgla. W trakcie wytwarzania łańcuchów węglowych
w miarę postępowania kondensacji, łańcuchy stają się dostatecznie długie, aby mogły się
zamknąć i wytworzyć pojedyńcze pierścienie. W dalszym procesie wzrostu łańcuchy
zwijają się w trwalsze wielooczkowe sieci. W najbardziej stabilnej odmianie węgla -
graficie - atomy są związane w nieskończoną sieć heksagonalną. Płaskie sieci (można je
sobie wyobrazić jako grafitowe arkusze) mają wiele niewysyconych wiązań. To stanowi
przyczynę przyłączania małych cząsteczek węgla i ich szybkiego wzrostu. Brak atomów
wysycających na brzegach takich arkuszy powoduje (zgodnie z prawem fizyki o dążeniu
każdego układu do minimum energii), że arkusz, mając tendencję do likwidowania
niewysyconych wiązań, po prostu zwija się. Powstają wówczas pięciokąty. Powoduje to
zakrzywianie się sieci i umożliwia zastąpienie dwóch niewysyconych wiązań przez jedno
zwykłe wiązanie węgiel- węgiel. Cała struktura fullerenów składa się z sieci
równobocznych pięciokątów i sześciokątów (ale tak, aby uniknąć styku dwóch
pięciokątów, gdyż miejsca te są niestabilne) i tworzy sferę pustą w środku. Ta postać
alotropowa węgla C
n
składa się z parzystej liczby atomów
n= 2 (16+m)
m =1, 2, 3...,
najczęściej C
60
i C
70
, największa zaobserwowana C
960
.
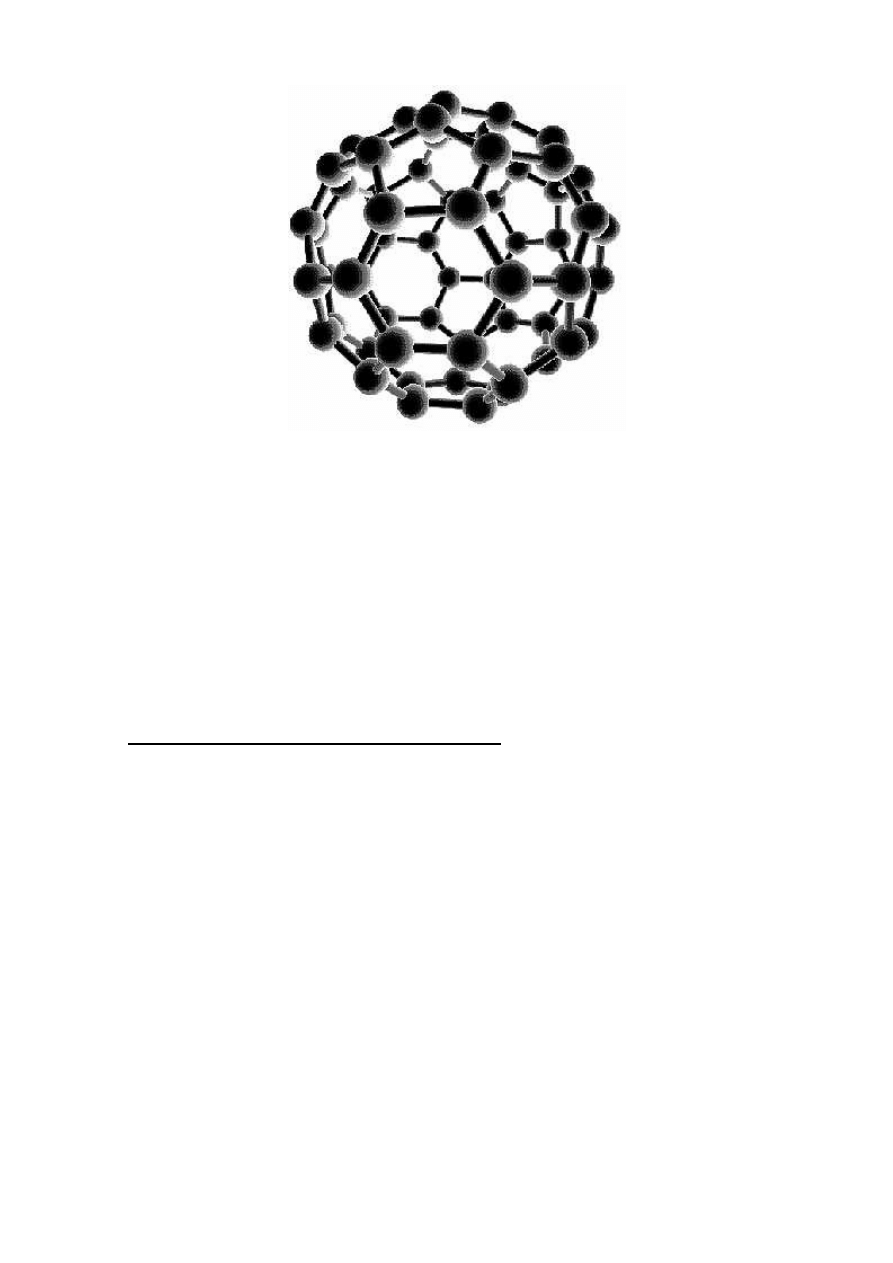
Rys. 7. Struktura fullerenu na przykładzie cząsteczki węgla C
60
.
Do tej pory stwierdzono, że trwałe są wszystkie klastery o parzystej liczbie atomów węgla
wiekszej od 32, przy czym najbardziej stabilna jest struktura C
60
(zawierająca 20
sześciokątów ) i C
70
(zawierająca 25 sześciokątów). Istnieje możliwość powstawania
struktury hiperfullerenowej tzn. takiej w której środku znajduje się cząsteczka C
60
,
otoczona kolejnymi fullerenami złożonymi z 240, 540 i 960 atomów. Proces budowy takiej
struktury może ciągnąć się bez ograniczeń. W efekcie mogą powstawać makroskopowe
cząsteczki o ikosaedrycznej (pięciokrotnej) symetrii.
Krystalizacja fullerenu polega na ułożenie cząsteczek C
60
(jak piłek futbolowych) w
regularną sieć płasko centrowaną. Do tej pory struktury te nie były jeszcze wytwarzane
przemysłowo, aczkolwiek dają one szerokie możliwości modyfikacji własności ciał
stałych. Kolor fullerenów jest żółty.
2.5. Defekty i dyslokacje struktury krystalicznej.
Defekty struktury krystalicznej mogą być: punktowe, liniowe (dyslokacje) i
płaszczyznowe (granice ziaren).
W
kryształach rzeczywistych miejsce przeznaczone na atom może być nie zajęte
(istnieje luka) lub atom może się znajdować w przestrzeni międzywęzłowej (defekt
międzywęzłowy). Te defekty punktowe przedstawiono na rysunku 8.
Dyslokację krawędziową można sobie wyobrazić jako skutek częściowego
wprowadzenia do wnętrza kryształu dodatkowej płaszczyzny atomów (rysunek 9).
Krawędź tej dodatkowej płaszczyzny stanowi linię faktycznej dyslokacji. W obszarze
otaczającym linię dyslokacji występuje wyraźne odkształcenie a płaszczyzny sieciowe
wyginają się.
Dyslokacja śrubowa występuje wtedy, gdy jedna część kryształu przesunie się
względem pozostałej o jeden odstęp międzypłaszczyznowy.
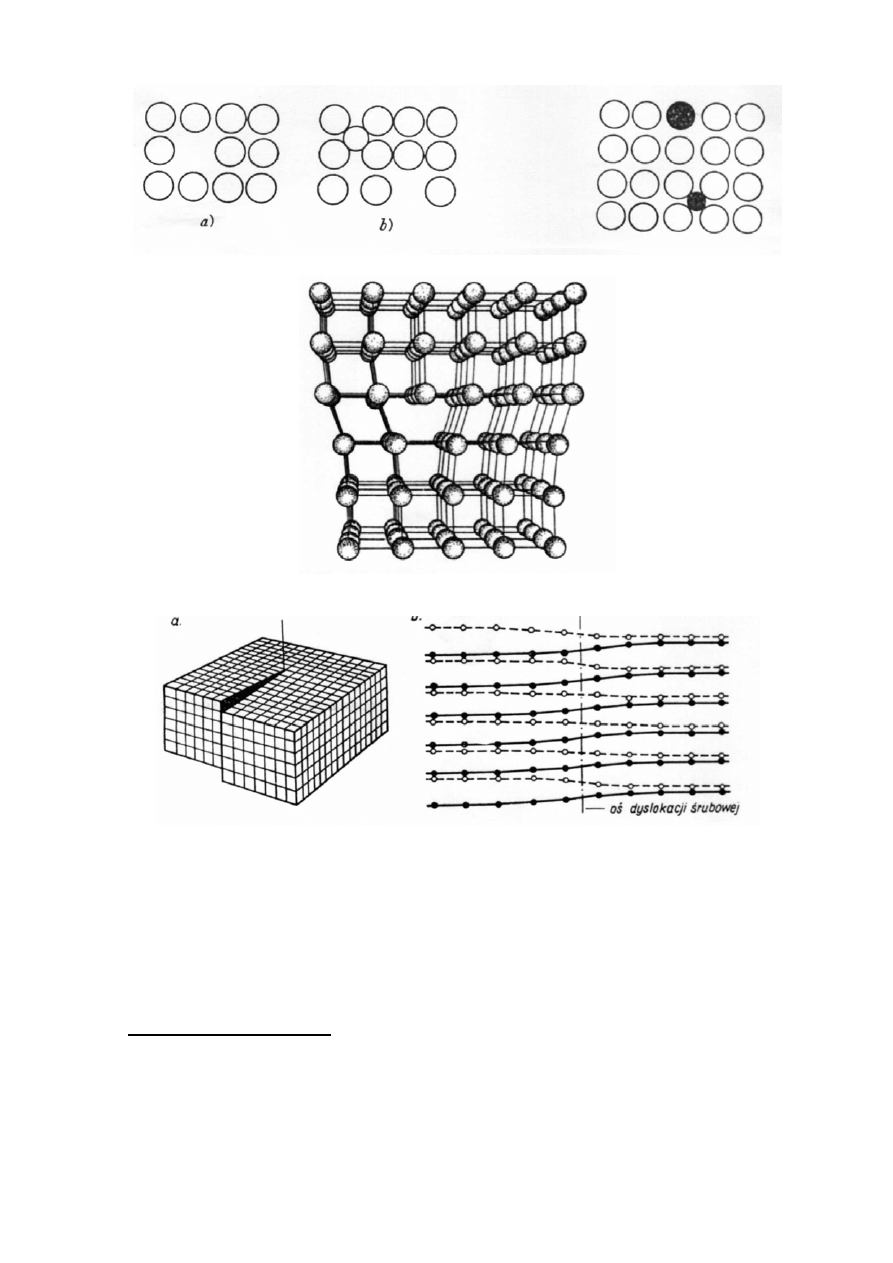
Rys. 8. Płaski schemat defektów punktowych w kryształach.
Rys. 9. Obraz dyslokacji krawędziowej.
Rys. 10. Dyslokacja śrubowa.
W miejscach, gdzie dyslokacje wychodzą na powierzchnię kryształu, pod wpływem
roztworów trawiących pojawiają się tzw. jamki trawienia. Odkształcenie i związane z tym
osłabienie wiązań atomowych pod wpływem dyslokacji powoduje bowiem ułatwienie w
tych obsszarach procesu trawienia.
Oprócz wymienionych wyżej defektów punktowych i liniowych mamy do czynienia
często z defektami płaszczyznowymi. Są nimi granice ziaren.
2.6. Ciała polikrystaliczne.
Najczęściej krystaliczne ciało stałe składa się z dużej liczby drobnych kryształów, czyli
ma budowę polikrystaliczną. Poszczególne kryształy nie przybierają prawidłowego dla
danej struktury kształtu lecz są mniej lub bardziej zdeformowane (zawierają opisane
defekty punktowe i liniowe). Zowią się one ziarnami lub krystalitami. Ich występowanie
jest związane z powstawaniem wielu centrów zarodkowania w trakcie procesu

krystalizacji. Na rysunku 11 przedstawiono schemat tworzenia się struktury
polikrystalicznej.
Rys. 11. Schemat powstawania struktury polikrystalicznej.
Z kolei z kształt ziaren wskazuje na kierunek ich wzrostu,czyli pośrednio na kierunek
gradientów temperaturowych, występujących podczas procesu krzepnięcia ciała stałego.
Na rysunku 12 przedstawiono schemat wzrostu pojedynczego dendrytu i układ ziaren w
wlewku.
Rys. 12 . Schemat powstawania dendrytu.
W
zależności od wzajemnej relacji między szybkością krystalizacji SK a szybkością
zarodkowania SZ mogą powstawać następujące struktury polikrystaliczne:
- gruboziarnista, gdy SK > SZ;
- dobnoziarnista, gdy SK < SZ.
Oceny wielkości ziaren przeprowadza się według skali ASTM lub normy PN-66/H-04507.
Do oznaczania wielkości ziarna metodą porównawczą służą wzorce rysunkowe 1-10
(rysunek 13), wykonane przy powiększeniu 100x i obejmujące pole widzenia o średnicy
0,8mm i powierzchni 0.9mm
2
.
Najczęściej orientacja przestrzenna sieci krystalicznej sasiednich ziaren jest
przypadkowa. Obszary kryształu, które różnią się wzajemną orientacją krystalograficzną
sieci nazywamy granicami ziaren. Jeżeli wzajemna dezorientacja dwóch obszarów
kryształu wynosi mniej niż 2 stopnie, to umownie granicę między tymi obszarami
nazywamy granicą niskokątową, a obszary oddzielone tą granicą - blokami mozaikowymi
lub podziarnami. W przypadku dezorientacji większej niż 2 stopnie mówimy o ziarnach i
granicach wysokokątowych. Budowa zarówno granic niskokątowych jak i
wysokokątowych (inaczej zwanych szerokokątowymi) jest związana z układem dyslokacji
i przedstawiono ją na rysunku 14. Strefa przejściowa między ziarnami ma wymiary rzędu
angsztremów.

Rys. 13. Wzorce do porównawczego określania wielkości ziaren.
Rys. 14. Obraz granic a) wąskokątowych b) szerokokątowych c) model granicy.
Wskutek obróbki plastycznej na zimno (np. walcowania) można uzyskać niemal
jednokierunkową orientację ziaren (tzw. tekstura walcowania - włóknista). Własności
mechaniczne, magnetyczne przy takiej jednokierunkowej orientacji ziarn będą zależne od
kierunku, w którym bada się określoną własność. Zjawisko to nazywa się anizotropią.
2.7. Kompozyty.
Kompozyty stanowią bardzo liczną grupę ciał stałych,a ich różnorodność ciągle
powiększa się. Zalicza się do nich:

1)
ceramikę: - materiały odporne na zużycie (Al
2
O
3
, ZrO
2
),
-
żaroodporne (Al
2
O
3
, SiC, Sialon),
- narzędziowe (j.w. + TiC, TiN, BN);
2) spieki metali i ich związków (np. ferryty);
3) tworzywa sztuczne wzmocnione włóknem szklanym, tkaniną (np. tekstolit), bądź
nawet tekturą (turbax);
4) kompozyty metalowe wzmacniane włóknem, włóknem nieciągłym, bądź też
otrzymywane drogą kierowanej krystalizacji stopów eutektycznych.
Właściwości ich związane są z:
- możliwością dowolnego doboru składu chemicznego w wyniku zmieszania
komponentów nierozpuszczających się wzajemnie, różniących się znacznie temperaturami
topnienia lub metali z niemetalami;
-
możliwości wytwarzania wyrobów porowatych o różnej gęstości przy tym samym
składzie chemicznym;
-
możliwością nasycenia porów metalami niskotopliwymi, polimerami lub olejami;
-
możliwością wytwarzania wyrobów z materiałów wysokotopliwych.
Struktura kompozytów zależy od rodzaju i procesów technologicznych ich wytwarzania.
Mogą one się składać z dwóch lub więcej komponentów o znacznie różniącej się
wytrzymałości i sprężystości. W grupach kompozytów 2
÷4 można wyróżnić dwie fazy:
-
fazę wzmacniającą ( wprowadzaną np. w postaci cienkich włókien kryształów lub
węglowych, bądź szkieletu z ciała trudnotopliwego np. wolframu w produkcji styków
elektrycznych);
-
osnowę ( w ciałach stosowanych jako materiały konstrukcyjne jest ona plastyczna lub
bardziej elastyczna, w technologii styków elektrycznych jej zadanie polega na
zapewnieniu odpowiedniej wartości przewodnictwa elektrycznego, np. srebro);
.
Rys. 15. Przykład struktury kompozytowej.
W ceramikach można wyróżnić trzy główne fazy: krystaliczną, bezpostaciową (szklistą) i
pory powietrzne. Na rysunku 15 przedstawiono przykład ciała kompozytowego
2.8.Tworzywa sztuczne.
Struktura tworzyw sztucznych wynika z metod ich syntezy:
- polimeryzacji - w trakcie tego procesu monomery łączą się ze sobą bez
wydzielenia produktów ubocznych (np. polietylen);
- polikondensacji - monomery łączą się ze sobą z wydzieleniem niskocząstecz-
kowych produktów ubocznych ;

- poliaddycji - dwie różne składowe tworzą makrocząsteczkę bez wydzielenia
produktu ubocznego i bez wzajemnego nasycania podwójnych wiązań węgla (np.
poliuretan).
Łańcuchy polimerów mogą być rozmieszczone chaotycznie lub wykazywać
uporządkowanie bliskiego zasięgu (rysunek 16). Krystaliczne postacie polimerów
powstają wówczas, gdy analogiczne cząsteczki monomerów w równolegle ułożonych
łańcuchach znajdują się na tej samej wysokości. Możemy otrzymać następujące typy sieci
przestrzennych:
-
układ jednoskośny (celuloza, polipropylen izotaktyczny);
-
układ trójskośny (poliuretan, politereftalan etylenu);
- romboedryczny (polistyren);
- rombowy (polietylen, polimetakrylan metylu).
Rys. 16. Typy uporządkowania łańcuchów polimerów.
We wszystkich przypadkach obserwuje się znaczne rozmiary komórki elementarnej.
Można je zaobserwować w postaci sferulitów (rysunek 16b). Przy obserwacji tworzyw
sztucznych przy powiększeniach mikroskopu optycznego ok. 100x można zobaczyć linie
rozpływu tworzywa podczas wlewania go do formy i pęcherze powietrzne.
2.WYKONANIE ĆWICZENIA.
2.1. Program ćwiczenia.
Wykonanie
ćwiczenia opiera się na mikroskopowych obserwacjach próbek różnych
ciał stałych oraz dokonaniu pomiarów, dotyczących wybranych aspektów budowy
strukturalnej ciał stałych.
2.2. Obserwacje struktur wybranych ciał stałych.
Za pomocą mikroskopu metalograficznego sprzężonego z kamerą cyfrową należy przeprowadzić
obserwacje i wykonać zdjęcia najbardziej charakterystycznych fragmentów struktury w próbkach:
5. szlif metalograficzny miedzi - zdjęcie struktury uwzględniające występowanie struktur
bliźniaczych (zalecany obiektyw x40);
6. szlify stopu potrójnego PbSnSb - obrazy struktury próbek stopu, krystalizującego z
różnymi prędkościami.
7. szlif metalograficzny stali niskowęglowej – struktura polikrystaliczna
8. szlif metalograficzny niklu – struktura polikrystaliczna
9. wolfram – struktura włóknista oraz po rekrystalizacji
10. Dwufazowa struktura kompozytowa wolfram-miedź (powiększenie 8 i 40 razy)
Zwrócić uwagę na charakterystyczne cechy obserwowanych struktur ( występowanie
dendrytów, linii płynięcia, kształtu kryształów, budowę fazy wzmacniającej w kompozycie
z tworzyw sztucznych).
W sprawozdaniu zamieścić wydrukowane obrazy struktur wraz z wnioskami,
dotyczącymi charakterystycznych cech budowy strukturalnej.
2.3. Pomiary wybranych wielkości charakterystycznych niektórych ciał stałych.
2.3.1.Pomiar wielkości ziaren i bliźniaków
Korzystając z programu Metilo wykonać pomiary wielkości ziaren oraz odległości
ziaren bliźniaczych dla obrazów struktur uzyskanych za pomocą mikroskopu
metalograficznego (pow. x8). Wykonać pięć pomiarów w różnych kierunkach, dla pięciu
różnych ziaren. Wykonać pomiary kilku szerokości odległości bliźniaczych. Wyniki
umieścić w tabeli:
Rodzaj próbki
Wielkość ziarna (µm)
Wielkość
bliźniaków (µm)
Nr.
1
2
3
4
Cu drobnoziarnista
(próbka 5.1)
5
1
2
3
4
Cu gruboziarnista
(próbka 5.2)
5
2.3.2. Pomiar wielkości ziaren – metoda porównawcza
Pomiar wielkości ziaren metodą porównawczą polega za porównaniu obrazów z
wzorcowymi siatkami ziarnistości. Pomiary wykonać za pomocą programu Met-ilo,
wczytując jako obraz pomocniczy kolejne siatki ziarnistości (szczegóły w instrukcji
dodatkowej). Pomiary wykonać dla próbek: 7, 8, 9. Wyniki zamieścić w tabeli:
Próbka Nr
siatki
Średnia wielkość
ziaren
7 (stal)
8 (nikiel)
9/1 9/2 (wolfram)
9/3 9/4 (wolfram)
2.3.3. Pomiar wielkości ziaren powstających w różnych warunkach
Dla potrójnego stopu PbSbSn dokonać pomiaru wielkości ziaren powstających przy
różnych szybkościach krystalizacji (trzy szybkości). Wyniki umieścić w tabeli:
Lp.
PbSbSn (6.1)
PbSbSn (6.2)
PbSbSn (6.3)
1
2
3
5
5
2.3.4. Określenie objętości względnej fazy
β (ziarna) rozmieszczonej losowo w
osnowie
α.
Pomiar składu procentowego wykonać dla próbki nr 10 dla dwóch różnych powiększeń.
Pomiar numeryczny wykonać przy pomocy programu Met-ilo, pomiar metodą siecznych
wykonać dla wydruków struktur krystalicznych (opis metody poniżej). Uzyskane wyniki
zamieścić w tabeli.
Powiększenie
mikroskopu
Pomiar numeryczny
Pomiar metodą
siecznych
Metalograficzny
obiektyw x40
Metalograficzny
obiektyw x8
2.3.4.1 Metoda siecznych pomiaru składu procentowego.
Określenie objętości względnej fazy
β (ziarna) rozmieszczonej losowo w osnowie α
metodą liniową polega na tym, że przez obraz mikrostruktury prowadzi się proste o
długości l (sieczne). Każda z prostych l przecina n
j
ziarn fazy
β, których granice odcinają
na prostych cięciwy l
ij
. Objętość względną V
v
fazy
β można wyrazić jako stosunek
zsumowanej długości cięciw do długości całkowitej wszystkich siecznych (rys. 17).
Estymatorem objętości względnej V
v
jest zatem ułamek L
L
, określony wzorem:
V
L
k
l
l
V
L
i
i j
j
≅
=
⋅
⋅
⋅
∑ ∑
1
gdzie j = 1, 2, ...k - numer siecznej ( kl = L - całkowita długość siecznych);
i = 1, 2, 3 - numer cięciwy (ziarna na siecznej)
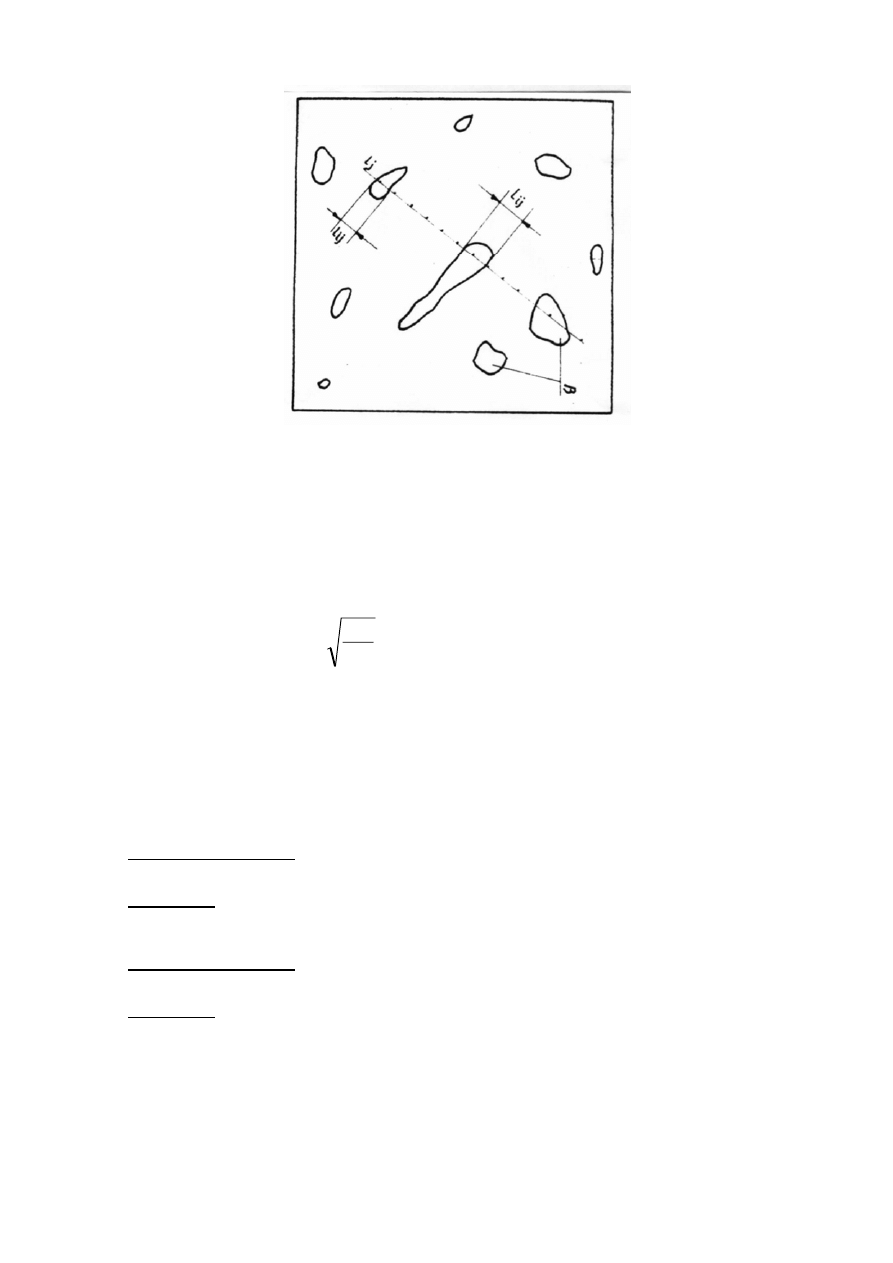
Rys.17. Zasada metody liniowej.
Przyjmując, że rozkład liczby ziarn fazy
β o określonej powierzchni w polu widzenia jest
rozkładem Poissona oraz, że wariancje względnej długości cięciw ziarn fazy i osnowy są
równe jedności, otrzymuje się następujące wyrażenie na błąd bezwzględny
δ
L
oszacowania
objętości względnej V
v
za pomocą L
L
:
δ
α
L
l
L
L
u
N
L
L
=
⋅
⋅
⋅
−
2
1
2
2
(
)
gdzie N
l
=
Σn
j
- całkowita liczba przeciętych ziarn, u
α
- współczynnik ufności, że
rzeczywisty błąd bezwzględny nie będzie przewyższał
δ
L
- przyjąć = 0.95.
Na podstawie powyższej metody ocenić zawartości faz w podanych szlifach
metalograficznych. Wyniki przedstawić w tabeli. Podać przykłady obliczeń i wnioski.
2.3.3.Uwagi i wnioski.
Literatura:
2.3.3.Uwagi i wnioski.
Literatura:
1. St. Szarras :" Budowa ciała stałego" - WNT W-wa 1975r.
2. C Kittel -"Wstęp do fizyki ciała stałego"
3. Ch. A. Wert, R.M. Thomson - Fizyka ciała stałego" - W-wa 1974, PWN
4. P.Wilkes - Fizyka ciała stałego
5. B.N. Buszmanow, J.A. Chromow -"Fizyka ciała stałego" WNT W-wa 1973.
6. R. Zallen - "Fizyka ciał amorficznych"- PWN W-wa 1994.
7. R. Fraś - Krystalizacja metali i stopów" - PWN W-wa 1992.
Wyszukiwarka
Podobne podstrony:
Ćwiczenia nr 6 (2) prezentacja
cwiczenie nr 7F
cwiczenie nr 2
Ćwiczenie nr 4
cwiczenia nr 5 Pan Pietrasinski Nieznany
cwiczenia nr 7
Cwiczenie nr 8 Teksty id 99954
Cwiczenia nr 2 RPiS id 124688 Nieznany
Cwiczenia nr 10 (z 14) id 98678 Nieznany
Ćwiczenie nr 1 (Access 2007)
cwiczenie nr 8F
Cwiczenie nr 2 Rysowanie precyzyjne id 99901
Cwiczenia nr 1 z l Zepoloych do
CWICZENIE NR 4 teoria
ćwiczenie nr 4
SPRAWOZDANIE Z CWICZENIA NR 4, Technologia zywnosci, semestr III, chemia zywnosci
Ćwiczenie nr 50b, sprawozdania, Fizyka - Labolatoria, Ćwiczenie nr50b
więcej podobnych podstron