
Badania struktury materiałów metodą elektronowej mikroskopii skaningowej i analizy rentgenowskiej w mikroobszarach
INŻYNIERIA MATERIAŁOWA
PAŃSTWOWA WYŻSZA SZKOŁA ZAWODOWA W TARNOWIE
METODY BADAŃ
BADANIA STRUKTURY MATERIAŁÓW METODĄ
ELEKTRONOWEJ MIKROSKOPII SKANINGOWEJ I
ANALIZY RENTGENOWSKIEJ W M1KROOBSZARACH
-1-

Badania struktury materiałów metodą elektronowej mikroskopii skaningowej i analizy rentgenowskiej w mikroobszarach
Wprowadzenie
Rozwój optyki elektronowej, elektroniki oraz potrzeba uzyskiwania dużych
powiększeń, niemożliwych do osiągnięcia w mikroskopii optycznej doprowadziły do
skonstruowania mikroskopu elektronowego, a w szczególności elektronowego
mikroskopu skaningowego. Elektronowy mikroskop skaningowy jest obecnie
najbardziej rozpowszechnionym typem mikroskopu elektronowego służącym do
obserwacji i badań mikrostruktury. Pierwszy jego egzemplarz wyprodukowany został
w firmie Cambridge Scientific Instruments w 1965 roku.
Istotną cechą wyróżniającą mikroskop skaningowy jest bardzo duża głębia
ostrości, umożliwiająca bezpośrednią obserwację rozwiniętych powierzchni
przełamów litych próbek. Zdolność rozdzielcza tego mikroskopu schodzi poniżej 10
nm. Zakres powiększeń możliwych do osiągnięcia – od 50 do 200000 razy.
Podstawy fizyczne mikroskopii skaningowej
W latach dwudziestych XX wieku stwierdzono, że elektrony tworzące strumień
poruszają, się w próżni po torach prostoliniowych a pola elektryczne i magnetyczne o
symetrii obrotowej mogą przekształcić rozbieżną wiązkę elektronów w wiązkę
zbieżną, skupiająca, się w punkcie. Wykazano w ten sposób, że strumień elektronów
zachowuje się w polu elektrycznym lub magnetycznym jak promienie świetlne w
soczewce, co stało się początkiem optyki elektronowej oraz prac nad wykorzystaniem
strumienia elektronów do generowania obrazów.
Wiązka elektronów skupiona do bardzo małych rozmiarów oddziaływuje z
powierzchnią materiału do głębokości zależnej od składu chemicznego (liczby
atomowej Z) i energii padających elektronów. Im większa energia i im mniejsza
liczba atomowa, tym głębsza penetracja elektronów w próbce. Elektrony
bombardujące, czyli pierwotne podlegają. zderzeniom sprężystym (zmiana toru bez
utraty energii, mająca miejsce podczas tzw. odbicia czy rozproszenia wstecznego) i
niesprężystym z atomami próbki. W wyniku zderzeń niesprężystych z elektronami
zewnętrznych powłok atomów na powierzchni próbki dochodzi do i przekazania
energii na tyle dużej aby przekroczyć wartość pracy wyjścia, przez co dostaje się
emisję elektronów wtórnych.
Elektrony wtórne stanowią ok. 90% wszystkich emitowanych z próbki
elektronów; charakteryzują się one stosunkowo niskimi energiami, znacznie poniżej
50eV. Elektrony odbite (wstecznie rozproszone), stanowiące ok. 3% wszystkich
emitowanych przez próbkę elektronów, mają wysoka, energię, na ogół w przedziale
50÷100eV, prawie równą energii elektronów padających (wielkość jej zależy od
parametrów pracy urządzenia). Jest jeszcze grupa elektronów o pośrednich
wielkościach energii. Pochodzą one z głębszych obszarów materiału i przed
-2-

Badania struktury materiałów metodą elektronowej mikroskopii skaningowej i analizy rentgenowskiej w mikroobszarach
opuszczeniem jego powierzchni podlegały wielokrotnym zderzeniom. Do grupy tej
należą także tzw. elektrony Auger, które zostały uwolnione z powłok położonych w
pobliżu jąder atomów budujących powierzchnię próbki.
Emisję elektronów wtórnych charakteryzuje tzw. współczynnik emisji wtórnej,
czyli stosunek liczby elektronów wtórnych do liczby elektronów padających na
próbkę, którego wartość zależy od energii elektronów pierwotnych, rodzaju materiału
i stanu powierzchni. Dla izolatorów wartość ta wynosi 2÷3, dla metali 0.7÷1.8.
Innym bardzo ważnym zjawiskiem obserwowanym gdy wiązka elektronów
działa na materiał jest emisja promieniowania rentgenowskiego charakterystycznego,
wykorzystywana w rentgenowskiej analizie spektralnej w mikroobszarach. Elektrony
wtórne generowane w głębszych obszarach próbki oraz elektrony pierwotne
penetrujące próbkę obsadzają powłoki elektronowe atomów uprzednio
zjonizowanych. Powrót tych atomów do stanu podstawowego (poprzez powrót
elektronów na podstawowe poziomy) jest związany z emisją promieniowania
rentgenowskiego charakterystycznego.
Zdolność rozdzielcza
Obszar, z którego emitowane są elektrony wtórne ma średnicę prawie równa,
średnicy plamki elektronów pierwotnych i niewielką grubość, w granicach 5÷50nm.
Wielkość tego obszaru określa tzw. zdolność rozdzielczą, czyli minimalna, odległość
pozwalająca, na rozróżnienie poszczególnych punktów. Z obszaru o kilkakrotnie
większej głębokości i średnicy emitowane są elektrony odbite, a obszar emisji
promieniowania rentgenowskiego jest jeszcze rozleglejszy.
Powiększenie
Powiększenie w mikroskopie skaningowym określane jest poprzez stosunek
wymiarów – liniowych uzyskanego na monitorze obrazu do rzeczywistych
wymiarów obserwowanego fragmentu próbki. Regulacji powiększenia dokonuje się
poprzez zmianę wielkości analizowanego obszaru próbki, co z kolei dokonuje się
zmieniając parametry odchylania wiązki za pomocą potencjometru. Operację tę
przeprowadza się szybko, w sposób ciągły lub skokowy. Nowoczesne mikroskopy
skaningowe pozwalają, na osiągnięcie powiększenia od 50x do 200000x. W
badaniach materiałów ceramicznych i pokrewnych pracuje się na ogół w zakresie
powiększeń 1000÷20000x.
Z uwagi na fakt, że w trakcie dokumentowania obserwacji mikroskopowych
wprowadza się powiększenie reprodukcji, uzyskując ilustracje o różnych rozmiarach,
w aparaturze pracującej obecnie do każdego obserwowanego obrazu dołączona jest
-3-
Badania struktury materiałów metodą elektronowej mikroskopii skaningowej i analizy rentgenowskiej w mikroobszarach
etykietka, na której, oprócz identyfikatora próbki (numeru), umieszczony jest
podpisany odcinek skali, dający dokładne wyobrażenie o wielkości obserwowanych
obiektów.
Zasada działania i budowa mikroskopu skaningowego
W elektronowym mikroskopie skaningowym badana powierzchnia próbki
penetrowana jest punkt po punkcie za pomocą wiązki elektronów pierwotnych.
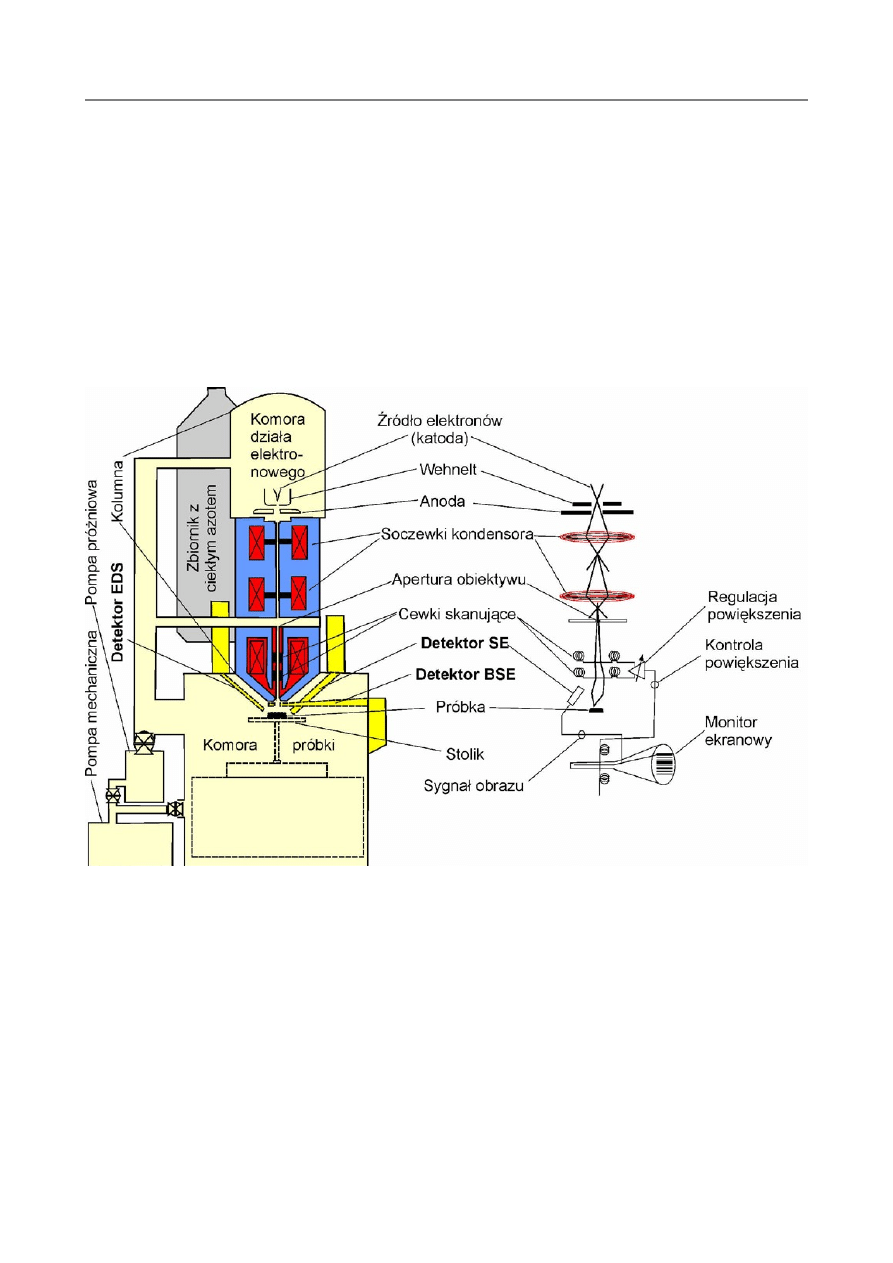
Elektrony emitowane są przez działo elektronowe (patrz rysunek 1).
Rys. 1. Schemat budowy elektronowego mikroskopu skaningowego (SEM)
Emiterem – katodą jest włókno wolframowe, osłaniane przez tzw. cylinder Wehnelta
– elektrodę sterująca, działa o potencjale ujemnym. Elektrony przyśpieszane są
następnie w polu elektrycznym między katoda. i anoda, (napięcie przyspieszające
rzędu 30÷50kV, wpływa na wielkość emisji), a następnie dążą w kierunku próbki w
kolumnie elektronooptycznej mikroskopu. Po drodze wiązka elektronów jest
ogniskowana przez zespół soczewek elektromagnetycznych do takich rozmiarów, aby
padając na próbkę działać na powierzchnię o średnicy 2÷5(10)nm. Rozmiar tej tzw.
plamki decyduje o zdolności rozdzielczej urządzenia. Układ ogniskujący składa się z
kondensora i obiektywu. Na wysokości soczewki obiektywu umieszczony jest zespół
-4-

Badania struktury materiałów metodą elektronowej mikroskopii skaningowej i analizy rentgenowskiej w mikroobszarach
cewek odchylających wiązkę w dwóch wzajemnie prostopadłych kierunkach. Dzięki
temu wiązka skanuje czyli omiata próbkę, tzn. przesuwa się po powierzchni próbki
punkt po punkcie, linia po linii. Ruch wiązki po powierzchni próbki jest sprzężony z
ruchem wiązki elektronów odtwarzających obraz próbki na ekranie monitora.
Działo elektronowe, soczewki elektromagnetyczne i urządzenia odchylające
rozmieszczone są w kolumnie elektronooptycznej mikroskopu, która w dolnej części
połączona jest z komorą próbek. Cały układ musi pracować w wysokiej próżni – ok.
10
-4
Pa, aby nie zakłócać biegu wiązki elektronów i z uwagi na bezpieczeństwo pracy
poszczególnych detali. Próżnię tę wytwarza system pomp. Działo elektronowe
wymaga chłodzenia wodą. Stolik z wgłębieniami na holdery do próbek może być z
zewnątrz manipulowany, tzn. przesuwany i przechylany, w celu ustawienia do
obserwacji odpowiedniego obiektu.
W mikroskopie skaningowym do odtwarzania obrazu powierzchni badanej
próbki wykorzystuje się emitowane pod działaniem padającej wiązki elektronów
elektrony wtórne. Elektrony te rejestrowane są przez detektor umiejscowiony w
komorze mikroskopu (licznik scyntylacyjny), do którego przyłożony jest wysoki
dodatni potencjał przyśpieszający (>10kV). Pojedyncze sygnały są wielokrotnie
wzmocnione i stanowią, prąd, którego zmieniające się od punktu do punktu natężenie
daje plamki o różnym stopniu jasności na ekranie monitora, odpowiadające
analizowanym miejscom powierzchni próbki. Wielkość emisji elektronów wtórnych
zależy od kąta padania wiązki elektronów pierwotnych na powierzchnię próbki, czyli
od (współrzędnych) położenia poszczególnych punktów na powierzchni próbki, a
więc od ukształtowania powierzchni próbki. Zależność ta stanowi podstawę
uzyskania obrazu morfologii powierzchni w mikroskopie skaningowym. Zmiana
wielkości „punktowych” sygnałów prądowych, odpowiadająca zróżnicowanej emisji
elektronów wtórnych uzyskanej w poszczególnych punktach nierównej, chropowatej
powierzchni próbki daje w efekcie kontrast na obrazie monitorowym, złożonym z
punktów o różnym natężeniu bieli – czerni (szarości), postrzegany przez oko ludzkie
jako na ogół mało „ziarnista” całość.
Preparatyka
Próbki przewodzące nie wymagają, specjalnej preparatyki przed obserwacjami
mikroskopowymi, oprócz odtłuszczenia powierzchni i osadzenia za pomocą
odpowiedniego kleju na podstawce (holderze) o średnicy 5÷15mm.
Kiedy wiązka elektronów bombarduje próbkę nieprzewodzącą następuje
kumulacja elektronów padających na powierzchni próbki (ładunek powierzchniowy),
przez co po zakłóceniu ulega zarówno emisja elektronów wtórnych jak i bieg wiązki
padającej w kolejnych operacjach skanowania. Może też dojść do przebicia
elektrycznego w próbce i miejscowego nagrzewania, co prowadzi do zniszczenia
-5-

Badania struktury materiałów metodą elektronowej mikroskopii skaningowej i analizy rentgenowskiej w mikroobszarach
preparatu. Aby uniknąć tych niepożądanych efektów pokrywa się powierzchnię
próbek nieprzewodzących warstewką materiału przewodzącego.: Jako materiał
pokryciowy stosowane są metale szlachetne, głównie złoto, z uwagi na łatwość emisji
elektronów, drobnoziarnistość, ułatwiająca, dokładne odwzorowanie powierzchni
pokrywanej i odporność na działanie czynników chemicznych. Warstwy maja,
grubość 0,01÷1nm, a pokrywanie odbywa się w napylarce próżniowej. W urządzeniu
tym pary metalu powstające w temperaturze łuku elektrycznego osadzają się na
próbkach. Próbki niemetaliczne pobrane do badań w postaci okruchów, przełamów,
proszków czy innych fragmentów większej całości, tak aby ich wielkość była
dopasowana do wielkości holderów, przykleja się uprzednio do powierzchni
holderów pastą przewodzącą. Próbki powinny być tak przygotowane aby nie ulegały
uszkodzeniu pod działaniem próżni i aby nie wydzielały składników, które mogłyby
zakłócić prace napylarki czy mikroskopu.
Podstawy mikroanalizy rentgenowskiej
Promieniowanie rentgenowskie
powstające podczas bombardowania
próbki wiązką elektronów, po detekcji, wzmocnieniu i przetworzeniu może
dostarczyć szeregu informacji dotyczących składu chemicznego, stanowiąc cenne
uzupełnienie obserwacji mikrostruktury. Mikroanaliza rentgenowska oparta jest na
prawie Moseleya, które stanowi, że długość fali promieniowania rentgenowskiego
jest funkcja, liczby atomowej Z i praktycznie nie zależy od fizycznego i chemicznego
stanu materiału.
Promieniowanie rentgenowskie składa się z widma ciągłego i
charakterystycznego pierwiastków wchodzących w skład próbki. Gdy elektrony
pierwotne ulegają, zderzeniom niesprężystym z jądrami atomów próbki badanej
tracą, w zależności od kąta zderzenia, różne ilości energii, emitowane w postaci
fotonów promieniowania ciągłego. Kształt widma promieniowania ciągłego zależy
od wielkości napięcia przyśpieszającego i charakteryzuje się pewna, minimalną
długością, fali. Gdy natomiast elektrony pierwotne lub wtórne o energii wyższej od
energii wiązania elektronów w atomach próbki badanej wybiją te elektrony na
wyższe poziomy energetyczne, czyli wprowadza, atomy w stan wzbudzenia, powrót
do stanu podstawowego będzie się odbywał poprzez przeskoki elektronów z dalszych
powłok na powłoki opróżnione, z wyemitowaniem kwantów promieniowania
charakterystycznego o ściśle określonych wielkościach energii, odpowiadających
różnicy poziomów energetycznych.
Przeskoki i emisje związane odpowiednio z powłokami K, L... noszą nazwy
serii widmowych K, L... itd. Poszczególne linie widmowe różnią, się natężeniem, co
wynika z prawdopodobieństwa przejść i wielkości energii. Najistotniejsze w
mikroanalizie są linie serii K, z uwagi na wysoka, energię wiązania elektronów w
-6-

Badania struktury materiałów metodą elektronowej mikroskopii skaningowej i analizy rentgenowskiej w mikroobszarach
pobliżu jądra i małe rozszczepienie. Im wyższy poziom energetyczny tym więcej linii
ze względu na postępujące rozszczepienie poziomów.
Generowanie promienia rentgenowskiego zachodzi w warstwie
przypowierzchniowej próbki, na głębokości uzależnionej od energii elektronów
padających i liczb atomowych pierwiastków budujących powierzchnię próbki.
Możliwość wzbudzenia poszczególnych linii promieniowania X jest dla każdego
pierwiastka charakteryzowana przez podanie krytycznej energii wzbudzania (energii
minimalnej); z wielkością tą związana jest głębokość krytyczna wzbudzenia. Obydwa
wymienione parametry – energia krytyczna i głębokość krytyczna wzbudzenia zależą
od krytycznej (minimalnej) wartości napięcia wzbudzającego. Wzrost napięcia ponad
wartość krytyczną daje wzrost wykrywalności i wzrost intensywności sygnału.
Jednakże, z uwagi na absorpcję promieniowania X przy wzroście napięcia (związanej
z wydłużeniem drogi promieniowania w materiale) oraz wzrost tła (promieniowania
charakterystycznego), stosuje się w praktyce kompromisowe wielkości, równe 2÷3
krotnej wartości wielkości krytycznej.
Spektroskopia promieniowania rentgenowskiego (analiza promieniowania
rentgenowskiego w mikroanalizatorach)
Spektroskopia promieniowania rentgenowskiego może być przeprowadzona
albo:
A – metodą dyspersji długości fal, albo
B – metodą dyspersji energii (rozdział uzyskanego od próbki sygnału na podstawie
długości fali lub wielkości energii).
Ad A: Długość fali wzbudzonego promieniowania charakterystycznego jest określana
dzięki zjawisku dyfrakcji promieni X na kryształach. Warunek wzmocnienia
promieni X ugiętych na krysztale podaje równanie Bragga:
nλ = 2dsinΘ
gdzie:
X – długość fali promieniowania X
d – odległość między płaszczyznami w krysztale
Θ – kąt odbłysku (kąt pomiędzy promieniem padającym a płaszczyzna, kryształu)
n – 1, 2, 3... rząd refleksu, tzn. krotność długości fali w różnicy dróg między
promieniami ugiętymi na sąsiednich płaszczyznach.
Gdy na kryształ pada wiązka promieniowania X o różnych długościach fal,
ulegają one dyfrakcji pod różnymi kątami Θ. W praktyce źródło promieniowania X
pozostaje nieruchome (próbka) – a kryształ, na którym dokonuje się dyfrakcja i
-7-

Badania struktury materiałów metodą elektronowej mikroskopii skaningowej i analizy rentgenowskiej w mikroobszarach
detektor promieniowania ugiętego wędrują po łuku będącym częścią, okręgu (tzw.
okręgu Rowlanda). Zmiana położenia kryształu (podczas jego mchu obrotowego)
powoduje, że w różnych jego położeniach spełniony jest warunek Bragga dla
promieni X o różnych długościach fali, charakterystycznych dla poszczególnych
pierwiastków budujących analizowaną próbkę. Kryształ dokonuje w ten sposób
stopniowej selekcji promieniowania o ściśle określonych długościach fali
(promieniowania monochromatycznego), a więc działa jak monochromator. Mierząc
kąty Θ można z równania Bragga wyznaczyć długość fali, a więc zidentyfikować
pierwiastki w próbce. Zwykle monochromator składa się z kilku kryształów, które
mają bardzo różne płaszczyzny ugięcia, aby była możliwość mierzenia szerokiego
zakresu długości fal.
Do detekcji promieniowania X stosuje się detektory jonizacyjne. Kwanty
promieniowania X dostają się przez okienko do licznika i wywołują w nim jonizację
gazu (mieszanina argonu z metanem). Powstałe wskutek tego elektrony i naładowane
dodatnio jony podążają odpowiednio do anody i katody stanowiąc impulsy prądowe
podlegające wielokrotnemu wzmocnieniu. Impulsy są zliczane przy pomocy
przelicznika sprzężonego z integratorem – miernikiem częstości impulsów (liczby
zliczeń w jednostce czasu), będącym odbiciem natężenia wiązki. Wskazania
integratora są podawane przez układ monitorujący i mogą. być rejestrowane za
pomocą. drukarki komputerowej.
Ad B: W metodzie dyspersji energii promieniowania X detekcję i rejestrację
promieniowania przeprowadza się na podstawie wielkości energii przypisanym
liniom charakterystycznym dla pierwiastków (od boru w górę układu okresowego).
Jako detektor służy w tym przypadku półprzewodnik (krzemowe złącze typu p-n
domieszkowane litem, zanurzone w ciekłym azocie). Wiązka fotonów
promieniowania X o energiach właściwych dla pierwiastków analizowanego obszaru
próbki pada na detektor i generuje pary elektron – dziura. Po przyłożeniu napięcia
ruch ładunków jest zamieniany na impulsy prądowe, które po wzmocnieniu,
przetworzeniu na impulsy napięciowe i rozdzieleniu w analizatorze dają, widmo
energetyczne – obszaru badanego złożone z pików odpowiadającym energiom
promieniowania X namierzonym w tym obszarze. Analizę takiego widma, tzn.
przypisanie poszczególnym pikom (pasmom) odpowiednich nazw pierwiastków
przeprowadza się za pomocą, programów komputerowych. Widmo przeanalizowane i
zaopatrzone w stosowne opisy jest przenoszone z monitora poprzez rejestrację za
pomocą, drukarki. W urządzeniach pracujących obecnie drukuje się jednocześnie
obraz morfologii badanej próbki.
Z punktu widzenia geometrii mikroanalizy wyróżnia się następujące techniki
pomiarowe:
a)
analiza jakościowa z całego analizowanego obrazu – identyfikacja
pierwiastków na pewnym obszarze wybranym na podstawie obserwacji pod
-8-

Badania struktury materiałów metodą elektronowej mikroskopii skaningowej i analizy rentgenowskiej w mikroobszarach
mikroskopem skaningowym,
b) analiza punktowa – określenie pierwiastków występujących w danym punkcie,
pomocne przy identyfikacji elementów mikrostruktury,
c) analiza zmian rozmieszczenia danego, wybranego pierwiastka wzdłuż linii,
d) powierzchniowe rozmieszczenie poszczególnych pierwiastków na badanej
powierzchni (mapa pierwiastka, tzw. mapping).
Poszczególne techniki b-d realizuje się poprzez skoncentrowanie wiązki
analizującej na wybranym punkcie (b), umożliwienie względnego ruchu próbki i
analizującej wiązki wzdłuż linii (c), analizę widm poszczególnych pierwiastków
oddzielnie, ale na całym wydzielonym obszarze badanym i przetworzenie impulsów
na obraz punktowy (d).
Przy stosowanym obecnie zapisie komputerowym wynik końcowy zawiera
obraz powierzchni próbki uzyskany za pomocą. SEM, z zaznaczonym, w zależności
od techniki, miejscem analizowanym lub dołączoną mapą (mapami).
-9-
Wyszukiwarka
Podobne podstrony:
Analiza spalin cz 3 Analizatory Nieznany (2)
zasady dzialania mikroskopu skaningowego
06 Lutomirski S i inni Analiza Nieznany
analizasygnalowiidentyfikacja2 Nieznany (2)
castorama i LM projekt analiza Nieznany
mikroskop 2 id 301809 Nieznany
ANOVA jednoczynnikowa analiza w Nieznany (2)
Egzamin Mikrostruktury id 15252 Nieznany
analizafinansowaprzedsiebiorstw Nieznany (2)
, chemia analityczna L, analiza Nieznany (2)
Instrukcja do cwiczen 'Analiza Nieznany
Analiza Matematyczna 2 Analiza0 Nieznany (2)
01 Ajdukiewicz C i inni Analiza Nieznany
Pomiary mikroskopowe id 374389 Nieznany
Analiza spalin cz 3 Analizatory Nieznany (2)
Teper Podstawy mikroskopii skaningowej
PODSTAWY MIKROSKOPII SKANINGOWEJ
analiza ryzyka bio id 61320 Nieznany
więcej podobnych podstron