
1. ALLOSTERIA – enzymy allosteryczne, modele oddziaływań, kinetyka, przykład
(30. KINETYKA SIGNOIDALNA – charakterystyka, znaczenie)
1. Allosteria to jeden z przykładów wpływu na sprawność katalityczną enzymu indukowaną przez
odwracalne wiązanie ligandów do samego białka enzymatycznego.
2. Enzymy allosteryczne to takie enzymy, których aktywność miejsca katalitycznego może być
regulowana przez efektory allosteryczne, znajdujące się w miejscu allosterycznym (miejsce na
podjednostce danego enzymu, które łączy się z ligandem modyfikującym wtórną strukturę enzymu
allosterycznego). Przyłączenie ligandu zmienia struktuę przestrzenną powodując albo hamowanie
(enzym traci aktywnośc katalityczną) lub aktywację (enzym po przyłączeniu efektora zyskuje
aktywność enzymatyczną).
3. Efektory nie są izosteryczne, lecz allosteryczne (dlatego, że zajmują inna przestrzeń) z substratem.
4.Hipotezę miejsc allosterycznych przedstawił Jacques Monod. Potwierdzają to badania
przeprowadzone za pomocą krystalografii rentgenowskiej i mutagenezy ukierunkowanej, wykazujące
istnienie odrębnych miejsc aktywnych i allosterycznych w przypadku różnorodnych enzymów.
5.Zmiany konformacyjne obejmujące miejsce katalityczne enzymu pod wpływem efektora
allosterycznego mogą wpływać na sprawność katalityczną enzymu (enzymy serii V-efekt allosteryczny
zmienia wartość Vmax bez wpływu na Kmpierwotnym skutkiem może być zmiana oreitnacji
przestrzennej łub ładunku reszt katalitycznych prowadząca do zmiejszenia Vmax), powinowactwo
enzymu do substratu (enzymy seri K-zmieniają Km, nie zmianiją Vmaxskutkiem zmian
konformacyjnych może być osłabienie wiązań między substratem i resztami aminokwasowymi w
miejscu wiążacym substrat.) lub też na obie właściwości.
6. Zmiany aloosteryczne często wykorzystywane są w hamowaniu przez sprzężenie zwrotne, bowiem
ostatni produkt szeregu przemian enzymatycznych może być inhibitorem allosterycznym do enzymu
dokonującego zazwyczaj pierwszej przemiany enzymatycznej w danym szlaku. Przykładem jest
karbamoilotransferaza asparaginianowa (ATCaza / CPSII) – katalizuje pierwszą reakcje szlaku
biosyntezy pirymidyn – enzym ten hamowany jest allosterycznie przez trifosforan cytydyny (CTP) oraz
aktywowany przez ATP. Wysoki poziom ATP może znieść jednak hamujący efekt CTP, umozliwiając
syntezę nukleotydów pirymidynowych w obecności podwyższonej ilości nukleotydów purynowych.
Hamowanie przez sprzężenie zwrotne może być bardziej skomplikowane i przebiegać w rozgałęzionym
szlaku biosyntezy obejmującym hamowanie na róznych poziomach tego szlaku.
7.Wykres zależności szybkości reakcji V od stężenie substratu S dla enzymów allosterycznych
przybiera kształt signoidalny w odróżnieniu od hiperbolicznego przewidzianego równianie Michaelisa-
Menten. Dzieje się tak dzięki kooperatywności wiązania cząsteczek substratu – jedno miejsce aktywne
enzymu może oddziaływać na inne miejsce aktywne tej samej czasteczki enzymu.
INHIBITOR ALLOSTERYCZNY – wiąze się w sposób uprzywilejowany z formą T, czyli przesuwa
równowagę w kierunku
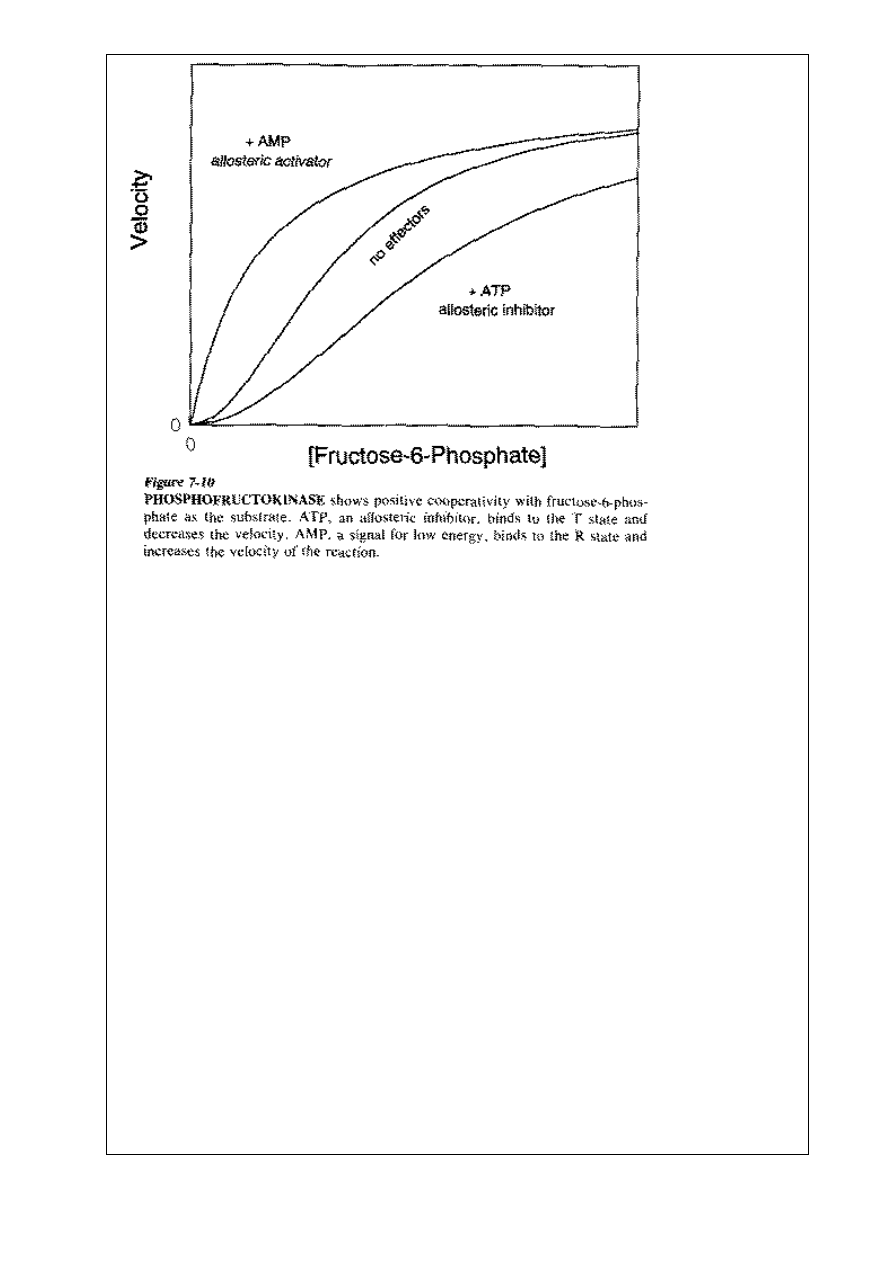
T.
AKTYWATOR ALLOSTERYCZNY – łączy się preferencyjnie z formą R, przesuwa rownowagę w
kierunku R.
EFEKT HOMOTROPOWY – odpowiada oddziaływaniom allosterycznym pomiędzy identycznymi
ligandami.
EFEKT HETEROTROPOWY – odpowiada oddziaływaniom allosterycznym pomiędzy róznymi
ligandami.
8.MODEL JEDNOPRZEJŚCIOWY (Monoda, Wyman, Changeux)
Zakładając, że enzym składa się z 2 podjsednostek (R – mająca duże powinowactwo do substratu / T –
małe powinowactwo). Formy R i T mogą przechodzić w siebie wzajemnie jednak warunkiem jest to, że
wszystkie podjednostki enzymu muszą występować w tej samej konformacji, tak aby została zachowana
symetria cząsteczki enzymu. Zakazana jest forma RT (istnieją formy RR i TT). Przy braku substratu
obie formy opisuje się jako To i Ro, a L to stosunek stężeń To do Ro.
W jednoprzejściowym modelu oddziaływań allosterycznych efekty mogą być: homotropowe – zawsze
dodanie (kooperatywne) lub heterotropowe – albo dodanie albo ujemne.
Liczba cząsteczek enzymu w formie R zwiększa się ze wzrostem stężenie substratu i dlatego wiązanie
substratu jest kooperatywne. Kiedy miejsca aktywne są w pełno osadzone, wtedy wszystkie cząsteczki
enzymu wystepują w formie R.
9. MODEL SEKWENCYJNY – kolejnych zdarzeń Koshlanda
każda z podjednostek enzymu występuje w jednej z form konformacyjnych (R lub T). Związanie
efektora allosterycznego powoduje zmianę konformacji podjednostki jednak nie ma wpływu na
konformacje drugiej jednostki. Zmiana konformacji podjsednostki natomiast powoduje zwiększenie
powinowactwa do substratu innych podjednostek tej samej czasteczki enzymu.
Wiązanie jest kooperatywne tylko wtedy kiedy powinowactwo RT o substratu jest większe od
powinowactwa TT.
Różnice z modelem jednoprzejściowym: - model sekwencyjny nie zakłada równowagi między stanami
T i R gdy brak substratu (przejście z T do R wymuszone jest wiązaniem substratu) – zmiana
konformacyjna z T do R wystepuje kolejno w róznych podjednostkach – forma RT jest zasadnicza (w
jednoprzejściowym niedozwolona) – brak konieczności symetrii podjednostek enzymu – oddziaływania
homotropowe mogą być dodatnie lub ujemne, a druga cząsteczka substratu może ulec silniejszemu lub
slabszemu wiązaniu niż pierwsza w zależności od odkształceń strukturalnych wprowadzonych przez
związanie pierwszej cząsteczki substratu.

10. Niektóre enzymy cechują się m.j. a niektóre m.s. a niektóre w ogóle nie stosują się do tego
schematu.
11.Dimer w formie R może wiązać 1 lub 2 cząsteczki substratu. Gdy brak substratu cały enzym
występuje w formie T. Dodatek substratu przesuwa konformacje do formy R, gdyż substrat wiąże się
tylko z R.
2. FOSFATAZA ALKALICZNA – izoenzymy, podział EC i kliniczny, występowanie, znaczenie
diagnostyczne, normy aktywności, katalizowana reakcja.
- reakcja: hydrolityczne odszczepienie reszty fosforanowej z organicznych estrów kwasu fosforanowego
przy pH>7.
- normy aktywności: w surowicy – 580/1400 nkat/l [ 35/84U/l]
- podział: Richtericha i Hessa: ekskrecyjne (wydalnicze) – enzymy żółci; EC:3.1.3.1
-izoenzymy: wątrobowy, kostny, jelitowy, łożyskowy
-występowanie: 1.tkanka kostna, 2.błona śluzowa jelita, 3.łożysko;4.niektóre komórki
nowotworów;5.nerki;6.wątroba [wewnątrz komórek znajduje się w jądrze, mitochondriach, lizosomach i
cytoplazmie]
-rozdzielanie izoenzymów:
a.elektroforeza(jelitowy<kostny<wątrobowy),
b.chromatografia,
c.metody immunologiczne (Ostase(Beckman)-immunoradiometryczne – miara ilościowa; metodra
Metra – wychwytuje izoenzym kostny (immunofiksacja) i mierzy jego aktywność
d.inaktywacja poszczególnych izoenzymów (1.kwaśne środowisko: k++,w+,j+,ł+/ 2.temperatura
k++(5min),w+,j+,ł-/ 3. wersenian sodu: k+,w+,j+,ł-/ 4. mocznik: k++,w++,j+,ł-/ 5. fenyloalanina: k-,w-
,j+,ł+/6ruchliwość elektroforetyczna pod wpł. Neuramidazy: kzm,wzm,jniezmieniona,łzm
-znaczenie diagnostyczne:
1.nowotwory niektóre – we krwi obecne specyficzne izoenzymy f. zasadowej (markery nowotworowe)
– Regan, Nagao, Kasahara -> stwierdza się je: rak płuc, żołądka, wątroby, nerek, gruczołu krokowego
(nie zawsze wystepuja i nie zawsze jest wzrost ich aktywności w chorobie nowotworowej)
2.choroby wątroby i dróg żółciowych przebiegające z zastojem żółci (żółtaczka mechaniczna, marskość
zaporowa wątroby)
3.choroby kości związane ze zniszczeniem i przebudową tkanki kostnej (1.włókniste torbielowate
zapalenie kości w przebiegu nadczynności przytarczyc, 2.zniekształcające zapalenie kości w chorobie
Pageta, 3. osteoblastyczne guzy kości, 4.zapalenie szpiku kostnego, 5. gruźlica kości, 6. długotrwałe
unieruchomienie, 7.niedobór witaminy D
4.izoenzym kostny jest markerem kościotworzenia (podobnie jak osteokalcyna)
5. fizjologiczny wzrost aktywności (dzieci – rozwój kości, kobiety pod koniec ciąży i 20 dni po
porodzie)
3.INHIBICJA NIEKOMPETYCYJNA, def., mechanizm działania, kinetyka reakcji
enzymatycznej (wykres podwójnych odwrotności)
4.PSA. Wyjaśnij skrót, budowa, występowanie, wykorzystanie w diagnostyce
1.Prostate Specyfic Antigen - antygen swoisty dla prostaty
2.Budowa:glikoproteina – monomer, kalikreina (proteaza serynowa); N-terminlna izoleucuna oraz C-
terminalna prolina
3.Występowanie: komórki wydzielnicze przewodów stercza (gruczoł krokowy)
4.Diagnostyka:jedyny marker wykrywania wczesnego nowotworu prostaty (inne markery pozwalają
monitorować jego rozwój). Wartość prawidłowa (w ng/ml zależna jest od wieku – im starszy mężczyzna
tym większa granica normy). Większa skuteczność diagnostyczna od PAP – izoenzym sterczowy
fosfatazy kwaśnej. Znaczenie diagnostyczne ma też oznaczenie frakcji wykrywalnej w stosunku do
całkowitego PSA.
5. Okres półtrwania 2-3 dni – po prostatectomii dopiero po kilku dniach zanika PSA w surowicy.
Nowotwór prostaty produkuje nawet 10x więcej PSA niż zdrowy organ.
6.Są 2 frakcje: wykrywalna wędrująca z alfa-2-M(wolna), niewykrywalna (z A1-związana)
5. TF – czynnik tkankowy, gdzie działa, w jakim celu.

1. Tromboplastyna (dawna nazwa – nieużywana), III czynnik krzepnięcia, czynnik tkankowy, CD142
2.Glikoproteina.
3.Wystepowanie: ściana naczyniowa, śródbłonek, monocyty.
Może występować w osoczu: pula latentna -> induktory -> pula aktywna. Główna przyczyna zespółu
rozsianego wykrzepiania wewnątrznaczyniowego(DIC).
4.Synteza: MOCYTY, MAKROFAGI (NIE MA SYNTEZY W TROMBOCYTACH!)
a. konstytutywna – w warstwie śródnabłonkowej i mięśniówce – indukcja krzepnięcia
b. synteza indukowana – 1.cytokinami(środbłonka i moncytów), 2.endotoksynami bakteryjnymi, 3.C5
dopełniacza
5.Funkcja-aktywator drogi zewnątrzpochodnej układu krzepnięcia(aktywacja czynnika VII
[prokonwertyna] do VIIa). Działa jako kofaktor czynnika VIIa, zwiekszając jego aktywność
enzymatyczną prowadzącą do aktywacji czynnika X. Współdziałanie czynnika VIIa i TF nazywane jest
zespołem czynnika tkankowego.
6.Makrofagi blaszki miażdżycowej obżerają się cholesterolem przy okazji tworzą duże ilości TF – po
przerwaniu blaszki miażdzycowej uwalnia się TF – wykrzepianie – zakrzep! Zawał lub udar danego
organu.
6. WPŁYW NA SPRAWNOŚC KATALITYCZNĄ ENZYMU, definicja, znaczenie, opisać
opierając się na przykładach + mechanizm
1.Poza zmiano ilości białka enzymatycznego (ograniczenie eskpresji genów, degradacja białek
enzymatycznych), która dotyczy np. hormonów sterydowych występuje tzw. wpływ na sprawnośc
katalityczną enzymu – czyli zmiana aktywności danego enzymu bez zmian jego ilości.
2.Znaczenie: znacznie szybsza odpowiedź organizmu na zapotrzebowanie danego enzymu niż w
przypadku zmian ilościowych tego białka enzymatycznego.
3.PRZYKŁADY:
1.Aktywacja proenzymu do enzymu ograniczona proteoliza (np. trypsynogen – trypsyna)
2..Interkonwersja enzymu [a.. jeden ze sposobów wpływu na aktywność katalityczna enzymu – zmiana
jego aktywności bez zmiany jego ilości.
b.. Zmiana formy aktywnej w nieaktywną dokonuje się pod wpływem dodania lub usnięcia grupy
fosforanowej (fosforylacja i defosforylacja – mofyfikacje posttranslacyjne). Fosforylowana może być
grupa –OH – tyrozyny, seryny i treoniny, atom N – argininy, histydyny i lizyny oraz atom tlenu
asparaginianu – powstają odpowiednio O- i N- pochodne. Proces ten katalizują kinazy(dołączają grupę
fosforanową) oraz fosfatazy (odszczepiają).
c.. Poprzez ten mechanizm działają wszystkie hormony, w których wtórnym przekaźnikiem jest cAMP.
Umożliwia to o wiele szybszą kontrolę działania aktywności enzymu niż działanie na poziomie
ekspresji genów.
d. przykład: syntaza glikogenowa aktywna w formie ufosforylowanej, oraz fosfataza glikogenowa
aktywna w formie nieufosforylowanej.]
3.Regulacje allosteryczne – zachodzą pod wpływem ligandów allosterycznych – aktywatorów (enzym
zyskuje aktywność) lub inhibitorów (enzym traci aktywność)
4.Kompartmentalizacja enzymów – enzymy znajdują się odpowiednich przedziałach komórkowych
gdzie pełnią określone funkcje i dostepnośc danego enzymu w przedziale decyduje, czy zachodzi dana
reakcja czy też nie. Przykład; synteza kwasy ALA – zachodzi w rybosomach, działa on w
mitochondriach- miejsca te oddzielone są błonami – aby doszło do syntezy porfiryn ALA musi być
przemieszczony.
Inny przykład:
CPS I – cykl mocznikowy
CPS II – w cytozolu, enzym w syntezie pirymidyn – enzymy maja oddzielen substraty i rozdzielone są
błonami.
Błony komórkowe umozliwiają regulacje dopływu substratu do poszczególnych enzymów, a także
wzajemne zazebianie się procesów tego samego przedziału komórkowego. Regulują przepływ:
koenzymów, inhibitorów, odpowiadają za szybkość odprowadzania produktów.
A.jądro –5’-nukleaza; Na/K zalezna ATP-aza
B.mitochondrium-błonazewnątrzena-oksydaza monoaminowa / przestrzeń miedyzbłonowa – kinaza
adenylowa / błona wewnętrzna – dehydrogenaza bursztynianowa / macierz – dehydrogenaza
glutaminowa
C.lizosomy – AcP, DNA-za, RNA-za
D.Aparat Golgiego – galaktozylotransferaza, pirofosfataza tiaminy

E.peroksysomy – katalaza, oksydaza D-aminokwasów
F.ER-glukozo-6-fosfataza
G.cytozol – dehydrogenaza mleczanowa i glukozo-6-fosforanowa
5.czynniki regulujące potencjał oksydoredukcyjny środowiska = aktywacja enzymów zawierających w
centrum katalitycznym grupy –SH. Mogą one ulec utlenieniu do –S-S- i enzym traci aktywność.
Aktywatorami takiego enzymu są substancje o niskim potencjale oksydoredukcyjnym (reduktory) –
papaina, dehydrogenaza alkholowa
6.Ujemne sprzężenie zwrotne
7.Kofaktory (koenzymy, gr. Prostetyczne, jony metali,aniony nieorganiczne)
8.pH, siła jonowa, T, aktywatory i inhibitory, potencjał
7.IZOENZYMY, definicja, metody rozdziału, znaczenie diagnostyczne, wykrywanie
1. Definicja: genetycznie uwarunkowane odmiany enzymu, występujące w organizmach tego samego
gatunku, niekiedy tej samej komórki, katalizujące tą samą reakcję, a różniące się strukturą molekularną i
niektórymi parametrami kinetyki enzymatycznej (Vmax, Km). Zazwyczaj różnią się składem
podjednostkowym lub sekwencja aminokwasową. Posiadają zróżnicowane właściwości: ruchliwość
elektroforetyczna, rozpuszczalność(różnicowanie tylko w próbie rivanolowej aminotransferaz),
odporność na inaktywację fizykochemiczną(izoenzym kostny AP ulega inaktywacji po 5 minutach
inkubacji w temp56C), wrażliwość na inhibitory(chamowanie izoenzymu sterczowego AcP L-
winianem), aktywność molekularna, stała Michaelisa – powinowactwo do substratów, zdolność do
katalizy reakcji, antygenowość(zdolność do powstawania kompleksu immunologicznego – ocenia się w
ten sposób stężenie danego izoenzymu.
2. Metody rozdziału:
A. Elektroforeza – najczęściej elektroforeza wysokonapięciowa albo elektroogniskowanie w gradiencie
pH.
B. Chromatografia – najczęsciej chromatografia powinowactwa albo HPLC (chromatografia
wysokonapięciowa)
C. Inaktywacja chemiczna – stosowanie swoistych inhibitorów.
D. inaktywacja fizyczna – najczęściej cieplna
E. Wykorzystanie właściwości katalitycznych – używanie analogów substratów, które są przekształcane
tylko przez jedną formę izoenzymu – używanie swoistych inhibitorów, które hamują konkretny
izoenzym
F. metody immunologiczne – najczęściej obecnie stosowane.
3. Znaczenie diagnostyczne – oznaczenie izoenzymu pozwala na wnioskowanie, że w organie z którego
pochodzi następuje proces chorobowy – izoenzym może być markerem diagnostycznym jeżeli zmiany
jego stężenia lub aktywności mają wartość predykcyjną. Oprócz rozpoznania danego procesu
chorobowego oznaczanie danego izoenzymu może służyć monitorowaniu rozwoju procesu
chorobowego. Dlatego dzieli się enzymy wg. Podziału klinicznego. Oznaczanie aktywności enzymów
ma szczególne znaczenie przy chorobach wątroby i mięśnia sercowego.
8.KOMPLEKSY WIELOENZYMATYCZNE – przykłady, budowa, stopnie organizacji,
znaczenie
1.Jest to kilka enzymów katalizujących następujące po sobie reakcje, łączących się w wyżej
zorganizowane struktury.
2.Znaczenie: przekazywanie związków pośrednich (intermediatorów) od jednego enzymu do drugiego
jest termodynamicznie korzystnie ponieważ nie dochodzi prawie do strat energii, co umownie zostało
nazwane kanałowaniem (channelingiem). Poza mniejszymi nakładami energetycznymi zwiększa się
dostępność substratu dla enzymu, rośnie wydajność całego procesu.
3.Stopnie organizacji:
A. poszczególne enzymy znajdują się w roztworze cytoplazmatycznym – nie są ze sobą w żaden sposób
związane (najprostszy układ). Cząsteczki substratu często dyfundują, więc łatwo przemieszczają się od
jednego enzymu do drugiego. Jest to najmniej korzystny układ bowiem dochodzi do rozproszenia dużej
ilości energii. Przykład – enzymy glikolizy; dehydrogenaza pirogronianowa (udział w rekacji
pomostowej)-jest to ukłąd 3 enzymów i 5 koenzymów
E1(tiamina),E2(kwaslipoilowy,CoA),E3(FAD,NAD+).
B. Poszczególne enzymy są fizycznie zasocjowane. Wystepuje tu łatwe przekazywanie intermediatów
(kanałowanie), a straty energii są znacznie niższe niż w pierwszym przypadku.Intermediaty nie
oddzielają się od kompleksu. Przykład: enzymy syntezy kwasów tłuszczowych.

C. Układy enzymów zasocjowane z błonami lub rybosomami. Zapobiega to rozproszeniu enzymów i
intermediatów w cytoplazmie i tym samym utrata energii jest bardzo mała. Poza tym zwiększa się
prawdopodobieństwo zderzenia substratu z centrum katalitycznym enzymu, co w rezultacie prowadzi do
znacznego przyspieszenia katalizowanej reakcji. Przykłady: rybosomy na RER, łańcuch oddechowy
(enzymy zasocjowane z wewnetrzną błoną mitochondrium)
9. JEDNOSTKI AKTYWNOŚCI ENZYAMTYCZNEJ
Aktywność enzymu- szybkość reakcji enzymatycznej mierzona w scisle określonychwarunkach i
wyrażana w następujących jednostkach:
1. KATAL (kat) – aktywnośc enzymu przekształcającego 1 mol substratu w czasie 1 sekundy, w
temperaturze 30C, w optymalnym pH i w warunkach reakcji rzedu zerowego (przy pełnym wysyceniu
enzymu substratem). Jest to jednostka bardzo duża dlatego stosuje się podwielokrotności katala.
Jednostka mol/s.
2. Międzynarodowa jednostka enzymatyczna (U/IU – aktywnośc enzymu rozkładającego 1 mikromol
substratu w czasie 1 minuty w temperaturze 30C w optymalnym PH i warunkach reakcji rzedu
zerowego. 1U=16,67x10-9kat. Czasem gdy substratem jest cukier lub inny polimer okreslenie 1 mol
substratu określa się 1 mol odpowiednich wiązań chemicznych. Gdy reakcja przebiega między 2
identycznymi cząsteczkami związku, za podstawę obliczeń przyjmuje się 2 mikromole tego związku.
Przy oznaczaniu aktywności biologicznej w roztworach wyniki przelicza się na 1000cm3 (stęzenie
aktywności enzymatycznej w roztworze).
3.Aktywność właściwa – liczba jednostek aktywności przypadająca na jednostkę masy białka (kat/kg,
U/mg)
4.Aktywnośc molekularna – określa reaktywnośc enzymu – jest to liczba cząsteczek substratu
przekształconych w produkty w czasie 1 minuty przez jedną cząsteczkę enzymu w optymalnych
warunkach. Dla enzymów mających kilka centrów katalitycznych mówi się o aktywności molekularnej
danego centrum katalitycznego. Jej pochodną jest aktywność molarna – kat/mol.
Umowne jednostki:
A. Bodanskiego – ilość uwolnionego fosforu z beta-glicerofosforanu sodu, przez fosfatazę znajdującą
się 100ml surowicy w ciągu 1h inkubacji w t=37C, w pH=8,6 dla AP i pH=5 dla AcP.
B.Kinga-Armstronga – ilośc mg fenolu uwolnionego z fenylofosforanu sodu, przez fosfatazę znajdujaca
się w 100 ml surowicy w t=37C w ciągu: 15 minut inkubacji w pH-8,6 dla AP i 60 minut w pH=4,9 dla
AcP
C.Caraway’a – taka aktywność która hydrolizuje 10mg skrobi w ciągu 30 minut do stopnia w którym
zanika reakcja z jodem, aktywnośc wyraża się do 100 ml materiału badanego.
10. INHIBICJA KOMPETYCYJNA, inhibitory jako leki
1.Acetylocholinesterazy: jaskra: neostygmina, prostygmina, fizostygmina / przełomy miasteniczne –
endrofonium / choroby Alzheimera – riwastygmina, donezapil
2. Dla fosfodiesterazy (metaloksantyny) – cGMP – cytrynian sildenafilu / cAMP – kofeina, teofilina,
teina, aminofilina
3.Dla oksydazy ksantynowej – allupurinol – leczenie dny moczanowej
11. INHIBITORY FOSFODIESTERAZY
1. enzym ten katalizuje rozkład cAMP i cGMP (do AMP i GMP), czyli wtórnych przekaźników.
2.Zablokowanie inhibitorem powoduje wzrost stężenia wtórnych przekaźników w komórce i
pozareceptorowego pobudzenia – brak zadziałania hormonu na receptor komórkowy.
3.Leki: inhibitory kompetycyjne bedące metaloksantynami – ich metabolizm prowadzi do powstania
kwasu moczowego (przeciwskazanie: hiperurynemia)
4.Przykłady:
-cytrynian sildanafilu (VIAGRA) – dla cGMP – leczenie impotencji i pierwotnego nadciśnienia
płucnego
-kofeina, teofilina, teina,, aminofilina
12. ENZYMY INDYKATOROWE, podział, występowanie, patogeneza zmian enzymatycznych w
surowicy
1.Fizjologicznie występują wewnątrzkomórkowo. Ich prawidłowe stężenie w osoczu jest niskie(raczej
nie mówi się o dolnej granicy normy). Stężenie wzrasta w wyniku uszkodzenia komórek(np. apoptoza –
przechodzą wtedy do krwioobiegu). Zwiększenie ich aktywności w osoczu może też wystąpić gdy

komórce brakuje energii aby zachować gradient – wtedy uciekają poza komórkę (wzrost
przepuszczalności błony). Niewielkie uszkodzenie komórki – enzymy cytoplazmatyczne uciekają,
rozległa martwica – uciekają enzymy mitochondriów i innych organelli – tzw. nekroenzymy.
2.Podział (wyłącznie do celów dydaktycznych):
A.narządowo specyficzne(mają raczej słabe właściwości diagnsotyczne poza CK):
-dla wątroby: dehydrogenaza etanolowa, dehydrogenaza sorbitolowa, transferaza
ornitynokarbamoilowa, aldolaza fruktozomonofosforanowa, argininaza
-dla mięśni poprzecznie prążkowanych: kinaza kreatynowa – CK, aldolaza fruktozodifosforanowa
B.narządowo niespecyficzne(łatwo je oznaczyć i wystepują w duzych ilościach): enzymy glikolizy,
enzymy cyklu Krebsa, enzymy cyklu pentozowego, enzymy przemiany bialkowej –
AspAT,GOT,aminotranferaza asparaginowa oraz AlAT,GPT,aminotransferaza alaninowa
13. KATAL, AKTYWNOŚĆ MOLEKULARNA – definicje.
1. Katal – jednostka aktywności enzymatycznej. Katal jest to taka aktywność, która przekształca 1 mol
substratów w czasie 1 sekundy w optymalnych warunkach reakcji przy stężeniu substratu
gwarantującym pełne wysycenie enzymu. Jednostka: mol/s. W praktyce stosuje się mikro-(-6),nano(-9)-
,pikokatale(-12). 1 kat=1mol/s=60mol/min=60x10^6umol/min=6x10^7IU
2. Aktywność molekularna – określa reaktywność enzymu – jest to liczba cząsteczek substratu
przekształconych w produkty, w czasie 1 minuty przez 1 cząsteczkę enzymu, w optymalnych
warunkach. Dla enzymów, które zawierają kilka centrów katalitycznych używa się określenia –
aktywność centrum katalitycznego – kcat, która równa się aktywności molekularnej podzielonej przez
liczbę centrów aktywnych w enzymie. Pochodna aktywności molekularnej to: aktywność molarna –
określana w kat/mol.
14. FOSFATAZA KWAŚNA – EC i podział kliniczny, izoenzymy, znaczenie diagnostyczne
0.norma w surowicy: 30-90nkat/l, 1,8-5,4IU/l
1.EC3.1.3.2 – hydrolaza, enzym ekskrecyjny (wydalniczy) – enzym płynu sterczowego (AcP)
2.Reakcja-hydrolityczne odszczepienie reszty ortofosforanowej z organicznych estrów kwasu
fosforowego. Enzym mało specyficzny. Optimum działania fosfatazy w pH<7.
3.Występowanie:1.gruczoł krokowy, 2.płytki krwi, 3.erytrocyty, 4.makrofagi, 5. wątroba, 6.śledziona,
7.nerki, 8.surowica, 9żółć, 10mocz(wydalana z moczem, u chłopców wzmożona aktywność).
4.Izoenzymy:
a.kostny-osteoklasty
b.nerkowy-hamowany przez formaldehyd, słabo przez fluorki i winian.
c.erytrocytarny-silnie hamowany przez formaldehyd, słabo przez winian
d.wątrobowy, -silnie hamowany przez formaldehyd, słabo przez winian
e.trombocytarny-silnie hamowany przez formaldehyd, słabo przez winian (podczas krzepnięcia wzrasta
aktywność fosfatazy kwasnej w surowicy – przyczyna: rozpadłe płytki)
f.sterczowy-silnie hamowana przez winian, nie przez formaldehyd. Wskaźnik istnienia raka gruczołu
krokowego (norma 1IU/l). Norma:25%całkowitej aktywności AcP.
g.śledzionowy
h.układu siateczkowo-środbłonkowego
5.Znaczenie diagnostyczne:
1.Rak gruczołu krokowego (lepszy marker to PSA).
2.zawał gruczołu krokowego oraz po zabiegach na prostacie wzrasta aktywność.
3.Stan zakrzepowo-zatorowy
4.Przełom hemolityczny
5.Choroby układu kostnego (Pageta, przezuty nowotworowe do kości)
6.Choroby wątroby(żółtaczka mechaniczna, marskość)
7.Ostre uszkodzenie nerek.
8.Choroby układu liforetikularnego z zajęciem watroby lub kości
9.Choroba reumatyczna
15. OKSYDOREDUKTAZY, klasyfikacja, katalizowane reakcje koenzymów, diagnostyka,
przykładmy (58.OKSYDAZY)
1. Pierwsza klasa enzymów wg. Podziału międzynarodowego – EC1.
2. Katalizują reakcje utleniania i redukcji.

3.Oksydoreduktazy przenoszą protony i elektrony z jednej cząsteczki (reduktor) na inną (utleniacz) ,
czyli akceptor według schematu: AH
2
+ B → A + BH
2
4.
Oksydoreduktazy obejmują:
A. Dehydrogenazy – odszczepiają atomy wodoru z substratu i przenoszą do na koenzym (głównie NAD
lub NADP-ulegają one redukcji) lub odwrotnie, a czasem na tlen (tzw. dehydrogenazy tlenowe). Atom
wodoru używany jest do hydroksylacji bądź transportowany do mitochondrium gdzie uczestniczy w
kaskadzie oksydacyjnej. Przykład dehydrogenaza alkoholowa (EC.1.1.1.1.) – etanol aldehyd octowy
(i odwrotnie, oraz inne alkhole), dehydrogenaza glutaminowa, pirogronianowa, jabłczanowa.
B. Hydroksylazy – katalizują rekacje hydroksylacji (kofaktorem hydroksylacji jest NADP) czyli
przyłączenie grupy –OH (hydroksylowej) do substratu. Hydroksylazy jednocześnie utleniają dwa
substraty (odrębne) w obecności tlenu czasteczkowego – jeden atom wchodzi między atomy C i H w
substracie tworząc –OH, a drugi do atomu wodoru drugiego substratu tworząc czasteczkę wody.
Przykłady: utlenianie związków sterydowych, hydroksylaza fenyloalaninowa (fenyloalanina->tyrozyny)
C. Peroksydazy – przeprowadzają rekacje rozkładu nadtlenku wodoru (donor + nadlenek woda +
utleniony donor) Przykład: peroksydaza glutationowa.
D. Oksygenazy – katalizują przyłączenie tlenu do związku organicznego. Dzielą się na dioksygenazy i
monooksygenazy. Przykład 3-monooksygenaza kinureninowa.
E. Oksydazy – aktywuja tlen czasteczkowy przez przeniesienie na niego niego elektronów, przez co
może on się łączyć z protonami tworząc cząsteczkę wody. Radzej powstawać może H2O2 (oksydazy
flawinowe). Przykład oksydaza monoaminowa (dezaimonacja D- i L- aminokwasów), oksydaza
ksantynowa
F. Reduktazy – katalizuja rekacje przeniesienia protonów i elektronów lub samych elektronów z
przenosników na dalsze układy redox.
5.Koenzymy: NAD, NADP, FMN, FAD/przenosą H+/, atomy metali, hemina komórkowa (e), kwas
liponowy(H+, gr. acylowe), ubichinon (Q)/H+/, plastochinon, difosfotiamina (gr. Aldehydowe)
6.Znaczenie w diagnostyce: katalaza – brak w osoczu, marker uszkodzenia wątroby, erytrocytów i
nerek; peroksydaza – redukowanie nadtlenku wodory i wodoronadtlenków fosfolipidów – wykrywanie:
reakcja benzydynowa; reakcja pseudoperoksydazowa hemoglobiny – wykrywanie utajonej krwi w kale;
oksydaza kwantynowa –utlenia aldehydy alifatyczne i aromatyczne oraz zasady purynowe (występuje w
mleku).
PRZYKŁAD WAŻNEGO KLINICZNIE
DEHYDROGENAZA MLECZANOWA (LDH) i a-HYDROKSYMAŚLANOWA (HBDH)
Dehydrogenaza mleczanowa – EC 1.1.1.27 – odwracalna przemiana kwasu pirogronowego do
mlekowego kończąca glikolityczny tor spalania glukozy. Enzym cytoplazmatyczny. Największa
aktywność; w wątrobie.
2.5 izoenzymów wędrujących z globulinami: LDH1=a1,LDH2=a2/LDH3=beta / LDH4 – szybkie
gamma / LDH5 – wolne gamma
3. Oba łańcuchy izomerów M(muscle) i H (bearth) mają podobną budowę i zbliżone właściwości
antygenowe, jednak róznią się składem aminokwasowym i wlaściwościami, w tym wrazliwością na
działanie dużego stężenia pirogronianu (w sercu hamowanie przez pirogronian umozliwia
wykorzystanie tego substratu w przemianach oksydacyjnych – brak hamowania w wątrobie umożliwia
przekształcanie w mleczan). Podjednostka M katalizuje przemiane pirogronianu w mleczan a
podjednostka H jest mniej swoista i katalizuje róznież przemianę a-ketomaslanu i a-hydroksymaslanu.
4.LDH1 (4xH) i LDH2 – przemiana oksydacyjna: serce, nerki, mózg.
5.LDH4/LDH5 – wątroba mięsnie szkieletowe
6. W surowicy: stały skład izoenzymów LDH w stanie zdrowia: LDH2>1,3>4,5.
7. Rozdział izoenzymów: chromatografia, immunologiczne metody, metoda termiczna – ogrzewanie w
60C (30minut) hamuje wszystkie frakcje poza sercowymi 1 i 2. 5 wytraca się acetonem, nei wytrąca się
rivanolem (1 odwrotnie). Wolno wedrujace izoenzymy LDH hamowane są przez: mocznik, szybsze
przez szczawiany.
8. Surowica ze śladami hemolizy wykazuje wzmoża aktywnośc LDH (izoenzymów 1 i 2). W surowicy
mrozonej LDH obniżone bo izoenzymy 4 i 5 są niestabilne w tej temp.
9. HBDH – LDH1 i 2 – szybko wędrujące frakcje związane z a-globulinami. Odwracalna reakcja
przemiany a-ketomaslanu do a-hydroksymaslanu. Wzrost przy zawale bo większa aktywność w sercu
niż w innych tkankach.
10.Znaczenie; diagnostyka chorób wątroby, zawał – wzost LDH1 i 2 (zwiększenie 1 do 2 powyżej
jedności; nowotwory, białaczki – LDH3; zapalenia, niedokrwistość megaloblastyczna, postępująca
dystrofia mięśniowa.

16.HOLOENZYM,APOENZYM ,KOENZYM ,GRUPA PROSTETYCZNA - definicja, funkcja
Holoenzym – katalitycznie aktywna, kompletna cząsteczka enzymu, zbudowana z części bialkowej i
niebiałkowej.
Apoenzym – jest to sama białkowa część enzymu.
Kofaktor – niebiałkowa część enzymu: koenzym lub grupa prostetyczna. Mogą nim być nukleotydy,
pochodne witamin, związki hemu, jony metali, pochodne cukrów,
Koenzym – drobnocząsteczkowy związek organiczny albo jon nieorganiczny, luźno złączony z częścią
białkową, który może być swobodnie od niego odłączony. Określa on rodzaj katalizowanej reakcji.
Koenzym może ulec zregenerowaniu jedynie w innej reakcji. Gdy nie następuje regeneracja koenzymu
to może się on wyczerpać. Koenzymy sprzegają rekacje i mogą być tzw. drugim substratem. Przykłady
NAD – regenerowany w łańcuchu oddechowym, ATP, witaminy, glutation.
Grupa prostetyczna – niebiałkowa część enzymu, która jest silnie połączona z apoenzymem, a usunięcie
grupy prostetycznej prowadzi do uszkodzenia enzymu. Grupa prostetyczna ulega zregenerowaniu w tej
samej reakcji. Przykłady FAD,FMN, grupa hemowa cytochromu, Cu w ceruloplazminie – usunięcie Cu
w tym enzymie odslania grupy tiolowe.
17.IZOENZYMY, ALLEOENZYMY, ENZYMY HYBRYDOWE.
1. Izoenzymy – enzymy wytworzone przez różne geny zajmujące różne loci, katalizujące tą samą
reakcje enzymatyczną. Loci te występować mogą na 1 chromosomie – amylaza (izoenzym trzustkowy i
ślinowy mają geny kodowane na tym samym chromosomie. Dehydrogenaza jabłczanowa – enzym cyklu
Krebsa – jej poszczególne izoenzymy są kodowane przez geny położone na różnych chromosmach.
2. Alleloenzymy (allelozymy) – enzymy kodowane przez rożne postacie alleliczne genu kodującego
dany enzym (w obrębie danego genu wystepuje duży polimorfizm) – jest jedno loci, ale wiele form
enzymatycznych. Przykład: dehydrogenaza glukozo-6-fosforanowa enzym cyklu pentozowego.
3.Enzymy hybrydowe – dotyczy enzymów oligomerycznych, których każda z podjednostek kodowana
jest przez inny gen, to w efekcie może być różna asocjacja, różny skład tych podjednostek i stąd może
wynikać polimorfizm tych enzymów. Przykład: dehydrogenaza mleczanowa, kinaza kretynowa.
18. ESTERAZY CHOLINOWE(ChE) / 21.ACETYLOCHOLINESTERAZY
1.hydrolazy EC,3.1.1.7/8.
2.Katalizowana reakcja: hydroliza estrów choliny
3.Wytwarzanie: wątroba i wydzielanie do krwi.
4. Podział ChE;
A. Acetylocholinesteraza AchE – esteraza cholinowa I (tzw. prawdziwa) – degraduje acetylocholine w
zakończeniach nerwowych (występuje również w erytrocytach, płucach, śledzionie, substancji szarej
OUN)-działanie bardziej specyficzne od BuChe,
B.Acylohydrolaza acylocholiny BuChE – esteraza cholinowa II, pseudoacetycholinesteraza,
butyrylocholinesteraza; w elektroforezie daje 7/12 frakcji (wiele form tego enzymu). Oznacza się ją w
osoczu. 0,3 do 0,5% populacji posiada gentoyp AA (zamiast EE) – enzym nie przejawia aktywności w
osoczu – pacjent pozostaje zwiotczony – oznaczyć trzeba liczbę dibukainową (inhibitor). Spadek tego
enzymu w ostrym zapaleniu wątroby, marskości wątroby, cukrzycy, nadczynności tarczycy, zawale
serca, hipoalbunemii.
5. Acetylocholinesteraza w centrum katalitycznym ma 2 miejsca wiązania: aminowe – łączenie ACH
przez cholinę, estrowe – łączenie przez resztę estrową, czyli acyl.
6. Efekty zablokowania cholinesterazy: brak degradacji acetylocholiny – przedłuzone pobudzenie
układu przywspółczulnego, zwężenie źrenicy, wzmozona perystaltyka, wzmorzone wydzielanie z
gruczołów egzokrynowych, zaburzenia termoregulacji, skurcz oskrzeli.
INHIBITORY:
Odwracalne:
1.fizostygmina,prostygmina,neostygmina – kompetycyjne, struktura podobne do ACH, po pewnym
czasie enzym rozkłąda fałszywy substrat, łacza się z miejscem aminowym jak i estrowym, leczenie
jaskry, obkurczenie mięsni i odslonięcie katu przesaczania – spadek cisnienia wewnątrzgałkowego.
2.endrofonium – tylko z miejscem aminowym
Nieodwracalne: fosfolina, fluorofosforan dwuizopropylowy (DFP), parathinon, nipapox – łączą się z
miejscem estrowym doprowadzając do nieodwracalnej fosforylacji tego miejsca – zmianie nie ulega
struktura enzymu – odłaczaja inhibitor: reaktywatory acetylocholinesterazy bo komórka sobie sama nie
poradzi – DFP i fsfolina – leczenie jaskry, DFP stosowany w biochemii – inaktywacji enzymów

wrazliwych na działnie reszt kwasy ortofosforowego.
Parathinon, nipapox (insektycydy) – podaje się: PAM, DAM, MINA – inaktywatory / albo atropiny
19. INHIBITORY FIBRYNOLIZY
1. HGRP – glikoproteina bogata w histydynę, tworzy odwracalny kompleks z plazminogenem; stężenie
obniża się w DIC, niewydolności wątroby, ostatnim trymestrze ciąży
2.d2-AP alfa2-antyplazmina; hamuje plazminę i plazminogen
Działające nieswoiście:
3.a2-M alfa2-makroglobulina 20% aktywności antyproteolitycznej osocza; hamuje plazminę
4.a1-PI a1-inhibitor proteaz hamuje elastazę i plazminę
5. AT-III – antytrombina III; 80% aktywności antyproteolitycznej osocza
6.PAI-1inhibitor aktywatora plazminogenu typu I; hamuje t-PA i u-PA; synteza w komórkach
śródbłonka, wątroby, megakariocytach, miocytach gładkich, niektórych komórkach nowotoworowych
7.PAI-2inhibitor aktywatora plazminogenu typu II; hamuje t-PA, u-PA; synteza monocyty i
makrofagi
8.WIBRONEKTYNA (białko tkanki łącznej) – stabilizuje PAI-1 i zmienia jego specyficzność, synteza
w tkance łącznej i płytkach krwi
9.Proteaza neksyn – synteza komórki śródbłonka, plytki krwi, inaktywuje plazminę, urokinazę,
trypsynę, trombinę
10.Lp(a) – lipoproteina a: - posiada w swojej strukturze apolipoproteinę a, która przypomina
plazminogen i TIP (tkankowy inhibitor ptoteaz) – bogata w cholesterol, ma precle duńskie (cringle-
strucles) – domeny przypominające strukturę plazminogenu i aktywatorów plazminy – hamuje
aktywnośc plazminogenu przez t-PA, u-Pa, streptokinaze – przez to zwiększa prawdopodobieństwo
procesu zakrzepowego i zatorowego
20. BIAŁKOWE CZYNNIKI KRZEPNIĘCIA, podział charakterystyka
1. Podział:
A.zespół protrombiny – II, VII, IX, X – produkcja; komórki śródmiąższowe wątroby, ulegają
modyfikacji posttranslacyjnej: gamma-karboksylacja zależna od witaminy K, enzym:karboksylaza.
Protrombina to marker funkcji wydzielnicznej wątroby.
B.wrażliwe na trombinę(trombina nie jest enzymem)-I, V, VIII, XIII(transglutaminaza). Czynniki V i
VIII posiadają naturalne inhibitory krzepnięcia.
C.czynniki kontaktu-XI,XII,prekalikreina, HWK-wysokoczasteczkowy kininogen (heigh molecular
weight kininogen)
2.Czynniki V(Aktywowany przez trombinę; Va to kofaktor reakcji aktywacji protrombiny przez
Xa),VIII(aktyw. Przez trombinę, VIIIa to kofaktor reakcji aktywacji czynnika X przez czynnik Ixa),
HMWK, III(wymaga obecności fosfolipidu, wystepuje na powierzchni śródbłonka, kofaktor dla
czynnika VIII) to kofaktory.
3.XIII – tiolowo-zależna transglutaminaza – aktywowana przez trombinę w obecności Ca2+. Stabilizuje
skrzep fibrynowy na zasadzie tworzenia poprzecznych wiązań kowalencyjnych.
4.Proenzymy/zygmogeny proteaz serynowych
XII.łączy się z powierzchniami o ładunku ujemnym uszkodzonej ściany, aktywowany przez
wysokoczasteczkowy kininogen i kalikreinę
XI.aktywowany przez XIIa
IX.aktywowany przez Xia w obecności Ca2+
VII.aktywowany przez trombinę w obecności jonów wapnia
X.uczynniany na powierzchni zaktywowanych płytek krwi przez kompleks tenazy(Ca2+, VIIIa, Ixa) jak
również przez czynnik VIIa w obecności TF i Ca2+
II.uczynniany na powierzchni zaktywowanych płytek krwi przez kompleks protrombinowy(Ca, Va,Xa)
25. KOMPLEKS TENAZY
1.Tenazowy kompleks aktywny.
2.Lokalizacja: aktywowane płytki krwi
3. Składa się z: JONY WAPNIA, FOSFOLIPIDY VIIIa, Ixa, X
4.Kompleks tenazy dzięki obecności proteazy serynowej IX-a rozkłada wiązanie między argininą i
izoleucyną (Arg-Ile) w cząsteczce czynnika X (56 kDa) tworząc dwułańcuchową proteazę serynową –
czynnika Xa.
5.Czynnik VIII to glikoproteina będąca kofaktorem służącym jako receptor dla czynników IX a oraz X

na powierzchni płytek krwi. Niekatywny czynnik VIII wiąże się z vWF.
26.ALFA-AMYLAZA, izoenzymy, metody rozdziału, znaczenie diagnostyczne, wykrywanie
0.normy – A.surowica:1,9-4,9nkat/l (60-160j.C.), B.mocz:1,9-9,8nkat/l(60-320j.C.)
1.rekacja: hydroliza skrobii i glikogenu (wielocukry należące do a-glukonów) {powinowactwo wzrasta
w miarę wydłużania łańcucha], w wyniku tego procesu powstają dekstryny, maltotrioza, maltoza,
koniec działalności – rozgałęzienie łańcucha (powstają dekstryny graniczne)
2.budowa: w centrum katalitycznym Ca2+ - stabilizuje enzym, odpowiada za jego aktywną konformację
(hamowanie amylazy: wiążace wapń związki – szczawian, cytrynian, wersenian, fluorki
3. występowanie: trzustka, ślinianki, wątroba, nerki, płuca, śledziona, serce, mięsnie szkieletowe, mózg
(śladowe ilości – wszystkie tkanki). Płyny ustrojowe: ślina i mocz (mniejsza aktywność – surowica,
mocz).
4.izoenzymy(w elektroforezie wędrują z gamma-globulinami) {surowica i mocz – izoenzymy P oraz S
w równych ilościach}:
A. trzustkowe: P1,P2,P3
B. z gruczołów ślinowych: S1, S2, S3
C. ze śluzówki jelita cienkiego P2
D. z gruczołu mlecznego: P2, S1, S2
E. komórki nabłonka kanalików Mullera jajników i jąder- O1(kobiety-laktacja, przed menstruacją), O2
5.izenzymy trzustkowe (najważniejsze z punktu widzenia diagnostycznego) – technika immunoinhibicji
(wykorzystanie przeciwciał hamujących izoenzymy S)
6.oznaczanie aktywności:
A. wg. Carawaya – taka aktywność enzymu która hydrolizuje 10mg skrobii w ciągu 30 minut do stopnia
w którym zanika reakcja z jodem [aktywność wyraża się w odniesieniu do 100ml materiału badanego]
B.metoda EPS (ethylidene protected substrate) – uzycie krótkich maltooligosacharydów (3 – 7 reszt
glukozy) z p-nitrofenolem i glukozydazy uwalniającej barwny nitrofenol. Substrat jest chroniany przed
działaniem glukozydazy poprzez zablokowanie cząsteczki blukozy na końcu redukujacym łańcucha
mostkiem etylidenowym. Barwnik może być uwolniony z substratu przez glukozydazę dopiero po
zadziałaniu a-amylazy.
7.znaczenie diagnostyczne:
-diagnostyka chorób trzustki (zwłaszcza ostre zapalenie trzustki-sok trzustkowy wydostaje się poza
komórki = samostrawienie trzustki) – po 20/30h max. w surowicy, 6/8h później w moczu, normalizacja
4/10 doba
-wzrost w moczu/surowicy(wraz z lipazą w surowicy): zatkanie przewodu trzustkowego lub żółciowego
wspólnego, zaostrzenie przewlekłego zapalenia trzustki, urazy trzustki, „ostry brzuch”
-wzrost samej a-amylazy:zapalenie ślinianek, zapalenie przewodów ślinianek, choroby jajników i jąder,
nowotwory wydzielające ektopowo amylazę
-wzrost w surowicy/spadek w moczu: niewydolność nerek, makroamylazemia (Ig (najczęściej IgA)
wiążą jeden z izoenzymów amylazy (P lub S) – większe kompelksy nie przesaczające się przez błonę
filtracyjną kłebuszka naczyniowego
-wzrost aktywności; zażywanie leków – kortykosteroidy, salicylany, sulfonamidy, tetracykliny
-obniżenie w surowicy i moczu: przewlekłe uszkodzenie trzustki: przewlekłe zapalenie,
mukowiscydoza, oraz wątroby. Ponad to w zatruciu barbituranami.
27. DROGI KRZEPNIĘCIA + 31. BADANIA
ZEWNĄTRZPOCHODNA:
1. Rozpoczyna się aktywacją czynnika III / TF – transmemranowa glikoproteina komórek ściany
naczyniowej, endotelium i monocytów. Synteza konstytutywna (podśródblonkowa i mięśniówka
naczyń) i synteza indukowana cytokinami, endotoksynami i układem dopełniacza. Nie występuje na
trombocytach – w osoczu tylko w razie uszkodzenia tkanek i aktywacji w/w komórek.
2.TF, Ca aktywują VII do VIIa który natomiast aktywuje czynnik X do Xa. Do doprowadza to
powstania trombiny i nastepnie włóknika – powstaje skrzep.
3.TFPI-tissue factor pathway inhibitor – inaktywuje czynnik X przez blok jego aktywacji oraz
zachamowanie powstawania VIIa.
4.Czynnik X wchodzi w skład protrombinazy: Xa, Va jako kofaktor, fosfolipidy, jony wapnia
WEWNĄTRZPOCHODNA:
1.Ujemnie naładowane kolagen, kwasy tłuszczowe, kalikreina. Fosfolipidy ujemnie naładowane
aktywują czynnik XII a ten kalikreine. Ta uwalnia HMWK od kinin I ten aktywuje pownownie XII

(poza tym XII aktywuje VII I fibrynolizę). XIIa aktywuje XI, a ten IX co doprowadza do aktywacji
czynnika X.
BADANIA:
Czas protrombinowy: do próbki dodaje się TF I jonów wapnia odpowiednio inkubujac I mierzac czas w
którym dochodzi do powstanai skrzepu. Jest miarą zewnątrzpochodnego układu krzepnięcia. Ulega
zwiększeniu przy uszkodzeniu: protrombiny, V, VII, X I w miejscym stopnu fibrynogenu. Oznacza się
go u pacjentów leczonych antykoagulantami. Oznacza się go w postaci czasu, wskaźnika Quicka
=Cpbadany/Cpkontrolny wyrażany w %, w postaci INR =Cpbadany/Cpkontrolny^ISI –
międzynarodowy współczynnik czułości tromboplastyny. 13-16sekund.
Czas kaolinowo-kefalinowy: czynniki XI I XII aktywawane są w zawiesinie kaolinu I kefaliny, po
podaniu CaCl2 mierzy się czas krzepnięcia. Miara sprawności wewnątrzpochodnego układu
krzepnięcia. Zamiast kaolinu aktywatorem może być celit lub kwas elagowy. Prawidlowy 26-37sek.
Przedłużenie czasu KK w: II, V, VIII, IX, XI, XII, hipofibrynogenia, niedobór czynnika Fletchera
(prekalikreiny). Oznacza sie w leczeniu heparyną.
28. syntaza/syntetaza, hydrolaza/hydroksylaza, oksydaza/oksygenaza – róznice
Syntazy – rodzaj liaz, katalizujący reakcjie niewymagające nakładu energii z ATP (lub innego związku,
działają w odwrotnym kierunku niż liazy prowadzac do syntezy wiązań C-C,C-O,C-S po przyłączeniu
zwiazku drobnocząsteczkowego (CO2, NH3, H2O, x-CHO) – np. syntaza tlenku azotu (indukowana).
Syntetazy – inaczej ligazy – katalizowanie syntezy nowych wiązań z udzialem energii pochodzacej z
ATP,GTP,itd. Koenzymy syntetaz: Atp, biotyna, tRNA
Syntezowanie wiązania C-O aminokwas + tRNA = ATP aminoacylo-t-RNA = AMP + PP
C-S R-OOOH + HS~Co +ATP acylo~CoA + AMP + PP
C-N pirogronian + CO2 + Atp + H2O szczawiocotan + ADP + P
C-C acylo~CoA + Co2 + H2O + Atp malonylo CoA + ADP + P
Hydrolazy – katalizują rozbicie wiązań przy udziale wody. W zalezności od rodzaju atakowanych
wiązań rozróżniamy: esterazy (acetylocholinesteraza – acetylocholina + H2O -> cholina + kw. Octowy),
fosfatazy (glukozo-6-fosfataza – D-glukozo-6-fosforan + H20 -> D-glukoza + H3PO4). Fosfodiesterazy,
sulfatazy, glikozydazy (a-amylaza), peptydazy, działające na wiązania bezwodników kwasowych –
ATPaza. Nie mają koenzymów!
Hydroksylazy – należą grupy oksydoreduktaz, dziają na 2 substraty, powodując ich utlenianie przez
przyłaczenie 1 atomu tlenu do jednego substratu, drugi tworzy wodę. Przykład hydroksylaza DOPA-
miny.
Oksygenazy – katalizują przyłączenie tlenu do związku organicznego. Dzielą się na dioksygenazy i
monooksygenazy. Przykład 3-monooksygenaza kinureninowa.
Oksydazy – aktywuja tlen czasteczkowy przez przeniesienie na niego niego elektronów, przez co może
on się łączyć z protonami tworząc cząsteczkę wody. Radzej powstawać może H2O2 (oksydazy
flawinowe). Przykład oksydaza monoaminowa (dezaimonacja D- i L- aminokwasów), oksydaza
ksantynowa
29.ENZYMY MNEMONICZNE
1.Enzymy mnemoczne działają na drodze mnemoniczności – stąd ich nazwa. (histeryczne, obdarzone
pamięcią). Enzymy monomeryczne kooperujące.
2.Mechanizm: do enzymu przyłącza się substrat – następuje zmiana konformacji enzymu – substrat się
odłącza – zmieniona konformacja enzymu utrzymuje się (enzym łatwiej przyjmuje nastepną czastęczkę
substratu
3.jest to tzw. PAMIĘĆ – czyli utrzymanie korzystnej konformacji w odpowiedzi na pierwszą cząsteczkę
subtratu. Po pewnym czasie enzym wraca do stanu przed związaniem ze substratem.
4.Przykład heksokinaza- transferaza, przemiana glukozy w glukozo-6-fosforan, kiedy jest wysycona to
glukokinaza (wątroba i inne organy) w hepatocytach przeprowadza tą samą rekację (Km glukokinazy
jest o wiele większe niż heksokinazy).
32. INHIBITORY PROTEAZ
1. To występujące w naturze bądź syntetyczne substancje hamujące aktywność enzymów
proteolitycznych.
2. Wyróżniamy wśród nich inhibitory specyficzne, powodujące inhibicję określonych proteaz lub typów

proteaz, a także niespecyficzne. Mogą one mieć charakter białkowy lub niebiałkowy. I.p. stosowane są
m.in. jako narzędzia badawcze w enzymologii oraz jako środki terapeutyczne w medycynie.
3. W praktyce laboratoryjnej w badaniach nad enzymami stosuje się: inhibitory proteaz serynowych,
inhibitory proteaz tiolowych, inhibitory metaloproteaz, inhibitory proteaz aspartylowych.
4. W klinicystyce wyróznia się inhibitory syntetyczne i naturalne, znajdujące zastosowanie z leczeniu
nadkrzepliwości i skaz krwotocznych.
A. syntetyczne – kwas E-aminokapronowy (EACA), kwas aminometylocykloheksylowy (AMCHA) i
jego izomer (AMA) – kwas p-aminometylobenzoesowy (PAMBA) – kwas delta-aminowalerianowy –
kwas delta-aminolewulinowy – kwas gamma-aminomasłowy – kwas omega-aminokapronowy
B.naturalne – trzystkowe (występują one również w tkance płucnej i przyusznicy_, kwaśny/zasadowy –
wyciągi z orzeszków ziemnych (flawonowy, leukoantycowy) – sojowy inhibitor trypsyny – inhibitor
trypsyny z białka jaja kurzego – inhibitor trypsyny z siary
33. INTERKONWERSJA
1. jeden ze sposobów wpływu na aktywność katalityczna enzymu – zmiana jego aktywności bez zmiany
jego ilości.
2. Zmiana formy aktywnej w nieaktywną dokonuje się pod wpływem dodania lub usnięcia grupy
fosforanowej (fosforylacja i defosforylacja – mofyfikacje posttranslacyjne). Fosforylowana może być
grupa –OH – tyrozyny, seryny i treoniny, atom N – argininy, histydyny i lizyny oraz atom tlenu
asparaginianu – powstają odpowiednio O- i N- pochodne. Proces ten katalizują kinazy(dołączają grupę
fosforanową) oraz fosfatazy (odszczepiają).
3. Poprzez ten mechanizm działają wszystkie hormony, w których wtórnym przekaźnikiem jest cAMP.
Umożliwia to o wiele szybszą kontrolę działania aktywności enzymu niż działanie na poziomie
ekspresji genów.
4. przykład: syntaza glikogenowa aktywna w formie ufosforylowanej, oraz fosfataza glikogenowa
aktywna w formie ufosforylowanej.
34. t-PA
1.tkankowy aktywator plazminogenu (tylko w obecności fibryny!) – proteaza serynowa
2.synteza: śródblonek (patologia: tez inne komórki go wydzielają) – uwalnia się do krwioobiegu – jeśli
nie zwiąże się z fibryną ulega inaktywacji lub tworzy kompleks z PAI-1 (plazminogen activator
inhibitor)
3.Po połączeniu z fibryną t-PA rozszczepia plazminogen – powstaje plazmina (rozkłada fibrynę –
rozpuszczenie skrzepu).
3. Jego stężenie w osoczu wykazuje rytm dobowy. Patologicznie rośnie w nowotworze i wstrząsie.
4.Zarówno plazmina jak i t-PA, nie mogąc pozostać związane z produktami degradacji fibryny
uwalniane sa do osocza gdzie związane są unieczynniane przez naturalne inhibitory.
5. Rekombinowany t-PA – alteplaza – rozkładanie zakrzepu odpowiedzialnego za patologię
36. INHIBITORY ANHYDRAZY WĘGLANOWEJ
1. Enzym katalizujący przemianę H2CO3 -> HCO3- + H+, utrzymywanie RKZ.
2.Wystepowanie: wiele narządów(np.nerki) oraz erytrocyty.
3.Inhibitory – leki o budowanie sulfoamidowej – ACETOZOLAMID (diuramid), metazolamid,
brynzolamid i dorzalamid – miejscowe leczenie jaskry
4. leki te stosuje się w leczeniu jaskry i choroby wysokościowej, są słabymi diuretykami (zwiekszenie
utraty wody, sodu i potasu z moczem)
37. ENDOENZYMY
1. enzymy wewnątrzkomórkowe, które spełniają swoją funkcje w komórkę w której powstały
2.Większośc enzymów ustroju biorących udział w przemianach anabolicznych i katabolicznych
3. Przemiany endoenzymów często układają się w długie szlaki metaboliczne i cykle, które powiązane
są ze sobą w ramach metabolizmy wewnątrz komórkowego.
4. Aktywność endoenzymów podlega złozonej regulacji gł. Przez związane ze zjawiskiem allosterii
mechanizmy sprzężenia zwrotnego, inhibicje niekompetycyjną. Itp.
5. Ważna dla endoenzymów jest kompartmentalizacja – dany enzym umiejscowiony jest w okreslonym
przedziale błonowym komórki (odpowiednie pH, jony, itp.)
6.Endoenzymy cytozolowe i mitochondrialne.
7. Do endoenzymów zalicza się enzymy indyktorowe wg. Podziału Richtericha i Hessa.

8.Wśród endoenzymów odnajduje się kompleksy wieloenzymatyczne; dehydrogenaza
pirogronianowa(mitochondrium) i inne.
38. AKTYWNOŚĆ WŁAŚCIWA
1. Określa stopień czystości preparatów enzymatycznych i jest to liczba jednostek standardowych
przypadająca na 1 miligram białka (IU/mg). Ewentualnie wyrażana w mikrokatalach na kilogram bialka
(ukat/kg).
40. TRAP
1.Winianooporna fosfataza kwaśna.
2.Wytwarzania:osteoklasty resorbujące kość
3.Jego funkcja biologiczna nie jest znana.
4.Wydaje się, że enzym ten mógłby stać się dobrym markerem aktywności osteoklastów, gdyby nie
trudności metodyczne.
5. W surowicy znajdują się inhibitory tego enzymu, jest on niestabilny i trudny do odróżnienia od
kwasowych fosfataz uwalnianych z tkanek pozakostnych.
45. ROLA METALI W KATALIZIE
1. Pełnią funkcje katalityczne jak i strukturalne centrum aktywnego wielu enzymów.
2.Pełnia funkcje aktywatorów lub inhibitorów katalizy enzymatycznej.
3.Są spoiwem między koenzymem, a apoenzymem lub enzymem a substratem.
4.Biorą udział w reakcjach oksydoredukcyjnych jako przenośniki elektronów.
5.Stabilizują strukturę apoenzymu, gdy występują poza miejscem katalitycznym i nie kontaktują się ze
substratem.
6. Jony metali cięzkich inaktywują enzymy – ołów, rtęć – łatwo wiążą się z grupami –SH centrum
aktywnego bądź w duzych stezeniach powodują denaturację częścia białkowej enzymu (apoenzymu)
7.Metaloenzymy – enzymy zawierające w centrach aktywnych jony metali decydujące o ich funkcji.
48. CZYNNIK VON WILLEBRANDA, budowa, znaczenie
1. Glikoproteina o budowie polimeru (różne polimery = różne czynniki vWF)
2. W procesie krzepnięcia biorą udział tylko duże polimery.
3. Wystepowanie: osocze, trombocyty, śródbłonek, macierz podśródbłonkowa
4. Stężenie w osoczu jest genetycznie uwarunkowane.
5.Wazopresyna działa na śródblonek powodując uwolnienie vWF.
6.Estrogeny działają na śródbłonek powodując syntezę vWF.
7.Funkcje: pierwotna – adhezja, agregacja płytek krwi umożliwiające powstanie czopa hemostatycznego
– wtórna-transport.
8.vWF jest markerem uszkodzenia śródbłonka.
9. Wzrost stężenia w osoczu sprzyja zakrzepicy tętniczej – jest to czynnik prognostyczny zawału, udaru,
zatorowości płucnej.
50. ENZYMY OLIGOMETYCZNE, kinetyka, przykłady
1. Oprócz enzymów monomerycznych (jednołańcuchowych-elastazy,trypsyna/wielołńcuchowych-
połączonych mostkami siarczkowymi-chymotrypsyna) wystepują enzymy oligomryczne mające budowę
jednostkową.
2. Oddziaływania między jednostkami mają charakter niekowalencyjny: hydrofobowe, jonowe,
wodorowe, siły van der Wallsa.
3 Dzielą się na homooligomery (zbydowane z takich samych jednostek – homodimery, homotermaery)
oraz heterooligomery zbudowane z róznych podjednostek.
4. Enzym oligomeryczny pod wpływem czynników fizycznych i chemicznych może dysocjować na
podjednostki.
5. Dysocjacja na podjednostki nie musi oznaczać utraty aktywności, a jedynie zmianę profilu – np.
dehydrogenaza aldehydu-3-fosfoglicerynowego – jako tetramer (aktywnośc dehydrogenazowa,
wynikająca z glikolizy oraz esterazowa – jako dimer – aktywność wyłącznie esterazowa.
6. Mozliwośc kooperacji między podjednostkami nosi nazwę allosterii.
51. UKŁAD DOPEŁNIACZA – drogi aktywacji, znaczenie kliniczne
1. Grupa ponad 20 białek enzyamtycznych osocza, aktywujących się nawzajem w sposób łańcuchowy –

tzn. aktywne składniki aktywują następne składniki układu dopełniacza.
2. Składniki dopełniacza produkują: nabłonek jelit, makrofagi, komórki wątroby).
3.Ukłąd dopełniacza wykazuje związek funkcjonalny z innymi układami enzymatycznymi: kinin,
plazminy, fibrynolitycznym, krzepnięcia. W zapaleniu czynniki aktywujące dopełniacz mogą
uruchamiać w/w układy.
4.Klasyczna droga aktywacji dopełniacza: Ig łaczą sie z przeciwciałem (M oraz G1, G3 I w mneijszym
stopniu G2). 3 stadia: rozpoznania: aktywacji: reakcji z bloną. C1 doemy 2 I 4 łańcucha cięzkiego Ig I
zostaje odłoniete miejsce wiązace składowe dopełniacza. C1 wiaze sie z IG w ocenosci Ca2+ dołacza
C1r – powstaje esteraza C1 – rozkłąda C2 I C4 na podjednostki a I b. Powstaje C4b2a zwiazny z błoną
lub komplekemsem a-p (która ma aktywnośc konwertazy C3). Z 3 zostaje uwoliony peptyd 3a –
powstaje C4b2a3b. Jeżeli stadia zachodziły na pow komórki to przyłaczane sa kolejno C6-9.
54. EKTOENZYMY
1. Enzymy związane ze śródbłonkiem (zakowtwiczone w jego błonie na fosforanie heparanu – dlatego
heparyna trawiaca ten zwiazek powoduje ich uwolnienie do światła naczynia krwionośnego). Naturalnie
w surowicy ich stężenie jest niskie.
2. Mogą być uwalniane z błony w wyniku subilizacji czynnikami lipofilnymi (np.kwasy żółciowe.
3. Przykłady:
A. lipaza lipoproteinowa (LPL) – śródbłonek mięsni
B. lipaza wątrobowa triglicerydowa (HTGL) – śródbłonek zarok wątroby
C.gamma-glutamylotranspeptydaza (GGTP) – alkohol odrywa GGTP z błon komórek wyścielających
kanaliki żółciowe
D.acylotransferaza lecytyna-cholesterol (LCAT)
E.fosfataza zasadowa (AP) – błony komórek wyścielające kanaliki żółciowe
4. W przypadku cholestazy do subilizacji i nastepuje wzrost AP i GGTP w osoczu.
55. ENZYMY EKSKRECYJNE, definicja, przykłady (x4)
1.Inna nazwa: wydalnicze.
2. Fizjologicznie wystepują w dużej ilości w wydzielinie narządów wydalniczych.
3.Fizjologicznie ich stężenie jest małe w osoczu, a jego wzrost spowodowany jest upośledzeniem
odpływu wydzieliny. Mówi się tutaj o górnej granicy normy w przypadku pataologii.
4.Podział:
a.)żółci – fosfataza zasadowa, GGTP, leucyloaminopeptydaza (LAP) / przy cholestazie wystepuje
uszkodzenie wątroby i dostają się do krwi – kamica dróg żółciowych, guz brodwaki Vatera, guz głowy
trzustki
b.)soku trzustkowego: a-amylaza, lipaza, DNA-za, RNA-za,trypsyna, chymotrypsyna
c.)ślinianek: a-amylaza
d.)płynu sterczowego: fosfataza kwaśna (izoenzym sterczowy)
e.)gruczołów wydzielniczych żołądka: pepsyna – znikome znaczenie diagnostyczne – w praktyce nie
oznacza się tego enzymu
60. WPŁYW TEMAPERATURY NA ENZYMY
1. Podczas badania aktywności danego enzymu ważne jest zachowanie stałej temperatury podczas
inkubacji.
2.Zazwyczaj podniesienie temperatury powoduje przyspieszenie szybkości reakcji – zwiększenie energii
kinetycznej reagujących cząsteczek.
3. Współczynnik temperaturowy – Q
10
przy wzroście temperatury o 10C szybkości wielu procesów
biologicznych, w tym szybkości enzymatycznej, podwaja się, czyli Q
10
=2. Gdy energia kinetyczna jest
zbyt duża, może dochodzić do rozerwania wiązań utrzymujących strukturę trzecio- i drugorzędową co
prowadzi do obniżenia aktywności.
4. Wykres aktywności względnej od temaperatury ma charakter paraboli.
5.Zmiana temperatury inkubacji o 1C może zmieniać szybkość reakcji o około 5-10% - dla przykłądu
aktywność amylazy oznaczana metodą kinetyczną w surowicy w temperaturze 25C wynosi 40U/l,
natomiast w temperaturze 37C wynosi 90U/l.
61. ENZYMY, KTÓRYCH OZNACZA SIĘ STĘŻENIE
1.CK-MB
2.Izoenzym sterczowy fosfatazy kwaśnej.

3.Izoenzym kostny fosfatazy zasadowej – immunologiczna metoda Ostase (Beckman)
64. INHIBITORY OKSYDAZY KSANTYNOWEJ
1. Enzym katalizujący reakcje: hipoksantyna ksantyna kwas moczowy
2. Inhibitor kompetycyjny ALLUPURINOL – strukturą przypomina związki ksantynowe.
3.Hiperurynemia – podwyższone stężenie kwasu moczowego we krwi; dochodzi do wytrącania kwasy
moczowego i moczanu monosodowego w tkankach. Objawy zapalenia stawów nazywane dną
moczanową. Gdy kryształy odkładają się w nerkach – kamica nerkowa.
4.Zespół reperfuzji – niedotlenione tkanki nagle otrzymują dopływ krwi –np. po operacjach
kardiochirurgicznych na sercu z zawałem – dochodzi do aktywacji oksydazy ksantynowej – produktem
ubocznych jest nadtlenek wodoru – wolne rodniki – stosowanie allupuronolu zapobiega niekorzystnemu
działaniu reaktywnych form tlenu (RFT).
65. CK-MB
1. Izoenzym kinazy kretynowej katalizującej odwracalną reakcje fosforylacji kretyny (kreatyna + ATP
fosfokreatyna + ADP). Izoenzym CPK-2 (muscle,brain).
2.charakterystyczny dla mięśnia sercowego i mięsni szkieletowych (jednak tylko kilka procent CPK w
mięsniu sercowym to izoenzym MB – znaczną większość stanowi MM; w mięsniach szkieletowych
występuje w jeszcze mniejszej ilości.
3.Aktywnośc izoenzymy MB nie przekracza 5% aktywności całkowitej CPK.
4. W surowicy wystepuje w 2 izoformach. CK-MB2 występuje w cytoplazmie komórek mięsnia
sercowego. Po uwolnieniu do osocza w wyniku działani osoczowej karboksypeptydazy dochodzi do
hydrolizy C-końcowej lizyny podjednostki M i przekształcenia izoformy w CK-MB1. Poszczególne
izoformy można rozdzielić metodą elektroforezy.
5.Zawartość podniesiona jest u noworodków po porodzie (do 1 miesiąca) i po nieiwelkim wysiłku
fizycznym. Patologicznie aktywność CPK wzrasta we wczesnej fazie zawału mięśnia sercowego –
wzrost aktywności obserwuje się po 6 godzinach, maksimum osiąga po 18-30 godzinach, normalizuje
się po 3 dobach – aby rozpoznać zawał mięśnia sercowego ważne jest obserwowanie dynamiki zmian.
Poza tym wzrost stężenie CK-MB wystepuje w zapaleniach, toksycznych oraz pourazowych chorobach
mięśnia sercowego (zewnętrzny masaż serca, defibrylacja).
6. W osoczu oznacza się stężenie a nie aktywnośći CK-MB dzięki użyciu odpowiednich przeciwciał
(metody immunologiczne), chromoogniskowaniem, HPLC, elektroforezą wysokonapięciową.
7.CK-MB jako enzymatyczny wskaźnik zawału mięśnia sercowego: mierzy się przyrost stężenia w
ciągu 2 godzin – jeżeli jest większy niż 1,6/ml wnioskuje się o stałej degradacji kardiomiocytów.
66. Czas protrombinowy (PT)
-służy do oceniania zewnątrzpochodnego układu krzepnięcia. Jego wartość jest zależna od
stężenia w osoczu krwi takich czynników krzepnięcia jak: czynnik II, czynnik V, czynnik VII,
czynnik X i fibrynogenu.
-Metoda oznaczenia
Do osocza cytrynianowego dodaje się preparat czynnika tkankowego (TF) i po inkubacji
mierzy się czas od dodania jonów wapnia do skrzepnięcia próbki.
-Czas protrombinowy możemy wyrazić jako:
-różnicę w sekundach pomiędzy PT osoby badanej i osocza kontrolnego
-procent aktywności protrombiny wyliczany z krzywej rozcieńczeń osocza prawidłowego
-procentowy wskaźnik czasu protrombinowego - wskaźnik Quicka
-współczynnik czasu protrombinowego wyrażony w sekundach
-międzynarodowy współczynnik znormalizowany – INR
-Wartości prawidłowe
13 - 17 sek.
0,9-1,3 INR (2-4 INR zakres terapeutyczny)
80%-120% wskaźnik Quicka
-Wydłużenie czasu protrombinowego
wrodzone niedobory czynników II, V, VII, X

przewlekłe choroby miąższu wątroby
leczenie antagonistami witaminy K
niedobory witaminy K
doustne środki antykoagulacyjne
DIC
znaczne niedobory fibrynogenu
dysfibrynogenemie
białaczka
mocznica
choroba Addisona-Biermera
niesteroidowe leki przeciwzapalne
zatrucie pochodnymi kumaryny (np. trutką na szczury)
-Skrócenie czasu protrombinowego
zakrzepica
trombofilia
zwiększona aktywność czynnika VII
okres okołoporodowy
67. APTT (ang. Activated Partial Thromboplastin Time
– czas częściowej tromboplastyny po aktywacji), czas kaolinowo-kefalinowy – podczas
badania jeden ze wskaźników krzepliwości krwi, ponieważ jest miarą aktywności
osoczowych czynników krzepnięcia XII, XI, IX i VIII.
-Tworzą one układ wewnątrzpochodny aktywacji protrombiny.
-APTT zależy także od czynników biorących udział w powstawaniu trombiny (protombiny,
czynnika X i V) i konwersji fibrynogenu do fibryny.
-Czas kaolinowo-kefalinowy powinien utrzymywać się w normie od 26,0 do 36,0 s.
-Badanie niezbędne w rozpoznawaniu i leczeniu skaz krwotocznych. Wydłużenie czasu
APTT może świadczyć o zespole antyfosfolipidowym, skrócenie zaś o nadkrzepliwości (nie
ma znaczenia diagnostycznego). Kontrolę APTT stosuje się również podczas leczenia
heparyną niefrakcjonowaną.
-Przyczyny przedłużenia APTT
niedobór czynników krzepnięcia: VIII (hemofilia A), IX (hemofilia B), XI (hemofilia C) oraz
czynnika X i protrombiny
afibrynogenemia, hipofibrynogenemia, dysfibrynogenemia
choroba von Willebranda
obecność antykoagulantu toczniowego
leczenie heparyną niefrakcjonowaną lub czasem w czasie leczenia doustnymi
antykoagulantami (antagonistami witaminy K)
zespół rozsianego krzepnięcia wewnątrznaczyniowego (DIC)
niedobór kininogenu wielkocząsteczkowego i prekalikreiny oraz czynnika XII
wrodzony lub nabyty niedobór czynnika V
przy uszkodzeniu wątroby lub niedoborze witaminy K
-Podstawowe zastosowanie
monitorowanie leczenia przeciwkrzepliwego (w trakcie leczenia powinno wynosić 60-90s)
diagnostyka wrodzonych i nabytych skaz krwotocznych
68. kinetyka sigmoidalna podr.
69. ligazy – podr.32
70. wpływ na sprawność katalityczną enzymu- podr.11

71. izoenzymy fosfatazy kwaśnej –podr.23
72. enzymy sekrecyjne- podr.16
73. inhibicja niekompetencyjna- podr
74. jony metali Pb, Ag, Hg- wykres L-B, znaczenie, mech
75. enzymy oznaczane w moczu (gdzie lepiej w osoczu czy moczu)
76. metody badań- dwupunktowa i kinetyczna – podr.33
77. metody oczyszczania enzymów z materiału biol.
78. Lizozym, muramidaza (ang. lysozyme)
- białko kationowe o ciężarze 14,4 kDa, które ma właściwości enzymu hydrolitycznego
rozkładającego peptydoglikan ściany komórkowej bakterii.
-Strukturę pierwszorzędową lizozymu stanowi łańcuch polipeptydowy złożony zaledwie ze
129 aminokwasów.
-Miejsce aktywne lizozymu stanowią dwie reszty aminokwasowe: Glutaminianu 35 i
Asparaginianu 52, oddalone od siebie w sekwencji aminokwasów lecz położone względnie
blisko siebie w przestrzeni.[1]
-Lizozym stanowi jeden z mechanizmów humoralnej, nieswoistej odporności (odpowiedź
immunologiczna). Został odkryty w 2. dekadzie XX wieku przez Alexandra Fleminga.
-Występuje w ziarnistościach granulocytów wielojądrowych, monocytów oraz makrofagów.
Znajduje się także w większości płynów tkankowych, oprócz: moczu, potu i płynu mózgowo-
rdzeniowego.
-Lizozym wywiera działanie przeciwbakteryjne poprzez destrukcyjny wpływ na ścianę
komórkową bakterii. Działanie lizozymu polega na rozrywaniu wiązań glikozydowych
pomiędzy cząsteczkami kwasu N-acetylomuraminowego (NAM) i N-acetyloglukozaminą
(NAG).
-Mechanizm działania lizozymu polega na hydrolizie wiązań β-1,4-glikozydowych pomiędzy
cząsteczkami NAM i NAG w taki sposób, że atom tlenu wiązania glikozydowego pozostaje
przy cząsteczce NAG podczas gdy cząsteczka NAM otrzymuje grupę OH z cząsteczki wody.
Proces ten biegnie zgodnie z mechanizmem substytucji nukleofilowej pierwszego rzędu SN1.
Substytucja SN1 biegnie przez utworzenie karbokationu, który ma strukturę płaską trójkątną a
atom węgla C1 cząsteczki NAM przyjmuje hybrydyzację sp2. Tylko takie odkształcenie
pierścienia glukopiranozowego pozwala na aktywność katalityczną lizozymu w jego miejscu
aktywnym. Działanie lizozymu jest najwyższe przy pH bliskim 5. Tylko w takim środowisku
reszta kwasu glutaninowego 35 jest w postaci protonowanej (-COOH)a reszta kwasu
asparaginowego 52 jest w postaci anionu (COO-).[1]
-Bakterie Gram ujemne (G-) są bardziej odporne na lizozym, ze względu na występowanie u
nich zewnętrznej błony komórkowej, ściana komórkowa jest w tym typie bakterii częściowo
rozłożona i taka forma nosi nazwę sferoplastu . Bakterie Gram dodatnie (G+) pozbawione
ściany komórkowej noszą nazwę protoplastu i wykazują wszystkie czynności życiowe.
Całkowite jej zniszczenie ma miejsce w roztworach hipotonicznych (gradient stężeń).
79.inhibitory enzymu konwertującego-podr
80. enzymy monomeryczne –podr
81. enzymy wskaźnikowe-podr (indykatorowe)

82. KM (jak inhibicja kompetycyjna i niekopmetycyjna na nią wpływa)-podr
Wyszukiwarka
Podobne podstrony:
opracowanie na kolosa z enzymów , SEMINARIUM 3-4
opracowanie na kolosa id 338294 Nieznany
WSPÓŁCZESNE KIERUNKI PEDAGOGICZNE - opracowanie na kolosa (1), współczesne kierunki pedagogiczne
opracowanie na kolosa
glikoliza, cykl crebsa opracowanie na kolosa
cukry opracowanie na kolosa
matbudy na kolosa, pytania opracowane na materialy, 1
Opracowanie pytań na kolosa Węgle i cykl Krebsa
OPRACOWANIA PYTAŃ NA KOLOSA
opracowane pytania na kolosa TW Nieznany
więcej podobnych podstron