
PROCESY UTLENIANIA I REDUKCJI
DEFINICJE PODSTAWOWE
HENRYK SCHOLL
Katedra Chemii Ogólnej i Nieorganicznej
Wydział Fizyki i Chemii
Uniwersytetu Łódzkiego
E mail: hscholl@chemul.uni.lodz.pl
1.
Podział i podstawowe definicje procesów utleniania i redukcji
a)
Procesy utleniania i redukcji (procesy redox) zachodzące w roztworach są
procesami homofazowymi zachodzącymi pomiędzy jonami lub cząsteczkami
różniącymi się wartościami standardowych potencjałów elektrochemicznych (E
0
)
oraz zmieniającymi się wartościami stężeń reagujących substancji

b) Reakcje utleniania i redukcji zachodzące na granicy faz elektroda/
roztwór elektrolitu z międzyfazową wymianą elektronów jest zatem
procesem heterofazowym.
c) Procesy redukcji
1.
Redukcja jonów lub kompleksów do niższych stopni utlenienia,
redukcja cząsteczek nieorganicznych lub organicznych - postać
zredukowana pozostaje w roztworze po zmianie stopnia utlenienia.
2.
Wydzielanie z roztworów lub na elektrodzie gazów, fazy metalicznej
lub amalgamatowej powstałych na drodze redukcji jonów lub
kompleksów.
3.
Redukcja nierozpuszczalnych związków lub warstw
powierzchniowych związana z utworzeniem fazy metalicznej lub
amalgamatu - jest to zwykle faza również nierozpuszczalna, ale o
układzie innym niż wyjściowy.
d) Procesy utleniania.
1. Utlenianie jonów, kompleksów lub innych cząstek do postaci
rozpuszczalnych o wyższym stopniu utlenienia.
2. Utlenianie metalu, ich tlenków lub soli, czy też ogólnie ujmując -
materiału elektrody prowadzące do utworzenia rozpuszczalnych jonów
lub kompleksów.
3. Utlenianie ich tlenków lub soli, czy też ogólnie ujmując materiału
elektrody z wytworzeniem nierozpuszczalnych warstewek tlenkowych lub
o składzie soli tego metalu.
Oczywiste jest, że mamy do czynienia ze sprzężeniem dwóch spośród
wyżej wymienionych reakcji zachodzących w układzie reduktor –
utleniacz lub katoda – anoda

2. KILKA POJĘĆ ELEMENTARNYCH O POTENCJAŁACH FAZY
Siła elektromotoryczna ogniwa (SEM) składa się z różnic potencjałów
występujących na wszystkich granicach faz, które z reguły (oprócz granicy
ciecz ciecz) znajdują się w warunkach równowagowych.
Równowaga podziału naładowanych cząstek między dwie fazy jest
uwarunkowana równością ich potencjałów chemicznych w obydwu fazach.
Wartość ujemna potencjału chemicznego jest miarą pracy, która musi być
wykonana w stałych warunkach, w celu przeprowadzenia jednego mola
nienaładowanych cząstek z wnętrza fazy w stan gazowy o nieskończenie
wielkim
rozcieńczeniu,
odpowiadającemu
umownemu
zeru
skali
potencjałów chemicznych (
i
=0).
Wydobycie mola naładowanych cząstek z wnętrza fazy wymaga nie tylko
pracy rozerwania wiązań chemicznych, lecz również pokonania sił
elektrycznych. Miarą tej pracy jest potencjał elektrochemiczny o zapisie:

e
= + zF
(1)
Drugi człon formalny strony powyższego równania jest pracą
elektrostatyczną
nieskończenie
powolnego
przeniesienia
niema-
terialnego ładunku zF z nieskończenie wielkiej odległości w próżni do
wnętrza rozważanej fazy - tj. do punktu o potencjale .
Stosunek tej pracy do przeniesionego ładunku nosi nazwę
wewnętrznego potencjału elektrycznego fazy ( ) lub inaczej - potencjału
Galvaniego
Rys.1. Ilustracja do rozważań o potencjałach fazowych.
Wewnętrzny potencjał elektryczny może się składać z dwóch udziałów:
•
potencjału zewnętrznego fazy (
) - potencjału Volty, 2) potencjału powierzchniowego
fazy (
).

Zewnętrzny potencjał elektryczny przewodnika definiuje się jako wartość
graniczną stosunku w/q dla q
0.
Jest to praca przeniesienia ładunku w pobliże przewodnika i zbliżenie do
punktu C leżącego poza działaniem "sił obrazu", pochodzących od
fikcyjnych ładunków wyindukowanych na powierzchni metalu przez
zbliżającą się cząstkę.
Odległość tę określa się wartością rzędu 10
-4
cm. Ładunek przenoszony z
tego punktu do wnętrza rozważanej fazy (punkt A) musi pokonać spadek
potencjału warstwy elektrochemicznej znajdującej się na powierzchni fazy.
Ta składowa potencjału wewnętrznego jest równa powierzchniowemu
potencjałowi elektrycznemu . Wobec tego:
= +
(2)

co w połączeniu z równaniem (1) daje:
e
= + zF + zF (3)
Należy również pamiętać, że to formalne ujęcie pomija składowe
elektryczne wchodzące w skład potencjału chemicznego, występującego np.
przy oddziaływaniu chmury jonowej na cząstkę.
Zmierzenie bezwzględnych wartości potencjałów wewnętrznych nie jest
możliwe w przypadku rozpatrywania dwóch sąsiadujących ze sobą faz.
Dołączając badany układ do jakiegokolwiek przyrządu pomiarowego
tworzy się dodatkową granicę faz i mierzy się sumę algebraiczną
istniejących różnic potencjałów na powierzchniach granicznych.
REASUMUJĄC
:
mierzy się zawsze różnicę potencjałów wewnętrznych na
końcach dwóch doprowadzeń - jednakowych chemicznie.
Niemierzalne są natomiast różnice potencjałów zewnętrznych i różnice tzw.
potencjałów rzeczywistych pomiędzy dwoma fazami:
= + zF
(4)
gdzie (-) jest pracą wyjścia elektronu z metalu.

2. Zdefiniowanie i sens fizyczny pojęcia „potencjał standardowy”
Reakcja przebiegająca w ogniwie jest sumą reakcji zachodzących na
obydwu elektrodach. Przepływ ładunku uwarunkowany jest przez:
1)
siły chemiczne zależne od powinowactwa reakcji (suma iloczynów
potencjałów
chemicznych
i
odpowiednich
współczynników
stechiometrycznych) równego zmniejszeniu entalpii swobodnej
układu (-G),
2) przepływ ładunku proporcjonalny do całkowitej wielokrotności stałej
Faraday’a.
Dla warunków równowagi mamy:
-
i
i
= -G - -nFE (5)
Przeprowadźmy analizę różnic potencjałowych wewnątrz ogniwa o
budowie:
Pt H
2
HCl
c
AgCl
(s)
Ag Pt

Oznaczmy i ponumerujmy poszczególne fazy z zaznaczonymi granicami ich
podziału:
Pt(e) H
2
r-r (H
+
, Cl
-
, Ag
+
AgCl (Ag
+
, Cl
-
) Ag (Ag
+
, e Pt’ (e)
1
2
3
4
5
1’
Siła elektromotoryczna ogniwa będzie równa sumie różnic wewnętrznych
potencjałów elektrycznych poszczególnych faz:
E = (
1’
-
5
) + (
5
-
4
) + (
4
-
3
) + (
3
-
2
) + (
2
-
1
) (5)
co w efekcie daje:
E = (
1’
-
1
) (6)
Oznacza to, że mierzymy wartość różnicy potencjałów wewnętrznych
ogniwa, które pracuje w warunkach bez poboru prądu ( I = 0)
W rozpatrywanym ogniwie zachodzi samorzutna reakcja odwracalna:
½ H
2
(2) + AgCl(4) H
+
(3) + Cl
-
(3) + Ag(5) (6)
Równowagi fazowe cząstek zdolnych do przechodzenia z jednej fazy do
drugiej są określone równaniami:
e
Cl
-
,
3
=
e
Cl
-
,
4
;
e
Ag
+
,
5
=
e
Ag+;4
;
e
e,1’
=
e
e,1
;
(7)
Wprowadzając formalne zależności:
e
AgCl,4
=
e
Ag+,
+
e
Cl-;4
;
e
Ag,5
=
e
e,1’
+
e
Ag+, 5
(8)
Wówczas dla reakcji elektrodowej:
2H
+
(3) + 2e H
2
(2)
(9)
mamy:
2
H
+
,3
+ 2
e
e,1
=
e
H2,2
(10)

Przechodząc do potencjałów chemicznych i uwzględniając definicję
termodynamiczną SEM ( E = -zF
i
lub w innej formie: E = -zF G)
otrzymamy:
FE = -
H+,3
-
Cl-, 3
-
Ag,5
-
AgCl,4
+
1/2
H2, 2 (11)
Dla stanu podstawowego (standardowego) mamy:
p
H2
= 1013 hPa, Ag jest w stanie metalicznym (a
Ag
= 1)
Wobec tego słuszne są zależności:
H2
,
2
=
H2
;
Ag,5
=
Ag
;
AgCl,4
=
AgCl
(12)
Ponieważ podstawowa formuła termodynamiczna opisująca wartość
potencjału chemicznego (potencjału Gibbsa, energii swobodnej), ma
postać:
H+ =
H+
0
+
RT ln a
H+
(13)
a równocześnie średni współczynnik roztworu elektrolitu typu 1 – 1 (tutaj:
kwasu solnego)oblicza się korzystając z równania :
a
HCl
= (a
H+
* a
Cl-
)
1/2
(14)
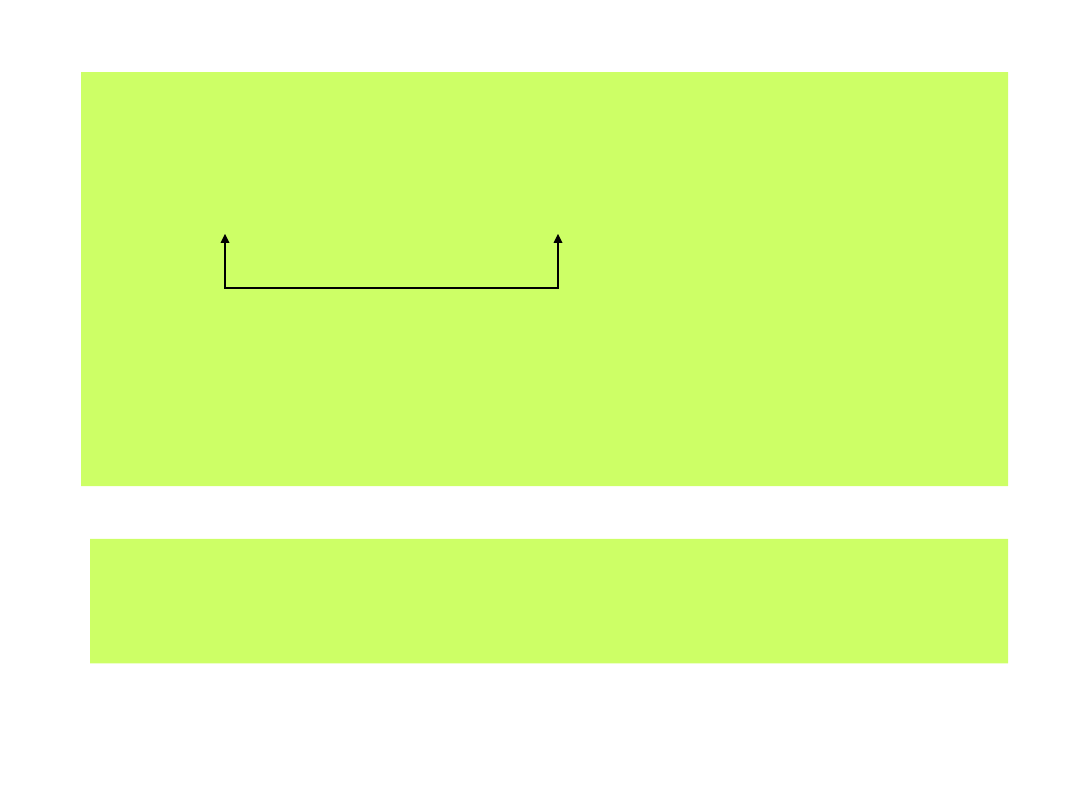
Równanie (11) przyjmie postać:
FE =
-
0
H+
-
0
Cl-
-
0
AgCl
+ ½
0
H2
– 2RT a
HCl
(15)
Suma wartości chemicznych potencjałów standardowych jest nową stałą
wielkością standardową:
E
0
= -
G
0
/ zF
(15)
Tym samym otrzymaliśmy równanie Nernsta:
E = E
0
[(0,059/ zF ] lg a
(16)

3. Elektrody odwracalne. Elektrody odniesienia
Konieczne jest zdefiniowanie powszechnie stosowanego pojęcia
elektrody odwracalnej.
Elektrodą taką nazywamy elektrodę, na której ustala się równowaga
odpowiedniego odwracalnego procesu elektrodowego z szybkością
wystarczającą dla celów praktycznych. Równowaga ta jest w istocie
równowagą podziału między metalem i roztworem cząstek naładowanych i
wspólnych dla obu faz. Pojęcie elektrody odwracalnej nie obejmuje
przypadku granicznego, gdy w jednej z faz stężenie cząstek określających
potencjał elektrody jest równe zeru.

I.
Elektrody pierwszego rodzaju
a) Odwracalne względem kationu
M
+
/M
.
Potencjał takiej elektrody jest określony zależnością:
(17)
W tym miejscu można mówić, że
dwie elektrody pierwszego
rodzaju
tworzące ogniwo, np.:
Ag AgNO
3
(m
1
) AgNO
3
(m
2
) Ag
tworzą w istocie
ogniwo stężeniowe
, którego SEM
określona jest wzorem:
(18)
Jeśli w ogniwie tym występuje dodatkowo potencjał
dyfuzyjny, to ogniwo takie nazywa się ogniwem
stężeniowym z przenoszeniem, gdzie t
+
i t
-
są odpowiednio
liczbami przenoszenia kationów i anionów
M
a
zF
RT
E
E
ln
0
2
,
1
,
2
,
1
,
ln
ln
a
a
F
RT
t
F
RT
t
a
a
F
RT
E
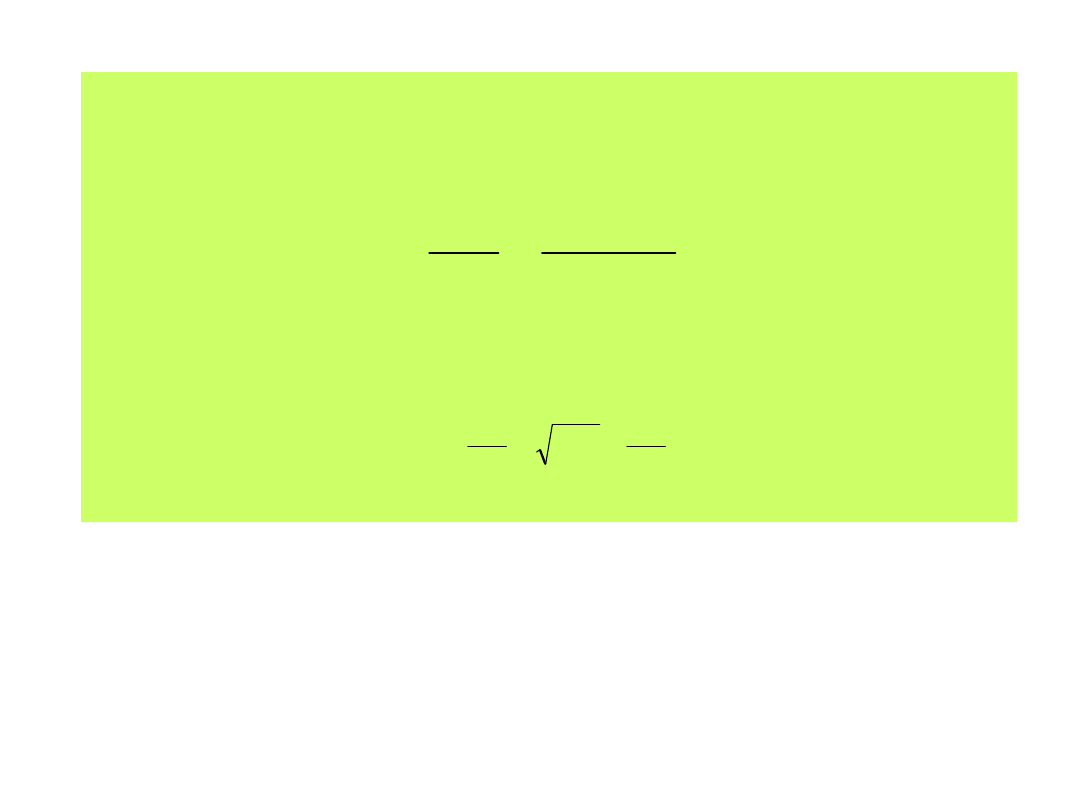
b)
Elektrody amalgamatowe: M
+
/M(Hg) o potencjale określonym
wzorem:
(19)
c) Elektrody gazowe typu H
+
/H
2
lub Cl
2
/Cl
-
o potencjałach
określonych równaniami:
(20)
)
(
ln
0
Hg
a
a
F
z
RT
E
E
M
H
H
H
H
a
F
RT
p
F
RT
E
ln
ln
2
2
,

II. Elektrody drugiego rodzaju.
Zbudowane są z metalu pokrytego trudno rozpuszczalną solą tego metalu i
układ ten zanurzony jest w roztworze elektrolitu o wspólnym anionie z
solą trudno rozpuszczalną. Należą do nich często stosowane i wygodne w
użyciu elektrody: kalomelowa Hg/Hg
2
Cl
2
/Cl
-
, siarczanowo-rtęciowa
Hg/Hg
2
SO
4
/SO
4
2-
, czy też chlorosrebrowa Ag/AgCl/Cl
-
. Potencjał tej
ostatniej elektrody jest np. określony wzorem:
(21)
wynikającym wprost ze wzoru Nernsta.
Cl
Ag
AgCl
Ag
AgCl
a
F
z
RT
E
E
ln
/
0
/

III. Elektrody redox
Są to elektrody oparte na reakcji podstawowej:
Ox + ne n
Red
Red
(22)
Budowa takiej elektrody zwykle sprowadza się do zanurzenia do
roztworu zawierającego substancję na dwóch różnych i trwałych
stopniach utlenienia nie reagującego z nią "metalu obojętnego", np. Au
lub Pt. Wartość potencjału elektrody redox zależy od aktywności form
utlenionej i zredukowanej, co opisuje równanie podstawowe:
(23)
W przypadku reakcji jednoelektronowej w temperaturze 298K,to po
wstawieniu odpowiednich wartości stałych fizycznych i po przeliczeniu
wartości logarytmów o podstawie naturalnej na wartości logarytmów o
podstawie dziesiętnej, wzór ten przyjmuje postać końcową:
(24)
n
n
n
n
M
M
M
M
a
a
zF
RT
E
E
1
1
ln
/
0
n
N
M
M
a
a
E
E
1
lg
059
,
0
0

Wartości potencjałów standardowych układów redox wyznacza się
korzystając z wartości zmian standardowych entalpii tworzenia będącej
wartością SEM odpowiedniego ogniwa:
zF
G
E
0
0
Warunki standardowe: ciśnienie gazów
p = 1013 hPa
temperatura
T = 298,15 K
aktywność jonów
a = 1
aktywność fazy stałej a = 1
stała Faraday’a F = 96 484 coul/mol
Symbol „z” oznacza liczbę elektronów przenoszonych w procesie
jednostkowym

Układem odniesienia potencjałów standardowych w roztworach
wodnych jest tzw.
Standardowa elektroda wodorowa
(angielski
skrót: she) tworząca półogniwo:
Pt H
2
H
+
a = 1
Potencjał standardowy tej elektrody zdefiniowany jako równy zero:
E
0
0,000 V
Wartość ta stanowi punkt odniesienia dla tzw. skali potencjałów
elektrodowych utworzonej poprzez pomiary (lub obliczenia) różnicy
potencjałów elektrycznych ogniw, w których jednym z półogniw stanowi
właśnie standardowa elektroda wodorowa
Praktyczne zastosowanie elektrody wodorowej jest ograniczone ze
względu na następujące trudności:
1)
stabilizacji wartości ciśnienia jednostkowego gazowego wodoru,
2)
korygowanie ciśnienia atmosferycznego i wilgotności otoczenia,
3)
jon wodorowy, wprawdzie najmniejszy rozmiarami wśród pozostałych
jonów, jest hydratowany otoczką sześciu cząsteczek wody,
4) nie można elektrody wodorowej stosować w rozpuszczalnikach
aprotycznych ze względu na chemiczne reakcje ich protonowania.
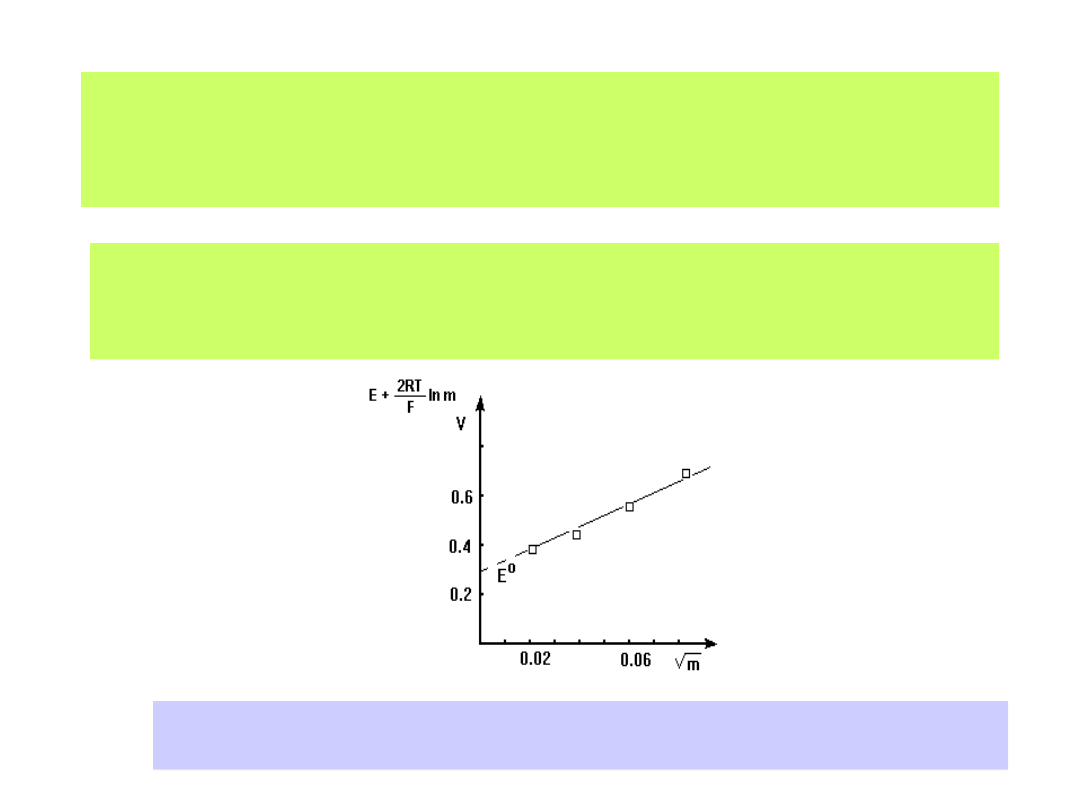
Wartość potencjału standardowego układów redox również wyznacza się
doświadczalnie budując odpowiednio zestawione ogniwo i mierząc
wartości SEM dla różnych stężeń roztworów elektrolitów. Graficzny zapis
zmierzonych SEM-ów ekstrapolujemy do wartości c
0:
Dla ogniwa:
Pt H
2
HCl AgCl Ag
otrzymamy wykres:
Ekstrapolacja graficzna wyników pomiarów SEM ogniwa Pt/H
2
/ HX/ AgX/Ag
pozwala na wyznaczenie wartości potencjału E
0
(m
1/2
0)
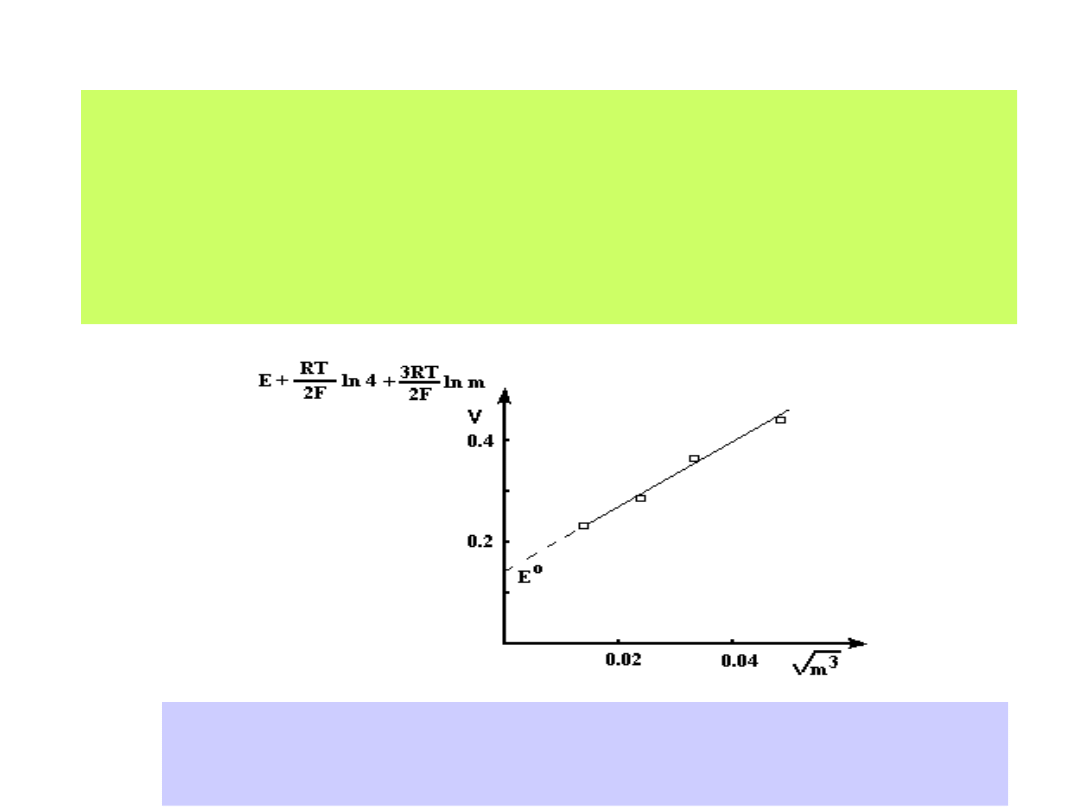
Natomiast dla ogniwa o układzie półogniw:
Zn ZnCl
2
(m) Hg
2
Cl
2
Hg
o reakcji podstawowej:
Zn +Hg
2
Cl
2
Zn
2+
+2Cl
-
+ 2Hg
mamy zależność graficzną w postaci:
Ekstrapolacja wyników pomiarów SEM ogniwa zawierającego elektrolit
typu 2 : 1 pozwala na wyznaczenie wartości potencjału standardowego
dla
m
0

WARTOŚCI STANDARDOWYCH POTENCJAŁÓW WYBRANYCH
(spośród ok. 900) UKŁADÓW REDOX
UKŁAD REDOX
REAKCJA REDOX
E
0
she
/V
Li
+
/ Li
H
(g)
/ OH
-
Na
+
/ Na
Mg(OH)
2
/ Mg
Mg
2+
/ M
Ti
2+
/ Ti
CdS /Cd
Si /H
2
O
Fe
2+
/ Fe
H
+
/ H
2
Cu
2+
/ Cu
Ag
2+
/Ag
Cl
2
/Cl
-
Li
+
+ e Li
H
(g)
+ OH
-
H
2
O + e
Na
+
+ e Na
Mg(OH)
2
+2e
Mg+2OH
-
Mg
2+
+ 2e Mg
Ti
2+
+ 2e Ti
CdS + 2e Cd + S
-2+
Si+H
2
O SiO
2
+4H
+
+4e
Fe
2+
+ 2e Fe
H
+
+ e ½ H
2
Cu
2+
+ 2e Cu
Ag
+
+ e Ag
Cl
2
+ 2e 2 Cl
-
-3,045
-2,930
- 2,714
-2,682
-2,404
-1,752
- 1,233
-0,842
-0,440
0,000 (def.)
+0,344
+0,800
+1,359

3. INFORMACJE O OGÓLNEJ CHARAKTERYSTYCE WARSTWY
ELEKTROCHEMICZNEJ
Pojęcia przedstawione w tej informacji są wprowadzeniem do
zagadnień o kluczowym znaczeniu dla elektrochemii. Ich rozwinięcie
przedstawić można jedynie zostanie w ramach wykładu specjalizacyj-
nego.
Na granicy rozdziału dwóch faz powstaje obszar, w którym natężenie pola
elektrycznego ma wartość różną od zera. Źródłem tego pola jest
nadmiarowy ładunek elektrycznie naładowanych cząstek - tj. jonów,
elektronów, zorientowanych dipoli itd.

Obszar ten, o nadmiarowym ładunku elektrycznym, nosi nazwę
warstwy
elektrochemicznej
,
dawniej zwaną - podwójną warstwą elektryczną.
Powstawanie warstwy elektrochemicznej jest zjawiskiem o charakterze
powszechnie spotykanym. Mamy więc do czynienia z warstwą
elektrochemiczną na granicach faz:
-ciecz / ciecz (dla niemieszających się cieczy)
-ciecz / powietrze (zwana swobodną powierzchnią roztworu)
-elektroda / gaz (w znikomym stopniu dotyczy elektrochemii)
-elektroda / roztwór (zasadnicza grupa zagadnień).
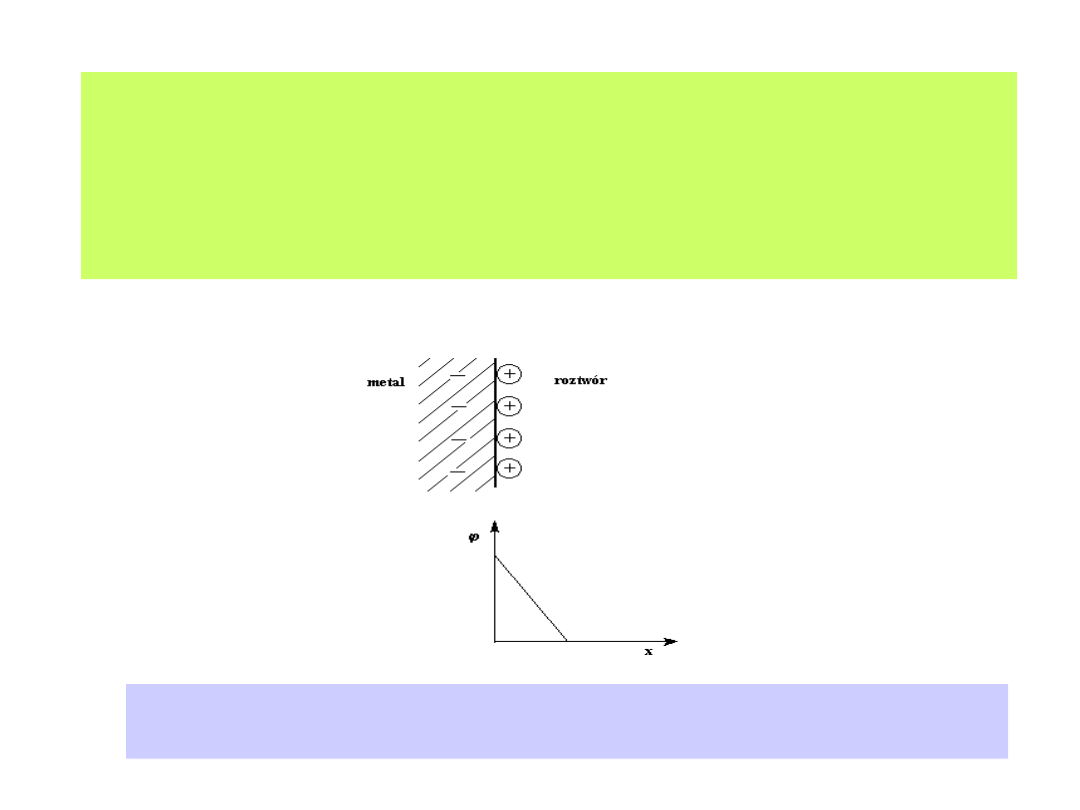
Pojęcie i propozycje modelu elektrycznego warstwy elektrochemicznej
datują się już z XIX wieku ( Helmholtz, Gouy, Chapman, Stern, Parsons i
inni). Początek prostym modelem warstwy elektrochemicznej dał
Helmholtz:
Model warstwy elektrochemicznej według propozycji Helmholtza.
Zwraca uwagę liniowy spadek potencjału wraz ze wzrostem odległości od elektrody.

Zgodnie z dzisiejszymi poglądami złożona warstwa elektrochemiczna może
być zilustrowana w następujący sposób:
Model warstwy elektrochemicznej w roztworze zawierającym powierzchniowo
czynne cząsteczki organiczne: a) - dipole wody, b) - zaadsorbowane molekuły
organiczne, c) - specyficznie zaadsorbowane aniony, d) - hydratowane jony
tworzące warstwę OHP
1
w odległości wynikającej z oddziaływań dipoli wody w
IHP, e) - OHP
2
odległa od elektrody w zależności od oddziaływań molekuł
organicznych, f) - hydratowane jony w warstwie dyfuzyjnej
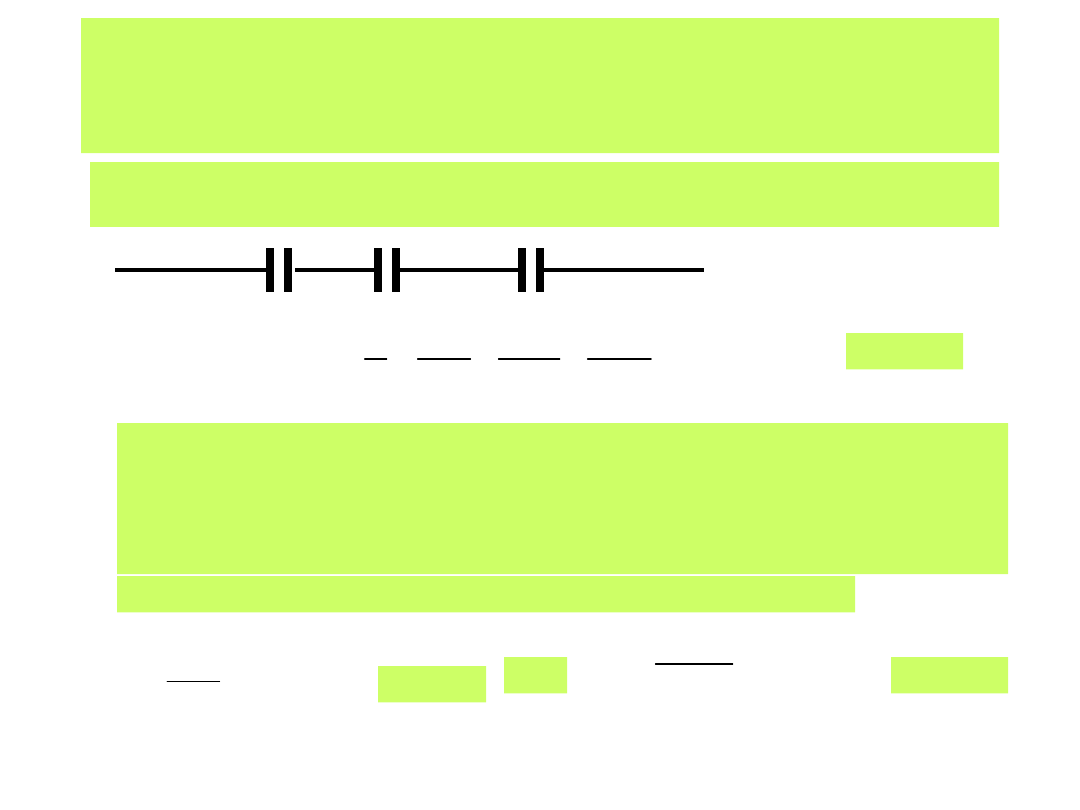
Taka budowa warstwy elektrochemicznej daje się w zastępczym obwodzie
elektrycznym zapisać jako szeregowe połączenie kondensatorów:
Metal IHP OHP
1
OHP
2
Opisuje to równanie
pojemności warstwy:
2
1
1
1
1
1
OHP
OHP
IHP
C
C
C
C
(26)
Wartości pojemności różniczkowej warstwy elektrochemicznej wyznacza
się doświadczalnie, a obecnie stosowane systemy aparatury pomiarowej
pozwalają na szybkie obliczanie jej wartości zgodnie z zastosowanymi
metodami pomiaru i oprogramowania komputerowego.
Gdzie z kolei pojemność różniczkowa opisana jest jako:
C
E
i
T
p
p
,
,
2
2
(26 a)
lub
C
E
T
p
p
m
,
,
)
(
(26 b)

4. PODSTAWY KINETYKI PROCESÓW ELEKTRODOWYCH
Jeżeli wynikiem procesu na rozważanej elektrodzie jest
przejście elektronu z elektrody do elektrolitu lub ładunku
dodatniego z elektrolitu do elektrody, to związany z tym prąd
elektryczny nazywa się
prądem katodowym
(I
k
). Prąd ten ma
znak ujemny.
Reakcja zachodząca jest
reakcją katodową lub reakcją
elektroredukcji.
Przypadek przeciwny - przejście ładunku dodatniego z elektrody
do elektrolitu lub odwrotne przejście elektronu z elektrolitu
charakteryzuje
prąd anodowy (I
a
)
o znaku dodatnim. Reakcja
zachodząca na tej elektrodzie jest
reakcją anodową lub
elektroutleniania.
Stosowana powszechnie gęstość prądu (i
a
, i
k
) dotyczy przeliczenia wartości
prądu na jednostkę powierzchni geometrycznej. Przeliczenie go na
rzeczywistą gęstość prądu (j
a
, j
k
) wymaga znajomości współczynnika
rozwinięcia powierzchni, co zwykle jest pomiarem dość skomplikowanym.
Głównym etapem elektrochemicznego procesu elektrodowego
jest bezpośrednia wymiana cząstek naładowanych między elektrodą a
roztworem. Kinetykę i mechanizm procesu elektrodowego rozpatruje się
w oparciu o
reakcję przejścia
(zwaną też reakcją przeniesienia ładunku
czy też wprost reakcją elektrodową).

Dokładniejsza
klasyfikacja
procesów
elektrodowych
uwzględnia
charakter przejścia ładunku pomiędzy elektrodą a substancją
elektroaktywną:
1. Przejście elektronu lub dziury między elektrodą a substancją
elektroaktywną.
2. Przejście jonu metalu z roztworu do elektrody lub odwrotnie.
3. Emisja elektronów z elektrody do roztworu z utworzeniem
solwatowanych elektronów, a następnie reakcja między solwatowanym
elektronem i obecnym w roztworze "zmiataczem" elektronów (ang.
scavenger), tj. rozpuszczalnikiem lub akceptorem elektronów.
Podstawowym warunkiem przejścia elektronu lub dziury z elektrody do
substancji elektroaktywnej (akceptora elektronów) jest jej obecność na
zewnętrznej płaszczyźnie Helmholtza (OHP) i obecność nieobsadzonego
termu zdolnego do przyjęcia elektronu.
Odpowiedni donorowy stan kwantowy elektronu w elektrodzie musi mieć
energię w przybliżeniu taką, jak nieobsadzony term w substancji „Ox”.
Podobnie jest w przypadku, gdy redukcja przebiega według mechanizmu
dziurowego, tj. wskutek przejścia dziury z substancji elektroaktywnej do
elektrody (Rys.)
Odwrotnie rzecz się ma w przypadku reakcji elektroredukcji - postać
„Red” musi mieć charakter elektronodonorowy i musi zawierać
obsadzony term o energii odpowiadającej nieobsadzonemu stanowi
kwantowemu w elektrodzie.
Przypomnijmy, że energia poziomu Fermiego (F) jest identyczna z
elektrochemicznym potencjałem elektronu w metalu. Zmieniając potencjał
wewnętrzny fazy o (polaryzacja elektrody E = ) można przesunąć
F
w
kierunku większych lub mniejszych energii odpowiednio ułatwiając lub
utrudniając proces elektrodowy.Ogólnie rzecz ujmując - schemat reakcji
elektrodowej wygląda nastąpująco:
Ox+ + [term obsadzony] Red + [term nieobsadzony]
Substancje elektroaktywne wchodzą z reguły w reakcję przejścia
elektronu, gdy zbliżają się na odległość zewnętrznej warstwy Helmholtza
(oznakowanej skrótem OHP). Niekiedy konieczna jest ich uprzednia
adsorpcja. Często na reakcję przejścia hamująco (inhibitująco) lub
przyspieszająco (stymulująco) działa adsorpcja produktów lub innych
cząstek z roztworu. Zmienia się wówczas struktura warstwy
elektrochemicznej i w konsekwencji obserwuje się zmianę szybkości
procesu elektrodowego.
Należy
pamiętać,
że
rzeczywisty
mechanizm
procesu
elektrodowego należy odróżnić od sumarycznej reakcji
zachodzącej na elektrodzie.
Występowanie gradientów stężenia reagentów elektroaktywnych w
otoczeniu elektrody (przestrzeni przyelektrodowej), obecność pola
elektrycznego i działanie sił mechanicznych uruchamiają procesy
transportu.
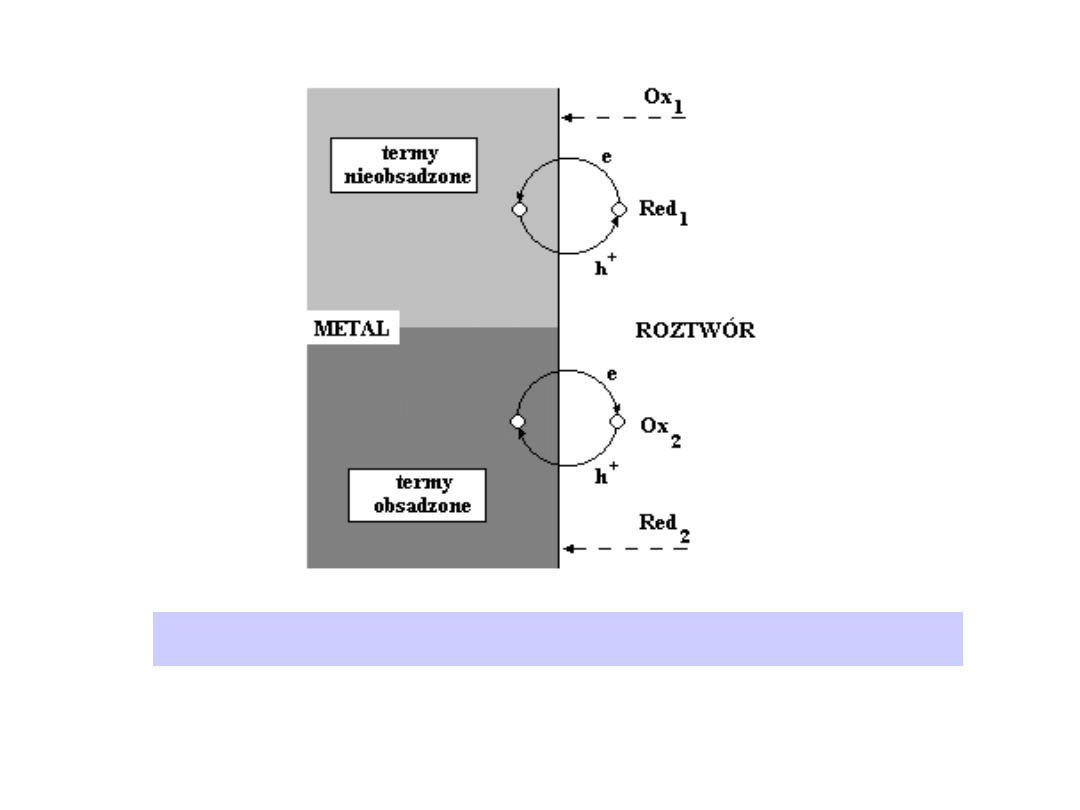
Ilustracja do rozważań o mechanizmie reakcji elektrodowej. Przejście
elektronu i przejście dziury
Substancje elektroaktywne wchodzą z reguły w reakcję
przejścia, gdy zbliżają się na odległość OHP.
Niekiedy konieczna jest ich uprzednia adsorpcja. Często na
reakcję przejścia hamująco (inhibitująco) lub przyspieszająco
(stymulująco) działa adsorpcja produktów lub innych cząstek z
roztworu. Zmienia się wówczas struktura warstwy
elektrochemicznej i w konsekwencji obserwuje się zmianę
szybkości procesu elektrodowego.
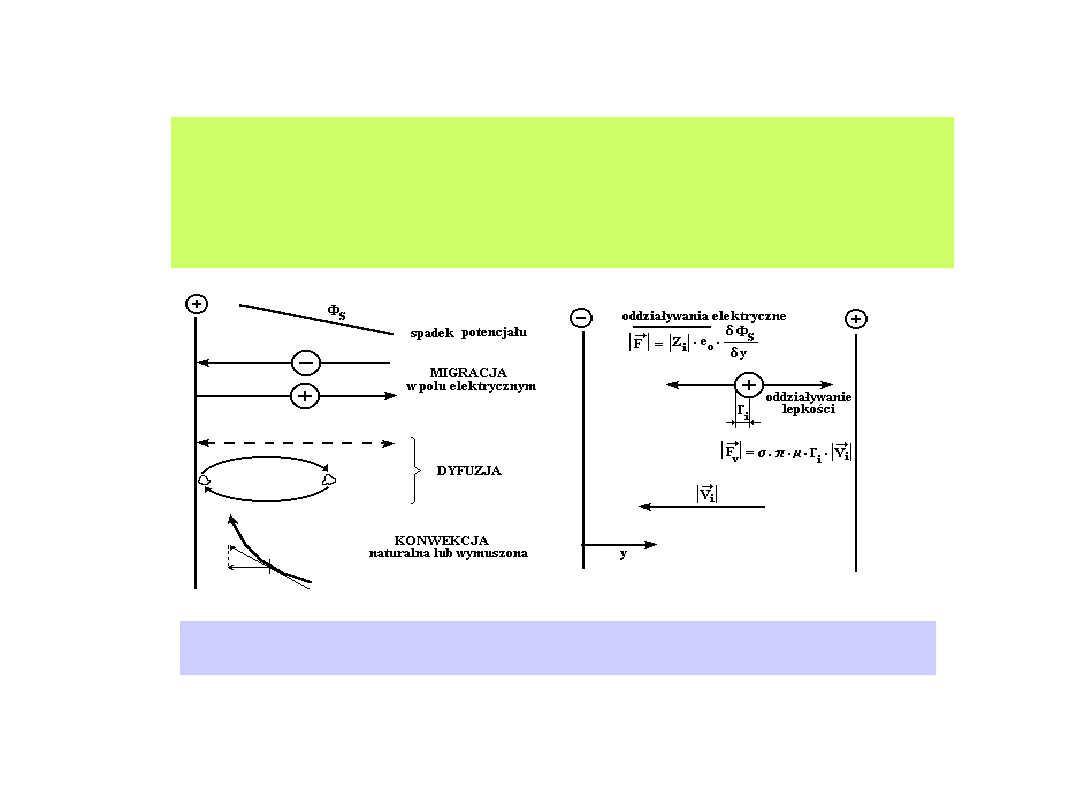
Występowanie gradientów stężenia reagentów
elektroaktywnych w otoczeniu elektrody (przestrzeni
przyelektrodowej), obecność pola elektrycznego i działanie sił
mechanicznych uruchamiają procesy transportu.
Transport jonowy w roztworze. a) - mechanizmy transportu substancji
jonowych, b) - siły działające na substancje jonowe.
Reakcja elektrodowa zwykle przebiega w potencjale innym niż potencjał
równowagowym. Charakteryzuje ją wówczas wartość
nadpotencjału
reakcji:
ox
red
c
RT
E
E
zF
c
RT
E
E
zF
zFk
j
)
(
exp
(
)
1
(
exp
0
0
0
gdzie k
0
– stała szybkości reakcji opisująca proces redox
- współczynnik przejścia elektronów. Jeśli = 0.5, to reakcja
elektrodowa jest reakcją doskonale odwracalną. Jest to też
prawdopodobieństwo energetycznie barierowego i
bezbarierowego przejścia elektronu
0
Re
0
0
ln
d
ox
c
c
zF
RT
E
E
Uwzględniając stężenia form Ox i Red wraz z potencjałem E
0
odnosimy
nadpotencjał do potencjału równowagowego reakcji poprzez wyrażenie
prądu wymiany:
(27)
(28)
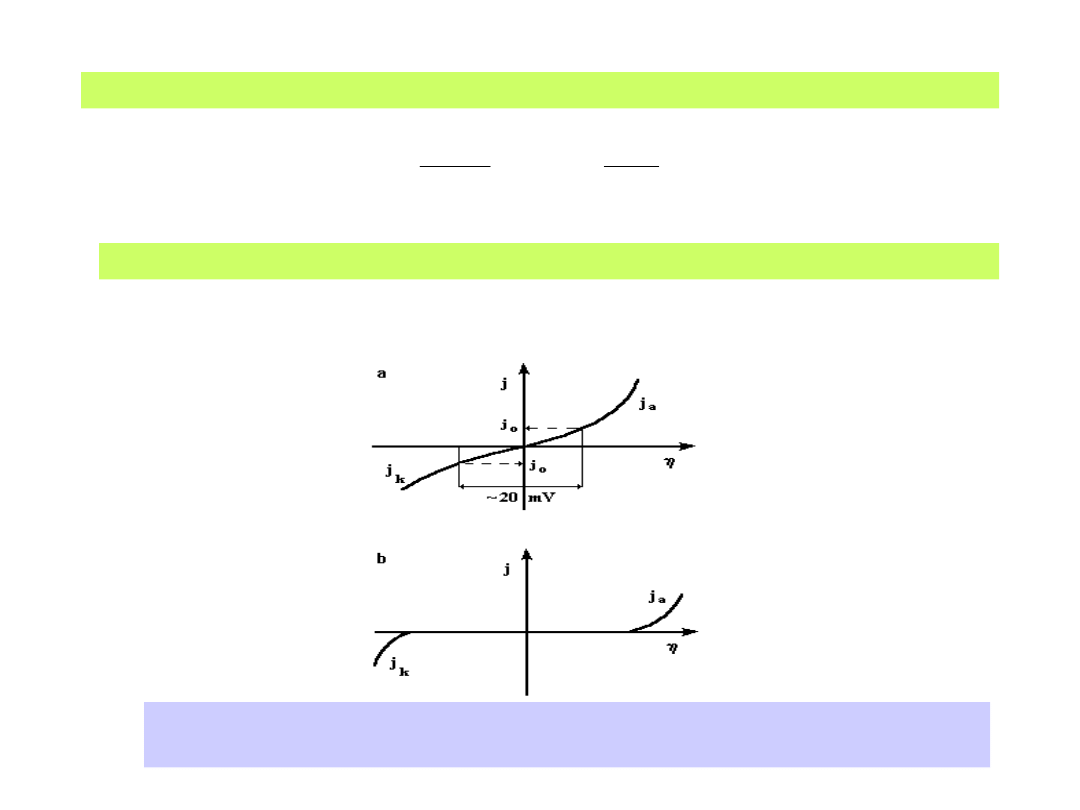
Równanie (28) można przekształcić i otrzymać je w postaci:
zF
RT
zF
RT
j
j
exp
)
1
(
exp
0
(29)
gdzie z kolei:
d
Ox
c
c
zFk
j
Re
1
0
0
(30)
Podstawowe krzywe polaryzacji elektrodowej: a) dla <<(RT/zF)
b) dla (RT/zF)

2. Jeśli nadpotencjał ma dużą wartość w porównaniu z RT/zF ,
wówczas równanie
(30)
po zlogarytmowaniu prowadzi do równania Tafela:
(31)
lub prościej je zapisując w postaci równania liniowego:
(32)
Omówione przypadki odnoszą się do odwracalnych (1) i
nieodwracalnych lub quasi-odwracalnych (2) reakcji pierwszego
rzędu. Oczywiste jest, że jak w klasycznej kinetyce chemicznej,
podobne rozważania obejmują także reakcje wyższych rzędów.
RT
zF
j
j
exp
0
j
zF
RT
j
zF
RT
lg
303
,
2
lg
303
,
2
0
j
b
a
lg
Równanie Tafela, opisujące kinetykę reakcji
zachodzącą w warunkach zewnętrznego źródła
elektronów (a głównie występujący w nim
współczynnik ) jest klasycznym przykładem
ilustrującym fakt, że proces elektrochemiczny
nie jest procesem czysto chemicznym i klasyczna
termodynamika reakcji chemicznych nie może być
stosowana.
Równanie to ponadto opisuje w swoisty sposób
istnienie bariery potencjału wynikającej z teorii
absolutnej szybkości reakcji chemicznych.
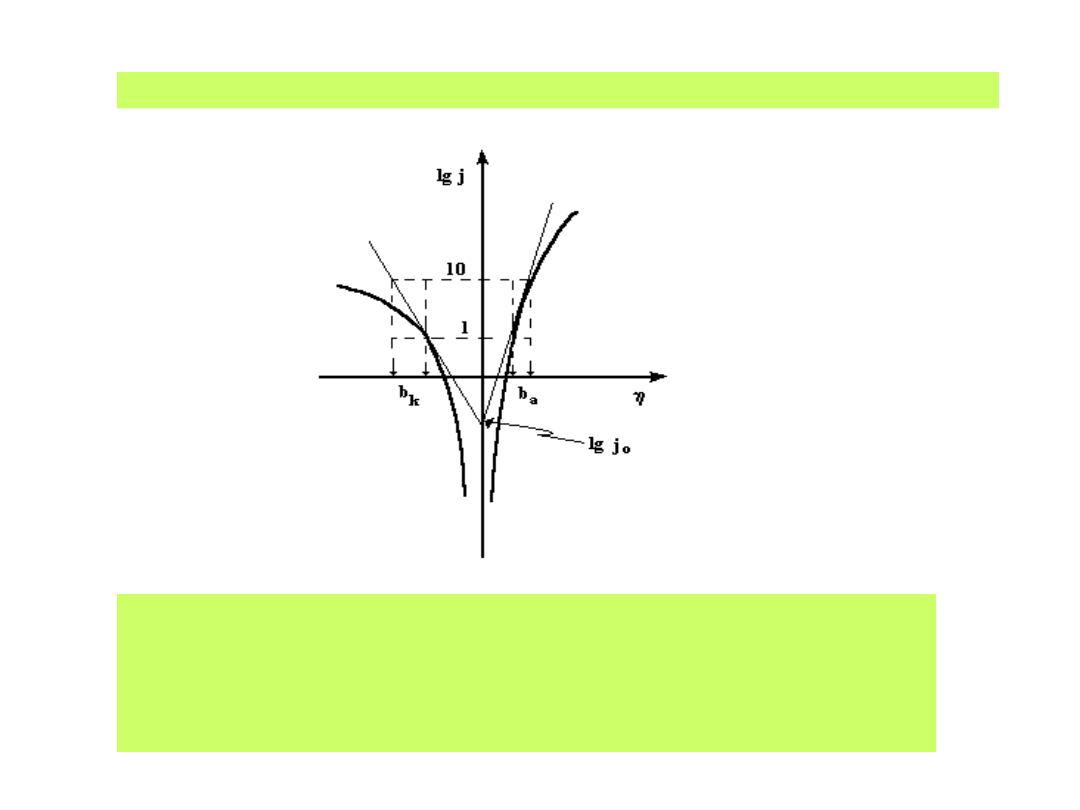
Zależności Tafela ilustruje wykres półlogarytmiczny:
Z którego wyznacza się wartość prądu wymiany j
0
oraz
współczynniki Tafela b
a
i b
k
charakteryzujące odwracalność reakcji
i liczby elektronów przenoszonych w procesach anodowych
i katodowych

5.KATODOWE ELEKTROSADZANIE METALI
EleElektrodowe osadzanie metali jest określeniem pochodzącym z lat 40 ubiegłego
stulecia,
wywywodzącym się z autokatalitycznego osadzania niklu przy nieobecności
zewnętrznego
źrźźródła prądu. Obecnie określenie to jest powszechnie stosowane.
O
Obejmuje różnorodne procesy elektroosadzania metali i stopów metali na dowolnym
p
Pdpodłożu z innego materiału przewodzącego.
Proces osadzania metalu powinien uwzględniać warunki zawarte w tabeli:
Składnik
Rola składnika w procesie
elektroosadzania metalu
1) Sól metalu (ML
m
n+
)
2) Czynnik redukujący (Red)
3) Bufor pH
4) Ligand kompleksujący (L)
5) Dodatki
Źródło jonów metalu osadzanego
Redukcja jonu metalu z jonu ML
n+
m
Regulacja wartości pH roztworu
Tworzenie jonu kompleksowego (ML
n+
m
)
stabilizuje skład kąpieli lub reguluje wielkość
osadzanych ziaren metalu

Ogólny schemat reakcji redukcji kompleksu do metalu można opisać
równaniem:
ML
m
n+
+ Red M + mL + Ox
n+
na którą składają się procesy cząstkowe:
redukcji:
M L
m
n+
+ ne M + mL
i utleniania:
Red +
ne + Ox
n+
Konieczna jest znajomość tzw. potencjału elektroosadzania, który
wyznaczamy na podstawie wykresów:
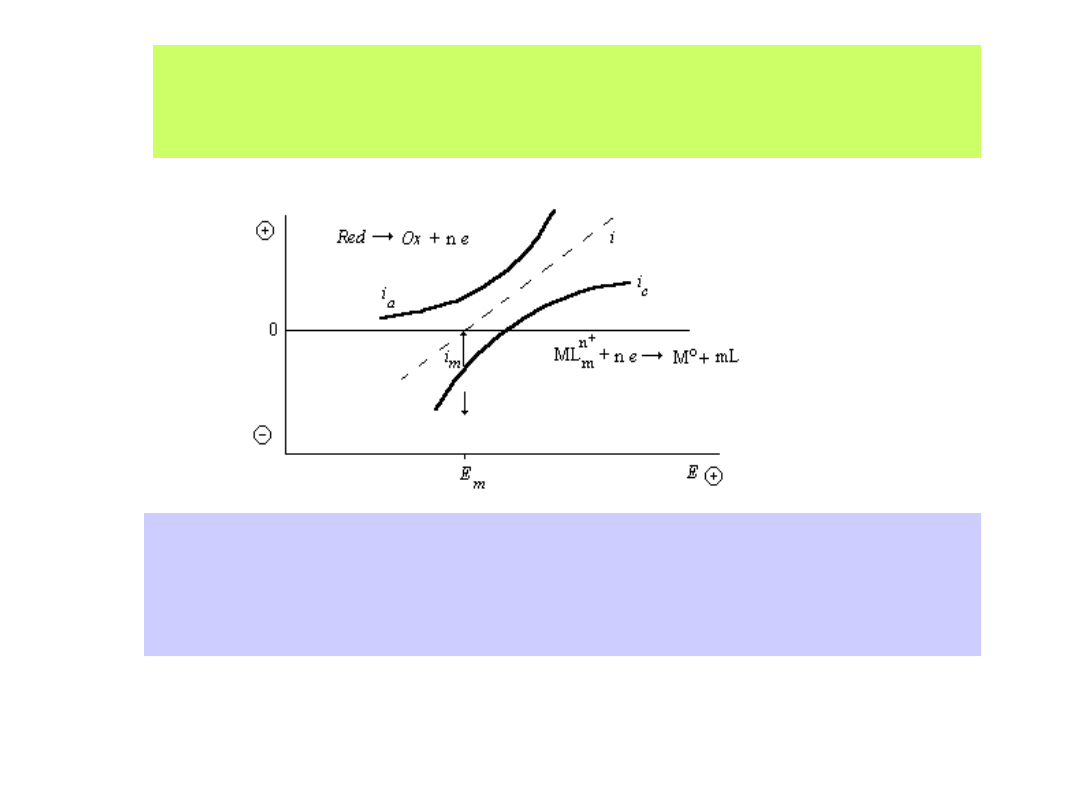
Konieczna jest też znajomość tzw. mieszanego potencjału
elektroosadzania metalu,
który wyznaczany jest na podstawie wykresu:
Wyznaczanie mieszanego potencjału elektroosadzania (E
m
):
i - sumaryczny prąd reakcji, i
a
- cząstkowy anodowy prąd reakcji,
i
k
- cząstkowy katodowy prąd reakcji, i
m
- prąd elektroosadzania w mieszanym
potencjale E
m
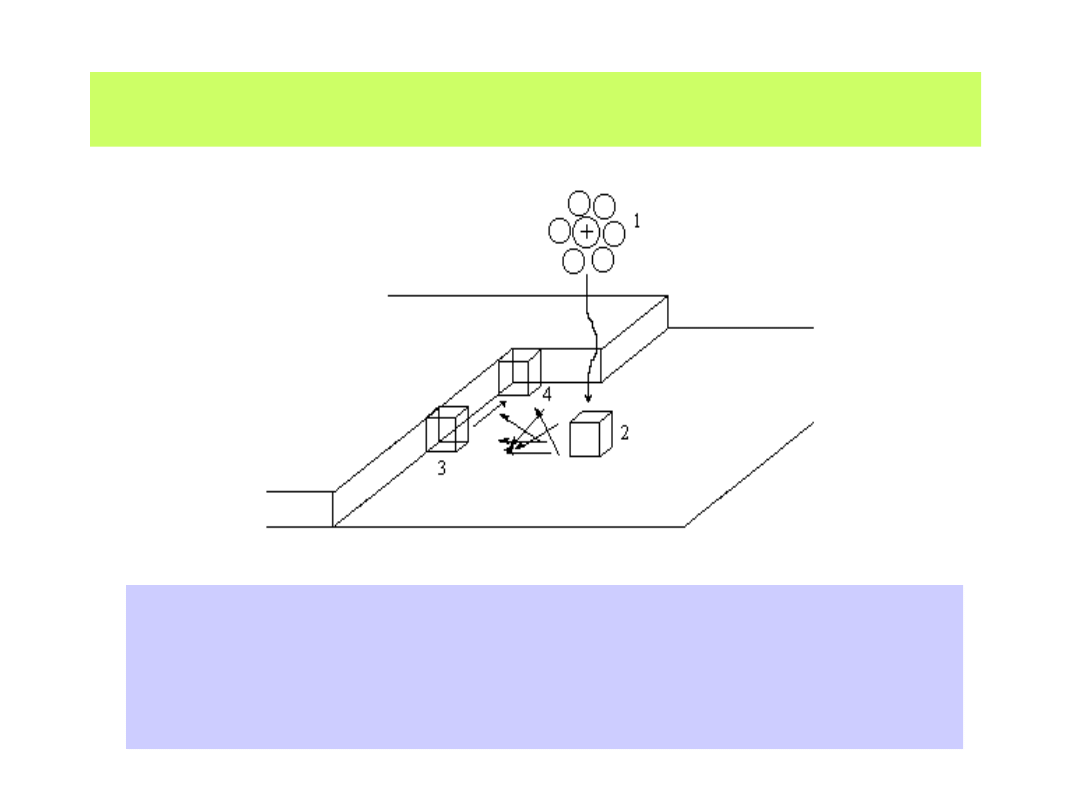
Kation zbliżający się do katody rozładowuje się i umieszcza się na niej
w sposób podany na poniższym schemacie
1 - jon zbliżający się do powierzchni traci otoczkę solwatacyjną, wymienia
elektron i w położeniu 2 tworzy tzw. adatom (atom zaadsorbowany);
3 - poprzez powierzchniową migrację dyfuzyjną atom wbudowuje się w sieć
krystaliczną, by przesunąć się w położenie 4 - o najmniejszej energii sieciowej
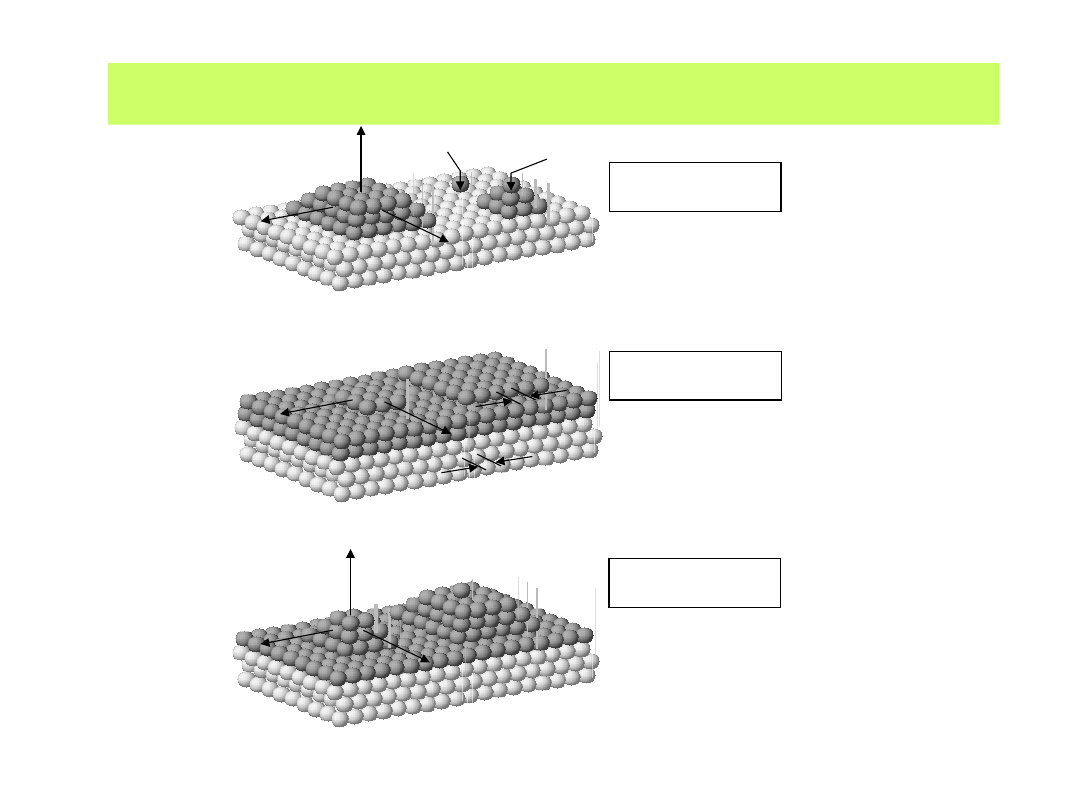
Dalszy przyrost warstw atomów metalu znalazł swój obraz w kilku
propozycjach modelowych:
Ψ
Me-Me
Ψ
Me-S
Ψ
Me-S
<< Ψ
Me-Me
model wzrostu "Volmer-Weber"
(wzrost 3D wysp)
a)
d
0
Me
d
0
S
Ψ
Me-S
>> Ψ
Me-Me
d
0
Me
≈ d
0
S
model wzrostu "Frank-van der
Merwe"
(wzrost warstwa na warstwie)
b
)
Ψ
Me-S
>> Ψ
Me-Me
d
0
Me
≠ d
0
S
model wzrostu "Stranski-
Krastanov"
(wzrost 3D wysp na wcześniej
osadzonej monowarstwie)
c)

Rzeczywiste obrazy powierzchni w skali atomowej (w
rozmiarach nanometrowych; 1 nm =10
-9
m, czyli 10
-6
mm można
obecnie doświadczalnie obserwować za pomocą systemów
pomiarowych wyposażonych w Elektrochemiczny Skaningowy
Mikroskop Tunelowy (ESTM) dla powierzchni przewodzących
prąd elektryczny lub w Mikroskop Sił Atomowych (AFM) dla
powierzchni nie przewodzących prądu.
Wyniki naszych prac można zobaczyć na posterach
prezentowanych na korytarzach Katedry

6. KATODOWE WYDZIELANIE WODORU
Katodowe wydzielanie wodoru opisuje sumaryczne równanie:
H
+
+ e ½ H
2
(33)
W rzeczywistości proces ten przebiega dwuetapowo, według
mechanizmu Volmera-Tafela lub według mechanizmu Volmera-
Heyrovskiego. W przypadku kwaśnego roztworu elektrolitu reakcje
Volmera, Tafela i Heyrovskiego opisują równania:
a) reakcja wydzielania wodoru jest
reakcją przeniesienia elektronu
,
zwana reakcją Volmera:
H
3
O
+
+ e H
ads
+ H
2
O
(34)
b) następuje reakcja rekombinacji zaadsorbowanych atomów wodoru,
zwana reakcją Tafela:
2 H
ads
H
2
(35)
c) następuje elektrochemiczna desorpcja zaadsorbowanego wodoru
(pochodzącego np. z reakcji Volmera) połączona z równoczesną
wymianą ładunku, zwana reakcją Heyrovsky'ego.
H
ads
+ H
3
O
+
+ e H
2
+ H
2
O(34)
Zależnie od rodzaju metalu zmienia się trwałość wiązań M - H,
odzwierciedlona w wartości entalpii swobodnej adsorpcji atomów
wodoru. Zatem natura metalu wywiera duży wpływ zarówno na
kinetykę, jak i na mechanizm procesu wydzielania wodoru na danej
elektrodzie. Eksperymentalnie uwidacznia się to w wartości prądu
wymiany i współczynnika przejścia elektronu .
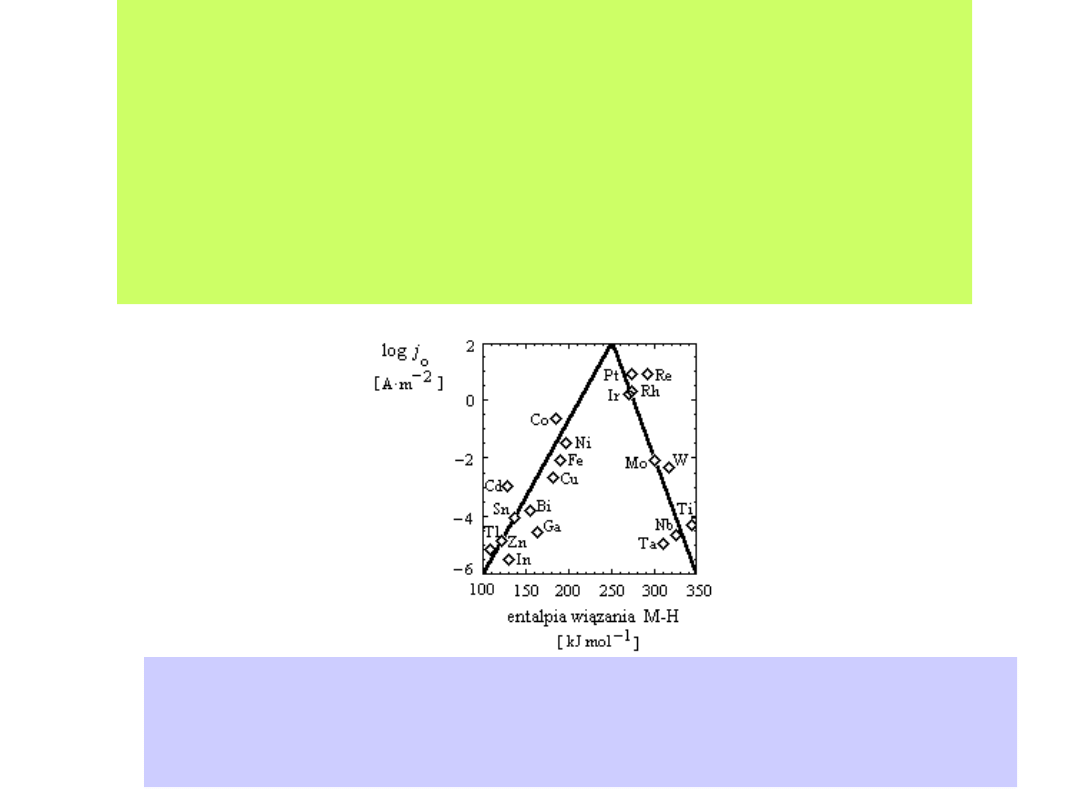
Ilustrują to tzw. krzywe wulkaniczne zaproponowane przez
Trasattiego
Wartości gęstości prądu wydzielania wodoru na różnych metalach z wodnych
roztworów elektrolitów w zależności od entalpii wiązania metal - wodór (kJ mol-1).
Wykres wykonany na podstawie pracy J. Trasatti, J. Electroanal. Chem., 39 (1972),
163

W przypadku metali o niskiej energii adsorpcji wodoru, jak Pb, Ag, Hg,
proces wydzielania wodoru jest limitowany przez reakcję Volmera.
Metale te charakteryzuje wysoki nadpotencjał wydzielania wodoru,
oczywiście z wodnych roztworów elektrolitów.
Im silniej wodór jest adsorbowany na powierzchni elektrody (np. Pt,
Ir, Rh, W), tym szybciej przebiega reakcja Volmera, a szybkość
wydzielania wodoru jest limitowana przez przebieg reakcji
Heyrovskiego lub reakcji Tafela. Elektrody takie charakteryzują się
niskim nadpotencjałem wydzielania wodoru.
We współczesnej technologii i w poszukiwaniach alternatywnych
źródeł energii stosowanie metali, ich stopów i tzw. kompozytów ma
wielkie znaczenie dla otrzymywania, magazynowania lub przesyłania
gazowego wodoru. Omawiane jest to w wykładzie o alternatywnych
źródłach energii.

7. ANODOWE WYDZIELANIE TLENU
Powróćmy do wielokrotnie sygnalizowanego procesu elektrodowego
wydzielania tlenu. Anodowe wydzielanie tlenu jest - mimo pozorów –
procesem wysoce złożonym.
Najlepiej został zbadany ten proces przebiegający na elektrodach z
metali szlachetnych Au, Pt, Rh). Reakcja przebiega z dużym
nadpotencjałem, spełniając równanie Tafela w zakresie potencjałów od
1,2 do 2,0 V
she
.
Etapem limitującym szybkość reakcji wydzielania tlenu jest
prawdopodobnie adsorpcja jonu hydroksylowego lub cząsteczki wody na
powierzchni metalu pokrytej tlenkami:
OH
-
ads
rds OH
ads
+ e (35)
lub H
2
O
ads
rds OH
ads
+ H
+
+ e (36)
Po nich następują szybkie reakcje: 2 OH
ads
O
ads
+ H
2
O (37)
2 O
ads
O
2
(38)

8. ANODOWE ROZTWARZANIE METALI
Anodowe utlenianie metali następuje w wyniku:
a)
samorzutnego utleniania (roztwarzania) metalu stanowiącego w
układzie elektrolizera elektrodę dodatnią, znajdującą się
w roztworze elektrolitu;
b) samorzutnego lub kontrolowanego roztwarzania metalu z
wytworzeniem trwałej warstewki pasywnej,
c) korozyjnego roztwarzania metali stanowiących materiał anodowy i
pod wpływem czynników zewnętrznych;
Korozji
, jej rodzajom i sposobom zapobiegania poświęcony jest inny
wykład;
d) kontrolowanego roztwarzania metalu przy niewielkich wartościach
gęstości prądu anodowego (elektropolerowanie), szczególnie
w nowoczesnych
nanotechnologiach i w elektronice;
e) kontrolowanego roztwarzania metalu w warunkach wymuszonego
anodowego prądu o wysokiej gęstości (rzędu kilkuset A /m
2
) i przy
turbulentnej wymianie elektrolitu (zwanego inaczej anodową obróbką
metali - ang. Electromachining.

9. REDUKCJA I UTLENIANIE ZWIĄZKÓW ORGANICZNYCH
ELEKTROSYNTEZA ORGANICZNYCH POŁĄCZEŃ
Pierwszą reakcję elektrodową związku organicznego wykonał
Faraday w XVIII wieku rowadząc elektrolizę roztworu kwasu octowego
dla udowodnienia, że i produkty gazowe (tutaj CO
2
) również są objęte
prawem zachowania masy.
Pierwszą elektrosyntezę przedłużania łańcucha alifatycznego
wykonał Kolbe w (XIX) wieku.
Dzisiaj metody elektroanalizy, elektroredukcji, elektroutleniania i
elektropolimeryzacji związków organicznych stosowane są powszechnie
ze względu na:
1)
selektywność reakcji wybranej grupy funkcyjnej;
2)
czystość produktów elektrosyntezy ze względu na brak produktów
reakcji reduktorów lub utleniaczy chemicznych (oczyszczanie
produktów końcowych);
3)
możliwość prowadzenia procesów elektrodowych w odpowiednio
4)
dobranych rozpuszczalnikach i na dowolnie wybranej elektrodzie.

K O N I E C
DZIĘKUJĘ ZA UWAGĘ
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
- Slide 23
- Slide 24
- Slide 25
- Slide 26
- Slide 27
- Slide 28
- Slide 29
- Slide 30
- Slide 31
- Slide 32
- Slide 33
- Slide 34
- Slide 35
- Slide 36
- Slide 37
- Slide 38
- Slide 39
- Slide 40
- Slide 41
- Slide 42
- Slide 43
- Slide 44
- Slide 45
- Slide 46
- Slide 47
- Slide 48
- Slide 49
- Slide 50
- Slide 51
Wyszukiwarka
Podobne podstrony:
w8 Procesy utleniania i redukcji
Procesy utleniania i redukcji, LICEUM różne, CHEMIA
PROCESY UTLENIANIA I REDUKCJI 2
PROCESY UTLENIANIA I REDUKCJI
W14S-Procesy utlenienia-redukcji, chemia, szkolka
W15 reakcje utlenienia redukcji
5 Reakcje utleniania i redukcji Reakcje w roztworach
CHEM OG, cw 4, REAKCJE UTLENIANIA I REDUKCJI
06 Sprawozdanie z reakcji utleniania i redukcji Naprawiony
Nowoczesne procesy utleniania
Cw10 Reakcje utleniania i redukcji, Polibuda, II semestr, fizyka, FIZA, lab, Chemia laborki, chemia
3.Reakcje utleniania i redukcji, Notatki AWF, Biochemia
3.Reakcje utleniania i redukcji, Zad
więcej podobnych podstron