Choroby genetyczne
spowodowane mutacją
punktową
Małgorzata Łupińska
Choroby wynikające z dziedziczenia
pojedynczych, zmutowanych alleli -
genopatie
Choroby związane z przemianami
aminokwasów aromatycznych
Choroby krwi
Choroby związane z przemianami
cukrów
Choroby związane z zaburzeniami
funkcji OUN
Inne
Choroby związane z
przemianami aminokwasów
aromatycznych:
-
Fenyloketonuria
-
Alkaptonuria
-
Albinizm
Fenyloketonuria
Fenyloketonuria jest chorobą genetyczną spowodowana
mutacją, położonego na
PAH
kodującego
. Choroba
dziedziczy się w
Mutacja
powoduje upośledzenie aktywności enzymatycznej PAH,
który katalizuje przekształcanie w organizmie aminokwasu
fenyloalaniny w inny aminokwas
. We krwi dziecka
chorego na fenyloketonurię zaczyna gromadzić się
fenyloalanina i produkty jej metabolizmu, przy względnym
niedoborze tyrozyny. Na skutek tego po pewnym czasie i
przy braku odpowiedniego leczenia może dojść do
uszkodzenia
.
Objawami nieleczonej choroby są: znacznego stopnia
upośledzenie rozwoju umysłowego
i motorycznego. Niedobór
jest przyczyną występowania jasnej karnacji skóry,
jasnych włosów i niebieskich tęczówek. Poza tym mogą
występować napady drgawkowe (
), hipotonia
mięśniowa, zaburzenia chodu, postawy, zesztywnienie
stawów. Do obrazu chorobowego dołącza charakterystyczny
"mysi" zapach oraz częste występowanie wysypek.
Fenyloketonuria
Przy wczesnym rozpoznaniu choroby (najlepiej zaraz po urodzeniu) i
odpowiednim leczeniu można zapobiec wystąpieniu objawów
choroby, dlatego w trzecim dniu po urodzeniu do 1998 roku
wszystkim noworodkom wykonywano to
, teraz
stosuje się badanie kolorymetryczne, umożliwiający bardziej
precyzyjne oznaczenie stężenia fenyloalaniny we krwi. Inna metodą
stosowaną niekiedy w diagnostyce fenyloketonurii jest badanie
metabolitów fenyloalaniny w moczu. Należy pamiętać, że
zwiększone stężenie fenyloalaniny we krwi może nie pojawić się
przed 3 dniem życia oraz że u wcześniaków stwierdza się
występowanie wyników fałszywie dodatnich (ze względu na
niedojrzałość układów enzymatycznych).
U chorego dziecka najpóźniej w ciągu 2 tygodni od urodzenia
wprowadza się odpowiednią
niskofenyloalaninową. Należy ograniczyć zawartość fenyloalaniny w
pokarmach i zastąpić ją tyrozyną (hydrolizaty kazeiny). Ponieważ
większość białek ma w swym składzie znaczną zawartość
fenyloalaniny jest to dieta niskobiałkowa z dużą ilością tyrozyny.
Powoduje to uniknięcie wzrostu stężenia fenyloalaniny we krwi i co
za tym idzie, wyeliminowanie jej szkodliwego wpływu fenyloalaniny
na tkankę mózgową. Przy rygorystycznym zachowywaniu diety
dziecko będzie rozwijać się prawidłowo, z drugiej strony nie
wprowadzenie lub zbyt późne wprowadzenie leczenia prowadzi do
nieodwracalnego uszkodzenia mózgu.
Fenyloketonuria
U chorego dziecka najpóźniej w ciągu 2 tygodni od
urodzenia wprowadza się odpowiednią
eliminacyjną niskofenyloalaninową. Należy
ograniczyć zawartość fenyloalaniny w pokarmach i
zastąpić ją tyrozyną (hydrolizaty kazeiny).
Ponieważ większość białek ma w swym składzie
znaczną zawartość fenyloalaniny jest to dieta
niskobiałkowa z dużą ilością tyrozyny. Powoduje
to uniknięcie wzrostu stężenia fenyloalaniny we
krwi i co za tym idzie, wyeliminowanie jej
szkodliwego wpływu fenyloalaniny na tkankę
mózgową. Przy rygorystycznym zachowywaniu
diety dziecko będzie rozwijać się prawidłowo, z
drugiej strony nie wprowadzenie lub zbyt późne
wprowadzenie leczenia prowadzi do
nieodwracalnego uszkodzenia mózgu.

Chłopczyk chory na
fenyloketonurię
Alkaptonuria – choroba niebieskich
pieluch
Alkaptonuria- jest to mutacja recesywna;
przyczyną jest zmiana jednego genu; jest to
choroba, która powoduje zaburzenia w procesie
; polega ona na tym, że
określone
oraz
tyrozyna) nie mogą być całkowicie rozłożone;
konsekwencją tego jest to, że w moczu
występuje kwas homogentyzynowy; w dalszej
kolejności kwas ten zaczyna się odkładać w
; nadaje im przy
tym charakterystyczny, brunatny kolor; duża
ilość kwasu homogentyzynowego znajduje się w
moczu, który ciemnieje na powietrzu.
Zapobieganie objawom – dieta uboga w tyrozynę
Gałka oczna
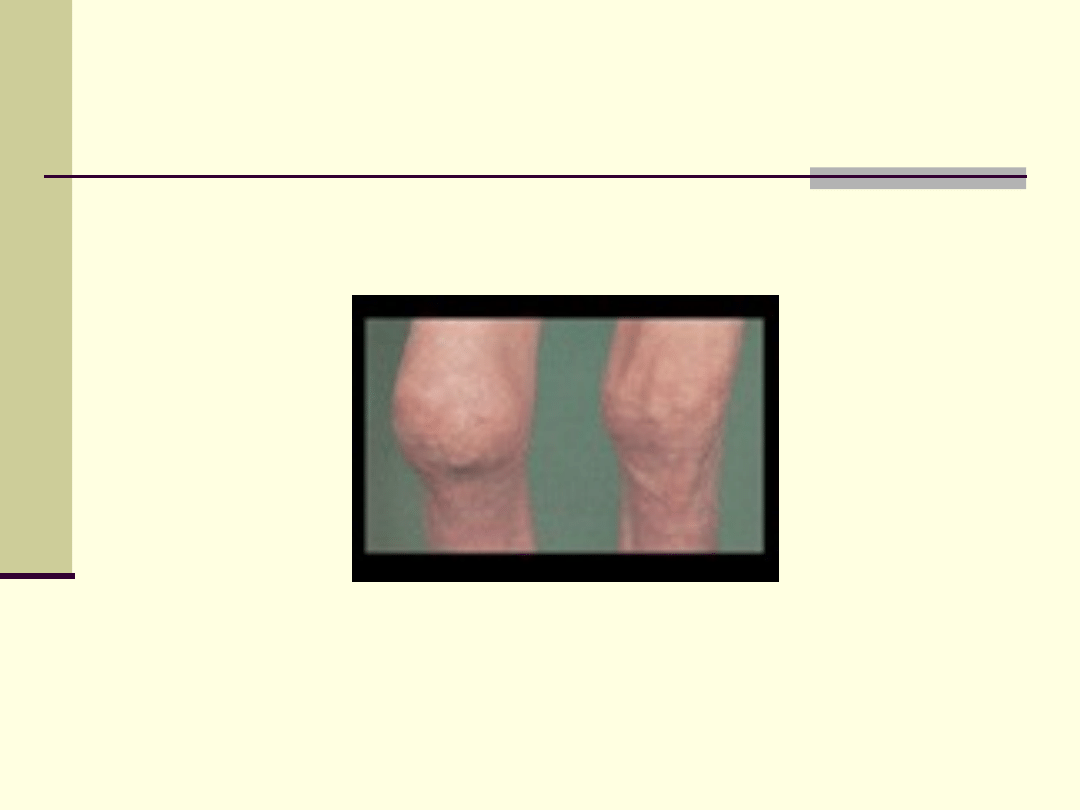
Kolana przy alkaptonurii
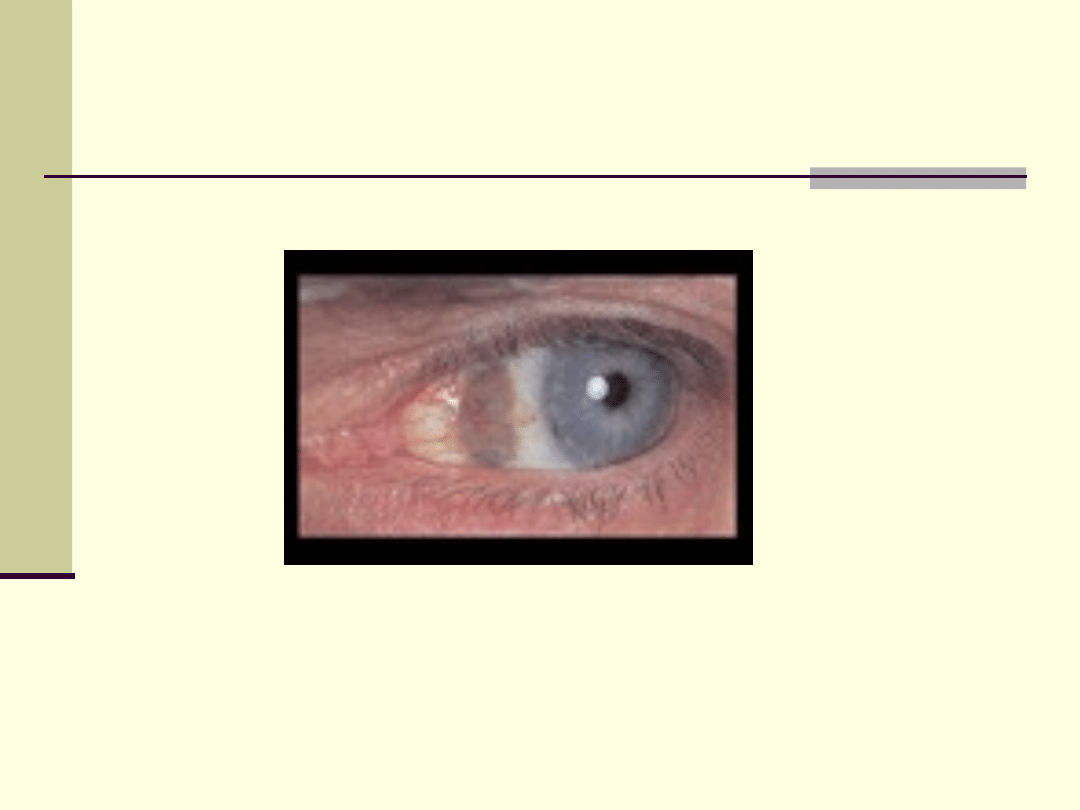
albinizm
Albinizm (bielactwo) - brak
w
, tworach skórnych,
i
(czerwone oczy lub rzadziej niebieskawe).
Osobnicy nie mający pigmentu w skórze, są
wrażliwi na działanie
,
mają bardzo jasną skórę, białe włosy, rzęsy i
brwi. Dziecko z albinizmem rodzi się różowawe,
co jest spowodowane prześwitywaniem
. Tęczówka ich
jest
bezbarwna, przeświecają przez nią naczynia
krwionośne, co nadaje oku czerwoną barwę.
Albinizm wywołany jest przez brak enzymu
przekształcającego prekursor
w barwnik melaninę. Warunkuje go
.

Dziewczynka z albinizmem
Choroby krwi:
-
Hemofilia
-
Anemia sierpowata
Anemia sierpowata
sierpowata- jest to choroba genetyczna;
efekt zamiany kwasu glutaminowego na walinę w
łańcuchu beta globiny; cechuje się tym, że w krwi
występują sierpowate krwinki czerwone; krwinki
tego rodzaju zawierają nieprawidłową
S; hemoglobina ta bardzo łatwo
wytrąca się w krwi żylnej; w konsekwencji wzrasta
ryzyko powstawania zakrzepów i zatorów;
choroba ta znacznie częściej występuje u
Murzynów i Mulatów - głównie w Afryce oraz
Ameryce; W obecnej chwili nie ma możliwości
leczenia przyczyny tej choroby. Jedyne co można
zrobić to transfuzja krwi. Schorzenie to można
rozpoznać około 3 miesiąca życia, na podstawie
mikroskopowego badania krwinek czerwonych
oraz chemicznego badania hemoglobiny.
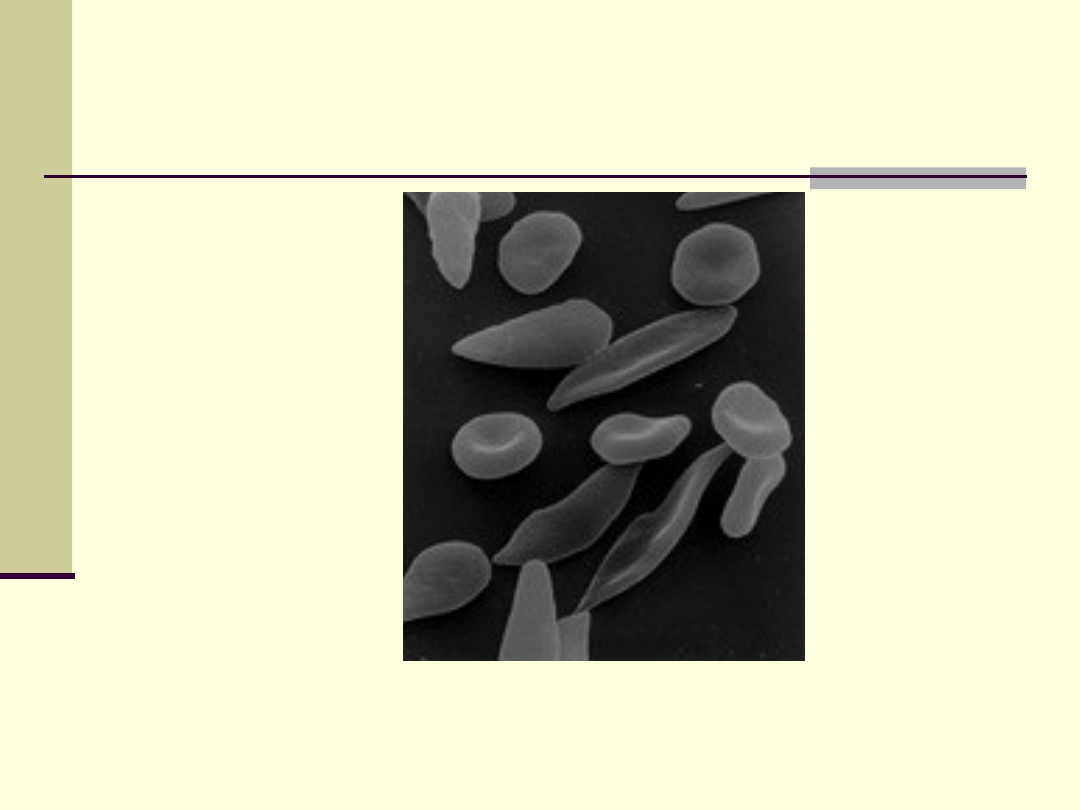
Wygląd erytrocytów
Korzyści niedokrwistości
sierpowatej
Choroba dziedziczy się w sposób autosomalny
(nie jest
) i recesywny, z allelem
kodominującym. Ten rodzaj dziedziczenia, polega
na tym, że nosiciele tylko jednej kopii wadliwego
genu (heterozygoty), w normalnych warunkach
nie mają objawów klinicznych, jednak ich
erytrocyty zawierają ok. 40% HbS.
Heterozygoty są również w dużym stopniu
odporne na
. Zjawisko takie nazywa się
przewagą heterozygot lub
.
Naddominacja powoduje, że na terenach
występowania malarii mutacja powodująca
anemię sierpowatą utrzymuje się w populacji.
Hemofilia
Hemofilia A i B są
.
Gen, którego mutacje powodują chorobę, znajduje
się na
.. Cecha ta dziedziczy się
, co oznacza, iż chorują jedynie osoby z
pełną
Typy hemofilii
- Hemofilia A - niedobór VIII czynnika krzepnięcia
krwi (czynnika antyhemolitycznego), "klasyczna
hemofilia"
- Hemofilia B - niedobór IX czynnika krzepnięcia krwi
(czynnika Christmasa), "choroba Christmasa"
- Hemofilia C - niedobór XI czynnika krzepnięcia krwi
(czynnika Rosenthala), (dotyczy
Aszkenazyjskich, choroba autosomalna recesywna)
Objawy hemofilii
Hemofilia objawia się występowaniem
, nawet po nieznacznych
(np.
usunięcie zęba) lub skaleczeniach, wylewami i
krwawieniami dostawowymi oraz do tkanek
miękkich. Wylewy dostawowe prowadzą do
przykurczów i zniekształceń
.
U chorych na hemofilię krwawienie może
pojawić się w każdym miejscu ciała. Czasem
jest ono widoczne na zewnątrz, a czasem nie.
Do krwawienia może dojść w wyniku urazu lub
zabiegu chirurgicznego. Czasem trudno jest
znaleźć jego uchwytną przyczynę.

Erytrocyty hemofilityka
Choroby związane z przemianami
cukrów
- galaktozemia
galaktozemia
Galaktozemia jest
(dziedziczoną w sposób
) objawiającą się niemożnością
przekształcenia
w
.
większości przypadków spowodowana jest brakiem
transferazy galaktozo-1-fosforanowej - z tej przyczyny osoby chore
nie mogą
galaktozy. Gromadząca się galaktoza
ulega redukcji do
- toksycznej substancji, której
podwyższony poziom prowadzi do tragicznych dla
konsekwencji.
Dzieci dotknięte galaktozemią źle się rozwijają, po spożyciu
wymiotują i cierpią na biegunkę, często mają powiększoną
, chorują na
. Zachodzi też możliwość rozwoju
i przedwczesnego
,
wywołanego uszkodzeniem wątroby.
Leczenie choroby polega na wczesnym wykluczeniu galaktozy z
, co powoduje cofnięcie się wszystkich objawów za wyjątkiem
opóźnienia w rozwoju umysłowym, które jest nieodwracalne.
Jednakże nawet pomimo właściwej diety zastosowanej przed
ukończeniem pierwszego miesiąca życia dziecka często dochodzi
do zaburzeń neurologicznych i obniżenia
.

Dziecko chore na
galaktozemię
Choroby związane z
zaburzeniami funkcji OUN
- Pląsawica Huntingtona
- Choroba Taya - Sachsa
Pląsawica Huntingtona
Pląsawica Huntingtona (choroba Huntingtona,
historyczna nazwa - taniec
-
przyczyną choroby jest
w
HD
kodującym
, położonym na
4. Objawami choroby są
niekontrolowane ruchy oraz upośledzenie
intelektualne. Nasilenie objawów postępuje w
czasie. Allel warunkujący pląsawicę Huntingtona
jest dominujący. Choroba występuje w Polsce u 1
na 15 000 osób. Objawia się między 35 a 50
rokiem życia. Polega na skurczu mięśni, silnych
bólach nóg i rąk oraz zaburzeniach umysłowych.
Choroba Taya - Sachsa
Wywołana jest przez recesywny gen
autosomalny. Objawia się uszkodzeniem
OUN, upośledzeniem umysłowym i
ślepotą. Najczęściej dzieci umierają
przed ukończeniem piątego roku życia.
Jej przyczyną jest brak enzymu
rozkładającego zbędne lipidy błony
neuronów w OUN
Inne choroby
-
Daltonizm
-
Mukowiscydoza
-
Chroba Duchenne`a
Daltonizm
Ślepota barw - zwana też zaburzeniem
rozpoznawania barw, u ludzi objawia się
niezdolnością do spostrzegania różnic pomiędzy
niektórymi lub wszystkimi barwami, które
normalnie są dostrzegane przez ludzi,
uwarunkowaną genetycznie, dziedziczoną
recesywnie w sprzężeniu z chromosomem X. Z
tego też powodu znacznie częściej dotyczy
mężczyzn (ok 1,5%) niż kobiet (ok. 0,5%). Ślepota
barw zwana jest też potocznie daltonizmem
(który tak naprawdę jest wadą polegająca na
nierozpoznawaniu barwy czerwonej i zielonej) –
nazwa ta pochodzi od angielskiego chemika Johna
Daltona, który w 1794 roku na własnym
przypadku opublikował jej opis.
Mukowiscydoza
Mukowiscydoza, inna nazwa
zwłóknienie torbielowate, choroba
ogólnoustrojowa o różnorodnej ekspresji
klinicznej. W klasycznej (pełnoobjawowej)
postaci objawia się skłonnością do
zapalenia oskrzeli i płuc, niewydolnością
części zewnątrzwydzielniczej trzustki,
niepłodnością mężczyzn oraz
podwyższonym stężeniem chlorków w
pocie.

Mukowiscydoza
W Wielkiej Brytanii każdy noworodek jest już objęty
badaniem przesiewowym w celu wykrycia takich chorób
jak fenyloketonuria i wrodzona niedoczynność tarczycy,
które występują rzadziej niż mukowiscydoza. Badanie to
polega na analizie krwi pobranej z nakłutej skóry na
pięcie dziecka. Ta sama kropla krwi mogłaby być
wykorzystana do badania w kierunku mukowiscydozy.
Mukowiscydoza
Jedna z najczęściej występujących
chorób genetycznych w rasie białej
( 1/2500 urodzeń).
Recesywny gen wywołujący tę chorobę
znajduje się w 7 chromosomie
Większość chorych umiera przed
ukończeniem 30 roku życia
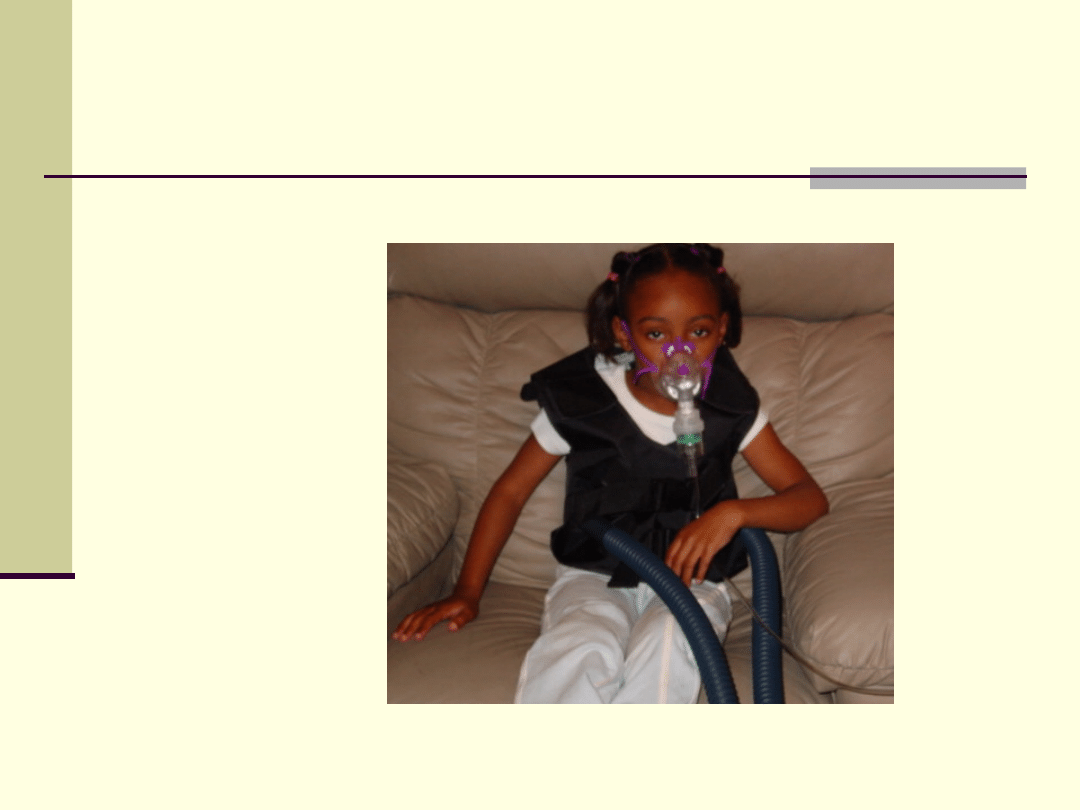
Dziewczynka chora na
mukowiscydozę
Choroba Duchenne`a
Dystrofia mięśniowa Duchenne’a to najczęstsza
dziedziczona recesywnie choroba sprzężona z
płcią. Występuje w jednym na 3300–3500 urodzeń
chłopców.
Choroba uwarunkowana jest allelem recesywnym
położonym na chromosomie X. Są to różne
mutacje genu kodującego dystrofinę. U chorych
zazwyczaj stwierdza się delecje jednego lub
więcej egzonów (60%), duplikacje (5-10%) i
mutacje punktowe (30-35%). Dystrofina wraz z
kompleksem innych białek łączy włókna aktynowe
z błoną komórkową, a także bierze udział w
przekazywaniu sygnałów w komórkach. Jej brak
powoduje uszkodzenia błony komórkowej i
nekrozę komórek mięśniowych. W DMD mutacje
powodują przesunięcie ramki odczytu i całkowity
brak dystrofiny w mięśniach,
Choroba Duchenne`a
Objawy
Choroba ma charakter postępujący. Pierwsze
objawy występują w wieku 3-8 lat. Obejmują
opóźniony rozwoju ruchowy, kaczkowaty chód i
kłopoty z bieganiem oraz chodzeniem po
schodach. Chorzy przy wstawaniu pomagają sobie
rękami.
W wieku 12 lat większość chorych nie jest już w
stanie samodzielnie chodzić. W niektórych
przypadkach chorobie towarzyszy upośledzenie
umysłowe. Często występuje kardiomiopatia.
Większość chorych umiera przed 30 rokiem życia,
najczęściej w wyniku niewydolności oddechowej
lub niewydolności krążenia.
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
- Slide 23
- Slide 24
- Slide 25
- Slide 26
- Slide 27
- Slide 28
- Slide 29
- Slide 30
- Slide 31
- Slide 32
- Slide 33
Wyszukiwarka
Podobne podstrony:
Choroby genetyczne spowodowane aberacjami chromosomowymi 2
Choroby genetyczne spowodowane aberacjami chromosomowymi
Choroby genetyczne. Mutacje punktowe, Biologia
więcej podobnych podstron