
NOWOTWORY
TKANEK MIĘKKICH I KOŚCI

Tkanki miękkie - struktury układu ruchu oraz
tkanek
podporowych trzewi i narządów miąższowych z
wyłączeniem układu szkieletowego
Stanowią ponad 50% ciężaru ciała
Mięsaki tkanek miękkich (MTM) stanowią 1%
wszystkich rejestrowanych nowotworów złośliwych
(ok.7% nowotworów u dzieci <15 rż)
W 1999 roku w Polsce 604 nowe zachorowania
(293 M + 302 K)
i 154 zgony z powodu MTM
(79 M i 75 K)

Mięsaki pochodzą głównie z mezodermy
Tkanki powstałe z mezodermy:
kości
tkanka łączna
opłucna
chrząstka
(włóknista i maziowa)
otrzewna
mięśnie
naczynia krwionośne
osierdzie

Etiologia: większości MTM nie jest znana
Czynniki predysponujące:
przewlekły obrzęk chłonny (np. po operacji Pateya i
radioterapii w raku piersi) – lymphangiosarcoma
tkanki uprzednio napromieniane (dawka 40 Gy) -
angiosarcoma, fibrohistiocytoma malignum (MFH),
fibrosarcoma, lymphangiosarcoma
ciało obce (dakronowe protezy naczyniowe, metal
stabilizujacy złamanie kości) - MFH, fibrosarcoma
Thorotrast (kontrast dawniej stosowany w
radiologii)

Genetic Predisposition to Soft Tissue Sarcoma
--------------------------------------------------------------------------------------------------------------
-
Sarcoma
Gene
Chromosome
Neurofibromatosis type I
Malignant peripheral
NF-1
17q11.2
(von Recklinghausen's
nerve sheath tumor
disease)
Retinoblastoma Soft tissue, osteogenic
Rb-1
13q14
Li-Fraumeni syndrome
Soft tissue, osteogenic
TP53
17p13
Gardner's syndrome
Fibrosarcoma, desmoid
APC
5q21
tumor
Werner's syndrome
Soft tissue
WRN 8p12
(adult progeria)
Gorlin's syndrome (nevoid
Fibrosarcoma, PTC
9q22.3
basal cell carcinoma
rhabdomyosarcoma
syndrome)
Carney's triad Gastrointestinal stromal
Unknown
Unknown
tumor

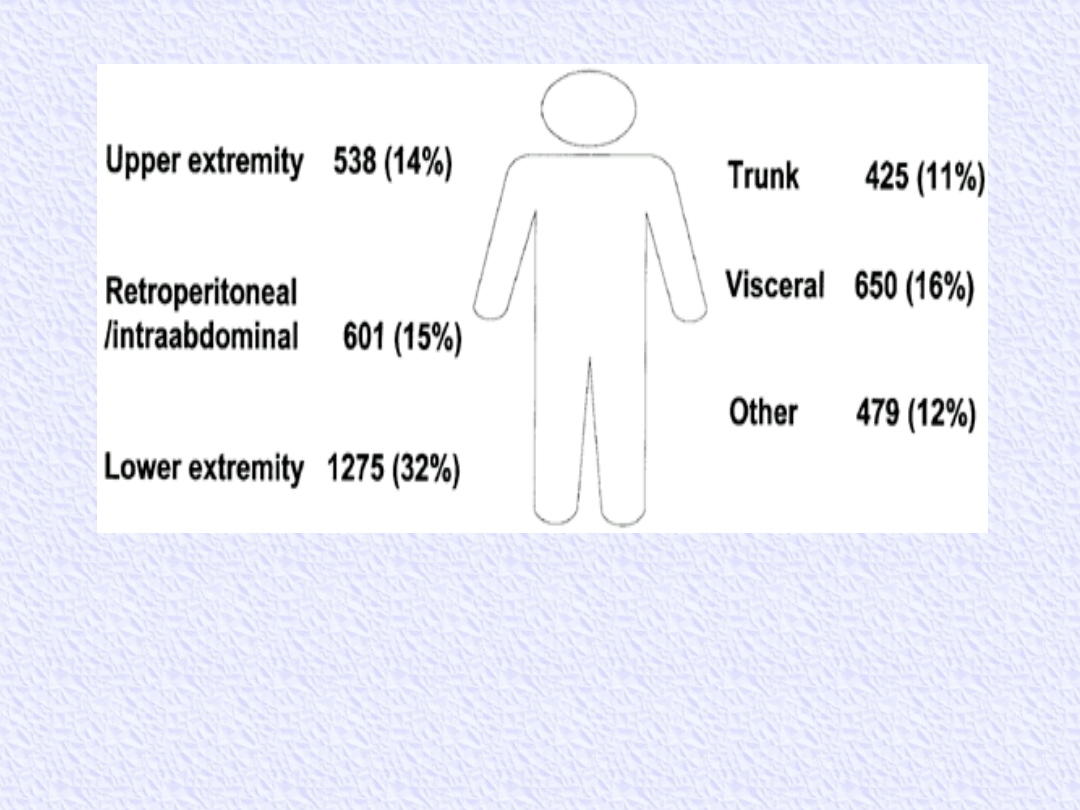
Distribution by site of soft tissue sarcomas in 3968 patients aged 16 or
older admitted to Memorial Sloan-Kettering Cancer Center between July
1982 and July 1999.
(ok. 50% mięsaków rozwija się na kończynach)

Objawy kliniczne MTM
Niebolesny guz, czasami rozlane zgrubienie o nieostrych
granicach
Szybki wzrost lub przyspieszenie wzrostu guza uprzednio
obserwowanego przez wiele miesięcy
Położenie z reguły podpowięziowe
Wielkość często powyżej 5 cm
Pojawienie się bólu/ parestezji (objaw późny)
Objawy paranowotworowe (hipoglikemia –
fibrosarcoma)

Historia naturalna i drogi szerzenia
•szczególna skłonność do szerzenia się
miejscowego wzdłuż struktur
anatomicznych (mięśnie, nerwy..),
wskutek czego rzeczywisty zasięg guza
zwykle przekracza widoczne jego
granice
•skłonność do dawania wczesnych
przerzutów odległych głównie do płuc
•przerzuty do okolicznych węzłów
chłonnych występują w przebiegu –
synovioma malignum i
rhabdomyosarcoma
•różne mięsaki cechuje różny przebieg

Historia naturalna
•Stopień złośliwości histologicznej ma w
MTM istotne znaczenie kliniczne
•Drugorzędowe znaczenie ma wielkość
guza
•U 80-100% chorych po prostym
wycięciu guza dochodzi do nawrotu
miejscowego
•U większości chorych nawrót miejscowy
współistnieje z rozsiewem.
•Wyleczenie chorych z przerzutami jest
rzadkością.
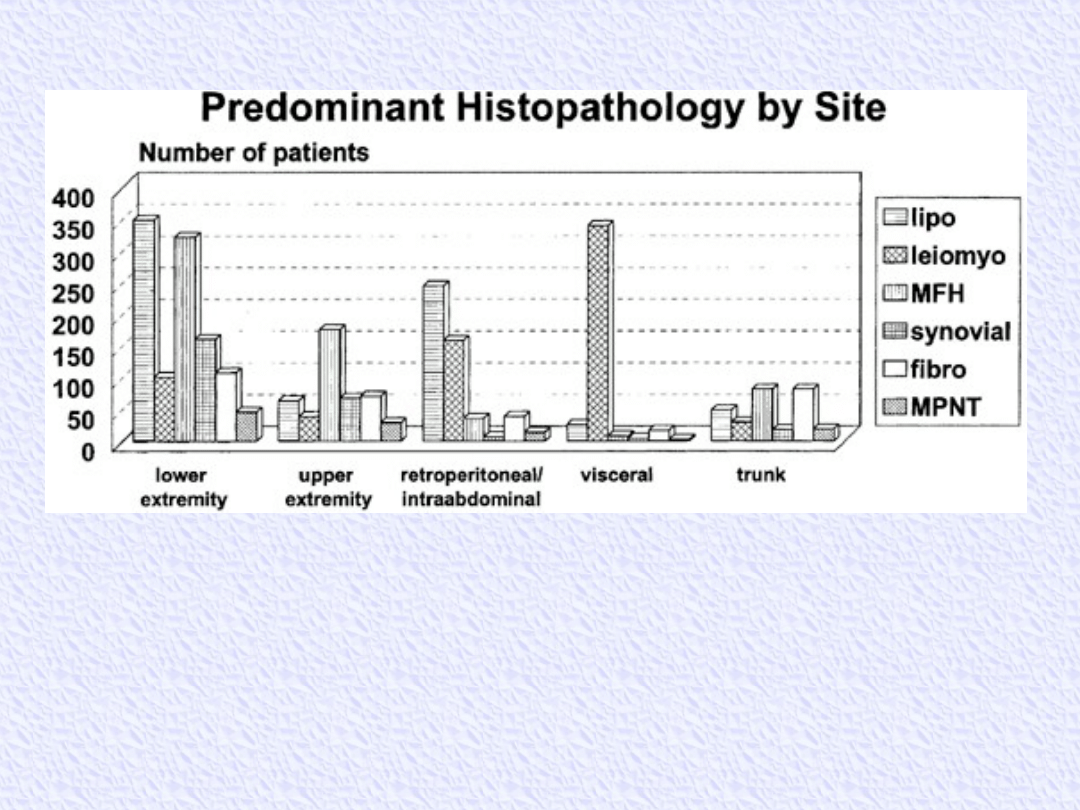
Predominant histology by site of soft tissue sarcomas in
3968 patients aged 16 or older admitted to Memorial Sloan-
Kettering Cancer Center between July 1982 and July 1999.
MFH, malignant fibroushistiocytoma; MPNT, malignant
peripheral nerve tumor.

Niekorzystne czynniki rokownicze w
MTM
•przerzuty odległe i do węzłów
chłonnych
•wysoki stopień złośliwości
histologicznej
•wielkość guza nowotworowego powyżej
5 cm
•naciek i/lub owrzodzenie skóry
•nieradykalny zabieg chirurgiczny

IA
G1-G2
T1A-1B
N0
M0
IB
G1-G2
T2A
N0
M0
IIA
G1-G2
T2B
N0
M0
IIB
G3-G4
T1A-1B
N0
M0
IIC
G3-G4
T2A
N0
M0
III
G3-G4
T2B
N0
M0
IVA
Każde G
Każde T
N1
M0
IVB Każde G
Każde T
Każde N
M1
G1 - dobrze zróżnicowany
G2 - średnio zróżnicowany
G3 - nisko zróżnicowany
G4 - niezróżnicowany
Stopnie zaawansowania klinicznego MTM
A - położenie
powierzchowne
(nadpowięziowe)
B - położenie głębokie
(podpowięziowe)
T1 - <= 5 cm
T2 > 5 cm
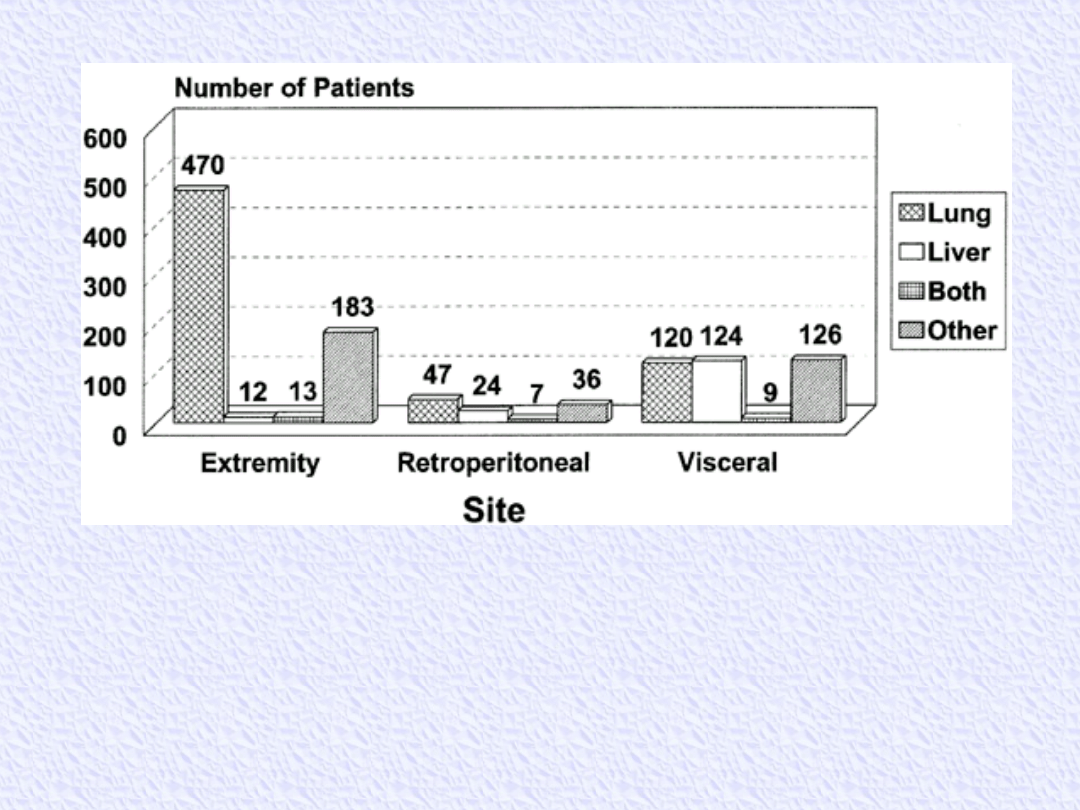
Common sites of metastasis that can guide investigation (from
1171 patients admitted to Memorial Sloan-Kettering Cancer
Center between July 1982 and July 1999). The primary site of
metastasis for extremity lesions is the lung.

Dignostyka MTM
USG
Biopsja “otwarta” – w miejscu cięcia
podczas planowego zabiegu
BAC
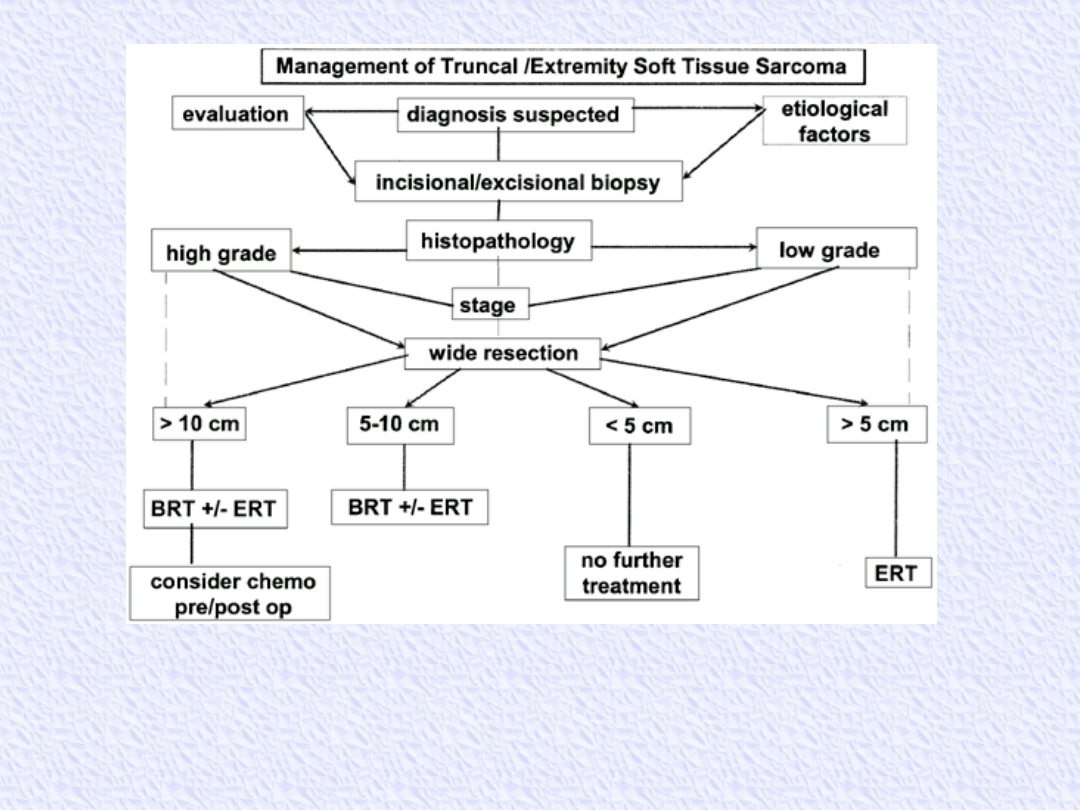
Management algorithm for extremity and superficial truncal
soft tissue sarcoma. BRT, brachytherapy; ERT, external-beam
radiation therapy.

MTM kończyn i tułowia
Leczenie chirurgiczne - najskuteczniejsze
operacja radykalna - wycięcie w jednym bloku z
guzem całego przedziału mięśniowego wraz z
pęczkami naczyniowo-nerwowymi
operacja z szerokim marginesem - celem jest
uzyskanie min. 2 cm zdrowej tkanki we
wszystkich kierunkach (coraz częściej
stosowana)
amputacja kończyny - obecnie tylko u 5%
pacjentów (u ok.. 95% możliwe operacje
oszczędzające kończynę)
Guzy poniżej 5 cm - całkowite wycięcie
chirurgiczne jest wystarczające, leczenie
uzupełniające w przypadku wznowy

Wskazania do amputacji
•naciek MTM na kość, główne naczynia krwionośne i
pnie nerwowe
•MTM pierwotnie pozaprzedziałowy lub
przekraczający granicę jednego przedziału
mięśniowego
•wznowa MTM po uprzednim radykalnym(np.
przedziałowym) wycięciu i radykalnej radioterapii
•amputacja paliatywna - wykonywana w stanach
zagrożenia życia

Radioterapia
Uzupełniająca pooperacyjna teleradioterapia
•Zmniejsza ryzyko nawrotu miejscowego
•Nie wpływa na całkowite przeżycie
•Odbywa się techniką zmniejszających się pól
•Pole obejmuje lożę po guzie, bliznę i spływ + 5-7
cm margines.
•Dawka 60-70 Gy zależnie od stopnia złośliwości
histologicznej, wielkości guza stanu marginesów i
lokalizacji
•Należy ochronić część obwodu kończyny, aby
uniknąć obrzęku

Radioterapia
Uzupełniająca brachyterapia - zalety
•krótszy czas leczenia
•znaczniejsze oszczędzenie tkanek zdrowych
(szybki spadek dawki z odległością)
•dokładniejsze wyznaczenie obszaru do
napromieniań w czasie zabiegu
•możliwość reirradiacji pozwalająca uniknąć
amputacji przy wznowie
Dawki brachyterapii:
jako samodzielna metoda po zabiegu: 45 Gy/ 4-6 dni
jako boost po teleterapii 15 - 20 Gy (z teleterapii 45 - 50 Gy)

Radioterapia
Teleterapia przedoperacyjna - zalety
•ograniczenie śródoperacyjnego rozsiewu komórek
nowotworowych
•mniejszy obszar napromieniany w porównaniu z
XRT pooperacyjną
•zmniejszenie się guza ułatwia późniejszy zabieg

Chemioterapia
Chemioterapia uzupełniająca (po
radykalnym leczeniu operacyjnym i
radioterapii)
- nie poprawia kontroli miejscowej, przeżycia
wolnego od nawrotu ani całkowitego przeżycia
Chemioterapia (doksorubicyna, ifosfamid,
dakarbazyna)
- na zastosowanie w leczeniu choroby
zaawansowanej i rozsianej

GIST (Gastrointestinal stromal tumors)
Nowotwory podścieliskowe przewodu
pokarmowego
Specyficzny marker diagnostyczny – c-Kit
(CD117) (ekspresję c-Kit kinazy tyrozynowej
wykazuje ponad 95% GIST)
-uprzednio – guzy mezenchymalne (FibroSa);
chemiooporne
-postaci łagodne, złośliwe
-najczęściej okolica żołądka

GLIVEC (imatinib)
Łączy się z receptorem kinazy
tyrozynowej
c-Kit
- w leczeniu nieoperacyjnych i/lub
przerzutowych GIST
-54% odpowiedzi częściowych
-28% stabilizacji choroby
-mediana czasu do odpowiedzi – 13 tygodni
-mediana czasu trwania odpowiedzi – nie
osiągnięto
Imatinib nie jest ściśle selektywny; hamuje także kinazę
tyrozynową Bcr-Abl oraz blokuje receptor płytkowego
czynnika wzrostu (PDGF), także przemiany komórkowe,
w których uczestniczą dwie powyższe kinazy

Pierwotne nowotwory złośliwe
kości:
W 1999 roku w Polsce
zarejestrowano:
430 nowych zachorowań (247 M +
183 K)
470 zgonów (247 M i 183 K)
Zachorowalność wyższa w
dzieciństwie (mięsak Ewinga; ok.
18%, mięsak kościopochodny; ok.
45%) niz w wieku dorosłym
(chrzęstniakomięsak; ok.15%)

Tkanki wchodzące w skład kości:
•chrzęstna
•kostna
•włóknista
•szpik kostny

Klasyfikacja pierwotnych nowotworów złośliwych kości
A. Osteosarcoma
B. Guzy okrągłokomórkowe
1. Guz Ewinga
2. PNET
C. Chondrosarcoma
D. Chłoniak nieziarniczy kości
E. Złośliwy włókniak histiocytarny
F. Inne:
1. Fibrosarcoma
2. Liposarcoma
3. Złosliwe guzy wielkokomórkowe
4. Haemangioendothelioma

Osteosarcoma - Kostniakomięsak
Jest to najczęstszy pierwotny nowotwór kości
Częstość występowania - 2,1 przyp. na 1 mln rocznie
Głównie dzieci i młodzi dorośli
Szczyt zachorowań przypada na drugą dekadę życia (60%
przyp)
85% pacjentów ma mniej niż 35 lat
U pacjentów powyżej 40 rż najczęściej jakiś stan
poprzedzający:
choroba Pageta
napromienianie kości
mnogie dziedziczne wyrośla kostne
wieloogniskowa dysplazja włóknista kości
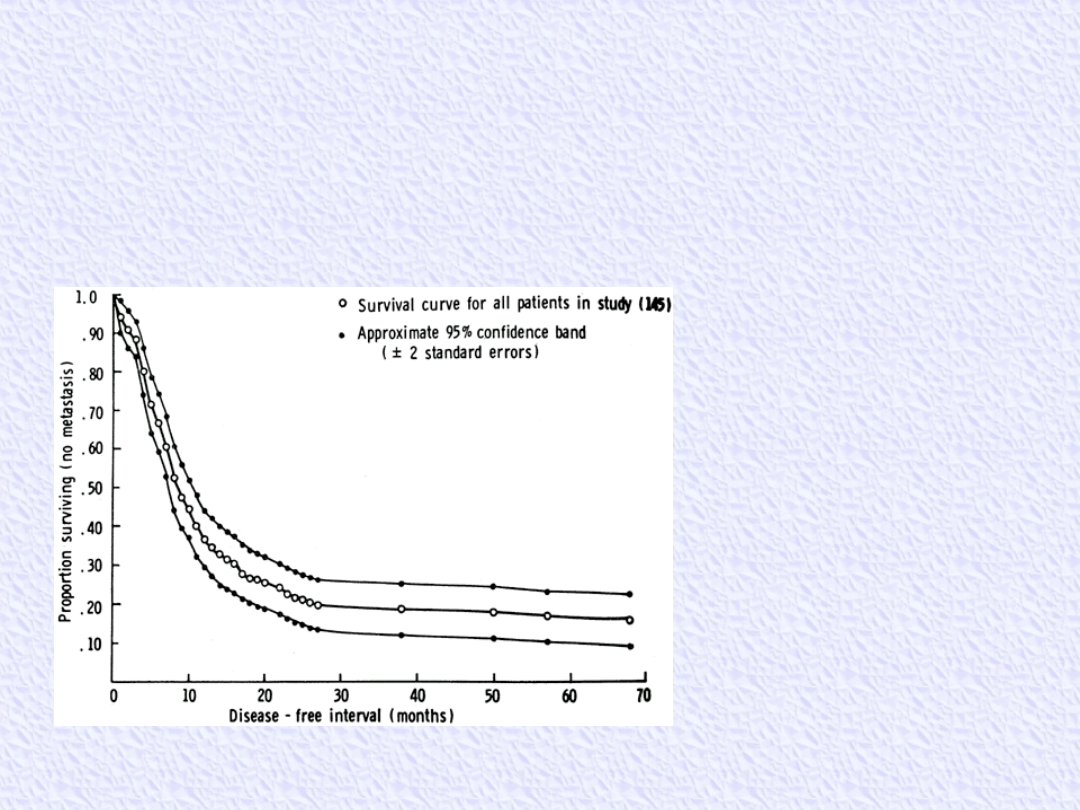
Przed erą chemioterapii uzupełniającej leczeniem stosowanym w
osteosarcoma była amputacja.
Przerzuty do płuc i innych kości pojawiały się najczęściej w okresie do
24 miesięcy
Całkowite przeżycie 2 lat wynosiło od 5% do 20%.
The historical survival curve
for 145 patients with
osteosarcoma treated by
surgery alone at Memorial
Sloan-Kettering Cancer
Center as reported by
Marcove and associates.
(From Marcove RC, Mike V,
Hajek JV, et al. Osteogenic
sarcoma under the age of
21. J Bone Joint Surg Am
1966;48:1, with
permission.)

Czynniki prognostyczne:
•lokalizacja nowotworu (guzy miednicy i szkieletu
osiowego rokowały gorzej, prawdopodobnie ze
względu na niedostępność i niecałkowite usunięcie
chirurgiczne, a także wyższy stopień
zaawansowania)
•wielkość guza
•martwica w wyniku przedoperacyjnej
chemioterapii
•podwyższony poziom FALK przed leczeniem

Zarys leczenia
Choroba zlokalizowana dotycząca kończyn
Leczenie przez wykwalifikowany zespół
wielodyscyplinarny
1. Podejrzenie kostniakomięsaka --> biopsja
wykonana przez ortopedę, specjalizującego się w
nowotworach kości
2. Leczenie chirurgiczne - wybór między amputacją a
leczeniem oszczędzającym
3. Chemioterapia - indukcyjna lub uzupełniająca
(aktywne leki - ADM, DDP, hdMTX, VCR)
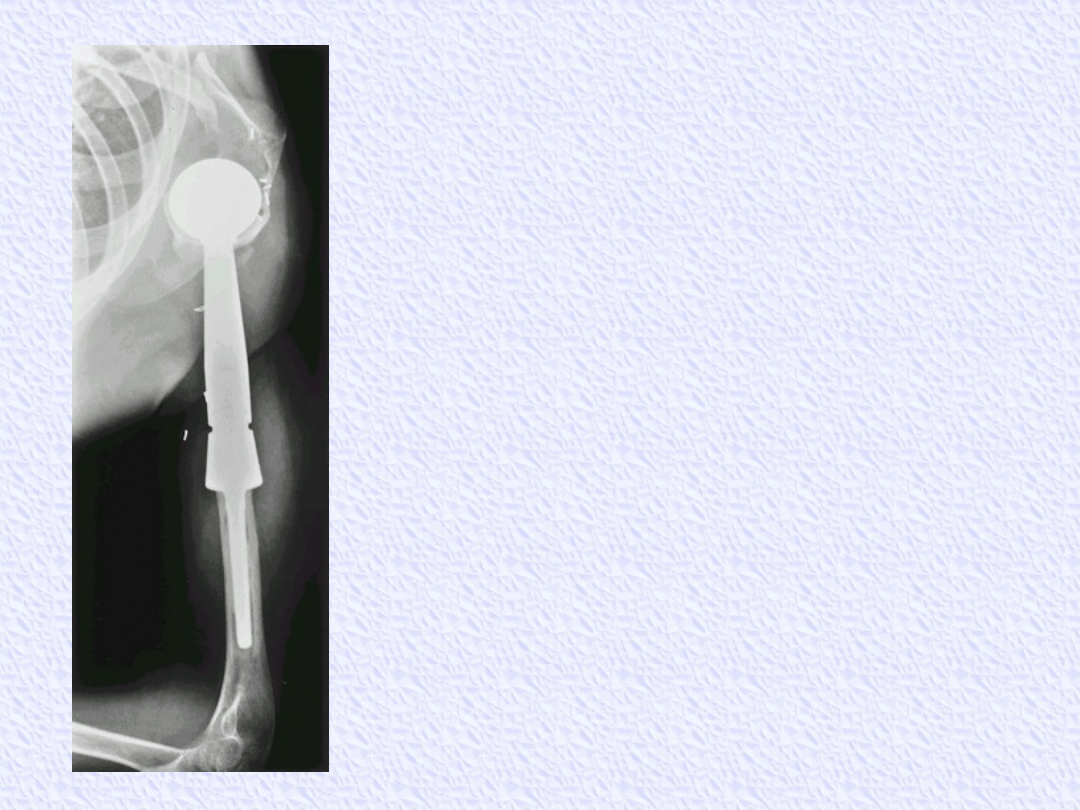
Osteosarcoma of the proximal
humerus. This patient
was placed in a shoulder splint
and given three cycles of
chemotherapy
in the hope of avoiding a
forequarter amputation. Due to
the good
clinical and radiographic
response, this patient
underwent a limb-sparing
resection (type V).
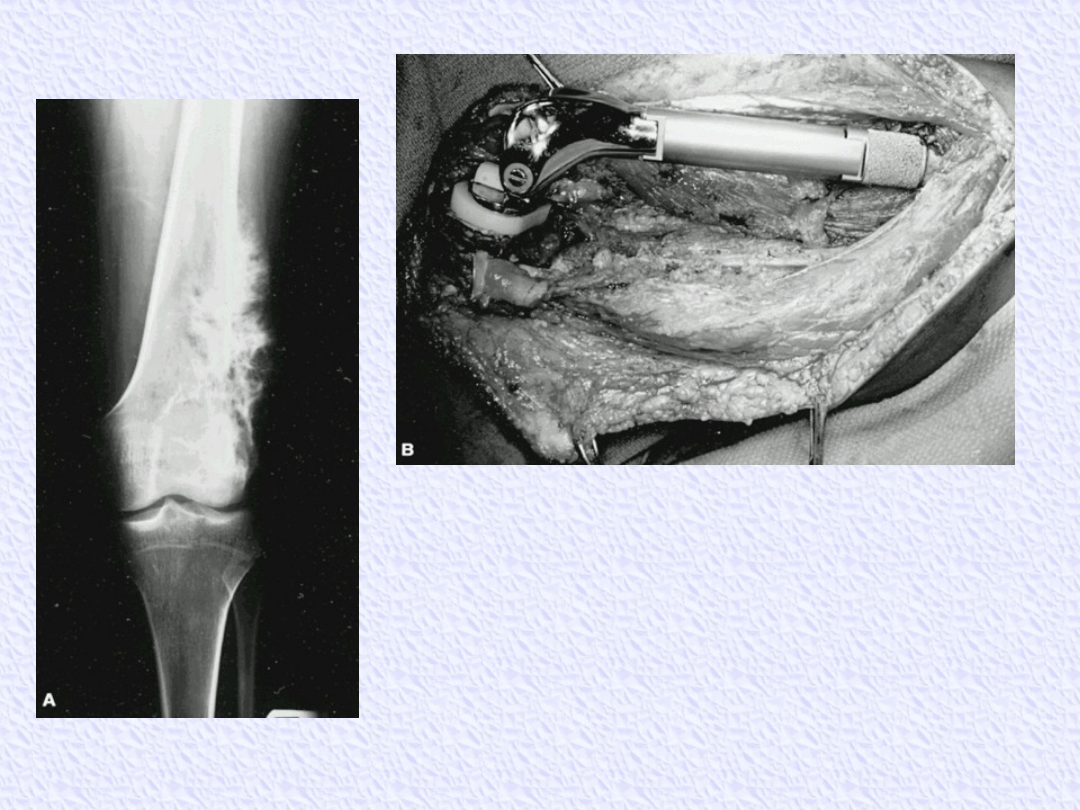
Osteosarcoma of the distal femur treated
by limb-sparing resection.
A: Plain radiograph of a distal femoral
osteosarcoma. B: Intraoperative
photograph shows a modular distal
femoral prosthesis.

Obecność przerzutów odległych przy rozpoznaniu
wiąże się ze złym rokowaniem, zwłaszcza przy
przerzutach pozapłucnych
Podejmuje się leczenie polegające na leczeniu
chirurgicznym guza pierwotnego oraz przerzutów i
chemioterapii w różnych kombinacjach (indukcyjna,
uzupełniająca, lub naprzemiennie z leczeniem
chirurgicznym)

Rola radioterapii w leczeniu
osteosarcoma
* guzy nieresekcyjne
* leczenie paliatywne (guzy nieresekcyjne np., w
kręgosłupie lub w miednicy; przerzuty do kości)

Chrzęstniakomięsak - chondrosarcoma
Drugi co do częstości złośliwy nowotwór kości
1/2 wszystkich chrzęstniakomięsaków powyżej 40 rż
Najczęstsze lokalizacje:
31% miednica
21% kość udowa
13% obręcz barkowa

Chondrosarcoma - czynniki prognostyczne
•stopień złośliwości histologicznej
•wielkość guza
•wiek - guzy u dzieci rokują gorzej niż u dorosłych
•lokalizacja (rokowanie lepsze dla guzów kości długich)
•stopień zaawansowania klinicznego

Leczenie kostniakomięsaków
Przede wszystkim chirurgiczne – radykalna
resekcja, techniki jak w osteosarcoma
Technika kriochirurgiczna polegająca na
wyłyżeczkowaniu guza i krioterapii pozostałej jamy
przy użyciu ciekłego azotu
Radioterapia -
w przypadkach guzów nieoperacyjnych bądź
nieresekcyjnych, zwłaszcza szkieletu osiowego,
obręczy barkowej i biodrowej, kości twarzy i czaszki
w leczeniu paliatywnym

Włókniakomięsak
* Rozwija się z włóknistych struktur kości i nie
wykazuje objawów wytwarzania tkanki kostnej lub
okostnawej.
* Osoby starsze
* Kości długie
* Moga rozwijać sie na podłożu ch. Pageta i w
następstwie RT
* Leczenie chirurgiczne – doszczętne wycięcie guza
* Rokowanie zależy od G i umiejscowienia
nowotworu (przeżycie u ok 30% chorych)

NOWOTWORY
OŚRODKOWEGO UKŁADU NERWOWEGO

Epidemiologia
Zachorowania i zgony w 1999
•na nowotwory mózgu
2559 nowych zachorowań (1320 M + 1239
K)
2099 zgonów (1320 M + 1239 K)
•na nowotwory rdzenia kręgowego, nerwów
czaszkowych i innych części OUN
123 nowe zachorowania (64 M + 59 K)
98 zgonów (74 M + 24 K)
•nowotwory opon mózgowych
69 nowych zachorowań (29 M + 40 K)
30 zgonów (14 M + 16 K)

Nowotwory OUN stanowia ok. 2-5%
nowotworów
Częściej u osób rasy białej
Wiek zachorowania - 2 szczyty 5-9 rż i 50-55
rż
Glejaki stanowią najczęstsze guzy lite przed
okresem dojrzewania
Medulloblastoma występuje znacznie częściej
u M, oponiaki u K
Niektóre występują rodzinnie (naczyniak
płodowy - haemangioblastoma)
Etiologia guzów mózgu nie jest znana

Klasyfikacja histopatologiczna WHO guzów mózgu
I. Nowotwory pochodzenia neuroepitelialnego
II. Nowotwory nerwów czaszkowych i rdzeniowych
nerwiak osłonkowy, schwannoma
nerwiakowłókniak
złośliwy nowotwór osłonek nerwów obwodowych
III. Nowotwory opon
IV. Chłoniak i nowotwory układu krwiotwórczego
(chłoniaki złośliwe, guz plazmatycznokomórkowy, mięsak szpikowy granulocytarny)
V. Nowotwory z pierwotnych komórek rozrodczych
(zarodczak, rak zarodkowy, guz zatoki endodermalnej,
rak kosmówkowy, potworniak)
VI. Torbiele i zmiany nowotworopodobne
VII. Nowotwory okolicy siodła tureckiego
gruczolak przysadki
rak przysadki
przewodziak czaszkogardłowy
VIII. Nowotwory rozprzestrzeniające się przez ciągłość
(
przyzwojak, struniak, chrzęstniak, rak)
IX. Nowotwory przerzutowe
X. Nowotwory niesklasyfikowane

I. Nowotwory pochodzenia
neuroepitelialnego
A. Pochodzenia astrocytarnego (gwiaździaki)
1. Astrocytoma G2
4. Astrocytoma pilocyticum G1
2. Astrocytoma anaplasticum G3
5. Xanthoastrocytoma pleomorphicum G1
3. Glioblastoma G4
6. Astocytoma gigantocellulare subependymale G1
B. Pochodzenia z gleju skąpowypustkowego (skąpodrzewiaki)
1. Oligodendroglioma G1-2
2. Oligodendroglioma anaplasticum
C. Pochodzące z wyściółki
1. Ependymoma G1-2
2. Ependymoma anaplasticum G3
D. Mieszane glejaki
1. Oligoastrocytoma mixtum G2
2. Oligoastrocytoma anaplasticum G3
E. Pochodzące ze splotu naczyniówkowego
1. Brodawczak splotu naczyniówkowego G1
2. Rak splotu naczyniówkowego G3-4
F. Pochodzenia neuroepitelialnego i o niepewnej histogenezie
1. Astroblastoma G3 2. Polar spongioblastoma G4 3. Gliomatosis cerebri G4
G. Pochodzenia neuronalnego i mieszanego neuronalno-glejowego
-
Gangliocytoma
H. Pochodzące z miąższu szyszynki
-
Pineocytoma, Pineoblastoma
I. Guzy zarodkowe
-
Medulloblastoma, Neuroblastoma, Ependymoblastoma, PNET

Table 43.2-1 Classification of Primary Intracranial Tumors by Cell of Origin
---------------------------------------------------------------------------------------------
Normal Cell Tumor
---------------------------------------------------------------------------------------------
Astrocyte
Astrocytomas, glioblastoma multiforme
Ependymocyte
Ependymoma, ependymoblastoma
Oligodendrocyte
Oligodendroglioma
Arachnoidal fibroblasts
Meningioma
Nerve cell or neuroblast retinoblastoma
Ganglioneuroma, neuroblastoma
External granular cell or neuroblast
Medulloblastoma
Schwann cell
Schwannoma (neurinoma)
Melanocyte
Melanotic carcinoma
Chorioid epithelial cell
Choroid plexus papilloma or carcinoma
Pituitary
Adenoma
Endothelial cell or stromal cell
Hemangioblastoma
Primitive germ cells
Germinoma, pinealoma, teratomas,
cholesteatoma
Pineal parenchymal cells
Pinealcytoma
Notochordal remnants
Chordoma
---------------------------------------------------------------------------------------------

Frequency of Primary Intracranial Central Nervous System (CNS) Tumors
--------------------------------------------------------------------------------------------------------
Histopathology
Primary Brain Tumors (%)
Gliomas (%)
--------------------------------------------------------------------------------------------------------
Glioblastoma multiforme
21.7
47
Malignant astrocytomas
16.6
36
All oligodendroglioma
3.1
6.7
All ependymomas
2.3
5.1
Low-grade astrocytoma
1.8
3.9
Meningioma and other
26.7
--
mesenchymal tumors
Pituitary
9.7
--
Nerve sheath (e.g., schwannoma) 7.3
--
CNS lymphoma
3.5
--
Medulloblastoma and other
1.7
--
primitive neuroectodermal tumors
All neuron and neuron/ glial tumors
1.0
--
Craniopharyngioma
1.0
--
Germ cell
0.5
--
Choroid plexus
0.3
--
Other tumors
2.7
--
-------------------------------------------------------------------------------------------------------

Objawy kliniczne - guzy mózgu
Ogólne:
1. Bóle głowy, nudności, wymioty
2. Zmiany osobowości, zaburzenia koncentracji
3. Spowolnienie funkcji psychomotorycznych
4. Wzrost ciśnienia śródczaszkowego
Ogniskowe:
1. Padaczka
2. Zespoły neurologiczne zależne od lokalizacji
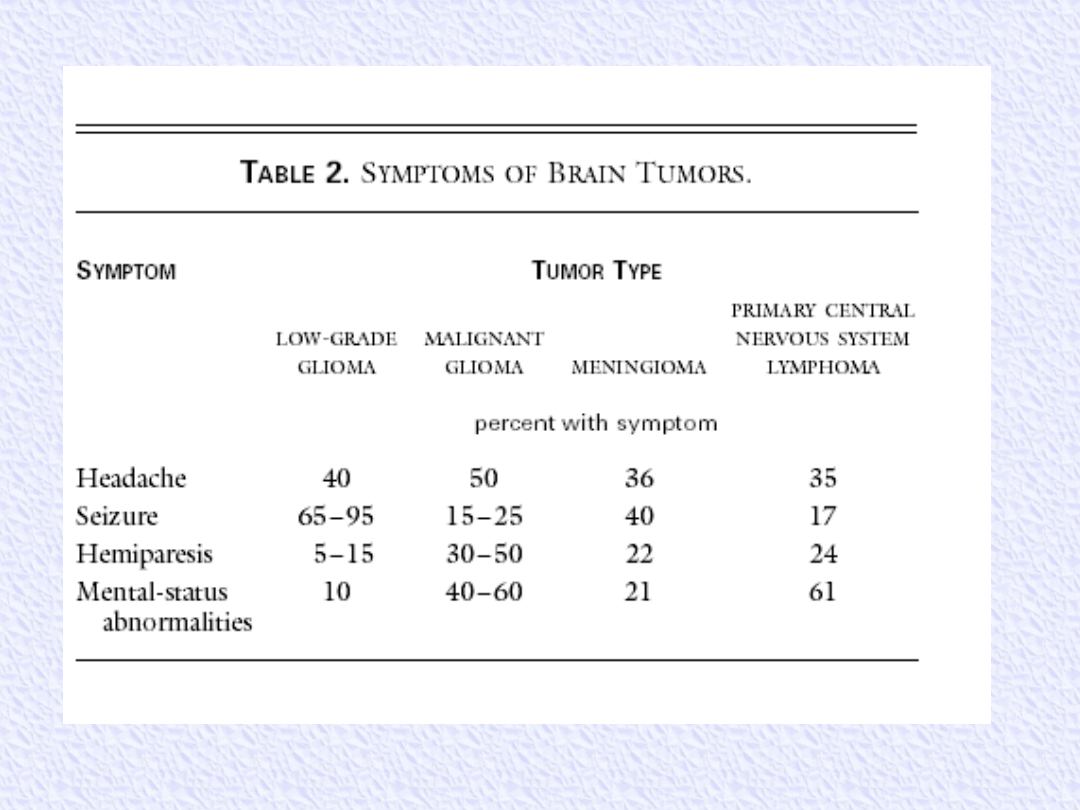
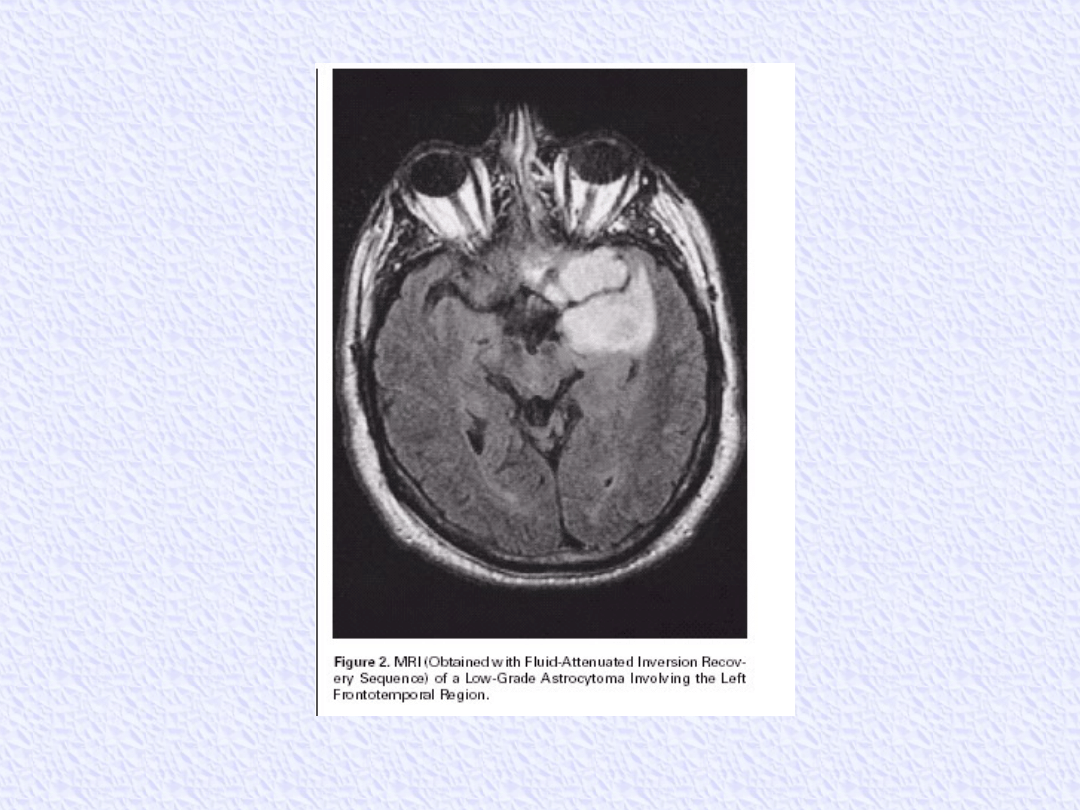
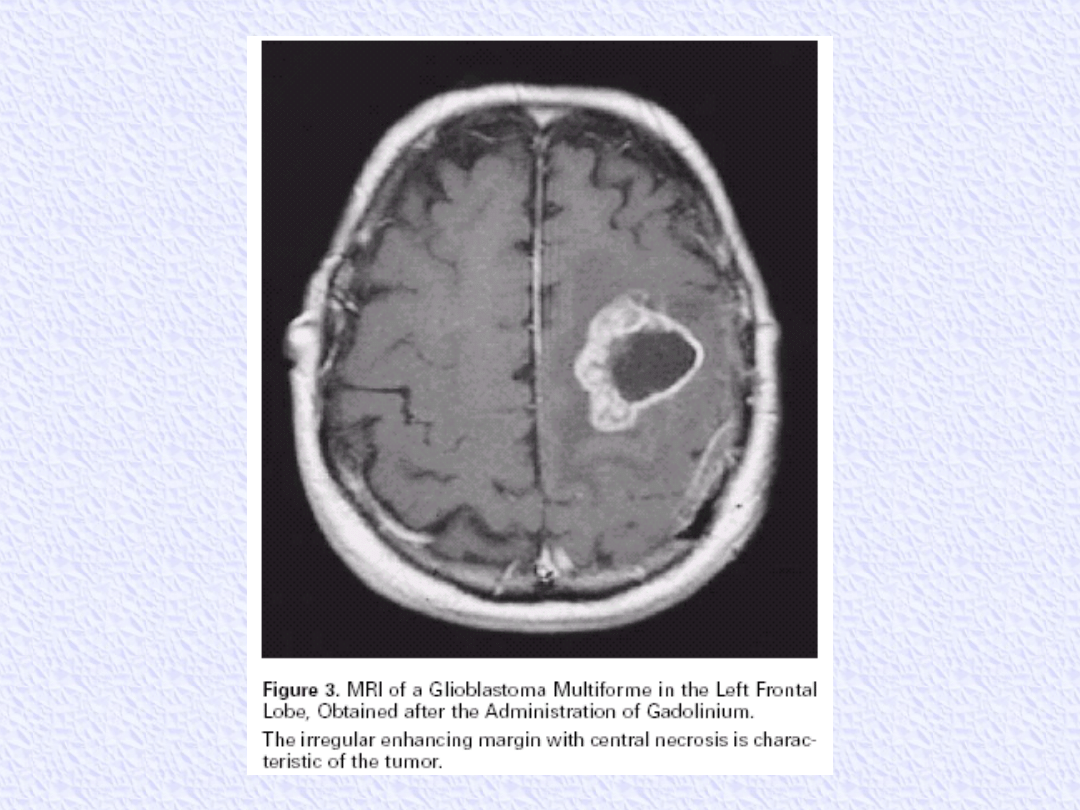

Nowotwory rdzenia kręgowego
Ból w miejscu lokalizacji guza (silniejszy w
nocy, wzmaga się przy kaszlu lub rozciąganiu)
Utrata/zaburzenie funkcji neurologicznych
poniżej miejsca lokalizacji guza

GLIOBLASTOMA MULTIFORME
Jest to najczęstszy pierwotny i najbardziej złośliwy
nowotwór mózgu
Etiologia nieznana
Zwykle gwałtowny przebieg, rokowanie złe
Szczyt zachorowań 5 i 6 dekada życia
Leczenie skojarzone - chirurgia + uzupełniająca
radioterapia (różne schematy, w tym
hiperfrakcjonowanie)
dawka na guz 60 Gy
radioterapia przedłuża życie

GLIOBLASTOMA MULTIFORME
Czynniki rokownicze
•Wiek
•Długośc trwania objawów
•Obecność objawów neurologicznych po zabiegu
•Stan ogólny
•Wielkość masy resztkowej po zabiegu

GLIOBLASTOMA MULTIFORME-
chemioradioterapia
Nowoczesne leczenie (leczenie skojarzone -
chirurgia + uzupełniająca radiochemioterapia z
temozolomidem)
III faza RT 60 Gy vs RT + Temodal 75mg/m2 p.o.
+ 6 cykli (150-200 mg/m2,
1-
5d, co 28d)
Wyniki:
med. przeżycia
12,1 vs 14,6 mies
2-letnie przeżycia całk.
10,4% vs 26,5%
Stupp NEJM 2005

GLEJAKI WYSOKO ZRÓŻNICOWANE
radioterapia przy niedoszczętnej resekcji
przy lokalizacji bezobjawowej, zwłaszcza w
tylnym dole czaszki obserwacja dopuszczalna
nawet po nieradykalnych zabiegach
Dawka 54 Gy po 1,8 Gy
Techniki konformalne i stereotaktyczne

Wyściółczaki
15% ryzyko rozsiewu do płynu mózgowo-rdzeniowego
zwłaszcza postacie anaplastyczne, zlokalizowane
podnamiotowo
Radioterapia:
wyściółczaki łagodne - napromienianie miejscowe (z
objęciem przylegającej komory) do 56 Gy
wyściółczaki złośliwe u dzieci
postacie złośliwe zlokalizowane podnamiotowo u
dorosłych
--> napromienianie osi m-r do 35 Gy

Rdzeniaki płodowe
85% przypadków przed 15 rokiem życia
Należy napromieniać oś mózgowo-rdzeniową do 36 Gy
Niekorzystne czynniki prognostyczne:
zakres resekcji guza <75%
rozsiew do płynu m-r
obecność przerzutów odległych
zajęcie pnia mózgu
wiek poniżej 4 lat
są wskazaniami do leczenia cytostatycznego
Przeżycie 5-letnie od 40% do 80%

Guzy zarodkowe
rozrodczakowe
nierozrodczakowe
Guzy zlokalizowane - radioterapia
24-30 Gy na układ komorowy
podwyższenie dawki na guz do 50 Gy (rozrodczaki) lub
60 Gy (nierozrodczaki)
Wskazania do napromieniania osi m-r:
kom neo w płynie m-r
obecność zmian przerzutowych w bad. MRI
Dawki 24 Gy (rozrodczaki) lub 30 Gy (nierozrodczaki)
Chemioterapia --> nierozrodczaki, guzy z domieszką
rozrodczaka, uogólnione rozrodczaki
Przeżycie 5-letnie:
rozrodczaki -100%
nierozrodczaki - 80%

OPONIAKI
NF2 --> oponiaki mnogie
Większość jest histologicznie łagodna
5% atypowe, 2% złośliwe
Jeżeli małe i bezobjawowe, wykryte przypadkowo
--> mogą być obserwowane
Leczenie z wyboru - operacyjne
Guzy podstawy czaszki często nieresekcyjne
(ważne dla życia struktury)
Przy wznowie, po resekcji uzupełniająca
radioterapia, która opóźnia odrost
Przeżycia 10-letnie dla zmian łagodnych - 70% -
80%
Przeżycia 5-letnie dla zmian złośliwych 0-30%
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
- Slide 23
- Slide 24
- Slide 25
- Slide 26
- Slide 27
- Slide 28
- Slide 29
- Slide 30
- Slide 31
- Slide 32
- Slide 33
- Slide 34
- Slide 35
- Slide 36
- Slide 37
- Slide 38
- Slide 39
- Slide 40
- Slide 41
- Slide 42
- Slide 43
- Slide 44
- Slide 45
- Slide 46
- Slide 47
- Slide 48
- Slide 49
- Slide 50
- Slide 51
- Slide 52
- Slide 53
- Slide 54
- Slide 55
- Slide 56
- Slide 57
- Slide 58
- Slide 59
- Slide 60
- Slide 61
Wyszukiwarka
Podobne podstrony:
SEM [1] 06 wzw
sem. 06- bad. neurolog u dzieci, Medycyna, Neurologia, 15 neurologia dziecieca
program I sem 06-07, Akademia Morska Szczecin Nawigacja, uczelnia, AM, AM, nawigacja, wykłady I sem
sem[1].06- Gruźlica, Medycyna, Interna, Pulmonologia
SEM [1] 06 wzw
06, Szkoła, Politechnika 1- 5 sem, chomikuj, 4 sem (graviora), dc word (graviora)
06, Politechnika Lubelska, Studia, semestr 5, Sem V, Sprawozdania, sprawozdania, Sprawozdania, Labor
instrukcja 06, sem 3, Podstawy elektrotechniki i elektroniki, Laboratoria, instrukcje do cwiczen 201
06.Edukacja zdrowotna a profilaktyka, Zdrowie publiczne, W. Leśnikowska - Ścigalska - ĆWICZENIA I se
ODL II sem termin1 14 06 25
ODL II sem termin3 06 25
Badanie 3-fazowego silnika klatkowego, Polibuda, IV semestr, SEM IV, Maszyny Elektryczne. Laboratori
Kolokwium 2 28.06.2010, sem 4, Astronautyka, kol
06 przewlekla niewydolność serca seminarium dr Szymanowska SEM 6 2
MB powtorka DYN sem V aktualizacja 06 IX
więcej podobnych podstron