
Genetyczne podłoże
niepełnosprawności
intelektualnej
i zespołów otępiennych
IV rok WL

Niepełnosprawność
intelektualna
W krajach UE 8,1% wydatków
ponoszonych na ochronę zdrowia dotyczy
opieki medycznej nad osobami z
niepełnosprawnością intelektualną
(11% dla mężczyzn, 6% dla kobiet).
W warunkach fizjologii intelekt człowieka
jest cechą kształtującą się wskutek
współdziałania czynników genetycznych i
środowiskowych

Klasyfikacja niepełnosprawności
intelektualnej wg ilorazu
inteligencji
NI w stopniu lekkim - 50-70
(80%)
NI w stopniu umiarkowanym - 35-49
NI w stopniu znacznym - 20-34
NI w stopniu głębokim - 0-19
(1%)
lekka
głęboka

Dlaczego sprawie
niepełnosprawności intelektualnej
należy poświęcić szczególną
uwagę?
Znaczna częstość występowania w populacji
(1-2% populacji ogólnej, 2-3% populacji
dziecięcej)
Trudna sytuacja rodziny
Konieczność właściwego poradnictwa
genetycznego
Łatwość popełnienia błędu w sztuce
dotyczącego rozpoznania przyczynowego i
porady genetycznej

Współczynniki korelacji
pomiędzy poziomem inteligencji
w rodzinie:
dzieci i rodziców
0.4
bliźniąt monozygotycznych
0.86
bliźniąt dizygotycznych 0.61
dzieci i rodziców w rodzinach adopcyjnych 0
Ostateczny IQ kształtuje się przy udziale
czynników środowiskowych
Bardzo ważna jest stymulacja rozwoju dziecka
już od pierwszych tygodni po urodzeniu

Różnice pomiędzy dwiema
grupami niepełnosprawności
intelektualnej
Grupa I
(konstytucj onalna)
Grupa I I
(patologiczna)
Stopień
niepełnosprawności
z reguły – lekka
(I Q>50)
z reguły – ciężka
(I Q<50)
wiek rozpoznania
szkolny
przedszkolny
Zmiany organiczne
w OUN
brak
obecne
Wpływ statusu
psychosocj ologicznego
rodziny
wyraźny
żaden lub niewielki
Częstość występow.
20-30/ 1000
3/ 1000

Przyczyny niepełnosprawności
intelektualnej u dzieci w wieku
szkolnym
I Q
<50
>50
Aberracj e chromosomowe
15%
5-10%
Choroby j ednogenowe
20-25%
5-10%
Wady OUN w zespołach wad
mnogich
10%
5%
Czynniki środowiskowe
30-35%
15%
nieznana
20%
60-65%
Im niższy IQ, tym większa szansa znalezienia przyczyny

Przyczyny NI powstałej w
okresie prenatalnym (70%
przyp.)
czynniki genetyczne (30-40% przypadków
o znanej etiologii)
np. Zespół Downa (~22%), FRAX (~6%)
cukrzyca matki
zatrucie ciążowe
zakażenia wewnątrzmaciczne (TORCH)
niedożywienie
fetopatia alkoholowa

Genetyczne przyczyny NI
Z. Downa - 1,3
FRAX - 0,25
DMD - 0,15
Z. Edwardsa - 0,125
TSC - 0,1
Fenyloketonuria -
0,067
Z. Cri du chat - 0,05
Galaktozemia - 0,017
Z. Huntera - 0,01
(częstość na 1000)

Wywiad dotyczący dziecka
Wiek, stan zdrowia, ekspozycja na szkodliwe
czynniki środowiskowe rodziców
Pokrewieństwo rodziców,
Przebieg ciąży (czynniki teratogenne) i
porodu,
Wrodzone wady rozwojowe,
Przebieg okresu noworodkowego,
Rozwój psychoruchowy dziecka,
Osobowość i zachowanie dziecka

Zaburzenia towarzyszące NI
Padaczka (występuje w 50% przypadków
ciężkiej NI)
Mózgowe porażenie dziecięce (20%)
Zaburzenia psychiatryczne (>50%) -
najczęściej nadpobudliwość ruchowa i autyzm
Dysfunkcje behawioralne (np. zaburzenia
łaknienia)
Choroby neurologiczne (np. wodogłowie)
Uszkodzenie wzroku/słuchu

Badanie fizykalne
Badanie fenotypu dziecka pod kątem
ewentualnych cech dysmorfii, małych
wrodzonych wad rozwojowych,
Elementy badania neurologicznego (ocena
chodu, mowy, napięcia mięśniowego),
Pomiary antropometryczne i ich analiza za
pomocą siatek centylowych, ocena symetrii
ciała,
Dokumentacja fotograficzna.

Badania laboratoryjne
U wszystkich pacjentów analiza kariotypu (GTG)
Podejrzenia zespołu mikrodelecji lub aberracji
subtelomerowych - FISH
W uzasadnionych przypadkach badanie molekularne w
kierunku znanych zespołów jednogenowych (np.FRAX,
HD, DMD, zespół Prader-Willi i inne),
Podejrzenie choroby metabolicznej - badanie krwi
metodą tandemowej spektrometrii masowej
Podejrzenie utajonej hiperfenyloalaninemii matczynej -
badania metaboliczne u matki

geny na chromosomie X
odpowiedzialnych za NI
Do tej pory zidentyfikowano ~20 genów na
chromosomie X, które odgrywają rolę w
patogenezie niepełnosprawności intelektualnej.
Badania za pomocą zaawansowanych metod
biologii molekularnej w rodzinach z XLMR
wskazują, że na chromosomie X istnieje ok. 100
genów, których mutacje mogą być
odpowiedzialne za wystąpienie
niepełnosprawności intelektualnej.

NI sprzężona z chromosomem X
FRAX -
gen FMR1
zespół Coffina i Lowry’ego - utratą funkcji
genu
kodujacego kinazę białka rybosomalnego S6
DMD -
gen dystrofiny
zespól Retta -
gen MPCP2
(Xq28) -Mutacje wykryto
u 96% pacjentek. Gen koduje białko, które jest
represorem transkrypcji działającym poprzez
wiązanie do zmetylowanego DNA.
choroba Pelizaeusa i Merzbachera (leukodystrofia)
-
gen PLP
- kontrolujący wzrost otoczki mielinowej.

Duża głowa
Podłużna twarz
Wydatna żuchwa
Duże uszy
Duże jądra
Gotyckie podniebienie
Nadmierna ruchomość stawów
FRAX
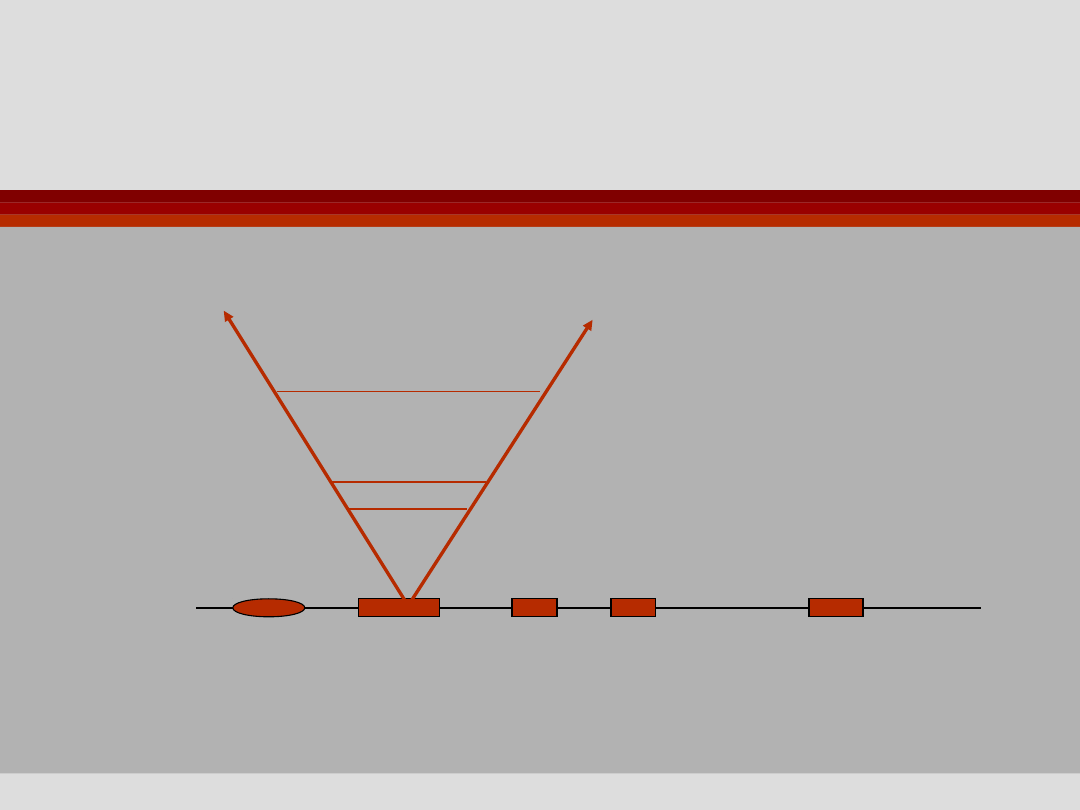
Mutacja dynamiczna
800 powt.
PEŁNA MUTACJA
200 powt.
Premutacja
60 powt.
“Szara strefa”
45 powt.
5 powt.
NORMA
Promotor Egzon 1 2 3 …. 17
Xq27.1

Upośledzenie umysłowe u mężczyzn
z FRA-X jest najbardziej stałym i najczęstszym
objawem, stwierdzanym u wszystkich męskich
nosicieli pełnej mutacji.
Iloraz inteligencji waha się pomiędzy 22-65,
najczęściej jest to upośledzenie umiarkowane,
nasilające się z wiekiem.
Deficyty w zakresie uwagi, mowy, języka i
komunikacji społecznej
W 25% przypadków - autyzm
FRAX

FRAX
Częstość występowania u mężczyzn ok. 1:4500
Dziedziczenie jako cecha dominująca sprzężona z
chromosomem X z niepełną penetracją u kobiet
Gen FMR1
Lokalizacja w chromosomie Xq27.3
Składa się z 17 eksonów
W 1 eksonie -sekwencja składająca się z
trójnukleotydowych powtórzeń CGG
tj...CGG CGG CGG CGG CGG...
Osoby zdrowe mają od 6- do około 50 powtórzeń

Premutacja w genie FMR1
Osoby posiadające od 50-200 powtórzeń CGG to
zdrowi nosiciele premutacji
Premutacja jest niestabilna – w czasie
gametogenezy może dojść do zwiększenia lub
zmniejszenia liczby powtórzeń. Tylko w trakcie
oogenezy premutacja może przejść w pełna
mutację ze wszystkimi skutkami klinicznymi
W 98% przypadków przyczyną choroby jest wzrost
liczby powtórzeń CGG powyżej 200.
Powoduje to inaktywację genu i brak białka FMRP,
które jest jego produktem

Kobiety nosicielki pełnej mutacji
w genie Fra X
50-70% wykazuje upośledzenie
umysłowe
zaburzenia zachowania
trudności w nauce

Locus Xp22.2-p22.1
Częstość około 1 na 40,000 do 50,000
urodzeń
gen RPS6KA3 (rybosomal protein S6 kinase).
Koduje białko uczestniczące w przekazywaniu
sygnałów.
Dziedziczenie XD. W większości przypadków
mężczyźni są bardziej dotknięci niż kobiety.
zespół Coffina i Lowry’ego

zespół Coffina i Lowry’ego
Niski wzrost,
Twarz o grubych rysach, wydatne czoło,
Hyperteloryzm, antymongoidalne
ustawienie szpar powiekowych,
Grube wargi,
Gruba przegroda nosa,
Brakujące uzębienie,
Duże dłonie, zwężające się czubki palców,
Głuchota czuciowo-nerwowa, padaczka,
Wady serca

zespół Coffina i Lowry’ego

Ryzyko powtórzenia choroby wynosi około 1%.
99% przypadków to mutacje sporadyczne.
Gen MECP2 jest odpowiedzialny za regulację
aktywności (wyłączanie) innych genów. Mutacje
prowadzą do zahamowania dojrzewania OUN.
Okres tworzenia synaps (koniec okresu
prenatalnego i pierwsze miesiące życia)
Zespół Retta

Zespół Retta
Wczesne zatrzymanie rozwoju
Niepowiększanie obwodu głowy (6-18 miesiąc)
Niepełnosprawność intelektualna głębszego
stopnia
Nieprawidłowy wzorzec oddychania
Utrata sprawności rąk
Napady padaczkowe
Stereotypie rąk
Autyzm
Skolioza
Uszkodzenie neuronu ruchowego
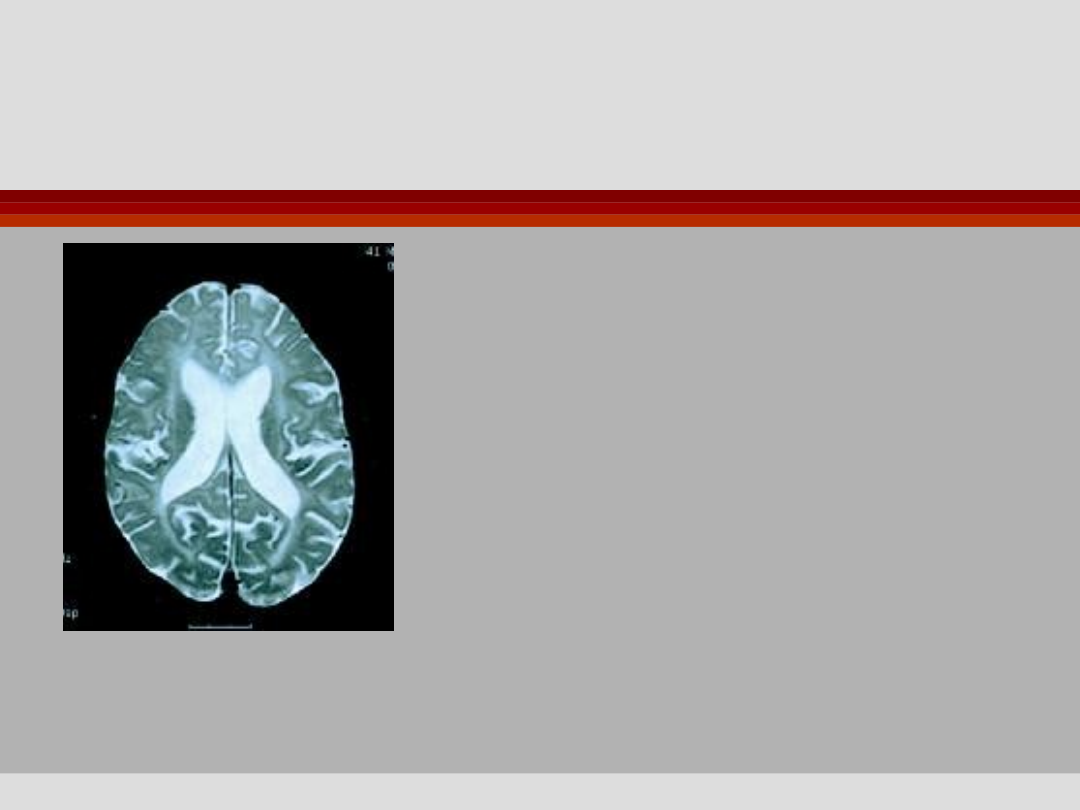
Choroba Pelizaeusa i
Merzbachera
Zajęcie istoty białej mózgu, pnia,
móżdżku
Produkty białkowe genu PLP
kodują 50% masy istoty białej
OUN
Postać klasyczna - dup PLP1,
(oczopląs od 1. r.ż., opóźnienie
rozwoju, ataksja, spowolniał a
mowa), przeżycia do 6. dekady.
Obraz MRI 41-letniego mężczyzny z duplikacją genu PLP
Zwiększenie wysycenia obrazu istoty białej, rozlane zmiany
atroficzne
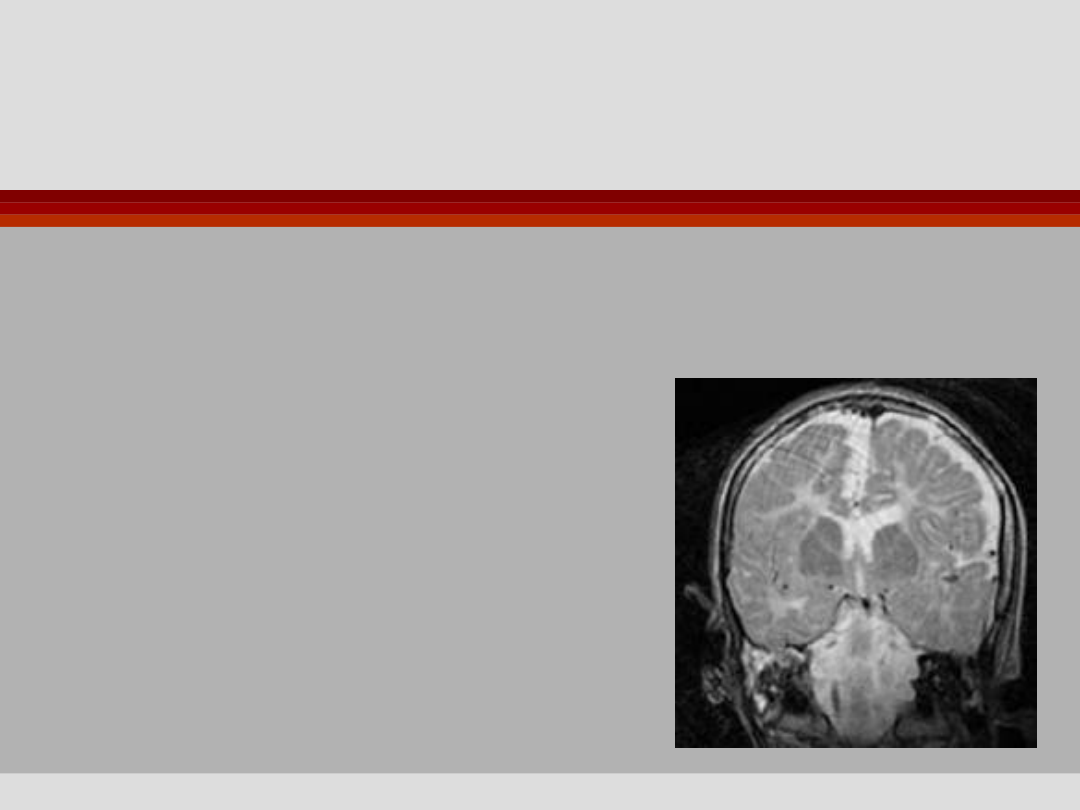
Choroba Pelizaeusa i
Merzbachera
Postać ciężka - mutacje
missence - drgawki, hipotonia
porażenie spastyczne - zgon w 1.
dekadzie z powodu
niewydolności oddechowej.
Obraz MRI 20-letniego
mężczyzny z mutacją Pro14Leu
genu PLP
Zwiększenie wysycenia obrazu
istoty białej, znaczna redukcja
objętości istoty białej

Malformacje mózgowe
Mogą być spowodowane błędem na
prawie każdym etapie rozwoju
embrionalnego OUN:
Kształtowania się cewy nerwowej
(przepuklina mózgowa)
Zaburzeń migracji neuronów
(lissecephalia)
Zaburzenia postmigracyjne (microgyria)

Choroby skórno-nerwowe
(fakomatozy)
stwardnienie guzowate
NF1
zespół Blocha i Salzbergera
zespół Ito

Małogłowie pierwotne
Pojawia się nie później niż w 32. tygodniu życia płodowego i
spowodowane jest zmniejszoną liczbą neuronów
powstających w czasie neuronogenezy
Nie występują cechy zespołu neurologicznego. W większości
przypadków wysokość i masa ciała są prawidłowe
Objętość mózgowia jest mniejsza niż u osób z prawidłowym
obwodem głowy, największej redukcji ulega kora mózgowa
Mutacje w genach:
mikrocefaliny
i
nieprawidłowego wrzeciona
w małogłowiu
(ASPM) (ang. Abnormal spindle in
microcephaly). Mutacje prowadzą do powstania skróconego
produktu białkowego

Pachygyria
Wady wynikające z zaburzonej migracji
neuroblastów (heterotopie) powodują
nieprawidłowe ukształtowanie zarówno zakrętów
płaszcza mózgu, jak i budowy kory mózgowej oraz
innych struktur mózgu.
Heterotopie zwykle towarzyszą wielu wadom
rozwojowym o.u.n., a często można je stwierdzić
również w zespole małogłowia i małomózgowia
pierwotnego prawdziwego (microcephalia vera).

Wadę tę przypisuje się głównie czynnikom
genetycznym, a przyjmuje się, że dziedziczy się ona
recesywnie autosomalnie. Klinicznie - oprócz małych
wymiarów czaszki (zwykle poniżej trzeciego odchylenia
standardowego), związanych z niedorozwojem
globalnym mózgu i z innymi wadami o.u.n. - może
występować opóźnienie w rozwoju psychoruchowym
(zwłaszcza zaburzenia rozwoju mowy) oraz padaczka.
Układ komorowy mózgu na ogół nie poszerza się
wyraźnie, co można wykazać w USG osiowej TK mózgu
lub w MR.
Pachygyria

Badania genetyczne nad
procesem uczenia się i
zapamiętywania
W 2000 roku psychiatra z Nowego Jorku Eric Kandel
otrzymał nagrodę Nobla za badania nad
genetycznymi uwarunkowaniami procesu
zapamiętywania.
Badania Kandela wpłynęły na powstanie nowego
podejścia do chorób umysłowych. Uwzględniło ono
postępy współczesnej biologii molekularnej i
genetyki, w tym epigenetyczną regulację działania
genów na terenie mózgu poprzez wpływ na nie
czynników społecznych (uczenie się).

Postępowanie z dziećmi z NI
oraz z ich rodzinami
Dzieci z zaburzeniami genetycznymi rozwijają się zgodnie
z ich własnymi normami, które w porównaniu z dziećmi
zdrowymi różnią się w mniejszym lub większym stopniu
Jeżeli rozwój dziecka przebiega inaczej niż oczekiwano,
wtedy do kompetencji wychowawczych powinno należeć
takie wspieranie rozwoju, (np. pedagogika integracyjna).
Postawienie diagnozy "upośledzony“ we wczesnym
dzieciństwie może mieć dramatyczny wpływ na
perspektywę kształtowania się zdolności intelektualnych.
Doszukiwanie się defektów i założenie braku życiowych
szans i aspiracji wpływa na brak stymulacji rozwoju
poprzez zwykłe działania wychowawcze rodziców.

Ocena fenotypu zachowania
Do udzielenia prognozy o możliwościach rozwojowych
dziecka niezbędna jest znajomość wiedza na temat
fenotypu zachowania dla określonego zespołu
genetycznego, na który składa się charakterystyczny
zbiór cech ruchowych, poznawczych, lingwistycznych i
społecznych.
Posługiwanie się subiektywnymi obserwacjami i często
opiniami rodziców wypełniających znormalizowane
kwestionariusze w badaniach naukowych
ukierunkowane na wyszukiwanie głównie negatywnych
form zachowania u dzieci mają swoje ograniczenia i
sankcjonują krzywdzące postrzeganie dzieci z NI.

Zasada oceny fenotypu
zachowania
z zastosowaniem elementów
pedagogiki Montessori
Pierwsza zasada głosi:
“pomóż mi to zrobić samemu“
i
uczy, że należy jedynie wspomagać dzieci w ich
samodzielnym wykonywaniu podjętych czynności
Druga zasada mówi:
“patrz na dziecko“,
czyli koncentruj
się na przebiegu bieżącego zadania - zabawy a
jednocześnie włączaj się do dialogu z dzieckiem
Metoda oceny fenotypu zachowania jest oparta na
bezpośredniej obserwacji i filmowaniu zachowania dzieci
w trakcie terapii zajęciowej, a następnie na szczegółowej
analizie uzyskanego materiału z obserwacji
zarejestrowanych kamerą

Genetyczne podłoże zespołów
otępiennych
Do tej pory opisano bardzo niewiele rodzin, w
których zespół otępienny występował zgodnie z
dziedziczeniem AD
Były to zazwyczaj przypadki wczesnego
zachorowania (przed 65 r.ż.)
W 95% przypadków zespoły otępienne wykazują
niewielki wpływ czynników genetycznych. Nawet
jeśli dwóch krewnych I stopnia choruje, to
ryzyko zachorowania jest niewiele podwyższone
w porównaniu do populacyjnego

Genetyczne podłoże choroby
Alzheimer’a o wczesnym
początku
Gen APP (amyloid precursor protein) na
chromosomie 21q21.2
presenilin 1 gene (PSEN1) 14q24.3
presenilin 2 gene (PSEN2) 1q31–q42 12p11
10q24
10p13
20p
19p13
7q36
9q22

Gen APOE apolipoproteiny E zlokalizowany na chromosomie
19q13 występuje w trzech formach: ApoE2, ApoE3, ApoE4.
60% populacji ma dwa allele APOE3 - pośrednie ryzyko
zachorowania - połowa tych osób zachoruje do 80. r.ż.
Allel APOE4 (w postaci heterozygotycznej występuje u 25%
populacji) niesie podwyższone ryzyko zachorowania na AD -
2,3-3 x większe w porównaniu do populacyjnego.
Homozygoty APOE4 (2% populacji) - 10x wzrost ryzyka
Allel APOE2 jest związany z obniżonym ryzykiem choroby.
1/6 populacji to heterozygoty, 1/200 to homozygoty -
najniższe ryzyko zachorowania
Genetyczne podłoże choroby
Alzheimer’a o późnym początku
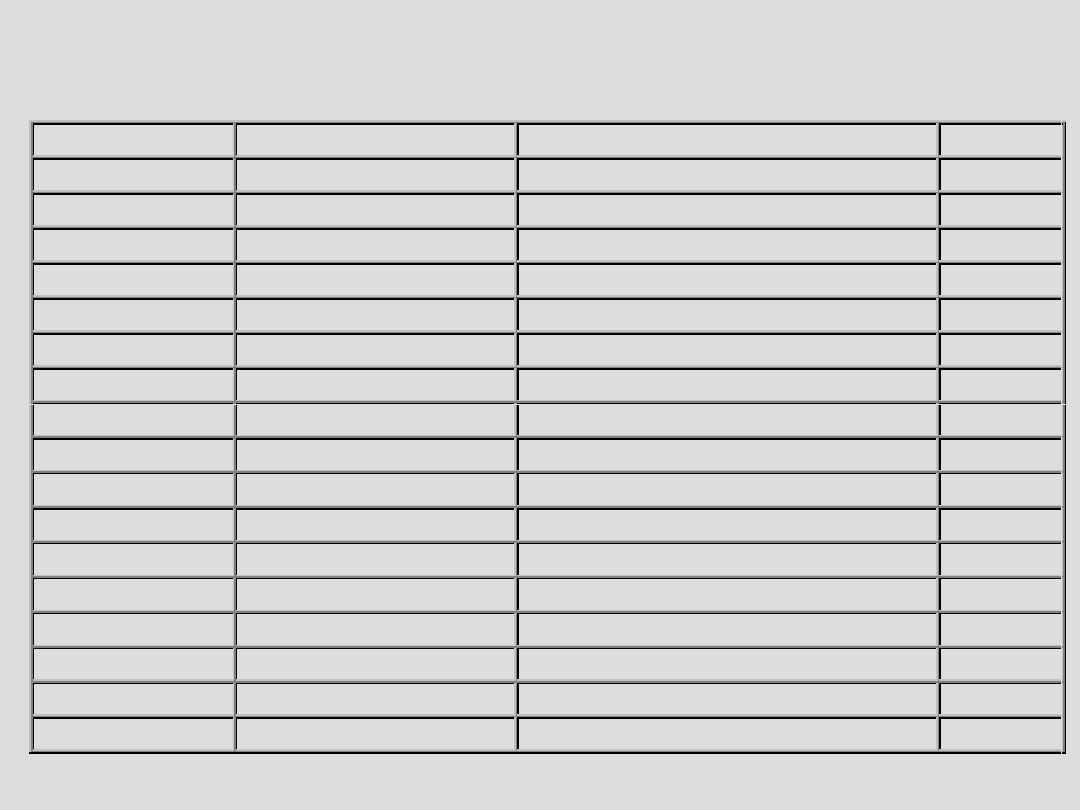
Gen
Locus
Choroba
Dziedz.
APP
21q21.2
Alzheimer’s disease
AD
PSEN1
14q24.3
Alzheimer’s disease
AD
PSEN2
1q31–q42
Alzheimer’s disease
AD
SNCA(PARK1)
4q21
Parkinson’s disease
AD
Parkin (PARK2) 6q25.2–q27
Parkinsonism (juvenile)
AR
SNCA (PARK4)
4q21 (locus triplication) Parkinson’s disease
AD
UCH-L1 (PARK5) 4p14
Parkinson’s disease
AD
PINK-1 (PARK6) 1p36
Parkinson’s disease
AR
DJ-1 (PARK7)
1p36
Parkinsonism
AR
LRRK2 (PARK8) 12q12
Parkinson’s disease
AD
SOD1 (ALS1)
21q21
Amyotrophic lateral sclerosis
AD, AR
Alsin (ALS2)
2q33
Amyotrophic lateral sclerosis
AR
SETX (ALS4)
9q34
Amyotrophic lateral sclerosis, juvenile AD
VAPB (ALS8)
20q13.33
Amyotrophic lateral sclerosis
AD
MAPT
17q21
ALS with FTD and Parkinsonism
AD
IT15
4p16.3
Huntington’s disease
AD
PRNP
20pter-p12
Prion diseases
AD
Geny związane z zespołami
otępiennymi
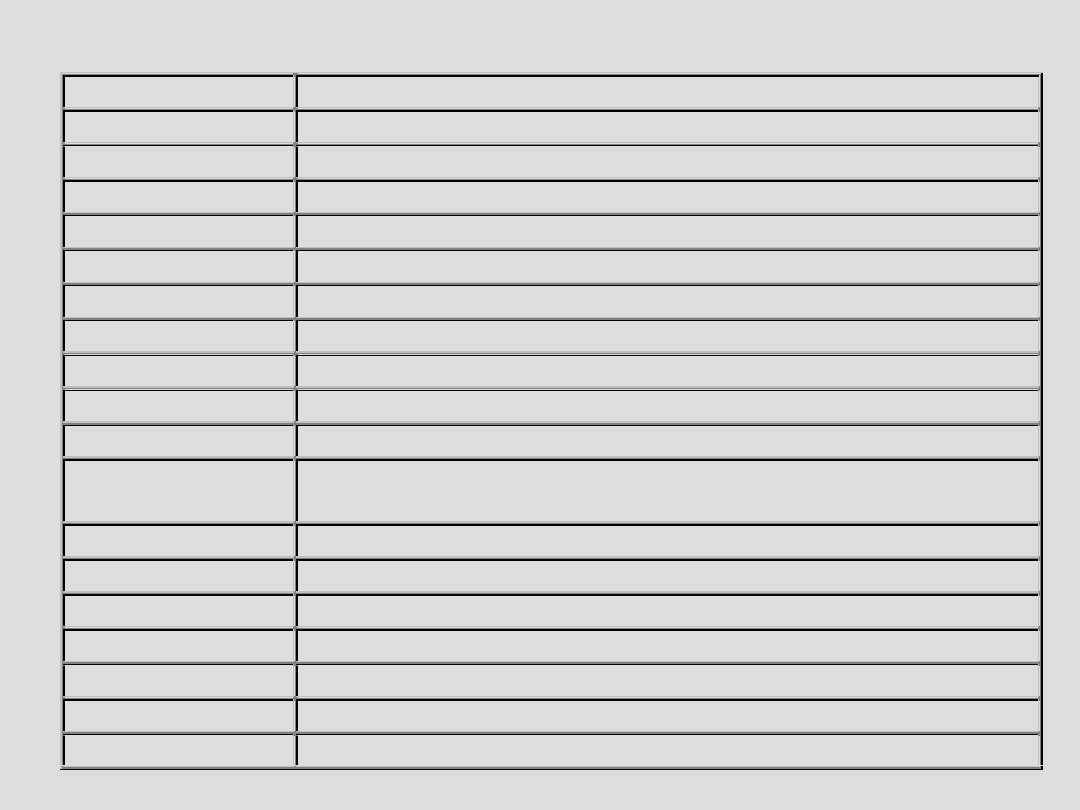
Susceptibility gene
Increased risk for
APOE (ε4-variant)
Alzheimer’s disease
VEGF
Alzheimer’s disease and Amyotrophic lateral sclerosis
MTHFR
Alzheimer’s disease (controversial results)
IL-1α
Alzheimer’s disease
GSTP1
Alzheimer’s disease, Parkinson’s disease
TF and HFE
Alzheimer’s disease (synergistic effect)
APOE(ε2-variant)
Parkinson’s disease
NAT2
Parkinson’s disease, Alzheimer’s disease
MAO-B
Parkinson’s disease (controversial results)
CYP2D6
Parkinson’s disease and Amyotrophic lateral sclerosis
PRNP (M129V
variant)
Creutzfeld-Jakob disease
Susceptibility gene Decreased risk for
APOE (ε2-variant)
Alzheimer’s disease
Susceptibility gene Associated with age at onset of
GSTO1 and GSTO2 Alzheimer’s disease, Parkinson’s disease,
APOE
Amyotrophic lateral sclerosis, Huntington’s disease (in males)
MAO-B
Amyotrophic lateral sclerosis
GluR6
Huntington’s disease
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
- Slide 23
- Slide 24
- Slide 25
- Slide 26
- Slide 27
- Slide 28
- Slide 29
- Slide 30
- Slide 31
- Slide 32
- Slide 33
- Slide 34
- Slide 35
- Slide 36
- Slide 37
- Slide 38
- Slide 39
- Slide 40
- Slide 41
- Slide 42
Wyszukiwarka
Podobne podstrony:
Bożenna Odowska, psychologia osób z ni
WARIANT C, FIR UE Katowice, SEMESTR IV, Ubezpieczenia, chomik, Ubezpieczenia (kate evening), Ubezpie
NI Spis tresci id 318044 Nieznany
01 02 Taikyoku Sono Ichi, Ni
NI 1 1 03 2009 r
Materiał wyrazowo obrazkowy lt d m mi n ni
NI MI SI RODZINNY DOM, TEKSTY
Uczenie się dzieci ni
Erich Von?niken Kosmiczne miasta w epoce kamiennej
GWSH - tur pielgrzymkowa, religie świata, Judaizm (Mozaizm) - religia wyznawana przez Żydów mająca n
miernictwo, ćwiczenie1, Model: Ni 42
biologia-tkanka nerwowa glejowa (2) , Tkanka glejowa, specjalna tkanka zwierzęca powstała z mezoderm
DGW 3, LABOLATORIUM ˙RODK˙W GA˙NICZYCH
Nihongo gramatyka, 85, Joshi -NI
fiz-magnetyzm ściąga, Źródłem pola magnetycznego są: 1 Magnesy naturalne Fe i jego stopy, Ni, Co)
więcej podobnych podstron